 Pingnian He
Pingnian He M. A. Hassan Talukder
M. A. Hassan Talukder Feng Gao
Feng Gao- Department of Cellular and Molecular Physiology, College of Medicine, The Pennsylvania State University, Hershey, PA, United States
Clinical and experimental evidence indicate that increased vascular permeability contributes to many disease-associated vascular complications. Oxidative stress with increased production of reactive oxygen species (ROS) has been implicated in a wide variety of pathological conditions, including inflammation and many cardiovascular diseases. It is thus important to identify the role of ROS and their mechanistic significance in microvessel barrier dysfunction under pathological conditions. The role of specific ROS and their cross talk in pathological processes is complex. The mechanisms of ROS-induced increases in vascular permeability remain poorly understood. The sources of ROS in diseases have been extensively reviewed at enzyme levels. This review will instead focus on the underlying mechanisms of ROS release by leukocytes, the differentiate effects and signaling mechanisms of individual ROS on endothelial cells, pericytes and microvessel barrier function, as well as the interplay of reactive oxygen species, nitric oxide, and nitrogen species in ROS-mediated vascular barrier dysfunction. As a counter balance of excessive ROS, nuclear factor erythroid 2 related factor 2 (Nrf2), a redox-sensitive cell-protective transcription factor, will be highlighted as a potential therapeutic target for antioxidant defenses. The advantages and limitations of different experimental approaches used for the study of ROS-induced endothelial barrier function are also discussed. This article will outline the advances emerged mainly from in vivo and ex vivo studies and attempt to consolidate some of the opposing views in the field, and hence provide a better understanding of ROS-mediated microvessel barrier dysfunction and benefit the development of therapeutic strategies.
Introduction
Oxidative stress with increased production of reactive oxygen species (ROS) has been implicated in a wide variety of pathological conditions, such as inflammation, atherosclerosis, ischemia-reperfusion injury, hypercholesterolemia, hypertension, diabetes, and heart failure (Mcquaid and Keenan, 1997; Lum and Roebuck, 2001; Harrison et al., 2003; Stocker and Keaney, 2004; Forstermann, 2008; Li and Forstermann, 2013; Kalogeris et al., 2016; Forstermann et al., 2017; Zhou et al., 2019). Growing evidence indicates that overproduction of ROS can lead to endothelial barrier dysfunction with subsequent increased vascular permeability, tissue damage, and organ dysfunction (Del Maestro et al., 1981; Cai and Harrison, 2000; Henricks and Nijkamp, 2001; Zhu and He, 2006; Hecquet et al., 2008; Zhou et al., 2009, 2013, 2019). However, the precise role played by each ROS and their products in normal and pathological processes is very complex, and the mechanisms of ROS-induced increased vascular permeability under inflammation and disease conditions remain largely unknown. This review will discuss the sources of ROS and antioxidants in the vasculature with focus on the effects and mechanisms of different ROS and ROS-derived reactive nitrogen species (RNS) on endothelial barrier function. The advantages and limitations of different experimental approaches used for the study of ROS-induced endothelial barrier function will also be discussed in this review.
Sources of ROS, Redox-Crosstalk, and Antioxidants in Vascular System
Reactive oxygen species are a collective term that refers to both oxygen-derived unstable free radicals, such as superoxide anion, hydroxyl radical, oxygen radical, NO, and non-free radicals, such as hydrogen peroxide (H2O2), hypochlorus acid, peroxynitrite (ONOO–), and ozone (Cai and Harrison, 2000; Madamanchi et al., 2005; Forstermann, 2008; Forstermann et al., 2017). ROS are ubiquitous and formed continuously in small amounts during the normal metabolism of cells and are normally inactivated by endogenous antioxidant defense mechanisms (Lum, 2001; Zweier and Talukder, 2006). The sources of different ROS and antioxidant enzymes in vascular diseases have been extensively reviewed by several authors (Cai and Harrison, 2000; Madamanchi et al., 2005; Mueller et al., 2005; Forstermann, 2008; Montezano and Touyz, 2012; Kalogeris et al., 2016; Forstermann et al., 2017). The major sources of ROS in ECs are from NADPH oxidase (Nox), xanthine oxidase (XO), mitochondria, and dysfunctional eNOS. Other potential vascular sources of ROS include cytochrome p450 mono-oxygenase, cyclo-oxygenase and lipoxygenase, NOS, and peroxidases (Forstermann, 2008). In addition to the ROS derived from activated ECs, the vascular endothelium is constantly exposed to various blood-borne agents and is the primary target for oxidants released from activated blood cells during inflammation. The activated neutrophils could release large amount of ROS to the circulation via membrane-bound NADPH oxidase during neutrophil respiratory burst, which provide the killing power for invading pathogens, but also cause host tissue injury and endothelial barrier dysfunction (Carden et al., 1990; Babior, 1999; Dahlgren and Karlsson, 1999; Kubes et al., 1990a; Madamanchi et al., 2005; He et al., 2006; Zhu and He, 2006; Forstermann, 2008). Importantly, there appears to be significant redox crosstalk between these pro-oxidant systems where ROS produced by one enzyme system can enhance the activity of other ROS-producing systems, leading to feed-forward processes with augmented ROS production and oxidative stress (Cai, 2005b; Forstermann, 2008; Daiber, 2010; Zinkevich and Gutterman, 2011; Daiber et al., 2017; Zhou et al., 2019). For instance, NADPH oxidase-derived ROS have been implicated in the activation of XO (Mcnally et al., 2003; Landmesser et al., 2007), mitochondrial ROS production (Wenzel et al., 2008; Kroller-Schon et al., 2014), uncoupling of eNOS (Landmesser et al., 2003), and NO/O2– derived ONOO– formation (Radi et al., 1991; Xia et al., 1996; Riobo et al., 2001; Pacher et al., 2007; Szabo et al., 2007; Zhou et al., 2019).
Reactive oxygen species in the cells and tissues are kept in balance by the anti-oxidant enzymes that regulate and often reduce the level of ROS in biological system. Vascular cells are equipped with several of these protective enzymes such as superoxide dismutase (SOD), catalase, glutathione peroxidase, thioredoxin, heme oxygenase, and paraoxonase (Forstermann, 2008). Several non-enzymatic antioxidants are also found in the biological systems such as endogenous glutathione, uric acid and bilirubin, and ingested antioxidant vitamins (Forstermann, 2008). While ROS at moderate and low concentrations have important signaling roles and beneficial effects under physiological conditions (Suzuki et al., 1997; Finkel, 1998; Kamata and Hirata, 1999; Rhee, 1999; Thannickal and Fanburg, 2000; Droge, 2002; Valko et al., 2007), at higher concentrations, especially with reduced antioxidant counter balance, ROS generate oxidative stress under pathophysiologic conditions causing potential damage to the biomolecules, disruption of cellular function and promoting disease progression (Yla-Herttuala, 1999; Stadtman and Levine, 2000; Lum, 2001; Madamanchi et al., 2005; Pacher et al., 2007; Szabo et al., 2007; Forstermann, 2008; Li et al., 2014). Although ample experimental evidence support the role of certain antioxidant enzymes or non-enzymatic antioxidants in ameliorating oxidative stress-mediated vascular damages, clinical studies so far have not been able to demonstrate conclusive results (Madamanchi et al., 2005; Landmesser et al., 2007; Forstermann, 2008; Boueiz and Hassoun, 2009). These led more studies to focus on the transcriptional regulations of antioxidant genes. Among varieties of redox-sensitive transcription factors, nuclear factor (erythroid 2–related) factor 2 (Nrf2) has recently been drawn attention and referred to as master antioxidant that regulates a wide variety of genes encoding detoxification enzymes and antioxidant proteins (Ma, 2013; Chen et al., 2015; Amin et al., 2019). Recent development of the role of Nrf2 in the regulation of redox homeostasis and its therapeutic prospects are further discussed in the later section.
Leukocyte-Dependent ROS-Mediated Microvascular Barrier Dysfunction
Acute inflammation is characterized by increased microvascular permeability to plasma proteins and leukocyte recruitment into inflammatory sites, which occurs mainly at post-capillary venules. The recruited granulocytes upon activation release various substances including ROS during respiratory bust. Due to its large quantity over other sources, ROS have been demonstrated to play a major role in the acute activation of endothelial cells, resulting in vascular leakages (Del Maestro et al., 1981; Zhu et al., 2005; Zhu and He, 2006; Granger and Kvietys, 2015). The leukocyte-dependent ROS-mediated microvascular barrier dysfunction was supported by experimental evidence that the blocking antibodies, induction of neutropenia, or the application of superoxide scavenger SOD reduced vascular damages in reperfusion of ischemic tissues and other inflammatory conditions (Korthuis et al., 1988; Carden et al., 1990; Kadambi and Skalak, 2000; Granger and Kvietys, 2015).
Role of Leukocyte Adhesion/Emigration in Microvessel Permeability
Leukocyte adhesion, as a necessary step leading to granulocytes migration from the vascular lumen to the interstitial space, has been indicated as a prerequisite for leukocyte to induce endothelial injury in studies conducted in autologous blood perfused organ or tissues (Carden et al., 1990; Kubes et al., 1990a). Supporting this notion, some in vitro studies reported that the adhesion process could directly trigger intracellular signals in ECs or protease release from neutrophils (Moll et al., 1998) resulting in increased EC permeability (Del Maschio et al., 1996). However, when vascular permeability and leucocyte/endothelium interactions were evaluated simultaneously in vasculature, the sites of the leucocyte adhesion and emigration were not always associated with vascular leakages (Hurley, 1964; Mcdonald, 1994; Baluk et al., 1995; Zeng et al., 2002). Multiple studies revealed temporal and spatial dissociations of leucocyte adhesion and/or emigration from vascular leakages, indicating that mechanisms other than the adhesion process could be critical to the vascular barrier dysfunction during inflammation (Harris et al., 1993; Kadambi and Skalak, 2000; Zeng et al., 2002; He, 2010).
Role of Neutrophil Released ROS in Microvessel Permeability
Whether leucocyte/endothelium interactions would be the cause of increased microvessel permeability has been controversial for decades (Carden et al., 1990; Kubes et al., 1990b; Baluk et al., 1997; Moll et al., 1998; Zeng et al., 2002; He, 2010). On one hand, studies reported leucocyte adhesion and emigration to be the critical event leading to tissue and organ dysfunction during inflammation, and the number of adherent leucocytes was considered as an index for the severity of tissue injury and vascular dysfunction (Tailor and Granger, 2003; Ishikawa et al., 2004). On the other hand, experimental evidence showed that the albumin leakage sites were often distinct from those of leucocyte adhesion and emigration, indicating independent mechanisms to be involved in the induction of leucocyte adhesion and increased microvessel permeability (Hurley, 1964; Suematsu et al., 1993; Baluk et al., 1998; Zeng et al., 2002). Studies conducted in intact microvessels by dissecting the processes clarified some of the debating issues derived from different experimental approaches. The experiments shown in Figure 1 demonstrated that the systemic application of TNF-α causes a significant number of leucocyte adhesion at vascular walls, but no increased microvessel permeability was observed (Zeng et al., 2002). However, increased permeability occurred after fMLP was applied to TNF-α-induced adherent leukocytes and the application of SOD blocked such increased permeability, demonstrating ROS-mediated permeability increases. Importantly, in the presence of fMLP, the magnitude of the permeability increases was directly correlated with the number of adherent of leukocytes, indicating that larger quantity of released ROS produced by higher number of adherent leukocytes results in higher magnitude of permeability increases (Zhu and He, 2006). Figure 1D basically shows ROS dose-dependent permeability increases in intact microvessels. These observations suggest that in the presence of activating stimuli for leukocyte respiratory burst, the number of adherent leukocytes could predict the severity of vascular dysfunction as others concluded (Tailor and Granger, 2003; Ishikawa et al., 2004).
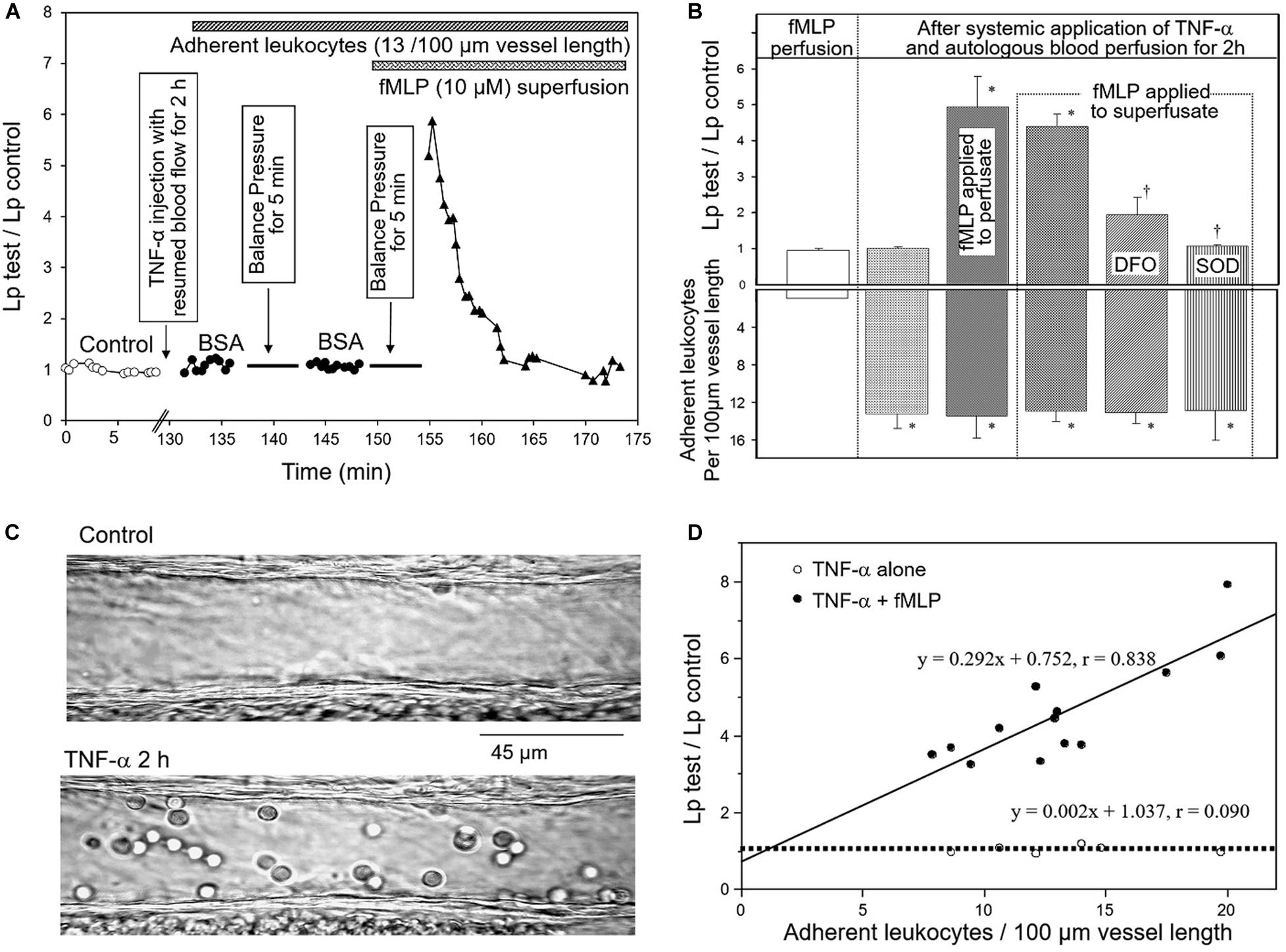
Figure 1. TNF-α-induced adherent leukocytes do not increase microvessel permeability until exposure to fMLP. (A) Lp measurements in an individually perfused rat mesenteric venule. The time course of the measured Lp shows that neither TNF-α-induced leukocyte adhesion alone nor stationary flow in the presence of adherent leukocytes (to allow accumulation of locally released agents) changed Lp. Transient increase in Lp only occurred after adherent leukocytes were exposed to fMLP (10 μmol/L). (B) Summary results showing relationship between number of adherent leukocytes (bottom graph) and changes in microvessel Lp (top graph). Significant increases in Lp only occurred after fMLP was applied to perfusate or superfusate, and was attenuated or prevented by pretreatment of the vessel with an iron chelator DFO or SOD, indicating the role of released ROS in increased Lp. (C) Images of individually perfused venules under control conditions and after TNF-α-induced leukocyte adhesion (2 h after systemically applied TNF-α). (D) The positive correlation between the increased Lp and the number of adherent leukocytes with fMLP stimulation (•) indicates that the quantity of fMLP-induced release of ROS correlated with the number of adherent leukocytes determines the magnitude of Lp increases. Modified and used with permission (Zhu and He, 2006).
Although neutrophil/endothelium interactions is an essential part of acute inflammatory responses, a significant portion of responsive leucocytes remains in the peripheral circulation during inflammation. It is well known that isolated neutrophils could release large quantities of ROS through respiratory burst in response to a variety of stimuli, which are detectable by chemiluminescence measurement (Condliffe et al., 1998b; Babior, 1999). In addition to ROS-mediated permeability increased through adherent leukocyte release of ROS, studies showed that fMLP- or C5a-activated non-adherent neutrophils (suspension in the perfusate) could also increase permeability in isolated coronary venules (Tinsley et al., 2002) and intact mesenteric venules (Zhu et al., 2005). These experimental evidence strongly support that ROS released from activated leukocytes, in either circulating or adherent form, constitute the major source of ROS in the plasma during inflammation and disease conditions, which could directly activate ECs lining the vascular walls (details discussed below), contributing to leukocyte-dependent increases in vascular permeability. These results also suggested that the physical interaction between leukocytes and endothelium through adhesion/emigration was not the direct cause of vascular leakages, and the adhesion process was also not the direct trigger for neutrophils to release ROS.
Neutrophil Priming and ROS Generation
Many cytokines and pro-inflammatory mediators have been reported as neutrophil primers that do not present the effector function on their own, but enable neutrophils to have enhanced responses to additional activating stimuli (Condliffe et al., 1998a; van Leeuwen et al., 2005; Zhu and He, 2006; El-Benna et al., 2016; Miralda et al., 2017). As shown in Figure 2, pre-exposure of neutrophils to a common priming cytokine, TNF-α, through systemic application in vivo did not induce ROS release as measured by chemiluminescence activity, but markedly augmented superoxide production in response to other mediators, such as fMLP (Zhu and He, 2006). The application of anti-TNF MoAb blocked TNF-α-mediated neutrophil priming (van Leeuwen et al., 2005). Under in vivo pathological conditions, systemically elevated cytokine levels or other priming agents such as chemoattractants, or microbial products could transform neutrophils into a primed state resulting in potentiated ROS release in responses to additional stimuli (Condliffe et al., 1998b; El-Benna et al., 2016; Miralda et al., 2017). Neutrophil priming with increased quantity of released ROS plays an important role in host defense against microbial pathogens, but also increases the risk for extensive tissue damage and endothelial barrier dysfunction. Severe clinical conditions such as adult respiratory distress syndrome (Parsons et al., 1989), organ failure, and mortality (Pinsky et al., 1993) are often associated with high plasma levels of inflammatory cytokines and/or endotoxin, and those stimuli-primed neutrophils with enhanced respiratory burst may explain the exacerbated tissue damages commonly observed in those conditions. Recent studies also suggest that primed neutrophils, in addition to enhanced oxidative burst, also involve activations of other neutrophil activities such as adhesion, surface receptor expression, etc (Miralda et al., 2017). Neutrophil priming and de-priming as well as the levels of the priming are complex processes which vary depending on the concentration of priming agents, exposure time, and the type of priming agents (Potera et al., 2016; Mcleish et al., 2017). The major priming agents, underlying mechanisms, and signaling pathways involved in priming neutrophil respiratory burst have been reviewed elsewhere in detail (Condliffe et al., 1998b; El-Benna et al., 2016; Miralda et al., 2017). In contrast to the priming agents that enhance the neutrophil responsiveness to stimuli, there are also cytokines that suppress neutrophil bacterial killing power with reduced ROS release. Multiple studies demonstrated that resistin, a cytokine commonly increased in chronic kidney diseases or sepsis, selectively inhibits neutrophil oxidative burst in response to stimuli and impair its bacterial killing capacity, resulting in immunosuppression (Cohen et al., 2008; Bonavia et al., 2017; Miller et al., 2019). A better understanding of the neutrophil priming in vivo under disease conditions may benefit the development of therapeutic strategies to minimize vascular damage, while enhancing its oxidative killing power at the bacterial battle field.
