 Sharon Negri
Sharon Negri Pawan Faris
Pawan Faris Roberto Berra-Romani
Roberto Berra-Romani Germano Guerra4
Germano Guerra4 Francesco Moccia
Francesco Moccia
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 21 January 2020
Sec. Vascular Physiology
Volume 10 - 2019 | https://doi.org/10.3389/fphys.2019.01618
This article is part of the Research TopicAdvances and Current Challenges in Calcium Signaling within the Cardiovascular SystemView all 8 articles
Vasculogenesis, angiogenesis and arteriogenesis represent three crucial mechanisms involved in the formation and maintenance of the vascular network in embryonal and post-natal life. It has long been known that endothelial Ca2+ signals are key players in vascular remodeling; indeed, multiple pro-angiogenic factors, including vascular endothelial growth factor, regulate endothelial cell fate through an increase in intracellular Ca2+ concentration. Transient Receptor Potential (TRP) channel consist in a superfamily of non-selective cation channels that are widely expressed within vascular endothelial cells. In addition, TRP channels are present in the two main endothelial progenitor cell (EPC) populations, i.e., myeloid angiogenic cells (MACs) and endothelial colony forming cells (ECFCs). TRP channels are polymodal channels that can assemble in homo- and heteromeric complexes and may be sensitive to both pro-angiogenic cues and subtle changes in local microenvironment. These features render TRP channels the most versatile Ca2+ entry pathway in vascular endothelial cells and in EPCs. Herein, we describe how endothelial TRP channels stimulate vascular remodeling by promoting angiogenesis, arteriogenesis and vasculogenesis through the integration of multiple environmental, e.g., extracellular growth factors and chemokines, and intracellular, e.g., reactive oxygen species, a decrease in Mg2+ levels, or hypercholesterolemia, stimuli. In addition, we illustrate how endothelial TRP channels induce neovascularization in response to synthetic agonists and small molecule drugs. We focus the attention on TRPC1, TRPC3, TRPC4, TRPC5, TRPC6, TRPV1, TRPV4, TRPM2, TRPM4, TRPM7, TRPA1, that were shown to be involved in angiogenesis, arteriogenesis and vasculogenesis. Finally, we discuss the role of endothelial TRP channels in aberrant tumor vascularization by focusing on TRPC1, TRPC3, TRPV2, TRPV4, TRPM8, and TRPA1. These observations suggest that endothelial TRP channels represent potential therapeutic targets in multiple disorders featured by abnormal vascularization, including cancer, ischemic disorders, retinal degeneration and neurodegeneration.
Blood vessel formation is an obligate requirement during embryonic development to nourish the cells of the rapidly expanding embryo with oxygen (O2) and nutrients and to remove their catabolic waste (Fischer et al., 2006; Udan et al., 2013). In addition, the circulatory vasculature serves to stabilize the body temperature and prevent pH unbalance, thereby contributing to maintain the homeostasis of the whole organism (Heinke et al., 2012). Furthermore, the vascular network drives the rapid communication among distant tissues mediated by cytokines and hormones and guides immune cells toward sites of inflammation or infection (Heinke et al., 2012; Udan et al., 2013). Finally, vascular endothelial cells emit instructive, angiocrine signals that regulate resident hematopoietic, epithelial, mesenchymal, cardiac and neuronal cells, as well as their corresponding populations of progenitor and stem cells, to finely orchestrate local metabolism and homeostasis and support organ regeneration independently on blood perfusion (Rafii et al., 2016). Neovessel formation also occurs throughout postnatal life, e.g., in uterine endometrium during the ovarian cycle, embryo implantation and placentation, during skeletal muscle growth and remodeling, during bone morphogenesis, and during wound healing after traumatic injury (Fischer et al., 2006; Chung and Ferrara, 2011). Therefore, insufficient vascularization or impaired perfusion due to vessel obstruction may lead to severe ischemic disorders, including acute myocardial infarction (AMI), stroke, peripheral artery disease (PAD), ischemic retinopathies, pre-eclampsia as well as to neurodegeneration (Fischer et al., 2006; Potente et al., 2011). On the other hand, aberrant vascularization is associated to life-threatening diseases, such as cancer, intraocular and inflammatory diseases, and pulmonary arterial hypertension (PAH) (Chung and Ferrara, 2011).
Vascular morphogenesis is a multistep process which requires endothelial cell to proliferate, migrate, align in the direction of blood flow, assemble into tubular structures, branch and anastomose with existing vasculature (Chung and Ferrara, 2011; Potente et al., 2011). Two distinct mechanisms, known as vasculogenesis and angiogenesis (Figure 1), are involved in the formation and maintenance of the vascular network both in the developing embryo and during postnatal life (Chung and Ferrara, 2011; Potente et al., 2011). Vasculogenesis consists in the de novo aggregation of circulating endothelial progenitor cells (EPCs), also referred to as angioblasts in the developing embryo, into functional vessels (Figure 1A). Subsequent expansion and remodeling of nascent capillary plexus requires the engagement of the angiogenic process, which may be distinguished into sprouting angiogenesis and intussusceptive angiogenesis (Figure 1B) (Fischer et al., 2006; Chung and Ferrara, 2011; Potente et al., 2011). Sprouting angiogenesis is activated when the balance between pro- and anti-angiogenic cues is tipped in favor of pro-angiogenic signals, such as vascular endothelial growth factor (VEGF), basic fibroblast growth factor (bFGF) and platelet derived growth factor (PDGF). Pro-angiogenic stimuli cause an increase in endothelial permeability, which leads to the extravasation of multiple plasma proteins (e.g., fibrinogen and fibronectin) that contribute to establish a provisional scaffold for migrating endothelial cells. Degradation of the basement membrane by matrix metalloproteinases (MMPs) released by the activated endothelium contributes to create the most suitable substrate for endothelial cell migration and to create the space necessary for tubule formation. Accordingly, the endothelial cell closest to VEGF adopts a migratory (non-proliferative) tip cell phenotype, thereby budding from the existing capillary toward the source of the stimulus. Adjacent endothelial cells experience lower VEGF levels, adopt a proliferative (non-migrating) stalk phenotype and trail behind the tip cell, thereby elongating the vessel sprout. Thereafter, the neovessel sprout comes in contact and fuses either with a neighboring angiogenic sprout or with a functional capillary, through a process known as anastomosis, which may involve, respectively, two or one tip cells. Therefore, sprouting endothelial cells assemble into a multicellular tube, which undergoes a complex remodeling leading to lumen formation, connection between parental vessels and functional blood flow (Fischer et al., 2006; Chung and Ferrara, 2011; Potente et al., 2011). Subsequently, naked endothelial cells become quiescent by adopting the “cobblestone”-like phalanx phenotype and the nascent vessel is further stabilized by the PDGF-dependent recruitment of mural cells, such as pericytes and vascular smooth muscle cells (VSMCs) (Potente et al., 2011). Microvascular growth may also be accomplished by intussusceptive angiogenesis, which consists in the insertion of a transcapillary pillar followed by the expansion of pillar diameter and consequent splitting of the existing capillary (Udan et al., 2013). It is now clear that EPCs play a crucial role in maintaining endothelial homeostasis and in restoring local blood perfusion upon an ischemic insult also in the adults (D’Alessio et al., 2015; Banno and Yoder, 2018). In addition, EPCs may be mobilized in peripheral circulation to sustain the angiogenic switch during the early phases of tumor growth (Moccia et al., 2015; Poletto et al., 2018). Finally, ischemic neovascularization may also impinge on arteriogenesis, including collateralization, which denotes the growth and remodeling of existing arterioles into larger vessels when a main artery is occluded (Heil et al., 2006).
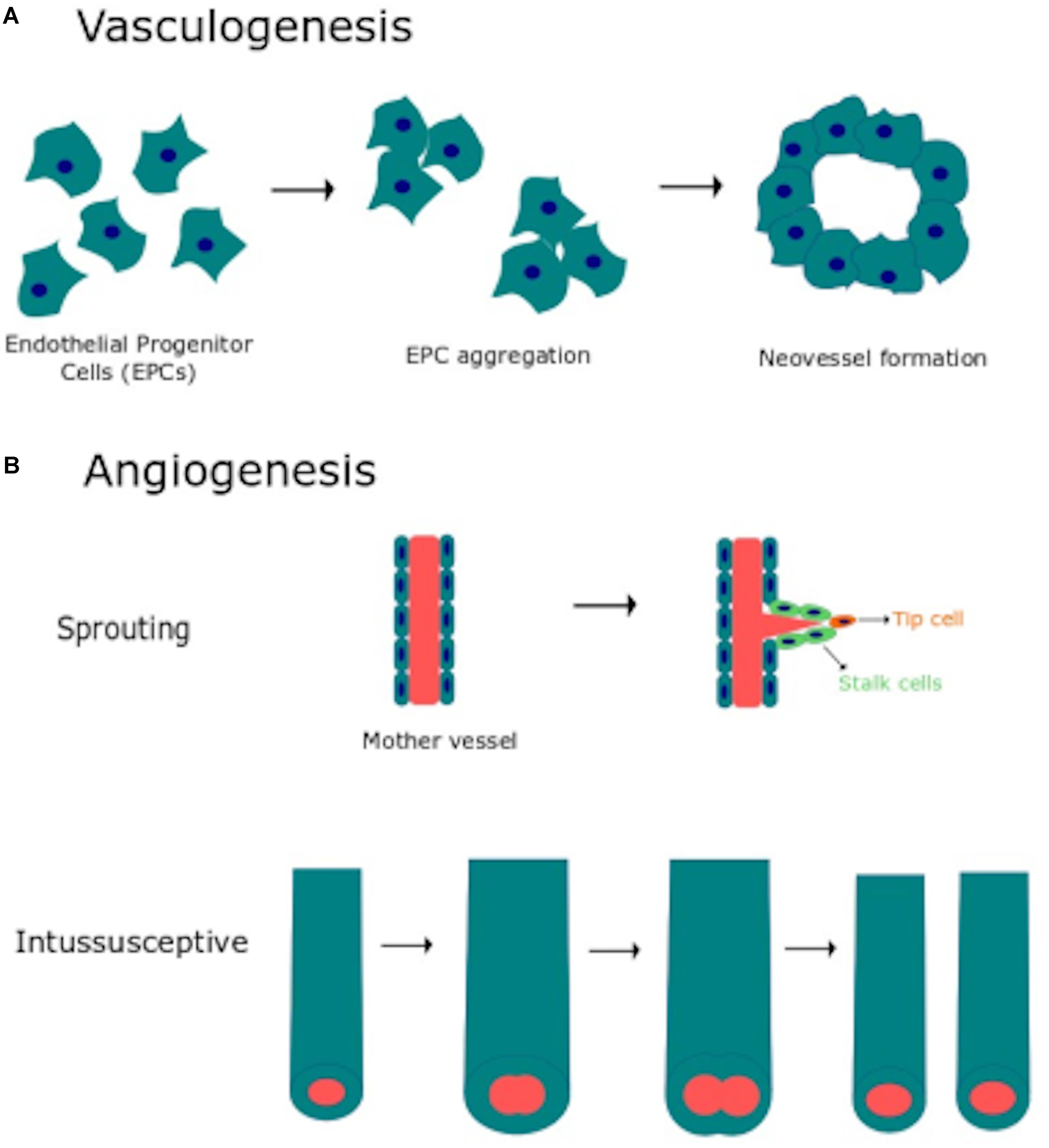
Figure 1. Vasculogenesis and angiogenesis are the main processes responsible for vascular remodeling. (A) Schematic representation of vasculogenesis, which consists in de novo aggregation of circulating endothelial progenitor cells (EPCs) into functional vessels. (B) Schematic representation of angiogenesis, the physiological process whereby capillaries give rise to neovessels to cope with oxygen and nutrient requirements. Angiogenesis may occur through two distinct mechanisms: sprouting angiogenesis and intussusceptive angiogenesis (see text for further details).
It has long been known that endothelial Ca2+ signals play a crucial role in angiogenesis and arteriogenesis (Simons et al., 2016). Multiple pro-angiogenic cues, such as VEGF, bFGF, PDGF, epidermal growth factor (EGF), stromal derived factor-1α (SDF-1α), and angiopoietins regulate endothelial cell behavior and fate through an increase in intracellular Ca2+ concentration ([Ca2+]i) (Moccia et al., 2003; Yokota et al., 2015; Noren et al., 2016; Savage et al., 2019). Likewise, VEGF and SDF-1α induce pro-angiogenic Ca2+ signals in endothelial colony forming cells (ECFCs) (Dragoni et al., 2011, 2013; Zuccolo et al., 2018; Moccia, 2020), which represent the only EPC subtype truly belonging to the vascular endothelial lineage (Medina et al., 2017; Poletto et al., 2018). Transient Receptor Potential (TRP) channels regulate numerous endothelial cell functions by mediating extracellular Ca2+ entry in response to chemical, mechanical and thermal stimuli (Moccia et al., 2012a; Earley and Brayden, 2015; Guerra et al., 2018; Smani et al., 2018; Thakore and Earley, 2019). Herein, we describe how endothelial TRP channels stimulate vascular remodeling by promoting angiogenesis, arteriogenesis and vasculogenesis through the integration of multiple environmental and intracellular cues. We then describe how remodeling of the endothelial blend of TRP channels could promote aberrant vascularization in solid tumors. Understanding how TRP channels regulate endothelial cell fate is indispensable to properly target them to rescue blood perfusion in ischemic disorders and interfere with the vascular network in i.e., cancer and intraocular disorders.
The TRP channel superfamily of non-selective cation channels comprises 28 members that are subdivided in six subfamilies by sequence homology: TRP Canonical (TRPC1-7), TRP Vanilloid (TRPV1-6), TRP Melastatin (TRPM1-8), TRP Ankyrin 1 (TRPA1), TRP Mucolipin (TRPM1-3), and TRP Polycystin (TRPP) (Gees et al., 2010; Earley and Brayden, 2015; Smani et al., 2018; Thakore and Earley, 2019). The TRPP subfamily consists of eight members, although only TRPP2, TRPP3 and TRPP5 display the molecular architecture and function of an ion channel (Gees et al., 2010). Moreover, TRPC2, which plays a crucial role in acrosomal reaction and pheromone detection in mice, is only a pseudo-gene in humans (Gees et al., 2010). Herein, we briefly survey the basic features of TRP channel structure and illustrate their main electrophysiological properties before focusing on the pro-angiogenic role of endothelial TRP channels.
The molecular topology of TRP channels comprises six transmembrane (TM1-6) α-helix segments, a reentrant pore loop between TM5 and TM6, and cytosolic NH2- and COOH-terminal tails. TRP channel subunits assemble into a tetrameric complex around a central cation conducting pathway lined by TM5, TM6 and their interconnecting pore loop (Gaudet, 2008). This structure is highly reminiscent of that described in voltage-gated K+ channels, although TRP channels lack a voltage-sensor in TM4 (Gaudet, 2008). The NH2- and COOH-terminal tails flanking the TM domain may greatly differ in length between different TRP subfamilies and may fulfill diverse functions, such as engaging subunit interactions and providing regulatory domains for cytosolic kinases and other cellular/cytoskeletal proteins as well as serving as binding sites for Ca2+ and Ca2+-dependent sensors, including calmodulin (CaM) and STIM1 (Gaudet, 2008; Birnbaumer, 2009; Gees et al., 2010). For instance, a 25 amino acid conserved TRP domain is located immediately distal to TM6 in TRPC, TRPM and TRPV members, while it is absent in other TRP subfamilies (Birnbaumer, 2009). The TRP domain includes two highly conserved sequences, known as TRP-box 1 and TRP-box 2, bordering a central region, which displays a higher degree of sequence variability (Venkatachalam and Montell, 2007). TRP-box 1 consists of three variations of the Glu-Trp-Lys-Phe-Ala-Arg (EWKFAR) motif and provides the binding site for phosphatidylinositol phosphates, such as phosphatidylinositol-4,5-bisphosphate (PIP2) (Birnbaumer, 2009), or is involved in subunit assembly (Venkatachalam and Montell, 2007). TRP-box 2 is a prolin-rich sequence, which is variable between different TRPC and TRPM subunits, while it is absent in TRPV channels (Venkatachalam and Montell, 2007). In addition, TRPC, TRPV and TRPA1 channels share multiple NH2-terminal ankyrin repeat domains (ARDs), which fulfill numerous functions, including cytoskeleton interaction and channel opening by membrane deformation (“spring hypothesis”), channel gating upon covalent modifications induced by extracellular ligands, and channel tetramerization (Gaudet, 2008). While TRPCs and TRPVs contain 3-4 ARDs, TRPA1 displays up to 14-19 ARDs at the NH2-terminal tail (Gees et al., 2010). Coiled coil domains, which are predicted to mediate subunit assembly and channel function, have been detected at the NH2- and COOH-termini of TRPCs, TRPVs, TRPMs, and TRPPs (Li M. et al., 2011). Moreover, coiled coil domains support the physical association between TRPC channels and the endoplasmic reticulum (ER) Ca2+ sensor, STIM1 (Lee et al., 2014). Intriguingly, the COOH-terminal tail of TRPM2, TRPM6 and TRPM7 displays functional enzymatic domains that may be involved in channel gating and catalyze specific cellular reactions. TRPM2 is featured by a NUDIX phosphohydrolase domain, which binds to and hydrolyses adenosine diphosphate ribose-ribose (ADPr) into 5-phosphate and adenosine monophosphate (AMP); ADPr, in turn, gates TRPM2, whereas AMP negatively modulates channel gating (Faouzi and Penner, 2014). TRPM6 and TRPM7 show an α-kinase domain, which consists in an atypical serine-threonine protein kinase targeting multiple targets, including myosin IIA, IIB, and IIC, elongation factor 2 kinase, annexin A1 (Fleig and Chubanov, 2014). Moreover, this COOH-terminal kinase may be proteolytically cleaved in a cell-specific manner, thereby translocating into the nucleus and regulating chromatin organization and gene expression through histone phosphorylation (Krapivinsky et al., 2014).
All TRP subunits may preferentially aggregate into homomeric cation channels (Lepage and Boulay, 2007), although a recent investigation failed to detect functional TRPC1 tetramers in a heterologous expression system (Storch et al., 2012). However, TRP channels may also assemble into heteromeric channels by combining with subunits belonging to either the same subfamily or different subfamilies (Ma et al., 2011a; Berrout et al., 2012; Storch et al., 2012). Heteromeric TRP channels have been widely reported in naïve cells, including vascular endothelial cells, and heterologous expression systems and present biophysical fingerprints and regulatory mechanisms distinct from their homomeric counterparts (Cheng et al., 2010). Subunit heteromerization has been extensively investigated in the TRPC subfamily. For instance, TRPC1 may assemble into functional heteromeric channels with TRPC3, TRPC4, or TRPC5 (Smani et al., 2018), while TRPC3, TRPC6, and TRPC7 may assemble into functional heteromeric channels in both naïve tissues (Goel et al., 2002) and heterologous expression systems (Hofmann et al., 2002). TRPV subunits may also associate into heterotetramers. For instance, any two members of TRPV1-4 subunits assembly into functional heteromeric channels that are exclusively located on the plasma membrane (Cheng et al., 2007), as well as several investigations demonstrated that TRPV5/TRPV6 subunits assemble into heteromeric channel complexes (Cheng et al., 2010). Furthermore, functional heterotetramers comprising TRP channel subunits from different subfamilies have widely been reported and include the following combinations: TRPC1/TRPP2 (Berrout et al., 2012), TRPC1/TRPV4 (Ma et al., 2011a), TRPC1/TRPV6 (Schindl et al., 2012), TRPV4/TRPC6 (Goldenberg et al., 2015), and TRPML3/TRPV5 (Guo et al., 2013).
