 Maicon Landim-Vieira1
Maicon Landim-Vieira1 Jamie R. Johnston1
Jamie R. Johnston1 Weizhen Ji2
Weizhen Ji2 Emily K. Mis2
Emily K. Mis2 Joshua Tijerino1Michele Spencer-Manzon2,3
Joshua Tijerino1Michele Spencer-Manzon2,3 Lauren Jeffries2
Lauren Jeffries2 E. Kevin Hall4
E. Kevin Hall4 David Panisello-Manterola2
David Panisello-Manterola2 Mustafa K. Khokha2,3
Mustafa K. Khokha2,3 Engin Deniz2
Engin Deniz2 P. Bryant Chase5
P. Bryant Chase5 Saquib A. Lakhani2*†
Saquib A. Lakhani2*† Jose Renato Pinto1*†
Jose Renato Pinto1*†- 1Department of Biomedical Sciences, College of Medicine, Florida State University, Tallahassee, FL, United States
- 2Pediatric Genomics Discovery Program, Department of Pediatrics, Yale School of Medicine, Yale University, New Haven, CT, United States
- 3Department of Genetics, Yale School of Medicine, Yale University, New Haven, CT, United States
- 4Department of Pediatrics, Yale School of Medicine, Yale University, New Haven, CT, United States
- 5Department of Biological Science, Florida State University, Tallahassee, FL, United States
Familial dilated cardiomyopathy (DCM), clinically characterized by enlargement and dysfunction of one or both ventricles of the heart, can be caused by variants in sarcomeric genes including TNNC1 (encoding cardiac troponin C, cTnC). Here, we report the case of two siblings with severe, early onset DCM who were found to have compound heterozygous variants in TNNC1: p.Asp145Glu (D145E) and p.Asp132Asn (D132N), which were inherited from the parents. We began our investigation with CRISPR/Cas9 knockout of TNNC1 in Xenopus tropicalis, which resulted in a cardiac phenotype in tadpoles consistent with DCM. Despite multiple maneuvers, we were unable to rescue the tadpole hearts with either human cTnC wild-type or patient variants to investigate the cardiomyopathy phenotype in vivo. We therefore utilized porcine permeabilized cardiac muscle preparations (CMPs) reconstituted with either wild-type or patient variant forms of cTnC to examine effects of the patient variants on contractile function. Incorporation of 50% WT/50% D145E into CMPs increased Ca2+ sensitivity of isometric force, consistent with prior studies. In contrast, incorporation of 50% WT/50% D132N, which had not been previously reported, decreased Ca2+ sensitivity of isometric force. CMPs reconstituted 50–50% with both variants mirrored WT in regard to myofilament Ca2+ responsiveness. Sinusoidal stiffness (SS) (0.2% peak-to-peak) and the kinetics of tension redevelopment (kTR) at saturating Ca2+ were similar to WT for all preparations. Modeling of Ca2+-dependence of kTR support the observation from Ca2+ responsiveness of steady-state isometric force, that the effects on each mutant (50% WT/50% mutant) were greater than the combination of the two mutants (50% D132N/50% D145E). Further studies are needed to ascertain the mechanism(s) of these variants.
Introduction
Pediatric cardiomyopathies, or diseases of the heart muscle in young patients, have an annual incidence of 1.1–1.5 per 100,000 and are the most common indication for heart transplantation in children (Lipshultz et al., 2003; Dipchand et al., 2013; Lee et al., 2017). Dilated cardiomyopathy (DCM), characterized by dilation and dysfunction of one or both ventricles, is the most common type, accounting for more than 50% of all pediatric cardiomyopathies (Lipshultz et al., 2003). Genetic variation in any of a number of proteins that affect cardiomyocyte function is an important cause of DCM (Hershberger et al., 2013; van der Velden and Stienen, 2019). Some of these variants appear to simply predispose toward cardiomyopathy, while others are a primary cause of cardiomyopathy (Burke et al., 2016).
Troponin plays a central role in contractile regulation in striated muscle. This protein complex is comprised of three distinct subunits: troponin I (TnI), troponin T (TnT), and troponin C (TnC) (Farah and Reinach, 1995). Cardiac Troponin C (cTnC), which is encoded by the TNNC1 gene and is abundantly expressed in cardiomyocytes, functions as a myofilament Ca2+ sensor and plays a critical role in regulating contraction (Li and Hwang, 2015). The tertiary structure of cTnC is dominated by the two globular halves of the molecule, the C- and N-domains, which are connected by a flexible linker (Takeda et al., 2003; Li and Hwang, 2015). The C-domain binds divalent cations (Ca2+ and/or Mg2+) at two EF-hand motifs – referred to as sites III and IV based on their location in the primary sequence – and has important structural and modulatory roles in the sarcomere (Holroyde et al., 1980; Marques et al., 2017; Veltri et al., 2017a). The N-domain contains an evolutionarily defunct site I and regulatory EF-hand site II that normally triggers contraction upon Ca2+ binding during systole (Holroyde et al., 1980).
Missense variants in cTnC have been associated with both dilated and hypertrophic cardiomyopathies (Mogensen et al., 2004; Landstrom et al., 2008; Willott et al., 2010; Pinto et al., 2011b; Parvatiyar et al., 2012), and knockout of cTnC in adult zebrafish results in a phenotype consistent with DCM (Ho et al., 2009). Here, we report the case of two siblings diagnosed with severe, early onset pediatric DCM associated with compound heterozygous variants in TNNC1. As a result of our attempts to understand the molecular basis of these pediatric DCM cases, we provide in vivo experimental evidence confirming that TNNC1 is a crucial gene for heart function, and further provide in vitro experimental evidence that altered contractile mechanics – unexpectedly – cannot always explain the pathogenicity of TNNC1 variants.
Experimental Procedures
Ethics
The human subjects research in this study was approved by the Institutional Review Board of Yale University School of Medicine. Xenopus were maintained and cared for in an aquatics facility in accordance with Yale University Institutional Animal Care and Use Committee protocols.
Sequencing Methods
Genomic DNA was isolated from either venous blood or saliva samples. Whole exome sequencing was performed by using IDT xGen capture kit followed by Illumina DNA sequencing (HiSeq 4000). Paired end sequence reads were converted to FASTQ format and were aligned to the reference human genome (hg19). SNVs and indels were called using a GATK pipeline and annotated using AnnoVar.
Genome Editing in Xenopus tropicalis
CRISPR/Cas9-mediated genome editing in Xenopus tropicalis tadpoles was used as previously described (Bhattacharya et al., 2015). Briefly, two non-overlapping, independent CRISPR sgRNAs targeting tnnc1 were designed to generate knockdowns.
CRISPR 1 oligo (targets exon 4):
CTAGCtaatacgactcactataGGTTCTTGGTCATGATGGTCgttt tagagctagaaTAGCAAG
and CRISPR 2 oligo (targets exon 5):
CTAGCtaatacgactcactataGGAGGAACTCATGCGAGATGgtt ttagagctagaaTAGCAAG.
The sgRNAs were synthesized using the EnGen sgRNA synthesis kit (NEB), and underwent subsequent purification and concentration using the RNA Clean & Concentrator-5 kit (Zymo). Individual sgRNAs were injected at 400 pg/embryo with 1.6 ng of Cas9 protein into single celled embryos according to standard methods (Ran et al., 2013). Uninjected control and CRISPR tadpoles were raised in 10 cm dishes until stage 42 to stage 45 of development. To visualize beating hearts, these tadpoles were embedded in low melt agarose in 1/9 X MR and images were obtained using a Thorlabs Telesto 1325 nm spectral domain optical coherence tomography system as previously described (Deniz et al., 2018).
Cloning, Expression and Purification of Recombinant Proteins
Wild-type (WT) and mutant (either D132N or D145E) human cTnC proteins were cloned, expressed, and purified as previously described (Landstrom et al., 2008; Dweck et al., 2010). Site-directed mutagenesis by PCR was used to engineer point mutations into the pET3-d vector. Sequences were verified prior to expression and purification of the cTnC mutants.
Calcium Solutions
Different Ca2+ solutions ranging from pCa 8.0 (relaxing) to 4.0 (activating) were calculated utilizing a pCa Calculator (Dweck et al., 2005). All solutions contained: 20 mM 3-[N-morpholino]propanesulfonic acid (MOPS), 7 mM ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), 15 mM phosphocreatine, 15 units mL–1 creatine phosphokinase, 2.5 mM MgATP2–, 1 mM free Mg2+, ionic strength maintained constant at 150 mM by adding Kpropionate (KPr), varying [Ca2+], pH 7.0. Anion of choice for the pCa solutions was propionate (e.g., CaPr2, MgPr2, and KPr). Solutions were prepared at room temperature (20–21°C).
Skinned Cardiac Muscle Preparation
Left ventricular papillary muscles were harvested from porcine hearts obtained from a local abattoir and dissected into small muscle bundles (Landstrom et al., 2008). Dissected cardiac tissue was exposed to pCa 8.0 relaxation solution (10–8 M free [Ca2+]) containing non-ionic detergent Triton X-100 (1% v:v). After incubating for 4 h at 4°C, skinned cardiac muscle preparations (CMPs) were transferred to 51% glycerol-relaxing pCa 8.0 solution (v:v), stored at −20°C, and utilized within 1 month. Strips of muscle, 0.8–1.0 mm in length and 0.15–0.25 mm in diameter, were mounted using aluminum foil T-clips to a force transducer on one end and a motor on the other end. CMPs were immersed in pCa 8.0 relaxation solution and the initial length (L0) was set at 2.1 μm sarcomere length measured by HeNe laser diffraction. Initial maximal Ca2+-activated tension was measured at pCa 4.0 (10–4 M free [Ca2+]).
Extraction of Native cTnC From CMPs and Reconstitution With Wild-Type or Mutant cTnCs
Native (endogenous) cTnC was extracted by incubating CMPs in 5 mM CDTA solution (pH 8.4) for ∼1.5 h at 21°C as previously described (Landstrom et al., 2008). To evaluate the efficacy of cTnC extraction, residual Ca2+-activated tension was measured by immersing the CMPs in pCa 4. CMPs exhibiting residual force >40% (n = 2) were excluded from analysis. CMPs with residual force ranging from 8.4 to 39.4% were reconstituted by incubation for ∼5 min total (five incubations of ∼1 min each) at 21°C with one of four conditions: drops of pCa 8.0 solution containing 1.0 mg mL–1 of either 100% WT cTnC, 50% WT/50% D132N cTnC mix, 50% WT/50% D145E cTnC mix or 50% D132N/50% D145E cTnC mix. Tension recovery was evaluated by comparing the reconstituted maximal tension at pCa 4.0 to the initial maximal Ca2+-activated tension measured prior to cTnC extraction (P/P0) and ranged from 81% to 123%.