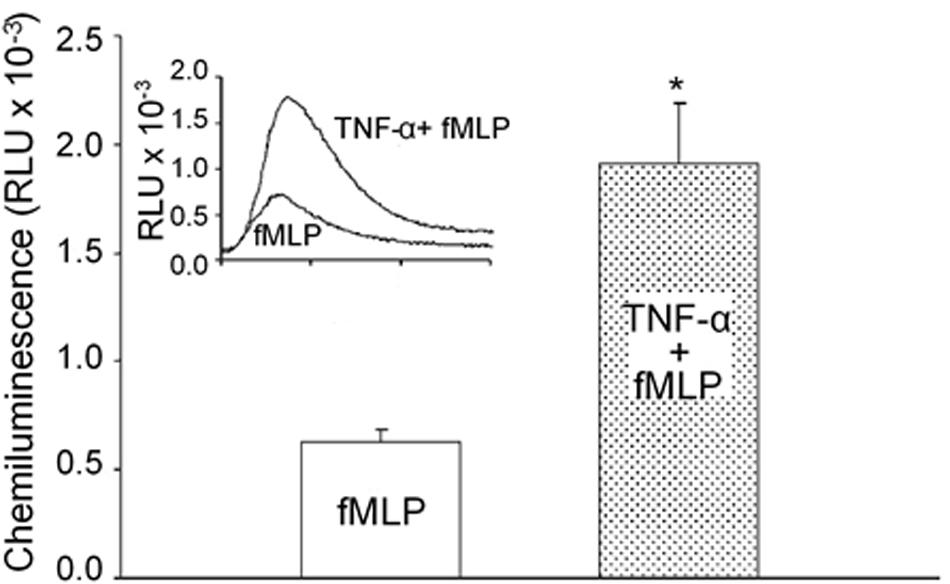
Figure 2. Priming effect of TNF-α on fMLP-stimulated neutrophil respiratory burst measured by chemiluminescence (CL). Preexposure of neutrophils to TNF-α through systemic application in vivo did not change basal CL but potentiated fMLP-stimulated CL by 3.1-fold. Bar graph shows magnitude difference in fMLP-stimulated CL between unprimed and TNF-α-primed neutrophils through in vivo application. Inset: representative individual time course from each group of experiments. Used with permission (Zhu and He, 2006). *Significant difference from fMLP stimulation alone.
Leukocyte-Derived Myeloperoxidase and Hypohalous Acids in Endothelial Dysfunction and Cardiovascular Diseases
Neutrophils in response to stimuli not only release ROS but also release myeloperoxidase (MPO), an enzyme closely linked to both inflammation and oxidative stress (Anatoliotakis et al., 2013; Ndrepepa, 2019). MPO is a member of the heme peroxidase-cyclooxygenase super family (Klebanoff, 2005; Davies et al., 2008; van der Veen et al., 2009; Davies, 2011). It is released by activated neutrophils, monocytes, and some tissue macrophages into extracellular spaces, where it catalyzes the conversion of hydrogen peroxide to hypohalous acids in the presence of halide and pseudo-halide ions (van Dalen et al., 1997; Hampton et al., 1998). This enzyme is abundantly expressed in granulocytes and accounts for approximately 5% of a neutrophil’s dry weight (Schultz and Kaminker, 1962; Davies, 2011). The major reactive species produced by MPO under physiological conditions are hypochlorous acid and hypothiocyanous acid. While hypohalous acids have potent antibacterial, antiviral, and antifungal properties and play key roles in the human immune system (Hampton et al., 1998; Barrett and Hawkins, 2012), an excessive recruitment and activation of leukocytes in the affected organ(s) under inflammatory conditions can amplify the inflammatory response and contribute to organ injury and dysfunction (Zhang C. et al., 2001; Sugiyama et al., 2004; Klebanoff, 2005; Lau and Baldus, 2006; Davies et al., 2008; Barrett and Hawkins, 2012). MPO-derived oxidants have been shown to damage vascular ECs, basement membrane and matrix components, and lead to leakage of plasma proteins, microvascular hemorrhage, and atherogenesis (Lush and Kvietys, 2000; Yang et al., 2006; Nicholls and Hazen, 2009; Wong et al., 2009; Tiyerili et al., 2016; Yu et al., 2019). MPO and its oxidant end products have been found in both human and experimental atherosclerotic lesions (Daugherty et al., 1994; van Leeuwen et al., 2008; La Rocca et al., 2009), and MPO-induced nitrotyrosine (NO2Tyr) formation in the vessel wall affects the matrix protein structure and function (Lau and Baldus, 2006). Some studies reported that MPO directly modulates endothelial NO production, NO bioavailability, and endothelium-dependent vasodilatation during acute inflammation (Zhang C. et al., 2001; Eiserich et al., 2002; Lau and Baldus, 2006). MPO-derived oxidants have also been found to inhibit sarco/endoplasmic reticulum Ca2+-ATPase activity, perturb Ca2+ homeostasis, resulting in the accumulation of cytosolic Ca2+ (Cook et al., 2012), caspase-3 activation and apoptosis of ECs in culture (Sugiyama et al., 2004). The direct effect of hypochlorous acid (HOCL) on endothelial permeability has been evaluated in cultured bovine pulmonary artery EC monolayers (Tatsumi and Fliss, 1994; Ochoa et al., 1997). Results showed that HOCl, either given directly or produced by conversion of H2O2 with MPO and Cl–, caused immediate changes in EC shape (cell retraction), electrical resistance, and increases in protein permeability (125I-labeled albumin clearance). The HOCl-mediated responses were shown to be faster and greater than those induced by H2O2 (Ochoa et al., 1997). The HOCl (10 to 100 μM)-induced increases in EC monolayer permeability occurred within 1–3 min, whereas H2O2-induced permeability increases in the same type of cultured ECs required higher concentration (50 to 400 μM) and longer exposure time (over 30 min), indicating that HOCI is a more potent oxidant than H2O2 to disrupt endothelial barrier function (Ochoa et al., 1997). Additionally, the MPO and MPO-catalyzed HOCI amplified high glucose-induced endothelial dysfunction in cell culture and rat aorta (Tian et al., 2017). Excessive production of hypohalous acids has been shown to be directly linked to many cardiovascular diseases (Zhang R. et al., 2001; Anatoliotakis et al., 2013; Ndrepepa, 2019) and circulating levels of MPO is clinically used as a biomarker to predict the health outcomes in those patients (Baldus et al., 2003; Brennan et al., 2003; Meuwese et al., 2007; Mocatta et al., 2007; Schindhelm et al., 2009). However, despite the growing evidence and interest in MPO-derived oxidant-induced cellular and molecular damage, the role of different MPO-derived species and mechanisms responsible for increased cardiovascular disease risk and patient outcomes are poorly defined and need further elucidation in detail (Davies, 2011; Barrett and Hawkins, 2012; Ndrepepa, 2019).
ROS-Mediated Microvessel Barrier Dysfunction
Reactive oxygen species are derived from molecular oxygen by sequential monovalent reductions and the essential step in this process is the univalent enzymatic reduction of oxygen to superoxide radical (Del Maestro et al., 1981; Dreher and Junod, 1995; Babior, 1999; Forstermann, 2008). Superoxide in the extracellular space can spontaneously dismutate into H2O2 and O2, and the simultaneous presence of superoxide, H2O2, and chelated metal catalysts in the extracellular space can result in the generation of more active hydroxyl radical. Superoxide is unstable with a lifetime of few milliseconds at neutral pH, and hydroxyl is extremely reactive and short-lived free radical produced in biological systems (Zweier and Talukder, 2006; Montezano and Touyz, 2012).
Among ROS, H2O2 is particularly important in microvascular injury because it is relatively stable and has longer half-life than superoxide, and it is easily diffusible within and between cells (Montezano and Touyz, 2012). In light of the sources of in vivo oxidative stress, in vitro oxidative stress can be produced by direct application of oxidants such as H2O2 or continuous generation of superoxide or H2O2 in enzymatic systems (Dreher and Junod, 1995). One of the most widely used models of oxidative stress is the hypoxanthine (HX)/XO enzyme system (HX/XO) where XO acting aerobically on HX produces superoxide and H2O2 (Del Maestro et al., 1981; Dreher and Junod, 1995). While there is no clear evidence for the effects of blockade of production or bioactivity of endogenous oxidants on basal microvascular permeability (Kvietys and Granger, 2012), the reduction of endogenous NO has been shown to cause an immediate increase in ICAM-1-mediated leukocyte adhesion (Xu et al., 2013; Gao et al., 2017). Both in vitro and in vivo studies demonstrated that exogenous administration of H2O2 or HX/XO system can directly cause oxidative stress and increase the permeability of endothelial monolayers (Shasby et al., 1985; Siflinger-Birnboim et al., 1992; Berman and Martin, 1993; Doan et al., 1994; Okayama et al., 1999), individually perfused intact microvessels (Zhu and He, 2006; Zhou et al., 2009, 2013), whole vascular beds (Del Maestro et al., 1981; Parks et al., 1984), and the isolated organs (Johnson et al., 1989; Barnard and Matalon, 1992; Seeger et al., 1995). The protective effect of antioxidants (such as superoxide dismutase, catalase, XO inhibitor) against the permeability increases in different experimental models (Del Maestro et al., 1981; Parks et al., 1984; Shasby et al., 1985; Kennedy et al., 1989; Okayama et al., 1999; Zhou et al., 2009) and clinical study (Bernard et al., 1997) further implicated the role of oxidative stress in endothelial barrier dysfunction. Importantly, some studies in cultured ECs and intact microvessels have demonstrated that superoxide and H2O2 exert different effects on the endothelial permeability (Berman and Martin, 1993; Mcquaid and Keenan, 1997; Okayama et al., 1999; Zhou et al., 2009) with different cellular mechanisms (Mcquaid et al., 1996; Okayama et al., 1999; Zhou et al., 2013). Details are discussed below.
Differential Effects of Superoxide and H2O2 on EC [Ca2+]i and Microvessel Permeability
A number of in vitro studies in cultured monolayers of ECs from different origin have demonstrated that H2O2 can disrupt endothelial barrier function in a concentration-dependent manner with variable magnitudes (Shasby et al., 1985; Siflinger-Birnboim et al., 1992, 1996; Berman and Martin, 1993; Doan et al., 1994), and oxidant-mediated endothelial barrier dysfunction could be partially reversed and can occur in the absence of cytotoxicity (Shasby et al., 1985; Mcquaid et al., 1996; Gupta et al., 1998). H2O2-induced permeability increases in cultured ECs (measured by different methods) were reported to be rapid (Mcquaid et al., 1996; Hecquet et al., 2008) or delayed (Berman and Martin, 1993; Okayama et al., 1999). Similarly, both early and delayed permeability increases were also reported for superoxide generated by HX-XO in cultured EC monolayers (Berman and Martin, 1993; Okayama et al., 1999). The results of these in vitro studies were quite different and could be related to different species or culture conditions of the ECs or to the method differences in permeability measurements.
In intact microvessel studies the inflammatory mediator-induced increases in microvessel permeability are mediated by transiently increased EC [Ca2+]i (Az-Ma et al., 1996; Zhu and He, 2005; Zhou and He, 2011). ROS has also been shown to induce increases in EC [Ca2+]i by many in vitro and in vivo studies with exogenously applied ROS as stimuli (Doan et al., 1994; Volk et al., 1997; Hecquet et al., 2008) which correlates with increased endothelial permeability (Shasby et al., 1985; Siflinger-Birnboim et al., 1996; Zhu and He, 2006; Zhou et al., 2009, 2013). Superoxide, H2O2, and hydroxyl radical have been shown to exert differential effects on [Ca2+]i in human ECs (Dreher and Junod, 1995). Importantly, it is reported that the toxicity of high levels of ROS could lead to a massive, steady influx of extracellular Ca2+, whereas low concentrations of ROS induce only transient Ca2+ changes, thus acting as signaling agonists (Volk et al., 1997). Certain in vitro studies reported rapid and transient increases in EC [Ca2+]i when ECs were exposed to H2O2 (Siflinger-Birnboim et al., 1996; Volk et al., 1997), similar to those found with applied superoxide derived by HX/XO, while others reported delayed increases in EC [Ca2+]i with H2O2 exposure (Dreher and Junod, 1995; Mcquaid et al., 1996; Okayama et al., 1999).
When the changes in microvessel permeability and endothelial [Ca2+]i were assessed under identical experimental conditions in individually perfused intact microvessels, H2O2 and superoxide demonstrated different effects on EC [Ca2+]i and microvessel permeability (Zhou et al., 2009). Figures 3A,B shows that superoxide, a potent reactive oxygen species (generated by HX/XO), induces immediate and transient increases in EC [Ca2+]i and microvessel permeability (measured by hydraulic conductivity, Lp) similar to inflammatory mediator-induced responses (Zhou et al., 2009). Whereas H2O2, a relatively stable reactive oxygen metabolite at a pathologically relevant concentration of 10 μM, induces delayed and progressive increases in EC [Ca2+]i and Lp in a time and concentration dependent manner (Figures 3C,D; Zhou et al., 2009, 2013, 2019). Importantly, the increased EC [Ca2+]i in either HX/XO or H2O2 perfused vessels were well correlated with the time course of the increases in microvessel Lp. Inhibiting Ca2+ influx by LaCl3 prevented the permeability increase induced by HX/XO or H2O2, thus indicating that Ca2+ influx plays an essential role in both superoxide- and H2O2-induced permeability increases. These findings also indicate that long-term exposure of H2O2 at a low concentration may cause accumulative lipid oxidation and membrane damage, resulting in Ca2+ overload and prolonged irreversible permeability increases (Zhou et al., 2013, 2019). Detailed mechanisms are discussed below. These reported long lasting effects of H2O2 could play a key role in ROS-mediated vascular dysfunction, which may resemble the pathogenesis of diabetes and other disease associated microvascular dysfunction.
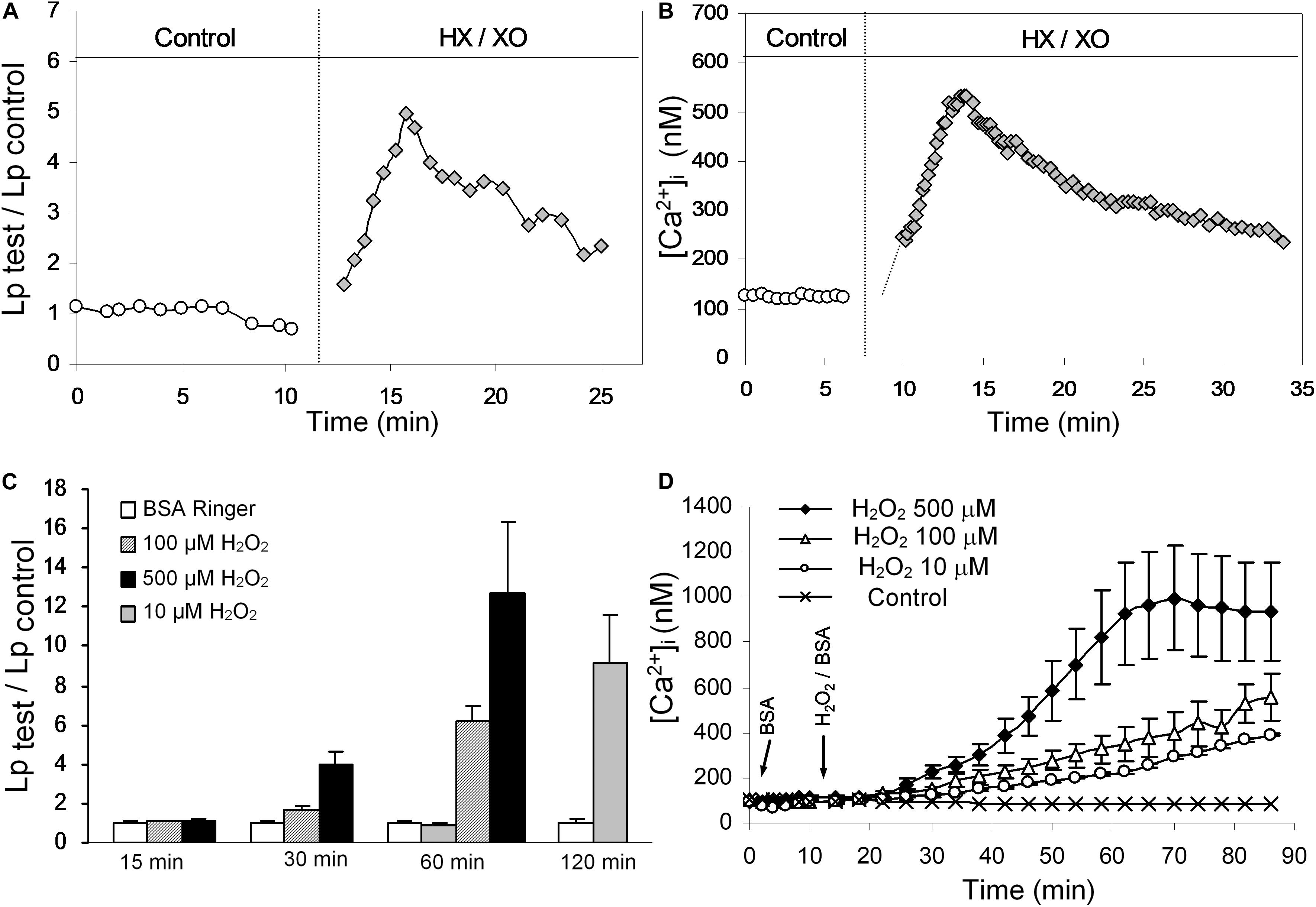
Figure 3. Results of individually perfused rat mesenteric venules demonstrating differential actions of superoxide and H2O2 on endothelial cell [Ca2+]i and microvessel permeability (measured by hydraulic conductivity, Lp). (A,B) Individual experiments showing transient increases in Lp and EC [Ca2+]i after exposure to superoxide generated by HX/XO. (C,D) In contrast to the immediate and transient effects of superoxide, H2O2 induced delayed and progressive increases in EC [Ca2+]i and Lp demonstrating a dose and time-dependent manner. Modified and used with permission (Zhou et al., 2009, 2013).
Methodology Differences in Exogenously Applied ROS
In most of the in vitro studies, the applied H2O2 or HX/XO to generate superoxide was often given to cultured cells as bolus, which causes an immediate reaction of the reactive agents with the cells and other components in the medium and lost the reactive activities quickly. Under those conditions, hundreds of μM or even mM concentrations of peroxide (far more than the pathological concentrations detected in the plasma) were usually needed to see the responses (Alexander et al., 2000; Lee et al., 2006; Jin et al., 2012; Suresh et al., 2015). Under in vivo conditions, the effective level of peroxide in the plasma that has direct effect on microvessel endothelial cells is the net amount after the produced ROS in the vasculature were reacted with the plasma antioxidant enzymes. The measured plasma peroxide levels in human with hypertension are close to 10 μM (Lacy et al., 2000) and in diabetic rats are about 10–15 μM (Xia et al., 2018). To better replicate the in vivo situations, the studies using individually perfused microvessels, conducted in the absence of blood cell components, directly perfused the vessel with a pathologically relevant net plasma H2O2 concentration and the changes in EC [Ca2+]i, microvessel permeability, and cell apoptosis status were assessed while the vessel was continuously exposed to a relatively constant concentration of ROS through the perfusion pipette with constantly refreshed perfusate (Zhou et al., 2013). Comparing to the commonly used bolus application of high concentration of reactive reagents to the tissue or cell culture medium, the continuous perfusion of a pathologically relevant concentration of ROS in intact microvessels closely mimicked the in vivo situation. These detailed technical differences may explain some of the differences in results.
H2O2-Induced Excessively Produced NO and Microvessel Permeability
The distinct time courses of superoxide and H2O2-induced increases in microvessel permeability indicate that these two species of ROS may evoke cellular damage via different signaling mechanisms (Zhou et al., 2009, 2013). It is well recognized that low levels of NO participate in a wide range of physiological processes, whereas large amounts of NO lead to nitrosative stress and endothelial dysfunction. Increased production of ROS has been shown to cause increases in microvessel permeability, tissue damage, and organ dysfunction (Del Maestro et al., 1981; Cai and Harrison, 2000; Zhou et al., 2009). A number of studies have suggested that NO regulates the interactions between oxidant-stimulated signaling pathways and endothelial barrier function and protects the cells from ROS-mediated deleterious effects (Clancy et al., 1992; Kubes et al., 1993; Kurose et al., 1995a; Wink et al., 1995; Okayama et al., 1999). For instance, NO has been shown to attenuate oxidant-induced endothelial barrier dysfunction in cultured ECs (Mcquaid et al., 1996; Gupta et al., 2001), in isolated perfused organs (Kavanagh et al., 1994; Poss et al., 1995), and in whole animal studies (Bloomfield et al., 1997; Mcelroy et al., 1997). However, there are also reports where NOS inhibitors do not modulate ROS-induced changes in endothelial barrier function or cell injury (Berman and Martin, 1993; Az-Ma et al., 1996) or even NO can exacerbate H2O2-induced endothelial permeability in cultured ECs (Mcquaid et al., 1996; Okayama et al., 1997). It is thus apparent that the roles of endogenous vs. exogenous or basal vs. excessive NO in ROS-mediated endothelial barrier function remain equivocal and warrants more evidence to determine the conditions where NO may act either as a proinflammatory or protective mediator in vivo.
