 Xuyuan Gao1,2,3
Xuyuan Gao1,2,3 Zhenya Tian2
Zhenya Tian2 Yan Zhang2Guangmei Chen2
Yan Zhang2Guangmei Chen2 Chao Ma2Zhenqi Tian2Shaowei Cui2Yongyue Lu1*Zhongshi Zhou2*
Chao Ma2Zhenqi Tian2Shaowei Cui2Yongyue Lu1*Zhongshi Zhou2*- 1College of Agriculture, South China Agricultural University, Guangzhou, China
- 2State Key Laboratory for Biology of Plant Diseases and Insect Pests, Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing, China
- 3Guangxi Key Laboratory of Biology for Crop Diseases and Insect Pests, Institute of Plant Protection, Guangxi Academy of Agricultural Sciences, Nanning, China
Increase in atmospheric CO2 directly affects the insect physiology and behavior, and indirectly affects the herbivorous insects by affecting their hosts. The increase in atmospheric CO2 is accompanied by an increase in temperature and heat waves. Ophraella communa LeSage is a natural enemy of Ambrosia artemisiifolia (common ragweed). The development and reproduction of this beetle is weakened upon eating common ragweed grown under stress conditions. As female behavior and physiology alter after mating, the reproductive tract of males is likely to modulate reproduction and development in this species. Herein, the transcriptional profiles of testes and accessory glands from male O. communa individuals feeding on common ragweed under conditions of high CO2 concentration and heat waves and that grown under ambient CO2 concentration were compared. Differentially expressed genes (DEGs) were identified between the same tissues from beetles fed on common ragweed grown under different stress conditions. There were 3, 2, 3, 1and 5 genes related to decomposition and transport of macromolecular substances, host location, stress response, reproduction, and poisonous food-utilization. No expected response was observed in the male reproductive tract, but some of the identified DEGs might control the development of the population. The results presented here should be helpful in guiding future studies on deciphering the indirect response of other organs to high CO2 concentration and heat waves, as well as the functions of seminal fluid proteins in O. communa.
Introduction
Global warming is increasing and has been one of the greatest challenges facing all organisms (Parmesan and Yohe, 2003). According to a special report of the Intergovernmental Panel on Climate Change (IPCC), the global average surface temperature has risen by 1.5°C above that in the pre-industrial era and is expected to keep on increasing for decades or centuries (Bongaarts, 2018). The warming climate has had a significant effect on organisms as well as on the ecosystems (Hughes, 2000; Parmesan and Yohe, 2003; Zvereva and Kozlov, 2006). In the future, extreme weather events are expected to increase further, heat waves (short-term extreme heat), defined as the extreme temperatures, may increase in frequency, severity, duration, or areal extent (Robinson, 2001; Della Marta et al., 2007; Griffiths and Bradley, 2007; Dosio, 2017). Heat waves are extremely high temperatures that can cause damage or death to humans and can damage plants in a short time (usually in a few days) (Hunt, 2007). In recent years, many countries and regions, such as those of Europe and the Americas have been reportedly hit by heat waves (Keellings et al., 2018; Trimmel et al., 2018; Rita et al., 2019). In China, heatwaves are experienced over a vast area (Gao et al., 2015). The greenhouse effect caused by elevated CO2 concentrations is one of the most common global warming phenomenon (Guo et al., 2009). As evidenced by data from the Mona Loa Mountain Meteorological Observatory in Hawaii, global atmospheric CO2 concentrations increased from 280 ppm before the Industrial Revolution to 401 ppm in 2016 and would further increase to ∼540–970 ppm (average 700 ppm) by the end of the 21st century (Zvereva and Kozlov, 2006). This indicates that heat waves will be particularly prominent in the context of climate warming.
Insects are poikilothermic animals. Their development, reproduction, physiology, and metabolism are significantly affected by global warming (Murray et al., 2013; Couture et al., 2015; Sentis et al., 2017; Ma et al., 2018). Increased CO2 concentrations are associated with substantial changes in the photosynthetic activity of plants (especially C3 plants), promote their growth and expansion (Tripathee, 2008), and increase the content of secondary defense substances, which indirectly affects the development and reproduction of herbivorous insects (Bidart-Bouzat and Imeh-Nathaniel, 2008; Murray et al., 2013). Heat waves are rare, extremely high temperature events that have a significant effect on plant growth, photosynthesis, physiology, and metabolism in the short term. The increase in CO2 concentration is always accompanied by an increase in temperature. Therefore, the comprehensive stress caused by high concentrations of CO2 and heat wave may increase the indirect effects on the growth, development, and population fecundity of herbivorous insects, whose phenotypes and genes would change accordingly.
Ambrosia artemisiifolia (common ragweed) originated in the Sonoran region of North America and is a C3 plant. As a malignant and invasive weed, it poses a serious threat to human health, agricultural production, and biodiversity (Vincent et al., 1992; Fumanal et al., 2005, 2007; Bohren, 2006; Bonini and Ceriotti, 2019). Ophraella communa LeSage (Coleoptera: Chrysomelidae) is derived from North America and its larvae and adults feeds on common ragweed leaves. This insect has been used as a natural enemy with good prospects for the control of common ragweed in the United States, Mexico, Canada, Japan, South Korea, and China (Palmer and Goeden, 1991; Sohn et al., 2002; Tamura et al., 2004; Zhou et al., 2010a, b). In China, O. communa has been widely used for effective control of common ragweed in Hunan, Hubei, Jiangxi, Guangdong, Guangxi, Fujian, and other provinces (Zhou et al., 2010a). However, we made an interesting observation in a previous study that the hottest days of the summer (colloquially referred to as the “dog days”) adversely effected the survival, development, and fecundity of O. communa and the population of this beetle showed an obvious decline. In contrast, the growth of common ragweed was the best during these days. In the same time, Bae et al. (2019) found the plant growth and the concentrations of alleochemicals in common ragweed significantly increased in elevated CO2. We have also noticed previously that the contents of secondary defense substances, such as tannins and phenols, in common ragweed leaves are significantly increased under the combined stress imposed by high CO2 concentration and heat waves. Further, we found that the fecundity and lifespans of control females (feeding on common ragweed under ambient CO2 concentration) were significantly decreased while mated with treated males (feeding on common ragweed under the stress of high CO2 concentration combined with the heat wave); the fecundity and lifespans of treated females did not show a clear change while mated with control males (unpublished work). Therefore, it is believed that by feeding on common ragweed growing under the stress imposed by a combination of high CO2 concentration and heat waves, the reproductive function of O. communa adults is significantly reduced, and the breeding ability of the population is reduced.
Male insects ejaculate sperm into the females during mating. In addition to sperm, a large number of seminal fluid proteins (SFPs) and non-protein small molecules are present in the seminal fluid. SFPs are the main effectors required for postmating physiological and behavioral changes in insect females. These changes include reduction in the likelihood of female re-mating, increase in female egg production, changes in female flight and feeding behavior, and alterations in female gene transcription and female genital structures (Avila et al., 2011). The SFPs are mainly produced in the male accessory glands, and the testes also secrete some of them (Avila et al., 2011). The function of insect SFPs has been studied most intensively in Drosophila (Wolfner, 2002, 2007; Ravi Ram and Wolfner, 2007). Studies on the function of SFPs have also been carried out in mosquitoes (Dottorini et al., 2007; Sirot et al., 2008; Rogers et al., 2009), bees (Collins et al., 2006; Baer et al., 2009), cockroaches (Braswell et al., 2006; Andrés et al., 2008), and red pirates (South et al., 2011). It is hypothesized that feeding on common ragweed grown under high CO2 concentration and heat wave stress has an indirect effect on the growth and reproduction of O. communa females, and the regulatory function of the SFPs may be an intrinsic reason for this effect. Nevertheless, no studies have been conducted on the SFPs of O. communa. Our understanding about the mechanisms underlying this relationship between herbivores and plants under conditions of elevated CO2 concentrations and heat waves is also very limited. Thus, the expressions of genes in the accessory glands and the testes (tissues of secreting SFPs) of O. communa males were compared. And the males were fed on common ragweed under elevated CO2 concentration and heat waves, while the males feeding on common ragweed under ambient CO2 concentration were as control. The results described herein would lay a good foundation for exploring the detailed functions of SFPs and might be helpful in explaining the mechanism underlying the indirect effects of CO2 and heat wave stress in the decline of the reproductive ability of O. communa.
