 Xinhao Wang
Xinhao Wang Hongpeng Zhang1
Hongpeng Zhang1- 1Department of Vascular and Endovascular Surgery, The First Medical Center of Chinese PLA General Hospital, Beijing, China
- 2Department of General Surgery, PLA No. 983 Hospital, Tianjin, China
- 3Department of Obstetrics, Zibo Central Hospital, Zibo, China
Aortic dissection (AD) is a fatal disease that accounts for a large proportion of aortic-related deaths and has an incidence of about 3–4 per 100,000 individuals every year. Recent studies have found that inflammation plays an important role in the development of AD, and that macrophages are the hub of inflammation in the aortic wall. Aortic samples from AD patients reveal a large amount of macrophage infiltration. The sites of macrophage infiltration and activity vary throughout the different stages of AD, with involvement even in the tissue repair phase of AD. Angiotensin II has been shown to be an important factor in the stimulation of macrophage activity. Stimulated macrophages can secrete metalloproteinases, inflammatory factors and other substances to cause matrix destruction, smooth muscle cell apoptosis, neovascularization and more, all of which destroy the aortic wall structure. At the same time, there are a number of factors that regulate macrophages to reduce the formation of AD and induce the repair of torn aortic tissues. The aim of this review is to take a close look at the roles of macrophages throughout the course of AD disease.
Introduction
Aortic dissection (AD) is a fatal disease that accounts for a large proportion of aortic-related deaths (Hiratzka et al., 2010). Epidemiological surveys have shown that the incidence of thoracic AD is 3–4 per 100,000 individuals per year (Olsson et al., 2006; Kochanek et al., 2011). AD is defined as blood flow that enters the aortic media through intimal tears, followed by formation of a true lumen (TL) and a false lumen (FL) with or without communication (Han et al., 2018). Computed tomography angiography (CTA) images of normal aorta and AD in patients from our center are shown in Figure 1. Clinically, AD can cause a series of serious complications, including aortic rupture and visceral ischemia. The majority of untreated patients with extended Stanford type A AD involving the ascending aorta die within 2 weeks (Chen et al., 1997). Clinical treatment options for AD include optimal medical treatment, aortic replacement, and thoracic endovascular aorta repair (Jia et al., 2013). However, the mortality rate for patients after treatment for AD remains high (Khayat et al., 2018). Surgical mortality for AD ranges from 10 to 35%, even at experienced medical centers (Nienaber and Clough, 2015). Unfortunately, because of its sudden and unpredictable nature, little is known about the pathological and molecular events that occur before and after the onset of AD, thus it is critical that we clarify the pathogenesis.
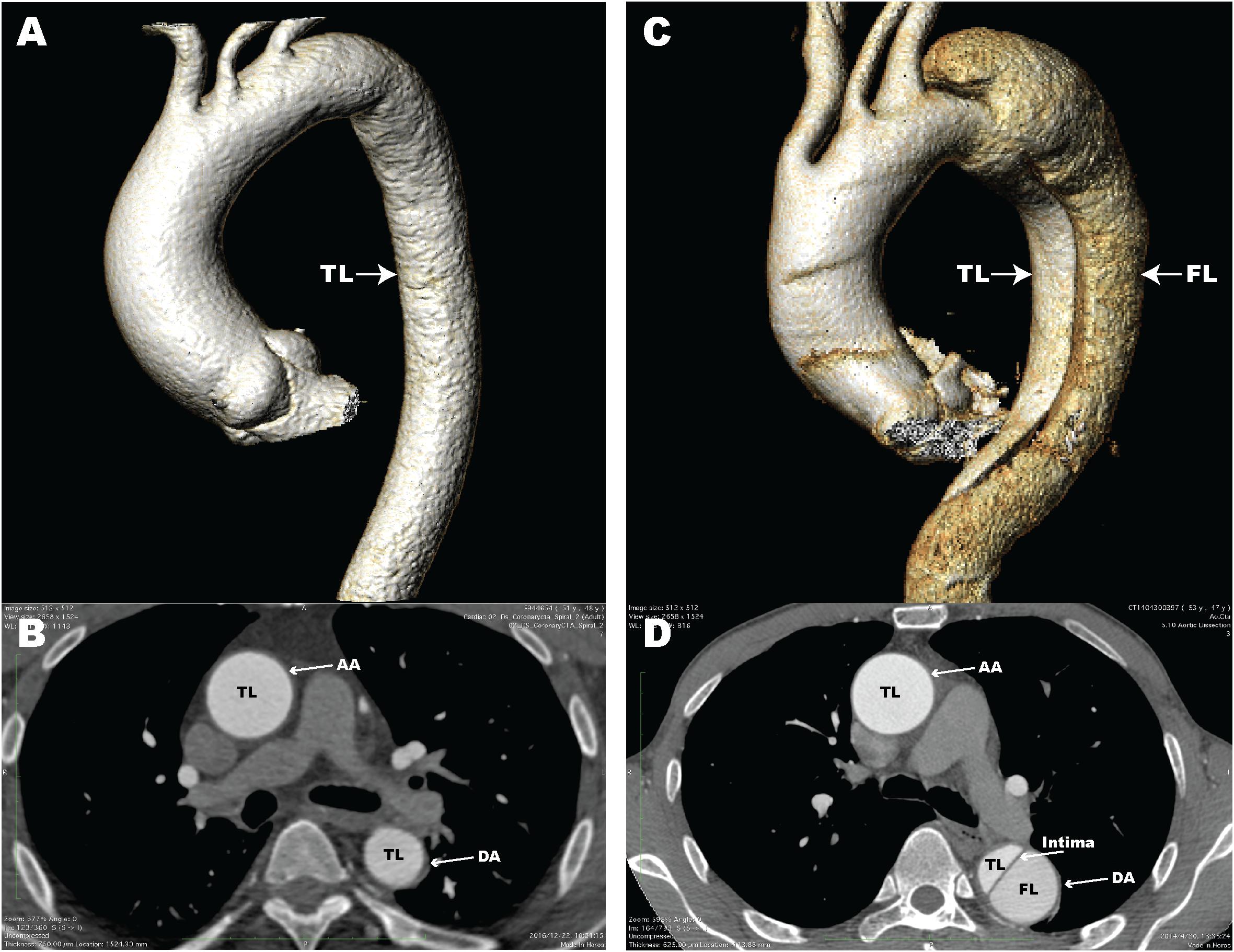
Figure 1. Computed tomography angiography (CTA) images of a normal aorta and an aortic dissection (AD). (A) 3D reconstructed CTA image of a normal aorta; (B) Axial CTA image of a normal aorta with blood flow only in the true lumen (TL); (C) 3D reconstructed CTA image from a patient with AD; (D) Axial CTA image of AD with blood flow in both the true lumen and false lumen (FL). The aortic intima is located between the TL and FL. AA, ascending aorta; DA, descending aorta.