Muscle Mechanics
pCa-Force: Ca2+-dependence of steady-state, isometric force was measured by incubating reconstituted CMPs in a series of Ca2+ solutions ranging from pCa 8.0 to 4.0 at 21°C. Normalized [Ca2+]-force data for each CMP were fit using non-linear least squares regression to a 2-parameter Hill equation to obtain parameter estimates for pCa50 and nHill, as described (Veltri et al., 2017a).
Kinetics of Tension Redevelopment (kTR): After force reached steady-state in each pCa solution, measurement of the rate of tension redevelopment was obtained by shortening CMPs by 20% L0, followed by rapid, 25% re-stretch, then release back to L0 (Gonzalez-Martinez et al., 2018). The apparent rate constant (kTR) was obtained from each tension recovery time course as described previously (Chase et al., 1994; Regnier et al., 1999). Conditions where steady-state, isometric force was below 15% of the maximal force were excluded from the kTR analysis because of the relatively low signal-to-noise at the lowest levels of isometric force. Estimations of the 3-state model parameters (f, g, and kOFF) were computed in MatLab as previously described (Gonzalez-Martinez et al., 2018) with the exceptions that force for each condition was assumed to be 1.0, and also because there are no direct measurements of Ca2+ binding to or dissocation from the mutant cTnCs, the value for kON for all simulations was assumed to be that derived from measurements on WT, 1.84 × 108 (Pinto et al., 2011a). In addition to estimates for parameters f, g, and kOFF, the modeling also provides an estimate for pCa50; the modified least-squares criterion is weighted such that the best fit, predicted pCa50 is essentially the same (well within experimental error) as that measured experimentally for the same condition.
Sinusoidal Stiffness
Sinusoidal stiffness (SS) was obtained by oscillating CMP length ∼0.2% L0 peak-to-peak at 100 Hz, and recording the length and force signals at a sampling rate of 1 kHz. SS measurements were performed and the data were analyzed as described previously (Gonzalez-Martinez et al., 2018).
Statistical Analyses
Statistical analyses including non-linear regression were performed using SigmaPlot v.12.0. Data were tested for significant statistical differences using one-way ANOVA with post hoc Student-Newman–Keuls test. Data are shown as mean ± S.E.
Results
Clinical Data
The proband was a female delivered full term following an uncomplicated pregnancy that included two normal fetal echocardiograms. She had initially been well without symptoms referable to the cardiovascular system. She demonstrated no feeding intolerance, premature fatigue, irritability, pallor, cyanosis, increased work of breathing, tachypnea, excessive diaphoresis, or lethargy. She had a good appetite and breast fed without difficulty. Screening echocardiography was done at 18 days of age due to an older sibling with neonatal cardiomyopathy of presumed idiopathic etiology. This showed borderline enlargement of the left ventricle with a left ventricular end diastolic diameter (LVEDD) of 2.18 cm (Z score of 0.8) and a low decreased M-mode left ventricular ejection fraction (LVEF) of 48%. Given the family history, she was admitted to the hospital briefly with a diagnosis of DCM for initiation of medical therapy with captopril, furosemide and aspirin. She was followed closely as an outpatient, with electrocardiogram having signs of left ventricular enlargement and serial echocardiograms demonstrating gradually worsening left ventricular function, necessitating a steady escalation of her medical management (Figure 1). Notably, right ventricular size and function remained normal throughout. By 7 months of age both dilation and function of her left ventricle had worsened with echocardiography showing LVEDD 3.40 cm (Z score of 4.09) and M-mode LVEF of 32%. At 13 months of age, echocardiography showed LVEDD 4.50 cm (Z score of 8.55) and M-mode LVEF of 33%. She was being managed with enalapril, carvedilol, digoxin, furosemide and aspirin, and had been listed for cardiac transplantation due to her severely depressed heart function. She suffered a cardiac arrest while awaiting a suitable donor and expired at 14 months of age.
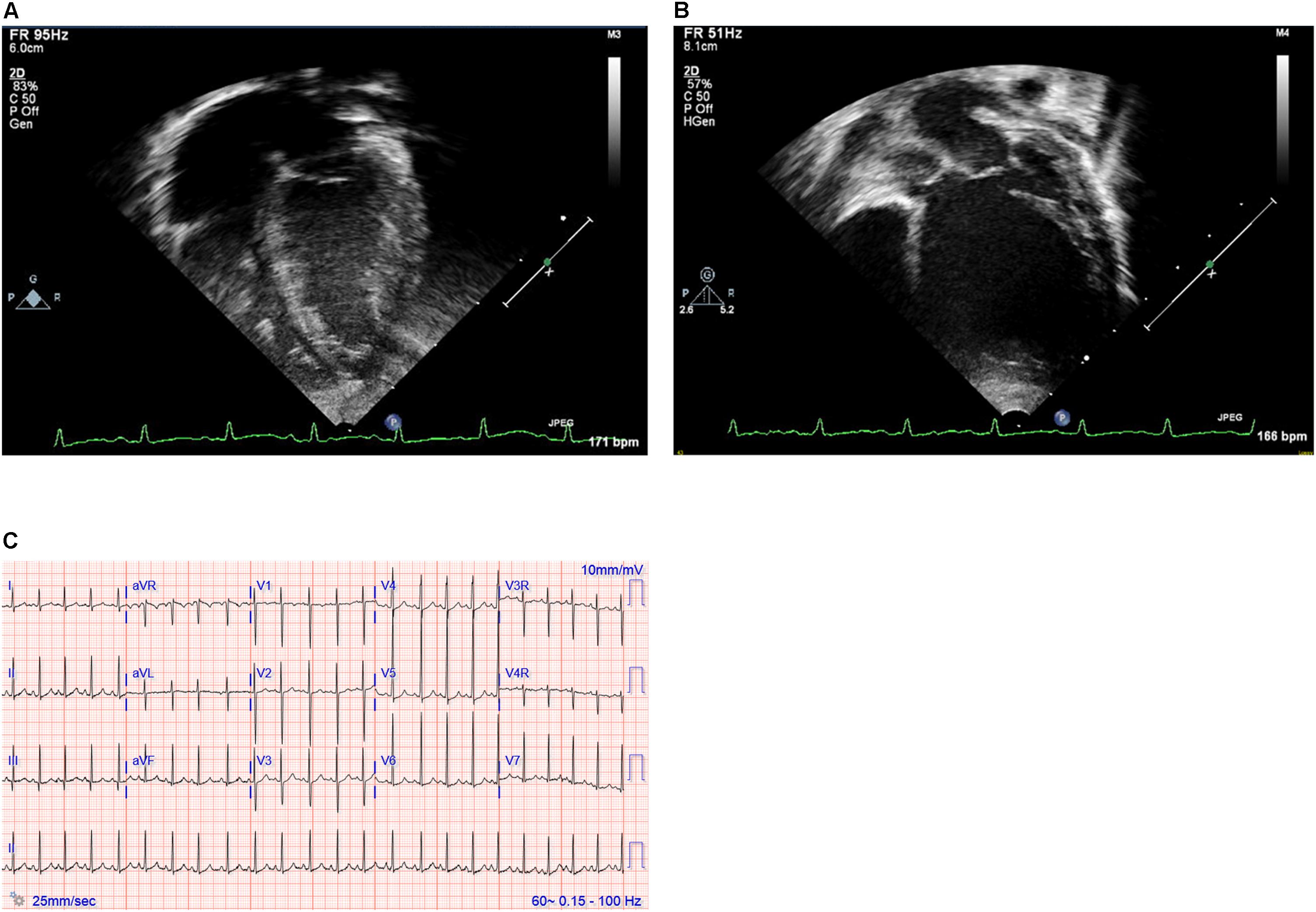
Figure 1. (A,B) Echocardiography images and (C) electrocardiogram. Still images obtained from echocardiogram of the proband at ages 2 weeks (A) and 12 months (B). Note particularly the progressive dilation and prominent rounding of the left ventricle (LV) in the later image. (C) Electrocardiogram at 19 months of age demonstrating sinus rhythm and left ventricular enlargement.
Family history was notable for an 11 year-old brother who presented in the neonatal period with poor feeding and growth, and was diagnosed with severe DCM. He was managed initially with medications then received a cardiac transplantation at 13 months of age. He has done well since, with good function of his transplanted heart, and without any signs of skeletal muscle problems. Both parents, currently in their early 40′s, have had normal echocardiograms and do not have any signs of cardiovascular disease. There is no other family history of cardiomyopathy, congenital heart defects or sudden unexpected cardiac death.
Genetic Testing
Due to the strong family history, the proband underwent genetic testing with whole exome sequencing, which identified two variants of uncertain significance, D132N (c.394G>A, p.Asp132Asn) and D145E (c.435C>A, p.Asp145Glu), in the TNNC1 gene (NM_003280.2). Subsequent analysis of the family (proband, brother and both parents) revealed that these variants were inherited in trans (Figures 2A,B). Both variants are located in the region of divalent cation binding site IV in the C-domain of cTnC (Figure 3). Both of these aspartate residues, as well as the surrounding amino acids, are highly conserved among vertebrates (Figure 3D). In the Genome Aggregation Database (gnomAD), D145E has a frequency of 1.28 × 10–4, whereas D132N has not been previously reported in a healthy population. These were also noted to be the only two rare (<0.5% minor allele frequency in the general population) single gene variants across the entire exome that were shared by both affected siblings, and thus no other rare or pathogenic variants were found in other known cardiomyopathy genes in either of the siblings. The proband was sequenced to a mean depth of 102 independent reads per targeted base and her affected brother and their unaffected parents were sequenced to a mean depth of 44–49X. Greater than 10X coverage was achieved for 98% of the proband’s exome and 94% for the rest of family members’ exomes. Greater than 20X coverage was achieved for 96% of the proband’s exome and 88% for her other family members.