Materials and Methods
Insect Collection and Dissection
Ophraella communa individuals were given different treatments of common ragweed since the egg stage. The eggs were produced on the same day by the same batch of adults raised in the laboratory. First, common ragweed seeds were sown in plastic nutrient bowls (Φ15 cm) with 3–5 seeds in each. The substrate was a 1:2 mixture of vermiculite: flower nutrient soil. Two groups—experimental and control—were set up. The seeds were germinated and grown in artificial climate chambers that can control temperature, humidity, photoperiod, light intensity, and CO2 concentration [Model: PRX-450-CO2 type; temperature range (°C): ∼0–65 ± 0.5°C; humidity range (RH): ∼25–95 ± 2%; produced by Shanghai COLIN Experimental Instrument Factory]. For the experiment group, the growth conditions were: 30 ± 1°C day temperature, 25 ± 1°C night temperature, 70 ± 5% relative humidity, 14-h L:10-h D photoperiod, 30,000 lux light intensity and 700 ppm CO2 concentration. When common ragweed grew to a height of about 10 cm, two robust seedlings were retained per bowl. As the plants grew to a height of about 30 cm, they were subjected to heat wave stress (40 ± 1°C) for 5 days; the daily treatment time was from 9:00 to 17:00 h; the temperature for the remaining time period was kept the same as the normal day temperature. After 5 days, the plants were returned to normal temperature and the other conditions remained unchanged. For the control group (Group CK), the day and night temperatures, photoperiod, and light intensity were the same as those of the experimental group, the CO2 concentration was 370 ppm. The plants grew under such conditions throughout the study. Thereafter, the adults of O. communa, aged 5–8 days, were attached to the plants to lay eggs, and the hatched larvae were directly raised on the same plant to the pupal and adult stages. One day after eclosion, the male adults were chosen to feed alone (still in the corresponding incubator) until they were sexually mature (2–4 days after eclosion). The unmated male adults were taken for anatomical examination. In particular, because it is not possible to tell the sex of O. communa at the larval or pupal stages, the males had to be identified in the adult stage.
Sample Preparation and RNA Extraction
The reproductive tract of O. communa males is mainly dominated by a pair of testes and male accessory glands (Supplementary Figure S1). In the present study, the testes (TE) and the male accessory glands (MAG) were sampled separately. For each tissue type, three independent replicates representing tissues pooled from 30 individuals were collected. The tissues of the male adults were dissected and washed in sterile ice-cold phosphate-buffered saline (PBS) treated with 0.1% diethylpyrocarbonate (DEPC), and were then immediately frozen and stored at −80°C for subsequent RNA extraction.
Total RNA was extracted with Trizol reagent (Life Technologies, United States) according to the manufacturer’s instructions. RNA quality was examined as follows: RNA degradation and contamination was monitored on 1% agarose gels; RNA purity was checked using the NanoPhotometer® spectrophotometer (IMPLEN, CA, United States); RNA concentration was measured using Qubit® RNA Assay Kit in Qubit® 2.0 Fluorometer (Life Technologies, CA, United States); RNA integrity was assessed using the RNA Nano 6000 Assay Kit with the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, United States).
cDNA Library Construction and RNA-Seq Analysis
The RNA of the required quality was sent to Beijing Genomics Institute in Wuhan for cDNA library construction and RNA- sequencing (RNA-seq). A total of 3 μg RNA per sample was used as the input material for RNA sample preparation. Sequencing libraries were generated using NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, United States) following manufacturer’s recommendations and index codes were added for attributing sequences to each sample. Briefly, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in NEBNext First Strand Synthesis Reaction Buffer (5X). First strand cDNA was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase (RNase H–) (NEB, United States). The synthesis of second strand cDNA was subsequently performed using DNA polymerase I, and it was treated with RNase H. The remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of the 3′-ends of the DNA fragments, NEBNext Adaptors with hairpin loop structure were ligated to prepare for hybridization. To select cDNA fragments, preferentially those that were 300 bp in length, the library fragments were purified with AMPure XP system (Beckman Coulter, Beverly, MA, United States). Thereafter, 3 μL USER Enzyme (NEB, United States) was mixed with size-selected, adaptor-ligated cDNA at 37°C for 15 min, followed by incubation for 5 min at 95°C before PCR. Subsequently, PCR was performed with Phusion High-Fidelity DNA polymerase, Universal PCR primers, and Index (X) primer. The amplified PCR products were purified (AMPure XP system) and the library quality was assessed on the Agilent Bioanalyzer 2100 system.
The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumina) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Hiseq platform (Hiseq X Ten, Illumina, Inc., San Diego, CA, United States) and paired-end reads (150 bp) were generated.
Data Processing, Assembly, and Annotation
Raw data (raw reads) in the fastq format were first processed through in-house perl scripts. In this step, clean data (clean reads) were obtained by removing the reads containing the adapter, reads containing ploy-N, and low quality reads from raw data. At the same time, Q20, Q30, GC-content, and sequence duplication levels of the clean data were calculated. All the downstream analyses were based on clean data of high quality. To obtain an integrated transcript set, all the clean data were put together and subjected to a combined assembly strategy. The transcript sequence obtained by splicing with Trinity was used as a reference sequence for subsequent analysis. The longest transcript of each gene was selected as Unigene for subsequent analysis. The left files (read 1 files) from all samples were pooled into one big left.fq file, and right files (read 2 files) into one big right.fq file. Transcriptome assembly was accomplished based on the left.fq and right.fq using Trinity (Grabherr et al., 2011) with min_kmer_cov set to 2 by default and all other parameters set default.
Seven databases were used to annotate the gene function; these were: Nr (NCBI non-redundant protein sequences, NCBI blast 2.2.28+, E-value = 1E-51); Nt (NCBI nucleotide sequences, NCBI blast 2.2.28+, E-value = 1E-52); Pfam (Protein family3, HMMER 3.0 package, hmmscan, E-value = 0.01); KOG/COG (Clusters of Orthologous Groups of proteins4, NCBI blast 2.2.28+, E-value = 1E-3); Swiss-Prot (a manually annotated and reviewed protein sequence database5, NCBI blast 2.2.28+, E-value = 1E-5); KO (KEGG Ortholog database6, KAAS (version r140224), KEGG Automatic Annotation Server, E-value = 1E-10] (Kanehisa et al., 2008); and GO (Gene Ontology7, Blast2GO v2.5, E-value = 1E-6) (Gotz et al., 2008).
Long non-coding RNAs (lncRNA) were predicted using CPC (Coding Potential Calculator8). In order to reduce the false positive rate in predicting lncRNA, the final results would be given on removing the transcript annotated by Swiss-Prot and Pfam database.
SNP and InDel Calling
Picard-tools v1.419 and samtools v0.1.18 (Li, 2011) were used to sort and remove duplicated reads, and merge the bam alignment results for each sample. GATK2 software (Mckenna et al., 2010) was used to perform SNP and InDel calling. The raw vcf files were filtered with the GATK standard filter method and other parameters [clusterWindowSize: 10; MQ0 ≥ 4 and (MQ0/(1.0∗DP)) > 0.1; QUAL < 10; QUAL < 30.0, QD < 5.0, HRun > 5], and only SNPs with a distance >5 were retained.
Analysis of Gene Expression Patterns
Gene expression levels were estimated by RSME (Li and Dewey, 2011) for each sample. RSEM counts bowtie’s results, clean data were mapped back onto the assembled transcriptome, and readcount for each gene was obtained from the mapping results. Read counts were converted to FPKM (expected number of Fragments Per Kilobase of transcript sequence per Millions base pair sequenced) values, and expression values were calculated in terms of FPKM (FPKM > 0.3) (Trapnell et al., 2010). For the same samples with biological replicates, differential expression analysis for the two conditions was performed using the DESeq R package10 (Anders and Huber, 2010). DESeq provides statistical routines for determining differential expression in the digital gene expression data using a model based on the negative binomial distribution. The resulting P-values were adjusted using the Benjamini and Hochberg’s approach for controlling the false discovery rate. Genes with an adjusted P-value < 0.05 estimated by DESeq were assigned as differentially expressed. Significance tests for each gene were conducted for “TE vs. TEck,” “MAG vs. MAGck.”
Gene Ontology Enrichment and KEGG Pathway Analyses
Gene Ontology (GO) enrichment analysis of the differentially expressed genes (DEGs) was implemented by the GOseq R packages based on Wallenius non-central hyper-geometric distribution (Young et al., 2010), which can adjust for gene length bias in DEGs. KEGG (Kanehisa et al., 2008) is a database resource for understanding high-level functions and utilities of the biological system, such as the cell, the organism, and the ecosystem, from molecular-level information, especially large-scale molecular datasets generated by genome sequencing and other high-throughput experimental technologies6. KOBAS (Mao et al., 2005) software was used to test the statistical enrichment of the DEGs in KEGG pathways.