While ascending ADs are clearly linked to inherited connective tissue diseases (Isselbacher, 2005), most ADs occur in the sixth decade of life in hypertensive populations without genetic susceptibility (Hagan et al., 2000). The main histological finding related to arterial wall weakening in AD is medial degeneration, which consists of profound degradation of the extracellular matrix (ECM) involving smooth muscle cell (SMC) depletion (Ramanath et al., 2009; Wang et al., 2012; An et al., 2017), elastic fiber fragmentation (Nakashima, 2010; Pratt and Curci, 2010; Roberts et al., 2011) and collagen degradation (Wang et al., 2006; Ren et al., 2014; Xu K. et al., 2018). However, many studies have demonstrated that the formation of AD is associated with aortic wall inflammation (Wu et al., 2013; Cifani et al., 2015; Niinimak et al., 2018). A recent study in a murine model demonstrated that indomethacin reduces the rates of AD by inhibiting macrophage accumulation (Tomida et al., 2019). Macrophages, which have both inflammatory and anti-inflammatory effects, are involved in the development of AD (Andreata et al., 2018; Visona et al., 2018; Xu Y. et al., 2018; Ye et al., 2018) as well as the complications of AD, such as acute AD-associated lung injury (Wu et al., 2016). Our laboratory focuses on identifying the factors that cause the accumulation of macrophages in the aortic wall, and how the effects of macrophages on the structure of the aortic wall lead to AD. Here, we review current knowledge about the role of macrophages in the formation of AD, including their upstream regulators and downstream effectors.
Evidence of Macrophage Participation in the Formation of Ad
Macrophages play a crucial role in aortic wall inflammation, and also are involved in AD. In an AD mouse model, T lymphocytes, macrophages and neutrophils simultaneously infiltrate the aorta when AD occurs, with macrophages being the most abundant cell type (Li et al., 2019). There is significant infiltration of macrophages into the tear section in AD patients (Xu Y. et al., 2018; Ye et al., 2018). Macrophage infiltration may be more severe in AD than in aortic aneurysm (Pisano et al., 2017). Half of the macrophages in the torn part of the AD mice model are BM-derived and half are non-BM-derived (Zou et al., 2019). Studies in AD cases classified as Stanford type A have shown that the aggregation of macrophages in the aortic media is critical for early AD formation (Cifani et al., 2015; Niinimak et al., 2018). However, the infiltration activity and location of macrophages vary among the different stages of AD. Overall, macrophages first accumulate in the aortic adventitia and infiltrate the media to promote a local inflammatory response after dissection. Compared with chronic AD (CAD), acute AD (AAD) is characterized by more severe inflammation in the media and adventitia, and more macrophage infiltration (Wu et al., 2013). Macrophages (CD68+) in the acute phase are concentrated in the hematoma, and the intima and media adjacent to the hematoma. In the subacute and early organizing phases, macrophages are mainly concentrated in the peripheral adipose tissue. The degree of macrophage infiltration is related to the repair process of the clot and adjacent vessel walls, and occurs in a time-dependent manner (Visona et al., 2018). This suggests that macrophages may be involved in not only AD formation but also AD remodeling. In fact, M1 macrophages are known to promote inflammation, while M2 macrophages eliminate inflammation and secrete and stabilize matrix components (Duffield, 2003). In the aortic repair stage, macrophages in the peripheral adipose tissue aggregate to participate in vascular wall repair.
Macrophages are also associated with some complications of AD, including FL rupture (Xu and Burke, 2013) and acute lung injury (Wu et al., 2016, 2017a,b). Therefore, macrophages play a vital role in AD.
Angiotensin Ii Regulates Macrophages to Cause the Onset of Ad
Studies have shown that Angiotensin II (Ang II) contributes to the development of AD in humans and experimental animals (Daugherty et al., 2000). Ang II is the main effector peptide of the renin-angiotensin system. It can induce vasoconstriction, hypertrophy, and extracellular remodeling through the Ang II type 1 receptor (AT1R) (Dimmeler et al., 1997; Schluter and Wenzel, 2008). The regulatory effect of Ang II on macrophages is also crucial in AD. During the onset of AD, Ang II can promote the infiltration of macrophages from the aortic adventitia to the media through a pathogenic pathway involving serum lipid composition (Tanaka et al., 2018). Weighted gene co-expression network analysis has identified FKBP11 as a key regulator in Ang II-induced AD. FKBP11 operates through a nuclear factor-kB-dependent pathway, and is hypothesized to promote macrophage infiltration and M1 differentiation (Wang et al., 2017). Ang II also promotes the infiltration of macrophages and the secretion of matrix metalloproteinases (MMPs) via the axis of Kruppel-like factor 6 and granulocyte macrophage-colony-stimulating factor (GM-CSF) (Son et al., 2015), as well as a disintegrin and metalloproteinase with thrombospondin motifs 1 (ADAMTS-1) (Gao et al., 2016), to cause local inflammation and tissue destruction. In addition, GM-CSF triggers the separation of the aortic wall (Son et al., 2015).
Several factors are known to enhance the regulation of macrophages by Ang II in AD. First, the rs12455792 variant of the SMAD4 gene has been shown to increase macrophage recruitment and the M1 type inflammatory response via activation of transforming growth factor-β signaling, and also to promote the vascular degeneration and pathological progress of thoracic AD (Wang Y. et al., 2018). Second, the T helper cell (Th)17-interleukin (IL)-17 axis, which is regulated by IL-6-signal transducer and activator of transcription (STAT)3 signaling, is a key regulator of Ang II-induced vascular inflammation. It works upstream of the macrophage recruitment induced by Ang II (Ju et al., 2013). Third, thrombospondin 1, which increases mRNA expression inducible nitric oxide synthase (iNOS) in macrophages treated with Ang II, might participate in AD by boosting differentiation of M1 macrophages and apoptosis of SMCs (Zeng et al., 2019). In addition, vascular SMC-specific E-prostanoid receptor 4 deletion exacerbates Ang II-induced AD by increasing macrophage infiltration (Xu et al., 2019). Also, the study by Ju et al. (2013) indicated that the acceleration of AD in Ang II-infused mice had nothing to do with the Ang II-associated increase in blood pressure (Ju et al., 2013). This suggests that simply lowering blood pressure without also reducing the amount of Ang II in serum might adversely affect efforts to control the incidence of AD.