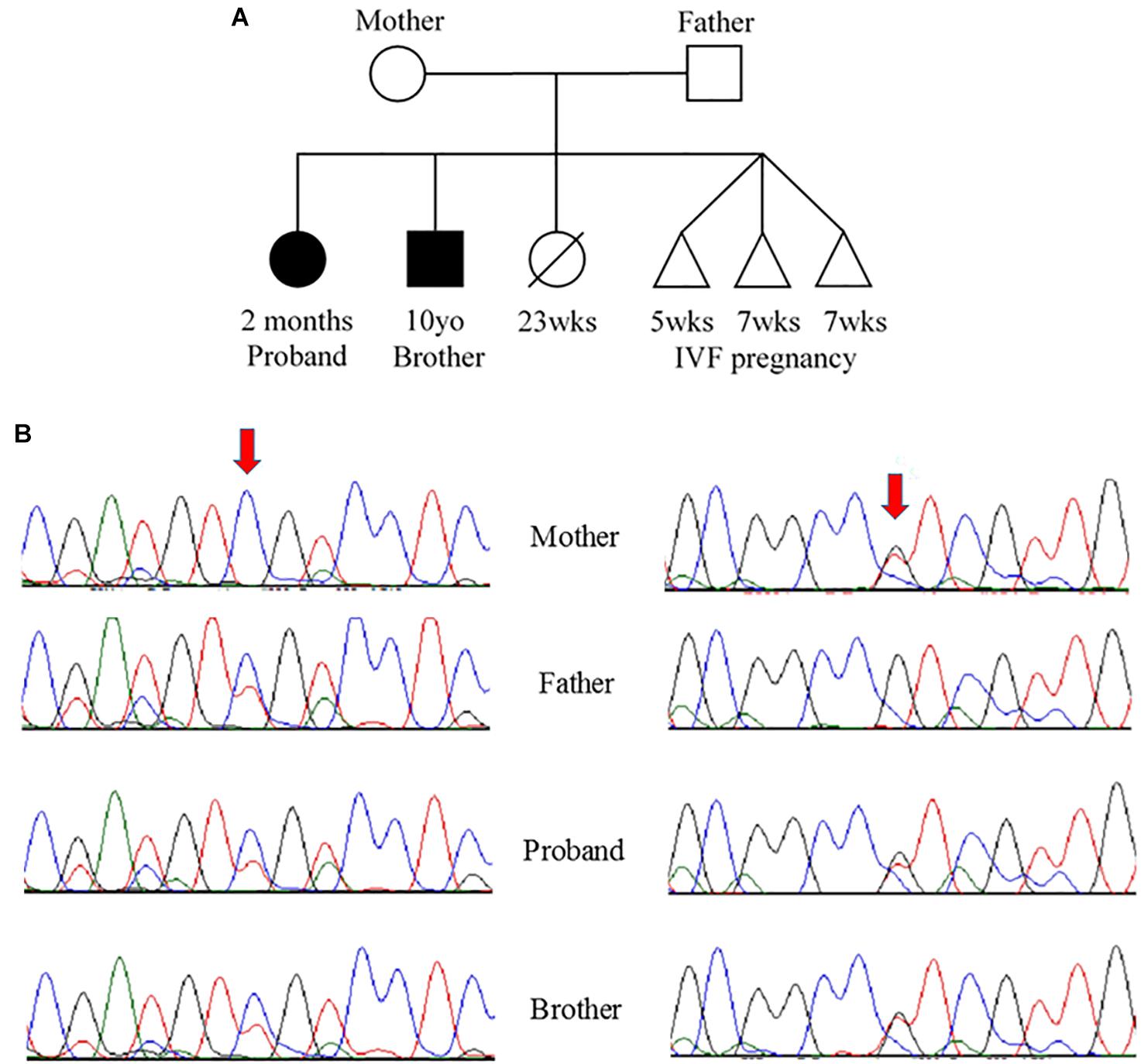
Figure 2. (A) Pedigree and (B) Sanger sequencing. (A) Family pedigree showing the proband and similarly affected brother (black shading). There was a prior loss of a severely premature live birth and three prior failed IVF pregnancies. (B) Sanger sequencing data from the four available family members. Red arrows indicate residue of interest. The left side shows residues around the c.394G>A change that results in D132N in one of the two alleles for TNNC1 for three of four individuals (mother is reference, other three are heterozygous for the variant). The right side shows residues around the c.435C>A change that results in D145E in one of the two alleles for TNNC1 for three of four individuals (father is reference, other three are heterozygous for the variant).
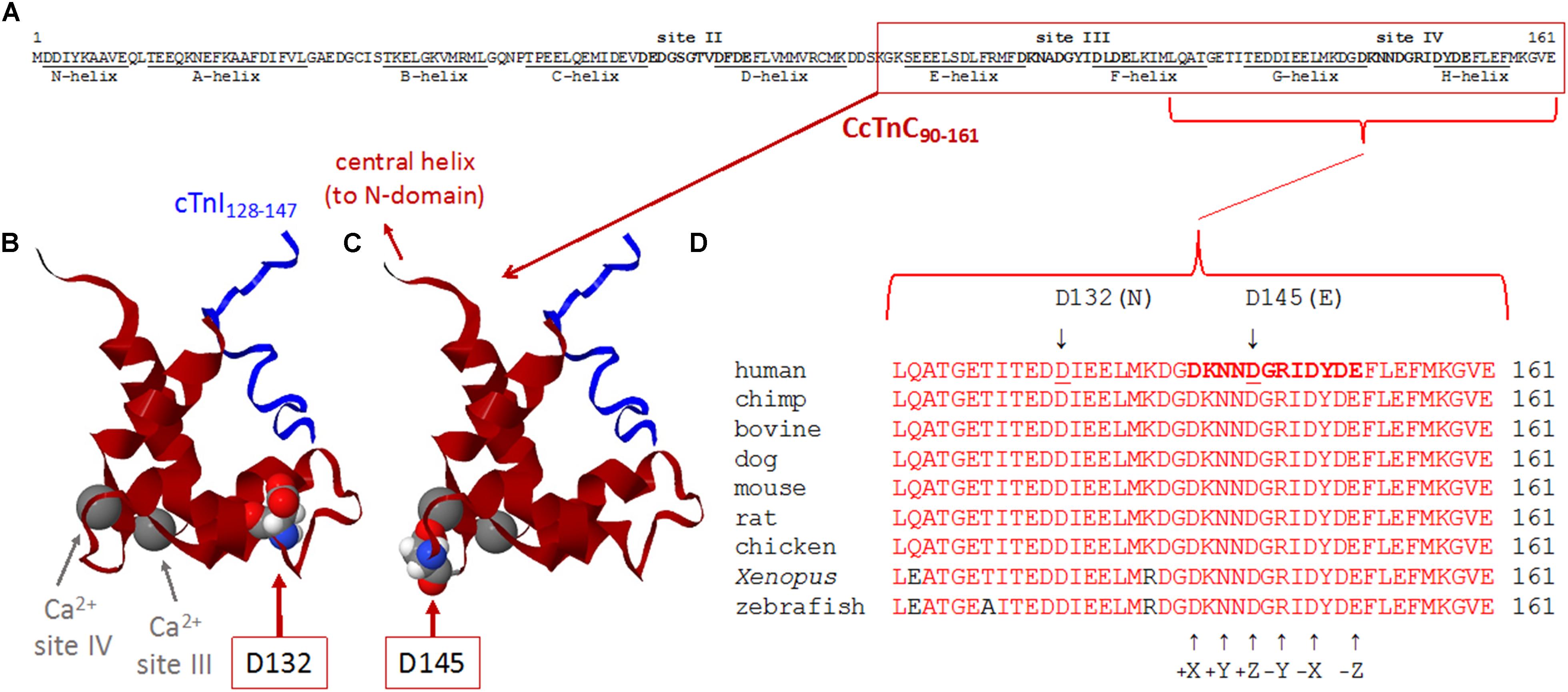
Figure 3. (A–D) Sequence and structure analyses of cTnC surrounding divalent cation-binding site IV. (A) Primary sequence of human cTnC (NP_003271.1). Divalent cation-coordinating regions (sites II, III, and IV) are highlighted in bold, and amino acids that comprise α-helical regions (N-helix and α-helices A–H) are underlined [annotated according to Li and Hwang (2015)]. Site II [EF-hand comprised of a-helices C–D and joining residues] is in the N-domain of cTnC and is the regulatory “trigger” site that activates contraction when Ca2+ binds during the systolic Ca2+ transient. Sites III and IV (EF-hands comprised of α-helices E–F or G–H, respectively, along with the respective residues that join each pair of α-helices) are in the C-domain and can bind Mg2+ or Ca2+. Note that all 161 amino acids of the primary sequence are intentionally shown as a single row to illustrate the relative sizes and locations of annotated regions. (B,C) Ribbon structures of the Ca2+-saturated C-domain of human cTnC in complex with a cardiac troponin I peptide (cTnI128–147), illustrating the locations of D132 and D145 (red arrows). NMR structure adapted from PDB 1OZS model 1 (Lindhout and Sykes, 2003) using MATLAB (ver. R2018b, The MathWorks, Inc.) molviewer. cTnC90–161 (dark red) and cTnI128–147 (blue) are shown as backbone ribbons, with space-filled atoms to highlight cTnC residues D132 and D145 (CPK colors). Ca2+ ions at sites III and IV are large gray spheres (gray arrows); note that, physiologically, it is likely that Mg2+ rather than Ca2+ would be bound at most sites III and IV in the sarcomeres of living cardiomyocytes. Labels in either panel (B) or (C) apply to the locations of structural elements in both panels. Note that only one affected amino acid is highlighted in each of panels (B,C), corresponding to the occurrence of only one of the two variants in each TNNC1 allele of the proband’s genome (Figure 2B). (D) Multiple alignments of residues around site IV demonstrate a remarkable degree of conservation of amino acids D132 and D145 across a wide range of species. Conserved amino acids are shown in red. Affected residues D132 and D145 are identified by underlines (human sequence) and arrows (above). D132 is within the G-helix (refer to panel A) that is part of site IV EF-hand. D145 is at the +Z location within the divalent cation-coordinating region of site IV; all 12 residues of this region are highlighted in bold in the human sequence (top sequence), as in panel (A), and the coordinating residues (+X, +Y, +Z, –Y, –X, and –Z) within site IV are annotated (arrows) below the aligned sequences.