Quantitative Real-Time PCR (qRT-PCR) Validation
To validate the results of differential expression obtained in the RNAseq analysis, 10 DEGs were analyzed by quantitative real-time PCR (qRT-PCR), with three biological replicates, including 4 DEGs in “MAG vs. MAGck” and 6 DEGs in “TE vs. TEck.” Total RNA was extracted from the samples mentioned above. Thereafter, 1,000 ng of total RNA was reverse transcribed in a 20 μL reaction containing 5× TransScript All-in-One SuperMix for Real time (TransGen Biotech, Beijing). All the specific primers were 18–20 bp and amplified 80–150 bp PCR products; the Tm for the primes ranged from 58 to 60°C. The primers are listed in Supplementary Table S1. A reference housekeeping gene (ribosomal protein L4, not yet published) was used for normalization. The reaction mixture (final volume of 20 μL) consisted of 1 μL cDNA, 10 μL SYBR Green Master Mix, 0.4 μL of each specific primer (10 μM), and 8.2 μL ddH2O. The thermal-cycler program was as follows: 5 min at 95°C for the initial denaturation, followed by 40 cycles of 10 s at 95°C and 34 s at 60°C. The 2–ΔΔCT method was used to estimate the changes in the expression between the two treatments (Livak and Schmittgen, 2001). The data were analyzed by the student t-test with SPSS 22.0 software (SPSS Inc., Chicago, IL, United States). The results were expressed as means ± SE.
Results
Establishment of the Male O. communa Transcriptome From the TE and MAG
The transcriptomes of TE and MAG from different treatment groups were sequenced in triplicates. After preprocessing the reads, a total of 72.3 GB of clean data was acquired. The Q20 and Q30 for all the samples were ≥96 and >91%, respectively. The GC content was about 40% (Supplementary Table S2). Because of the absence of a reference genome, the obtained clean reads were assembled to get a reference sequence for subsequent analysis. Overall, 67,205 unigenes were generated from the transcriptome, which included 21,928 coding and 45,277 long non-coding genes. The mean length of the unigenes was 768 bp, with N50 and N90 lengths of 1,468 bp and 274 bp, respectively (Table 1). Sequence length distribution analysis showed that 52.01% of the unigenes were between 301 and 2,000 bp in size, and unigenes less than 301 bp and greater than 2,000 bp accounted for 39.09 and 8.9% of the total number, respectively (Supplementary Figure S2).
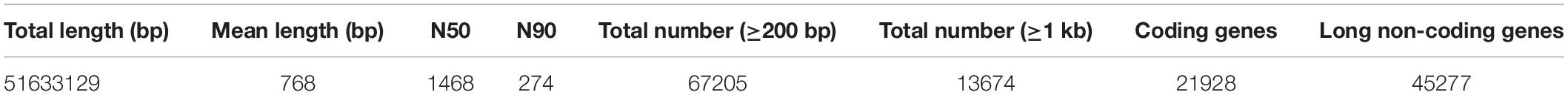
Table 1. Statistics of the final assembly and prediction of coding genes.
Gene Function Annotation
The transcriptome gene function was annotated using seven databases (Supplementary Table S3). A total of 28,369 unigenes (42.21% of the total unigenes) were annotated in at least one of the databases. Maximum number of genes (34.4%) were annotated in the NR database (Supplementary Table S3): most of the sequences (41.2%) matched with those of the model organism, Tribolium castaneum, followed by those of Dendroctonus ponderosae (12.8%), Lasius niger (4.2%), Acyrthosiphon pisum (3.8%), Vollenhovia emeryi (2.5%); the remaining 35.5% of unigenes gave BLAST hits with sequences from other species (Supplementary Figure S3). Besides, several unigenes were annotated using the sequences in the NT, KO, SwissProt, and PFAM databases (Supplementary Table S3).
After the GO annotation of genes, they were grouped into three main categories, namely “Biological process,” “Cellular process,” and “Molecular function” (Supplementary Figure S4). Among the processes, based on the KOG database, 10.72% of the unigenes were categorized into 26 different functional groups (Supplementary Figure S5). After KO annotation of the genes, they were classified according to the KEGG metabolic pathways they participated in. These genes belonged to five branches (“Cellular Processes,” Environmental Information Processing,” “Genetic Information Processing,” “Metabolism,” and “Organismal Systems”) (Supplementary Figure S6).
Putative SNPs and InDels
A total of 203411 SNPs and 31254 InDels were identified in this study. The distribution of SNPs in each sample is shown in Supplementary Table S4. The number and percentage of SNPs in each coding and non-coding region of each sample were calculated. Additionally, the number and percentage of synonymous and non-synonymous mutations per sample were identified. These molecular markers, SNPs and InDels, might be useful for further studies to detect the migration and diffusion of O. communa. The identity of the predicted molecular markers should be validated in future research to ensure their accuracy.
Gene Expression Analysis Across Different Treatments
For identifying DEGs among the tissues in the different treatment groups, the FPKM conversion for each read count was performed, and the unigenes with FPKM values above 0.3 were selected. The results showed that 10,296 unigenes were expressed in all the samples, and there were several unigenes specifically expressed in each sample (on account of 3 replicates) of TE, TEck, MAG, and MAGck groups (Supplementary Table S5). The overall view of gene expression in each sample is presented in the FPKM boxplot (Supplementary Figure S7). The discreteness of gene expression indicates the degree of difference between samples. Venn diagrams were used to present the number of common and unique differential genes between different combinations of comparisons (Figure 1). The relationship between qvalue and log2 (foldchange) of the unigenes and the significantly up- and down-regulated DEGs between two tissues were revealed using the volcano plot (Figure 2). Unexpectedly, only nine and five DEGs were identified in the comparisons of TE with TEck and of MAG with MAGck, respectively (Figure 1). Among the nine DEGs, seven were up-regulated and two were down-regulated; among the five DEGs, one was up-regulated and four were down-regulated (Figure 2).

Figure 1. Venn diagram for the differentially expressed genes identified in different comparisons.
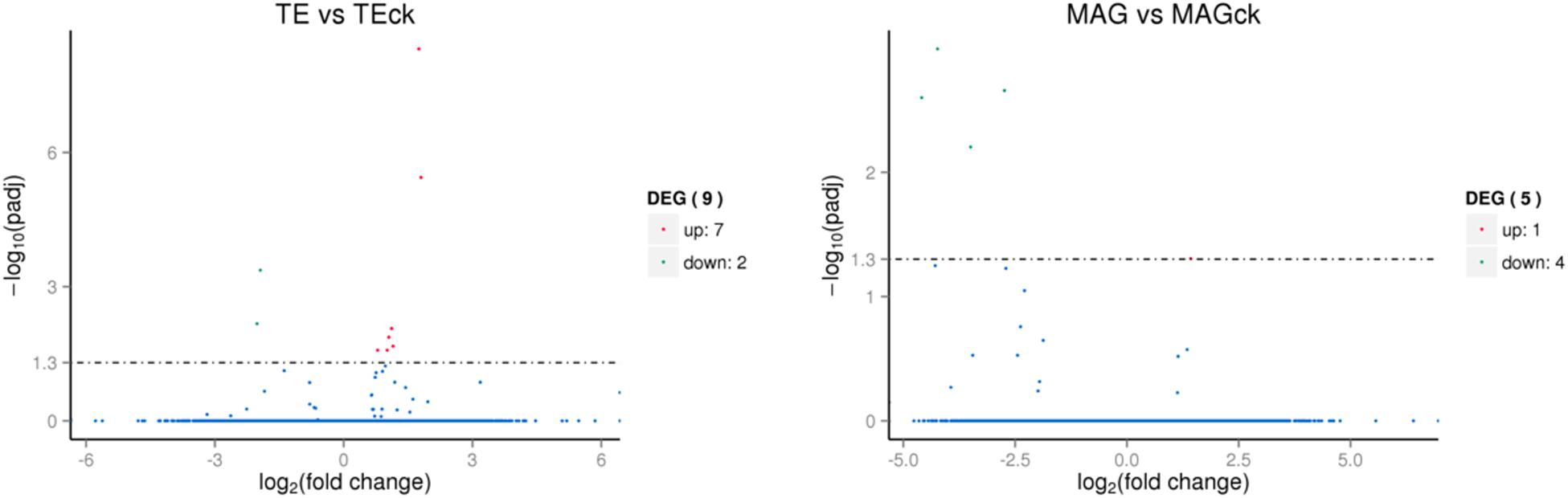
Figure 2. Significantly up- and down-regulated differentially-expressed genes (DEGs) between the tissues. The abscissa represents the fold-change in the expression of genes in different samples; the ordinate represents the statistical significance of the change in gene expression; the smaller the corrected p-value, the larger is the -log10 (corrected p-value), that is, more remarkable is the difference. The scatter points in the figure represent individual genes, the blue dots indicate genes with no significant differences, the red dots indicate up-regulated genes with significant differences, and the green dots indicate down-regulated genes with significant differences.