H2O2-induced increases in EC NO production have been indicated to play distinct roles in different vasculature (Cai, 2005a). H2O2 is considered an important vasodilator in large arteries or arterioles through its activation on eNOS and increased NO production (Yang et al., 1999; Cai et al., 2003). Whereas in venules, the H2O2-induced excessive NO was linked to nitrosative stress and endothelial dysfunction (Yuan et al., 1993; Baluk et al., 1997; Zhu and He, 2005; Hatakeyama et al., 2006; Zhou and He, 2010, 2011; Mundi et al., 2017; Zhou et al., 2019). H2O2-induced increases in EC NO and the relationship between NO and EC [Ca2+]i were characterized in detail from studies conducted in intact microvessels and presented in Figure 4 (Zhou et al., 2013). It was demonstrated that H2O2 at 10 μM, a concentration close to the plasma level in human disease conditions (Lacy et al., 2000), induced an immediate and long lasting increased NO production, which was characterized by two phases. The initial phase with highest NO production rate occurred immediately upon H2O2 application, which was correlated with increased eNOS Ser1177 phosphorylation in the absence of elevated [Ca2+]i and did not cause a corresponding increase in permeability. The second phase of increased NO occurred after 20 min of H2O2 perfusion when EC [Ca2+]i started to increase, and was associated with Ca2+-dependent Thr495 dephosphorylation and blocked by LaCl3, a blocker for Ca2+ influx (Figure 4; Zhou et al., 2013). Comparing platelet activating factor (PAF)-induced NO production that occurred downstream from transient increases in EC [Ca2+]i and was required for increases in microvessel permeability, the magnitude of H2O2-induced NO was about 6 times of PAF-induced NO (Figure 5). Most importantly, H2O2-induced NO lasted 50 min, much longer than that of PAF-induced NO (6 min). This large amount of NO, instead of causing an immediate increase in microvessel permeability that occurred in PAF-stimulated microvessels (Zhu and He, 2005; Zhou and He, 2010), lead to caspase activation, delayed and cumulative increases in EC [Ca2+]i, vascular cell apoptosis, and progressively increased microvessel permeability (Figures 3, 6). The results also showed that the H2O2-induced caspase activation not only results in increases in EC [Ca2+]i, but the increased [Ca2+]i can further activate caspases. Blockade of Ca2+ influx by LaCl3 significantly attenuated H2O2-induced caspase activation. These observations suggested that H2O2-induced NO-mediated [Ca2+]i overload and cell apoptosis can be augmented through a positive feedback mechanism. Importantly, inhibition of NOS prevented all of the H2O2-induced events including caspase activation, cell apoptosis, increases in endothelial [Ca2+]i and microvessel permeability, indicating a crucial role of NO in H2O2-mediated vascular barrier dysfunction. However, the H2O2-induced largely increased NO (1st phase), in the absence of increased EC [Ca2+]i, did not increase microvessel permeability, suggesting excessive NO alone is not sufficient to increase microvessel permeability. By linking the timing of H2O2-induced increases in microvessel Lp (Figure 3C) with the superimposed time courses of H2O2-induced changes in EC NO and [Ca2+]i shown in Figure 4B, the Lp increase only occurred after EC [Ca2+]i was significantly elevated. Therefore, these studies evidently indicate that it is NO-mediated EC [Ca2+]i overload that causes increases in permeability. The mechanisms involved in the excessive NO-mediated impairment of EC Ca2+ homeostasis are discussed in the flowing section with the focus on the interplay between ROS, NO, and reactive nitrogen species in vascular barrier dysfunction.
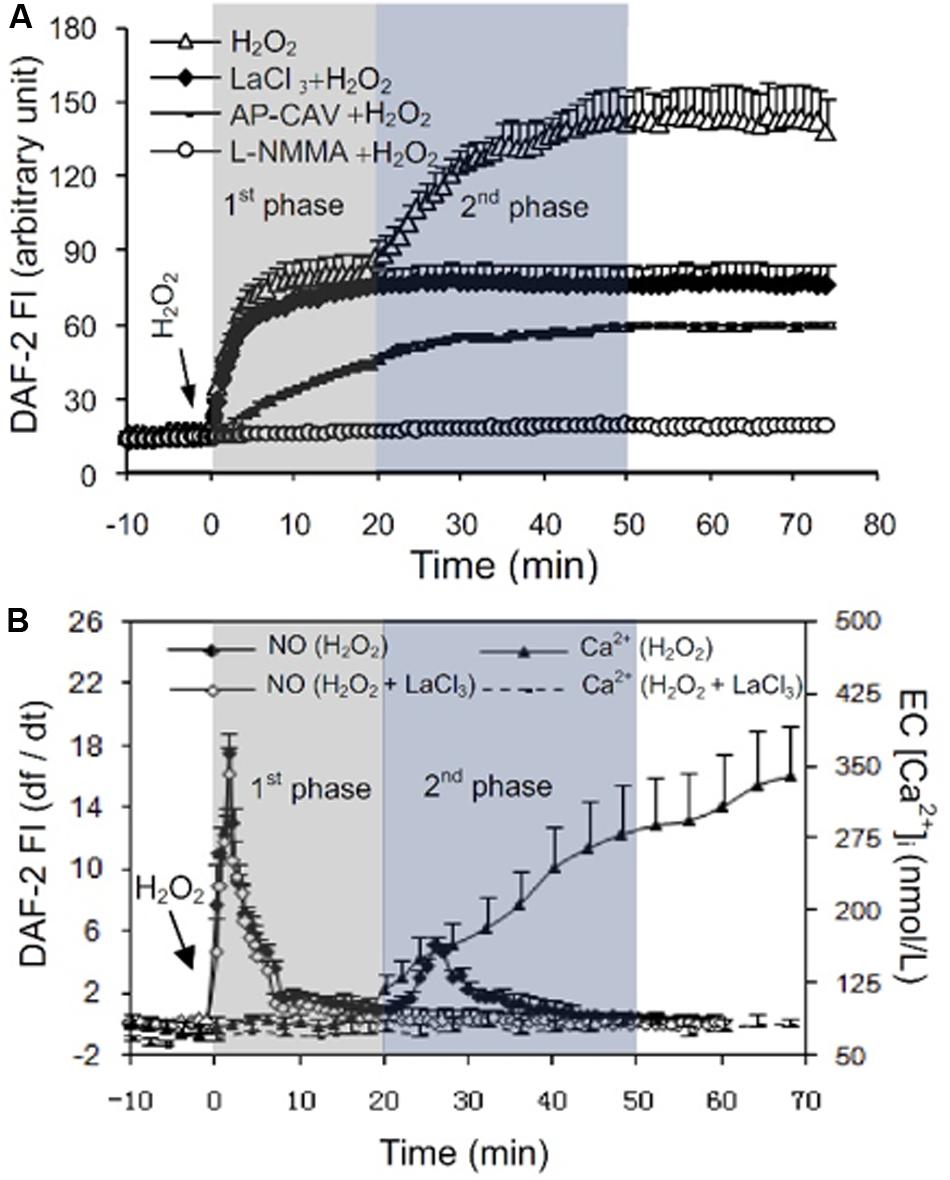
Figure 4. Measurements of H2O2-induced EC NO in DAF-2 DA loaded rat mesenteric venules. (A) Perfusion of vessels with H2O2 (10 μmol/L) increases EC NO via an initial Ca2+ influx-independent (1st) phase followed by a 2nd Ca2+-dependent phase. The application of NOS inhibitors NG-monomethyl-L-arginine (L-NMMA) or AP-CAV prevent or reduce H2O2-induced NO in both phases, and blocking Ca2+ influx with LaCl3 only prevented the 2nd phase of NO production. (B) Superimposed time courses of NO production rate (df/dt, calculated from the cumulative DAF FI curve, left Y-axis) with the changes in EC [Ca2+]i (right Y-axis) in H2O2-perfused vessels demonstrate that the H2O2-induced increases in EC [Ca2+]i only started to increase during the 2nd phase of increased NO, but increased progressively thereafter. Used with permission (Zhou et al., 2013).
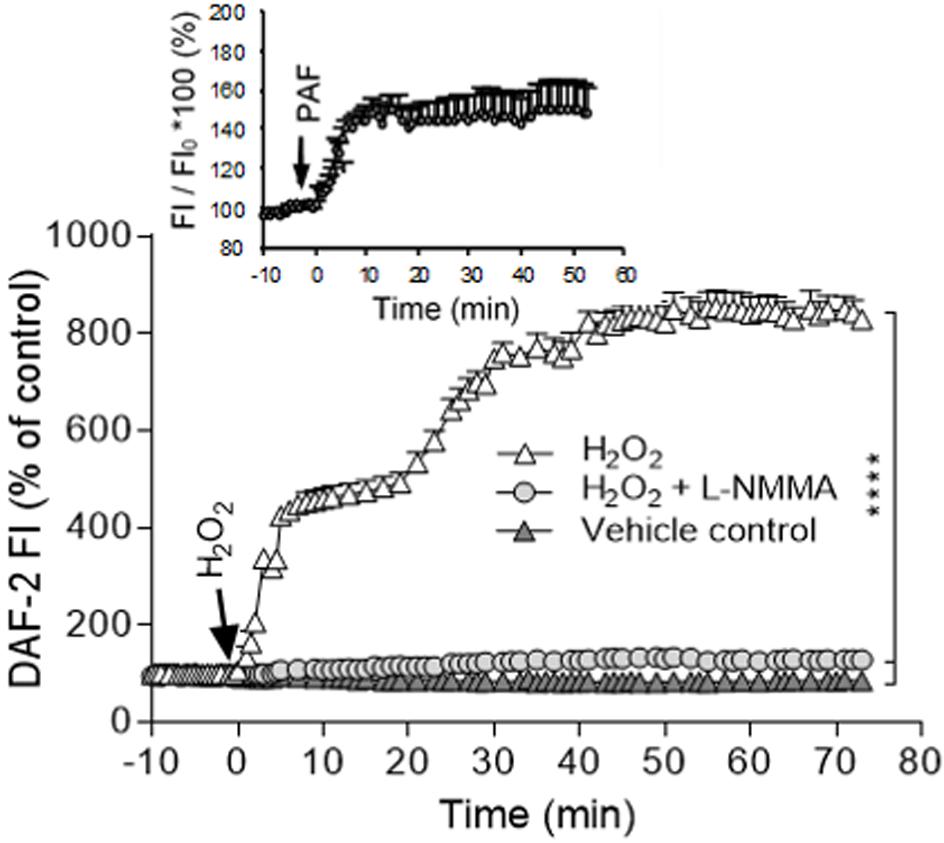
Figure 5. Comparison of PAF and H2O2-induced NO production in rat mesenteric microvessels. NO was measured in DAF-2 DA loaded microvessels. When the vessel was exposed to PAF, the cumulative change of DAF-2 FI at plateau level was about 1.5 times of that of the control and lasted about 6 min (plateau indicates no further increased NO production). Whereas in H2O2 (10 μM) perfused microvessels, the maximum change of DAF-2 FI was about nine times of that of the control, about six times higher than that of PAF-induced NO production. Importantly, the increased NO lasted about 50 min. Modified and used with permission (Zhu and He, 2005; Zhou et al., 2019).
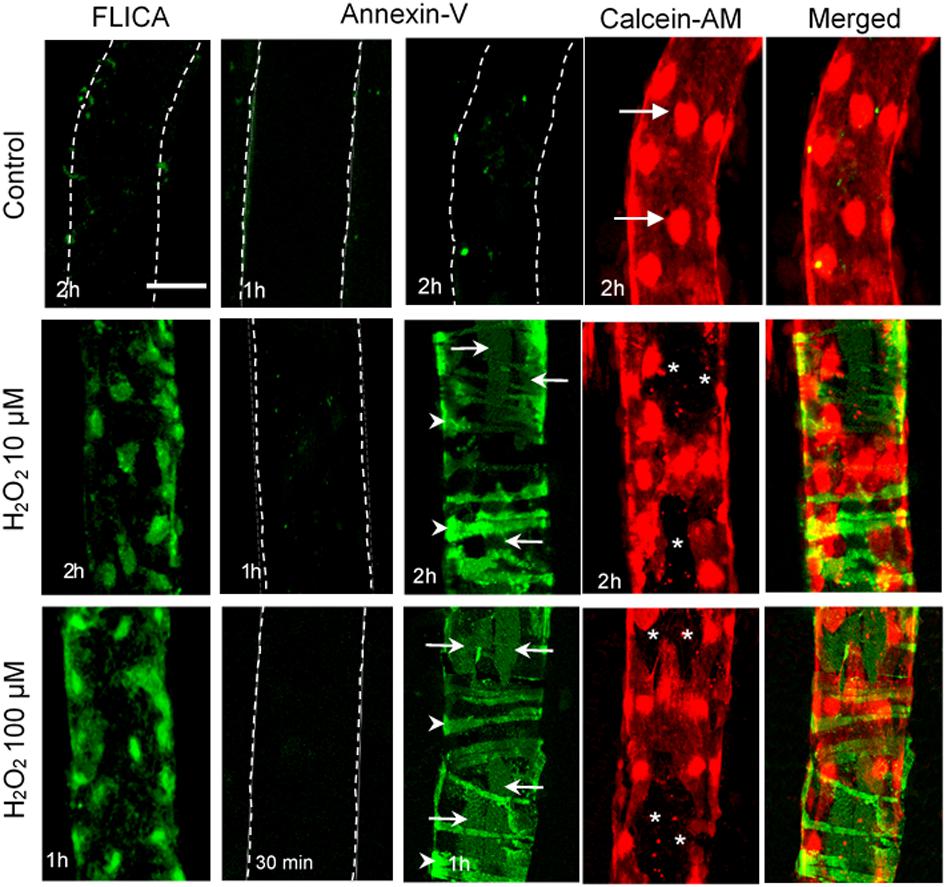
Figure 6. H2O2-induced vascular cell apoptosis. Representative confocal images of FLICA (indicator for activated caspase 3/7), Alexa488-Annexin-V (labeling externalized phosphatidylserine, PS), Calcein-AM (live cell staining), and merged images of Annexin-V with Calcein-AM in H2O2-perfused rat mesenteric venules. These images illustrate that H2O2 induces time and dose dependent caspase activation and vascular cell apoptosis. Importantly, pericytes start apoptosis earlier than endothelial cells (ECs) in the same vessel segment (i.e., with identical H2O2 concentration and exposure time), suggesting their higher vulnerability to ROS than ECs. Arrows indicate ECs and arrowheads indicate pericytes. *ECs not stained with Calcein-AM. Used with permission (Zhou et al., 2013).
ROS, NO, ONOO–, and Microvessel Permeability
Increased production of ONOO– has been implicated to be associated with inflammation and various cardiovascular diseases (Eschwege et al., 1999; Knepler et al., 2001; Wu and Wilson, 2009). ONOO–, a highly reactive oxidant produced by the reaction of superoxide with NO, is capable of oxidizing lipid membranes, nucleic acids, and metabolic enzymes (Beckman et al., 1990; Beckman and Koppenol, 1996). A compelling body of evidence supports a role of ONOO–-mediated damage at inflammatory site (Salvemini et al., 1998). ONOO– has been reported to cause disruption of endothelial cytoskeletal proteins (Knepler et al., 2001; Neumann et al., 2006) or activate upstream signaling cascades (Vepa et al., 1997), leading to EC barrier dysfunction. In cultured ECs, ONOO– has been shown to increase basal and agonist-stimulated Akt- and AMP-activated Ser1179 phosphorylation of eNOS, but decreased NO production and bioactivity (Zou et al., 2002). In addition, while ONOO–-induced accumulation of cGMP in ECs could regulate EC shape and function, activation of poly(ADP-ribose) synthetase (PARS) by ONOO– is reported to suppress mitochondrial respiration and cause endothelial dysfunction (Szabo et al., 1997). These findings suggest that ONOO– can potentially alter endothelial function through complex mechanisms.
H2O2, in addition to inducing NO production, also increases superoxide production, which has been considered as important feed-forward mechanisms for ROS-mediated pathogenesis of cardiovascular diseases (Cai, 2005b; Zinkevich and Gutterman, 2011). In vitro studies reported that the H2O2-induced superoxide production was prevented by the inhibition of p22phox with siRNA and knockout of gp91phox, the two transmembrane subunits of NAD(P)H oxidase, in cultured vascular endothelial cells (Li et al., 2001; Djordjevic et al., 2005). To further elucidate the underlying mechanism for H2O2-induced NO-dependent delayed increases in EC [Ca2+]i, cell apoptosis, and progressively increased microvessel permeability, additional studies were conducted in intact microvessels to explore the potential involvement of NO-derived other form of oxidant agent (Zhou et al., 2019). Consistent with others report, the study first demonstrated increased production of superoxide in both ECs and pericytes in H2O2 perfused vessels, which was inhibited by DPI, a commonly used NAD(P)H oxidase inhibitor, supporting NAD(P)H oxidase as the primary source for the H2O2-induced superoxide production (Zhou et al., 2019). This increased superoxide was not only concurrent but also in close proximity with H2O2-induced markedly increased NO, thus resulting in spontaneous generation of ONOO–. The direct evidence of H2O2-induced ONOO– formation is demonstrated with an extensive NO-dependent tyrosine nitration in H2O2 perfusion microvessels (Figure 7). Nitrotyrosine is a well-recognized biomarker for ONOO–, which commonly served as an indication of systemic nitroxidative stress (Radi, 2013; Zhou et al., 2019). The direct effects of ONOO– in intact microvessels and its role in H2O2-induced permeability increase were further characterized in a recent study conducted in intact microvessels. The study showed that the NO-derived ONOO– plays an important role in H2O2-induced increases in microvessel permeability and the application of an endogenous ONOO– scavenger, uric acid, prevents H2O2-induced increases in endothelial [Ca2+]i and microvessel permeability. The application of exogenous ONOO– in microvessels also showed delayed dose- and time-dependent increases in EC [Ca2+]i and permeability, a pattern similar to that induced by H2O2 (Figure 8). Most importantly, exogenous ONOO– activates eNOS and further increases NO production, resulting in amplified ONOO– formation and severe tissue damage. These findings suggest that H2O2-induced excessive NO-derived ONOO– is the primary mediator of H2O2-induced cell injury and progressively increased microvascular permeability. The fact that ONOO– could cause further activation of eNOS with amplified NO constitutes a self-promoted augmentation of nitroxidative stress, which results in progressively increased permeability. The signaling cascade and the sequential events found in H2O2-perfused intact microvessel are illustrated in Figure 9 (Zhou et al., 2019), which provide mechanistic insights into a better understanding of the pathogenesis of oxidant-induced RNS-mediated microvascular dysfunction.
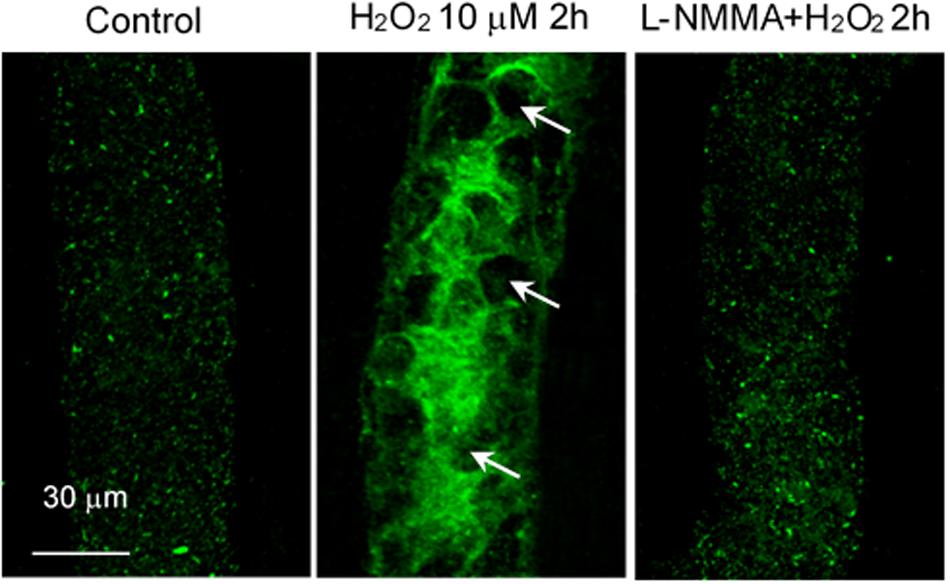
Figure 7. H2O2 induced NO-dependent tyrosine nitration in rat mesenteric venules. Confocal images showing the fluorescent immunostaining of nitrotyrosine in vessels perfused with control (Ringer solution containing 1% BSA, left), H2O2 (10 μmol/L, middle, arrows indicate the nucleus of ECs without nitrated tyrosine formation), and H2O2 in the presence of L-NMMA (2 mmol/L, right). Used with permission (Zhou et al., 2019).