TNNC1 in Xenopus
To assess the consequences of altered cTnC in an in vivo system, we began by using CRISPR/Cas9-mediated gene editing to knockout TNNC1 in Xenopus. Loss of the TNNC1 gene did not prevent the early stages of development, and resulted in a dramatic cardiac phenotype in tadpoles consistent with DCM (Figure 4 and Supplementary Video S1). Notably, the knockout tadpoles demonstrated ventricular dilation, wall thinning and almost imperceptible cardiac motion. With the ultimate goal of introducing the two mutations separately and together, we then attempted to rescue this cardiomyopathy phenotype by expression of human cTnC in depleted frog embryos. Unpredictably, we were unable to obtain expression of cTnC in the tadpole hearts despite employing various techniques injecting either TNNC1 mRNA or TNNC1 plasmid DNA under control of a CMV promoter (Table 1).
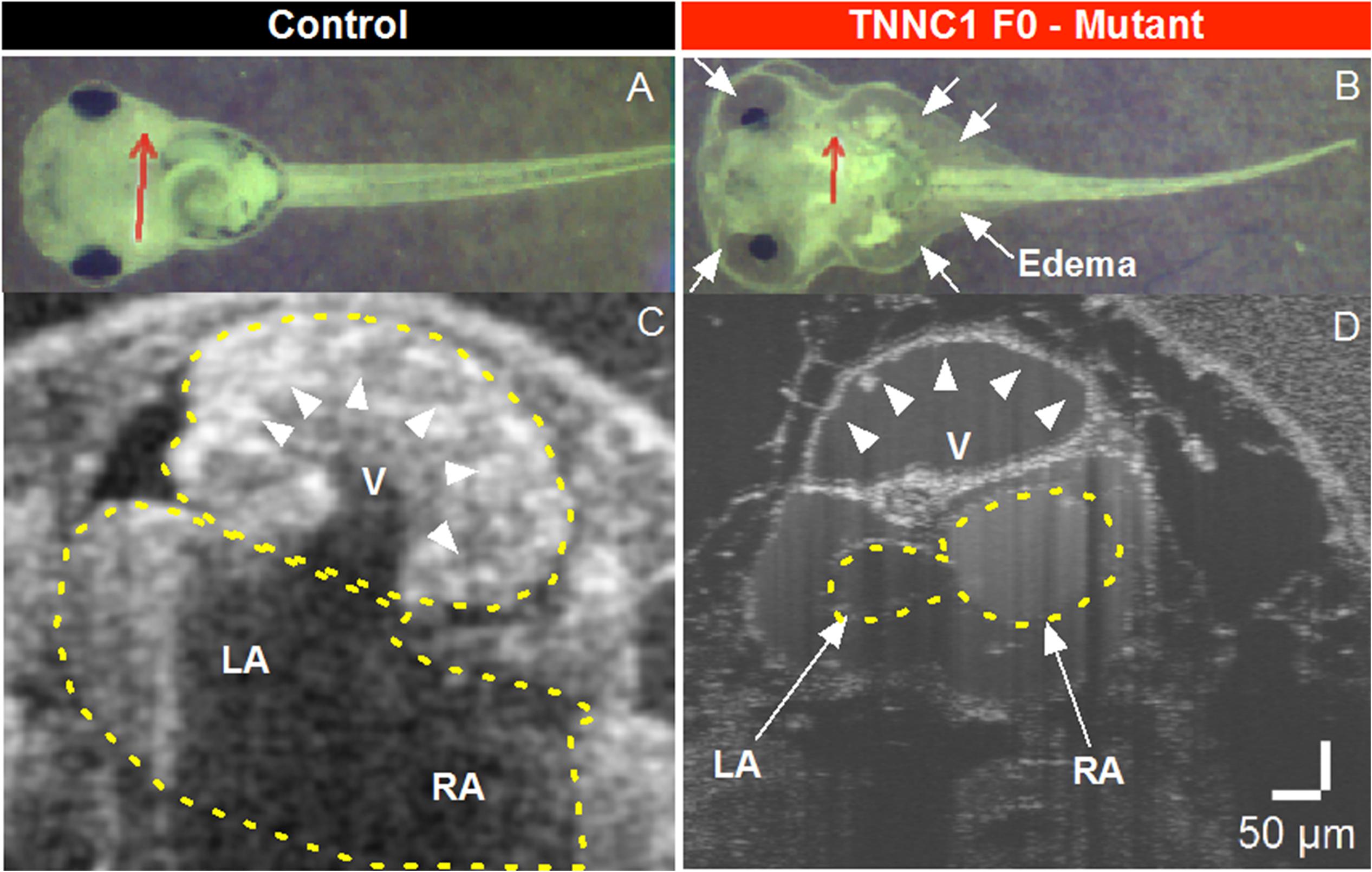
Figure 4. (A,B) Brightfield microscopy and (C,D) Optical Coherence Tomography (OCT) imaging of Control and TNNC1 F0 knockout tadpoles. As compared to control tadpoles (A), TNNC1 knockout tadpoles (B) show normal gross morphology, but significant total body edema (white arrows) secondary to severe dilated cardiomyopathy leading to congestive heart failure. Red arrows indicate plane of OCT images in (C,D). OCT images demonstrate normal size and wall thickness of cardiac chambers in control tadpole (C), with severe dilation and enlargement of the ventricle in TNNC1 knockout tadpole (D), indicated with white arrowheads. V = ventricle, LA = left atrium, RA = right atrium.
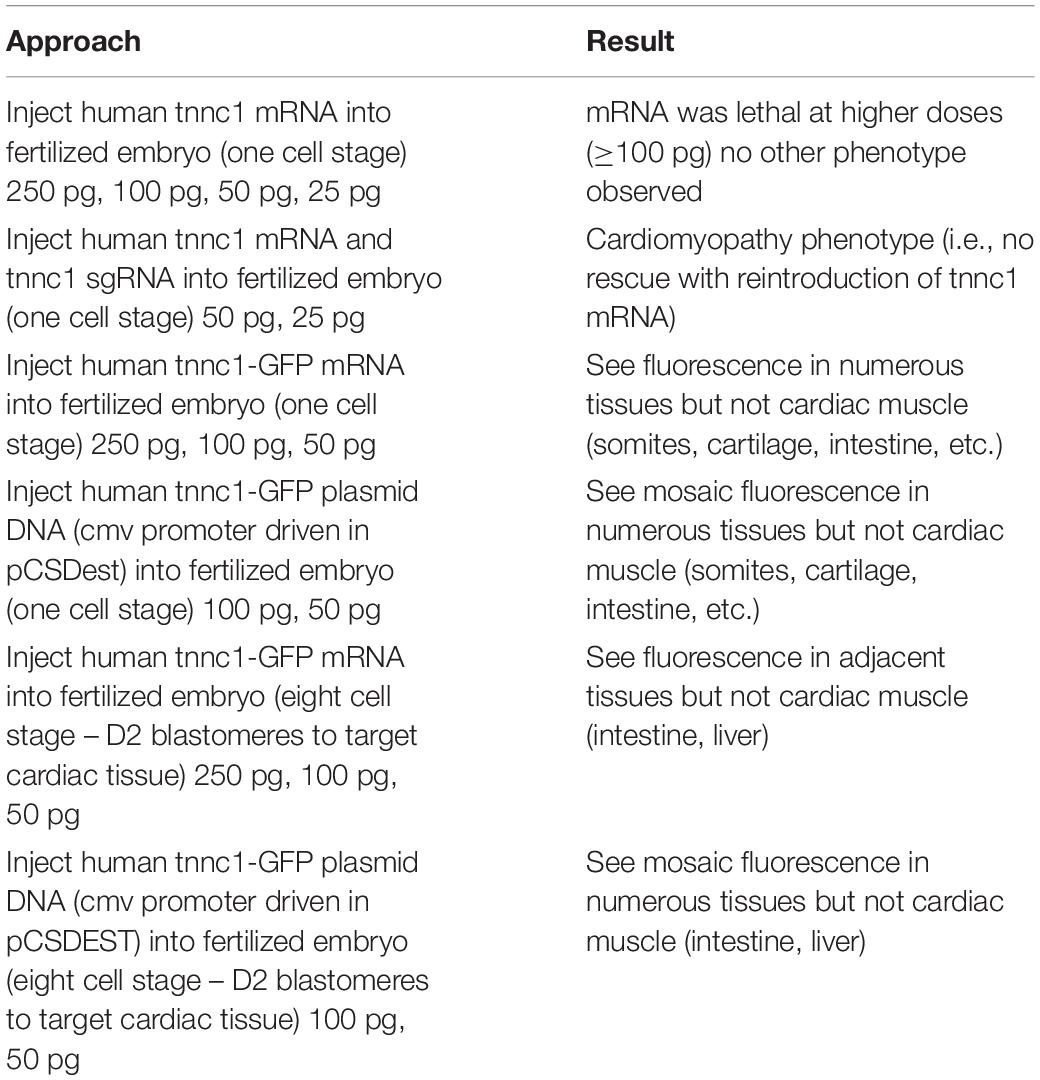
Table 1. Attemps to rescue TNNC1 knockout in Xenopus tropicalis.
Muscle Mechanics
It was previously shown that cTnC-depleted CMPs reconstituted with cTnC-D145E increased Ca2+ sensitivity of steady-state isometric force generation (Landstrom et al., 2008; Pinto et al., 2011a; Veltri et al., 2017a). However, to our knowledge, the effects of cTnC-D132N or mixtures of these cTnCs have not been previously reported. Considering that both siblings were found to be compound heterozygous for D145E and D132N in cTnC, we sought to explore their potential functional significance in vitro.
Native, endogenous cTnC was extracted from porcine CMPs using CDTA incubations and reconstituted with recombinant expressed human cTnC-WT (control) or mixtures of cTnCs (section “Experimental Procedures”). The range of average of residual tension post-extraction was 18% ∼ 35%, with individual values ranging from 8.4 to 39.4%, indicating that the majority of native cTnC had been extracted from the myofilaments (Table 2, where force values are reported as specific force). Upon reconstitution, the average tension recovery was at least 87%, indicating that the majority of troponin molecules had been functionally reconstituted with exogenous cTnC and that both WT and mutant recombinant cTnCs were competent for Ca2+-activation of contraction.