This study focused on the indirect effects of the elevated atmospheric CO2 levels associated with heat waves on the male reproductive tract of O. communa. The transcriptional changes in the TE vs. TEck and MAG vs. MAGck comparisons could reflect these effects. Among the DEGs, one odorant binding protein gene related to host plants that was up-regulated. Three DEGs related to the adjustment of energy metabolism were annotated, among which the unigene encoding putative sugar transporter 2 in the MAG vs. MAGck comparison was down-regulated and the other two DEGs encoding putative glycosyl hydrolase and alpha-esterase in the TE vs. TEck comparison were up-regulated. There were three stress-related DEGs including two kinds of predicted facilitated trehalose transporters, Tret1 (down-regulated) and an extracellular Cu/Zn-superoxide dismutase (up-regulated). A putative juvenile hormone, epoxide hydrolase 2 (up-regulated) related to reproduction was also identified. Six DEGs were not annotated using the NR database; however, the unigene, Oc27982_g1, was identified as defensin-related using PFAM; the unigene, Oc40935_g1, was annotated as acyltransferase using PFAM, which plays important roles in gene expression, metabolism, and signaling; the unigene, Oc45230_g1, was also annotated as an odorant binding protein gene using PFAM; the unigene, Oc41705_g2, was annotated as multidrug resistance-associated protein using SwissProt, PFAM, and KOG, indicating that this gene could be detoxification-related; the unigene, Oc37599_g1, was annotated as a transporter in other databases, which may be detoxification-related; the unigene, Oc37929_g1, was annotated as aldose reductase using SwissProt, which might play a role in the protective response to feeding on common ragweed exposed to heat wave and elevated CO2 concentrations (Table 2). The function of Oc27982_g1, Oc40935_g1, Oc41705_g2, Oc37599_g1, and Oc37929_g1 were all related to poisonous food-utilization.
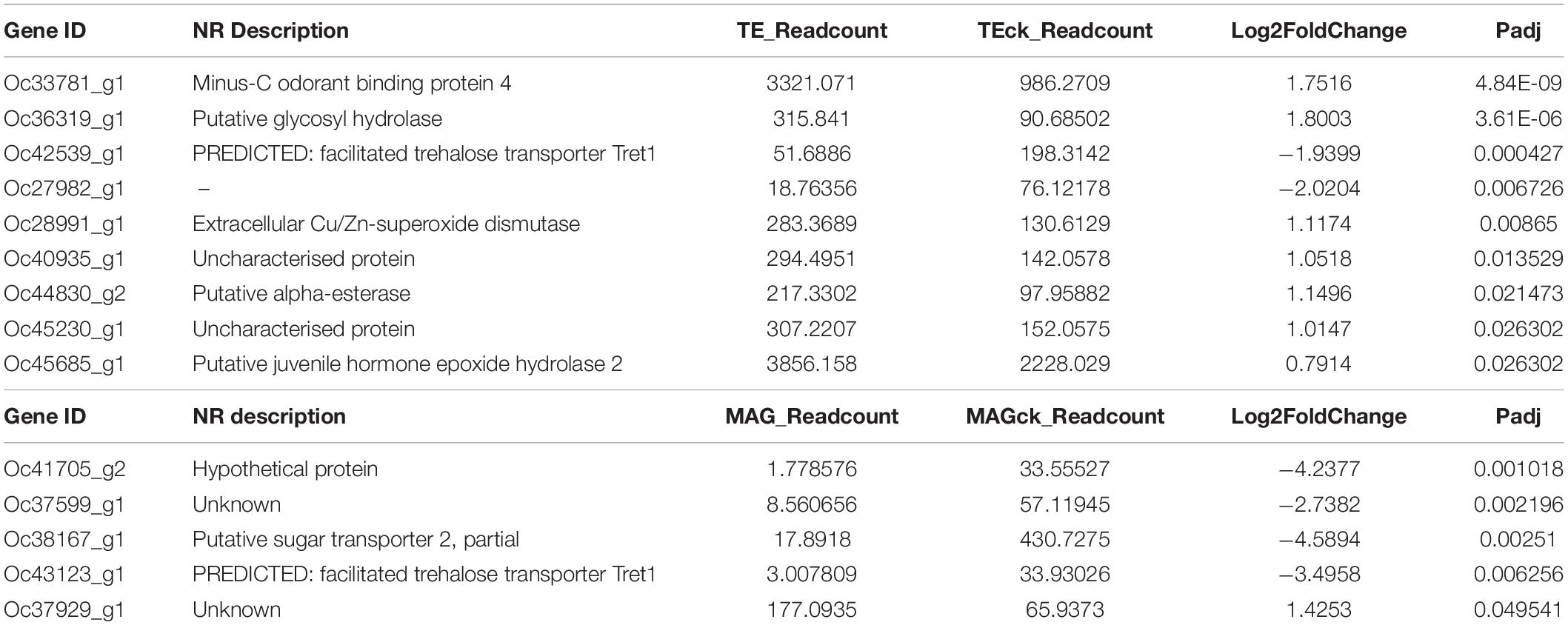
Table 2. Differentially expressed genes identified in the TE vs. TEck and MAG vs. MAGck comparisons.
KEGG Enrichment of DEGs
All the DEGs were mapped to the terms for enrichment analysis in the KEGG database. In the comparison of TE vs. TEck, 3 DEGs (Oc28991_g1, Oc45685_g1, and Oc44830_g2) were involved in 9 pathways, which were up-regulated. Oc28991_g1 was enriched in “prion diseases,” “amyotrophic lateral sclerosis (ALS),” “peroxisome” and “huntington’s disease.” Oc45685_g1 was enriched in “metabolism of xenobiotics by cytochrome P450,” “chemical carcinogenesis,” and “bile secretion.” Oc44830_g2 was enriched in “glycerophospholipid metabolism” and “cholinergic synapse.” In the comparison of MAG vs. MAGck, only Oc37929_g1 (up-regulated) was enriched in “fructose and mannose metabolism” “pentose and glucuronate interconversions” “glycerolipid metabolism,” and “galactose metabolism” pathways (Figures 3A,B).
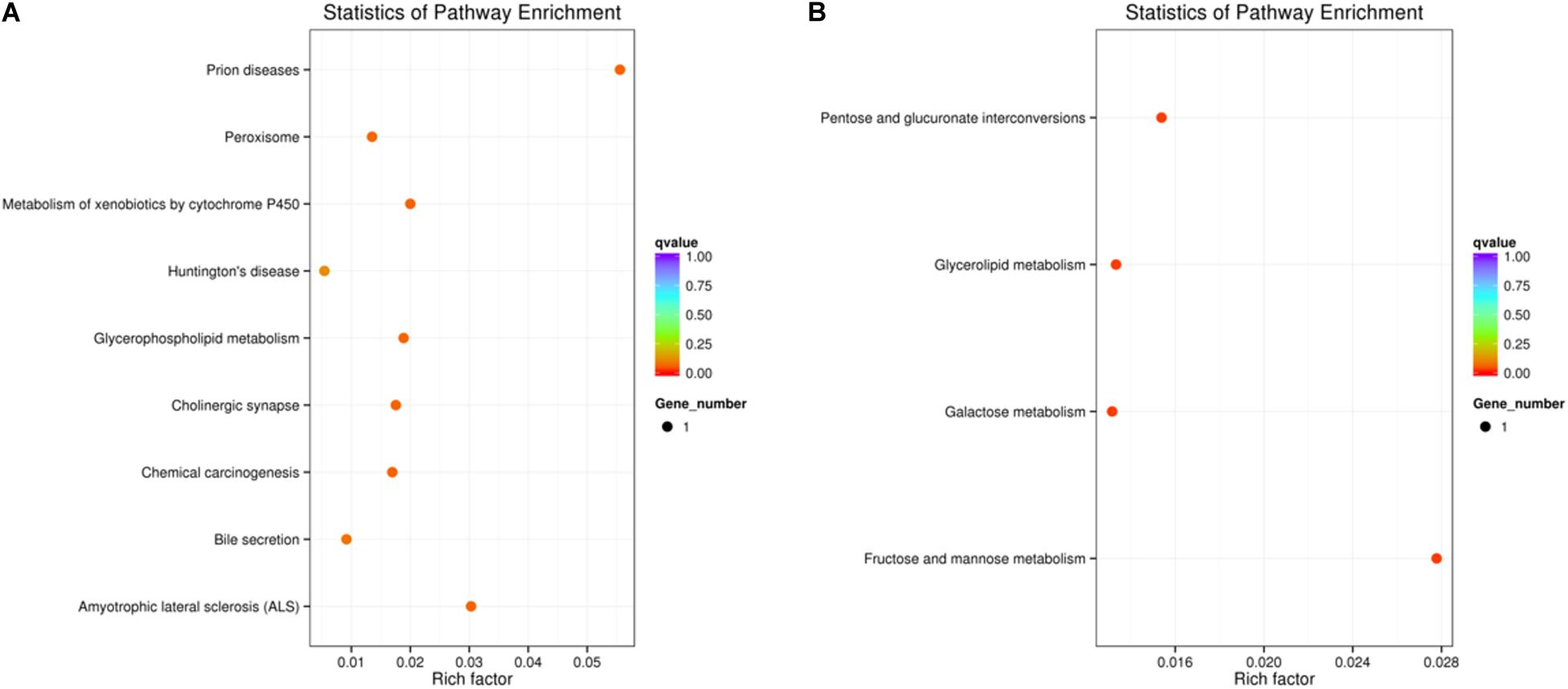
Figure 3. Scatter plot for KEGG pathway enrichment of differentially expressed genes. The vertical axis represents the path name and the horizontal axis represents the Rich factor corresponding to the path. The size of the q-value is represented by the color of the dot. The smaller the q-value is, the closer the color is to red. The number of differential genes contained in each route is represented by the size of the dot. (A) TE vs. TEck. (B) MAG vs. MAGck.