TRP channels are polymodal cellular sensors that may be gated by a plethora of chemical and physical stimuli, including intracellular second messengers [e.g., diacylglycerol (DAG), arachidonic acid (AA), ADPr and hydrogen peroxide (H2O2)], intracellular ions (e.g., an increase in cytosolic H+ and Ca2+ and a decrease in cytosolic Mg2+), dietary agonists (e.g., capsaicin, menthol, and allylisothiocyanate or AITC), synthetic ligands (e.g., 4α-phorbol-12,13-didecanoate), gasotransmitters [e.g., nitric oxide (NO)], proteins (e.g., G proteins), mechanical perturbation (e.g., membrane stretch, osmotic swelling and laminar shear stress), and temperature fluctuations (Venkatachalam and Montell, 2007; Gees et al., 2010; Earley and Brayden, 2015; Guerra et al., 2018; Samanta et al., 2018; Smani et al., 2018; Thakore and Earley, 2019). In addition, some members of the TRPC subfamily may be recruited by depletion of the ER Ca2+ store via direct binding with STIM1 and/or Orai1 or with inositol-1,4,5-trisphosphate (InsP3) receptors (InsP3Rs) (Xi et al., 2008; Di Buduo et al., 2014; Lee et al., 2014; Ambudkar et al., 2017).
TRP channels are permeable to monovalent (i.e., Na+ and K+) and divalent (e.g., Ca2+ and Mg2+) cations, although their relative permeability to Ca2+ and Na+ (PCa/PNa) may widely change among the different subunits. For instance, TRPV5 and TRPV6 display the largest PCa/PNa (>100), while TRPM4 and TRPM5 are virtually impermeable to Ca2+ (PCa/PNa < 0.01), although they are gated by an increase in [Ca2+]i (Gees et al., 2010). The PCa/PNa of the remaining TRP channels is situated within these extremes of the selectivity spectrum, ranging from 0.5 (TRPM2) up to 20 (TRPV1) (Gees et al., 2010). Nevertheless, TRPM6 and TRPM7 are more permeable to Mg2+, whereas TRPV1, TRPML1, and TRPP3 display a high permeability to H+. The resting membrane potential (VM) of vascular endothelial cells, which may scatter between −88 and −20 mV (Voets et al., 1996; Moccia et al., 2002), is more negative than the reversal potential for Na+ and Ca2+ (ENa = ECa ≈ +60 mV), but more positive than the reversal potential for K+ (EK = −88 mV). Therefore, in response to extracellular stimulation, TRP channels conduct inward Na+ (and Ca2+ or Mg2+) currents, which exert a profound impact on intracellular Ca2+ dynamics. Accordingly, Ca2+ entry through Ca2+-permeable TRP channels may directly increase the [Ca2+]i, whereas Na+ influx induces membrane depolarization, thereby activating voltage-gated Ca2+ channels in excitable cells and modulating the driving-force for Ca2+ entry in non-excitable cells (Gees et al., 2010; Curcic et al., 2019). The functional impact exerted by Ca2+-permeable TRP channels on intracellular Ca2+ homeostasis depends, therefore, on their “fractional Ca2+ current,” that has not been estimated for all TRP channels. For instance, apart from TRPV5 and TRPV6 that mediate real Ca2+ currents, TRPA1 and TRPM3 exhibit fractional Ca2+ currents of ∼20%, while TRPV1 shows a fractional Ca2+ current of ∼5% (Gees et al., 2010; Earley and Brayden, 2015). The capability of TRP channels to generate a substantial increase in [Ca2+]i has actually been fully appreciated in vascular endothelial cells loaded with the Ca2+-sensitive fluorophore, Fluo-3 or Fura-2, or expressing genetically encoded Ca2+ indicators, such as GCaMP, by using high-speed confocal and total internal reflection (TIRF) microscopy (Earley and Brayden, 2015; Thakore and Earley, 2019). A recent series of investigations characterized the Ca2+ entry events through unitary or clusters of functionally coupled endothelial TRP channels, also known as TRP Ca2+ sparklets. Ca2+ sparklets were measured from both primary endothelial cells and intact endothelium upon stimulation of TRP channels displaying sufficient Ca2+ permeability and unitary conductance (Sonkusare et al., 2012; Pires et al., 2015; Sullivan et al., 2015), such as TRPV1, TRPV4, and TRPA1. It has been estimated that the amount of extracellular Ca2+ entering vascular endothelial cells through individual TRPV4 Ca2+ sparklets exceeds by ∼100 times that recorded during voltage-gated L-type Ca2+ sparklets. Of note, the unitary amplitudes of TRPA1 and TRPV3 Ca2+ sparklets are, respectively, two- and threefold greater that those recorded for TRPV4 Ca2+ sparklets (Sonkusare et al., 2012; Pires et al., 2015; Sullivan et al., 2015). This finding confirms that some TRP channels are able to generate a large increase in local Ca2+ concentration, which could be remain spatially confined to the submembranal space or propagate to the bulk cytosol via the Ca2+-dependent recruitment of InsP3Rs and ryanodine receptors (RyRs) (Earley and Brayden, 2015; Thakore and Earley, 2019).
Endothelial cells line the inner lumen of blood vessels and are, therefore, exposed to a myriad of chemical (e.g., growth factors and chemokines) and physical (e.g., laminar shear stress and pulsatile stretch) cues, which must be properly perceived for the maintenance of tissue homeostasis (Moccia et al., 2012a,2019b). TRP channels represent the most suitable signal transduction pathway whereby vascular endothelial cells integrate such a vast array of extracellular stimuli due to the versatility of their gating mechanisms. Accordingly, endothelial TRP channels serve as polymodal cellular sensors as, with the exception of TRPC3, TRPC6 and TRPM4, they are sensitive to multiple signaling pathways (Earley and Brayden, 2015; Smani et al., 2018; Thakore and Earley, 2019).
Most mammalian TRP isoforms were reported in vascular endothelial cells, including: TRPC1-7; TRPV1-4; TRPA1; TRPP1-2; and TRPM1-8 except TRPM5 (Moccia et al., 2012a; Earley and Brayden, 2015; Smani et al., 2018; Thakore and Earley, 2019). However, the pattern of TRP channel distribution may vary across the vascular tree and in different animal species. For instance, TRPC3 is widely expressed in human, but not bovine, pulmonary artery endothelial cells, whereas TRPC1 is present in mouse, but not rat, aortic endothelial cells (Moccia et al., 2012a). Finally, mouse, but not human, brain microvascular endothelial cells selectively express TRPC1-6 channels (Brown et al., 2008; Zuccolo et al., 2019). Nevertheless, conflicting reports about the expression of specific TRP isoforms in the same endothelial cell type suggest that cell culture conditions and expression detection techniques may affect our knowledge of endothelial TRP channel distribution. Notably, the blend of ion channels endowed to endothelial cells may be remarkably different in vitro and may be further affected by serial passages of cultured cells (Moccia et al., 2012a; Earley and Brayden, 2015; Smani et al., 2018; Thakore and Earley, 2019). It is, therefore, mandatory that the expression of each TRP isoform along the vascular tree is confirmed in vivo. As reported in other cell types, naïve TRP channels may consist of heteromeric subunits also in vascular endothelial cells. The following endothelial TRP channel complexes were reported: TRPC1-TRPC4 (Cioffi et al., 2012; Sundivakkam et al., 2012; Greenberg et al., 2017), TRPC1-TRPV4 (Ma et al., 2011a, b), TRPC3-TRPC4 (Poteser et al., 2006), TRPV1-TRPV4 (O’Leary et al., 2019), TRPP2-TRPC1 (Berrout et al., 2012), and TRPC1-TRPP2-TRPV4 (Du et al., 2014). As anticipated earlier, heteromerization expands the range of gating properties and biophysical properties of endothelial TRP channels, thereby boosting their impact on the regulation of endothelial cell functions.
TRP channels regulate virtually all endothelial cell functions by generating either a spatially restricted Ca2+ domain around the cytosolic mouth of the channel pore or a global increase in [Ca2+]i (Curcic et al., 2019; Thakore and Earley, 2019). TRP channels mediate Ca2+ entry in vascular endothelial cells exposed to a myriad of autacoids and hormones, including acetylcholine, ATP, bradykinin, erythropoietin, histamine, and angiotensin II, or to mechanical stimuli, such as pulsatile stretch and laminar shear stress (Moccia et al., 2012a; Earley and Brayden, 2015; Smani et al., 2018; Thakore and Earley, 2019). In addition, TRP channels modulate endothelial VM by conducting a depolarizing inward current, carried by Na+ and Ca2+ (with the exception of the Ca2+-impermeable TRPM4), thereby causing a positive shift in VM that could be boosted by the Ca2+-dependent recruitment of TRPM4 (yet to be demonstrated) (Moccia et al., 2012a; Earley and Brayden, 2015; Smani et al., 2018; Thakore and Earley, 2019). Furthermore, TRP channels-induced membrane depolarization could activate endothelial voltage-gated channels, although this issue remains highly controversial (Moccia et al., 2012a). Notably, human umbilical vein endothelial cells (HUVECs) may express NaV1.5 and NaV1.7, which represent the pore-forming α-subunits of, respectively, cardiac and neuronal voltage-gated Na+ channels (Andrikopoulos et al., 2011b). Likewise, CaV3.1 T-type voltage-gated Ca2+ channels are expressed in pulmonary microvascular endothelial cells (Zheng et al., 2019) as well as L- and R-type voltage-gated Ca2+ current were recorded in several types of endothelial cells (Moccia et al., 2012a). Alternately, when coupled to the Ca2+-activated intermediate- and small-conductance K+ channels (IKCa and SKCa, respectively), TRP channels may result in endothelial-dependent hyperpolarization (EDH). EDH may in turn be electrotonically propagated to adjacent VSMCs, thereby promoting vasodilation and increasing local blood flow in mesenteric arteries and cerebral microcirculation (Earley and Brayden, 2015; Guerra et al., 2018; Moccia et al., 2019a; Thakore and Earley, 2019).
We refer the reader to a number of recent reviews that provide a comprehensive description of the functional role played by endothelial TRP channels (Earley and Brayden, 2015; Smani et al., 2018; Thakore and Earley, 2019). Herein, we briefly recall that endothelial TRP channels may either mediate short-term responses, e.g., vasodilation or increase in vascular permeability, or support long-lasting processes, e.g., gene expression, proliferation and migration. For instance, acetylcholine recruits TRPC4 to induce NO release and vasodilation in mouse aortic endothelial cells (Freichel et al., 2001), whereas it impinges on TRPV4 to trigger NO- and EDH-dependent vasorelaxation in mouse small mesenteric arteries (Zhang et al., 2009). Likewise, direct activation of TRPV4 with the specific agonist GSK1016790A (GSK) induces vasodilation in rat pulmonary artery by promoting NO release and EDH activation (Addison et al., 2016). Moreover, dietary manipulation may result in robust vasodilation, thereby increasing local blood flow, in several vascular beds. For instance, the plant-derived flavone apigenin stimulates TRPV4 to induce EDH-dependent vasodilation in rat mesenteric arteries (Ma et al., 2012), whereas carvacrol, a pungent ingredient of oregano, and AITC, which is abundant in mustard oil, recruit the EDH pathway by, respectively, stimulating TRPV3 (Pires et al., 2015) and TRPA1 (Earley et al., 2009) in mouse cerebrovascular endothelial cells. Finally, a heteromeric channel formed by TRPC1, TRPV4 and TRPP2 mediates flow-induced vasodilation in mesenteric arteries (Du et al., 2014), whereas laminar shear stress activates the TRPP1-TRPP2 complex to induce NO release and vasodilation in mouse aorta (AbouAlaiwi et al., 2009). Multiple TRP channels, including TRPC1, TRPC3, TRPC4, TRPC6, TRPV4, TRPM2, and TRPM4, may also modulate vascular permeability (Earley and Brayden, 2015; Thakore and Earley, 2019). For instance, TRPC1 and TRPC4 mediate thrombin-induced permeability increase in HUVEC monolayers (Paria et al., 2006) and mouse lung vasculature (Tiruppathi et al., 2002), respectively. Likewise, TRPC3 overexpression/hyperactivation has been associated to the exaggerated increase in blood-brain barrier (BBB) permeability observed in response to status epilepticus (Ryu et al., 2013). Furthermore, TRPM2 mediates H2O2-induced endothelial hyperpermeability in human pulmonary artery endothelial cells (Hecquet et al., 2008), whereas TRPC6 sustains endotoxin-induced disruption of pulmonary vascular barrier (Tauseef et al., 2012) and pulmonary ischemia-reperfusion evoked edema (Weissmann et al., 2012) in mice. Endothelial TRPM4 is up-regulated in a rodent model of spinal cord injury, thereby resulting in capillary fragmentation with secondary hemorrhage, which further exacerbates the damage to surrounding tissues (Gerzanich et al., 2009). Finally, systemic activation of endothelial TRPV4 with GSK is responsible for the dramatic increase in microvascular permeability and hemorrhage reported in the lung, intestine, and kidney (Willette et al., 2008). In agreement with this observation, lung injury induced by high vascular pressure is mediated by TRPV4-mediated Ca2+ entry in capillary endothelial cells (Jian et al., 2008). Extracellular Ca2+ entry through TRP channels may also lead to the recruitment of a number of Ca2+-dependent transcription factors, including nuclear factor-kappaB (NF-κB) (Thippegowda et al., 2010), activating protein-1 (AP-1; i.e., c-Fos and c-Jun) (Fantozzi et al., 2003), and nuclear factor of activated T cells (NFAT) (Zhu et al., 2019). Endothelial TRP channels could also serve as sensors of oxidative stress and local temperature changes. For instance, H2O2 induces an aberrant increase in [Ca2+]i by activating TRPM2 in pulmonary artery endothelial cells and after ischemia-reperfusion injury in mouse brain microcirculation (Hecquet et al., 2008; Pires and Earley, 2017). Likewise, oxidative stress is detected by a heteromeric channel formed by TRPC3 and TRPC4 in porcine aortic endothelial cells (Poteser et al., 2006), and by TRPA1 (Sullivan et al., 2015) and TRPV3 (Yoshida et al., 2006) in mouse cerebrovascular endothelial cells, while it is unclear whether TRPC5 and TRPV4 are sensitive to redox signaling (Pires and Earley, 2017). It is worth noting that a recent report suggested that redox regulation of the endothelial TRPA1 under hypoxic conditions attenuates ischemic damage following brain stroke by inducing cerebral artery relaxation (Pires and Earley, 2018). Finally, TRPV4 is able to detect a moderate increase in temperature (from 19 to 38°C) in mouse aortic endothelial cells (Watanabe et al., 2002), whereas TRPV1 mediates Ca2+ entry when heating from room temperature to over 40°C (Mergler et al., 2010). This sensitivity has been related to temperature-dependent changes in NO release, i.e., in vascular tone, and in endothelial permeability (Watanabe et al., 2002; Mergler et al., 2010). Besides endothelial cells, within the vascular wall, TRP channels are also largely expressed in VSMCs, thereby regulating multiple functions, including proliferation, migration, contractility, and mechanosensation (Inoue et al., 2006; Earley and Brayden, 2015; Randhawa and Jaggi, 2015).
Endothelial TRP channels have long been associated to angiogenesis and vascular remodeling due to their ability to engage intracellular signaling pathways associated to endothelial cell proliferation, migration, adhesion, tubulogenesis, and permeability (Moore et al., 1998; Fantozzi et al., 2003; Cheng et al., 2006). Endothelial TRP channels may control angiogenesis by mediating Ca2+ entry in response to extracellular growth factors, such as VEGF and bFGF, that are liberated in peripheral circulation upon an ischemic insult or by growing tumors (Moccia et al., 2019b). Alternately, TRP channels sense cellular stress factors, such as an increase in ROS production or a decrease in Mg2+ levels, or an increase in laminar shear stress, which ultimately stimulates arteriogenesis (Moccia and Guerra, 2016). Finally, endothelial TRP channels could promote vascular remodeling following stimulation with synthetic agonists.
Endothelial TRPC channels sustain angiogenesis by sustaining the Ca2+ response to pro-angiogenic cues, including VEGF, bFGF, and insulin-like growth factor 1 (IGF-1), and/or by detecting subtle changes in the composition of the local microenvironment, and/or in response to small molecule drugs (Smani et al., 2018; Moccia et al., 2019b). While the mechanisms whereby TRPC3, TRPC4, TRPC5 and TRPC6 control angiogenesis has been largely understood, the role of TRPC1 is still shrouded in mystery.
An increase in endothelial [Ca2+]i is indispensable to sustain the pro-angiogenic effect of growth factors and chemokines (Moccia et al., 2012b, 2019b; Smani et al., 2018). Growth factors bind to their cognate receptor tyrosine kinase (RTKs), thereby stimulating phospholipase C-γ1 (PLCγ1) to cleave phosphatidylinositol 4,5-bisphosphate (PIP2), a minor phospholipid component of the plasma membrane, into the intracellular second messengers, InsP3 and DAG (Moccia et al., 2019b). InsP3, in turn, triggers endogenous Ca2+ release by gating InsP3Rs, which are Ca2+-permeable, non-selective cation channels embedded in ER membrane (Moccia et al., 2019b). VEGF-induced InsP3-dependent ER Ca2+ release may require the concomitant mobilization of endo-lysosomal Ca2+ through two-pore channel 2 (TPC2) (Favia et al., 2014), while it is barely supported by RyRs, as recently demonstrated in human aortic endothelial cells (HAECs) (Evangelista et al., 2012). InsP3-induced intracellular Ca2+ release results in a dramatic fall in ER Ca2+ concentration ([Ca2+]ER) that is detected by STIM1, a single-pass transmembrane protein that senses [Ca2+]ER (Moccia et al., 2012b). Once activated, STIM1 oligomerizes and translocates toward peripheral ER-plasma membrane junctions (≈ 20 nm), known as puncta, where it tethers and gates Orai1, the pore-forming subunits of the Ca2+ release-activated Ca2+ (CRAC) channels (Moccia et al., 2019b), thereby inducing store-operated Ca2+ entry (SOCE). The pro-angiogenic role of SOCE has been mainly evaluated upon endothelial cell stimulation with VEGF, the master regulator of vascular remodeling (Moccia et al., 2019b). The physical association between STIM1 and Orai1 mediate VEGF-induced SOCE, proliferation and in vitro tubulogenesis in HUVECs (Abdullaev et al., 2008; Li J. et al., 2011) and HAECs (Evangelista et al., 2012). Of note, Li J. et al. (2011) failed to detect VEGF-evoked inward Ca2+ currents in HUVECs, which is consistent with the notion that the CRAC current (ICRAC) may fall below the resolving power of whole-cell patch clamp recording systems.