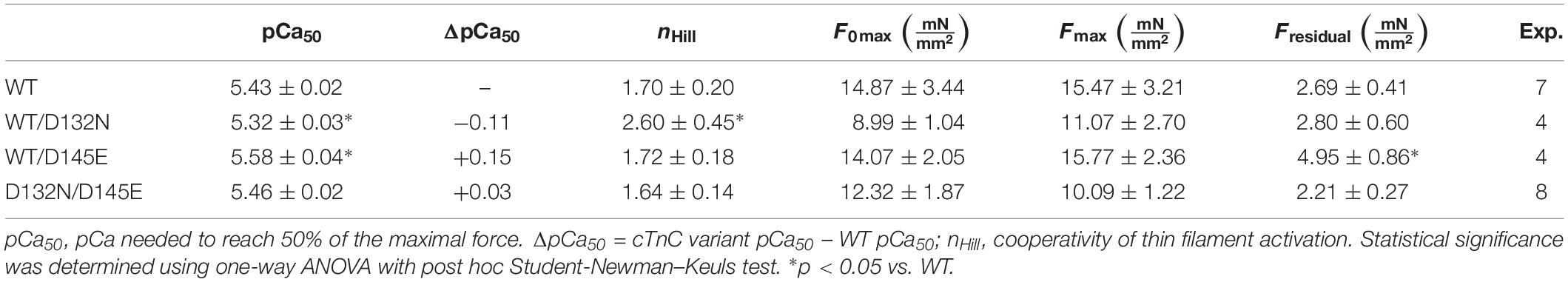
Table 2. Hill equation parameter estimates for Ca2+ dependence of steady-state isometric force obtained from porcine CMPs reconstituted with exogenous cTnC WT or variants depicted in Figure 5.
CMPs reconstituted with a mixture of 50% WT and 50% D132N variant (WT/D132N) displayed significantly reduced myofilament Ca2+ sensitivity of force generation (pCa50 = 5.32 ± 0.03), with a rightward shift of 0.11 pCa units compared with CMPs reconstituted with 100% WT cTnC (pCa50 = 5.43 ± 0.02) (Table 2 and Figures 5A,B). Consistent with previous reports that compared 100% D145E with 100% WT (Landstrom et al., 2008; Pinto et al., 2009; Veltri et al., 2017a), we found that reconstitution with a mixture of 50% WT and 50% D145E (WT/D145E) significantly increased myofilament Ca2+ sensitivity of tension (pCa50 = 5.58 ± 0.04), with a leftward shift of 0.15 pCa units (Table 2 and Figures 5A,B). Interestingly, we observed no significant difference in myofilament Ca2+ sensitivity of tension upon reconstitution of CMPs with a mixture of 50% D132N and 50% D145E (D132N/D145E) (Table 2 and Figures 5A,B). Although the D132N variant in 50%:50% combination with either WT or D145E tended to reduce maximal tension recovery, the differences were not statistically significant (Table 2 and Figure 5B). The Hill coefficient (nHill) is an indicator of cooperativity of thin filament activation. Compared to WT (nHill = 1.70 ± 0.20), only the WT/D132N mixture exhibited a significant change – an increase – in the apparent cooperativity (nHill = 2.60 ± 0.45) (Table 2).
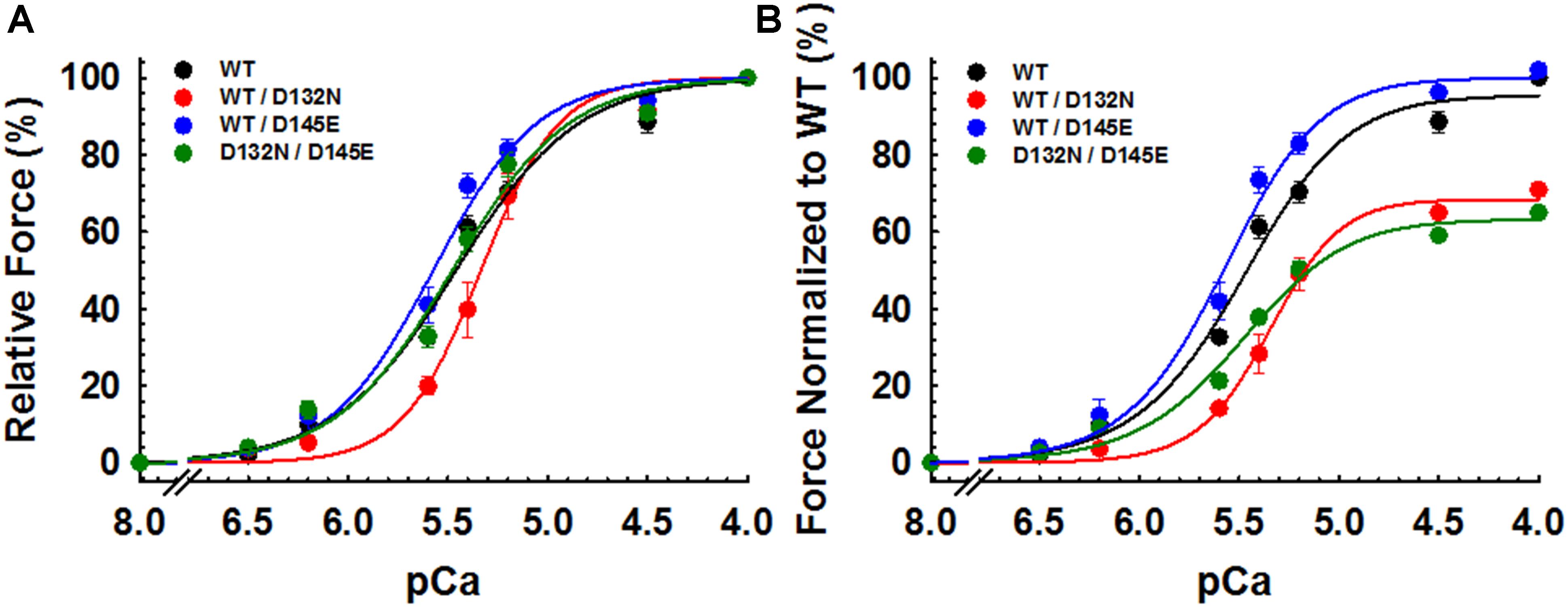
Figure 5. Ca2+ dependence of steady-state isometric tension in porcine CMPs reconstituted with exogenous cTnC WT (100% WT) or variants (50% WT/50% D132N, 50% WT/50% D145E, or 50% D132N/50% D145E). (A) Relative steady-state isometric force as a function of pCa. The force values were normalized to the maximal steady-state isometric force in the same preparation. (B) Normalized steady-state isometric force as a function of pCa. The force values were normalized to the maximal steady-state isometric force generated by WT. Data are shown as mean ± S.E. and best fit parameter estimates from non-linear least squares regression on the Hill equation are summarized in Table 2 (n = 4–8).
Because we observed no significant difference in myofilament Ca2+ sensitivity of tension for the 50%:50% combination of the two variant cTnCs (D132N/D145E) compared with 100% WT (Table 2 and Figures 5A,B), we investigated two additional indices of contractile function: SS and kinetics of tension redevelopment (kTR). SS is used as a metric of the overall number of cross-bridges, whereas kTR is used a metric of the kinetics of cross-bridge cycling at maximal Ca2+-activation and also informs about thin filament regulatory unit dynamics at submaximal Ca2+-activation. We found no significant difference in maximum SS values when comparing CMPs reconstituted with the variants (WT/D132N; WT/D145E; D132N/D145E) to those reconstituted with 100% WT (Table 3 and Figures 6A,B), suggesting that force per cross-bridge was unchanged. In addition, there was little or no difference in any of the relationships between SS and isometric force (Figure 6B).
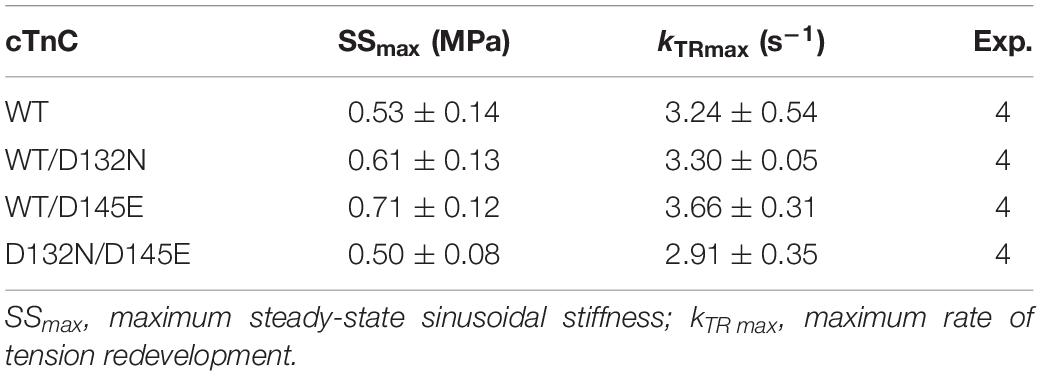
Table 3. Maximal sinusoidal stiffness (SSmax) and rate of tension redevelopment (kTR max) measured in porcine CMPs reconstituted with exogenous cTnC WT or variants depicted in Figures 6, 7, respectively.