Validation of Transcriptome Data Using qPCR
Among these 10 DEGs, the gene expression patterns analyzed by qPCR were consistent with the results of the FPKM method (Figure 4), indicating that the transcriptome analysis results are reliable.
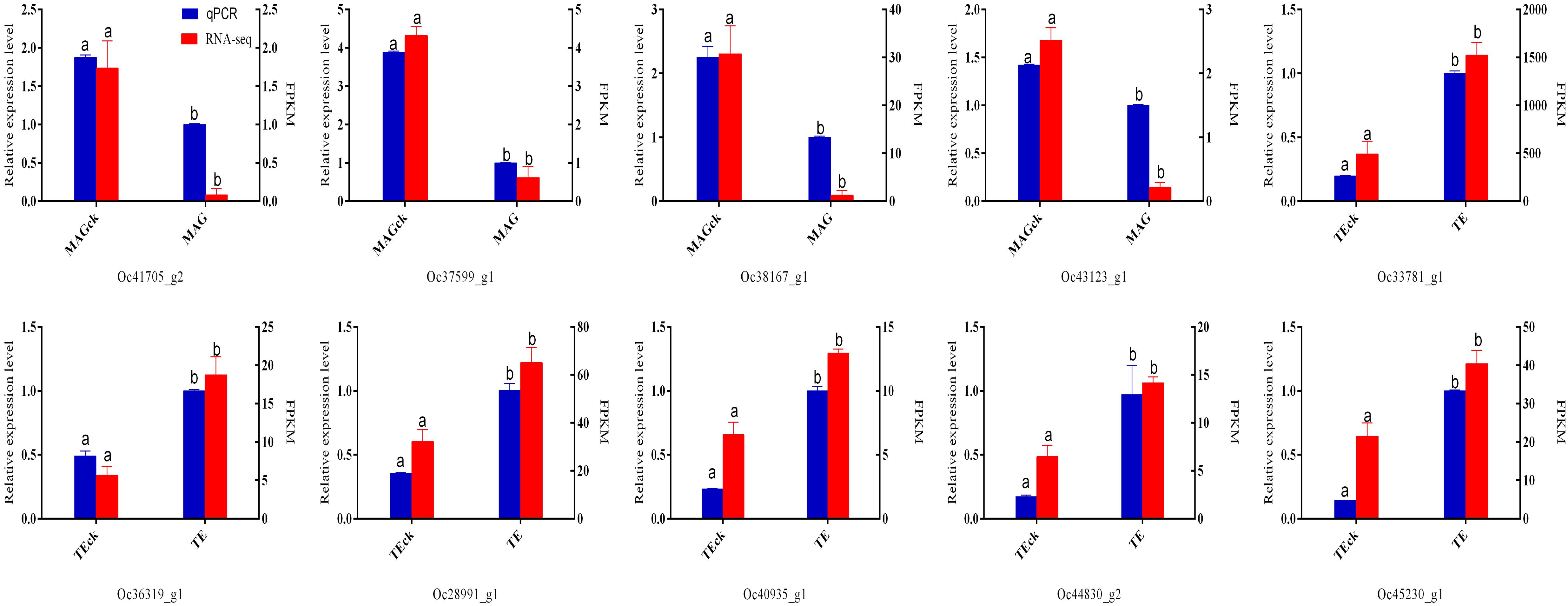
Figure 4. Expression levels of differentially expressed genes as analyzed by RNA-seq and qPCR.
Seminal Fluid Proteins of Other Insects Reported in This Study
Recently, many seminal fluid proteins were identified in insects, but most of them have no names or their functions remain unclear. Based on the annotation of the transcriptome, the unigenes were investigated, which were annotated as the usual seminal fluid proteins (Table 3). Only one unigene was annotated for “protein outspread,” “prophenoloxidase,” and “unitz-like protease inhibitor.” There were more than two unigenes annotated for other seminal fluid proteins, which need further confirmation.

Table 3. The unigenes annotated as the usual seminal fluid proteins in other insects.
Discussion
The use of high-throughput transcriptome technologies enables to investigate the genetic responses that organisms deploy in alternative environments (Roy et al., 2010). So far, RNA-seq was employed to evaluate the changes induced by different treatments (Zheng et al., 2013; Alfonso-Parra et al., 2016; Wu et al., 2019). In the present study, the RNA-seq data was analyzed to explain the effect of climate change on O. communa. The expression profiles between TE and TEck, and between MAG and MAGck represent the differences in the reproductive tract of males upon feeding common ragweed exposed to different conditions, which simulate the closest rearing conditions prevalent in nature. Based on the statistical analysis, differently expressed genes were identified.
Two DEGs were annotated as putative sugar transporter 2 (Oc38167_g1) and putative glycosyl hydrolase (Oc36319_g1). Oc44830_g2 was enriched in “glycerophospholipid metabolism” and “cholinergic synapse” pathways. The proteins participate in the decomposition and transport of macromolecular substances. This suggests that differentially expressed energy metabolism-related genes may be involved in regulation of reproduction. Odorant binding proteins are well-known to facilitate the transfer of apolar odorants in the aqueous antennal perilymph during pheromone reception (Vogt et al., 1991; Shen et al., 2019). Among the DEGs identified for the TE vs. TEck comparison, two odorant binding proteins (Oc33781_g1 and Oc45230_g1) were identified. They may be related to the response of O. communa to the host plant exposed to high CO2 concentration and heat waves. Similarly, two odorant binding proteins were reported to affect the taste perception and host-plant preference in Drosophila sechellia (Matsuo et al., 2007). However, little is known about the regulatory mechanism for the insects’ post mating behavior. A DEG, Oc45685_g1, was identified as putative juvenile hormone epoxide hydrolase 2. Juvenile hormone (JH) is an essential hormone for insects. It is called “insect growth regulator” (Staal, 1975) and has central roles in the regulation of insect development and reproduction (Laufer et al., 1987). The juvenile hormone, epoxide hydrolase, regulates insect JH titre along with JH esterase (Touhara and Prestwich, 1993). Oc45685_g1 was up-regulated in TE with respect to its level in TEck, indicating that the treatment to males may affect their development and reproduction. Indeed, in a study on T. castaneum, researchers found that the MAG size and SFP amount in JH-deficient males decreased and they acted with less vigor in the mating behavior, there was poor sperm transfer, and less production of eggs and offspring (Parthasarathy et al., 2009).
Trehalose plays a much more complex role in insect physiology than just being an energy store (Jungreis, 1980). The trehalose concentration can regulate taste receptors and the central nervous system, which affects the insects’ food selection and feeding behavior (Thompson Nelson, 2003). It can also act as a stabilizer of proteins to protect insects from harsh environments (Wyatt, 1967). The two DEGs, trehalose transporter Tret1 (Oc42539_g1 and Oc43123_g1), which were down-regulated, might be responsible for the fact that the population showed an obvious declining trend in the hottest days. Superoxide dismutase (SOD) is a catalyser in the disproportionation which reduces oxygen to generate oxygen and hydrogen peroxide. SOD has been widely discovered in the cells of various organisms. It can specifically scavenge superoxide anion and protect the body from oxidative damage (Xu, 2013). Cu/Zn-superoxide dismutase is one enzyme of SOD. Mittapalli et al. (2007) suggested that the induced expression of superoxide dismutase gene, in the midgut of Mayetiola destructor, caused a protective response to oxidative damage from the food. In this study, Oc28991_g1, encoding Cu/Zn-superoxide dismutase, may also play an important role in protective response to oxidative damage while feeding the beetles on the weed. Different expressions of these three DEGs help to mitigate the indirect stresses created by high CO2 concentration combined with the heat waves.
For O. communa, successful mating is necessary for producing the offspring. Numerous SFPs are major effectors for a range of female postmating responses related to fertility. They are transferred to females with sperm during mating (Avila et al., 2011). Therefore, identification of SFPs is the primary step to understand the changes in reproduction. Many researchers predicted the insect SFPs using comparative transcriptomics. McGraw et al. (2008) examined gene expression profiles of whole female Drosophila melanogaster at four time points following copulation to explore the function of SFPs. Gotoh et al. (2017) identified enriched genes in the sperm-storage organ relative to those in the body of queen Crematogaster osakensis to identify the putative SFPs. SFPs are the products of male reproductive tract secretory tissues, not only including accessory glands (Avila et al., 2011); however, in most of the studies, the accessory gland has been considered as the only organ that secretes SFPs, and these have been defined as accessory gland proteins (Acps) (Chapman and Davies, 2004; South et al., 2011; Yu et al., 2016). For rigorous experiment, the male accessory glands and testes were both sampled. If all the DEGs in this study were transferred to females as SFPs, they may be involved in changing female likehood of remating, increasing ovulation and egg-laying rates, changing female flight and feeding behavior, inducing antimicrobial activities, and modulating sperm storage parameters. An increasing number of SFPs have been identified in insects and the function of these identified SFPs include roles in processes, such as odorant binding, and stress response, in addition to some functions that remain unknown. For example, Sirot et al. (2008) and Xu et al. (2013) found odorant binding proteins were SFPs of Aedes aegypti. In the present study, two DEGs encoding odorant binding proteins were also revealed. Certainly, the proteins from the male reproductive tract may alter the sperm production and quality except for being effectors as SFPs. The formation of sperm involves a series of molecular and spectacular morphological changes in insect testes (Vedelek et al., 2018). Thus, reduction of sperm motility and viability may also contribute the effect of the reproduction of females.