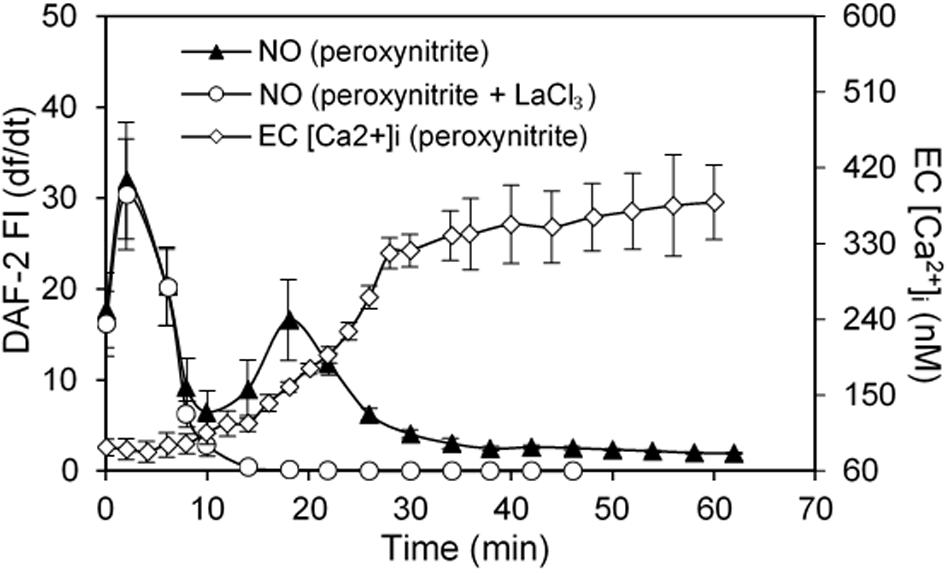
Figure 8. ONOO– induced NO production and increases in EC [Ca2+]i in perfused rat mesenteric venules. Perfusion of vessels with ONOO– (50 μM) increased NO in 2 phases followed by increases in EC [Ca2+]i. The NO production rate (df/dt, left Y axis) derived from cumulative DAF-2 FI in ONOO–-perfused vessels is superimposed with the time course of increased EC [Ca2+]i (right Y axis). The application of LaCl3 (50 μM) did not affect ONOO–-induced initial NO production but prevented the 2nd phase of NO production, indicating the 2nd phase of NO is Ca2+influx-dependent. Importantly, EC [Ca2+]i only started to increase after 20 min of ONOO– perfusion, but increased progressively thereafter, a pattern similar to that observed in H2O2-perfused vessels. Used with permission (Zhou et al., 2019).
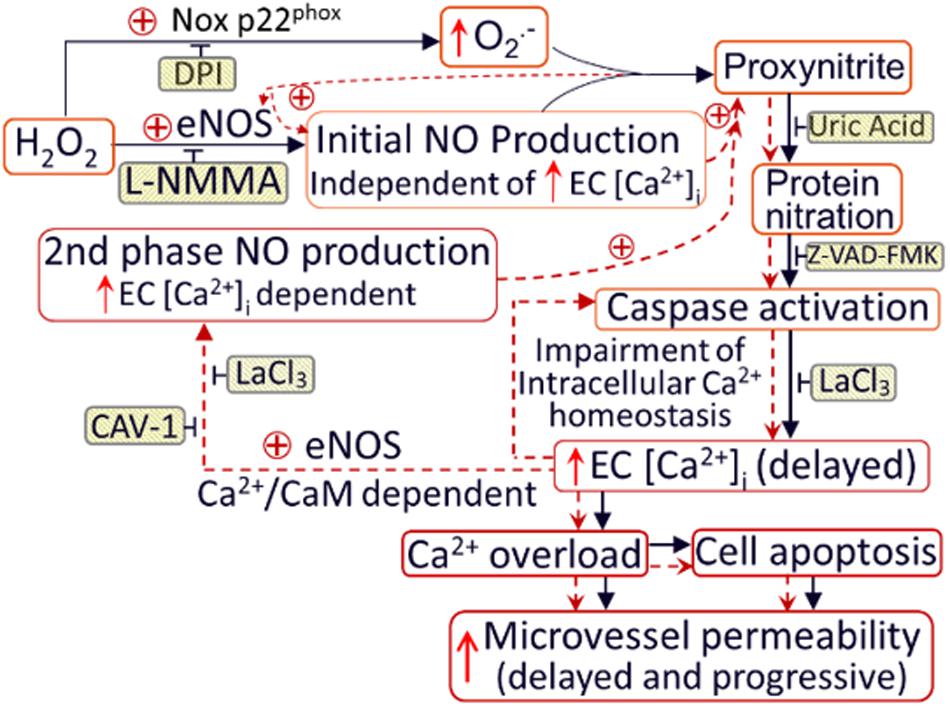
Figure 9. Schematic diagram illustrating the mechanisms of H2O2-induced microvessel barrier dysfunction. H2O2 at pathophysiological levels activate eNOS and NADPH oxidase in the endothelium and pericytes, leading to increased formation of NO and O2–. The temporal and spatial proximity of largely produced NO and O2– promote the formation of ONOO–, a more potent oxidant, leading to ONOO–-mediated lipid peroxidation, protein tyrosine nitration, subsequent impairment of intracellular Ca2+ homeostasis, and vascular cell apoptosis. Both NO-derived ONOO– and the delayed increases in EC [Ca2+]i are able to further activate eNOS, resulting in amplified NO production and NO-dependent downstream cascade. These positive feedback loops, indicated by red dotted lines, play an important role in sustained production of ONOO–, and eventually ONOO–-mediated endothelial cell Ca2+ overload, vascular cell apoptosis, and progressively increased microvessel permeability. The application of inhibitors (shaded boxes) blocks the downstream events. DPI (diphenylene iodonium, NADPH oxidase inhibitor); eNOS: endothelial nitric oxide synthase; CAV-1: Caveolin-1 (eNOS inhibitor); z-VAD-FMK: caspase inhibitor. Used with permission (Zhou et al., 2019).
Role of Basal No in Microvessel Barrier Function
Basal NO and agonist-induced excessive NO have been shown to play different roles in the regulation of microvessel permeability (Zhu and He, 2005; Zhou and He, 2010; Xu et al., 2013). As discussed above, ROS- or inflammatory mediator-induced excessive NO contributes to increased vascular permeability through different mechanisms (Moncada et al., 1991; Yuan et al., 1993; Mayhan, 1994; Wu et al., 1996; He et al., 1997; Zhu and He, 2005; Hatakeyama et al., 2006; Sanchez et al., 2009; Zhou and He, 2011), whereas basal NO was reported to play a critical role in preventing leukocyte adhesion (Furchgott and Vanhoutte, 1989; Kubes et al., 1991; Moncada et al., 1991) and the adhesion and aggregation of platelets (Radomski et al., 1990a, b). Reduced basal NO synthesis or NO bioavailability has been reported in various disease conditions including hypertension, diabetes, hypercholesterolemia, and reperfusion injury, which was commonly linked to endothelial dysfunction and vascular inflammation (Moncada and Higgs, 2006; Potenza et al., 2009). Genetic deletion of eNOS or over expression of caveolin-1, an endogenous inhibitor of eNOS, has been shown to cause increased leukocyte adhesion and macrophage infiltration, and accelerated atherosclerosis in apolipoprotein E (apoE) deficient mice (Sasaki et al., 2003; Fernandez-Hernando et al., 2009; Atochin and Huang, 2010; Fritzsche et al., 2010; Ponnuswamy et al., 2012). Those studies indicated an important role of basal NO in maintaining vascular function and preventing vascular inflammation, but the mechanisms were not well defined. Although the animals with genetic deletion of eNOS generated a constant reduction of NO systemically, such condition may trigger complex adaptation and compensatory effects in the vascular system. Therefore, the inflammatory manifestation observed in those animals may not unequivocally represent the role of basal NO.
Some in vivo studies demonstrated that the application of NOS inhibitor through either systemic injection or superfusion to the vascular beds induced increases in microvessel permeability (Kubes and Granger, 1992; Kurose et al., 1995b). However, reduced basal NO in the presence of blood would cause leukocyte and platelet adhesion, which may confound the direct effect of basal NO on permeability. When studies were conducted in individually perfused vessels in the absence of blood components in the vessel lumen, reduced basal NO production by NOS inhibitor or caveolin-1 peptide, AP-CAV, showed no effect on basal microvessel permeability (Xu et al., 2013). The increased permeability observed in whole vascular bed studies could be attributed to the reduced basal NO-induced increases in adherent leukocytes. In the presence of any inflammatory stimulus, the adherent leukocytes could be activated to release ROS and thus increase the permeability. The mechanisms of reduced basal NO-induced leukocyte adhesion were demonstrated recently (Xu et al., 2013; Gao et al., 2017). Those studies showed that when blood flow was resumed in vessels that were pretreated with NOS inhibitor or AP-CAV to reduce endothelial basal NO, large amounts of leukocytes quickly adhered to rat venules and the adhesion was demonstrated to be EC ICAM-1 mediated (Figure 10; Xu et al., 2013). Most importantly, the ICAM-1-mediated adhesion occurred very quickly (less than 30 min), which rules out the possibility of de novo protein synthesis and translocation. Such rapid changes in ICAM-1 adhesiveness predicts a NO-dependent regulation of the adhesive binding avidity of constitutively expressed endothelial ICAM-1. This prediction was further validated by an additional in vitro cell culture study showing that the reduced basal NO-induced increases in EC adhesive binding were mediated by Src-dependent ICAM-1 activation through its phosphorylated state (Gao et al., 2017).
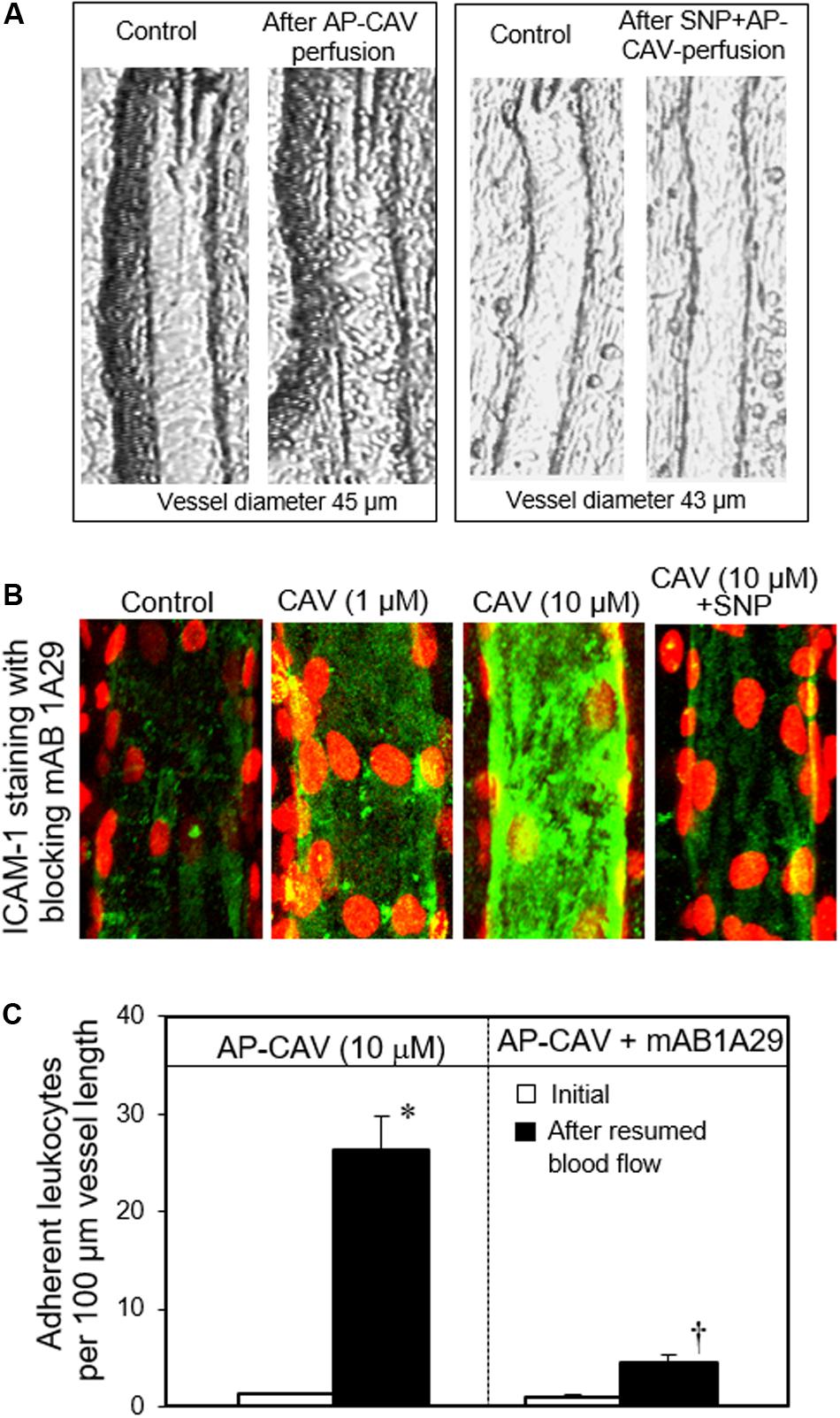
Figure 10. Perfusion of rat mesenteric venules with AP-CAV, an endogenous NOS inhibitor, induced basal NO-dependent ICAM-1 mediated leukocyte adhesion. Intact venules were perfused by AP-CAV for 30 min followed by resuming blood flow in the same vessel for 10 min. Leukocyte adhesion was quantified when each vessel was recannulated with BSA-Ringer solution. (A) Video images of a perfused venule under control conditions and after AP-CAV (10 μM)-induced leukocyte adhesion, and the administration of a NO donor, sodium nitroprusside (SNP), abolished AP-CAV-induced leukocyte adhesion. (B) AP-CAV induced dose-dependent increases in the binding affinity between EC ICAM-1 and its blocking antibody mAb1A29. Confocal images of mAb1A29 (green) and vascular cell nuclei (red) immunofluorescence co-staining under control conditions, after AP-CAV perfusion, and after adding SNP to AP-CAV perfused vessels. (C) Perfusion of vessels with ICAM-1 inhibitory antibody, mAb1A29, significantly attenuated AP-CAV induced leukocyte adhesion. * and † indicate a significant increase and decrease from the control, respectively. Used with permission (Xu et al., 2013).
Other in vitro studies proposed that the inhibition of eNOS-derived NO does not directly contribute to EC conversion to a pro-adhesive phenotype but rather interferes with the generation of ROS by NADPH oxidase and results ROS-mediated inflammatory response (Niu et al., 1994; Kvietys and Granger, 2012). In contrast to this proposal, the timing and pattern of leukocyte adhesion observed in intact microvessels (Xu et al., 2013) strongly support a direct association between basal NO and adhesive states of ECs. Importantly, if the increased adhesiveness of ECs is ROS mediated, an increased permeability should be observed. Instead, the results showed that basal NO reduction-induced adherent leukocytes do not cause permeability increase unless additional stimuli (such as fMLP) are applied to trigger neutrophil respiratory burst (Xu et al., 2013). Taken together, these studies demonstrated that basal NO is essential for maintaining the non-adhesive state of endothelium through regulation of the adhesive binding of endothelial ICAM-1.
Role of ROS in Pericyte Loss and Microvessel Permeability
The albuminal side of endothelium is surrounded by a morphologically distinct cell termed pericyte. Pericyte was first described as perivascular contractile cell that wrap around the ECs of microvessels (Sims, 1986). They are also known as Rouget cells named after their discoverer, or referred to as mural cells because of their perivascular position, or as microvascular smooth muscle cells (SMCs) because of their contractile nature (Armulik et al., 2011; Stapor et al., 2014; van Dijk et al., 2015; Ferland-McCollough et al., 2017).
Pericyte Structure and Function in Microvessels
Pericytes are found on pre-capillary arterioles, capillaries, post-capillary venules, and collecting venules of many organs, but they are more extensive in postcapillary values (Sims, 1986; Shepro and Morel, 1993). They are embedded within the endothelial basement membrane (BM) and have a cell body with a prominent nucleus, a small amount of cytoplasm with extending long processes along the longitudinal axis of the blood vessels, and usually span many ECs (Armulik et al., 2011). The morphology of pericyte is also reported to be very distinct in different organs ranging from a typical flattened or elongated, stellate-shaped cell with multiple cytoplasmic processes encircling a large albuminal vessel area in central nervous system (CNS) to that of a mesangial cell of kidney glomerulus as rounded, compact, and covering a minimal albuminal vessel area (Armulik et al., 2005; Hartmann et al., 2015). The pericyte coverage of the endothelial albuminal surface varies at different organs and vascular beds, and it reflects the relative ratio between the ECs to pericytes (Sims, 1986). The highest pericyte coverage of microvessels is reported in CNS and retina with EC-to-pericyte ratio of 1:1 to 3:1 forming blood-brain barrier and blood-retinal barrier, and the lower EC-to-pericyte ratio of 100:1 in skeletal muscle (Sims, 1986; Shepro and Morel, 1993). Pericytes and ECs not only share a common BM, they are directly connected to each other through the interruptions in the BM (Rucker et al., 2000). The intimate relationship between albuminal pericytes and luminal ECs through the BM is not only the physical communications; it involves several signaling mechanisms that regulate pericyte-EC interactions and communication. The signaling pathways have been extensively reviewed by several authors (Armulik et al., 2005; von Tell et al., 2006; Gaengel et al., 2009; Armulik et al., 2011; Hill et al., 2014; Warmke et al., 2016). Investigations with genetic mouse models have showed that pericytes and ECs are interdependent, as a result a primary defect in one cell type have inevitable consequences on the other cell (Armulik et al., 2005; Gaengel et al., 2009). Because of its intimate relationship with endothelium and BM, pericytes have recently come into focus as an emerging key factor in the regulation of diverse vascular functions both in health and disease. Briefly, pericytes are functionally associated with the regulation of vascular contraction and blood flow (Shepro and Morel, 1993; Rucker et al., 2000; Bergers and Song, 2005; van Dijk et al., 2015), vessel permeability (Edelman et al., 2006; Yuan and He, 2012), vascular stability (von Tell et al., 2006), BM organization (Stratman and Davis, 2012), and angiogenesis (Gerhardt and Betsholtz, 2003; Bergers and Song, 2005).
Role of Pericytes in the Regulation of Microvessel Permeability
Vascular endothelial cells have been recognized as the principal barrier for fluid and solute exchange between circulating blood and surrounding tissues. During inflammation, the increased permeability mainly occurs at venules. The majority of the venular wall is wrapped by perivascular cells, mainly pericytes (Murfee et al., 2005). The prime location of pericytes suggests that they may serve as an additional barrier at the vessel walls that restricts macromolecule leakage when permeability is increased. Additionally, based on its contractile nature, it may also actively involve the regulation of microvascular permeability. The role of pericytes in microvessel barrier function has been controversial. Some of the studies reported that the contraction of pericytes prompted the gap formation between endothelial cells (Kelley et al., 1988). While other in vivo studies indicated that pericytes might protect endothelial junctions upon histamine exposure (Sims et al., 1990). In a microvascular lung pericyte/EC co-culture study, addition of pericytes is reported to increase the permeability barrier compared to EC alone (Dente et al., 2001). A study using a pericyte deficient mouse model revealed that the lack of pericytes increased the permeability of BBB to water and a range of solute tracers (Armulik et al., 2010; Daneman et al., 2010). Another in vivo evidence derived from inflamed vessels demonstrated that pericyte F-actin bundles and pericyte processes completely covered largely separated VE-cadherin and open endothelial gaps at the peak of the permeability increases (Yuan and He, 2012). Figure 11 illustrated details with confocal images and ultrastructural micrograph. These in vivo observations suggest that pericytes, instead of promoting endothelial gap formation, play an important role in lessening a potentially higher degree of leakage when endothelial barrier is impaired. In vessels going through inflammation-induced vascular remodeling, the EM structure of the vascular walls showed proliferated perivascular cells and expanded extracellular matrix, which not only serve as a secondary barrier that lessen the vascular leakage but also provide a supporting structure to stabilize the microvessel wall during stimulation-induced EC cytoskeleton contractions (Yuan and He, 2012).