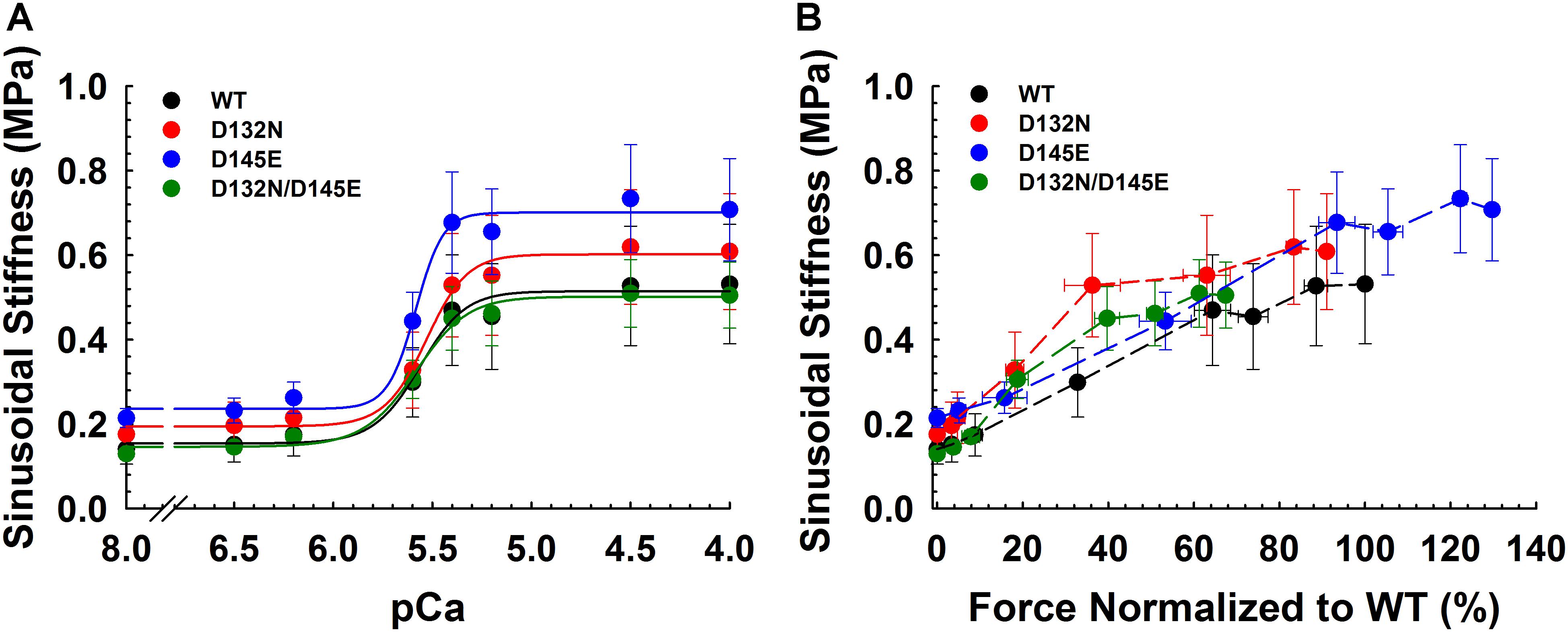
Figure 6. Sinusoidal stiffness analysis in porcine CMPs reconstituted with exogenous cTnC WT (100% WT) or variants (50% WT/50% D132N, 50% WT/50% D145E, or 50% D132N/50% D145E). (A) Ca2+ dependence of steady-state sinusoidal stiffness. (B) Normalized force vs. steady-state sinusoidal stiffness. The force values were normalized to the maximal steady-state isometric force generated by WT. (B) Dashed lines were drawn to connect the points. Data are shown as mean ± S.E. and maximum SS values are summarized in Table 3.
Maximum kTR values were not significantly different when comparing CMPs reconstituted with the variants (WT/D132N; WT/D145E; D132N/D145E) to those reconstituted with 100% WT (Table 3 and Figures 7A,B). This result informs us that, according to Brenner (1988), the sum of the sum of the rates of cross-bridge attachment and detachment (f+g) is approximately constant for all four conditions. To explore the possibility that the variants may have altered cross-bridge attachment (f) and detachment (g) rates even though the sum remained constant, we used a 3-state model of muscle regulation to evaluate the Ca2+-dependence of kTR (Landesberg and Sideman, 1994; Hancock et al., 1997; Loong et al., 2013; Gonzalez-Martinez et al., 2018). The force-kTR data for CMPs containing the mutants (WT/D132N, WT/D145E or D132N/D145E) appear to be less curvilinear than for WT (Figure 7B). The fits of the 3-state model capture the curvature of the data from CMPs containing mutants, but does not fully capture the greater curvature of the WT data (Figure 7B). Best fit values for f, g, and kOFF corresponding to the lines in Figure 7B are reported in Table 4. The 3-state model’s four parameters (f, g, kON, and kOFF) are all lumped parameters that primarily inform about cross-bridge cycling (f and g) and thin filament regulatory unit dynamics including Ca2+ binding to and dissociation from cTnC (kON and kOFF). Computational modeling indicates that kOFF is, as expected, generally faster as Ca2+ sensitivity decreases (Table 4), although this relationship is modulated by additional factors because kOFF reflects more than just Ca2+ dissociation from cTnC. Furthermore, the modeling indicates that f was slower than WT and g was faster for both WT/D132N and WT/D145E (Table 4). For the two mutants together (D132N/D145E), f and g changed in the same directions as for the individual variants (WT/D132N and WT/D145E), but the magnitudes of the changes were less; in other words, the combination of the two variants appeared to partially ameliorate the effects of the individual variants, as was also found for Ca2+ sensitivity of steady state isometric force (Table 2 and Figures 5A,B).
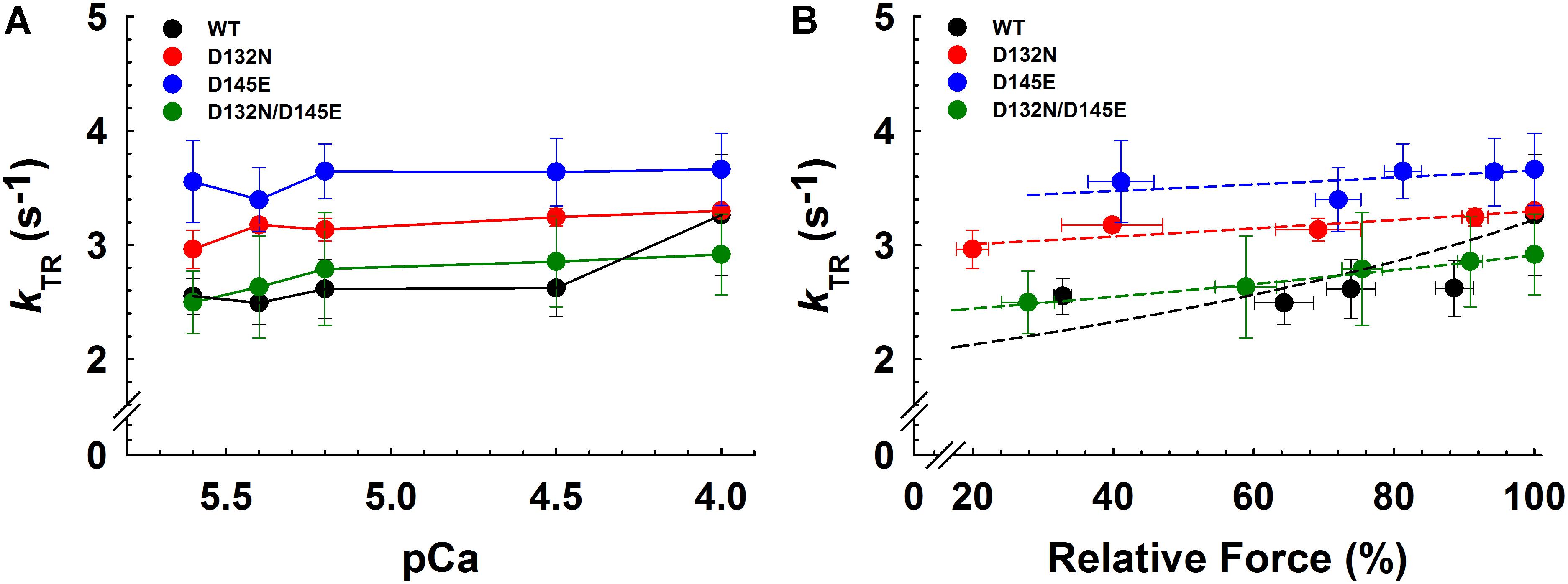
Figure 7. Rate of tension redevelopment (kTR) analysis in porcine CMPs reconstituted with exogenous cTnC WT (100% WT) or variants (50% WT/50% D132N, 50% WT/50% D145E, or 50% D132N/50% D145E). (A) kTR as a function of increasing Ca2+ concentration. Solid lines were drawn to connect the points. (B) Relative force vs. kTR. The force values were normalized to the maximal steady-state isometric force for each preparation. Data are shown as mean ± S.E. and maximum kTR values are summarized in Table 3. Dashed lines were obtained from fits to the 3-state model, which take into account the changes in Ca2+ sensitivity (Table 2 and Figures 5A,B) as described in section “Experimental Procedures”; best fit parameter estimates are summarized in Table 4.
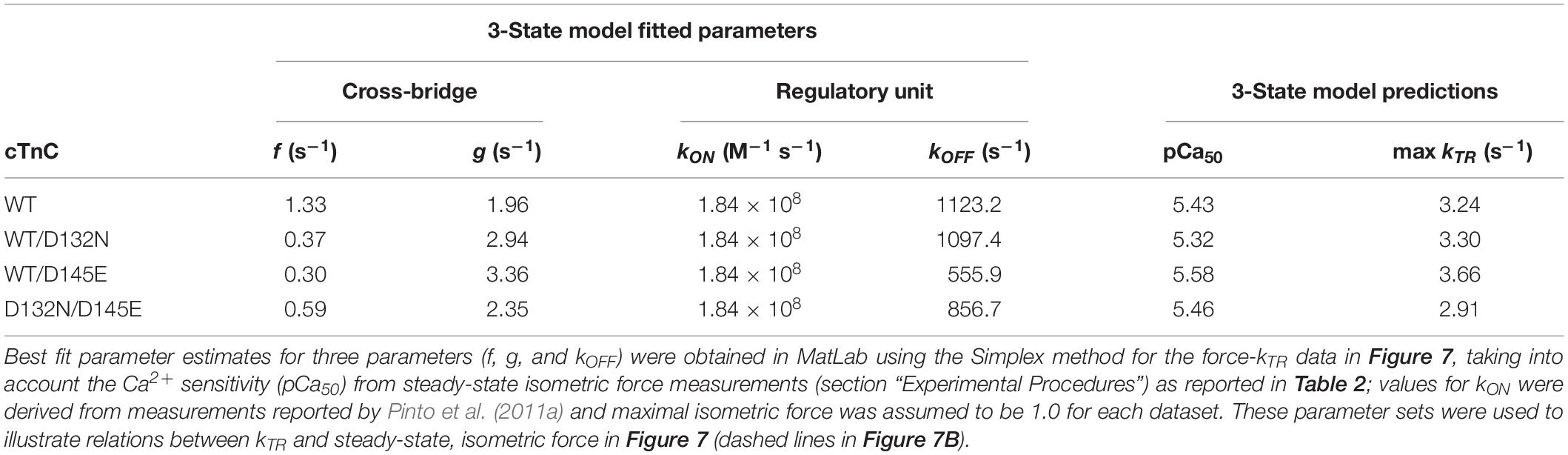
Table 4. Optimized parameter estimates and predictions from the 3-state model for force-kTR data depicted in Figure 7.