Although SFPs evolve rapidly, the protein classes represented in seminal fluid are constrained (Mueller et al., 2005). We investigated all the unigenes in our data, using several common SFPs as keywords, and found that they all matched one or more unigenes (Table 3). We could not tell if any of the unigenes was an SFP gene, but the genes encoding SFPs of O. communa were certainly included in this transcriptome. In future research, the function of some representative SFPs could directly be carried out based on the existing data resources. The present study provides a new perspective on revealing the functions of SFPs. In addition, Avila et al. (2011) raised an important point that Acp (SFP) genes evolve de novo, perhaps from long non-coding RNA. In this study, 45277 long non-coding genes were predicted, which provide novel and broad avenues for further research.
Generally, many DEGs can be identified by comparing the transcriptomes (Wissing et al., 2014; Lin et al., 2018). However, 14 DEGs were identified in the TE vs. TEck and MAG vs. MAGck comparisons. The reasons for the decline in population could be multifaceted. The females’ post-mating response caused by SFPs and sperm motility and numbers is only one aspect, which mainly affects the number of offspring. The impact on females themselves has not been investigated. Female specific proteins, containing vitellin (VN) and vitellogenin (VG), are main nutrition resource in early stage of life for oviparous insects (Swevers et al., 2005; Zhao et al., 2018). VG proteins become the major VN proteins (Hagedorn and Kunkel, 1979). Therefore, the VG gene expression level in females is key role in determining reproductive potential. A follow-up research would be needed to confirm it. Besides, feeding on common ragweed exposed to high CO2 concentration and heat wave may affect other organs of O. communa, such as olfactory organ and midgut, which play a direct role in the development and survival of the first generation. Insects depend on olfaction to estimate the overall situation of habitats, involving individual aspects, such as the spotting of food, host, mate or prey, and group communication aspects, such as aggregation and avoidance (Fan et al., 2011). The midgut of insects is responsible for digestion and nutrient uptake and, in particular, for detoxification and oxidative stress response (Hakim et al., 2010). Therefore, under the same experimental conditions, DEGs in other tissues may also contribute to the changes in the population. The analysis of DEGs between the testes and accessory glands revealed the indirect response of the reproductive tract to high CO2 and heat waves at another level.
Conclusion
In this study, we assembled the transcriptome of the testes and accessory glands from male O. communa, and investigated transcriptional changes under the premise of feeding different common ragweed, that were separately grown under high CO2 concentration and heat waves or under ambient CO2 concentration. The results indicated that the reproductive tract of males presented an indirect response to high CO2 concentration and heat waves. The DEGs related to decomposition and transport of macromolecular substances, host location, and stress response may trigger a series of post-copulatory events that detrimentally affect the development and growth of the population. Subsequently, along with future studies on females and other organs of O. communa, the molecular mechanism of the indirect response to high CO2 concentration and heat waves can be perfectly elucidated. And along with proteomic studies on mated females, SFPs of O. communa can be specifically identified.
Data Availability Statement
The raw RNA sequencing data were deposited in the NCBI Sequence Read Archive (SRA) with the accession number PRJNA580170 (Temporary Submission ID: SUB6480492).
Ethics Statement
The animal study was reviewed and approved by Ethics of Animal Experiment of Institute of Plant Protection, Chinese Academy of Agricultural Sciences.
Author Contributions
ZZ and YL designed the experiments. XG, ZhenyT, ZhenqT, SC, and GC performed the experiments. XG, YZ, and CM analyzed the data. XG and ZZ wrote the manuscript.
Funding
This research was funded by National Natural Science Foundation of China (Nos. 31972340 and 31672089).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Editage (www.editage.cn) for its linguistic assistance during the preparation of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2020.00417/full#supplementary-material
FIGURE S1 | Testes and accessory glands from male Ophraella communa.
FIGURE S2 | Sequence-length distribution of unigenes assembled from transcripts.
FIGURE S3 | Species distribution map on alignment with non-redundant (NR) database.
FIGURE S4 | Gene ontology annotation of the unigenes.
FIGURE S5 | KOG classification chart for the unigenes.
FIGURE S6 | Classification of KEGG metabolic pathways. (A) Cellular processes; (B) environmental information processing; (C) genetic information processing; (D) metabolism; (E) organismal systems.
FIGURE S7 | FPKM distribution of the tissues. The abscissa shows the sample name, the ordinate is log10(FPKM+1). MAG and TE: the accessory gland and testis from Ophraella communa males fed on ragweed exposed to elevated CO2 concentrations and heat wave. MAGck and TEck: the accessory gland and the testis from O. communa males fed on ragweed exposed to the ambient CO2 concentration.
TABLE S1 | Primers used in qRT-PCR.
TABLE S2 | Summary of the output quality of sequencing data.
TABLE S3 | Statistics of the final assembly and prediction of coding genes.
TABLE S4 | Distribution of single nucleotide polymorphisms (SNPs) in each sample.
TABLE S5 | Statistical table of FPKM values for all the samples.
Footnotes
- ^ https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastp&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome
- ^ https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome
- ^ http://pfam.xfam.org/
- ^ http://www.ncbi.nlm.nih.gov/COG/
- ^ http://www.ebi.ac.uk/uniprot/
- ^ http://www.genome.jp/kegg/
- ^ http://www.geneontology.org/
- ^ http://cpc2.cbi.pku.edu.cn
- ^ http://broadinstitute.github.io/picard
- ^ https://www.huber.embl.de/users/anders/DESeq/
References
Alfonso-Parra, C., Ahmed-Braimah, Y. H., Degner, E. C., Avila, F. W., Villarreal, S. M., Pleiss, J. A., et al. (2016). Mating-induced transcriptome changes in the reproductive tract of female Aedes aegypti. PLoS Negl. Trop. Dis. 10:e0004451. doi: 10.1371/journal.pntd.0004451
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
Andrés, J. A., Maroja, L. S., and Harrison, R. G. (2008). Searching for candidate speciation genes using a proteomic approach: seminal proteins in field crickets. Proc. R. Soc. B Biol. Sci. 275, 1975–1983. doi: 10.1098/rspb.2008.0423
Avila, F. W., Sirot, L. K., LaFlamme, B. A., Rubinstein, C. D., and Wolfner, M. F. (2011). Insect seminal fluid proteins: identification and function. Annu. Rev. Entomol. 56, 21–40. doi: 10.1146/annurev-ento-120709-144823
Bae, J., Byun, C., Ahn, Y. G., Choi, J. H., and Kang, H. (2019). Effect of elevated atmospheric carbon dioxide on the allelopathic potential of common ragweed. J. Ecol. Environ. 43:21. doi: 10.1186/s41610-019-0116-5
Baer, B., Heazlewood, J. L., Taylor, N. L., Eubel, H., and Millar, A. H. (2009). The seminal fluid proteome of the honeybee Apis mellifera. Proteomics 9, 2085–2097. doi: 10.1002/pmic.200800708
Bidart-Bouzat, M. G., and Imeh-Nathaniel, A. (2008). Global change effects on plant chemical defenses against insect herbivores. J. Integr. Plant Biol. 50, 1339–1354. doi: 10.1111/j.1744-7909.2008.00751.x
Bohren, C. (2006). Ambrosia artemisiifolia L.-in Switzerland: concerted action to prevent further spreading. Nachr. Dtsch. Pflanzenschutzd. 58, 304–308.