Nevertheless, earlier work suggested that TRPC1 contributed to VEGF-induced SOCE and store-operated Ca2+ current (known as ISOC) in HUVECs (Jho et al., 2005). The ISOC displays strikingly distinct biophysical features as compared to ICRAC, including a linear current-to-voltage relationship, a reversal potential (Erev) approximately equal to 0 mV, and a single-channel conductance in the pS range (Moccia et al., 2019b). Accordingly, Jho et al. (2005) detected a VEGF-induced inward Ca2+-permeable current, which clearly resembled the TRPC1-mediated current previously described in VSMCs with a contractile phenotype (Shi et al., 2017) and in human salivary gland cells (Cheng et al., 2011). Moreover, a recent report confirmed that VEGF-induced SOCE in HUVECs was affected by genetic silencing of TRPC1 (Kusaba et al., 2010). Notably, Abdullaev et al. (2008) also reported that genetic deletion of TRPC1 (and TRPC4) did not affect the ICRAC development and amplitude, but retarded cell growth in HUVECs. Several hypotheses have been put forward to explain this puzzling controversy. For instance, the intracellular recording solution employed in Abdullaev et al. (2008) and Li J. et al. (2011) has been widely employed to record the ICRAC rather than the ISOC (Cs+, which is poorly permeable through Orai1 channels, as main intracellular cation). Notably, Jho et al. (2005) reported that VEGF-induced ISOC was sensitive to 1 μM La3+, which selectively targets Orai1 rather than TRPC1 (Prakriya and Lewis, 2015). In addition, STIM1 may also interact with TRPC1 and TRPC4 in vascular endothelial cells (Ma et al., 2011a; Sundivakkam et al., 2012). Therefore, one could speculate that Orai1-mediated extracellular Ca2+ entry triggers TRPC1 insertion into the plasma membrane, followed by STIM1-dependent recruitment, as demonstrated in human salivary gland cells (Cheng et al., 2011). Alternately, it has been shown that the endothelial Orai1 may assemble and determine the Ca2+-selectivity of a heteromeric complex also involving TRPC1, TRPC4 and, possibly, TRPV4 (Brough et al., 2001; Cioffi et al., 2005; Ma et al., 2011a; Cioffi et al., 2012; Xu et al., 2015). Therefore, the controversial contribution of TRPC1 (and TRPC4) to VEGF-induced endothelial SOCE may reflect multiple factors, including cell source, cell passage, culture medium, substrate of adhesion, experimental protocols, detection methods and so on (Beech, 2012). We refer the reader to a number of excellent review articles dealing with the ongoing controversy regarding the molecular make-up of endothelial SOCE (Blatter, 2017; Groschner et al., 2017).
Whatever the gating mechanism, TRPC1 and TRPC4 do play a role in angiogenesis, as more widely illustrated in the next paragraphs (Figure 2).
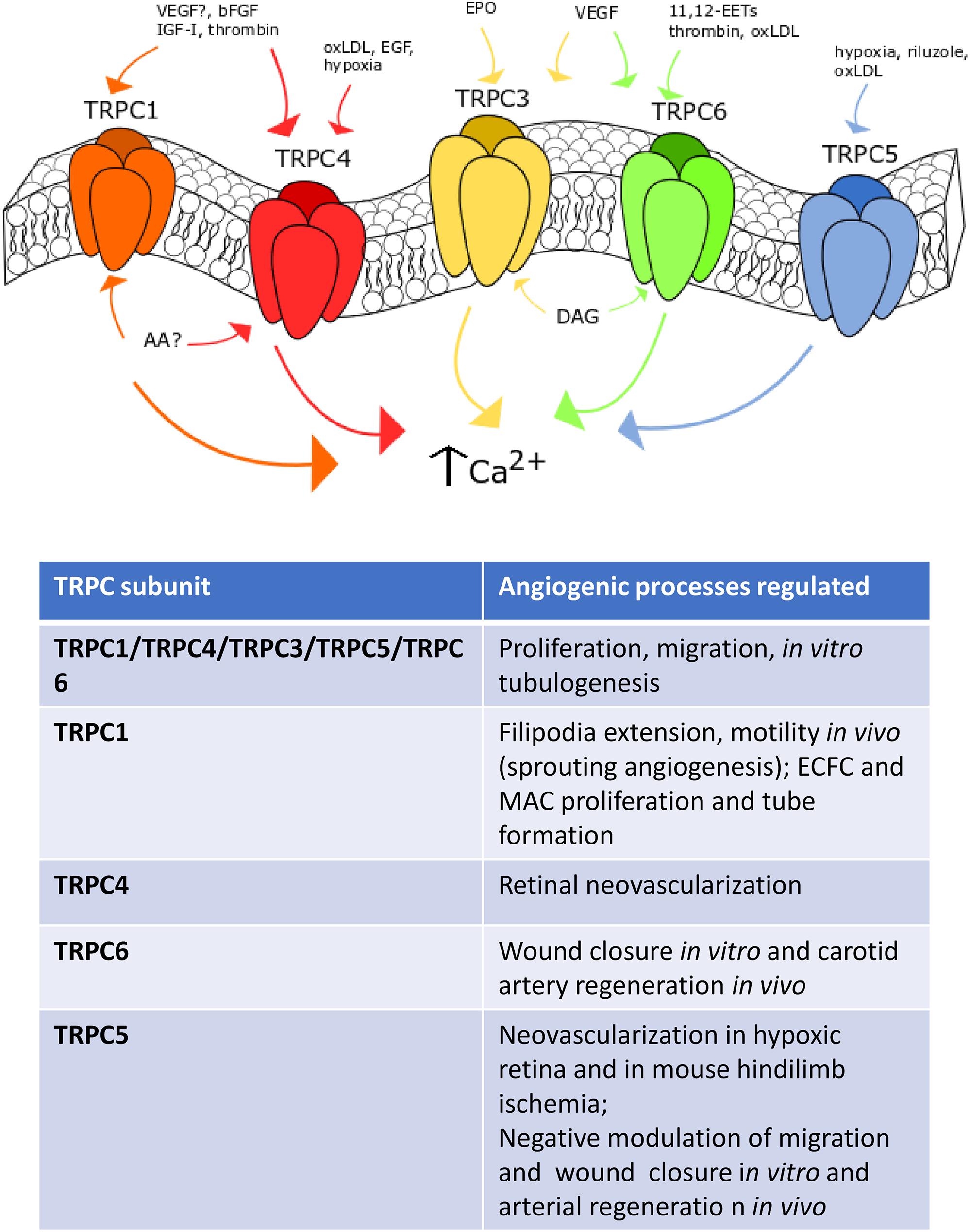
Figure 2. TRPC channels in angiogenesis. Representation of the main TRPC channels involved in angiogenesis. TRPC1 mediates angiogenesis after VEGF- (still debated), IGF-, bFGF- and thrombin-induced stimulation. TRPC4 is sensitive to EGF, oxLDL and hypoxia. TRPC3 responds to erythropoietin (EPO) and VEGF stimulation, such as TRPC6, which can be also activated by oxLDL, thrombin and 14,15-EETs. Finally, TRPC5 is sensitive to hypoxia, riluzole and oxLDL-dependent activation.
As recently reviewed in Moccia et al. (2019b), bFGF evokes extracellular Ca2+ entry in bovine aortic endothelial cells (BAECs) by binding to FGF receptor-1 (FGFR-1) and stimulating cytosolic phospholipase A2, phospholipase D (PLD) and PLC to release AA, which in turn recruits a heteromeric TRPC1/TRPC4 channel (Antoniotti et al., 2002, 2003, 2006). Blocking AA production and preventing Ca2+ influx with carboxyamidotriazole (CAI) inhibit BAEC proliferation (Antoniotti et al., 2003), thereby suggesting that the heteromeric TRPC1/TRPC4 channel underlies the pro-angiogenic effect of bFGF. Interestingly, IGF-I elicits extracellular Ca2+ influx by engaging the same Ca2+-permeable route as that stimulated by bFGF in BAECs (Munaron and Fiorio Pla, 2000). In agreement with these observations, genetic silencing of TRPC1 by means of antisense morpholino oligonucleotides prevent filopodia extension, motility and proliferation in endothelial tip cells of zebrafish larvae (Yu et al., 2010). TRPC1 was found to drive sprouting angiogenesis by mediating VEGF-induced extracellular signal-regulated kinases (ERK) in intersegmental vessels (Yu et al., 2010). Endothelial Ca2+ oscillations sustain VEGF-induced filopodia elongation and endothelial cell migration during angiogenesis in zebrafish (Savage et al., 2019), but the role of TRPC1 in this signaling remains to be elucidated. While these studies provide the evidence that TRPC1 sustains angiogenesis, other reports argue against this conclusion. For instance, genetic silencing of TRPC1 did not affect the spontaneous Ca2+ transients that drive in vitro tubulogenesis in EA.hy926 cells (Antigny et al., 2012), a widely employed HUVECs-derived cell line. In addition, vascular development was not impaired in TRPC1-deficient mice, while endothelium-dependent hyperpolarization was exacerbated (Schmidt et al., 2010). Further studies are, therefore, required to confirm the pro-angiogenic role of TRPC1.
Unlike TRPC1, genetic deletion of TRPC4 via specific small interfering RNAs (siRNAs; siTRPC4, in this case) impaired spontaneous Ca2+ oscillations and tube formation in EA.hy926 cells (Antigny et al., 2012). Consistently, TRPC4 mediate VEGF-induced neovascularization in hypoxic retina (Song et al., 2015). Oxygen depletion increased TRPC4 expression in mouse retina, whereas the intravitreal infusion of a specific siTRPC4 impaired the subsequent process of retinal neovascularization (Song et al., 2015). Accordingly, genetic deletion of TRPC4 prevented VEGF-induced migration and tube formation by interfering with the ERK and Akt signaling pathways in human retinal microvascular endothelial cells (Song et al., 2015). Earlier work carried out on human pulmonary artery endothelial cells (PAECs) revealed that hypoxia leads to TRPC4 up-regulation through the AP-1 transcription factors and that TRPC4 was able to mediate SOCE by carrying the ISOC (Fantozzi et al., 2003). Furthermore, TRPC4 could play a crucial role in oxidized low-density lipoprotein (oxLDL)-induced in vitro angiogenesis. OxLDL-induced proliferation, migration and tube formation in human coronary artery endothelial cells (HCAECs) were impaired by transfecting the cells with a specific siTRPC4 (Qin et al., 2016). TRPC4 induced angiogenesis by promoting VEGF release and inducing the nuclear translocation of the Ca2+-sensitive transcription factor, NF-κB (Qin et al., 2016). It has, therefore, been suggested that TRPC4 might provide an alternative molecular target to treat atherosclerotic neovascularization. Of note, TRPC4 could be preferentially recruited by EGF to the plasma membrane in proliferating endothelial cell clusters, which establish immature cell-to-cell contacts (Chen et al., 2016). TRPC4 is then retrieved from the plasma membrane upon formation of mature endothelial barriers and is relocated within junctional complexes, where it is barely available to physiological stimuli (Graziani et al., 2010). This phenotype-dependent subcellular localization of TRPC4 could further help understand why some reports identified this channel as a pore-forming subunit of endothelial ISOC (Fantozzi et al., 2003; Sundivakkam et al., 2012) or ICRAC (Freichel et al., 2001; Cioffi et al., 2005), while others failed (Abdullaev et al., 2008).
While the role played by TRPC1 and TRPC4 channels in VEGF-induced SOCE is controversial, there is no doubt that TRPC3 and TRPC6 may sustain VEGF-induced extracellular Ca2+ entry and angiogenesis in vascular endothelial cells (Figure 2). TRPC3 and TRPC6 are gated by the PLCγ1 metabolite, DAG, in a protein kinase C (PKC)-independent manner (Moccia et al., 2018b). An earlier report suggested that a heteromeric TRPC3/TRPC6 channel was involved in VEGF-induced Ca2+ influx in human microvascular endothelial cells (HMVECs) (Cheng et al., 2006). Subsequently, the same group demonstrated that VEGF-induced extracellular Ca2+ entry, proliferation and tube formation were impaired in HMVECs overexpressing a dominant negative TRPC6 construct (Hamdollah Zadeh et al., 2008). Similarly, TRPC3 and TRPC6 could sustain VEGF-induced extracellular Ca2+ entry in HUVECs. Pharmacological (by means of selective inhibitors) and genetic (by using selective siRNAs) manipulation demonstrated that TRPC3-mediated Na+ entry could switch the Na+/Ca2+ exchanger (NCX) in the reverse (Ca2+ entry) mode (Andrikopoulos et al., 2011a, 2017). The subsequent increase in [Ca2+]i was sufficient to recruit PKC-α and engage the ERK signaling pathway, thereby promoting VEGF-induced proliferation and tube formation in HUVECs (Andrikopoulos et al., 2011a, 2017). TRPC6 may also sustain VEGF-induced Ca2+ entry in HUVECs (Ge et al., 2009). Likewise, genetic deletion of TRPC6 with a specific siRNA reduced VEGF-induced extracellular Ca2+ entry, proliferation and tube formation in vitro as well as angiogenesis in vivo (Ge et al., 2009). However, TRPC6 was not involved in the pro-angiogenic Ca2+ response to bFGF (Ge et al., 2009). Interestingly, a selective siTRPC3 was able to impair spontaneous Ca2+ oscillations and assembly into a bidimensional tubular network in EA.hy926 cells, while genetic deletion of TRPC6 was without consequence (Antigny et al., 2012). Additional investigations demonstrated that TRPC3 and TRPC6 may drive endothelial cell proliferation, motility and tube formation in response to pro-angiogenic cues other than VEGF. For instance, erythropoietin-induced intracellular Ca2+ signals and migration in EA.hy926 cells were impaired by inhibiting TRPC3-mediated Ca2+ influx with Pyr3 and a blocking antibody selectively targeting TRPC3 (Maltaneri et al., 2018). In addition, the Ca2+ response to erythropoietin was sensitive to pharmacological blockade of L-type voltage-gated Ca2+ channels (VGCCs) with diltiazem and amlodipine (Maltaneri et al., 2018). It should, however, be pointed out that CaV1 subunits, which mediate L-type Ca2+ currents, are yet to be identified in vascular endothelial cells (Moccia et al., 2012a) and that a recent report demonstrated that endothelial SOCE is also sensitive to a number of drugs targeting L-type VGCCs (Mergler et al., 2003). TRPC6, in turn, mediates (±)-11,12-EET- and thrombin-induced migration and tube formation in HUVECs (Ding et al., 2014) and human PAECs (Kini et al., 2010). Interestingly, thrombin-induced TRPC6-mediated extracellular Ca2+ entry is tightly regulated by the dual lipid-protein phosphatase, phosphatase and tensin homologue (PTEN). PTEN was found to physically interact with TRPC6 through its tail domain (residues 394-403), thereby promoting the membrane translocation of TRPC6 and Ca2+ influx upon exposure to thrombin (Kini et al., 2010). Conversely, TRPC6 can be activated by lysophosphatidylcholine, the most abundant lysophospholipid in oxLDL, to inhibit wound closure in cultured mouse aortic endothelial cells and carotid artery regeneration in a mouse model of hypercholesterolemia (Rosenbaum et al., 2015). Therefore, the role of TRPC6 in vascular remodeling could be tightly modulated by local microenvironment and be dampened under pro-atherosclerotic conditions.
TRPC5 is also able to modulate angiogenesis in several vascular beds (Figure 2) (Smani et al., 2018). TRPC5 mediates extracellular Ca2+ entry downstream PLC activation, although the gating role of STIM proteins and ER Ca2+ store emptying remains to be fully elucidated (Zholos, 2014; Moccia, 2018). A number of chemical mediators may activate TRPC5 by acting on the extracellular side. These include, but are not limited to, NO, H+, Pb2+, lanthanides, reduced thioredoxin, and Ca2+, which may also act intracellularly (Zholos, 2014). Finally, TRPC5 is sensitive to membrane stretch and to cold temperatures (37°C to 25°C) (Zholos, 2014). Earlier work showed that TRPC5 may negatively affect cell migration in BAECs. According to these studies, TRPC6-mediated Ca2+ influx recruits myosin light chain kinase (MLCK) in an ERK- and NADPH oxidase-dependent manner, thereby inducing TRPC5 to translocate toward the plasma membrane and inhibit BAEC motility (Chaudhuri et al., 2008, 2017). Recently, however, TRPC5 was found to sustain intracellular Ca2+ oscillations and tube formation in EA.hy926 cells (Antigny et al., 2012). Furthermore, it has been shown that genetic deletion of TRPC5 with a selective siRNA impaired hypoxia-induced capillary sprouting and tube formation in primary mouse intestinal mesenteric vascular endothelial cells (Zhu et al., 2019). Consistently, hypoxia-induced retinal neovascularization was compromised in a mouse model deficient of TRPC5 (Zhu et al., 2019). Finally, genetic knockdown of TRPC5 prevented vascular revascularization in a mouse model of hindlimb ischemia at 14 days post-intervention. Intriguingly, vessel density and blood flow recovery were enhanced by over-expressing TRPC5 upon injection of an adeno-associated virus construct encoding for TRPC5 or by administrating riluzole, the first medication approved by the Food and Drug Administration to treat amyotrophic lateral sclerosis (Zhu et al., 2019). As widely reviewed in Bellingham (2011), riluzole may inhibit voltage-gated Na+ and Ca2+ channels, whereas it can stimulate Ca2+-dependent and voltage-independent two-pore K+ currents. However, recent studies revealed that riluzole may also activate TRPC5 with no requirement for PLC engagement (Richter et al., 2014a, b). In agreement with these observations, riluzole elicited non-selective cation currents and extracellular Ca2+ entry also in mouse mesenteric vascular endothelial cells (Zhu et al., 2019). TRPC5-mediated Ca2+ entry stimulated angiogenesis by promoting the nuclear translocation of NFAT isoform c3 (NFATc3) and angiopoietin 1 expression (Zhu et al., 2019). Similar to TRPC6, however, TRPC5 activation by oxidized lipids may prevent endothelial wound closure in vitro and regeneration of endothelial monolayers in vivo (Rosenbaum et al., 2015).
The TRPV subfamily encompasses 6 isoforms, TRPV1-6. However, only the TRPV isoforms 1-4 (TRPV1-4) play clear roles in vascular function (Earley and Brayden, 2015), while TRPV5 and TRPV6 channels are mainly located in the luminal membrane of renal and intestinal epithelium (Gees et al., 2010). TRPV1-4 are non-selective cation channels, which are featured by a permeability ratio PCa/PNa between ∼1 and ∼15 (Gees et al., 2010). Conversely, TRPV5 and TRPV6 represent a separate group, are less similar (22–24% identity) to other TRP isoforms and are highly Ca2+-selective (Gees et al., 2010). TRPV1-4 channels serve as polymodal Ca2+-entry pathways being activated by multiple cues, such as temperature increases, mechanical stimuli and chemical agonists (Gees et al., 2010; Earley and Brayden, 2015). They are known to regulate a plethora of endothelial functions, including vascular tone and permeability, inflammation, and vascular remodeling (Earley and Brayden, 2015; Guerra et al., 2018; Smani et al., 2018; Moccia et al., 2019a; Thakore and Earley, 2019). Herein, we will focus on TRPV1 and TRPV4, which are the most important endothelial TRPV isoforms involved in angiogenesis and arteriogenesis (Figure 3). Furthermore, TRPV1 and TRPV4 channels may control vascular remodeling by promoting VSMC proliferation and migration under pathological conditions, such as pulmonary hypertension (Inoue et al., 2006; Randhawa and Jaggi, 2015).