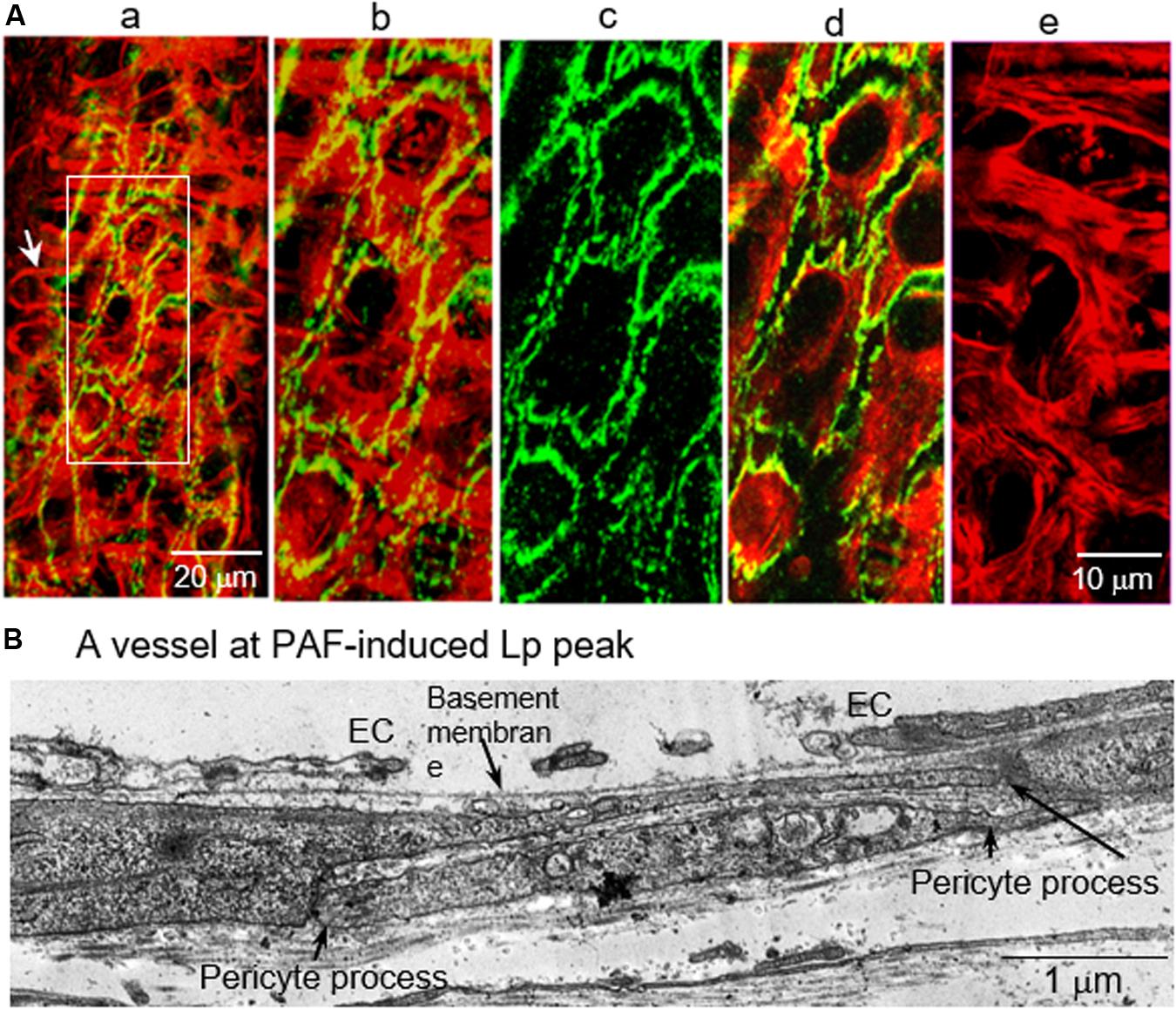
Figure 11. Confocal and electron micrographs of rat mesenteric venules demonstrating the positioning of pericyte processes in relation to open endothelial junctions at PAF-induced peak increase of Lp. (A) Confocal images: Image a is a partial projection of the lower half of the vessel wall with dual staining of F-actin (red) and VE-cadherin (green). An arrow indicates a typical pericyte structure: a single nucleus with multiple cellular processes. Details of a local region of the vessel wall (rectangular box) are displayed in images b to e with a higher magnification. Image b is a dual channel image projection of both endothelial and pericyte layers demonstrating that all of the open VE-cadherin is covered by aggregated pericyte actin bundles. Image c shows large separations of endothelial VE-cadherin. Image d is a dual channel image projection at the endothelial cell layer showing the changes in endothelial F-actin and VE-cadherin. Image e shows the aggregated pericyte F-actin bundles (a projection of image sections blow endothelial cells). Micrograph (B) shows the structural changes in vascular wall at the PAF-induced Lp peak. It displays a wide open endothelial gap with intact basement membrane. Consistent with confocal images, two extended overlapping pericyte processes provide a complete coverage of the endothelial gap. These images suggest a role of pericytes in stabilizing the vascular wall and lessening an otherwise greater magnitude of leakage while endothelial barrier is impaired. Modified and used with permission (Yuan and He, 2012).
ROS-Mediated Pericyte Apoptosis
As pericytes are abundant in microvasculature and have been linked to a wide array of vascular functions, alterations in pericyte coverage or pericyte dysfunctions can contribute to multiple vascular pathologies. Accordingly, pericyte loss has been demonstrated in the pathogenesis of several microvascular complications under disease conditions (Armulik et al., 2005; Hill et al., 2014; van Dijk et al., 2015; Warmke et al., 2016; Ferland-McCollough et al., 2017). The most characterized and established microvascular complications with pericyte loss are associated with diabetes, such as diabetic retinopathy, diabetic nephropathy, and diabetic neuropathy (van Dijk et al., 2015; Warmke et al., 2016; Ferland-McCollough et al., 2017). Figure 12 illustrates the progression of retina vessels from hyperglycemia and oxidative stress-induced loss of pericytes and endothelial cells, increased vascular permeability, to the development of proliferative retinopathy (Ferland-McCollough et al., 2017). Loss of pericyte number and coverage has also been reported in neurodegeneration and Alzheimer’s disease (Sagare et al., 2013; Sengillo et al., 2013). Ischemic stroke within CNS is reported to cause microvascular pericyte loss during ischemia/reperfusion (Hill et al., 2014). However, the mechanisms involved in pericyte loss under those pathological conditions remain unclear. In all of the diseases mentioned above, oxidative stress plays an important role and ROS-induced cell injury could be the main cause leading to cell apoptosis. An early indicator of apoptosis is the translocation of phosphatidylserine (PS) from the inner to the outer leaflet of the plasma membrane, which can be detected by Annexin V, a phospholipid-binding protein that has high affinity for PS. As that shown in Figures 3, 6, perfusion of microvessels with H2O2 at 10 μM for 2 h caused caspase activation, cell apoptosis, followed by EC Ca2+ overload and increased microvessel permeability (Zhou et al., 2013). Importantly, when the apoptotic cells in H2O2-perfused vessels were illustrated by Annxin-V staining (Figure 6), pericytes undergo apoptosis earlier than most of the ECs, albeit both of them were exposed to the same concentration of H2O2 with identical exposure time. This observation indicates that pericyte are more vulnerable to oxidative stress than ECs. This may also explain the early loss of microvascular pericytes that was commonly found in diabetic patients and animals as that shown in Figure 12. The loss of pericytes in retina microvessels could be the main cause for aneurism formation in diabetic retinopathy and the increased plasma levels of ROS could play an important role (Shojaee et al., 1999).
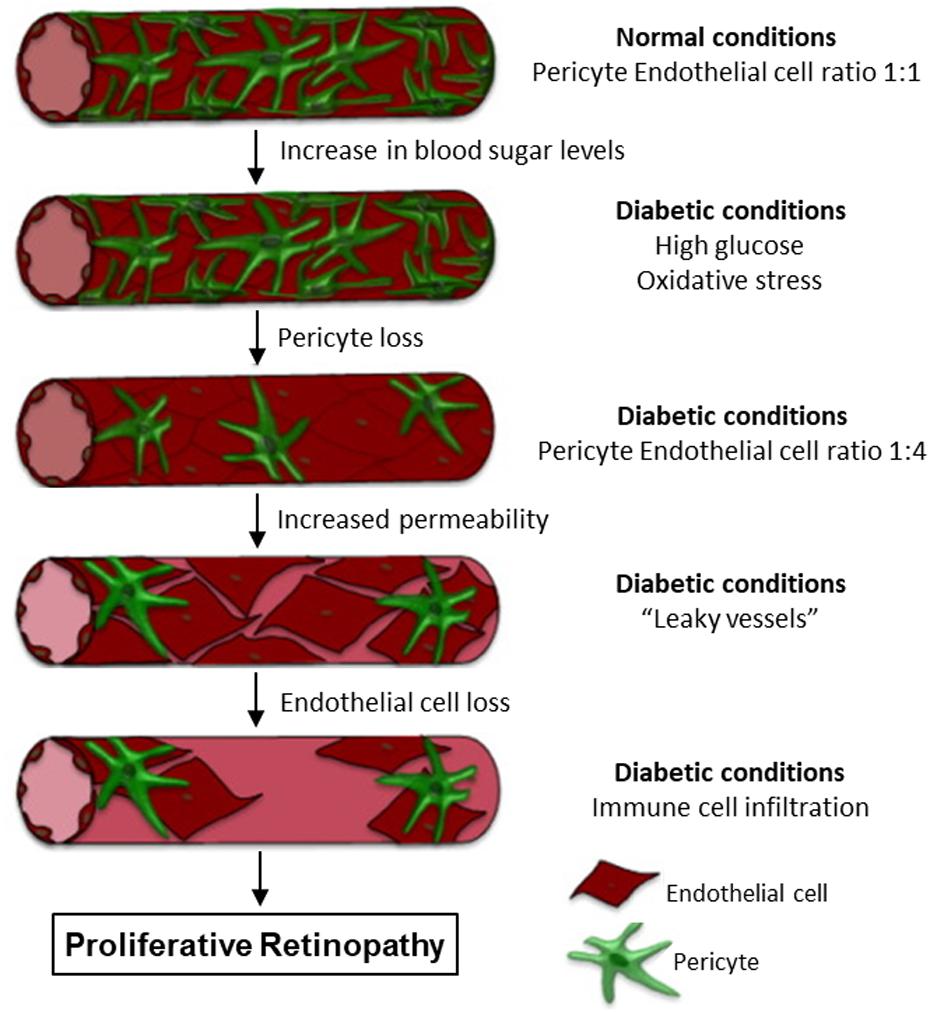
Figure 12. Retina vascular progression to proliferative retinopathy under diabetic conditions. Hyperglycemia and oxidative stress cause pericyte apoptosis and increases in microvessel permeability. The loss of pericytes and endothelial cells will further increase fluid and protein leakage in the retina and immune cell infiltration. These intra-ocular vascular changes will then contribute to the development of proliferative retinopathy. Modified and used with permission (Ferland-McCollough et al., 2017).
In summary, in recent years our knowledge in pericyte characterization, functions, and its involvement in pathological conditions has evolved significantly, but there are far more questions than answers. The multifaceted role of pericytes thus emphasizes the need of further studies in intact microvasculature with novel in vivo and ex vivo experimental approaches to fully understand the specific pericyte function in health and disease, and to further explore their regenerative potential and the signaling pathways affected vascular function in different pathological states.
Oxidative Stress and Antioxidant Counter Balance
It is now well recognized that the state of oxidative stress in the vascular wall is developed following an imbalance between ROS and antioxidant enzymes where excess cellular production of ROS outperforms the capacity of endogenous antioxidant defense mechanisms. Growing evidence from basic and clinical studies suggest that increased oxidative stress plays a pivotal role in the initiation and progression of many cardiovascular diseases (Madamanchi et al., 2005; Landmesser et al., 2007; Forstermann, 2008; Boueiz and Hassoun, 2009). While animal studies supported the potential role of antioxidants in preventing oxidative stress-induced vascular disorders (Modig and Sandin, 1988; Bernard, 1991; Leff et al., 1993), clinical trials with various antioxidants have been disappointing and failed to demonstrate conclusive results (Madamanchi et al., 2005; Landmesser et al., 2007; Forstermann, 2008; Boueiz and Hassoun, 2009). While several factors have been identified for the lack of clinical efficacy of antioxidants (Madamanchi et al., 2005; Forstermann, 2008; Boueiz and Hassoun, 2009), it has become essential to have a better understanding of ROS-mediated signaling mechanisms, their localization, and the integration of both ROS-dependent transcriptional and signaling pathways in vascular pathophysiology (Madamanchi et al., 2005). Consequently, the field of oxidative stress has recently been widened significantly and it is often viewed as an imbalance of genes encoding antioxidant enzymes and proteins, and how those genes are regulated under disease conditions with increased ROS production.
Antioxidant enzymes such as SOD, catalase, and glutathione peroxidase are able to break down ROS promptly to other less reactive and non-reactive downstream products. These proteins play key roles in counter balance of oxidative stress-induced cell damage. The regulation of cellular redox homeostasis mainly occurs at the transcription level. Cells activate transcription of protective antioxidant genes under oxidative stress via varieties of redox-sensitive transcription factors, of which Nrf2 has been recognized as a master regulator of antioxidative responses (Motohashi and Yamamoto, 2004; Amin et al., 2019). Under normal physiological conditions, Nrf2 is located in the cytosol at a low concentration and is the target for proteasomal degradation by its interaction with a cytosolic regulatory protein Kelch-like ECH-associated protein (Keap1) (Nguyen et al., 2009). With increased ROS, Nrf2 dissociates from Keap1 and translocates to the nucleus where it binds to the promoters of genes containing the cis-acting antioxidant response element (ARE). Its binding to ARE at nucleus promotes the transcription of a wide variety of antioxidant genes (Ma, 2013). Many ARE genes are involved in detoxification enzymes such as glutathione-S-transferases, heme oxygenase-1, and NAD(P)H dehydrogenase (Nguyen et al., 2009) and hence the Nrf2 activity has been viewed as a key indicator of cellular antioxidant capacity. Since many factors may affect Nrf2 activity such as its dissociation from Keap1, translocation to nucleus, its efficiency in binding to ARE due to enhanced nuclear export and degradation, exactly how Nrf2 activity is regulated under different pathological condition-induced oxidative stress remains to be uncovered. The Nrf2/Keap1-ARE signaling pathways and biochemical aspect of their interactions under different disease conditions have been reviewed elsewhere (Chen et al., 2015; Amin et al., 2019).
Nrf2 dysfunction or reduced Nrf2 activity has been found in certain oxidative stress-associated conditions such as aging-related vascular inflammation (Usatyuk and Natarajan, 2004; Ungvari et al., 2011b), obesity-induced neurovascular dysfunction (Tarantini et al., 2018), and the disturbed flow-mediated atheroprone regions of the vasculature (Mcsweeney et al., 2016). Genetic deletions of Nrf2 in mice and rats are viable under normal conditions, but they have shown higher sensitivity to increased oxidant stress-induced vascular damages and organ dysfunction (Chan and Kwong, 2000; Chan et al., 2001; Chanas et al., 2002; Ungvari et al., 2011a; Priestley et al., 2016). High fat diet-induced increases in vascular ROS levels were greater in Nrf2–/– than in Nrf2+/+ mice (Ungvari et al., 2011a). Nrf2 deficient rats lead to the development of salt-induced oxidative stress, endothelial dysfunction, and microvessel rarefaction in normotensive rats (Priestley et al., 2016). A recent study conducted in individually perfused microvessels showed that Nrf2 deficient rats have higher plasma H2O2 levels than WT rats under both normal and STZ-induced diabetic conditions. Compared to WT rats, microvessels in Nrf2 deficient rats showed significantly higher basal permeability (measured by hydraulic conductivity, Lp) as well as increased permeability responses to PAF in both normal and STZ-induced diabetic rats (Xia et al., 2018). Importantly, the magnitude of the Lp increases mirrored the plasma H2O2 levels where higher ROS levels correlated with higher permeability increases when exposed to an inflammatory mediator, PAF. These studies indicate an important antioxidant role of Nrf2 under both normal and pathological conditions.
The important role of Nrf2 in the regulation of antioxidant responses led many studies to search pharmacological activators or inducers of Nrf2 to enhance Nrf2-driven antioxidant defense (Shih et al., 2005; Chen et al., 2015; Sharma et al., 2017). The induction of NRF2-driven antioxidant response in whole animal showed reduced ischemic damage from stroke and mitochondrial stress (Shih et al., 2005), and increased expression of Nrf2-driven genes protected the blood brain barrier after brain injury (Zhao et al., 2007). To date, increasing number of chemical compounds and plant natural compounds have been recognized to have antioxidant effect mediated by Nrf2-ARE related pathways from in vivo and in vitro studies (Chen et al., 2015). Some of the Nrf2 inducer supplements also showed promising anti-aging and anti-oxidant effects in clinical studies (Ghanim et al., 2011; Pergola et al., 2011). However, the conclusive evidence of their ability in improving clinical outcomes has been limited and warrants further investigation.
Current studies indicate that increased ROS and/or reduced antioxidant defense play important roles in priming endothelial cells and transitioning them into a phenotype with higher susceptibility to additional inflammatory stimuli or pathological conditions. Importantly, these studies open up a novel area of future investigations where direct upregulation of Nrf2 antioxidant defense system could prove to be a highly valuable therapeutic strategy against conditions associated with increased oxidative stress.
Summary
Clinical and experimental evidence indicate that increased vascular permeability contributes to many disease-associated vascular complications. It is thus important to identify the critical factors and underlying mechanisms involved in permeability increases. Increased production of ROS has been implicated in the pathogenesis of many cardiovascular diseases. As those outlined in this review, each species of ROS plays different roles in the regulation of vascular function. Importantly, one species of ROS can further amplify redox signaling, resulting in augmented oxidative and nitroxidative stress. Increasing number of studies recognize that H2O2, a relatively stable reactive oxygen metabolite, plays an important role in ROS-mediated vascular barrier dysfunction. For decades, increased ROS-induced vascular dysfunction has often been associated with reduced NO production through eNOS uncoupling. However, emerging evidence revealed that the H2O2-induced EC [Ca2+]i overload, cell apoptosis, and progressively increased microvessel permeability were mediated by H2O2-induced excessive NO and NO-derived peroxynitrite. The interplay of ROS, NO, and RNS followed by a self-promoted amplification of nitroxidative stress contributes to ROS-mediated microvascular dysfunction, including early stage of pericyte loss in microvessels. The schematic diagram shown in Figure 13 summarizes the underlying mechanisms involved in leukocyte-dependent and ROS-mediated microvessel barrier dysfunction. Future studies on molecular mechanisms and gene regulations of ROS overproduction, targeted interference of the amplification cascade, or enhancement of the antioxidant would benefit the development of therapeutic strategies to alleviate ROS-mediated vascular barrier dysfunction, which underlies a variety of cardiovascular diseases.
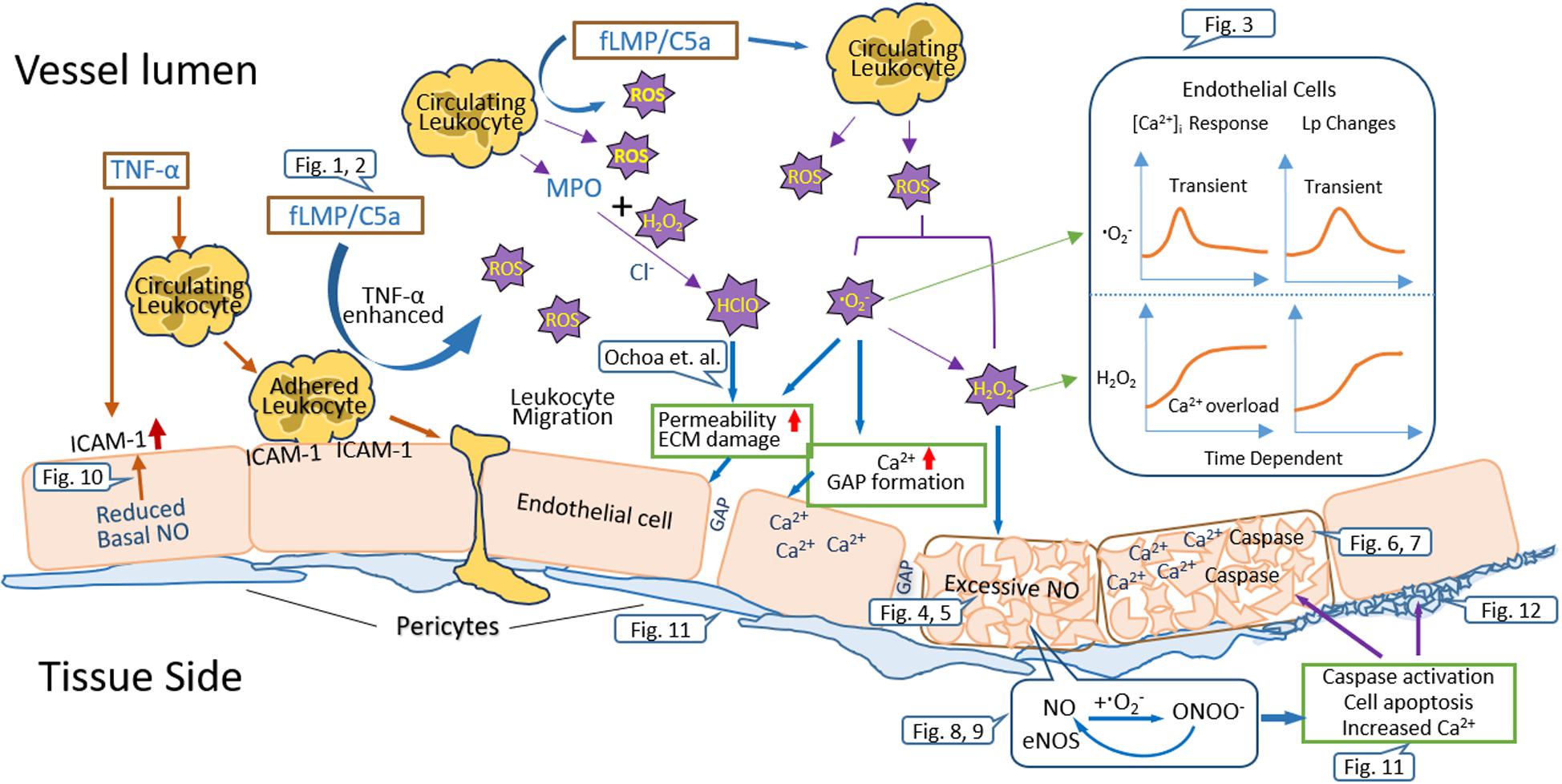
Figure 13. Schematic diagram summarizing the underlying mechanisms involved in leukocyte-dependent and ROS-mediated microvessel barrier dysfunction.