Bongaarts, J. (2018). Intergovernmental panel on climate change special report on global warming of 1.5°C Switzerland: IPCC, 2018. Popul. Dev. Rev. 45, 251–252. doi: 10.1111/padr.12234
Bonini, M., and Ceriotti, V. (2019). Ragweed story: from the plant to the patient. Aerobiologia 36, 45–48. doi: 10.1007/s10453-019-09571-5
Braswell, W. E., Andres, J. A., Maroja, L. S., Harrison, R. G., Howard, D. J., and Swanson, W. J. (2006). Identification and comparative analysis of accessory gland proteins in Orthoptera. Genome 49, 1069–1080. doi: 10.1139/g06-061
Chapman, T., and Davies, S. J. (2004). Functions and analysis of the seminal fluid proteins of male Drosophila melanogaster fruit flies. Peptides 25, 1477–1490. doi: 10.1016/j.peptides.2003.10.023
Collins, A. M., Caperna, T. J., Williams, V., Garrett, W. M., and Evans, J. D. (2006). Proteomic analyses of male contributions to honey bee sperm storage and mating. Insect Mol. Biol. 15, 541–549. doi: 10.1111/j.1365-2583.2006.00674.x
Couture, J. J., Meehan, T. D., Kruger, E. L., and Lindroth, R. L. (2015). Insect herbivory alters impact of atmospheric change on northern temperate forests. Nat. Plants 1:15016. doi: 10.1038/nplants.2015.16
Della Marta, P. M., Haylock, M. R., Luterbacher, J., and Wanner, H. (2007). Doubled length of western European summer heat waves since 1880. J. Geophys. Res. Atmos. 112:D15103. doi: 10.1029/2007JD008510
Dosio, A. (2017). Projection of temperature and heat waves for Africa with an ensemble of CORDEX regional climate models. Clim. Dyn. 49, 493–519. doi: 10.1007/s00382-016-3355-5
Dottorini, T., Nicolaides, L., Ranson, H., Rogers, D. W., Crisanti, A., and Catteruccia, F. (2007). A genome-wide analysis in Anopheles gambiae mosquitoes reveals 46 male accessory gland genes, possible modulators of female behavior. Proc. Natl. Acad. Sci. U.S.A. 104, 16215–16220. doi: 10.1073/pnas.0703904104
Fan, J., Francis, F., Liu, Y., Chen, J. L., and Cheng, D. F. (2011). An overview of odorant-binding protein functions in insect peripheral olfactory reception. Genet. Mol. Res. 10, 3056–3069. doi: 10.4238/2011.December.8.2
Fumanal, B., Chauvel, B., and Bretagnolle, F. (2005). “Demography of an allergenic European invasive plant: Ambrosia artemisiifolia,” in Proceedings of the International Symposium “Plant Protection and Plant Health in Europe, Braunschweig, 9–11.
Fumanal, B., Chauvel, B., and Bretagnolle, F. (2007). Estimation of pollen and seed production of common ragweed in France. Ann. Agric. Environ. Med. 14, 233–236.
Gao, J., Sun, Y., Liu, Q., Zhou, M., Lu, Y., and Li, L. (2015). Impact of extreme high temperature on mortality and regional level definition of heat wave: a multi-city study in China. Sci. Total Environ. 505, 535–544. doi: 10.1016/j.scitotenv.2014.10.028
Gotoh, A., Shigenobu, S., Yamaguchi, K., Kobayashi, S., Ito, F., and Tsuji, K. (2017). Transcriptome profiling of the spermatheca identifies genes potentially involved in the long-term sperm storage of ant queens. Sci. Rep. 7:5972. doi: 10.1038/s41598-017-05818-8
Gotz, S., Garcia-Gomez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. doi: 10.1093/nar/gkn176
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Griffiths, M. L., and Bradley, R. S. (2007). Variations of twentieth-century temperature and precipitation extreme indicators in the northeast United States. J. Clim. 20, 5401–5417. doi: 10.1175/2007jcli1594.1
Guo, K., Hao, S. G., Sun, O. J., and Kang, L. (2009). Differential responses to warming and increased precipitation among three contrasting grasshopper species. Glob. Change Biol. 15, 2539–2548. doi: 10.1111/j.1365-2486.2009.01861.x
Hagedorn, H. H., and Kunkel, J. G. (1979). Vitellogenin and vitellin in insects. Annu. Rev. Entomol. 24, 475–505. doi: 10.1146/annurev.en.24.010179.002355
Hakim, R. S., Baldwin, K., and Smagghe, G. (2010). Regulation of midgut growth, development, and metamorphosis. Annu. Rev. Entomol. 55, 593–608. doi: 10.1146/annurev-ento-112408-085450
Hughes, L. (2000). Biological consequences of global warming: is the signal already apparent? Trends Ecol. Evol. 15, 56–61. doi: 10.1016/S0169-5347(99)01764-4
Hunt, B. G. (2007). A climatology of heat waves from a multimillennial simulation. J. Clim. 20, 3802–3821. doi: 10.1175/Jcli4224.1
Jungreis, A. M. (1980). Hemolymph as a Dynamic Tissue. Insect Biology in the Future. Cambridge, MA: Academic Press, 273–294.
Kanehisa, M., Araki, M., Goto, S., Hattori, M., Hirakawa, M., Itoh, M., et al. (2008). KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484. doi: 10.1093/nar/gkm882
Keellings, D., Bunting, E., and Engstrom, J. (2018). Spatiotemporal changes in the size and shape of heat waves over North America. Clim. Change 147, 165–178. doi: 10.1007/s10584-018-2140-3
Laufer, H., Landau, M., Homola, E., and Borst, D. W. (1987). Methyl farnesoate: its site of synthesis and regulation of secretion in a juvenile crustacean. Insect Biochem. 17, 1129–1131. doi: 10.1016/0020-1790(87)90134-X
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. doi: 10.1093/bioinformatics/btr509
Lin, Y., Wang, Y., Li, B., Tan, H., Li, D., Li, L., et al. (2018). Comparative transcriptome analysis of genes involved in anthocyanin synthesis in blueberry. Plant Physiol. Biochem. 127, 561–572. doi: 10.1016/j.plaphy.2018.04.034
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 25, 402–408. doi: 10.1006/meth.2001
Ma, C. S., Wang, L., Zhang, W., and Rudolf, V. H. W. (2018). Resolving biological impacts of multiple heat waves: interaction of hot and recovery days. Oikos 127, 622–633. doi: 10.1111/oik.04699
Mao, X., Cai, T., Olyarchuk, J. G., and Wei, L. (2005). Automated genome annotation and pathway identification using the KEGG orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793. doi: 10.1093/bioinformatics/bti430
Matsuo, T., Sugaya, S., Yasukawa, J., Aigaki, T., and Fuyama, Y. (2007). Odorant-binding proteins OBP57d and OBP57e affect taste perception and host-plant preference in Drosophila sechellia. PLoS Biol. 5:e118. doi: 10.1371/journal.pbio.0050118
McGraw, L. A., Clark, A. G., and Wolfner, M. F. (2008). Post-mating gene expression profiles of female Drosophila melanogaster in response to time and to four male accessory gland proteins. Genetics 179, 1395–1408. doi: 10.1534/genetics.108.086934
Mckenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Mittapalli, O., Neal, J. J., and Shukle, R. H. (2007). Antioxidant defense response in a galling insect. Proc. Natl. Acad. Sci. U.S.A. 104, 1889–1894. doi: 10.1073/pnas.0604722104
Mueller, J. L., Ravi Ram, K., McGraw, L. A., Bloch Qazi, M. C., Siggia, E. D., Clark, A. G., et al. (2005). Cross-species comparison of Drosophila male accessory gland protein genes. Genetics 171, 131–143. doi: 10.1534/genetics.105.043844
Murray, T. J., Ellsworth, D. S., Tissue, D. T., and Riegler, M. (2013). Interactive direct and plant-mediated effects of elevated atmospheric [CO2] and temperature on a eucalypt-feeding insect herbivore. Glob. Change Biol. 19, 1407–1416. doi: 10.1111/gcb.12142
Palmer, W. A., and Goeden, R. D. (1991). The host range of Ophraella communa Lesage (Coleoptera: Chrysomelidae). Coleopt. Bull. 45, 115–120. doi: 10.1016/1049-9644(91)90106-A
Parmesan, C., and Yohe, G. (2003). A globally coherent fingerprint of climate change impacts across natural systems. Nature 421, 37–42. doi: 10.1038/nature01286
Parthasarathy, R., Tan, A., Sun, Z., Chen, Z., Rankin, M., and Palli, S. R. (2009). Juvenile hormone regulation of male accessory gland activity in the red flour beetle, Tribolium castaneum. Mech. Dev. 126, 563–579. doi: 10.1016/j.mod.2009.03.005
Ravi Ram, K., and Wolfner, M. F. (2007). Seminal influences: Drosophila Acps and the molecular interplay between males and females during reproduction. Integr. Comp. Biol. 47, 427–445. doi: 10.2307/4540173
Rita, A., Camarero, J. J., Nole, A., Borghetti, M., Brunetti, M., Pergola, N., et al. (2019). The impact of drought spells on forests depends on site conditions: the case of 2017 summer heat wave in southern Europe. Glob. Change Biol. 26, 851–863. doi: 10.1111/gcb.14825
Rogers, D. W., Baldini, F., Battaglia, F., Panico, M., Dell, A., Morris, H. R., et al. (2009). Transglutaminase-mediated semen coagulation controls sperm storage in the malaria mosquito. PLoS Biol. 7:e1000272. doi: 10.1371/journal.pbio.1000272
Roy, S., Ernst, J., Kharchenko, P. V., Kheradpour, P., Negre, N., Eaton, M. L., et al. (2010). Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330, 1787–1797. doi: 10.1126/science.1198374
Sentis, A., Hemptinne, J. L., and Brodeur, J. (2017). Non-additive effects of simulated heat waves and predators on prey phenotype and transgenerational phenotypic plasticity. Glob. Change Biol. 23, 4598–4608. doi: 10.1111/gcb.13674
Shen, J., Hu, L., Dai, J., Chen, B., Zhong, G., and Zhou, X. (2019). Mutations in pheromone-binding protein3 contribute to pheromone response variations in Plutella xylostella (L.) (Lepidoptera: Plutellidae). Pest Manag. Sci. 75, 2034–2042. doi: 10.1002/ps.5325
Sirot, L. K., Poulson, R. L., McKenna, M. C., Girnary, H., Wolfner, M. F., and Harrington, L. C. (2008). Identity and transfer of male reproductive gland proteins of the dengue vector mosquito, Aedes aegypti: potential tools for control of female feeding and reproduction. Insect Biochem. Mol. Biol. 38, 176–189. doi: 10.1016/j.ibmb.2007.10.007
Sohn, J. C., An, S. L., Lee, J. E., and Park, K. T. (2002). Notes on exotic species, Ophraella communa LeSage (Coleoptera: Chrysomelidae) in Korea. Korean J. Appl. Entomol. 41, 145–150.