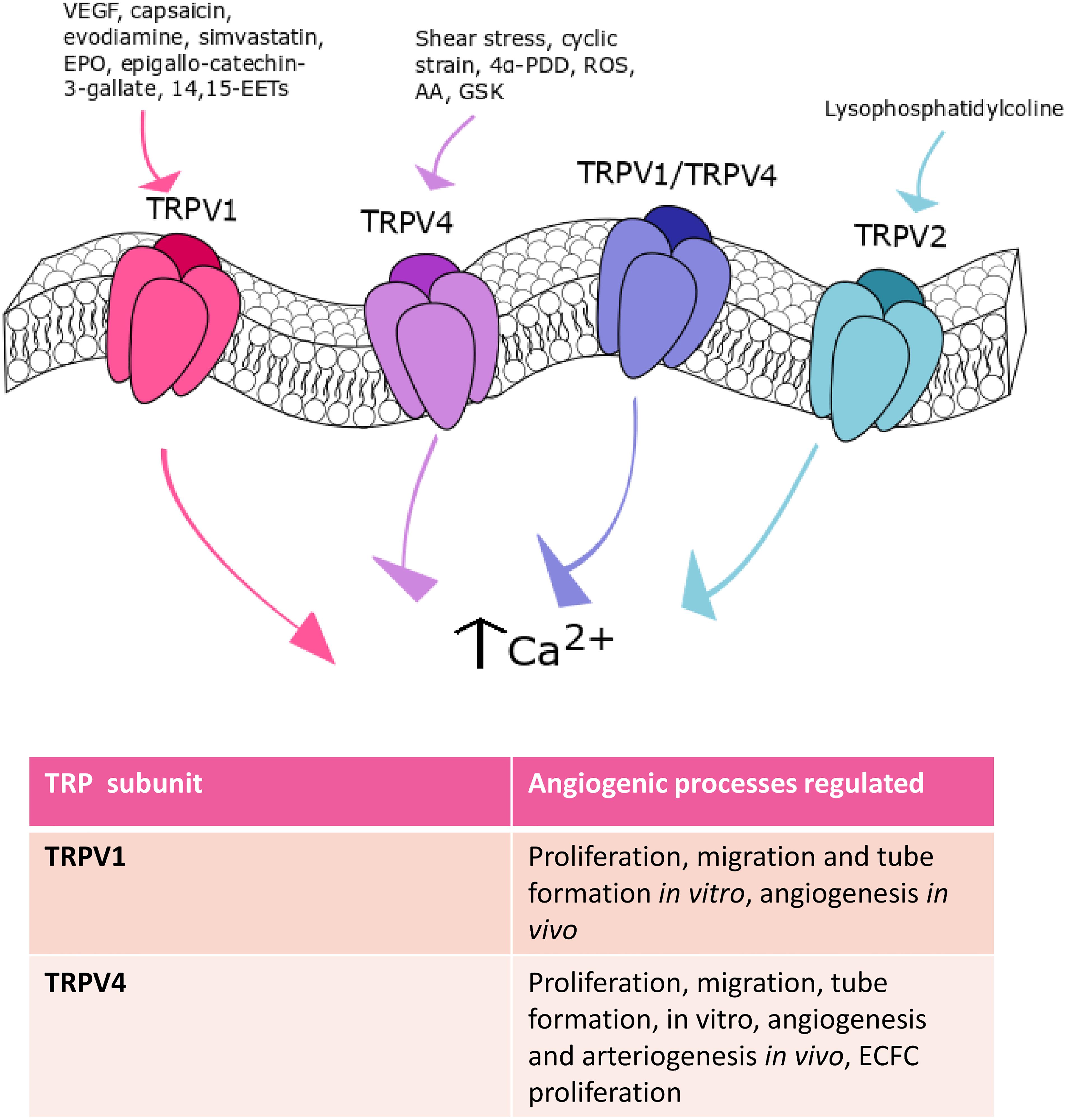
Figure 3. TRPV channels in angiogenesis. TRPV1, TRPV2, and TRPV4 are the main TRPV isoforms that mediate angiogenesis. TRPV1 is involved in angiogenesis in response to: VEGF, capsaicin, evodiamine, simvastatin, EPO, epigallocatechin-3-gallate, and 14,15-EETs. Shear stress-, cyclic strain-, 4α- PDD-, ROS-, AA- and GSK-mediated TRPV4 activation participate to angiogenesis. Finally, TRPV2 stimulation with lysophosphatidylcoline can play a role in this process.
TRPV1, also known as the vanilloid receptor 1 (VR1), was the first mammalian TRPV family member to be discovered and the most extensively studied (Gees et al., 2010). TRPV1 is an archetypal polymodal channel as it is activated by distinct physical and chemical stimuli, such as noxious heat (>42°C) and extracellular protons, plants-derived products (e.g., capsaicin and gingerol), hydrogen sulfide (H2S), and tarantula spider-derived vanillotoxins (Bevan et al., 2014). Multiple evidences demonstrated that endothelial TRPV1 may stimulate angiogenesis (Smani et al., 2018; Thakore and Earley, 2019), although earlier work suggested that capsaicin, the most widely employed TRPV1 agonist, actually exerts an anti-angiogenic effect (Min et al., 2004). This study showed that capsaicin was able to inhibit VEGF-induced DNA synthesis, proliferation, migration and tube formation in HUVECs. In addition, capsaicin was found to impair VEGF-induced vascularization in Matrigel plugs in vivo (Min et al., 2004). This study, however, did not assess TRPV1 contribution in the anti-angiogenic effect of capsaicin. Furthermore, a subsequent investigation revealed that intraperitoneal administration of evodiamine, a TRPV1 agonist, induced angiogenesis in Matrigel plugs in vivo (Ching et al., 2011). TRPV1-mediated extracellular Ca2+ entry promoted PI3K/Akt/CaMKII signaling, which in turn led to: (1) TRPV1 phosphorylation, (2) physical interaction between eNOS and TRPV1; (3) eNOS phosphorylation and, ultimately, (4) NO release (Ching et al., 2011). Accordingly, evodiamine failed to induce angiogenesis when Matrigel plugs were implanted in eNOS- or TRPV1-deficient mice (Ching et al., 2011). Of note, it has long been known that NO induces endothelial cell proliferation, migration, and tube formation (Mancardi et al., 2011). Moreover, TRPV1 induced AMP-activated protein kinase (AMPK) phosphorylation, which favored the formation of a TRPV1-eNOS complex, thereby boosting NO release and angiogenesis both in Matrigel plugs and in a mouse model of hindlimb ischemia (Ching et al., 2012). In addition, TRPV1 mediates simvastatin-, erythropoietin-, epigallocatechin-3-gallate- and 14,15-EET-induced angiogenesis. Simvastatin, a 3-hydroxy-3-methylglutaryl-CoA reductase blocker that is employed to treat hypercholesterolaemia and cardiovascular disorders, stimulated in vivo angiogenesis by promoting NO release through the assembly of the TRPV1-Akt-CaMKII-AMPK-eNOS complex described above (Su et al., 2014b). The same signaling pathway is recruited to promote tube formation in vitro and vascular development in vivo by erythropoietin (Yu et al., 2017) and epigallocatechin-3-gallate, the main catechin present in the green tea (Guo et al., 2015). Finally, pharmacological (with capsazepine) and genetic (with a selective siRNA) inhibition of TRPV1 prevented 14,15-EET-induced Ca2+ influx, NO release, in vitro tube formation and in vivo angiogenesis in HMECs (Su et al., 2014a). A recent investigation demonstrated that TRPV1 may assemble into a heteromeric complex with TRPV4 in mouse retinal microvascular endothelial cells (RMECs) (O’Leary et al., 2019). The TRPV1/TRPV4 heteromeric channel did not promote VEGF-induced extracellular Ca2+ entry and sprouting angiogenesis, but it was per se able to mediate Ca2+ influx and drive RMEC proliferation, motility and capillary-like tube formation (O’Leary et al., 2019). While these studies hinted at extracellular Ca2+ entry as the main mechanism whereby TRPV1 induces angiogenesis, an alternative signaling mode has been described in EA.hy926 cells (Hofmann et al., 2014). Herein, Hofmann et al. (2014) demonstrated that TRPV1 mediated the uptake of the endogenous cannabinoid, anandamide, in a Ca2+-independent manner. The intracellular transport of anandamide, in turn, promoted EA.hy926 cell proliferation and tube formation (Hofmann et al., 2014), which is consistent with the emerging notion that TRP channels drive angiogenesis independently on their ability to conduct extracellular Ca2+ (Abdullaev et al., 2008). These studies strongly suggest that the pharmacological stimulation of TRPV1 represents a promising pathway to induce angiogenesis in the presence of cardiovascular risk factors (i.e., hypercholesterolaemia) or upon an ischemic insult.
Early work revealed that endothelial TRPV1 may also be responsible for vascular remodeling observed in rat model of arteriovenous fistula (AVF) created at the femoral artery and vein (Chen et al., 2010). The following increase in blood flow led to the shear stress-induced activation of TRPV1, which in turn caused venodilation, wall thickening and vascular remodeling in fistula veins. These effects were exacerbated by the concomitant up-regulation of endothelial TRPV1. It was demonstrated that TRPV1 recruited CaMKII to phosphorylate eNOS and induce NO-dependent MMP2 activation (Chen et al., 2010). The pharmacological blockade of TRPV1 prevented the increase in shear stress and vascular remodeling observed upon AVF formation by inhibiting the signaling pathways engaged by extracellular Ca2+ entry (Chen et al., 2010). These findings suggest that endothelial TRPV1 could be also activated by mechanical deformation of the plasma membrane, as previously demonstrated in retinal ganglion cells (Sappington et al., 2009) and in the mechanosensitive fibers of renal pelvis (Feng et al., 2008).
Originally, TRPV4 was identified as a mechanosensor or osmosensor, activated by hypotonic osmotic stress and shear stress (Shibasaki, 2016). Subsequently, TRPV4 was found to be heat-sensitive, being activated by temperatures greater than 27 °C (Shibasaki, 2016). TRPV4 can be also gated by synthetic agonists, such as the phorbol ester 4α-phorbol 12,13-didecanoate (4α-PDD), by acidic pH and citrate and, finally, by endogenous agonists, such as the endocannabinoid anandamide, AA, and its hydrolytic products 5,6-EETs and 14,15-EETs (White et al., 2016). TRPV4 channels display higher selectivity to Ca2+ than Na+ ions and mediate outward rectifier ionic currents (White et al., 2016). Early work demonstrated that mechanical stimulation of TRPV4 by cyclic strain induced bovine capillary endothelial cells to realign perpendicular to the direction of the strain upon PI3K-dependent β1 integrin recruitment, stress fiber and focal adhesion rearrangement (Thodeti et al., 2009). Interestingly, the same group recently demonstrated that genetic silencing of TRPV4 increased the cell surface expression and phosphorylation at Y1175 of VEGFR2 in HMECs, thereby stimulating VEGF-induced migration (Kanugula et al., 2019). The emerging role played by TRPV4 in angiogenesis was further corroborated by Hatano et al. (2013), who found that TRPV4 activation by 4α-PDD induced proliferation in human brain capillary endothelial cells. In agreement with this observation, the intra-tail injection of 4α-PDD at days 1-4 in rats undergoing transient brain ischemia decreased infarct volume by ≈50% and significantly reduced neurological deficit by promoting brain angiogenesis and neurogenesis (Chen et al., 2018). TRPV4 activation was shown to induce eNOS phosphorylation, to stimulate VEGF levels in the ischemic area and to up-regulate VEGFR2 expression (Chen et al., 2018). TRPV4-mediated extracellular Ca2+ entry has been implicated also in neovascular diseases, such as PAH (Suresh et al., 2018) and cancer (see Section Remodeling of Endothelial TRP Channels Promote Aberrant Tumor Vascularization). Accordingly, preliminary work demonstrated that oxidative stress was able to dismantle endothelial permeability by stimulating the Src kinase Fyn to activate TRPV4 in human and mouse lung microvascular endothelial cells (Suresh et al., 2015). Recently, it has been demonstrated that the higher levels of cytosol- and mitochondria-derived ROS led to the constitutive opening of TRPV4, thereby increasing resting [Ca2+]i and promoting aberrant endothelial cell proliferation and migration (Suresh et al., 2018). This observation strongly suggests that endothelial TRPV4 is involved in PAH pathogenesis.
As mentioned earlier, arteriogenesis refers to the remodeling of pre-existing arterio-arteriolar anastomoses to completely developed and functional arteries (Fischer et al., 2006). Earlier work provided the evidence that TRPV4 transcription was increased by the elevated shear stress driving collateral vessel growth after femoral artery ligature (FAL) in rats (Troidl et al., 2009). Furthermore, 4α-PDD, a TRPV4 agonist (see Paragraph TRPV4 in Angiogenesis), stimulated proliferation in porcine aortic endothelial cells in vitro and promoted collateral vessel growth and local tissue perfusion after FAL in vivo (Troidl et al., 2009). Of note, the infusion of 4α-PDD failed to stimulate arteriogenesis and rescue blood supply in a mouse model KO for TRPV4 (Troidl et al., 2009). Subsequently, the same group confirmed the role of TRPV4 in arteriogenesis also in a porcine model of hindlimb-ischemia (Troidl et al., 2010). In addition, a number of Ca2+-dependent transcription factors were found to mediate the pro-arteriogenic effect of TRPV4 in pigs, including NFAT, myocyte enhancer factor 2C and DREAM (Troidl et al., 2010). Finally, endothelial TRPV4 expression was also increased in cerebral collateral circulation after bilateral carotid artery ligature (BCL) (Schierling et al., 2011). Similar to FAL (Troidl et al., 2009), 4α-PDD treatment led to enhanced collateral growth in vivo by inducing endothelial cell proliferation (Schierling et al., 2011). These observations reinforce the tenet that the pharmacological stimulation of TRPV4 provides an alternative therapeutic strategy to restore blood perfusion in damaged or obstructed vessels.
TRPV1 and TRPV4 could induce vascular remodeling by also stimulating proliferation and migration in pulmonary artery VSMCs. Early work demonstrated that TRPV1 and TRPV4 were expressed, mediated extracellular Ca2+ entry and supported migration through cytoskeletal remodeling in these cells (Martin et al., 2012). Subsequently, it has been shown that chronic hypoxia increased TRPV1 and TRPV4 expression in pulmonary artery VSMCs, thereby increasing the Ca2+ response to mechanical stimulation, the rate of cell motility, and the nuclear translocation of NFATc4 (Parpaite et al., 2016). In agreement with these observations, TRPV4 protein was up-regulated in a rat model of PAH, which led to an increase in membrane swelling- and 4α-PDD-induced extracellular Ca2+ entry in VSMCs (Yang et al., 2012). Furthermore, TRPV4 overexpression caused a significant elevation in resting [Ca2+]i, which ultimately resulted in an increased myogenic tone. Of note, development of PAH, vascular remodeling and right ventricle hypertrophy induced by chronic hypoxia were prevented in a mouse model deficient of TRPV4 (Yang et al., 2012). These observations, therefore, hint at TRPV4 as a crucial player in both endothelial dysfunction (see Paragraph TRPV4 in Angiogenesis) and vascular remodeling in PAH. Nevertheless, a recent investigation confirmed that TRPV1 protein was up-regulated in pulmonary artery VSMCs harvested from subjects affected by idiopathic PAH (IPAH). Moreover, capsaicin- and membrane swelling-induced Ca2+ entry were enhanced and led to an increase in CREB phosphorylation, which was associated to the higher proliferation rate of IPAH VSMCs (Song et al., 2017). The role of TRPV4 in PAH remains to be confirmed in human samples.
The TRPM subfamily consists of 8 members, TRPM1-8, named based upon the first member, i.e., TRPM1, that has been first identified as tumor suppressor in melanoma (Gees et al., 2010). TRPM channels may be classified in three separate groups based on their sequence homology: TRPM1/TRPM3, TRPM4/TRPM5; TRPM6 and TRPM7; TRPM2 and TRPM8, which bear scarce amino-acid sequence similarity (Ramsey et al., 2006). TRPM channels are featured by a variable permeability to Ca2+, which ranges from the Ca2+ impermeable channels TRPM4 and TRPM to the highly Ca2+ permeable TRPM3, TRPM6 and TRPM7 (Gees et al., 2010). Herein, we focus on TRPM2, TRPM4 and TRPM7 (Figure 4), which represent the sole TRPM members involved in angiogenesis (Smani et al., 2018).
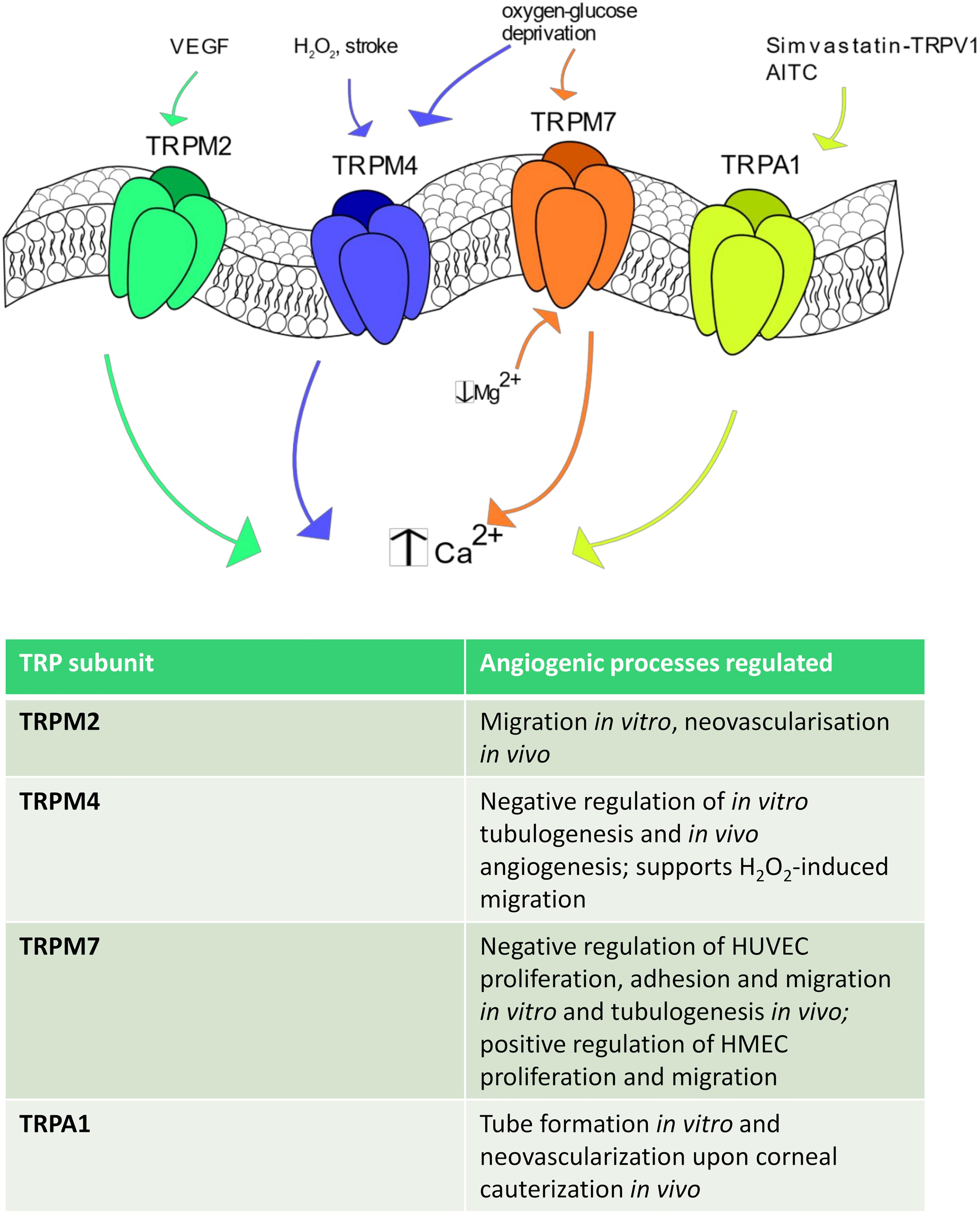
Figure 4. TRPM and TRPA1 channels in angiogenesis. Some TRPM isoforms are shown to be involved in angiogenesis. In particular, TRPM2 stimulates angiogenesis after VEGF stimulation, whereas TRPM4 is activated by H2O2, stroke and oxygen-glucose deprivation. TRPM7 regulates endothelial cell proliferation and tube formation being sensitive to a reduction in intracellular Mg2+ concentration and to oxygen-glucose deprivation. Finally, TRPA1 promotes angiogenesis in response to AITC and upon simvastatin-dependent TRPV1 activation.