Macrophages Secrete Multiple Factors Involved in Ad Formation
Studies have shown that macrophages secrete a variety of factors associated with AD, including metalloproteinases, ILs, vascular endothelial growth factor (VEGF), and others. These factors cause further macrophage recruitment, vascular SMC apoptosis and elastic fiber degradation, ultimately leading to AD.
Metalloproteinases That Play an Important Role in AD
Metalloproteinases include an extensive zinc-dependent collagenases and elastases, which belong to the superfamily of metzincins, and are closely related to inflammation and tissue damage. Each metalloproteinase has multiple expression profiles that are unique to different tissue types and occur during inflammation, each exerting specific functions (Visse and Nagase, 2003). Several are secreted by macrophages and participate in the formation of AD. The most important of these are the MMPs and the ADAMTS enzymes.
Matrix metalloproteinases are able to regulate inflammation and tissue remodeling when in balance with tissue inhibitors of metalloproteinases (TIMPs), and this regulation plays an important role in AD. An imbalance between MMPs and their TIMPs can cause AD artery wall remodeling and degradation of the exogenous matrix (Cifani et al., 2015). Studies have shown that a variety of the MMPs that are elevated in AD – namely MMP-8, MMP-9 and MMP-12 – are mainly secreted by macrophages. Among these, MMP-9 is also closely related to acute lung injury caused by AD (Wu et al., 2017b), and targeted depletion of macrophages suppresses AD together with spatial regulation of MMP-9 in the aorta (Li et al., 2019). While MMP-3 itself is not secreted by macrophages, it is also significantly elevated in AD patients and can activate MMP-8 secreted by macrophages. This amplification cascade induces a widespread degradation of the aortic wall (Gronski et al., 1997). At the same time, studies have shown that MMP-12 is a biomarker for some types of AD, and that it can even play a role in identifying patients at greater risk of AD (Proietta et al., 2014; Cifani et al., 2015; Liu et al., 2018). Taken together, these data on MMPs support the importance of macrophages in the development of AD.
The ADAMTS enzymes play important roles in many vascular diseases and, similar to MMPs, are linked to tissue destruction and inflammation. The ADAMTS family comprise key extracellular metalloproteinases involved in ECM turnover (Gao et al., 2016; Ren et al., 2017; Wang S. et al., 2018). ADAMTS-1 and ADAMTS-4 were found to be related to AD, with increased expression levels in macrophages (Ren et al., 2013). Increased levels of circulating ADAMTS-1 have been correlated with the presence of accumulated ADAMTS-1-positive macrophages in aortic tissues in AAD patients (Ren et al., 2013; Gao et al., 2016; Wang S. et al., 2018). In experiments on elderly mice fed with Ang II, the incidence of AD was 42%, and the macrophages and neutrophils that infiltrated the aortic media were found to have elevated ADAMTS-1. The aortic tissue from AAD mice exhibited enhanced expression of ADAMTS-1, and ADAMTS1-immunoreactive macrophages infiltrated the intima, media and adventitia in dissected aortic walls (Gao et al., 2016). The incidence and rupture rates of β-aminopropionitrile-induced AD in ADAMTS-1 knockout mice were significantly lower than those in ADAMTS-1flox/flox mice (Wang S. et al., 2018). ADAMTS-4 has also been shown to be directly associated with AD in a mouse model, causing vascular SMC apoptosis, elastic fiber destruction, and versican degradation in the aortic wall. ADAMTS-4–/– mice had a reduced incidence of AD (Ren et al., 2017). Also, a recent study established that metabolic reprogramming in macrophages has a pivotal role in hypoxia-inducible factor-1α-ADAM17 signaling activation and furthers the development of AD (Lian et al., 2019).
Effects of ILs on the Aortic Wall
The ILs comprise a family of important proinflammatory cytokines that are thought to be to secreted mainly by macrophages (Shimizu et al., 2006; Golledge et al., 2008). Many ILs participate in the formation of AD. Studies have shown that IL-6, IL-8 (Proietta et al., 2014), IL-11 (Xu Y. et al., 2018), IL-12 (Ye et al., 2018), IL-16 (Fan et al., 2017) and IL-18 (Hu et al., 2019) are elevated in the serum of AD patients, and may represent biomarkers for the diagnosis of AD. Genes related to IL-3 are highly expressed in AD patients, and in vitro experiments have shown that IL-3 can increase MMP-12 expression in macrophages via the pathway involving the c-JUN N-terminal kinase and extracellular signal-related kinase 1/2 pathway by binding to the IL-3β receptor (Liu et al., 2018). IL-6 might be required for macrophage activation in the early vascular inflammation that leads to AD (Tieu et al., 2011). Previous studies have shown that IL-6 signaling is mediated by macrophage activation in Ang II-induced vascular disease (Schuett et al., 2009), and that the IL-6-STAT3 model pathway regulates the downstream Th7-IL-17 axis and upregulates monocyte macrophage activity (Ju et al., 2013). IL-18 may promote M1 macrophage differentiation and increase macrophage-induced apoptosis of SMCs (Hu et al., 2019). Taken together, it appears that macrophages secrete ILs to promote local inflammation of the aortic wall, while receiving regulatory signals from ILs to further expand the inflammatory response, for the ultimate promotion of AD.
VEGF-Mediated Neoangiogenesis Also Contributes to AD Formation
Research from Del Porto et al. (2014) has demonstrated that VEGF-mediated neoangiogenesis plays an important role in ascending aortic wall remodeling. VEGF was mainly found in pro-inflammatory macrophages, and in the endothelial cells that constitute the neovessel walls spreading throughout the tunica media (Del Porto et al., 2014). Neoangiogenesis has been shown to promote inflammation (Reinders et al., 2003) and trigger matrix degradation (Galis et al., 1994) and, therefore, participate in progression and destabilization of atherosclerotic lesions (Slevin et al., 2009). Taken together, it appears that VEGF release and neoangiogenesis may participate in the progression of aortic wall injury, via both inflammation and matrix degradation. In addition, the growth of structurally altered vessels (Melter et al., 2000) that are prone to rupture and bleeding may represent the starting point of the delamination of the aortic media.
In summary, macrophages can secrete a variety of substances involved in AD and related conditions. Macrophages utilize these substances to cause elastic fiber degradation, vascular SMC apoptosis, and neovascularization. This series of changes weakens the aortic wall and creates conditions that are ripe for the development of AD.
Macrophage-Associated Factors That Protect Against Ad
Substances such as Ang II can regulate macrophages through a series of signal transductions, increasing the incidence of AD. At the same time, there are many protective factors that regulate macrophages in ways that can reduce the incidence of AD.