South, A., Sirot, L. K., and Lewis, S. M. (2011). Identification of predicted seminal fluid proteins in Tribolium castaneum. Insect Mol. Biol. 20, 447–456. doi: 10.1111/j.1365-2583.2011.01083.x
Staal, G. B. (1975). Insect growth regulators with juvenile hormone activity. Annu. Rev. Entomol. 20, 417–460. doi: 10.1146/annurev.en.20.010175.002221
Swevers, L., Raikhel, A. S., Sappington, T. W., Shirk, P., and Latrou, K. (2005). “Vitellogenesis and post-vitellogenic maturation of the insect ovarian follicle,” in Comprehensive Molecular Insect Science, eds L. I. Gilbert, K. Iatrou, and S. S. Gill (London: Elsevier), 87–155. doi: 10.1016/B0-44-451924-6/00093-4
Tamura, Y., Hattori, M., Konno, K., Kono, Y., Honda, H., Ono, H., et al. (2004). Triterpenoid and caffeic acid derivatives in the leaves of ragweed, Ambrosia artemisiifolia L. (Asterales: Asteraceae), as feeding stimulants of Ophraella communa LeSage (Coleoptera: Chrysomelidae). Chemoecology 14, 113–118. doi: 10.1007/s00049-004-0269-1
Thompson Nelson, S. (2003). Trehalose – the insect ‘blood’ sugar. Adv. Insect Physiol. 31, 205–285. doi: 10.1016/S0065-2806(03)31004-5
Touhara, K., and Prestwich, G. D. (1993). Juvenile hormone epoxide hydrolase. Photoaffinity labeling, purification, and characterization from tobacco hornworm eggs. J. Biol. Chem. 268, 19604–19609. doi: 10.1111/j.1432-1033.1993.tb18209.x
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Trimmel, H., Weihs, P., Leidinger, D., Formayer, H., Kalny, G., and Melcher, A. (2018). Can riparian vegetation shade mitigate the expected rise in stream temperatures due to climate change during heat waves in a human-impacted pre-alpine river? Hydrol. Earth Syst. Sci. 22, 437–461. doi: 10.5194/hess-22-437-2018
Tripathee, R. (2008). Effect of CO2 on the Response of C and N Relations to a Heat wave in Sunflower and Corn. Doctoral dissertation, University of Toledo, Toledo, OH.
Vedelek, V., Bodai, L., Grézal, G., Kovács, B., Boros, I. M., Laurinyecz, B., et al. (2018). Analysis of Drosophila melanogaster testis transcriptome. BMC Genomics 19:697. doi: 10.1186/s12864-018-5085-z
Vincent, G., Deslauriers, S., and Cloutier, D. (1992). Problems and eradication of Ambrosia artemisiifolia L. in Quebec in the urban and suburban environments. Allerg. Immunol. 24, 84–89.
Vogt, R. G., Prestwich, G. D., and Lerner, M. R. (1991). Odorant-binding-protein subfamilies associate with distinct classes of olfactory receptor neurons in insects. J. Neurobiol. 22, 74–84. doi: 10.1002/neu.480220108
Wissing, M. L., Kristensen, S. G., Andersen, C. Y., Mikkelsen, A. L., Host, T., Borup, R., et al. (2014). Identification of new ovulation-related genes in humans by comparing the transcriptome of granulosa cells before and after ovulation triggering in the same controlled ovarian stimulation cycle. Hum. Reprod. 29, 997–1010. doi: 10.1093/humrep/deu008
Wolfner, M. F. (2002). The gifts that keep on giving: physiological functions and evolutionary dynamics of male seminal proteins in Drosophila. Heredity 88, 85–93. doi: 10.1038/sj.hdy.6800017
Wolfner, M. F. (2007). “SPERM”(seminal proteins (are) essential reproductive modulators): the view from Drosophila. Soc. Reprod. Fertil. Suppl. 65, 183–199.
Wu, Q., Wang, J., Mao, S., Xu, H., Wu, Q., Liang, M., et al. (2019). Comparative transcriptome analyses of genes involved in sulforaphane metabolism at different treatment in Chinese kale using full-length transcriptome sequencing. BMC Genomics 20:377. doi: 10.1186/s12864-019-5758-2
Wyatt, G. R. (1967). The biochemistry of sugars and polysaccharides in insects. Adv. Insect Physiol. 4, 287–360. doi: 10.1016/S0065-2806(08)60210-6
Xu, J. (2013). Research progress in superoxide dismutase and its application. Sci. Technol. Food Ind. 34, 387–391.
Xu, J., Baulding, J., and Palli, S. R. (2013). Proteomics of Tribolium castaneum seminal fluid proteins: identification of an angiotensin-converting enzyme as a key player in regulation of reproduction. J. Proteomics 78, 83–93. doi: 10.1016/j.jprot.2012.11.011
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A. (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11:R14. doi: 10.1186/gb-2010-11-2-r14
Yu, B., Li, D. T., Lu, J. B., Zhang, W. X., and Zhang, C. X. (2016). Seminal fluid protein genes of the brown Planthopper, Nilaparvata lugens. BMC Genomics 17:654. doi: 10.1186/s12864-016-3013-7
Zhao, J., Sun, Y., Xiao, L., Tan, Y., Jiang, Y., and Bai, L. (2018). Vitellogenin and vitellogenin receptor gene expression profiles in Spodoptera exigua are related to host plant suitability. Pest Manag. Sci. 74, 950–958. doi: 10.1002/ps.4794
Zheng, W., Ma, L., Zhao, J., Li, Z., Sun, F., and Lu, X. (2013). Comparative transcriptome analysis of two rice varieties in response to rice stripe virus and small brown Planthoppers during early interaction. PLoS One 8:e82126. doi: 10.1371/journal.pone.0082126
Zhou, Z. S., Guo, J. Y., Chen, H. S., and Wan, F. H. (2010a). Effect of humidity on the development and fecundity of Ophraella communa (Coleoptera: Chrysomelidae). Biocontrol 55, 313–319. doi: 10.1007/s10526-009-9242-9
Zhou, Z. S., Guo, J. Y., Chen, H. S., and Wan, F. H. (2010b). Effects of temperature on survival, development, longevity, and fecundity of Ophraella communa (Coleoptera: Chrysomelidae), a potential biological control agent against Ambrosia artemisiifolia (Asterales: Asteraceae). Environ. Entomol. 39, 1021–1027. doi: 10.1603/EN09176
Keywords: Ophraella communa LeSage, carbon dioxide, heat wave, transcriptome, seminal fluid proteins
Citation: Gao X, Tian Z, Zhang Y, Chen G, Ma C, Tian Z, Cui S, Lu Y and Zhou Z (2020) Transcriptome Analysis of Ophraella communa Male Reproductive Tract in Indirect Response to Elevated CO2 and Heat Wave. Front. Physiol. 11:417. doi: 10.3389/fphys.2020.00417
Received: 15 January 2020; Accepted: 06 April 2020;
Published: 05 May 2020.
Edited by:
Fernando Ariel Genta, Oswaldo Cruz Foundation (Fiocruz), BrazilReviewed by:
Dong Wei, Southwest University, ChinaCan Li, Guiyang University, China
Frank William Avila, University of Antioquia, Colombia
Copyright © 2020 Gao, Tian, Zhang, Chen, Ma, Tian, Cui, Lu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongyue Lu, bHV5b25neXVlQHNjYXUuZWR1LmNu; Zhongshi Zhou, emhvdXpob25nc2hpQGNhYXMuY24=