Author Contributions
All authors contributed to the writing, editing, and the effort is reflected by the author order and had final approval of the submitted version.
Funding
This work was supported by the NIH National Institute of Diabetes and Digestive and Kidney Diseases DK097391, and National Heart, Lung, and Blood Institute grants HL130363 and HL144620.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alexander, J. S., Alexander, B. C., Eppihimer, L. A., Goodyear, N., Haque, R., Davis, C. P., et al. (2000). Inflammatory mediators induce sequestration of VE-cadherin in cultured human endothelial cells. Inflammation 24, 99–113. doi: 10.1023/a:1007025325451
Amin, K. N., Bhakkiyalakshmi, E., Ravichandran, J., Sarada, D. V. L., and Mohanram, R. K. (2019). The pivotal role of nuclear factor erythroid 2-related factor 2 in diabetes-induced endothelial dysfunction. Pharmacol. Res. 7:104601. doi: 10.1016/j.phrs.2019.104601
Anatoliotakis, N., Deftereos, S., Bouras, G., Giannopoulos, G., Tsounis, D., Angelidis, C., et al. (2013). Myeloperoxidase: expressing inflammation and oxidative stress in cardiovascular disease. Curr. Top. Med. Chem. 13, 115–138. doi: 10.2174/1568026611313020004
Armulik, A., Abramsson, A., and Betsholtz, C. (2005). Endothelial/pericyte interactions. Circ. Res. 97, 512–523.
Armulik, A., Genove, G., and Betsholtz, C. (2011). Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 21, 193–215. doi: 10.1016/j.devcel.2011.07.001
Armulik, A., Genove, G., Mae, M., Nisancioglu, M. H., Wallgard, E., Niaudet, C., et al. (2010). Pericytes regulate the blood-brain barrier. Nature 468, 557–561.
Atochin, D. N., and Huang, P. L. (2010). Endothelial nitric oxide synthase transgenic models of endothelial dysfunction. Pflugers. Arch. 460, 965–974. doi: 10.1007/s00424-010-0867-4
Az-Ma, T., Fujii, K., and Yuge, O. (1996). Self-limiting enhancement by nitric oxide of oxygen free radical-induced endothelial cell injury: evidence against the dual action of NO as hydroxyl radical donor/scavenger. Br. J. Pharmacol. 119, 455–462. doi: 10.1111/j.1476-5381.1996.tb15694.x
Baldus, S., Heeschen, C., Meinertz, T., Zeiher, A. M., Eiserich, J. P., Munzel, T., et al. (2003). Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation 108, 1440–1445. doi: 10.1161/01.CIR.0000090690.67322.51
Baluk, P., Bertrand, C., Geppetti, P., Mcdonald, D. M., and Nadel, J. A. (1995). NK1 receptors mediate leukocyte adhesion in neurogenic inflammation in the rat trachea. Am. J. Physiol. 268, L263–L269. doi: 10.1152/ajplung.1995.268.2.L263
Baluk, P., Bolton, P., Hirata, A., Thurston, G., and Mcdonald, D. M. (1998). Endothelial gaps and adherent leukocytes in allergen-induced early- and late-phase plasma leakage in rat airways. Am. J. Pathol. 152, 1463–1476.
Baluk, P., Hirata, A., Thurston, G., Fujiwara, T., Neal, C. R., Michel, C. C., et al. (1997). Endothelial gaps: time course of formation and closure in inflamed venules of rats. Am. J. Physiol. 272, L155–L170. doi: 10.1152/ajplung.1997.272.1.L155
Barnard, M. L., and Matalon, S. (1992). Mechanisms of extracellular reactive oxygen species injury to the pulmonary microvasculature. J. Appl. Physiol. 72, 1724–1729. doi: 10.1152/jappl.1992.72.5.1724
Barrett, T. J., and Hawkins, C. L. (2012). Hypothiocyanous acid: benign or deadly? Chem. Res. Toxicol. 25, 263–273. doi: 10.1021/tx200219s
Beckman, J. S., Beckman, T. W., Chen, J., Marshall, P. A., and Freeman, B. A. (1990). Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 87, 1620–1624. doi: 10.1073/pnas.87.4.1620
Beckman, J. S., and Koppenol, W. H. (1996). Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am. J. Physiol. 271, C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424
Bergers, G., and Song, S. (2005). The role of pericytes in blood-vessel formation and maintenance. Neuro. Oncol. 7, 452–464. doi: 10.1215/S1152851705000232
Berman, R. S., and Martin, W. (1993). Arterial endothelial barrier dysfunction: actions of homocysteine and the hypoxanthine-xanthine oxidase free radical generating system. Br. J. Pharmacol. 108, 920–926. doi: 10.1111/j.1476-5381.1993.tb13487.x
Bernard, G. R. (1991). N-acetylcysteine in experimental and clinical acute lung injury. Am. J. Med. 91, 54S–59S. doi: 10.1016/0002-9343(91)90284-5
Bernard, G. R., Wheeler, A. P., Arons, M. M., Morris, P. E., Paz, H. L., Russell, J. A., et al. (1997). A trial of antioxidants N-acetylcysteine and procysteine in ARDS. The Antioxidant in ARDS Study Group. Chest 112, 164–172. doi: 10.1378/chest.112.1.164
Bloomfield, G. L., Holloway, S., Ridings, P. C., Fisher, B. J., Blocher, C. R., Sholley, M., et al. (1997). Pretreatment with inhaled nitric oxide inhibits neutrophil migration and oxidative activity resulting in attenuated sepsis-induced acute lung injury. Crit. Care Med. 25, 584–593. doi: 10.1097/00003246-199704000-00006
Bonavia, A., Miller, L., Kellum, J. A., and Singbartl, K. (2017). Hemoadsorption corrects hyperresistinemia and restores anti-bacterial neutrophil function. Intensive Care Med. Exp. 5:36. doi: 10.1186/s40635-017-0150-5
Boueiz, A., and Hassoun, P. M. (2009). Regulation of endothelial barrier function by reactive oxygen and nitrogen species. Microvasc. Res. 77, 26–34. doi: 10.1016/j.mvr.2008.10.005
Brennan, M. L., Penn, M. S., Van Lente, F., Nambi, V., Shishehbor, M. H., Aviles, R. J., et al. (2003). Prognostic value of myeloperoxidase in patients with chest pain. N. Engl. J. Med. 349, 1595–1604.
Cai, H. (2005a). Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences. Cardiovasc. Res. 68, 26–36. doi: 10.1016/j.cardiores.2005.06.021
Cai, H. (2005b). NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ. Res. 96, 818–822. doi: 10.1161/01.RES.0000163631.07205.fb
Cai, H., and Harrison, D. G. (2000). Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ. Res. 87, 840–844. doi: 10.1161/01.res.87.10.840
Cai, H., Li, Z., Davis, M. E., Kanner, W., and Harrison, D. G. (2003). Akt-dependent phosphorylation of serine 1179 and mitogen-activated protein kinase kinase/extracellular signal-regulated kinase 1/2 cooperatively mediate activation of the endothelial nitric-oxide synthase by hydrogen peroxide. Mol. Pharmacol. 63, 325–331. doi: 10.1124/mol.63.2.325
Carden, D. L., Smith, J. K., and Korthuis, R. J. (1990). Neutrophil-mediated microvascular dysfunction in postischemic canine skeletal muscle. Role of granulocyte adherence. Circ. Res. 66, 1436–1444. doi: 10.1161/01.res.66.5.1436
Chan, J. Y., and Kwong, M. (2000). Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochim. Biophys. Acta 1517, 19–26. doi: 10.1016/s0167-4781(00)00238-4
Chan, K., Han, X. D., and Kan, Y. W. (2001). An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc. Natl. Acad. Sci. U.S.A. 98, 4611–4616. doi: 10.1073/pnas.081082098
Chanas, S. A., Jiang, Q., Mcmahon, M., Mcwalter, G. K., Mclellan, L. I., Elcombe, C. R., et al. (2002). Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem. J. 365, 405–416. doi: 10.1042/BJ20020320
Chen, B., Lu, Y., Chen, Y., and Cheng, J. (2015). The role of Nrf2 in oxidative stress-induced endothelial injuries. J. Endocrinol. 225, R83–R99. doi: 10.1530/JOE-14-0662
Clancy, R. M., Leszczynska-Piziak, J., and Abramson, S. B. (1992). Nitric oxide, an endothelial cell relaxation factor, inhibits neutrophil superoxide anion production via a direct action on the NADPH oxidase. J. Clin. Invest. 90, 1116–1121. doi: 10.1172/JCI115929
Cohen, G., Ilic, D., Raupachova, J., and Horl, W. H. (2008). Resistin inhibits essential functions of polymorphonuclear leukocytes. J. Immunol. 181, 3761–3768. doi: 10.4049/jimmunol.181.6.3761
Condliffe, A. M., Hawkins, P. T., Stephens, L. R., Haslett, C., and Chilvers, E. R. (1998a). Priming of human neutrophil superoxide generation by tumour necrosis factor-alpha is signalled by enhanced phosphatidylinositol 3,4,5-trisphosphate but not inositol 1,4,5-trisphosphate accumulation. FEBS Lett. 439, 147–151. doi: 10.1016/s0014-5793(98)01358-1
Condliffe, A. M., Kitchen, E., and Chilvers, E. R. (1998b). Neutrophil priming: pathophysiological consequences and underlying mechanisms. Clin. Sci. 94, 461–471. doi: 10.1042/cs0940461
Cook, N. L., Viola, H. M., Sharov, V. S., Hool, L. C., Schoneich, C., and Davies, M. J. (2012). Myeloperoxidase-derived oxidants inhibit sarco/endoplasmic reticulum Ca2+-ATPase activity and perturb Ca2+ homeostasis in human coronary artery endothelial cells. Free Radic. Biol. Med. 52, 951–961. doi: 10.1016/j.freeradbiomed.2011.12.001
Dahlgren, C., and Karlsson, A. (1999). Respiratory burst in human neutrophils. J. Immunol. Methods 232, 3–14.
Daiber, A. (2010). Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta 1797, 897–906. doi: 10.1016/j.bbabio.2010.01.032
Daiber, A., Di Lisa, F., Oelze, M., Kroller-Schon, S., Steven, S., Schulz, E., et al. (2017). Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 174, 1670–1689. doi: 10.1111/bph.13403
Daneman, R., Zhou, L., Kebede, A. A., and Barres, B. A. (2010). Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468, 562–566. doi: 10.1038/nature09513
Daugherty, A., Dunn, J. L., Rateri, D. L., and Heinecke, J. W. (1994). Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Invest. 94, 437–444. doi: 10.1172/JCI117342
Davies, M. J. (2011). Myeloperoxidase-derived oxidation: mechanisms of biological damage and its prevention. J. Clin. Biochem. Nutr. 48, 8–19. doi: 10.3164/jcbn.11-006FR
Davies, M. J., Hawkins, C. L., Pattison, D. I., and Rees, M. D. (2008). Mammalian heme peroxidases: from molecular mechanisms to health implications. Antioxid. Redox Signal. 10, 1199–1234. doi: 10.1089/ars.2007.1927
Del Maestro, R. F., Bjork, J., and Arfors, K. E. (1981). Increase in microvascular permeability induced by enzymatically generated free radicals. I. In vivo study. Microvasc. Res. 22, 239–254. doi: 10.1016/0026-2862(81)90095-9
Del Maschio, A., Zanetti, A., Corada, M., Rival, Y., Ruco, L., Lampugnani, M. G., et al. (1996). Polymorphonuclear leukocyte adhesion triggers the disorganization of endothelial cell-to-cell adherens junctions. J. Cell Biol. 135, 497–510. doi: 10.1083/jcb.135.2.497
Dente, C. J., Steffes, C. P., Speyer, C., and Tyburski, J. G. (2001). Pericytes augment the capillary barrier in in vitro cocultures. J. Surg. Res. 97, 85–91. doi: 10.1006/jsre.2001.6117
Djordjevic, T., Pogrebniak, A., Belaiba, R. S., Bonello, S., Wotzlaw, C., Acker, H., et al. (2005). The expression of the NADPH oxidase subunit p22phox is regulated by a redox-sensitive pathway in endothelial cells. Free Radic. Biol. Med. 38, 616–630. doi: 10.1016/j.freeradbiomed.2004.09.036
Doan, T. N., Gentry, D. L., Taylor, A. A., and Elliott, S. J. (1994). Hydrogen peroxide activates agonist-sensitive Ca(2+)-flux pathways in canine venous endothelial cells. Biochem. J. 297(Pt 1), 209–215. doi: 10.1042/bj2970209
Dreher, D., and Junod, A. F. (1995). Differential effects of superoxide, hydrogen peroxide, and hydroxyl radical on intracellular calcium in human endothelial cells. J. Cell Physiol. 162, 147–153. doi: 10.1002/jcp.1041620118
Droge, W. (2002). Free radicals in the physiological control of cell function. Physiol. Rev. 82, 47–95. doi: 10.1152/physrev.00018.2001
Edelman, D. A., Jiang, Y., Tyburski, J., Wilson, R. F., and Steffes, C. (2006). Pericytes and their role in microvasculature homeostasis. J. Surg. Res. 135, 305–311.
Eiserich, J. P., Baldus, S., Brennan, M. L., Ma, W., Zhang, C., Tousson, A., et al. (2002). Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science 296, 2391–2394. doi: 10.1126/science.1106830
El-Benna, J., Hurtado-Nedelec, M., Marzaioli, V., Marie, J. C., Gougerot-Pocidalo, M. A., and Dang, P. M. (2016). Priming of the neutrophil respiratory burst: role in host defense and inflammation. Immunol. Rev. 273, 180–193. doi: 10.1111/imr.12447
Eschwege, P., Paradis, V., Conti, M., Holstege, A., Richet, F., Deteve, J., et al. (1999). In situ detection of lipid peroxidation by-products as markers of renal ischemia injuries in rat kidneys. J. Urol. 162, 553–557.
Ferland-McCollough, D., Slater, S., Richard, J., Reni, C., and Mangialardi, G. (2017). Pericytes, an overlooked player in vascular pathobiology. Pharmacol. Ther. 171, 30–42. doi: 10.1016/j.pharmthera.2016.11.008
Fernandez-Hernando, C., Yu, J., Suarez, Y., Rahner, C., Davalos, A., Lasuncion, M. A., et al. (2009). Genetic evidence supporting a critical role of endothelial caveolin-1 during the progression of atherosclerosis. Cell Metab. 10, 48–54. doi: 10.1016/j.cmet.2009.06.003
Forstermann, U. (2008). Oxidative stress in vascular disease: causes, defense mechanisms and potential therapies. Nat. Clin. Pract. Cardiovasc. Med. 5, 338–349. doi: 10.1038/ncpcardio1211
Forstermann, U., Xia, N., and Li, H. (2017). Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ. Res. 120, 713–735. doi: 10.1161/CIRCRESAHA.116.309326
Fritzsche, C., Schleicher, U., and Bogdan, C. (2010). Endothelial nitric oxide synthase limits the inflammatory response in mouse cutaneous leishmaniasis. Immunobiology 215, 826–832. doi: 10.1016/j.imbio.2010.05.022
Furchgott, R. F., and Vanhoutte, P. M. (1989). Endothelium-derived relaxing and contracting factors. FASEB J. 3, 2007–2018. doi: 10.1161/01.hyp.19.5.442
Gaengel, K., Genove, G., Armulik, A., and Betsholtz, C. (2009). Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler. Thromb. Vasc. Biol. 29, 630–638. doi: 10.1161/ATVBAHA.107.161521
Gao, F., Lucke-Wold, B. P., Li, X., Logsdon, A. F., Xu, L. C., Xu, S., et al. (2017). Reduction of endothelial nitric oxide increases the adhesiveness of constitutive endothelial membrane ICAM-1 through src-mediated phosphorylation. Front. Physiol. 8:1124. doi: 10.3389/fphys.2017.01124
Gerhardt, H., and Betsholtz, C. (2003). Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 314, 15–23.
Ghanim, H., Sia, C. L., Korzeniewski, K., Lohano, T., Abuaysheh, S., Marumganti, A., et al. (2011). A resveratrol and polyphenol preparation suppresses oxidative and inflammatory stress response to a high-fat, high-carbohydrate meal. J. Clin. Endocrinol. Metab. 96, 1409–1414. doi: 10.1210/jc.2010-1812
Granger, D. N., and Kvietys, P. R. (2015). Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. 6, 524–551. doi: 10.1016/j.redox.2015.08.020
Gupta, M. P., Ober, M. D., Patterson, C., Al-Hassani, M., Natarajan, V., and Hart, C. M. (2001). Nitric oxide attenuates H(2)O(2)-induced endothelial barrier dysfunction: mechanisms of protection. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L116–L126. doi: 10.1152/ajplung.2001.280.1.L116
Gupta, M. P., Steinberg, H. O., and Hart, C. M. (1998). H2O2 causes endothelial barrier dysfunction without disrupting the arginine-nitric oxide pathway. Am. J. Physiol. 274, L508–L516. doi: 10.1152/ajplung.1998.274.4.L508
Hampton, M. B., Kettle, A. J., and Winterbourn, C. C. (1998). Inside the neutrophil phagosome: oxidants, myeloperoxidase, and bacterial killing. Blood 92, 3007–3017.
Harris, N. R., Benoit, J. N., and Granger, D. N. (1993). Capillary filtration during acute inflammation: role of adherent neutrophils. Am. J. Physiol. 265, H1623–H1628. doi: 10.1152/ajpheart.1993.265.5.H1623
Harrison, D., Griendling, K. K., Landmesser, U., Hornig, B., and Drexler, H. (2003). Role of oxidative stress in atherosclerosis. Am. J. Cardiol. 91, 7A–11A.
Hartmann, D. A., Underly, R. G., Grant, R. I., Watson, A. N., Lindner, V., and Shih, A. Y. (2015). Pericyte structure and distribution in the cerebral cortex revealed by high-resolution imaging of transgenic mice. Neurophotonics 2:41402. doi: 10.1117/1.NPh.2.4.041402
Hatakeyama, T., Pappas, P. J., Hobson, R. W. II, Boric, M. P., Sessa, W. C., and Duran, W. N. (2006). Endothelial nitric oxide synthase regulates microvascular hyperpermeability in vivo. J. Physiol. 574, 275–281. doi: 10.1113/jphysiol.2006.108175
He, P. (2010). Leucocyte/endothelium interactions and microvessel permeability: coupled or uncoupled? Cardiovasc. Res. 87, 281–290. doi: 10.1093/cvr/cvq140
He, P., Liu, B., and Curry, F. E. (1997). Effect of nitric oxide synthase inhibitors on endothelial [Ca2+]i and microvessel permeability. Am. J. Physiol. 272, H176–H185. doi: 10.1152/ajpheart.1997.272.1.H176
He, P., Zhang, H., Zhu, L., Jiang, Y., and Zhou, X. (2006). Leukocyte-platelet aggregate adhesion and vascular permeability in intact microvessels: role of activated endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 291, H591–H599. doi: 10.1152/ajpheart.01228.2005
Hecquet, C. M., Ahmmed, G. U., Vogel, S. M., and Malik, A. B. (2008). Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ. Res. 102, 347–355. doi: 10.1161/CIRCRESAHA.107.160176
Henricks, P. A., and Nijkamp, F. P. (2001). Reactive oxygen species as mediators in asthma. Pulm. Pharmacol. Ther. 14, 409–420. doi: 10.1006/pupt.2001.0319
Hill, J., Rom, S., Ramirez, S. H., and Persidsky, Y. (2014). Emerging roles of pericytes in the regulation of the neurovascular unit in health and disease. J. Neuroimmun. Pharmacol. 9, 591–605. doi: 10.1007/s11481-014-9557-x
Hurley, J. V. (1964). Acute inflammation: the effect of concurrent leucocytic emigration and increased permeability on particle retention by the vascular wall. Br. J. Exp. Pathol. 45, 627–633.