The suppressor of cytokine signaling 3 (Socs3) gene in macrophages may play a critical role in protecting the aorta from AD. In wild-type mice, focal medial disruption of the aorta rarely causes AD development. However, Socs3 deletion in macrophages increased proliferation and inflammation, biased differentiation of macrophages toward a tissue-destructive phenotype, and dysregulated the differentiation of vascular SMCs. These findings may be clinically relevant, as immunofluorescence staining and imaging cytometry analysis of human AD tissue suggested that adventitial macrophage STAT3 in the aortic wall was activated in regions adjacent to the dissected lesion and at risk of destruction (Ohno-Urabe et al., 2018). Thus, Socs3 in macrophages appears to act as a protector in stressed aorta, relieving excessive inflammation and triggering the progression of tissue repair, including proper modulation of vascular SMC function.
Glucocorticoids promote vascular remodeling by reducing tumor necrosis factor (TNF)-α secretion and increasing the levels of uncombined soluble TNF receptor II (TNF-sRII) to inhibit AD formation. Increased glucocorticoids reduce the marginalization, extravasation and local activation of macrophages, thereby inhibiting MMP-2 secretion and thus protecting collagen degradation. These findings indicate that glucocorticoids and TNF-sRII may be interesting targets for future AD intervention (Zhang et al., 2018).
Micro RNA (miR)-320 was shown to be significantly downregulated in dissected aortic tissue. MiR-320 may participate in post-transcriptional processing of several MMPs. In fact, overexpressed miR-320 was able to inhibit MMP secretion. A recent study indicated that low miR-320 expression leads to insufficient suppression of MMP secretion, leading to higher expression of MMPs, aggravated ECM destruction, and increased risk of AD (Liao et al., 2018).
Administration of a CD31 agonist peptide greatly reduced the incidence of AD in ApoE–/– mice, and was found bind to wound-associated leukocytes, including macrophages (Fornasa et al., 2012; Andreata et al., 2018). In addition, CD31 signaling in macrophages facilitates aortic remodeling and healing after dissection. An analysis of AD and intramural hematoma samples from patients undergoing surgical treatment revealed that the CD31 expression was lost by the M1 macrophages that densely penetrated into the acute aortic wall lesion sites, with CD31 re-expression accompanied the appearance of M2 and the disappearance of M1 macrophages at the valid aortic wall healing sites (Fornasa et al., 2012; Andreata et al., 2018). M1 macrophages are considered to play a necessary role in early AD proinflammation, and the evidence also supports the view that the transition of wound-related macrophages from the proinflammatory M1 to the pro-reparative M2 phenotype plays a key role in driving inflammation resolution and promoting wound healing (Mantovani et al., 2013; Andreata et al., 2018). These studies suggest that macrophages play an important role in the early stages of AD, and an irreplaceable role in the remodeling and healing of AD. In other words, macrophage activity may persist throughout the course of AD disease.
The macrophage class A1 scavenger receptor (SR-A1)-Tyro3 axis in macrophages reduces AD damage by promoting efferocytosis and suppressing inflammation. SR-A1 deficiency augments AD in mice, facilitates vascular inflammation and apoptosis, and inhibits macrophage efferocytosis. Furthermore, these effects of SR-A1 deficiency can be attenuated by activation of the Tyro3 pathway (Zhang et al., 2019). The beneficial effects of the SR-A1-Tyro3 axis in AD may facilitate the development of a unified mechanism of inflammation and exocytosis in macrophages.
Sestrin-2 (SESN2) is an important antioxidant protein that is mainly secreted by macrophages. The expression of Sestrin-2 is significantly higher in the aorta and plasma of AD patients than in healthy donors. In co-cultures of macrophages and SMCs, the overexpression of SESN2 in macrophages significantly reduced apoptosis of Ang II-induced SMCs; this effect was reversed by NRF2 silencing. In other words, Sestrin-2 may reduce Ang II-induced SMC apoptosis and participate in AD through the NRF2 pathway (Xiao et al., 2019).
Conclusion and Outlook
AD is a disease in which the structure of the aortic wall is degraded, eventually causing the intima to rupture, with blood entering the media to form a FL (Han et al., 2018). Macrophages respond mainly to Ang II, and the recruitment, activity and secretion of macrophages in the aortic wall trigger and maintain inflammation there (Dimmeler et al., 1997; Daugherty et al., 2000; Schluter and Wenzel, 2008). Proinflammatory cytokines and metalloproteinases are the main secretions involved in this process. Macrophages have been shown to cause vascular SMC apoptosis, elastic fiber degradation (Ren et al., 2017) and neovascularization (Del Porto et al., 2014) leading to the destruction and separation of the aortic wall in both humans and mice. M2 polarization also plays an important role in the repair and remodeling process after the dissection has occurred, with M2 macrophages inhibiting the development of inflammation and promoting the repair of local tissues (Fornasa et al., 2012; Mantovani et al., 2013). This feature is critical to the stability of AD. In general, macrophages play a crucial role in AD and are potential targets for the prevention and treatment of this disease. And, the summarize diagram of this review was shown in Figure 2.