Discussion
Given the central role of cTnC in cardiomyocyte contraction, it is little surprise that a growing number of variants in the TNNC1 gene have been identified in genetic screens for familial cardiomyopathies, both hypertrophic and dilated. Here, we report two siblings with severe, early onset DCM who were found to be compound heterozygous for two TNNC1 variants: D145E inherited from the healthy mother and D132N inherited from the healthy father. Considering that myofilament dysfunction typically correlates with severity of cardiomyopathy, we anticipated that the combination of the two variants (D132N/D145E) would have adverse consequences for Ca2+ regulation of contractility. However, in contrast to what might be expected from the family pedigree, our functional data indicate that these two variant cTnC proteins when present as a mixture of one variant with WT (as in the mother and father) significantly altered Ca2+ regulation of steady-state isometric force in opposite directions, while the combination of the two variants (as in the affected siblings) did not.
Dissonance Between Clinical Outcomes and Mechanical Measurements
Although multiple in silico predictors of variant effects – including SIFT, PolyPhen2 and CADD – predict that each of these variants is likely to be detrimental, they are both classified as variants of uncertain significance in ClinVar. The D132N variant has not been reported in the Genome Aggregation Database (gnomAD). The population frequency of the D145E variant is reported as 0.0001228 in gnomAD, and the Atlas of Cardiac Genetic Variations classifies this uncommon variant as “unlikely to be pathogenic.” This classification of D145E is consistent with individuals such as the proband’s mother, who is heterozygous and asymptomatic. The available evidence, therefore, suggests that D145E could possibly be a risk factor for cardiomyopathy in some cases, but does not appear to be disease-causing when present in the heterozygous state. The two affected siblings presented here, in contrast to the parents, have D145E in trans with the D132N variant. When viewed in this light, the D145E population frequency is well within an expected range for a recessive disease.
To assess the combined effects of two potentially dysfunctional TNNC1 alleles, we performed knockout experiments in Xenopus, revealing a dramatic DCM phenotype in tadpoles (Figure 4 and Supplementary Video S1) that is consistent with a previous report in zebrafish (Ho et al., 2009). Our multiple attempts to rescue this phenotype with human cTnC protein expression (Table 1) were unsuccessful, however, as the protein could be found expressed in other tissues but not in the heart (data not shown). The reason for this remains unclear to us at this time, though the fact that we have extensive experience obtaining wide expression – including in the heart – of multiple other proteins in tadpoles, suggests that this may be an issue specific to TNNC1.
Interestingly, the D145E variant was first described in a patient with hypertrophic cardiomyopathy (HCM) and it was the only variant found in a screen of fifteen HCM-susceptibility genes (Landstrom et al., 2008). In contrast, a subsequently described proband with DCM also had a rare variant in the MYBPC3 gene, which was hypothesized to combine with cTnC D145E to manifest as DCM (Hershberger et al., 2010; Pinto et al., 2011b). Based on these opposing findings, it was suggested that this second patient may have initially developed HCM that rapidly progressed to DCM (Freeman et al., 2001; Fujino et al., 2001, 2002; Nanni et al., 2003). The proband described here, however, was followed closely since birth given her brother’s diagnosis, and she never had any echocardiographic or other evidence of HCM, suggesting that this combination of cTnC alleles results purely in DCM. Of significance is another example of compound heterozygosity where the D145E variant was again associated with inherited cardiomyopathy (Ploski et al., 2016). In this case, two pediatric patients were diagnosed with autosomal recessive restrictive cardiomyopathy which was fatal during infancy. Sanger sequencing revealed that both of the affected individuals were compound heterozygous for genetic variants in TNNC1 corresponding to D145E and A8V in the cTnC protein.
Variants in TNNC1 can affect skeletal muscle function because cTnC is also expressed in slow skeletal muscle. The D145E mutant was reported to increase slow skeletal muscle ATPase activity at low and high calcium concentrations in reconstituted myofibrils (Veltri et al., 2017b). Although it is known that D145E does not affect myofilament calcium sensitivity in slow skeletal skinned fibers (Veltri et al., 2017b), it is unclear whether D132N has any effect on slow skeletal muscle function. Importantly, no skeletal muscle weakness was reported in the patient’s history.
Implications for cTnC Function in the Cardiac Thin Filament
Both variants are located in the C-domain of cTnC (Figure 3). The missense D132N variant is located within the G-helix that is part of EF-hand site IV, and D145E is one of the divalent cation (Ca2+ or Mg2+) coordinating residues (+Z position) in site IV. While the regulatory site for Ca2+ binding is site II in the N-domain of cTnC, our prior results indicate that there is communication between the C- and N-domains (Badr et al., 2016) such that changes in the C-domain can influence the regulatory N-domain (Marques et al., 2017; Veltri et al., 2017a). The results of this study demonstrated that the D145E variant (WT/D145E) increases myofilament Ca2+ sensitivity even when WT is also present, while the D132N mutation (WT/D132N) had the opposite effect of decreasing Ca2+ sensitivity (Table 2 and Figures 5A,B) even though the two affected residues are relatively close in the primary sequence and the 3D structure (Figure 3). In general, myofilament incorporation of troponin variants that cause opposing effects on thin filament Ca2+ responsiveness are expected to normalize the myofilament Ca2+ sensitivity (Li et al., 2010; Alves et al., 2014, 2017; Dieseldorff Jones et al., 2018), and that was what was found in this study. Compared to WT, the 50%:50% mix of D132N/D145E showed similar myofilament Ca2+ sensitivity and Hill coefficient (nHill) values, the latter generally reflecting cooperative processes associated with isometric force generation. While incorporation of the two mutants appeared to normalize opposite effects of the two variants on Ca2+ dependence of isometric force, what is difficult to reconcile is the severe DCM pathology associated with the presence of the two mutations.
A paradigm associating changes in myofilament Ca2+ sensitivity commonly found in HCM (increased) and DCM (decreased) is generally well-accepted (Willott et al., 2010; van der Velden and Stienen, 2019). This study, however, presents a unique clinical case in which we observed no significant changes in myofilament Ca2+ sensitivity due to a combination of variants in patients with DCM. How can this finding be explained? After depleting native cTnC from CMPs, each available unfilled cTnC site was presumably reconstituted, depending on the experiment, with either exogenous WT, WT/D132N mix, WT/D145E mix, or D132N/D145E mix in an attempt to mimic what is expected to be found in the patients (D132N/D145E) or the parents (WT/D132N or WT/D145E), along with the control condition (100% WT). Previous studies showed that D145E cTnC dissociates more slowly from the thin filament compared to WT cTnC indicating that D145E has a higher binding affinity for the thin filament (Marques et al., 2017), although the functional data (Table 2) do not suggest that this difference in affinity influenced the results.
As with our finding, there has been some variation in prior reports on variants in TNNC1 with regard to the hypothesis that HCM is always associated with increased Ca2+ sensitivity and DCM with decreased Ca2+ sensitivity. The first DCM-associated variant identified in TNNC1, G159D (Mogensen et al., 2004), has been explored by several groups with conflicting findings. It was initially reported that G159D did not alter myofilament Ca2+ sensitivity in Tn-exchanged skinned rat trabeculae (Biesiadecki et al., 2007). In contrast, both cTnC-depleted skinned porcine papillary muscle and in vitro regulated thin filaments reconstituted with cTnC-G159D exhibited reduced myofilament Ca2+ sensitivity (Robinson et al., 2007; Dweck et al., 2008). Conversely, G159D exhibited increased myofilament Ca2+ sensitivity in both human skinned ventricular myocytes obtained from a patient bearing G159D, and thin filaments reconstituted with skeletal muscle actin, human cardiac tropomyosin, and troponin complex containing cTnC G159D (Dyer et al., 2009).
Loss of the lusitropic response (cardiac muscle relaxation) is considered a common feature of DCM and HCM (Wang et al., 2012; Dweck et al., 2014; Messer and Marston, 2014). In the case of thin filament mutants, this can be partially attributed to uncoupling of cTnI phosphorylation from modulation of Ca2+ sensitivity (Memo et al., 2013; Messer and Marston, 2014). Studies using TnC-extracted porcine papillary muscle fibers reconstituted with cTnC DCM-associated mutants (Y5H, M103I or I148V) exhibited diminished or abolished PKA-mediated Ca2+ desensitization (Pinto et al., 2011b). Although we did not test for PKA-mediated myofilament Ca2+ desensitization in our current study, we speculate that the presence of both mutant proteins could interfere with cTnI phosphorylation levels and functional consequences. This could cause a loss of downstream β-adrenergic stimulation which is crucial to effect the lusitropic response and diastolic ventricular filling.