Ishikawa, M., Cooper, D., Arumugam, T. V., Zhang, J. H., Nanda, A., and Granger, D. N. (2004). Platelet-leukocyte-endothelial cell interactions after middle cerebral artery occlusion and reperfusion. J. Cereb. Blood Flow Metab. 24, 907–915. doi: 10.1097/01.WCB.0000132690.96836.7F
Jin, B. Y., Lin, A. J., Golan, D. E., and Michel, T. (2012). MARCKS protein mediates hydrogen peroxide regulation of endothelial permeability. Proc. Natl. Acad. Sci. U.S.A. 109, 14864–14869. doi: 10.1073/pnas.1204974109
Johnson, A., Phillips, P., Hocking, D., Tsan, M. F., and Ferro, T. (1989). Protein kinase inhibitor prevents pulmonary edema in response to H2O2. Am. J. Physiol. 256, H1012–H1022. doi: 10.1152/ajpheart.1989.256.4.H1012
Kadambi, A., and Skalak, T. C. (2000). Role of leukocytes and tissue-derived oxidants in short-term skeletal muscle ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 278, H435–H443. doi: 10.1152/ajpheart.2000.278.2.H435
Kalogeris, T., Baines, C. P., Krenz, M., and Korthuis, R. J. (2016). Ischemia/Reperfusion. Compr. Physiol. 7, 113–170.
Kavanagh, B. P., Mouchawar, A., Goldsmith, J., and Pearl, R. G. (1994). Effects of inhaled NO and inhibition of endogenous NO synthesis in oxidant-induced acute lung injury. J. Appl. Physiol. 76, 1324–1329. doi: 10.1152/jappl.1994.76.3.1324
Kelley, C., D’amore, P., Hechtman, H. B., and Shepro, D. (1988). Vasoactive hormones and cAMP affect pericyte contraction and stress fibres in vitro. J. Muscle Res. Cell Motil. 9, 184–194. doi: 10.1007/BF01773740
Kennedy, T. P., Rao, N. V., Hopkins, C., Pennington, L., Tolley, E., and Hoidal, J. R. (1989). Role of reactive oxygen species in reperfusion injury of the rabbit lung. J. Clin. Invest. 83, 1326–1335. doi: 10.1172/JCI114019
Knepler, J. L. Jr., Taher, L. N., Gupta, M. P., Patterson, C., and Pavalko, F. (2001). Peroxynitrite causes endothelial cell monolayer barrier dysfunction. Am. J. Physiol. Cell Physiol. 281, C1064–C1075. doi: 10.1152/ajpcell.2001.281.3.C1064
Korthuis, R. J., Grisham, M. B., and Granger, D. N. (1988). Leukocyte depletion attenuates vascular injury in postischemic skeletal muscle. Am. J. Physiol. 254, H823–H827. doi: 10.1152/ajpheart.1988.254.5.H823
Kroller-Schon, S., Steven, S., Kossmann, S., Scholz, A., Daub, S., Oelze, M., et al. (2014). Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid. Redox Signal. 20, 247–266.
Kubes, P., and Granger, D. N. (1992). Nitric oxide modulates microvascular permeability. Am. J. Physiol. 262, H611–H615. doi: 10.1152/ajpheart.1996.271.4.H1702
Kubes, P., Kanwar, S., Niu, X. F., and Gaboury, J. P. (1993). Nitric oxide synthesis inhibition induces leukocyte adhesion via superoxide and mast cells. FASEB J. 7, 1293–1299. doi: 10.1096/fasebj.7.13.8405815
Kubes, P., Suzuki, M., and Granger, D. N. (1990a). Modulation of PAF-induced leukocyte adherence and increased microvascular permeability. Am. J. Physiol. 259, G859–G864. doi: 10.1152/ajpgi.1990.259.5.G859
Kubes, P., Suzuki, M., and Granger, D. N. (1990b). Platelet-activating factor-induced microvascular dysfunction: role of adherent leukocytes. Am. J. Physiol. 258, G158–G163. doi: 10.1152/ajpgi.1990.258.1.G158
Kubes, P., Suzuki, M., and Granger, D. N. (1991). Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc. Natl. Acad. Sci. U.S.A. 88, 4651–4655. doi: 10.1073/pnas.88.11.4651
Kurose, I., Wolf, R., Grisham, M. B., Aw, T. Y., Specian, R. D., and Granger, D. N. (1995a). Microvascular responses to inhibition of nitric oxide production. Role of active oxidants. Circ. Res. 76, 30–39. doi: 10.1161/01.res.76.1.30
Kurose, I., Wolf, R., Grisham, M. B., and Granger, D. N. (1995b). Effects of an endogenous inhibitor of nitric oxide synthesis on postcapillary venules. Am. J. Physiol. 268, H2224–H2231. doi: 10.1152/ajpheart.1995.268.6.H2224
Kvietys, P. R., and Granger, D. N. (2012). Role of reactive oxygen and nitrogen species in the vascular responses to inflammation. Free Radic. Biol. Med. 52, 556–592. doi: 10.1016/j.freeradbiomed.2011.11.002
La Rocca, G., Di Stefano, A., Eleuteri, E., Anzalone, R., Magno, F., Corrao, S., et al. (2009). Oxidative stress induces myeloperoxidase expression in endocardial endothelial cells from patients with chronic heart failure. Basic Res. Cardiol. 104, 307–320. doi: 10.1007/s00395-008-0761-9
Lacy, F., Kailasam, M. T., O’connor, D. T., Schmid-Schonbein, G. W., and Parmer, R. J. (2000). Plasma hydrogen peroxide production in human essential hypertension: role of heredity, gender, and ethnicity. Hypertension 36, 878–884. doi: 10.1161/01.hyp.36.5.878
Landmesser, U., Dikalov, S., Price, S. R., Mccann, L., Fukai, T., Holland, S. M., et al. (2003). Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Invest. 111, 1201–1209. doi: 10.1172/JCI14172
Landmesser, U., Spiekermann, S., Preuss, C., Sorrentino, S., Fischer, D., Manes, C., et al. (2007). Angiotensin II induces endothelial xanthine oxidase activation: role for endothelial dysfunction in patients with coronary disease. Arterioscler. Thromb. Vasc. Biol. 27, 943–948. doi: 10.1161/01.ATV.0000258415.32883.bf
Lau, D., and Baldus, S. (2006). Myeloperoxidase and its contributory role in inflammatory vascular disease. Pharmacol. Ther. 111, 16–26. doi: 10.1016/j.pharmthera.2005.06.023
Lee, K. S., Kim, S. R., Park, S. J., Park, H. S., Min, K. H., Lee, M. H., et al. (2006). Hydrogen peroxide induces vascular permeability via regulation of vascular endothelial growth factor. Am. J. Respir. Cell Mol. Biol. 35, 190–197. doi: 10.1165/rcmb.2005-0482OC
Leff, J. A., Wilke, C. P., Hybertson, B. M., Shanley, P. F., Beehler, C. J., and Repine, J. E. (1993). Postinsult treatment with N-acetyl-L-cysteine decreases IL-1-induced neutrophil influx and lung leak in rats. Am. J. Physiol. 265, L501–L506. doi: 10.1152/ajplung.1993.265.5.L501
Li, H., and Forstermann, U. (2013). Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr. Opin. Pharmacol. 13, 161–167. doi: 10.1016/j.coph.2013.01.006
Li, H., Horke, S., and Forstermann, U. (2014). Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 237, 208–219.
Li, W. G., Miller, F. J. Jr., Zhang, H. J., Spitz, D. R., Oberley, L. W., (2001). H(2)O(2)-induced O(2) production by a non-phagocytic NAD(P)H oxidase causes oxidant injury. J. Biol. Chem. 276, 29251–29256. doi: 10.1074/jbc.M102124200
Lum, H. (2001). Lysophospholipids in the regulation of endothelial barrier function. Am. J. Physiol. Lung Cell. Mol. Physiol. 281, L1335–L1336. doi: 10.1152/ajplung.2001.281.6.L1335
Lum, H., and Roebuck, K. A. (2001). Oxidant stress and endothelial cell dysfunction. Am. J. Physiol. Cell. Physiol. 280, C719–C741. doi: 10.1152/ajpcell.2001.280.4.C719
Lush, C. W., and Kvietys, P. R. (2000). Microvascular dysfunction in sepsis. Microcirculation 7, 83–101.
Ma, Q. (2013). Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 53, 401–426. doi: 10.1002/iub.2066
Madamanchi, N. R., Vendrov, A., and Runge, M. S. (2005). Oxidative stress and vascular disease. Arterioscler. Thromb. Vasc. Biol. 25, 29–38.
Mayhan, W. G. (1994). Nitric oxide accounts for histamine-induced increases in macromolecular extravasation. Am. J. Physiol. 266, H2369–H2373. doi: 10.1152/ajpheart.1994.266.6.H2369
Mcdonald, D. M. (1994). Endothelial gaps and permeability of venules in rat tracheas exposed to inflammatory stimuli. Am. J. Physiol. 266, L61–L83. doi: 10.1152/ajplung.1994.266.1.L61
Mcelroy, M. C., Wiener-Kronish, J. P., Miyazaki, H., Sawa, T., Modelska, K., Dobbs, L. G., et al. (1997). Nitric oxide attenuates lung endothelial injury caused by sublethal hyperoxia in rats. Am. J. Physiol. 272, L631–L638. doi: 10.1152/ajplung.1997.272.4.L631
Mcleish, K. R., Merchant, M. L., Creed, T. M., Tandon, S., Barati, M. T., Uriarte, S. M., et al. (2017). Frontline science: tumor necrosis factor-alpha stimulation and priming of human neutrophil granule exocytosis. J. Leukoc. Biol. 102, 19–29. doi: 10.1189/jlb.3HI0716-293RR
Mcnally, J. S., Davis, M. E., Giddens, D. P., Saha, A., Hwang, J., Dikalov, S., et al. (2003). Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol. Heart Circ. Physiol. 285, H2290–H2297. doi: 10.1152/ajpheart.00515.2003
Mcquaid, K. E., and Keenan, A. K. (1997). Endothelial barrier dysfunction and oxidative stress: roles for nitric oxide? Exp. Physiol. 82, 369–376. doi: 10.1113/expphysiol.1997.sp004032
Mcquaid, K. E., Smyth, E. M., and Keenan, A. K. (1996). Evidence for modulation of hydrogen peroxide-induced endothelial barrier dysfunction by nitric oxide in vitro. Eur. J. Pharmacol. 307, 233–241. doi: 10.1016/0014-2999(96)00271-3
Mcsweeney, S. R., Warabi, E., and Siow, R. C. (2016). Nrf2 as an endothelial mechanosensitive transcription factor: going with the flow. Hypertension 67, 20–29. doi: 10.1161/HYPERTENSIONAHA.115.06146
Meuwese, M. C., Stroes, E. S., Hazen, S. L., Van Miert, J. N., Kuivenhoven, J. A., Schaub, R. G., et al. (2007). Serum myeloperoxidase levels are associated with the future risk of coronary artery disease in apparently healthy individuals: the EPIC-Norfolk prospective population study. J. Am. Coll. Cardiol. 50, 159–165. doi: 10.1016/j.jacc.2007.03.033
Miller, L., Singbartl, K., Chroneos, Z. C., Ruiz-Velasco, V., Lang, C. H., and Bonavia, A. (2019). Resistin directly inhibits bacterial killing in neutrophils. Intensive Care Med. Exp. 7:30. doi: 10.1186/s40635-019-0257-y
Miralda, I., Uriarte, S. M., and Mcleish, K. R. (2017). Multiple phenotypic changes define neutrophil priming. Front. Cell. Infect. Microbiol. 7:217. doi: 10.3389/fphys.2017.0217
Mocatta, T. J., Pilbrow, A. P., Cameron, V. A., Senthilmohan, R., Frampton, C. M., Richards, A. M., et al. (2007). Plasma concentrations of myeloperoxidase predict mortality after myocardial infarction. J. Am. Coll. Cardiol. 49, 1993–2000. doi: 10.1016/j.jacc.2007.02.040
Modig, J., and Sandin, R. (1988). Haematological, physiological and survival data in a porcine model of adult respiratory distress syndrome induced by endotoxaemia. Effects of treatment with N-acetylcysteine. Acta Chir. Scand. 154, 169–177.
Moll, T., Dejana, E., and Vestweber, D. (1998). In vitro degradation of endothelial catenins by a neutrophil protease. J. Cell Biol. 140, 403–407. doi: 10.1083/jcb.140.2.403
Moncada, S., and Higgs, E. A. (2006). The discovery of nitric oxide and its role in vascular biology. Br. J. Pharmacol. 147(Suppl. 1), S193–S201. doi: 10.1038/sj.bjp.0706458
Moncada, S., Palmer, R. M., and Higgs, E. A. (1991). Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 43, 109–142.
Montezano, A. C., and Touyz, R. M. (2012). Reactive oxygen species and endothelial function–role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin. Pharmacol. Toxicol. 110, 87–94. doi: 10.1111/j.1742-7843.2011.00785.x
Motohashi, H., and Yamamoto, M. (2004). Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 10, 549–557. doi: 10.1016/j.molmed.2004.09.003
Mueller, C. F., Laude, K., Mcnally, J. S., and Harrison, D. G. (2005). ATVB in focus: redox mechanisms in blood vessels. Arterioscler. Thromb. Vasc. Biol. 25, 274–278. doi: 10.1161/01.ATV.0000153515.72375.3b
Mundi, S., Massaro, M., Scoditti, E., Carluccio, M. A., Van Hinsbergh, V. W. M., Iruela-Arispe, M. L., et al. (2017). Endothelial permeability, Ldl deposition, and cardiovascular risk factors - a review. Cardiovasc. Res. 114, 35–52. doi: 10.1093/cvr/cvx226
Murfee, W. L., Skalak, T. C., and Peirce, S. M. (2005). Differential arterial/venous expression of NG2 proteoglycan in perivascular cells along microvessels: identifying a venule-specific phenotype. Microcirculation 12, 151–160. doi: 10.1080/10739680590904955
Ndrepepa, G. (2019). Myeloperoxidase - A bridge linking inflammation and oxidative stress with cardiovascular disease. Clin. Chim. Acta 493, 36–51. doi: 10.1016/j.cca.2019.02.022
Neumann, P., Gertzberg, N., Vaughan, E., Weisbrot, J., Woodburn, R., Lambert, W., et al. (2006). Peroxynitrite mediates TNF-alpha-induced endothelial barrier dysfunction and nitration of actin. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L674–L684. doi: 10.1152/ajplung.00391.2005
Nguyen, T., Nioi, P., and Pickett, C. B. (2009). The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 284, 13291–13295. doi: 10.1074/jbc.R900010200
Nicholls, S. J., and Hazen, S. L. (2009). Myeloperoxidase, modified lipoproteins, and atherogenesis. J. Lipid Res. 50(Suppl.), S346–S351. doi: 10.1194/jlr.R800086-JLR200
Niu, X. F., Smith, C. W., and Kubes, P. (1994). Intracellular oxidative stress induced by nitric oxide synthesis inhibition increases endothelial cell adhesion to neutrophils. Circ. Res. 74, 1133–1140. doi: 10.1161/01.res.74.6.1133
Ochoa, L., Waypa, G., Mahoney, J. R. Jr., Rodriguez, L., and Minnear, F. L. (1997). Contrasting effects of hypochlorous acid and hydrogen peroxide on endothelial permeability: prevention with cAMP drugs. Am. J. Respir. Crit. Care Med. 156, 1247–1255. doi: 10.1164/ajrccm.156.4.96-10115
Okayama, N., Grisham, M. B., Kevil, C. G., Eppihimer, L. A., Wink, D. A., and Alexander, J. S. (1999). Effect of reactive oxygen metabolites on endothelial permeability: role of nitric oxide and iron. Microcirculation 6, 107–116.
Okayama, N., Kevil, C. G., Correia, L., Jourd’heuil, D., Itoh, M., Grisham, M. B., et al. (1997). Nitric oxide enhances hydrogen peroxide-mediated endothelial permeability in vitro. Am. J. Physiol. 273, C1581–C1587. doi: 10.1152/ajpcell.1997.273.5.C1581
Pacher, P., Beckman, J. S., and Liaudet, L. (2007). Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 87, 315–424. doi: 10.1152/physrev.00029.2006
Parks, D. A., Shah, A. K., and Granger, D. N. (1984). Oxygen radicals: effects on intestinal vascular permeability. Am. J. Physiol. 247, G167–G170. doi: 10.1152/ajpgi.1984.247.2.G167
Parsons, P. E., Worthen, G. S., Moore, E. E., Tate, R. M., and Henson, P. M. (1989). The association of circulating endotoxin with the development of the adult respiratory distress syndrome. Am. Rev. Respir. Dis. 140, 294–301. doi: 10.1164/ajrccm/140.2.294
Pergola, P. E., Raskin, P., Toto, R. D., Meyer, C. J., Huff, J. W., Grossman, E. B., et al. (2011). Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 365, 327–336. doi: 10.1056/NEJMoa1105351
Pinsky, M. R., Vincent, J.-L., Deviere, J., Alegre, M., Kahn, R. J., and Dupont, E. (1993). Serum cytokine levels in human septic shock. Relation to multiple-system organ failure and mortality. Chest 103, 565–575. doi: 10.1378/chest.103.2.565
Ponnuswamy, P., Schrottle, A., Ostermeier, E., Gruner, S., Huang, P. L., Ertl, G., et al. (2012). eNOS protects from atherosclerosis despite relevant superoxide production by the enzyme in apoE mice. PLoS One 7:e30193. doi: 10.1371/journal.pone.0030193
Poss, W. B., Timmons, O. D., Farrukh, I. S., Hoidal, J. R., and Michael, J. R. (1995). Inhaled nitric oxide prevents the increase in pulmonary vascular permeability caused by hydrogen peroxide. J. Appl. Physiol. 79, 886–891. doi: 10.1152/jappl.1995.79.3.886
Potenza, M. A., Gagliardi, S., Nacci, C., Carratu, M. R., and Montagnani, M. (2009). Endothelial dysfunction in diabetes: from mechanisms to therapeutic targets. Curr. Med. Chem. 16, 94–112. doi: 10.2174/092986709787002853
Potera, R. M., Jensen, M. J., Hilkin, B. M., South, G. K., Hook, J. S., Gross, E. A., et al. (2016). Neutrophil azurophilic granule exocytosis is primed by TNF-alpha and partially regulated by NADPH oxidase. Innate Immun. 22, 635–646. doi: 10.1177/1753425916668980
Priestley, J. R., Kautenburg, K. E., Casati, M. C., Endres, B. T., Geurts, A. M., and Lombard, J. H. (2016). The NRF2 knockout rat: a new animal model to study endothelial dysfunction, oxidant stress, and microvascular rarefaction. Am. J. Physiol. Heart Circ. Physiol. 310, H478–H487. doi: 10.1152/ajpheart.00586.2015
Radi, R. (2013). Protein tyrosine nitration: biochemical mechanisms and structural basis of functional effects. Acc. Chem. Res. 46, 550–559. doi: 10.1021/ar300234c
Radi, R., Beckman, J. S., Bush, K. M., and Freeman, B. A. (1991). Peroxynitrite oxidation of sulfhydryls. The cytotoxic potential of superoxide and nitric oxide. J. Biol. Chem. 266, 4244–4250.
Radomski, M. W., Palmer, R. M., and Moncada, S. (1990a). An L-arginine/nitric oxide pathway present in human platelets regulates aggregation. Proc. Natl. Acad. Sci. U.S.A. 87, 5193–5197. doi: 10.1073/pnas.87.13.5193
Radomski, M. W., Palmer, R. M., and Moncada, S. (1990b). Characterization of the L-arginine:nitric oxide pathway in human platelets. Br. J. Pharmacol. 101, 325–328. doi: 10.1111/j.1476-5381.1990.tb12709.x
Rhee, S. G. (1999). Redox signaling: hydrogen peroxide as intracellular messenger. Exp. Mol. Med. 31, 53–59. doi: 10.1038/emm.1999.9
Riobo, N. A., Clementi, E., Melani, M., Boveris, A., Cadenas, E., Moncada, S., et al. (2001). Nitric oxide inhibits mitochondrial NADH:ubiquinone reductase activity through peroxynitrite formation. Biochem. J. 359, 139–145. doi: 10.1042/0264-6021:3590139
Rucker, H. K., Wynder, H. J., and Thomas, W. E. (2000). Cellular mechanisms of CNS pericytes. Brain Res. Bull. 51, 363–369.