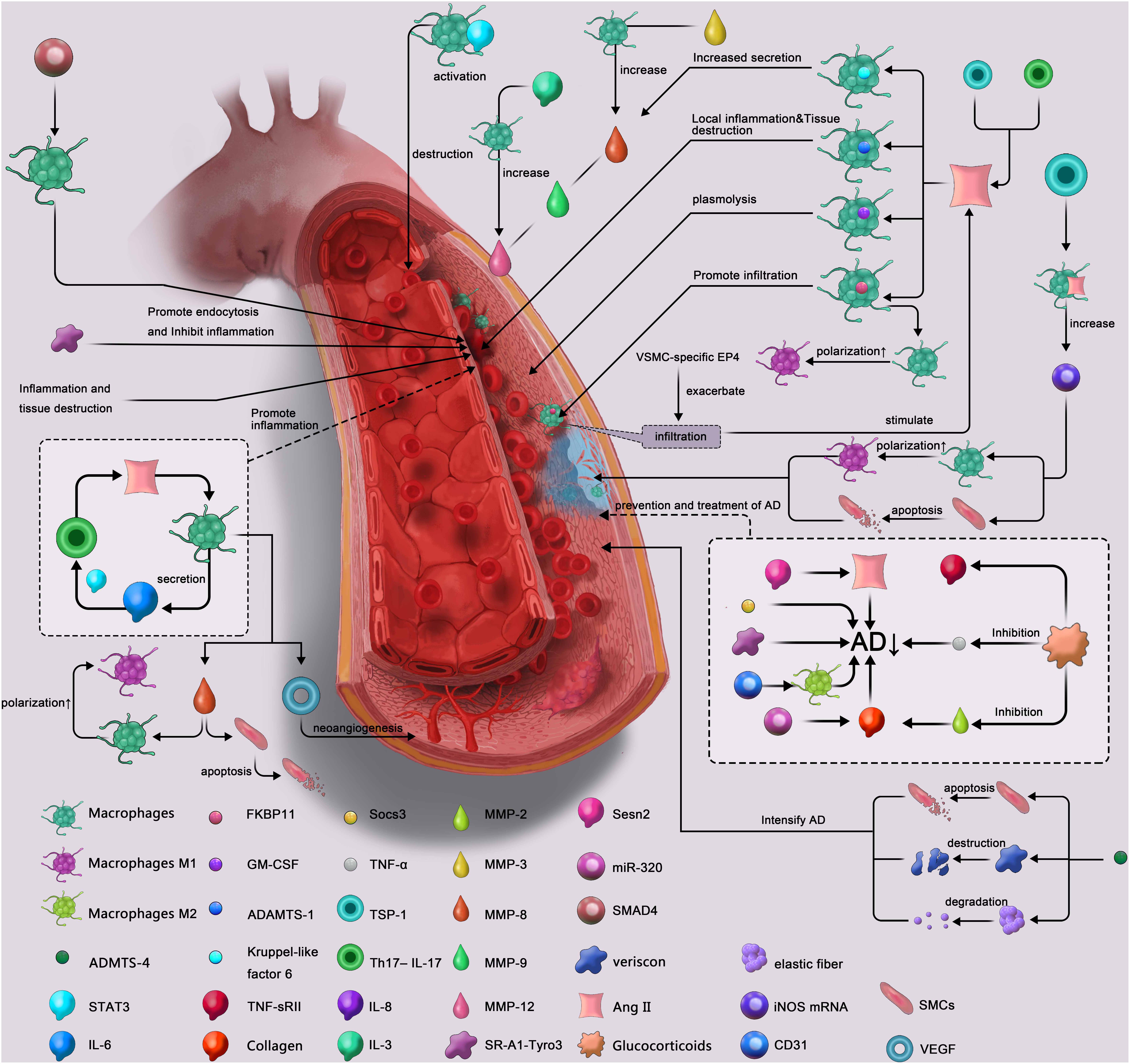
Figure 2. Summarize diagram of the role of macrophage in aortic dissection. AD↓, reduces the incidence of aortic dissection; ADAMTS, a disintegrin and metalloproteinase with thrombospondin motifs; STAT, signal transducer and activator of transcription; IL, Interleukin; GM-CSF, granulocyte macrophage-colony-stimulating factor; TNF-sR II, tumor necrosis factor receptor II; Socs 3, suppressor of cytokine signaling 3; TNF, tumor necrosis factor; TSP-1, thrombospondin 1; Th17-IL-17, T helper cell 17-interleukin -17; MMP, matrix metalloproteinase; SR-A1-Tyro3, A1 scavenger receptor-Tyro3 axis; miRNA, microRNA; Ang II, angiotensin II; iNOS mRNA, inducible nitric oxide synthase mRNA; SMCs, smooth muscle cells; VEGF, vascular endothelial growth factor.
Increasingly, AD-related studies are shifting their focus to aortic inflammation and the role of macrophages in AD formation and repair (Fornasa et al., 2012; Andreata et al., 2018). Thus far, efforts at understanding the contribution of inflammation and macrophages to AD have relied upon analyses of clinical samples and mechanistic studies in animal models. However, because of the contingency of lethality of AD disease, it is rare to obtain preoperative samples or imaging data in patients, and most experiments in animals are designed to wait for the AD to occur before material is collected for analysis. Monitoring of the dynamic changes in inflammation and macrophage activity throughout the development of AD would allow us to distinguish between the two possibilities of accumulated inflammation that eventually leads to AD, and intense inflammation that is triggered by AD. Currently, there is only one medical case report with documented abnormal inflammatory activity in the aortic wall that gradually increased in the 5 years leading up to the patient’s eventual AD (Tahara et al., 2016). Because the patient in that case received repeated combined positron emission tomography (PET)/computed tomography, it is possible that technologies such as PET and 4D magnetic resonance imaging could play a crucial role in revealing the mechanisms of AD, dynamic changes in inflammation and macrophage activity.
Author Contributions
HZ contributed in interpretation of data of the work and critical revision of the work. AM contributed in analysis of data of the work, figure drawing, and critical revision of the work. All authors above have approved the version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Ye Wu and Yangyang Ge for their suggestions for this review.
References
An, Z., Liu, Y., Song, Z. G., Tang, H., Yuan, Y., and Xu, Z. Y. (2017). Mechanisms of aortic dissection smooth muscle cell phenotype switch. J. Thorac. Cardiovasc. Surg. 154:e6.