TRPM2 is equally permeable to Na+ and Ca2+ (PCa/PNa ≈ 0.7–0.9) and is gated by the intracellular second messenger ADPr, which binds to its COOH-terminal NUDIX domain, H2O2 and compounds that generate reactive oxygen/nitrogen species (Faouzi and Penner, 2014; Thakore and Earley, 2019). A recent investigation revealed that VEGF induced Ca2+ influx through TRPM2 in a ROS-dependent manner in human pulmonary artery endothelial cells (Mittal et al., 2015). Furthermore, extracellular Ca2+ entry induced TRPM2 to assemble with c-Src and VE-cadherin at adherens junctions, thereby causing VE-cadherin internalization and stimulating endothelial cell migration in vitro (Mittal et al., 2015). Accordingly, VEGF failed to promote neovascularization of Matrigel plugs in TRPM2 KO mice as well as blood flow recovery was severely impaired in a TRPM2 KO mice model of hindlimb ischemia (Mittal et al., 2015). Finally, microvascular density and restoration of post-ischemic blood flow were severely compromised in a TRPM2 KO mouse model (Mittal et al., 2015). For all these studies, the TRPM2 KO mice were provided by GlaxoSmithKline and were devoid of side effects (Mittal et al., 2015).
As mentioned earlier, TRPM4, as well as its companion TRPM5, represents a peculiar TRP channel as it is permeable only to monovalent cation, although it is gated by an increase in [Ca2+]i (Gees et al., 2010). It has been shown that oxygen-glucose deprivation was able to increase TRPM4 expression and impair in vitro tubulogenesis in HUVECs. Nevertheless, pharmacological blockade of TRPM4 with 9-phenanthrol restored their pro-angiogenic behavior (Loh et al., 2014). Notably, cerebral stroke induced in rats by permanent occlusion of the middle cerebral artery resulted in endothelial TRPM4 up-regulation and remarkable loss of endothelial integrity within the penumbra region (Loh et al., 2014). However, local injection of a siTRPM4 prevented vascular dysfunction and increased capillary density, which hints at TRPM4 as a negative modulator of angiogenesis (Loh et al., 2014). Accordingly, the brain infarct volume and recovery of motor function were enhanced by genetic deletion of TRPM4, although the protective effect of TRPM4 silencing on motor function was only transient and ceased at day 5 post-intervention (Loh et al., 2014). It should, however, be pointed out that TRPM4 was also found to enhance H2O2-induced HUVEC depolarization and migration (Sarmiento et al., 2015). Therefore, the role played by TRPM4 in angiogenesis could depend on the surrounding context and/or the endothelial cell type, as noticed for other endothelial TRP isoforms.
TRPM7 provides a constitutively open pathway for Mg2+, Zn2+, and Ca2+, and is regulated by multiple signals, including PIP2 hydrolysis, decrease in free cytosolic Mg2+ levels and cell volume changes (Fleig and Chubanov, 2014). Earlier work confirmed that TRPM7 was gated by a drop in intracellular Mg2+ levels in HUVECs and revealed that genetic deletion with a specific siRNA increased HUVEC proliferation by enhancing ERK phosphorylation, eNOS expression and NO release (Inoue and Xiong, 2009). Conversely, genetic deletion of TRPM7 stimulated HUVEC adhesion to the substrate, migration in the wound healing assay and tubulogenic activity in Matrigel (Zeng et al., 2015). In agreement with the previous finding, TRPM7 silencing promoted ERK phosphorylation (Zeng et al., 2015). A subsequent investigation reported that a selective siTRPM7 reduced HMEC proliferation by inducing cell cycle arrest in the G0/G1 and G1 phases and impaired HMEC migration, while it did not affect in vitro tube formation (Baldoli and Maier, 2012). A more recent investigation disclosed that TRPM7 negatively modulates migration and tube formation in brain microvascular endothelial cells both after oxygen-glucose deprivation in vitro and after cerebral stroke in vivo (Yang et al., 2018). This study further revealed that TRPM7 was down-regulated by the adipose-derived stem cells (ADSCs)-derived exosomal microRNA-181b-5p (181-bExos) (Yang et al., 2018). Notably, TRPM7 expression was decreased, while endothelial cell migration and tube formation were enhanced, by the ADSCs-derived 181-bExos (Yang et al., 2018). Overall, it appears that endothelial TRPM channels might not provide the best means to stimulate therapeutic angiogenesis. Accordingly, while the pro-angiogenic role of TRPM2 remains to be confirmed also by future investigations, TRPM4 and TRPM7 emerge as negative modulator of the angiogenic process. Notably, the observation that their expression levels are increased under hypoxic conditions strongly suggest that TRPM4 and TRPM7 play a crucial role in limiting the revascularization process of ischemic tissues.
TRPA1 displays higher permeability to Ca2+ than Na+ (PCa/PNa = 7.9), presents a significant fractional Ca2+ current, and is activated by dietary compounds, such as allicin and AITC, mechanical deformation, and thermal stimuli (Thakore and Earley, 2019). Interestingly, the pro-angiogenic effect of simvastatin was prevented also in TRPA1 null mice, in which the simvastatin-induced formation of the TRPV1-Akt-CaMKII-AMPK-eNOS complex was also compromised (Su et al., 2014b). This finding led to the hypothesis that TRPA1 is the final effector of simvastatin-induced TRPV1 signaling. Conversely, the direct stimulation of TRPA1 with AITC did not induced HUVEC proliferation and migration neither was involved in the pro-angiogenic response to VEGF (Usui-Kusumoto et al., 2019). However, this investigation also reported that TRPA1 activation by VEGF on surrounding cells promoted tube formation when HUVEC were grown on a feeder monolayer of fibroblasts and to promote stromal neovascularization upon corneal cauterization (Usui-Kusumoto et al., 2019).
EPCs are released in peripheral circulation throughout postnatal life to replace damaged/senescent endothelial cells, to rescue the vascular network and restore local blood supply upon an ischemic insult, or to support tumor growth and vascularization in solid malignancies (Yoder, 2012; Moccia and Guerra, 2016; Poletto et al., 2018). The term EPC actually encompasses two distinct cellular populations that belong to diverse lineages and exploit different mechanisms to support vasculogenesis. These, respectively, include myeloid angiogenic cells (MACs) and endothelial colony forming cells (ECFCs) (Medina et al., 2017). As discussed in a recent Consensus Statement on Nomenclature, MACs are myeloid cells (Medina et al., 2017), which are mobilized from bone marrow and support neovascularization by liberating paracrine signals that stimulate local angiogenesis and/or recruit ECFCs to the neovessel site. Conversely, ECFCs are truly endothelial precursors that are mobilized from vascular niches, sustain angiogenesis in a paracrine manner and provide the building blocks for neovessel formation. As illustrated below, TRP channels may regulate proliferation, tubulogenesis and neovessel formation in both human ECFCs and rodent MACs (Moccia and Guerra, 2016).
A thorough transcriptomic analysis revealed that peripheral blood-derived human ECFCs (PB-ECFCs) expressed only TRPC1 and TRPC4, while umbilical cord blood-derived ECFCs were also endowed with TRPC3 (Sanchez-Hernandez et al., 2010). Genetic silencing by means of specific siRNAs subsequently disclosed that TRPC1 interacts with STIM1 and Orai1 to generate SOCE in ECFCs (Li J. et al., 2011; Lodola et al., 2012). It is not clear whether Orai1 and TRPC1 form separate Ca2+-permeable channels, each of them being gated by STIM1, or whether they assemble into a supermolecular complex, which possibly involves TRPC4. Nevertheless, depletion of the ER Ca2+ pool failed to induce a detectable store-operated Ca2+ current in ECFCs (Li J. et al., 2011; Moccia et al., 2019b), which suggests that Orai1 and TRPC1 tightly interact to mediate an ICRAC-like conductance, as described in mouse PAECs (Cioffi et al., 2012). SOCE may be recruited, respectively, by stromal derived factor-1α (SDF-1α) to promote ECFC migration in vitro and neovessel formation in vivo (Zuccolo et al., 2018) and by VEGF to stimulate ECFC proliferation and tube formation (Dragoni et al., 2011, 2015b). Of note, SOCE engages the ERK 1/2 and PI3K/Akt signaling pathways to drive SDF-1α-induced ECFC motility (Zuccolo et al., 2018), whereas the pro-angiogenic response to VEGF involves the Ca2+-sensitive transcription factor, NF-κB (Dragoni et al., 2011). Similar to human ECFCs, STIM1, TRPC1 and Orai1 interact to mediate SOCE and support in vitro angiogenesis (proliferation, motility and the formation) in rodent MACs (Kuang et al., 2010, 2012; Wang et al., 2015). Furthermore, genetic knockdown of TRPC1 prevented neovessel formation in Matrigel plugs in vivo by impairing CaM-dependent eNOS activation (Du et al., 2018).
TRPC3, in turn, is selectively expressed in UCB-ECFCs (Sanchez-Hernandez et al., 2010), where it is physiologically gated by DAG and triggers VEGF-induced intracellular Ca2+ oscillations (Dragoni et al., 2013). Genetic (with specific siRNAs) and pharmacological (with Pyr3) manipulation revealed that TRPC3-mediated extracellular Ca2+ entry triggers the dynamic interplay between InsP3Rs and SOCE which shapes the spiking Ca2+ response to VEGF, thereby promoting UCB-ECFC proliferation (Dragoni et al., 2013). It has been suggested that TRPC3 involvement is responsible for the higher frequency of VEGF-induced intracellular Ca2+ oscillations in UCB-ECFCs, which is associated to their higher proliferative potential (Moccia et al., 2018a). This observation led to the hypothesis that the exogenous insertion of TRPC3 might rejuvenate the reparative phenotype of senescent/aging UCB-ECFCs, thereby improving the efficacy of autologous cell-based therapy in ischemic patients (Moccia et al., 2018a).
A series of independent investigations reported that, besides TRPC1, TRPC3 and TRPC4, human ECFCs also express TRPM7 (Baldoli and Maier, 2012), TRPV1 (Hofmann et al., 2014), and TRPV4 (Dragoni et al., 2015a). Genetic silencing of TRPM7 with a specific siRNA did not affect ECFCs’ proliferation rate (Baldoli and Maier, 2012), while selective stimulation of TRPV4 with GSK only exerted a weak effect on PB-ECFC growth (Dragoni et al., 2015a). Nevertheless, TRPV4-mediated extracellular Ca2+ entry supported AA-evoked intracellular Ca2+ signals, NO release and PB-ECFC proliferation (Dragoni et al., 2015a). This feature could become therapeutically relevant as AA is massively released in circulation during myocardial ischemia and upon brain damage (Moccia et al., 2018a). Unlike TRPV4, TRPV1 activation is per se sufficient to induce ECFC proliferation. An earlier investigation confirmed that TRPV1 drove UCB-ECFC proliferation by mediating anandamide uptake in a Ca2+-independent manner, as also reported in EA.hy926 cells (Hofmann et al., 2014). More recently, however, Lodola et al. (2019) revealed that optical excitation of PB-ECFCs with visible light (520 nm) induced TRPV1-mediated membrane depolarization when the cells were plated on the photo-sensitive polymer Poly(3-hexyl-thiophene) (P3HT). Visible light stimulation was able to promote TRPV1 opening by increasing ROS levels and local temperature within the cleft between the photo-sensitive substrate and cell surface, although a contribution of local pH was not ruled out. Notably, optical excitation of TRPV1 promoted PB-ECFC proliferation and tube formation by inducing the nuclear translocation of p65 NF-κB (Lodola et al., 2019). The pro-angiogenic effect of TRPV1 activation was prevented by buffering intracellular Ca2+ levels with BAPTA, thereby suggesting that Ca2+ influx through TRPV1 was necessary to induce angiogenesis (Lodola et al., 2019). It has, therefore, been suggested that optoceutical stimulation of TRPV1-mediated intracellular Ca2+ signals could represent an alternative strategy to improve the therapeutic outcome of regenerative medicine of ischemic disorders.
Recent pieces of evidence suggested that dysregulation of the endothelial Ca2+ machinery is crucial to support neovascularization and resistance to anti-angiogenic and chemotherapeutic drugs in a growing number of malignancies (Ma et al., 2014; Moccia, 2018; Scarpellino et al., 2019). Herein, we focus on the mechanisms whereby aberrant expression and/or activity of endothelial TRP channels impact on tumor vascularization.
TRPV4 has been the first endothelial TRP channel to be clearly associated to malignant angiogenesis. Accordingly, TRPV4 expression was remarkably enhanced in breast tumor-derived endothelial cells (B-TECs) and TRPV4 activation with AA or 4αPDD induced migration in B-TECs, but not control human cardiac microvascular endothelial cells (Pla et al., 2012) Conversely, AA-induced BTEC migration was prevented upon cell transfection with a short hairpin RNA selectively targeting TRPV4 (shTRPV4). Notably, AA-induced intracellular Ca2+ entry was larger in migrating, rather than non-migrating, B-TECs, and the cell surface expression of TRPV4 was significantly increased by a brief (10 min) pre-incubation with AA itself (Pla et al., 2012). Furthermore, earlier work demonstrated that AA was able to promote extracellular Ca2+ entry in B-TECs during the early steps of the tubulogenic process, but not within an established capillary-like network (Fiorio Pla et al., 2008). These data, therefore, strongly indicate that the up-regulation of endothelial TRPV4 channels plays a key role during the early phases of angiogenesis in breast cancer (BC).
Conversely, TRPV4 was down-regulated in prostate adenocarcinoma-derived endothelial cells (A-TECs), which displayed enhanced motility due to their lower mechanosensitivity toward extracellular matrix (ECM) stiffness. This, in turn, resulted in excessive tumor growth and vascularization in a mouse model deficient of TRPV4 and subcutaneously injected with Lewis Lung Carcinoma (LLC) cells (Adapala et al., 2016). Notably, tumor vasculature in TRPV4 KO mice presented a greater percentage of hyper-permeable, pericyte-free and dilated microvessels (Adapala et al., 2016), which are known to temper the therapeutic outcome of anticancer treatments (Moccia, 2018). A subsequent report disclosed that TRPV4 down-regulation caused a significant reduction in VE-cadherin expression at cell-to-cell contacts, which further contributed to increase vascular leakage (Cappelli et al., 2019). Overexpression or pharmacological stimulation of TRPV4 with GSK were, however, sufficient to normalize aberrant capillary-like tubules in vitro by restoring their mechanosensitivity toward ECM stiffness through the blockade of basal Rho activity (Thoppil et al., 2015; Adapala et al., 2016). Moreover, the daily intraperitoneal delivery of GSK improved pericyte coverage in TRPV4 KO mice xenografted with LLC cells and favored cisplatin-induced tumor shrinkage (Adapala et al., 2016). In addition, GSK-induced TRPV4-mediated extracellular Ca2+ influx inhibited the ERK 1/2 pathway, thereby decreasing A-TEC proliferation in vitro and reducing LLC growth in vivo (Thoppil et al., 2015). These findings, therefore, suggest that, depending on the tumor type, TRPV4 may be inhibited or activated to interfere with malignant vascularization.
A recent investigation showed that endothelial TRP channels are remodeled also in prostate cancer (PCa). Bernardini et al. (2019) demonstrated that TRPA1, TRPV2 and TRPC3 were up-regulated in three different endothelial cell lines established from PCa patients and in the endothelial cells selectively lining tumor vessels in vivo. Pharmacological (with selective agonists) and genetic (with specific siRNAs) manipulation further revealed that: (1) TRPA1 supported PCa-derived endothelial cell migration; (2) TRPC3 promoted PCa-derived endothelial cell chemoattraction toward tumor microenvironment; and (3) TRPV2 induced capillary-like formation in vitro and vascular development in a mouse model of postnatal retina in vivo (Bernardini et al., 2019). The pro-angiogenic effect of TRP channels on PCa-derived endothelial cells was associated to a robust increase in intracellular Ca2+ activity as compared to non-tumor endothelium (Bernardini et al., 2019). Therefore, the pharmacological blockade of a number of selective TRP isoforms could represent a promising treatment for PCa patients. Conversely, endothelial TRPM8 down-regulation was found to regulate angiogenesis independently on Ca2+ signaling. Accordingly, TRPM8 was remarkably down-regulated in B-TECs as compared to HMECs and HUVECs, where it was mainly located within the ER (Genova et al., 2017). Genova et al. (2017) showed that TRPM8 inhibited migration and tube formation in normal endothelial cells by trapping Rap1 intracellularly, thereby preventing its relocation toward the plasma membrane, which is required to activate β1-integrin signaling. As a consequence, TRPM8 down-regulation in B-TECs is likely to accelerate vascular growth, thereby suggesting that TRPM8 activation (e.g., by icilin or menthol) could represent an efficient strategy to treat breast cancer.
TRPC1 may be heavily remodeled in tumor-derived ECFCs. For instance, TRPC1 and SOCE were up-regulated in renal cellular carcinoma (RCC)- (Lodola et al., 2012) and primary myelofibrosis (PMF)-derived ECFCs (Dragoni et al., 2014), but not in BC- and infantile hemangioma (IH)-derived ECFCs (Zuccolo et al., 2016; Lodola et al., 2017). Notably, pharmacological blockade of SOCE with BTP2 impaired proliferation and/or tube formation in RCC-, BC- and IH-derived ECFCs (Lodola et al., 2012, 2017), but not in PMF-derived ECFCs (Dragoni et al., 2014). As VEGF failed to induce pro-angiogenic Ca2+ oscillations in tumor-derived ECFCs (Lodola et al., 2012, 2017; Dragoni et al., 2015b; Zuccolo et al., 2016), a feature which might underpin the primary and secondary resistance to anti-VEGF drugs, TRPC1 (and SOCE) inhibition could represent a promising strategy to interfere with tumor vascularization (Moccia, 2018).
Endothelial TRP channels serve a crucial role in vascular remodeling by regulating angiogenesis, arteriogenesis and vasculogenesis. The remarkable heterogeneity in their gating mechanisms, which can be further increased by the propensity of some isoforms to assemble into heteromeric complexes, render TRP channels the most versatile Ca2+ entry pathway in vascular endothelial cells, ECFCs and MACs. For instance, TRPC3 and TRPC6 mediate the pro-angiogenic response to VEGF, whereas TRPV4 senses the increase in laminar shear stress upon vascular occlusion, thereby promoting arteriogenesis. Nevertheless, endothelial TRP channels may also dampen angiogenesis, as reported for TRPM4 in HUVECs and TRPM7 in brain microvascular endothelial cells. Of course, a number of issues are yet to be clarified in order to fully appreciate and exploit the therapeutic potential of endothelial TRP channels. For instance, it is still unclear whether and how TRPC1 is recruited by VEGF in vascular endothelial cells and how it interacts with STIM1 and Orai1 to mediate VEGF-induced intracellular Ca2+ oscillations in ECFCs. Furthermore, while the role of endothelial TRP channels has been mainly investigated in vitro, the measurement of TRP channels-mediated endothelial Ca2+ signals in vivo remains a challenging issue. Recent investigations exploited a zebrafish model engineered to selectively express an endothelial genetic Ca2+ indicator (Yokota et al., 2015; Savage et al., 2019). Likely, these observations will pave the way for future studies aiming at assess the role of endothelial TRP channels in vivo. The sensitivity of endothelial TRPV channels to a moderate to large increase in local temperature also represents a puzzling issue (Watanabe et al., 2002; Mergler et al., 2010; Lodola et al., 2019). Accordingly, heat therapy induced a robust angiogenic switch in the myocardium of both healthy (Gong et al., 2006) and hypertensive (Ihori et al., 2016), whereas it rescued the capillary network and attenuated myocardial damage in a rat model of AMI (Sobajima et al., 2011). The contribution of thermosensitive TRPV1 and TRPV4 channels to heat-induced neovascularization requires to be evaluated. This hypothesis is corroborate by the recent evidence according to which TRPV1 may integrate local heat and ROS production to stimulate ECFC proliferation and tube formation (Lodola et al., 2019). An additional facet of endothelial TRP signaling that deserves further attention is its involvement in the abnormal vascularization that features multiple diseases, such as cancer, ischemic disorders and retinal degeneration. While remodeling of endothelial TRP channels has been largely appreciated, it is still unclear whether and which endothelial TRP isoforms are dysregulated, e.g., in coronary microcirculation of individuals suffering from AMI, atherosclerosis or hypertension. Exploring this issue, although technically challenging due to the difficulty of harvesting human endothelial samples, will permit to explore the therapeutic potential of endothelial TRP channels. For instance, pre-clinical trials already showed that stimulating TRPC5 with riluzole could represent a promising strategy to treat PAD, while the intra-myocardial/coronary injection of P3HT nanoparticles coupled to optical pacing of the heart could stimulate circulating ECFCs to rescue coronary circulation upon TRPV1 activation. Conversely, TRPV4 could be inhibited or stimulated to interfere with or normalize tumor vasculature in, respectively, BC and LLC.