Sagare, A. P., Bell, R. D., Zhao, Z., Ma, Q., Winkler, E. A., Ramanathan, A., et al. (2013). Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 4:2932. doi: 10.1038/ncomms3932
Salvemini, D., Wang, Z. Q., Stern, M. K., Currie, M. G., and Misko, T. P. (1998). Peroxynitrite decomposition catalysts: therapeutics for peroxynitrite-mediated pathology. Proc. Natl. Acad. Sci. U.S.A. 95, 2659–2663. doi: 10.1073/pnas.95.5.2659
Sanchez, F. A., Rana, R., Kim, D. D., Iwahashi, T., Zheng, R. F., Lal, B. K., et al. (2009). Internalization of eNOS and NO delivery to subcellular targets determine agonist-induced hyperpermeability. Proc. Natl. Acad. Sci. U.S.A. 106, 6849–6853. doi: 10.1073/pnas.0812694106
Sasaki, M., Bharwani, S., Jordan, P., Elrod, J. W., Grisham, M. B., Jackson, T. H., et al. (2003). Increased disease activity in eNOS-deficient mice in experimental colitis. Free Radic. Biol. Med. 35, 1679–1687. doi: 10.1016/j.freeradbiomed.2003.09.016
Schindhelm, R. K., Van Der Zwan, L. P., Teerlink, T., and Scheffer, P. G. (2009). Myeloperoxidase: a useful biomarker for cardiovascular disease risk stratification? Clin. Chem. 55, 1462–1470. doi: 10.1373/clinchem.2009.126029
Schultz, J., and Kaminker, K. (1962). Myeloperoxidase of the leucocyte of normal human blood. I. Content and localization. Arch. Biochem. Biophys. 96, 465–467. doi: 10.1016/0003-9861(62)90321-1
Seeger, W., Hansen, T., Rossig, R., Schmehl, T., Schutte, H., Kramer, H. J., et al. (1995). Hydrogen peroxide-induced increase in lung endothelial and epithelial permeability–effect of adenylate cyclase stimulation and phosphodiesterase inhibition. Microvasc. Res. 50, 1–17. doi: 10.1006/mvre.1995.1033
Sengillo, J. D., Winkler, E. A., Walker, C. T., Sullivan, J. S., Johnson, M., and Zlokovic, B. V. (2013). Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 23, 303–310. doi: 10.1111/bpa.12004
Sharma, A., Rizky, L., Stefanovic, N., Tate, M., Ritchie, R. H., Ward, K. W., et al. (2017). The nuclear factor (erythroid-derived 2)-like 2 (Nrf2) activator dh404 protects against diabetes-induced endothelial dysfunction. Cardiovasc. Diabetol. 16:33. doi: 10.1186/s12933-017-0513-y
Shasby, D. M., Lind, S. E., Shasby, S. S., Goldsmith, J. C., and Hunninghake, G. W. (1985). Reversible oxidant-induced increases in albumin transfer across cultured endothelium: alterations in cell shape and calcium homeostasis. Blood 65, 605–614.
Shih, A. Y., Li, P., and Murphy, T. H. (2005). A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J. Neurosci. 25, 10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005
Shojaee, N., Patton, W. F., Hechtman, H. B., and Shepro, D. (1999). Myosin translocation in retinal pericytes during free-radical induced apoptosis. J. Cell Biochem. 75, 118–129.
Siflinger-Birnboim, A., Goligorsky, M. S., Del Vecchio, P. J., and Malik, A. B. (1992). Activation of protein kinase C pathway contributes to hydrogen peroxide-induced increase in endothelial permeability. Lab. Invest. 67, 24–30.
Siflinger-Birnboim, A., Lum, H., Del Vecchio, P. J., and Malik, A. B. (1996). Involvement of Ca2+ in the H2O2-induced increase in endothelial permeability. Am. J. Physiol. 270, L973–L978. doi: 10.1152/ajplung.1996.270.6.L973
Sims, D. E., Miller, F. N., Donald, A., and Perricone, M. A. (1990). Ultrastructure of pericytes in early stages of histamine-induced inflammation. J. Morphol. 206, 333–342. doi: 10.1002/jmor.1052060310
Stapor, P. C., Sweat, R. S., Dashti, D. C., Betancourt, A. M., and Murfee, W. L. (2014). Pericyte dynamics during angiogenesis: new insights from new identities. J. Vasc. Res. 51, 163–174. doi: 10.1159/000362276
Stocker, R., and Keaney, J. F. (2004). Role of oxidative modifications in atherosclerosis. Physiol. Rev. 84, 1381–1478.
Stratman, A. N., and Davis, G. E. (2012). Endothelial cell-pericyte interactions stimulate basement membrane matrix assembly: influence on vascular tube remodeling, maturation, and stabilization. Microsc. Microanal. 18, 68–80. doi: 10.1017/S1431927611012402
Suematsu, M., Schmid-Schonbein, G. W., Chavez-Chavez, R. H., Yee, T. T., Tamatani, T., Miyasaka, M., et al. (1993). In vivo visualization of oxidative changes in microvessels during neutophil activation. Am. J. Physiol. 264, H881–H891. doi: 10.1152/ajpheart.1993.264.3.H881
Sugiyama, S., Kugiyama, K., Aikawa, M., Nakamura, S., Ogawa, H., and Libby, P. (2004). Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arterioscler. Thromb. Vasc. Biol. 24, 1309–1314. doi: 10.1161/01.ATV.0000131784.50633.4f
Suresh, K., Servinsky, L., Reyes, J., Baksh, S., Undem, C., Caterina, M., et al. (2015). Hydrogen peroxide-induced calcium influx in lung microvascular endothelial cells involves TRPV4. Am. J. Physiol. Lung Cell. Mol. Physiol. 309, L1467–L1477. doi: 10.1152/ajplung.00275.2015
Suzuki, Y. J., Forman, H. J., and Sevanian, A. (1997). Oxidants as stimulators of signal transduction. Free Radic. Biol. Med. 22, 269–285. doi: 10.1016/s0891-5849(96)00275-4
Szabo, C., Cuzzocrea, S., Zingarelli, B., O’connor, M., and Salzman, A. L. (1997). Endothelial dysfunction in a rat model of endotoxic shock. Importance of the activation of poly (ADP-ribose) synthetase by peroxynitrite. J. Clin. Invest. 100, 723–735. doi: 10.1172/JCI119585
Szabo, C., Ischiropoulos, H., and Radi, R. (2007). Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 6, 662–680. doi: 10.1038/nrd2222
Tailor, A., and Granger, D. N. (2003). Hypercholesterolemia promotes P-selectin-dependent platelet-endothelial cell adhesion in postcapillary venules. Arterioscler. Thromb. Vasc. Biol. 23, 675–680. doi: 10.1161/01.ATV.0000056742.97580.79
Tarantini, S., Valcarcel-Ares, M. N., Yabluchanskiy, A., Tucsek, Z., Hertelendy, P., Kiss, T., et al. (2018). Nrf2 deficiency exacerbates obesity-induced oxidative stress, neurovascular dysfunction, blood-brain barrier disruption, neuroinflammation, amyloidogenic gene expression, and cognitive decline in mice, mimicking the aging phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 73, 853–863. doi: 10.1093/gerona/glx177
Tatsumi, T., and Fliss, H. (1994). Hypochlorous acid and chloramines increase endothelial permeability: possible involvement of cellular zinc. Am. J. Physiol. 267, H1597–H1607. doi: 10.1152/ajpheart.1994.267.4.H1597
Thannickal, V. J., and Fanburg, B. L. (2000). Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 279, L1005–L1028.
Tian, R., Ding, Y., Peng, Y. Y., and Lu, N. (2017). Myeloperoxidase amplified high glucose-induced endothelial dysfunction in vasculature: role of NADPH oxidase and hypochlorous acid. Biochem. Biophys. Res. Commun. 484, 572–578. doi: 10.1016/j.bbrc.2017.01.132
Tinsley, J. H., Ustinova, E. E., Xu, W., S. Y., and Yuan, S. Y. (2002). Src-dependent, neutrophil-mediated vascular hyperpermeability and β-catenin modification. Am. J. Physiol. Cell. Physiol. 283, C1745–C1751. doi: 10.1152/ajpcell.00230.2002
Tiyerili, V., Camara, B., Becher, M. U., Schrickel, J. W., Lutjohann, D., Mollenhauer, M., et al. (2016). Neutrophil-derived myeloperoxidase promotes atherogenesis and neointima formation in mice. Int. J. Cardiol. 204, 29–36. doi: 10.1016/j.ijcard.2015.11.128
Ungvari, Z., Bailey-Downs, L., Gautam, T., Jimenez, R., Losonczy, G., Zhang, C., et al. (2011a). Adaptive induction of NF-E2-related factor-2-driven antioxidant genes in endothelial cells in response to hyperglycemia. Am. J. Physiol. Heart Circ. Physiol. 300, H1133–H1140. doi: 10.1152/ajpheart.00402.2010
Ungvari, Z., Bailey-Downs, L., Sosnowska, D., Gautam, T., Koncz, P., Losonczy, G., et al. (2011b). Vascular oxidative stress in aging: a homeostatic failure due to dysregulation of NRF2-mediated antioxidant response. Am. J. Physiol. Heart Circ. Physiol. 301, H363–H372. doi: 10.1152/ajpheart.01134.2010
Usatyuk, P. V., and Natarajan, V. (2004). Role of mitogen-activated protein kinases in 4-hydroxy-2-nonenal-induced actin remodeling and barrier function in endothelial cells. J. Biol. Chem. 279, 11789–11797. doi: 10.1074/jbc.M311184200
Valko, M., Leibfritz, D., Moncol, J., Cronin, M. T., Mazur, M., and Telser, J. (2007). Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 39, 44–84. doi: 10.1016/j.biocel.2006.07.001
van Dalen, C. J., Whitehouse, M. W., Winterbourn, C. C., and Kettle, A. J. (1997). Thiocyanate and chloride as competing substrates for myeloperoxidase. Biochem. J. 327(Pt 2), 487–492. doi: 10.1042/bj3270487
van der Veen, B. S., De Winther, M. P., and Heeringa, P. (2009). Myeloperoxidase: molecular mechanisms of action and their relevance to human health and disease. Antioxid. Redox Signal. 11, 2899–2937. doi: 10.1089/ars.2009.2538
van Dijk, C. G., Nieuweboer, F. E., Pei, J. Y., Xu, Y. J., Burgisser, P., Van Mulligen, E., et al. (2015). The complex mural cell: pericyte function in health and disease. Int. J. Cardiol. 190, 75–89. doi: 10.1016/j.ijcard.2015.03.258
van Leeuwen, H. J., Van Der Tol, M., Van Strijp, J. A., Verhoef, J., and Van Kessel, K. P. (2005). The role of tumour necrosis factor in the kinetics of lipopolysaccharide-mediated neutrophil priming in whole blood. Clin. Exp. Immunol. 140, 65–72. doi: 10.1111/j.1365-2249.2005.02748.x
van Leeuwen, M., Gijbels, M. J., Duijvestijn, A., Smook, M., Van De Gaar, M. J., Heeringa, P., et al. (2008). Accumulation of myeloperoxidase-positive neutrophils in atherosclerotic lesions in LDLR-/- mice. Arterioscler. Thromb. Vasc. Biol. 28, 84–89. doi: 10.1161/ATVBAHA.107.154807
Vepa, S., Scribner, W. M., and Natarajan, V. (1997). Activation of protein phosphorylation by oxidants in vascular endothelial cells: identification of tyrosine phosphorylation of caveolin. Free Radic. Biol. Med. 22, 25–35. doi: 10.1016/s0891-5849(96)00241-9
Volk, T., Hensel, M., and Kox, W. J. (1997). Transient Ca2+ changes in endothelial cells induced by low doses of reactive oxygen species: role of hydrogen peroxide. Mol. Cell Biochem. 171, 11–21. doi: 10.1023/a:1006886215193
von Tell, D., Armulik, A., and Betsholtz, C. (2006). Pericytes and vascular stability. Exp. Cell Res. 312, 623–629.
Warmke, N., Griffin, K. J., and Cubbon, R. M. (2016). Pericytes in diabetes-associated vascular disease. J. Diabetes Complicat. 30, 1643–1650.
Wenzel, P., Mollnau, H., Oelze, M., Schulz, E., Wickramanayake, J. M., Muller, J., et al. (2008). First evidence for a crosstalk between mitochondrial and NADPH oxidase-derived reactive oxygen species in nitroglycerin-triggered vascular dysfunction. Antioxid. Redox Signal 10, 1435–1447. doi: 10.1089/ars.2007.1969
Wink, D. A., Cook, J. A., Pacelli, R., Liebmann, J., Krishna, M. C., and Mitchell, J. B. (1995). Nitric oxide (NO) protects against cellular damage by reactive oxygen species. Toxicol. Lett. 82-83, 221–226. doi: 10.1016/0378-4274(95)03557-5
Wong, N. D., Gransar, H., Narula, J., Shaw, L., Moon, J. H., Miranda-Peats, R., et al. (2009). Myeloperoxidase, subclinical atherosclerosis, and cardiovascular disease events. JACC Cardiovasc. Imaging 2, 1093–1099. doi: 10.1016/j.jcmg.2009.05.012
Wu, F., and Wilson, J. X. (2009). Peroxynitrite-dependent activation of protein phosphatase type 2A mediates microvascular endothelial barrier dysfunction. Cardiovasc. Res. 81, 38–45. doi: 10.1093/cvr/cvn246
Wu, H. M., Huang, Q., Yuan, Y., and Granger, H. J. (1996). VEGF induces NO-dependent hyperpermeability in coronary venules. Am. J. Physiol. 271, H2735–H2739.
Xia, X., Lapenna, K. B., Zhang, Y., and He, P. (2018). Nrf2 deficiency exacerbates oxidative stress and microvessel susceptibility to inflammation in diabetic rats. FASEB J. 32, 708–706.
Xia, Y., Dawson, V. L., Dawson, T. M., Snyder, S. H., and Zweier, J. L. (1996). Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. Proc. Natl. Acad. Sci. U.S.A. 93, 6770–6774. doi: 10.1073/pnas.93.13.6770
Xu, S., Zhou, X., Yuan, D., Xu, Y., and He, P. (2013). Caveolin-1 scaffolding domain promotes leukocyte adhesion by reduced basal endothelial nitric oxide-mediated ICAM-1 phosphorylation in rat mesenteric venules. Am. J. Physiol. Heart Circ. Physiol. 305, H1484–H1493. doi: 10.1152/ajpheart.00382.2013
Yang, J., Cheng, Y., Ji, R., and Zhang, C. (2006). Novel model of inflammatory neointima formation reveals a potential role of myeloperoxidase in neointimal hyperplasia. Am. J. Physiol. Heart Circ. Physiol. 291, H3087–H3093. doi: 10.1152/ajpheart.00412.2006
Yang, Z., Zhang, A., Altura, B. T., and Altura, B. M. (1999). Hydrogen peroxide-induced endothelium-dependent relaxation of rat aorta involvement of Ca2+ and other cellular metabolites. Gen. Pharmacol. 33, 325–336. doi: 10.1016/s0306-3623(99)00019-1
Yu, H., Wang, M., Wang, D., Kalogeris, T. J., Mchowat, J., Ford, D. A., et al. (2019). Chlorinated Lipids Elicit Inflammatory Responses in vitro and in vivo. Shock 51, 114–122. doi: 10.1097/SHK.0000000000001112
Yuan, D., and He, P. (2012). Vascular remodeling alters adhesion protein and cytoskeleton reactions to inflammatory stimuli resulting in enhanced permeability increases in rat venules. J. Appl. Physiol. 113, 1110–1120. doi: 10.1152/japplphysiol.00102.2012
Yuan, Y., Granger, H. J., Zawieja, D. C., Defily, D. V., and Chilian, W. M. (1993). Histamine increases venular permeability via a phospholipase C-NO synthase-guanylate cyclase cascade. Am. J. Physiol. 264, H1734–H1739. doi: 10.1152/ajpheart.1993.264.5.H1734
Zeng, M., Zhang, H., Lowell, C., and He, P. (2002). Tumor necrosis factor-alpha-induced leukocyte adhesion and microvessel permeability. Am. J. Physiol. Heart Circ. Physiol. 283, H2420–H2430. doi: 10.1152/ajpheart.00787.2001
Zhang, C., Patel, R., Eiserich, J. P., Zhou, F., Kelpke, S., Ma, W., et al. (2001). Endothelial dysfunction is induced by proinflammatory oxidant hypochlorous acid. Am. J. Physiol. Heart Circ. Physiol. 281, H1469–H1475. doi: 10.1152/ajpheart.2001.281.4.H1469
Zhang, R., Brennan, M. L., Fu, X., Aviles, R. J., Pearce, G. L., Penn, M. S., et al. (2001). Association between myeloperoxidase levels and risk of coronary artery disease. JAMA 286, 2136–2142. doi: 10.1001/jama.286.17.2136
Zhao, J., Moore, A. N., Redell, J. B., and Dash, P. K. (2007). Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J. Neurosci. 27, 10240–10248. doi: 10.1523/JNEUROSCI.1683-07.2007
Zhou, X., and He, P. (2010). Endothelial [Ca2+]i and caveolin-1 antagonistically regulate eNOS activity and microvessel permeability in rat venules. Cardiovasc. Res. 87, 340–347. doi: 10.1093/cvr/cvq006
Zhou, X., Qian, Y., Yuan, D., Feng, Q., and He, P. (2019). H2O2-induced microvessel barrier dysfunction: the interplay between reactive oxygen species, nitric oxide, and peroxynitrite. Physiol. Rep. 7:e14206. doi: 10.14814/phy2.14206
Zhou, X., Wen, K., Yuan, D., Ai, L., and He, P. (2009). Calcium influx-dependent differential actions of superoxide and hydrogen peroxide on microvessel permeability. Am. J. Physiol. Heart Circ. Physiol. 296, H1096–H1107. doi: 10.1152/ajpheart.01037.2008
Zhou, X., Yuan, D., Wang, M., and He, P. (2013). H2O2-induced endothelial NO production contributes to vascular cell apoptosis and increased permeability in rat venules. Am. J. Physiol. Heart Circ. Physiol. 304, H82–H93. doi: 10.1152/ajpheart.00300.2012
Zhou, X. P., and He, P. N. (2011). Temporal and spatial correlation of platelet-activating factor-induced increases in endothelial [Ca2+](i), nitric oxide, and gap formation in intact venules. Am. J. Physiol. Heart Circ. Physiol. 301, H1788–H1797. doi: 10.1152/ajpheart.00599.2011
Zhu, L., Castranova, V., and He, P. (2005). fMLP-stimulated neutrophils increase endothelial [Ca2+]i and microvessel permeability in the absence of adhesion: role of reactive oxygen species. Am. J. Physiol. Heart Circ. Physiol. 288, H1331–H1338. doi: 10.1152/ajpheart.00802.2004
Zhu, L., and He, P. (2005). Platelet-activating factor increases endothelial [Ca2+]i and NO production in individually perfused intact microvessels. Am. J. Physiol. Heart Circ. Physiol. 288, H2869–H2877. doi: 10.1152/ajpheart.01080.2004
Zhu, L., and He, P. (2006). fMLP-stimulated release of reactive oxygen species from adherent leukocytes increases microvessel permeability. Am. J. Physiol. Heart Circ. Physiol. 290, H365–H372. doi: 10.1152/ajpheart.00812.2005
Zinkevich, N. S., and Gutterman, D. D. (2011). ROS-induced ROS release in vascular biology: redox-redox signaling. Am. J. Physiol. Heart Circ. Physiol. 301, H647–H653. doi: 10.1152/ajpheart.01271.2010
Zou, M. H., Hou, X. Y., Shi, C. M., Nagata, D., Walsh, K., and Cohen, R. A. (2002). Modulation by peroxynitrite of Akt- and AMP-activated kinase-dependent Ser1179 phosphorylation of endothelial nitric oxide synthase. J. Biol. Chem. 277, 32552–32557. doi: 10.1074/jbc.M204512200
Keywords: reactive oxygen species, reactive nitrogen species, microvessel permeability, pericyte, Nrf-2
Citation: He P, Talukder MAH and Gao F (2020) Oxidative Stress and Microvessel Barrier Dysfunction. Front. Physiol. 11:472. doi: 10.3389/fphys.2020.00472
Received: 13 October 2019; Accepted: 17 April 2020;
Published: 27 May 2020.
Edited by:
Santosh Kumar, The University of Iowa, United StatesReviewed by:
Marcel Bonay, INSERM U1179 Handicap Neuromusculaire: Physiopathologie, Biothérapie et Pharmacologie Appliquées (END-ICAP), FranceStefania Marzocco, University of Salerno, Italy
Jingyan Han, Boston University, United States
Copyright © 2020 He, Talukder and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pingnian He, cGluZ2hlQHBlbm5zdGF0ZWhlYWx0aC5wc3UuZWR1