Andreata, F., Syvannarath, V., Clement, M., Delbosc, S., Guedj, K., Fornasa, G., et al. (2018). Macrophage CD31 signaling in dissecting aortic aneurysm. J. Am. Coll. Cardiol. 72, 45–57. doi: 10.1016/j.jacc.2018.04.047
Chen, K., Varon, J., Wenker, O. C., Judge, D. K., Fromm, R. E. Jr., and Sternbach, G. L. (1997). Acute thoracic aortic dissection: the basics. J. Emerg. Med. 15, 859–867. doi: 10.1016/s0736-4679(97)00196-0
Cifani, N., Proietta, M., Tritapepe, L., Di Gioia, C., Ferri, L., Taurino, M., et al. (2015). Stanford-A acute aortic dissection, inflammation, and metalloproteinases: a review. Ann. Med. 47, 441–446. doi: 10.3109/07853890.2015.1073346
Daugherty, A., Manning, M. W., and Cassis, L. A. (2000). Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J. Clin. Invest. 105, 1605–1612. doi: 10.1172/jci7818
Del Porto, F., di Gioia, C., Tritapepe, L., Ferri, L., Leopizzi, M., Nofroni, I., et al. (2014). The multitasking role of macrophages in Stanford type A acute aortic dissection. Cardiology 127, 123–129. doi: 10.1159/000355253
Dimmeler, S., Rippmann, V., Weiland, U., Haendeler, J., and Zeiher, A. M. (1997). Angiotensin II induces apoptosis of human endothelial cells. Protective effect of nitric oxide. Circ. Res. 81, 970–976. doi: 10.1161/01.res.81.6.970
Duffield, J. S. (2003). The inflammatory macrophage: a story of Jekyll and Hyde. Clin. Sci. 104, 27–38. doi: 10.1042/cs1040027
Fan, F. D., Xu, Z. J., Zhou, Q., and Wang, D. J. (2017). Expression profiles and clinical implication of plasma chemokines in patients with stanford type a aortic dissection. Zhonghua Xin Xue Guan Bing Za Zhi 45, 318–322. doi: 10.3760/cma.j.issn.0253-3758.2017.04.012
Fornasa, G., Clement, M., Groyer, E., Gaston, A. T., Khallou-Laschet, J., Morvan, M., et al. (2012). A CD31-derived peptide prevents angiotensin II-induced atherosclerosis progression and aneurysm formation. Cardiovasc. Res. 94, 30–37. doi: 10.1093/cvr/cvs076
Galis, Z. S., Muszynski, M., Sukhova, G. K., Simon-Morrissey, E., Unemori, E. N., Lark, M. W., et al. (1994). Cytokine-stimulated human vascular smooth muscle cells synthesize a complement of enzymes required for extracellular matrix digestion. Circ. Res. 75, 181–189. doi: 10.1161/01.res.75.1.181
Gao, Y., Wu, W., Yu, C., Zhong, F., Li, G., Kong, W., et al. (2016). A disintegrin and metalloproteinase with thrombospondin motif 1 (ADAMTS1) expression increases in acute aortic dissection. Sci. China Life Sci. 59, 59–67. doi: 10.1007/s11427-015-4959-4
Golledge, J., Tsao, P. S., Dalman, R. L., and Norman, P. E. (2008). Circulating markers of abdominal aortic aneurysm presence and progression. Circulation 118, 2382–2392. doi: 10.1161/circulationaha.108.802074
Gronski, T. J. Jr., Martin, R. L., Kobayashi, D. K., Walsh, B. C., Holman, M. C., Huber, M., et al. (1997). Hydrolysis of a broad spectrum of extracellular matrix proteins by human macrophage elastase. J. Biol. Chem. 272, 12189–12194. doi: 10.1074/jbc.272.18.12189
Hagan, P. G., Nienaber, C. A., Isselbacher, E. M., Bruckman, D., Karavite, D. J., Russman, P. L., et al. (2000). The international registry of acute aortic dissection (IRAD): new insights into an old disease. JAMA 283, 897–903.
Han, L., Dai, L., Zhao, Y. F., Li, H. Y., Liu, O., Lan, F., et al. (2018). CD40L promotes development of acute aortic dissection via induction of inflammation and impairment of endothelial cell function. Aging 10, 371–385. doi: 10.18632/aging.101394
Hiratzka, L. F., Bakris, G. L., Beckman, J. A., Bersin, R. M., Carr, V. F., and Casey, D. E. (2010). 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease. A report of the american college of cardiology foundation/american heart association task force on practice guidelines, american association for thoracic surgery, american college of radiology, american stroke association, society of cardiovascular anesthesiologists, society for cardiovascular angiography and interventions, society of interventional radiology, society of thoracic surgeons, and society for vascular medicine. J. Am. Coll. Cardiol. 55:e0027-29.
Hu, H., Zhang, G., Hu, H., Liu, W., Liu, J., Xin, S., et al. (2019). Interleukin-18 expression increases in the aorta and plasma of patients with acute aortic dissection. Mediators Inflamm. 2019:8691294. doi: 10.1155/2019/8691294
Jia, X., Guo, W., Li, T. X., Guan, S., Yang, R. M., Liu, X. P., et al. (2013). The results of stent graft versus medication therapy for chronic type B dissection. J. Vasc. Surg. 57, 406–414. doi: 10.1016/j.jvs.2012.08.064
Ju, X., Ijaz, T., Sun, H., Ray, S., Lejeune, W., Lee, C., et al. (2013). Interleukin-6-signal transducer and activator of transcription-3 signaling mediates aortic dissections induced by angiotensin II via the T-helper lymphocyte 17-interleukin 17 axis in C57BL/6 mice. Arterioscler. Thromb. Vasc. Biol. 33, 1612–1621. doi: 10.1161/ATVBAHA.112.301049
Khayat, M., Cooper, K. J., Khaja, M. S., Gandhi, R., Bryce, Y. C., and Williams, D. M. (2018). Endovascular management of acute aortic dissection. Cardiovasc. Diagn. Ther. 8, S97–S107. doi: 10.21037/cdt.2017.10.07
Kochanek, K. D., Xu, J., Murphy, S. L., Minino, A. M., and Kung, H. C. (2011). Deaths: final data for 2009. Natl. Vital Stat. Rep. 60, 1–116.
Li, X., Liu, D., Zhao, L., Wang, L., Li, Y., Cho, K., et al. (2019). Targeted depletion of monocyte/macrophage suppresses aortic dissection with the spatial regulation of MMP-9 in the aorta. Life Sci. [Epub ahead of print].