FM and SN drafted and finally edited the manuscript. PF, RB-R, and GG significantly contributed to revise the manuscript. All the authors read and approved the final version of the manuscript.
FM acknowledges financial support from: Italian Ministry of Education, University and Research (MIUR): Dipartimenti di Eccellenza Program (2018–2022) – Department of Biology and Biotechnology “L. Spallanzani”, University of Pavia; and by Fondo Ricerca Giovani from the University of Pavia.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdullaev, I. F., Bisaillon, J. M., Potier, M., Gonzalez, J. C., Motiani, R. K., and Trebak, M. (2008). Stim1 and orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 103, 1289–1299. doi: 10.1161/01.res.0000338496.95579.56
AbouAlaiwi, W. A., Takahashi, M., Mell, B. R., Jones, T. J., Ratnam, S., Kolb, R. J., et al. (2009). Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ. Res. 104, 860–869. doi: 10.1161/circresaha.108.192765
Adapala, R. K., Thoppil, R. J., Ghosh, K., Cappelli, H. C., Dudley, A. C., Paruchuri, S., et al. (2016). Activation of mechanosensitive ion channel TRPV4 normalizes tumor vasculature and improves cancer therapy. Oncogene 35, 314–322. doi: 10.1038/onc.2015.83
Addison, M. P., Singh, T. U., Parida, S., Choudhury, S., Kasa, J. K., Sukumaran, S. V., et al. (2016). NO synthase inhibition attenuates EDHF-mediated relaxation induced by TRPV4 channel agonist GSK1016790A in the rat pulmonary artery: role of TxA2. Pharmacol. Rep. 68, 620–626. doi: 10.1016/j.pharep.2016.01.003
Ambudkar, I. S., De Souza, L. B., and Ong, H. L. (2017). TRPC1, Orai1, and STIM1 in SOCE: friends in tight spaces. Cell Calcium 63, 33–39. doi: 10.1016/j.ceca.2016.12.009
Andrikopoulos, P., Baba, A., Matsuda, T., Djamgoz, M. B., Yaqoob, M. M., and Eccles, S. A. (2011a). Ca2+ influx through reverse mode Na+/Ca2+ exchange is critical for vascular endothelial growth factor-mediated extracellular signal-regulated kinase (ERK) 1/2 activation and angiogenic functions of human endothelial cells. J. Biol. Chem. 286, 37919–37931. doi: 10.1074/jbc.m111.251777
Andrikopoulos, P., Fraser, S. P., Patterson, L., Ahmad, Z., Burcu, H., Ottaviani, D., et al. (2011b). Angiogenic functions of voltage-gated Na+ channels in human endothelial cells: modulation of vascular endothelial growth factor (VEGF) signaling. J. Biol. Chem. 286, 16846–16860. doi: 10.1074/jbc.m110.187559
Andrikopoulos, P., Eccles, S. A., and Yaqoob, M. M. (2017). Coupling between the TRPC3 ion channel and the NCX1 transporter contributed to VEGF-induced ERK1/2 activation and angiogenesis in human primary endothelial cells. Cell. Signal. 37, 12–30. doi: 10.1016/j.cellsig.2017.05.013
Antigny, F., Girardin, N., and Frieden, M. (2012). Transient receptor potential canonical channels are required for in vitro endothelial tube formation. J. Biol. Chem. 287, 5917–5927. doi: 10.1074/jbc.m111.295733
Antoniotti, S., Fiorio Pla, A., Barral, S., Scalabrino, O., Munaron, L., and Lovisolo, D. (2006). Interaction between TRPC channel subunits in endothelial cells. J. Recept. Signal Transduct. Res. 26, 225–240. doi: 10.1080/10799890600784050
Antoniotti, S., Fiorio Pla, A., Pregnolato, S., Mottola, A., Lovisolo, D., and Munaron, L. (2003). Control of endothelial cell proliferation by calcium influx and arachidonic acid metabolism: a pharmacological approach. J. Cell. Physiol. 197, 370–378. doi: 10.1002/jcp.10359
Antoniotti, S., Lovisolo, D., Fiorio Pla, A., and Munaron, L. (2002). Expression and functional role of bTRPC1 channels in native endothelial cells. FEBS Lett. 510, 189–195. doi: 10.1016/s0014-5793(01)03256-2
Baldoli, E., and Maier, J. A. (2012). Silencing TRPM7 mimics the effects of magnesium deficiency in human microvascular endothelial cells. Angiogenesis 15, 47–57. doi: 10.1007/s10456-011-9242-0
Banno, K., and Yoder, M. C. (2018). Tissue regeneration using endothelial colony-forming cells: promising cells for vascular repair. Pediatr. Res. 83, 283–290. doi: 10.1038/pr.2017.231
Beech, D. J. (2012). Orai1 calcium channels in the vasculature. Pflugers. Arch. 463, 635–647. doi: 10.1007/s00424-012-1090-2
Bellingham, M. C. (2011). A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade? CNS Neurosci. Ther. 17, 4–31. doi: 10.1111/j.1755-5949.2009.00116.x
Bernardini, M., Brossa, A., Chinigo, G., Grolez, G. P., Trimaglio, G., Allart, L., et al. (2019). Transient receptor potential channel expression signatures in tumor-derived endothelial cells: functional roles in prostate cancer angiogenesis. Cancers 11:E956.
Berrout, J., Jin, M., and O’neil, R. G. (2012). Critical role of TRPP2 and TRPC1 channels in stretch-induced injury of blood-brain barrier endothelial cells. Brain Res. 1436, 1–12. doi: 10.1016/j.brainres.2011.11.044
Birnbaumer, L. (2009). The TRPC class of ion channels: a critical review of their roles in slow, sustained increases in intracellular Ca(2+) concentrations. Annu. Rev. Pharmacol. Toxicol. 49, 395–426. doi: 10.1146/annurev.pharmtox.48.113006.094928
Blatter, L. A. (2017). Tissue specificity: SOCE: implications for Ca2+ Handling in Endothelial Cells. Adv. Exp. Med. Biol. 993, 343–361. doi: 10.1007/978-3-319-57732-6_18
Brough, G. H., Wu, S., Cioffi, D., Moore, T. M., Li, M., Dean, N., et al. (2001). Contribution of endogenously expressed Trp1 to a Ca2+-selective, store-operated Ca2+ entry pathway. FASEB J. 15, 1727–1738. doi: 10.1096/fj.01-0108com
Brown, R. C., Wu, L., Hicks, K., and O’neil, R. G. (2008). Regulation of blood-brain barrier permeability by transient receptor potential type C and type v calcium-permeable channels. Microcirculation 15, 359–371. doi: 10.1080/10739680701762656
Cappelli, H. C., Kanugula, A. K., Adapala, R. K., Amin, V., Sharma, P., Midha, P., et al. (2019). Mechanosensitive TRPV4 channels stabilize VE-cadherin junctions to regulate tumor vascular integrity and metastasis. Cancer Lett. 442, 15–20. doi: 10.1016/j.canlet.2018.07.042
Chaudhuri, P., Colles, S. M., Bhat, M., Van Wagoner, D. R., Birnbaumer, L., and Graham, L. M. (2008). Elucidation of a TRPC6-TRPC5 channel cascade that restricts endothelial cell movement. Mol. Biol. Cell 19, 3203–3211. doi: 10.1091/mbc.e07-08-0765
Chaudhuri, P., Rosenbaum, M. A., Birnbaumer, L., and Graham, L. M. (2017). Integration of TRPC6 and NADPH oxidase activation in lysophosphatidylcholine-induced TRPC5 externalization. Am. J. Physiol. Cell Physiol. 313, C541–C555.
Chen, C. K., Hsu, P. Y., Wang, T. M., Miao, Z. F., Lin, R. T., and Juo, S. H. (2018). TRPV4 activation contributes functional recovery from ischemic stroke via angiogenesis and neurogenesis. Mol. Neurobiol. 55, 4127–4135.
Chen, F., Zhu, L., Cai, L., Zhang, J., Zeng, X., Li, J., et al. (2016). A stromal interaction molecule 1 variant up-regulates matrix metalloproteinase-2 expression by strengthening nucleoplasmic Ca2+ signaling. Biochim. Biophys. Acta 1863, 617–629. doi: 10.1016/j.bbamcr.2016.01.007
Chen, Y. S., Lu, M. J., Huang, H. S., and Ma, M. C. (2010). Mechanosensitive transient receptor potential vanilloid type 1 channels contribute to vascular remodeling of rat fistula veins. J. Vasc. Surg. 52, 1310–1320. doi: 10.1016/j.jvs.2010.05.095
Cheng, H. W., James, A. F., Foster, R. R., Hancox, J. C., and Bates, D. O. (2006). VEGF activates receptor-operated cation channels in human microvascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 26, 1768–1776. doi: 10.1161/01.atv.0000231518.86795.0f
Cheng, K. T., Liu, X., Ong, H. L., Swaim, W., and Ambudkar, I. S. (2011). Local Ca(2)+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca(2)+ signals required for specific cell functions. PLoS Biol. 9:e1001025. doi: 10.1371/journal.pbio.1001025
Cheng, W., Sun, C., and Zheng, J. (2010). Heteromerization of TRP channel subunits: extending functional diversity. Protein Cell 1, 802–810. doi: 10.1007/s13238-010-0108-9
Cheng, W., Yang, F., Takanishi, C. L., and Zheng, J. (2007). Thermosensitive TRPV channel subunits coassemble into heteromeric channels with intermediate conductance and gating properties. J. Gen. Physiol. 129, 191–207. doi: 10.1085/jgp.200709731
Ching, L. C., Chen, C. Y., Su, K. H., Hou, H. H., Shyue, S. K., Kou, Y. R., et al. (2012). Implication of AMP-activated protein kinase in transient receptor potential vanilloid type 1-mediated activation of endothelial nitric oxide synthase. Mol. Med. 18, 805–815. doi: 10.2119/molmed.2011.00461
Ching, L. C., Kou, Y. R., Shyue, S. K., Su, K. H., Wei, J., Cheng, L. C., et al. (2011). Molecular mechanisms of activation of endothelial nitric oxide synthase mediated by transient receptor potential vanilloid type 1. Cardiovasc. Res. 91, 492–501. doi: 10.1093/cvr/cvr104
Chung, A. S., and Ferrara, N. (2011). Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol. 27, 563–584. doi: 10.1146/annurev-cellbio-092910-154002
Cioffi, D. L., Wu, S., Alexeyev, M., Goodman, S. R., Zhu, M. X., and Stevens, T. (2005). Activation of the endothelial store-operated ISOC Ca2+ channel requires interaction of protein 4.1 with TRPC4. Circ. Res. 97, 1164–1172. doi: 10.1161/01.res.0000193597.65217.00
Cioffi, D. L., Wu, S., Chen, H., Alexeyev, M., St Croix, C. M., Pitt, B. R., et al. (2012). Orai1 determines calcium selectivity of an endogenous TRPC heterotetramer channel. Circ. Res. 110, 1435–1444. doi: 10.1161/circresaha.112.269506
Curcic, S., Schober, R., Schindl, R., and Groschner, K. (2019). TRPC-mediated Ca(2+) signaling and control of cellular functions. Semin. Cell Dev. Biol. 94, 28–39. doi: 10.1016/j.semcdb.2019.02.001
D’Alessio, A., Moccia, F., Li, J. H., Micera, A., and Kyriakides, T. R. (2015). Angiogenesis and Vasculogenesis in Health and Disease. Biomed. Res. Int. 2015:126582.
Di Buduo, C. A., Moccia, F., Battiston, M., De Marco, L., Mazzucato, M., Moratti, R., et al. (2014). The importance of calcium in the regulation of megakaryocyte function. Haematologica 99, 769–778. doi: 10.3324/haematol.2013.096859
Ding, Y., Fromel, T., Popp, R., Falck, J. R., Schunck, W. H., and Fleming, I. (2014). The biological actions of 11,12-epoxyeicosatrienoic acid in endothelial cells are specific to the R/S-enantiomer and require the G(s) protein. J. Pharmacol. Exp. Ther. 350, 14–21. doi: 10.1124/jpet.114.214254
Dragoni, S., Guerra, G., Fiorio Pla, A., Bertoni, G., Rappa, A., Poletto, V., et al. (2015a). A functional transient receptor potential vanilloid 4 (TRPV4) channel is expressed in human endothelial progenitor cells. J. Cell. Physiol. 230, 95–104. doi: 10.1002/jcp.24686
Dragoni, S., Reforgiato, M., Zuccolo, E., Poletto, V., Lodola, F., Ruffinatti, F. A., et al. (2015b). Dysregulation of VEGF-induced proangiogenic Ca2+ oscillations in primary myelofibrosis-derived endothelial colony-forming cells. Exp. Hematol. 43:e1013.
Dragoni, S., Laforenza, U., Bonetti, E., Lodola, F., Bottino, C., Berra-Romani, R., et al. (2011). Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells 29, 1898–1907. doi: 10.1002/stem.734
Dragoni, S., Laforenza, U., Bonetti, E., Lodola, F., Bottino, C., Guerra, G., et al. (2013). Canonical transient receptor potential 3 channel triggers vascular endothelial growth factor-induced intracellular Ca2+ oscillations in endothelial progenitor cells isolated from umbilical cord blood. Stem Cells Dev. 22, 2561–2580. doi: 10.1089/scd.2013.0032
Dragoni, S., Laforenza, U., Bonetti, E., Reforgiato, M., Poletto, V., Lodola, F., et al. (2014). Enhanced expression of Stim, Orai, and TRPC transcripts and proteins in endothelial progenitor cells isolated from patients with primary myelofibrosis. PLoS One 9:e91099. doi: 10.1371/journal.pone.0091099
Du, J., Ma, X., Shen, B., Huang, Y., Birnbaumer, L., and Yao, X. (2014). TRPV4, TRPC1, and TRPP2 assemble to form a flow-sensitive heteromeric channel. FASEB J. 28, 4677–4685. doi: 10.1096/fj.14-251652
Du, L. L., Shen, Z., Li, Z., Ye, X., Wu, M., Hong, L., et al. (2018). TRPC1 deficiency impairs the endothelial progenitor cell function via inhibition of calmodulin/eNOS pathway. J. Cardiovasc. Transl. Res. 11, 339–345. doi: 10.1007/s12265-018-9798-9
Earley, S., and Brayden, J. E. (2015). Transient receptor potential channels in the vasculature. Physiol. Rev. 95, 645–690. doi: 10.1152/physrev.00026.2014
Earley, S., Gonzales, A. L., and Crnich, R. (2009). Endothelium-dependent cerebral artery dilation mediated by TRPA1 and Ca2+-Activated K+ channels. Circ. Res. 104, 987–994. doi: 10.1161/circresaha.108.189530
Evangelista, A. M., Thompson, M. D., Weisbrod, R. M., Pimental, D. R., Tong, X., Bolotina, V. M., et al. (2012). Redox regulation of SERCA2 is required for vascular endothelial growth factor-induced signaling and endothelial cell migration. Antioxid. Redox. Signal. 17, 1099–1108. doi: 10.1089/ars.2011.4022
Fantozzi, I., Zhang, S., Platoshyn, O., Remillard, C. V., Cowling, R. T., and Yuan, J. X. (2003). Hypoxia increases AP-1 binding activity by enhancing capacitative Ca2+ entry in human pulmonary artery endothelial cells. Am. J. Physiol. Lung. Cell Mol. Physiol. 285, L1233–L1245.
Favia, A., Desideri, M., Gambara, G., D’alessio, A., Ruas, M., Esposito, B., et al. (2014). VEGF-induced neoangiogenesis is mediated by NAADP and two-pore channel-2-dependent Ca2+ signaling. Proc. Natl. Acad. Sci. U.S.A. 111, E4706–E4715.
Feng, N. H., Lee, H. H., Shiang, J. C., and Ma, M. C. (2008). Transient receptor potential vanilloid type 1 channels act as mechanoreceptors and cause substance P release and sensory activation in rat kidneys. Am. J. Physiol. Renal Physiol. 294, F316–F325.
Fiorio Pla, A., Grange, C., Antoniotti, S., Tomatis, C., Merlino, A., Bussolati, B., et al. (2008). Arachidonic acid-induced Ca2+ entry is involved in early steps of tumor angiogenesis. Mol. Cancer Res. 6, 535–545. doi: 10.1158/1541-7786.mcr-07-0271
Fischer, C., Schneider, M., and Carmeliet, P. (2006). Principles and therapeutic implications of angiogenesis, vasculogenesis and arteriogenesis. Handb. Exp. Pharmacol. 176, 157–212. doi: 10.1007/3-540-36028-x_6
Freichel, M., Suh, S. H., Pfeifer, A., Schweig, U., Trost, C., Weissgerber, P., et al. (2001). Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4-/- mice. Nat. Cell Biol. 3, 121–127. doi: 10.1038/35055019
Gaudet, R. (2008). TRP channels entering the structural era. J. Physiol. 586, 3565–3575. doi: 10.1113/jphysiol.2008.155812
Ge, R., Tai, Y., Sun, Y., Zhou, K., Yang, S., Cheng, T., et al. (2009). Critical role of TRPC6 channels in VEGF-mediated angiogenesis. Cancer Lett. 283, 43–51. doi: 10.1016/j.canlet.2009.03.023
Gees, M., Colsoul, B., and Nilius, B. (2010). The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2:a003962. doi: 10.1101/cshperspect.a003962
Genova, T., Grolez, G. P., Camillo, C., Bernardini, M., Bokhobza, A., Richard, E., et al. (2017). TRPM8 inhibits endothelial cell migration via a non-channel function by trapping the small GTPase Rap1. J. Cell Biol. 216, 2107–2130. doi: 10.1083/jcb.201506024
Gerzanich, V., Woo, S. K., Vennekens, R., Tsymbalyuk, O., Ivanova, S., Ivanov, A., et al. (2009). De novo expression of Trpm4 initiates secondary hemorrhage in spinal cord injury. Nat. Med. 15, 185–191. doi: 10.1038/nm.1899
Goel, M., Sinkins, W. G., and Schilling, W. P. (2002). Selective association of TRPC channel subunits in rat brain synaptosomes. J. Biol. Chem. 277, 48303–48310. doi: 10.1074/jbc.m207882200
Goldenberg, N. M., Wang, L., Ranke, H., Liedtke, W., Tabuchi, A., and Kuebler, W. M. (2015). TRPV4 Is required for hypoxic pulmonary vasoconstriction. Anesthesiology 122, 1338–1348. doi: 10.1097/aln.0000000000000647
Gong, B., Asimakis, G. K., Chen, Z., Albrecht, T. B., Boor, P. J., Pappas, T. C., et al. (2006). Whole-body hyperthermia induces up-regulation of vascular endothelial growth factor accompanied by neovascularization in cardiac tissue. Life Sci. 79, 1781–1788. doi: 10.1016/j.lfs.2006.06.025
Graziani, A., Poteser, M., Heupel, W. M., Schleifer, H., Krenn, M., Drenckhahn, D., et al. (2010). Cell-cell contact formation governs Ca2+ signaling by TRPC4 in the vascular endothelium: evidence for a regulatory TRPC4-beta-catenin interaction. J. Biol. Chem. 285, 4213–4223. doi: 10.1074/jbc.m109.060301
Greenberg, H. Z. E., Carlton-Carew, S. R. E., Khan, D. M., Zargaran, A. K., Jahan, K. S., and Vanessa, et al. (2017). Heteromeric TRPV4/TRPC1 channels mediate calcium-sensing receptor-induced nitric oxide production and vasorelaxation in rabbit mesenteric arteries. Vascul. Pharmacol. 9, 53–62. doi: 10.1016/j.vph.2017.08.005
Groschner, K., Shrestha, N., and Fameli, N. (2017). Cardiovascular and hemostatic disorders: SOCE in cardiovascular cells: emerging targets for therapeutic intervention. Adv. Exp. Med. Biol. 993, 473–503. doi: 10.1007/978-3-319-57732-6_24
Guerra, G., Lucariello, A., Perna, A., Botta, L., De Luca, A., and Moccia, F. (2018). The role of endothelial Ca(2+) signaling in neurovascular coupling: a view from the lumen. Int. J. Mol. Sci. 19:E938.