Lian, G., Li, X., Zhang, L., Zhang, Y., Sun, L., Zhang, X., et al. (2019). Macrophage metabolic reprogramming aggravates aortic dissection through the HIF1alpha-ADAM17 pathway. eBioMed. 49:41. doi: 10.1016/j.ebiom.2019.09.041
Liao, M., Zou, S., Bao, Y., Jin, J., Yang, J., Liu, Y., et al. (2018). Matrix metalloproteinases are regulated by MicroRNA 320 in macrophages and are associated with aortic dissection. Exp. Cell Res. 370, 98–102. doi: 10.1016/j.yexcr.2018.06.011
Liu, C., Zhang, C., Jia, L., Chen, B., Liu, L., Sun, J., et al. (2018). Interleukin-3 stimulates matrix metalloproteinase 12 production from macrophages promoting thoracic aortic aneurysm/dissection. Clin. Sci. 132, 655–668. doi: 10.1042/CS20171529
Mantovani, A., Biswas, S. K., Galdiero, M. R., Sica, A., and Locati, M. (2013). Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 229, 176–185. doi: 10.1002/path.4133
Melter, M., Reinders, M. E., Sho, M., Pal, S., Geehan, C., Denton, M. D., et al. (2000). Ligation of CD40 induces the expression of vascular endothelial growth factor by endothelial cells and monocytes and promotes angiogenesis in vivo. Blood 96, 3801–3808. doi: 10.1182/blood.v96.12.3801.h8003801_3801_3808
Nakashima, Y. (2010). Pathogenesis of aortic dissection: elastic fiber abnormalities and aortic medial weakness. Ann. Vasc. Dis. 3, 28–36. doi: 10.3400/avd.ctiia09001
Nienaber, C. A., and Clough, R. E. (2015). Management of acute aortic dissection. Lancet 385, 800–811. doi: 10.1016/S0140-6736(14)61005-9
Niinimak, E., Pynnonen, V., Kholova, I., Paavonen, T., and Mennander, A. (2018). Neovascularization with chronic inflammation characterizes ascending aortic dissection. Anatol. J. Cardiol. 20, 289–295. doi: 10.14744/AnatolJCardiol.2018.42223
Ohno-Urabe, S., Aoki, H., Nishihara, M., Furusho, A., Hirakata, S., Nishida, N., et al. (2018). Role of macrophage Socs3 in the pathogenesis of aortic dissection. J. Am. Heart Assoc. 7, e007389. doi: 10.1161/JAHA.117.007389
Olsson, C., Thelin, S., Stahle, E., Ekbom, A., and Granath, F. (2006). Thoracic aortic aneurysm and dissection: increasing prevalence and improved outcomes reported in a nationwide population-based study of more than 14,000 cases from 1987 to 2002. Circulation 114, 2611–2618. doi: 10.1161/circulationaha.106.630400
Pisano, C., Balistreri, C. R., Ricasoli, A., and Ruvolo, G. (2017). Cardiovascular disease in ageing: an overview on thoracic aortic aneurysm as an emerging inflammatory disease. Mediators Inflamm. 2017:1274034. doi: 10.1155/2017/1274034
Pratt, B., and Curci, J. (2010). Arterial elastic fiber structure. Function and potential roles in acute aortic dissection. J. Cardiovasc. Surg. 51, 647–656.
Proietta, M., Tritapepe, L., Cifani, N., Ferri, L., Taurino, M., and Del Porto, F. (2014). MMP-12 as a new marker of Stanford-A acute aortic dissection. Ann. Med. 46, 44–48. doi: 10.3109/07853890.2013.876728
Ramanath, V. S., Oh, J. K., Sundt, T. M. III, and Eagle, K. A. (2009). Acute aortic syndromes and thoracic aortic aneurysm. Mayo Clin. Proc. 84, 465–481. doi: 10.1016/S0025-6196(11)60566-1
Reinders, M. E., Sho, M., Robertson, S. W., Geehan, C. S., and Briscoe, D. M. (2003). Proangiogenic function of CD40 ligand-CD40 interactions. J. Immunol. 171, 1534–1541. doi: 10.4049/jimmunol.171.3.1534
Ren, P., Hughes, M., Krishnamoorthy, S., Zou, S., Zhang, L., Wu, D., et al. (2017). Critical role of adamts-4 in the development of sporadic aortic aneurysm and dissection in mice. Sci. Rep. 7:12351. doi: 10.1038/s41598-017-12248-z
Ren, P., Zhang, L., Xu, G., Palmero, L. C., Albini, P. T., Coselli, J. S., et al. (2013). ADAMTS-1 and ADAMTS-4 levels are elevated in thoracic aortic aneurysms and dissections. Ann. Thorac. Surg. 95, 570–577. doi: 10.1016/j.athoracsur.2012.10.084
Ren, Z., Wang, Z., Hu, Z., Hu, X., Zhang, H., Wu, H., et al. (2014). Decreased expression of P54(nrb) /NonO correlates with collagen deposition and fibrosis in human aortic dissection. Histopathology 65, 570–580. doi: 10.1111/his.12434
Roberts, W. C., Vowels, T. J., Kitchens, B. L., Ko, J. M., Filardo, G., Henry, A. C., et al. (2011). Aortic medial elastic fiber loss in acute ascending aortic dissection. Am. J. Cardiol. 108, 1639–1644. doi: 10.1016/j.amjcard.2011.09.005
Schluter, K. D., and Wenzel, S. (2008). Angiotensin II: a hormone involved in and contributing to pro-hypertrophic cardiac networks and target of anti-hypertrophic cross-talks. Pharmacol. Ther. 119, 311–325. doi: 10.1016/j.pharmthera.2008.05.010
Schuett, H., Luchtefeld, M., Grothusen, C., Grote, K., and Schieffer, B. (2009). How much is too much? Interleukin-6 and its signalling in atherosclerosis. Thromb. Haemost. 102, 215–222. doi: 10.1160/TH09-05-0297
Shimizu, K., Mitchell, R. N., and Libby, P. (2006). Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 26, 987–994. doi: 10.1161/01.atv.0000214999.12921.4f
Slevin, M., Krupinski, J., and Badimon, L. (2009). Controlling the angiogenic switch in developing atherosclerotic plaques: possible targets for therapeutic intervention. J. Angiogenes. Res. 1:4. doi: 10.1186/2040-2384-1-4
Son, B. K., Sawaki, D., Tomida, S., Fujita, D., Aizawa, K., Aoki, H., et al. (2015). Granulocyte macrophage colony-stimulating factor is required for aortic dissection/intramural haematoma. Nat. Commun. 6:6994. doi: 10.1038/ncomms7994
Tahara, N., Hirakata, S., Okabe, K., Tahara, A., Honda, A., Igata, S., et al. (2016). FDG-PET/CT images during 5 years before acute aortic dissection. Eur. Heart J. 37:1933. doi: 10.1093/eurheartj/ehw075
Tanaka, H., Iida, Y., Iwaki, T., Suzuki, Y., Sano, H., Miyajima, C., et al. (2018). Elevated plasma levels of LDL cholesterol promote dissecting thoracic aortic aneurysms in angiotensin ii-induced Mice. Ann. Vasc. Surg. 48, 204–213. doi: 10.1016/j.avsg.2017.10.006
Tieu, B. C., Ju, X., Lee, C., Sun, H., Lejeune, W., Recinos, A., et al. (2011). Aortic adventitial fibroblasts participate in angiotensin-induced vascular wall inflammation and remodeling. J. Vasc. Res. 48, 261–272. doi: 10.1159/000320358
Tomida, S., Aizawa, K., Nishida, N., Aoki, H., Imai, Y., Nagai, R., et al. (2019). Indomethacin reduces rates of aortic dissection and rupture of the abdominal aorta by inhibiting monocyte/macrophage accumulation in a murine model. Sci. Rep. 9:10751. doi: 10.1038/s41598-019-46673-z
Visona, S. D., de Boer, O. J., Mackaaij, C., de Boer, H. H., Pertiwi, K. R., and de Winter, R. W. (2018). Immunophenotypic analysis of the chronological events of tissue repair in aortic medial dissections. Cardiovasc. Pathol. 34, 9–14. doi: 10.1016/j.carpath.2018.01.009
Visse, R., and Nagase, H. (2003). Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res. 92, 827–839. doi: 10.1161/01.res.0000070112.80711.3d
Wang, L., Zhang, J., Fu, W., Guo, D., Jiang, J., and Wang, Y. (2012). Association of smooth muscle cell phenotypes with extracellular matrix disorders in thoracic aortic dissection. J. Vasc. Surg. 56:1709e1. doi: 10.1016/j.jvs.2012.05.084
Wang, S., Liu, Y., Zhao, G., He, L., Fu, Y., Yu, C., et al. (2018). Postnatal deficiency of ADAMTS1 ameliorates thoracic aortic aneurysm and dissection in mice. Exp. Physiol. 103, 1717–1731. doi: 10.1113/EP087018
Wang, T., He, X., Liu, X., Liu, Y., Zhang, W., Huang, Q., et al. (2017). Weighted gene Co-expression network analysis identifies FKBP11 as a key regulator in acute aortic dissection through a nf-kb dependent pathway. Front. Physiol. 8:1010. doi: 10.3389/fphys.2017.01010
Wang, X., LeMaire, S. A., Chen, L., Shen, Y. H., Gan, Y., Bartsch, H., et al. (2006). Increased collagen deposition and elevated expression of connective tissue growth factor in human thoracic aortic dissection. Circulation 114, I200–I205.
Wang, Y., Yin, P., Chen, Y. H., Yu, Y. S., Ye, W. X., Huang, H. Y., et al. (2018). A functional variant of SMAD4 enhances macrophage recruitment and inflammatory response via TGF-beta signal activation in Thoracic aortic aneurysm and dissection. Aging 10, 3683–3701. doi: 10.18632/aging.101662
Wu, D., Choi, J. C., Sameri, A., Minard, C. G., Coselli, J. S., Shen, Y. H., et al. (2013). Inflammatory cell infiltrates in acute and chronic thoracic aortic dissection. Aorta 1, 259–267. doi: 10.12945/j.aorta.2013.13-044
Wu, Z., Chang, J., Ren, W., Hu, Z., Li, B., and Liu, H. (2017a). Bindarit reduces the incidence of acute aortic dissection complicated lung injury via modulating NF-kappaB pathway. Exp. Ther. Med. 14, 2613–2618. doi: 10.3892/etm.2017.4830
Wu, Z., Ruan, Y., Chang, J., Li, B., and Ren, W. (2016). Angiotensin II is related to the acute aortic dissection complicated with lung injury through mediating the release of MMP9 from macrophages. Am. J. Transl. Res. 8, 1426–1436.
Wu, Z., Wang, Z., Xu, P., Zhang, M., Cheng, L., and Gong, B. (2017b). A novel finding: macrophages involved in inflammation participate in acute aortic dissection complicated with acute lung injury. Curr. Mol. Med. 17, 568–579. doi: 10.2174/1566524018666180222123518
Xiao, T., Zhang, L., Huang, Y., Shi, Y., Wang, J., Ji, Q., et al. (2019). Sestrin2 increases in aortas and plasma from aortic dissection patients and alleviates angiotensin II-induced smooth muscle cell apoptosis via the Nrf2 pathway. Life Sci. 218, 132–138. doi: 10.1016/j.lfs.2018.12.043
Xu, H., Du, S., Fang, B., Li, C., Jia, X., Zheng, S., et al. (2019). VSMC-specific EP4 deletion exacerbates angiotensin II-induced aortic dissection by increasing vascular inflammation and blood pressure. Proc. Natl. Acad. Sci. U.S.A. 116, 8457–8462. doi: 10.1073/pnas.1902119116
Xu, K., Xu, C., Zhang, Y., Qi, F., Yu, B., Li, P., et al. (2018). Identification of type IV collagen exposure as a molecular imaging target for early detection of thoracic aortic dissection. Theranostics 8, 437–449. doi: 10.7150/thno.22467
Xu, L., and Burke, A. (2013). Acute medial dissection of the ascending aorta: evolution of reactive histologic changes. Am. J. Surg. Pathol. 37, 1275–1282. doi: 10.1097/PAS.0b013e318294adc3
Xu, Y., Ye, J., Wang, M., Wang, Y., Ji, Q., Huang, Y., et al. (2018). Increased interleukin-11 levels in thoracic aorta and plasma from patients with acute thoracic aortic dissection. Clin. Chim. Acta 481, 193–199. doi: 10.1016/j.cca.2018.03.014
Ye, J., Wang, M., Jiang, H., Ji, Q., Huang, Y., Liu, J., et al. (2018). Increased levels of interleukin-22 in thoracic aorta and plasma from patients with acute thoracic aortic dissection. Clin. Chim. Acta 486, 395–401. doi: 10.1016/j.cca.2017.10.033
Zeng, T., Yuan, J., Gan, J., Liu, Y., Shi, L., Lu, Z., et al. (2019). Thrombospondin 1 Is increased in the aorta and plasma of patients with acute aortic dissection. Can. J. Cardiol. 35, 42–50. doi: 10.1016/j.cjca.2018.11.008
Zhang, L., Zhou, J., Jing, Z., Xiao, Y., Sun, Y., Wu, Y., et al. (2018). Glucocorticoids regulate the vascular remodeling of aortic dissection Via the p38 MAPK-HSP27 pathway mediated by soluble TNF-RII. eBioMed. 27, 247–257. doi: 10.1016/j.ebiom.2017.12.002
Zhang, Z., Jiang, Y., Zhou, Z., Huang, J., Chen, S., Zhou, W., et al. (2019). Scavenger receptor A1 attenuates aortic dissection via promoting efferocytosis in macrophages. Biochem. Pharmacol. 168, 392–403. doi: 10.1016/j.bcp.2019.07.027
Keywords: aortic dissection, macrophage, inflammation, Ang II, aortic wall
Citation: Wang X, Zhang H, Cao L, He Y, Ma A and Guo W (2020) The Role of Macrophages in Aortic Dissection. Front. Physiol. 11:54. doi: 10.3389/fphys.2020.00054
Received: 30 August 2019; Accepted: 21 January 2020;
Published: 05 February 2020.
Edited by:
Yi Zhu, Tianjin Medical University, ChinaReviewed by:
Suowen Xu, University of Rochester, United StatesSivareddy Kotla, The University of Texas MD Anderson Cancer Center, United States
Copyright © 2020 Wang, Zhang, Cao, He, Ma and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Guo, guoweiplagh@sina.com