Guo, B. C., Wei, J., Su, K. H., Chiang, A. N., Zhao, J. F., Chen, H. Y., et al. (2015). Transient receptor potential vanilloid type 1 is vital for (-)-epigallocatechin-3-gallate mediated activation of endothelial nitric oxide synthase. Mol. Nutr. Food Res. 59, 646–657. doi: 10.1002/mnfr.201400699
Guo, Z., Grimm, C., Becker, L., Ricci, A. J., and Heller, S. (2013). A novel ion channel formed by interaction of TRPML3 with TRPV5. PLoS One 8:e58174. doi: 10.1371/journal.pone.0058174
Hamdollah Zadeh, M. A., Glass, C. A., Magnussen, A., Hancox, J. C., and Bates, D. O. (2008). VEGF-mediated elevated intracellular calcium and angiogenesis in human microvascular endothelial cells in vitro are inhibited by dominant negative TRPC6. Microcirculation 15, 605–614. doi: 10.1080/10739680802220323
Hatano, N., Suzuki, H., Itoh, Y., and Muraki, K. (2013). TRPV4 partially participates in proliferation of human brain capillary endothelial cells. Life Sci. 92, 317–324. doi: 10.1016/j.lfs.2013.01.002
Hecquet, C. M., Ahmmed, G. U., Vogel, S. M., and Malik, A. B. (2008). Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ. Res. 102, 347–355. doi: 10.1161/circresaha.107.160176
Heil, M., Eitenmuller, I., Schmitz-Rixen, T., and Schaper, W. (2006). Arteriogenesis versus angiogenesis: similarities and differences. J. Cell Mol. Med. 10, 45–55. doi: 10.1111/j.1582-4934.2006.tb00290.x
Heinke, J., Patterson, C., and Moser, M. (2012). Life is a pattern: vascular assembly within the embryo. Front. Biosci. 4, 2269–2288. doi: 10.2741/e541
Hofmann, N. A., Barth, S., Waldeck-Weiermair, M., Klec, C., Strunk, D., Malli, R., et al. (2014). TRPV1 mediates cellular uptake of anandamide and thus promotes endothelial cell proliferation and network-formation. Biol. Open 3, 1164–1172. doi: 10.1242/bio.20149571
Hofmann, T., Schaefer, M., Schultz, G., and Gudermann, T. (2002). Subunit composition of mammalian transient receptor potential channels in living cells. Proc. Natl. Acad. Sci. U.S.A. 99, 7461–7466. doi: 10.1073/pnas.102596199
Ihori, H., Nozawa, T., Sobajima, M., Shida, T., Fukui, Y., Fujii, N., et al. (2016). Waon therapy attenuates cardiac hypertrophy and promotes myocardial capillary growth in hypertensive rats: a comparative study with fluvastatin. Heart Vessels 31, 1361–1369. doi: 10.1007/s00380-015-0779-5
Inoue, K., and Xiong, Z. G. (2009). Silencing TRPM7 promotes growth/proliferation and nitric oxide production of vascular endothelial cells via the ERK pathway. Cardiovasc. Res. 83, 547–557. doi: 10.1093/cvr/cvp153
Inoue, R., Jensen, L. J., Shi, J., Morita, H., Nishida, M., Honda, A., et al. (2006). Transient receptor potential channels in cardiovascular function and disease. Circ. Res. 99, 119–131. doi: 10.1161/01.res.0000233356.10630.8a
Jho, D., Mehta, D., Ahmmed, G., Gao, X. P., Tiruppathi, C., Broman, M., et al. (2005). Angiopoietin-1 opposes VEGF-induced increase in endothelial permeability by inhibiting TRPC1-dependent Ca2 influx. Circ. Res. 96, 1282–1290. doi: 10.1161/01.res.0000171894.03801.03
Jian, M. Y., King, J. A., Al-Mehdi, A. B., Liedtke, W., and Townsley, M. I. (2008). High vascular pressure-induced lung injury requires P450 epoxygenase-dependent activation of TRPV4. Am. J. Respir. Cell Mol. Biol. 38, 386–392. doi: 10.1165/rcmb.2007-0192oc
Kanugula, A. K., Adapala, R. K., Midha, P., Cappelli, H. C., Meszaros, J. G., Paruchuri, S., et al. (2019). Novel noncanonical regulation of soluble VEGF/VEGFR2 signaling by mechanosensitive ion channel TRPV4. FASEB J. 33, 195–203. doi: 10.1096/fj.201800509r
Kini, V., Chavez, A., and Mehta, D. (2010). A new role for PTEN in regulating transient receptor potential canonical channel 6-mediated Ca2+ entry, endothelial permeability, and angiogenesis. J. Biol. Chem. 285, 33082–33091. doi: 10.1074/jbc.m110.142034
Krapivinsky, G., Krapivinsky, L., Manasian, Y., and Clapham, D. E. (2014). The TRPM7 chanzyme is cleaved to release a chromatin-modifying kinase. Cell 157, 1061–1072. doi: 10.1016/j.cell.2014.03.046
Kuang, C. Y., Yu, Y., Guo, R. W., Qian, D. H., Wang, K., Den, M. Y., et al. (2010). Silencing stromal interaction molecule 1 by RNA interference inhibits the proliferation and migration of endothelial progenitor cells. Biochem. Biophys. Res. Commun. 398, 315–320. doi: 10.1016/j.bbrc.2010.06.088
Kuang, C. Y., Yu, Y., Wang, K., Qian, D. H., Den, M. Y., and Huang, L. (2012). Knockdown of transient receptor potential canonical-1 reduces the proliferation and migration of endothelial progenitor cells. Stem Cells Dev. 21, 487–496. doi: 10.1089/scd.2011.0027
Kusaba, T., Okigaki, M., Matui, A., Murakami, M., Ishikawa, K., Kimura, T., et al. (2010). Klotho is associated with VEGF receptor-2 and the transient receptor potential canonical-1 Ca2+ channel to maintain endothelial integrity. Proc. Natl. Acad. Sci. U.S.A. 107, 19308–19313. doi: 10.1073/pnas.1008544107
Lee, K. P., Choi, S., Hong, J. H., Ahuja, M., Graham, S., Ma, R., et al. (2014). Molecular determinants mediating gating of transient receptor potential canonical (TRPC) channels by stromal interaction molecule 1 (STIM1). J. Biol. Chem. 289, 6372–6382. doi: 10.1074/jbc.m113.546556
Lepage, P. K., and Boulay, G. (2007). Molecular determinants of TRP channel assembly. Biochem. Soc. Trans. 35, 81–83. doi: 10.1042/bst0350081
Li, J., Cubbon, R. M., Wilson, L. A., Amer, M. S., Mckeown, L., Hou, B., et al. (2011). Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ. Res. 108, 1190–1198. doi: 10.1161/circresaha.111.243352
Li, M., Yu, Y., and Yang, J. (2011). Structural biology of TRP channels. Adv. Exp. Med. Biol. 704, 1–23. doi: 10.1007/978-94-007-0265-3_1
Lodola, F., Laforenza, U., Bonetti, E., Lim, D., Dragoni, S., Bottino, C., et al. (2012). Store-operated Ca2+ entry is remodelled and controls in vitro angiogenesis in endothelial progenitor cells isolated from tumoral patients. PLoS One 7:e42541. doi: 10.1371/journal.pone.0042541
Lodola, F., Laforenza, U., Cattaneo, F., Ruffinatti, F. A., Poletto, V., Massa, M., et al. (2017). VEGF-induced intracellular Ca2+ oscillations are down-regulated and do not stimulate angiogenesis in breast cancer-derived endothelial colony forming cells. Oncotarget 8, 95223–95246.
Lodola, F., Rosti, V., Tullii, G., Desii, A., Tapella, L., Catarsi, P., et al. (2019). Conjugated polymers optically regulate the fate of endothelial colony-forming cells. Sci. Adv. 5:eaav4620. doi: 10.1126/sciadv.aav4620
Loh, K. P., Ng, G., Yu, C. Y., Fhu, C. K., Yu, D., Vennekens, R., et al. (2014). TRPM4 inhibition promotes angiogenesis after ischemic stroke. Pflugers. Arch. 466, 563–576. doi: 10.1007/s00424-013-1347-4
Ma, X., Chen, Z., Hua, D., He, D., Wang, L., Zhang, P., et al. (2014). Essential role for TrpC5-containing extracellular vesicles in breast cancer with chemotherapeutic resistance. Proc. Natl. Acad. Sci. U.S.A. 111, 6389–6394. doi: 10.1073/pnas.1400272111
Ma, X., Cheng, K. T., Wong, C. O., O’neil, R. G., Birnbaumer, L., Ambudkar, I. S., et al. (2011a). Heteromeric TRPV4-C1 channels contribute to store-operated Ca(2+) entry in vascular endothelial cells. Cell Calcium 50, 502–509. doi: 10.1016/j.ceca.2011.08.006
Ma, X., Nilius, B., Wong, J. W., Huang, Y., and Yao, X. (2011b). Electrophysiological properties of heteromeric TRPV4-C1 channels. Biochim. Biophys. Acta 1808, 2789–2797. doi: 10.1016/j.bbamem.2011.07.049
Ma, X., He, D., Ru, X., Chen, Y., Cai, Y., Bruce, I. C., et al. (2012). Apigenin, a plant-derived flavone, activates transient receptor potential vanilloid 4 cation channel. Br. J. Pharmacol. 166, 349–358. doi: 10.1111/j.1476-5381.2011.01767.x
Maltaneri, R. E., Schiappacasse, A., Chamorro, M. E., Nesse, A. B., and Vittori, D. C. (2018). Participation of membrane calcium channels in erythropoietin-induced endothelial cell migration. Eur. J. Cell Biol. 97, 411–421. doi: 10.1016/j.ejcb.2018.06.002
Mancardi, D., Pla, A. F., Moccia, F., Tanzi, F., and Munaron, L. (2011). Old and new gasotransmitters in the cardiovascular system: focus on the role of nitric oxide and hydrogen sulfide in endothelial cells and cardiomyocytes. Curr. Pharm. Biotechnol. 12, 1406–1415. doi: 10.2174/138920111798281090
Martin, E., Dahan, D., Cardouat, G., Gillibert-Duplantier, J., Marthan, R., Savineau, J. P., et al. (2012). Involvement of TRPV1 and TRPV4 channels in migration of rat pulmonary arterial smooth muscle cells. Pflugers. Arch. 464, 261–272. doi: 10.1007/s00424-012-1136-5
Medina, R. J., Barber, C. L., Sabatier, F., Dignat-George, F., Melero-Martin, J. M., Khosrotehrani, K., et al. (2017). Endothelial progenitors: a consensus statement on nomenclature. Stem Cells Transl. Med. 6, 1316–1320. doi: 10.1002/sctm.16-0360
Mergler, S., Dannowski, H., Bednarz, J., Engelmann, K., Hartmann, C., and Pleyer, U. (2003). Calcium influx induced by activation of receptor tyrosine kinases in SV40-transfected human corneal endothelial cells. Exp. Eye Res. 77, 485–495. doi: 10.1016/s0014-4835(03)00154-4
Mergler, S., Valtink, M., Coulson-Thomas, V. J., Lindemann, D., Reinach, P. S., Engelmann, K., et al. (2010). TRPV channels mediate temperature-sensing in human corneal endothelial cells. Exp. Eye Res. 90, 758–770. doi: 10.1016/j.exer.2010.03.010
Min, J. K., Han, K. Y., Kim, E. C., Kim, Y. M., Lee, S. W., Kim, O. H., et al. (2004). Capsaicin inhibits in vitro and in vivo angiogenesis. Cancer Res. 64, 644–651.
Mittal, M., Urao, N., Hecquet, C. M., Zhang, M., Sudhahar, V., Gao, X. P., et al. (2015). Novel role of reactive oxygen species-activated Trp melastatin channel-2 in mediating angiogenesis and postischemic neovascularization. Arterioscler. Thromb. Vasc. Biol. 35, 877–887. doi: 10.1161/atvbaha.114.304802
Moccia, F. (2018). Endothelial Ca(2+) signaling and the resistance to anticancer treatments: partners in crime. Int. J. Mol. Sci. 19:E217.
Moccia, F. (2020). Calcium signaling in endothelial colony forming cells in health and disease. Adv. Exp. Med. Biol. 1131, 1013–1030. doi: 10.1007/978-3-030-12457-1_40
Moccia, F., Berra-Romani, R., Baruffi, S., Spaggiari, S., Adams, D. J., Taglietti, V., et al. (2002). Basal nonselective cation permeability in rat cardiac microvascular endothelial cells. Microvasc. Res. 64, 187–197. doi: 10.1006/mvre.2002.2430
Moccia, F., Berra-Romani, R., and Rosti, V. (2018a). Manipulating intracellular Ca2+ signals to stimulate therapeutic angiogenesis in cardiovascular disorders. Curr. Pharm. Biotechnol. 19, 686–699. doi: 10.2174/1389201019666180808165309
Moccia, F., Lucariello, A., and Guerra, G. (2018b). TRPC3-mediated Ca(2+) signals as a promising strategy to boost therapeutic angiogenesis in failing hearts: the role of autologous endothelial colony forming cells. J. Cell. Physiol. 233, 3901–3917. doi: 10.1002/jcp.26152
Moccia, F., Berra-Romani, R., and Tanzi, F. (2012a). Update on vascular endothelial Ca2+ signalling: a tale of ion channels, pumps and transporters. World J. Biol. Chem. 3, 127–158.
Moccia, F., Bonetti, E., Dragoni, S., Fontana, J., Lodola, F., Romani, R. B., et al. (2012b). Hematopoietic progenitor and stem cells circulate by surfing on intracellular Ca2+ waves: a novel target for cell-based therapy and anti-cancer treatment? Curr. Signal. Transd. Ther. 7, 161–176. doi: 10.2174/157436212800376672
Moccia, F., Berra-Romani, R., Tritto, S., Signorelli, S., Taglietti, V., and Tanzi, F. (2003). Epidermal growth factor induces intracellular Ca2+ oscillations in microvascular endothelial cells. J. Cell. Physiol. 194, 139–150. doi: 10.1002/jcp.10198
Moccia, F., and Guerra, G. (2016). Ca(2+) signalling in endothelial progenitor cells: friend or foe? J. Cell Physiol. 231, 314–327. doi: 10.1002/jcp.25126
Moccia, F., Negri, S., Faris, P., and Berra-Romani, R. (2019a). Targeting the endothelial Ca 2+ tool kit to rescue endothelial dysfunction in obesity associated-hypertension. Curr. Med. Chem. doi: 10.2174/0929867326666190905142135 [Epub ahead of print].
Moccia, F., Negri, S., Shekha, M., Faris, P., and Guerra, G. (2019b). Endothelial Ca(2+) signaling, angiogenesis and vasculogenesis: just what it takes to make a blood vessel. Int. J. Mol. Sci. 20:E3962.
Moccia, F., Zuccolo, E., Poletto, V., Cinelli, M., Bonetti, E., Guerra, G., et al. (2015). Endothelial progenitor cells support tumour growth and metastatisation: implications for the resistance to anti-angiogenic therapy. Tumour Biol. 36, 6603–6614. doi: 10.1007/s13277-015-3823-2
Moore, T. M., Brough, G. H., Babal, P., Kelly, J. J., Li, M., and Stevens, T. (1998). Store-operated calcium entry promotes shape change in pulmonary endothelial cells expressing Trp1. Am. J. Physiol. 275, L574–L582.
Munaron, L., and Fiorio Pla, A. (2000). Calcium influx induced by activation of tyrosine kinase receptors in cultured bovine aortic endothelial cells. J. Cell. Physiol. 185, 454–463. doi: 10.1002/1097-4652(200012)185:3<454::aid-jcp17>3.0.co;2-a
Noren, D. P., Chou, W. H., Lee, S. H., Qutub, A. A., Warmflash, A., Wagner, D. S., et al. (2016). Endothelial cells decode VEGF-mediated Ca2+ signaling patterns to produce distinct functional responses. Sci. Signal. 9:ra20. doi: 10.1126/scisignal.aad3188
O’Leary, C., Mcgahon, M. K., Ashraf, S., Mcnaughten, J., Friedel, T., Cincola, P., et al. (2019). Involvement of TRPV1 and TRPV4 channels in retinal angiogenesis. Invest. Ophthalmol. Vis. Sci. 60, 3297–3309.
Paria, B. C., Bair, A. M., Xue, J., Yu, Y., Malik, A. B., and Tiruppathi, C. (2006). Ca2+ influx induced by protease-activated receptor-1 activates a feed-forward mechanism of TRPC1 expression via nuclear factor-kappaB activation in endothelial cells. J. Biol. Chem. 281, 20715–20727. doi: 10.1074/jbc.m600722200
Parpaite, T., Cardouat, G., Mauroux, M., Gillibert-Duplantier, J., Robillard, P., Quignard, J. F., et al. (2016). Effect of hypoxia on TRPV1 and TRPV4 channels in rat pulmonary arterial smooth muscle cells. Pflugers. Arch. 468, 111–130. doi: 10.1007/s00424-015-1704-6
Pires, P. W., and Earley, S. (2017). Redox regulation of transient receptor potential channels in the endothelium. Microcirculation 24.
Pires, P. W., and Earley, S. (2018). Neuroprotective effects of TRPA1 channels in the cerebral endothelium following ischemic stroke. eLife 7:e35316.
Pires, P. W., Sullivan, M. N., Pritchard, H. A., Robinson, J. J., and Earley, S. (2015). Unitary TRPV3 channel Ca2+ influx events elicit endothelium-dependent dilation of cerebral parenchymal arterioles. Am. J. Physiol. Heart Circ. Physiol. 309, H2031–H2041.
Pla, A. F., Ong, H. L., Cheng, K. T., Brossa, A., Bussolati, B., Lockwich, T., et al. (2012). TRPV4 mediates tumor-derived endothelial cell migration via arachidonic acid-activated actin remodeling. Oncogene 31, 200–212. doi: 10.1038/onc.2011.231
Poletto, V., Rosti, V., Biggiogera, M., Guerra, G., Moccia, F., and Porta, C. (2018). The role of endothelial colony forming cells in kidney cancer’s pathogenesis, and in resistance to anti-VEGFR agents and mTOR inhibitors: a speculative review. Crit. Rev. Oncol. Hematol. 132, 89–99. doi: 10.1016/j.critrevonc.2018.09.005
Potente, M., Gerhardt, H., and Carmeliet, P. (2011). Basic and therapeutic aspects of angiogenesis. Cell 146, 873–887. doi: 10.1016/j.cell.2011.08.039
Poteser, M., Graziani, A., Rosker, C., Eder, P., Derler, I., Kahr, H., et al. (2006). TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J. Biol. Chem. 281, 13588–13595. doi: 10.1074/jbc.m512205200
Prakriya, M., and Lewis, R. S. (2015). Store-operated calcium channels. Physiol. Rev. 95, 1383–1436.
Qin, W., Xie, W., Xia, N., He, Q., and Sun, T. (2016). Silencing of transient receptor potential channel 4 alleviates oxLDL-induced angiogenesis in human coronary artery endothelial cells by inhibition of VEGF and NF-kappaB. Med. Sci. Monit. 22, 930–936. doi: 10.12659/msm.897634
Rafii, S., Butler, J. M., and Ding, B. S. (2016). Angiocrine functions of organ-specific endothelial cells. Nature 529, 316–325. doi: 10.1038/nature17040
Ramsey, I. S., Delling, M., and Clapham, D. E. (2006). An introduction to TRP channels. Annu. Rev. Physiol. 68, 619–647.
Randhawa, P. K., and Jaggi, A. S. (2015). TRPV4 channels: physiological and pathological role in cardiovascular system. Basic Res. Cardiol. 110:54.
Richter, J. M., Schaefer, M., and Hill, K. (2014a). Clemizole hydrochloride is a novel and potent inhibitor of transient receptor potential channel TRPC5. Mol. Pharmacol. 86, 514–521. doi: 10.1124/mol.114.093229
Richter, J. M., Schaefer, M., and Hill, K. (2014b). Riluzole activates TRPC5 channels independently of PLC activity. Br. J. Pharmacol. 171, 158–170. doi: 10.1111/bph.12436
Rosenbaum, M. A., Chaudhuri, P., and Graham, L. M. (2015). Hypercholesterolemia inhibits re-endothelialization of arterial injuries by TRPC channel activation. J. Vasc. Surg. 62:e1042.
Ryu, H. J., Kim, J. E., Kim, Y. J., Kim, J. Y., Kim, W. I., Choi, S. Y., et al. (2013). Endothelial transient receptor potential conical channel (TRPC)-3 activation induces vasogenic edema formation in the rat piriform cortex following status epilepticus. Cell Mol. Neurobiol. 33, 575–585. doi: 10.1007/s10571-013-9931-x
Samanta, A., Hughes, T. E. T., and Moiseenkova-Bell, V. Y. (2018). Transient receptor potential (TRP) channels. Subcell. Biochem. 87, 141–165.
Sanchez-Hernandez, Y., Laforenza, U., Bonetti, E., Fontana, J., Dragoni, S., Russo, M., et al. (2010). Store-operated Ca(2+) entry is expressed in human endothelial progenitor cells. Stem Cells Dev. 19, 1967–1981. doi: 10.1089/scd.2010.0047
Sappington, R. M., Sidorova, T., Long, D. J., and Calkins, D. J. (2009). TRPV1: contribution to retinal ganglion cell apoptosis and increased intracellular Ca2+ with exposure to hydrostatic pressure. Invest. Ophthalmol. Vis. Sci. 50, 717–728.
Sarmiento, D., Montorfano, I., Cerda, O., Caceres, M., Becerra, A., Cabello-Verrugio, C., et al. (2015). Increases in reactive oxygen species enhance vascular endothelial cell migration through a mechanism dependent on the transient receptor potential melastatin 4 ion channel. Microvasc. Res. 98, 187–196. doi: 10.1016/j.mvr.2014.02.001
Savage, A. M., Kurusamy, S., Chen, Y., Jiang, Z., Chhabria, K., Macdonald, R. B., et al. (2019). tmem33 is essential for VEGF-mediated endothelial calcium oscillations and angiogenesis. Nat. Commun. 10:732.
Scarpellino, G., Genova, T., Avanzato, D., Bernardini, M., Bianco, S., Petrillo, S., et al. (2019). Purinergic calcium signals in tumor-derived endothelium. Cancers 11:E766.
Schierling, W., Troidl, K., Apfelbeck, H., Troidl, C., Kasprzak, P. M., Schaper, W., et al. (2011). Cerebral arteriogenesis is enhanced by pharmacological as well as fluid-shear-stress activation of the Trpv4 calcium channel. Eur. J. Vasc. Endovasc. Surg. 41, 589–596. doi: 10.1016/j.ejvs.2010.11.034
Schindl, R., Fritsch, R., Jardin, I., Frischauf, I., Kahr, H., Muik, M., et al. (2012). Canonical transient receptor potential (TRPC) 1 acts as a negative regulator for vanilloid TRPV6-mediated Ca2+ influx. J. Biol. Chem. 287, 35612–35620. doi: 10.1074/jbc.m112.400952
Schmidt, K., Dubrovska, G., Nielsen, G., Fesus, G., Uhrenholt, T. R., Hansen, P. B. et al. (2010). Amplification of EDHF-type vasodilatations in TRPC1-deficient mice. Br. J. Pharmacol. 161, 1722–1733. doi: 10.1111/j.1476-5381.2010.00985.x
Shi, J., Miralles, F., Kinet, J. P., Birnbaumer, L., Large, W. A., and Albert, A. P. (2017). Evidence that Orai1 does not contribute to store-operated TRPC1 channels in vascular smooth muscle cells. Channels 11, 329–339. doi: 10.1080/19336950.2017.1303025
Shibasaki, K. (2016). TRPV4 ion channel as important cell sensors. J. Anesth. 30, 1014–1019. doi: 10.1007/s00540-016-2225-y
Simons, M., Gordon, E., and Claesson-Welsh, L. (2016). Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 17, 611–625. doi: 10.1038/nrm.2016.87
Smani, T., Gomez, L. J., Regodon, S., Woodard, G. E., Siegfried, G., Khatib, A. M., et al. (2018). TRP channels in angiogenesis and other endothelial functions. Front. Physiol. 9:1731. doi: 10.3389/fphys.2018.01731
Sobajima, M., Nozawa, T., Shida, T., Ohori, T., Suzuki, T., Matsuki, A., et al. (2011). Repeated sauna therapy attenuates ventricular remodeling after myocardial infarction in rats by increasing coronary vascularity of noninfarcted myocardium. Am. J. Physiol. Heart Circ. Physiol. 301, H548–H554.
Song, H. B., Jun, H. O., Kim, J. H., Fruttiger, M., and Kim, J. H. (2015). Suppression of transient receptor potential canonical channel 4 inhibits vascular endothelial growth factor-induced retinal neovascularization. Cell Calcium 57, 101–108. doi: 10.1016/j.ceca.2015.01.002
Song, S., Ayon, R. J., Yamamura, A., Yamamura, H., Dash, S., Babicheva, A., et al. (2017). Capsaicin-induced Ca(2+) signaling is enhanced via upregulated TRPV1 channels in pulmonary artery smooth muscle cells from patients with idiopathic PAH. Am. J. Physiol. Lung. Cell Mol. Physiol. 312, L309–L325.
Sonkusare, S. K., Bonev, A. D., Ledoux, J., Liedtke, W., Kotlikoff, M. I., Heppner, T. J., et al. (2012). Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 336, 597–601. doi: 10.1126/science.1216283
Storch, U., Forst, A. L., Philipp, M., Gudermann, T., Mederos, Y., and Schnitzler, M. (2012). Transient receptor potential channel 1 (TRPC1) reduces calcium permeability in heteromeric channel complexes. J. Biol. Chem. 287, 3530–3540. doi: 10.1074/jbc.m111.283218
Su, K. H., Lee, K. I., Shyue, S. K., Chen, H. Y., Wei, J., and Lee, T. S. (2014a). Implication of transient receptor potential vanilloid type 1 in 14,15-epoxyeicosatrienoic acid-induced angiogenesis. Int. J. Biol. Sci. 10, 990–996. doi: 10.7150/ijbs.9832
Su, K. H., Lin, S. J., Wei, J., Lee, K. I., Zhao, J. F., Shyue, S. K., et al. (2014b). The essential role of transient receptor potential vanilloid 1 in simvastatin-induced activation of endothelial nitric oxide synthase and angiogenesis. Acta Physiol. 212, 191–204. doi: 10.1111/apha.12378
Sullivan, M. N., Gonzales, A. L., Pires, P. W., Bruhl, A., Leo, M. D., Li, W., et al. (2015). Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci. Signal. 8:ra2. doi: 10.1126/scisignal.2005659
Sundivakkam, P. C., Freichel, M., Singh, V., Yuan, J. P., Vogel, S. M., Flockerzi, V., et al. (2012). The Ca(2+) sensor stromal interaction molecule 1 (STIM1) is necessary and sufficient for the store-operated Ca(2+) entry function of transient receptor potential canonical (TRPC) 1 and 4 channels in endothelial cells. Mol. Pharmacol. 81, 510–526. doi: 10.1124/mol.111.074658
Suresh, K., Servinsky, L., Jiang, H., Bigham, Z., Yun, X., Kliment, C., et al. (2018). Reactive oxygen species induced Ca(2+) influx via TRPV4 and microvascular endothelial dysfunction in the SU5416/hypoxia model of pulmonary arterial hypertension. Am. J. Physiol. Lung. Cell Mol. Physiol. 314, L893–L907.
Suresh, K., Servinsky, L., Reyes, J., Baksh, S., Undem, C., Caterina, M., et al. (2015). Hydrogen peroxide-induced calcium influx in lung microvascular endothelial cells involves TRPV4. Am. J. Physiol. Lung. Cell Mol. Physiol. 309, L1467–L1477.
Tauseef, M., Knezevic, N., Chava, K. R., Smith, M., Sukriti, S., Gianaris, N., et al. (2012). TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J. Exp. Med. 209, 1953–1968. doi: 10.1084/jem.20111355
Thakore, P., and Earley, S. (2019). Transient receptor potential channels and endothelial cell calcium signaling. Compr. Physiol. 9, 1249–1277. doi: 10.1002/cphy.c180034
Thippegowda, P. B., Singh, V., Sundivakkam, P. C., Xue, J., Malik, A. B., and Tiruppathi, C. (2010). Ca2+ influx via TRPC channels induces NF-kappaB-dependent A20 expression to prevent thrombin-induced apoptosis in endothelial cells. Am. J. Physiol. Cell Physiol. 298, C656–C664.
Thodeti, C. K., Matthews, B., Ravi, A., Mammoto, A., Ghosh, K., Bracha, A. L., et al. (2009). TRPV4 channels mediate cyclic strain-induced endothelial cell reorientation through integrin-to-integrin signaling. Circ. Res. 104, 1123–1130. doi: 10.1161/circresaha.108.192930
Thoppil, R. J., Adapala, R. K., Cappelli, H. C., Kondeti, V., Dudley, A. C., Gary Meszaros, J., et al. (2015). TRPV4 channel activation selectively inhibits tumor endothelial cell proliferation. Sci. Rep. 5:14257.
Tiruppathi, C., Freichel, M., Vogel, S. M., Paria, B. C., Mehta, D., Flockerzi, V., et al. (2002). Impairment of store-operated Ca2+ entry in TRPC4(-/-) mice interferes with increase in lung microvascular permeability. Circ. Res. 91, 70–76. doi: 10.1161/01.res.0000023391.40106.a8
Troidl, C., Nef, H., Voss, S., Schilp, A., Kostin, S., Troidl, K., et al. (2010). Calcium-dependent signalling is essential during collateral growth in the pig hind limb-ischemia model. J. Mol. Cell Cardiol. 49, 142–151. doi: 10.1016/j.yjmcc.2010.03.021
Troidl, C., Troidl, K., Schierling, W., Cai, W. J., Nef, H., Mollmann, H., et al. (2009). Trpv4 induces collateral vessel growth during regeneration of the arterial circulation. J. Cell Mol. Med. 13, 2613–2621. doi: 10.1111/j.1582-4934.2008.00579.x
Udan, R. S., Culver, J. C., and Dickinson, M. E. (2013). Understanding vascular development. Wiley Interdiscip. Rev. Dev. Biol. 2, 327–346. doi: 10.1002/wdev.91
Usui-Kusumoto, K., Iwanishi, H., Ichikawa, K., Okada, Y., Sumioka, T., Miyajima, M., et al. (2019). Suppression of neovascularization in corneal stroma in a TRPA1-null mouse. Exp. Eye Res. 181, 90–97. doi: 10.1016/j.exer.2019.01.002
Voets, T., Droogmans, G., and Nilius, B. (1996). Membrane currents and the resting membrane potential in cultured bovine pulmonary artery endothelial cells. J. Physiol. 497(Pt 1), 95–107. doi: 10.1113/jphysiol.1996.sp021752
Wang, L. Y., Zhang, J. H., Yu, J., Yang, J., Deng, M. Y., Kang, H. L., et al. (2015). Reduction of store-operated Ca(2+) entry correlates with endothelial progenitor cell dysfunction in atherosclerotic mice. Stem Cells Dev. 24, 1582–1590. doi: 10.1089/scd.2014.0538
Watanabe, H., Vriens, J., Suh, S. H., Benham, C. D., Droogmans, G., and Nilius, B. (2002). Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J. Biol. Chem. 277, 47044–47051. doi: 10.1074/jbc.m208277200
Weissmann, N., Sydykov, A., Kalwa, H., Storch, U., Fuchs, B., Mederos, Y., et al. (2012). Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat. Commun. 3:649.
White, J. P., Cibelli, M., Urban, L., Nilius, B., Mcgeown, J. G., and Nagy, I. (2016). TRPV4: molecular conductor of a diverse orchestra. Physiol. Rev. 96, 911–973. doi: 10.1152/physrev.00016.2015
Willette, R. N., Bao, W., Nerurkar, S., Yue, T. L., Doe, C. P., Stankus, G., et al. (2008). Systemic activation of the transient receptor potential vanilloid subtype 4 channel causes endothelial failure and circulatory collapse: Part 2. J. Pharmacol. Exp. Ther. 326, 443–452. doi: 10.1124/jpet.107.134551
Xi, Q., Adebiyi, A., Zhao, G., Chapman, K. E., Waters, C. M., Hassid, A., et al. (2008). IP3 constricts cerebral arteries via IP3 receptor-mediated TRPC3 channel activation and independently of sarcoplasmic reticulum Ca2+ release. Circ. Res. 102, 1118–1126. doi: 10.1161/circresaha.108.173948
Xu, N., Cioffi, D. L., Alexeyev, M., Rich, T. C., and Stevens, T. (2015). Sodium entry through endothelial store-operated calcium entry channels: regulation by Orai1. Am. J. Physiol. Cell Physiol. 308, C277–C288.
Yang, X. R., Lin, A. H., Hughes, J. M., Flavahan, N. A., Cao, Y. N., Liedtke, W., et al. (2012). Upregulation of osmo-mechanosensitive TRPV4 channel facilitates chronic hypoxia-induced myogenic tone and pulmonary hypertension. Am. J. Physiol. Lung. Cell Mol. Physiol. 302, L555–L568.
Yang, Y., Cai, Y., Zhang, Y., Liu, J., and Xu, Z. (2018). Exosomes secreted by adipose-derived stem cells contribute to angiogenesis of brain microvascular endothelial cells following oxygen-glucose deprivation in vitro through MicroRNA-181b/TRPM7 Axis. J. Mol. Neurosci. 65, 74–83. doi: 10.1007/s12031-018-1071-9
Yoder, M. C. (2012). Human endothelial progenitor cells. Cold Spring Harb. Perspect. Med. 2:a006692.
Yokota, Y., Nakajima, H., Wakayama, Y., Muto, A., Kawakami, K., Fukuhara, S., et al. (2015). Endothelial Ca 2+ oscillations reflect VEGFR signaling-regulated angiogenic capacity in vivo. eLife 4:e08817.
Yoshida, T., Inoue, R., Morii, T., Takahashi, N., Yamamoto, S., Hara, Y., et al. (2006). Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat. Chem. Biol. 2, 596–607. doi: 10.1038/nchembio821
Yu, P. C., Gu, S. Y., Bu, J. W., and Du, J. L. (2010). TRPC1 is essential for in vivo angiogenesis in zebrafish. Circ. Res. 106, 1221–1232. doi: 10.1161/circresaha.109.207670
Yu, Y. B., Su, K. H., Kou, Y. R., Guo, B. C., Lee, K. I., Wei, J., et al. (2017). Role of transient receptor potential vanilloid 1 in regulating erythropoietin-induced activation of endothelial nitric oxide synthase. Acta Physiol. 219, 465–477. doi: 10.1111/apha.12723
Zeng, Z., Inoue, K., Sun, H., Leng, T., Feng, X., Zhu, L., et al. (2015). TRPM7 regulates vascular endothelial cell adhesion and tube formation. Am. J. Physiol. Cell Physiol. 308, C308–C318.
Zhang, D. X., Mendoza, S. A., Bubolz, A. H., Mizuno, A., Ge, Z. D., Li, R., et al. (2009). Transient receptor potential vanilloid type 4-deficient mice exhibit impaired endothelium-dependent relaxation induced by acetylcholine in vitro and in vivo. Hypertension 53, 532–538. doi: 10.1161/hypertensionaha.108.127100
Zheng, Z., Chen, H., Xie, P., Dickerson, C. A., King, J. A. C., Alexeyev, M. F., et al. (2019). alpha1G T-type calcium channel determines the angiogenic potential of pulmonary microvascular endothelial cells. Am. J. Physiol. Cell Physiol. 316, C353–C364.
Zhu, Y., Gao, M., Zhou, T., Xie, M., Mao, A., Feng, L., et al. (2019). The TRPC5 channel regulates angiogenesis and promotes recovery from ischemic injury in mice. J. Biol. Chem. 294, 28–37. doi: 10.1074/jbc.ra118.005392
Zuccolo, E., Bottino, C., Diofano, F., Poletto, V., Codazzi, A. C., Mannarino, S., et al. (2016). Constitutive store-operated Ca(2+) entry leads to enhanced nitric oxide production and proliferation in infantile hemangioma-derived endothelial colony-forming cells. Stem Cells Dev. 25, 301–319. doi: 10.1089/scd.2015.0240
Zuccolo, E., Di Buduo, C., Lodola, F., Orecchioni, S., Scarpellino, G., Kheder, D. A., et al. (2018). Stromal cell-derived factor-1alpha promotes endothelial colony-forming cell migration through the Ca(2+)-dependent activation of the extracellular signal-regulated Kinase 1/2 and phosphoinositide 3-Kinase/AKT pathways. Stem Cells Dev. 27, 23–34. doi: 10.1089/scd.2017.0114
Keywords: endothelial cells, endothelial colony forming cells, Ca2+ signaling, TRPC, TRPV, TRPM
Citation: Negri S, Faris P, Berra-Romani R, Guerra G and Moccia F (2020) Endothelial Transient Receptor Potential Channels and Vascular Remodeling: Extracellular Ca2 + Entry for Angiogenesis, Arteriogenesis and Vasculogenesis. Front. Physiol. 10:1618. doi: 10.3389/fphys.2019.01618
Received: 14 November 2019; Accepted: 23 December 2019;
Published: 21 January 2020.
Edited by:
Pasquale Pagliaro, University of Turin, ItalyReviewed by:
Astrid Parenti, University of Florence, ItalyCopyright © 2020 Negri, Faris, Berra-Romani, Guerra and Moccia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Moccia, ZnJhbmNlc2NvLm1vY2NpYUB1bmlwdi5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.