Alternative possibilities exist because the two mutations are not present in the same protein. In the affected siblings, the two variant proteins are expected to be distributed randomly in thin filaments, and differences in function might affect communication between adjacent regulatory units when they contain different cTnCs. The lack of change in nHill for D132N/D145E (Table 2 and Figures 5A,B) suggests this might not be a plausible explanation, although the Hill coefficient represents the overall response and does not inform about highly localized effects when two variant cTnCs are present. Mechanical kinetics provides information beyond steady-state force measurement, and cooperative interactions among regulatory units do not appear to be required for fast rates of tension redevelopment, kTR (Chase et al., 1994). Both the force-kTR data (Figure 7B) and the 3-state model analyses of those force-kTR relations (Table 4) suggest that the presence of either variant (WT/D132N or WT/D145E) shifts the force-kTR relation, and alters contractile kinetics (kTR in Figure 7B, and f and g in Table 4) in the same direction. However, both the kinetic data (Figure 7B) and modeling (Table 4) support the conclusion from analysis of the steady-state force-pCa data (Table 2 and Figures 5A,B) that the combination of the two variants, D132N/D145E, in thin filaments may partially ameliorate the effects of either mutant on contractile function. This is because D132N/D145E yields results more similar to 100% WT than either WT/D132N or WT/D145E (Table 4 and Figure 7B). Thus it appears to be necessary to seek alternative explanations, beyond changes in biomechanical function, to explain the deleterious effects of the two variants in combination in the proband and her brother.
cTnC Function Beyond the Sarcomere
We and others have previously suggested that troponin subunits may participate in cellular functions that extend beyond contractile regulation in the sarcomere (Asumda and Chase, 2012; Wu et al., 2015; Zhang et al., 2016; Pinto et al., 2017). In fact, cTnC has been previously detected in cardiomyocyte nuclei, as well as the nuclei of certain cancer cells (Johnston et al., 2018). In addition, cTnC has been identified in the mitochondrial compartment of some cancer cell lines. Therefore, a potential mechanistic explanation for the severe disease phenotype associated with troponin mutants may be attributed to perturbations in its putative nuclear/mitochondrial function, whereby gene expression or bioenergetics could be altered by cTnC variants such as those reported here.
In summary, we report the association of compound heterozygous variants in TNNC1 in siblings with early onset familial DCM. We provide functional evidence that both variants separately lead to abnormal myofilament Ca2+ sensitivity in which the variants D132N and D145E are, respectively, associated with reduced or increased myofilament Ca2+ sensitivity relative to WT, while these effects compensated for no change relative to WT when the two variants were combined. Analyses of activation-dependence of kTR suggest that both variants have similar effects on contractile kinetics, although there was no significant change in maximal kTR, and that the combination of the two variants again partially ameliorated the effects of the individual variants. None of the observed changes in mechanics, however, can explain the clinical outcomes in this family. Further studies are needed to understand precisely how these variants combine to result in a severe DCM phenotype.
Disclosure
SL is part owner of Qiyas Higher Health, a startup company unrelated to this work.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The studies involving human participants were reviewed and approved by the Institutional Review Board of Yale University School of Medicine. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. The animal study was reviewed and approved by the Yale University Institutional Animal Care and Use Committee.
Author Contributions
JP and SL conceptualized the study and oversaw project administration. ML-V, JJ, WJ, EM, JT, MS-M, LJ, EH, DP-M, MK, and ED contributed to the data curation. PC contributed to computational data analysis and modeling. ML-V, JJ, JT, EM, PC, SL, and JP wrote the manuscript. All authors reviewed and approved the final version of the manuscript.
Funding
JP was supported by the National Institutes of Health (grant no. HL128683). The Yale PGDP was supported by the Yale New Haven Hospital and Sara and Jeffery Buell.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to this family for sharing their story. The authors also thank Bradley’s Country Store, Tallahassee, FL, United States, for generously supplying porcine hearts, and Monica Konstantino for patient recruitment.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2019.01612/full#supplementary-material
VIDEO S1 | Optical Coherence Tomography imaging showing contractility of tadpole hearts. Control heart (top panel) with good systolic contractility. Two views of TNNC1-F0 mutant hearts (middle and lower panels) showing minimal motion and lack of effective contractility.
References
Alves, M. L., Dias, F. A., Gaffin, R. D., Simon, J. N., Montminy, E. M., Biesiadecki, B. J., et al. (2014). Desensitization of myofilaments to Ca2+ as a therapeutic target for hypertrophic cardiomyopathy with mutations in thin filament proteins. Circ. Cardiovasc. Genet. 7, 132–143. doi: 10.1161/CIRCGENETICS.113.000324
Alves, M. L., Warren, C. M., Simon, J. N., Gaffin, R. D., Montminy, E. M., Wieczorek, D. F., et al. (2017). Early sensitization of myofilaments to Ca2+ prevents genetically linked dilated cardiomyopathy in mice. Cardiovasc. Res. 113, 915–925. doi: 10.1093/cvr/cvx068
Asumda, F. Z., and Chase, P. B. (2012). Nuclear cardiac troponin and tropomyosin are expressed early in cardiac differentiation of rat mesenchymal stem cells. Differentiation 83, 106–115. doi: 10.1016/j.diff.2011.10.002
Badr, M. A., Pinto, J. R., Davidson, M. W., and Chase, P. B. (2016). Fluorescent protein-based Ca2+ sensor reveals global, divalent cation-dependent conformational changes in cardiac troponin C. PLoS One 11:e0164222. doi: 10.1371/journal.pone.0164222
Bhattacharya, D., Marfo, C. A., Li, D., Lane, M., and Khokha, M. K. (2015). CRISPR/Cas9: an inexpensive, efficient loss of function tool to screen human disease genes in Xenopus. Dev. Biol. 408, 196–204. doi: 10.1016/j.ydbio.2015.11.003
Biesiadecki, B. J., Kobayashi, T., Walker, J. S., John Solaro, R., and de Tombe, P. P. (2007). The troponin C G159D mutation blunts myofilament desensitization induced by troponin I Ser23/24 phosphorylation. Circ. Res. 100, 1486–1493. doi: 10.1161/01.res.0000267744.92677.7f
Brenner, B. (1988). Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc. Natl. Acad. Sci. U.S.A. 85, 3265–3269. doi: 10.1073/pnas.85.9.3265
Burke, M. A., Cook, S. A., Seidman, J. G., and Seidman, C. E. (2016). Clinical and mechanistic insights into the genetics of cardiomyopathy. J. Am. Coll. Cardiol. 68, 2871–2886. doi: 10.1016/j.jacc.2016.08.079
Chase, P. B., Martyn, D. A., and Hannon, J. D. (1994). Isometric force redevelopment of skinned muscle fibers from rabbit activated with and without Ca2+. Biophys. J. 67, 1994–2001. doi: 10.1016/S0006-3495(94)80682-4
Deniz, E., Mis, E. K., Lane, M., and Khokha, M. K. (2018). CRISPR/Cas9 F0 screening of congenital heart disease genes in Xenopus tropicalis. Methods Mol. Biol. 1865, 163–174. doi: 10.1007/978-1-4939-8784-9_12
Dieseldorff Jones, K. M., Koh, Y., Weller, R. S., Turna, R. S., Ahmad, F., Huke, S., et al. (2018). Pathogenic troponin T mutants with opposing effects on myofilament Ca2+ sensitivity attenuate cardiomyopathy phenotypes in mice. Arch. Biochem. Biophys. 661, 125–131. doi: 10.1016/j.abb.2018.11.006
Dipchand, A. I., Kirk, R., Edwards, L. B., Kucheryavaya, A. Y., Benden, C., Christie, J. D., et al. (2013). The registry of the international society for heart and lung transplantation: sixteenth official pediatric heart transplantation report–2013; focus theme: age. J. Heart Lung Transplant. 32, 979–988. doi: 10.1016/j.healun.2013.08.005
Dweck, D., Hus, N., and Potter, J. D. (2008). Challenging current paradigms related to cardiomyopathies. Are changes in the Ca2+ sensitivity of myofilaments containing cardiac troponin C mutations (G159D and L29Q) good predictors of the phenotypic outcomes? J. Biol. Chem. 283, 33119–33128. doi: 10.1074/jbc.M804070200
Dweck, D., Reyes-Alfonso, A. Jr., and Potter, J. D. (2005). Expanding the range of free calcium regulation in biological solutions. Anal. Biochem. 347, 303–315. doi: 10.1016/j.ab.2005.09.025
Dweck, D., Reynaldo, D. P., Pinto, J. R., and Potter, J. D. (2010). A dilated cardiomyopathy troponin C mutation lowers contractile force by reducing strong myosin-actin binding. J. Biol. Chem. 285, 17371–17379. doi: 10.1074/jbc.M109.064105
Dweck, D., Sanchez-Gonzalez, M. A., Chang, A. N., Dulce, R. A., Badger, C. D., Koutnik, A. P., et al. (2014). Long term ablation of protein kinase a (PKA)-mediated cardiac troponin I phosphorylation leads to excitation-contraction uncoupling and diastolic dysfunction in a knock-in mouse model of hypertrophic cardiomyopathy. J. Biol. Chem. 289, 23097–23111. doi: 10.1074/jbc.M114.561472
Dyer, E. C., Jacques, A. M., Hoskins, A. C., Ward, D. G., Gallon, C. E., Messer, A. E., et al. (2009). Functional analysis of a unique troponin c mutation, GLY159ASP, that causes familial dilated cardiomyopathy, studied in explanted heart muscle. Circ. Heart Fail 2, 456–464. doi: 10.1161/CIRCHEARTFAILURE.108.818237
Farah, C. S., and Reinach, F. C. (1995). The troponin complex and regulation of muscle contraction. FASEB J. 9, 755–767. doi: 10.1096/fasebj.9.9.7601340
Freeman, K., Colon-Rivera, C., Olsson, M. C., Moore, R. L., Weinberger, H. D., Grupp, I. L., et al. (2001). Progression from hypertrophic to dilated cardiomyopathy in mice that express a mutant myosin transgene. Am. J. Physiol. Heart Circ. Physiol. 280, H151–H159. doi: 10.1152/ajpheart.2001.280.1.H151
Fujino, N., Shimizu, M., Ino, H., Okeie, K., Yamaguchi, M., Yasuda, T., et al. (2001). Cardiac troponin T Arg92Trp mutation and progression from hypertrophic to dilated cardiomyopathy. Clin. Cardiol. 24, 397–402. doi: 10.1002/clc.4960240510
Fujino, N., Shimizu, M., Ino, H., Yamaguchi, M., Yasuda, T., Nagata, M., et al. (2002). A novel mutation Lys273Glu in the cardiac troponin T gene shows high degree of penetrance and transition from hypertrophic to dilated cardiomyopathy. Am. J. Cardiol. 89, 29–33. doi: 10.1016/s0002-9149(01)02158-0
Gonzalez-Martinez, D., Johnston, J. R., Landim-Vieira, M., Ma, W., Antipova, O., Awan, O., et al. (2018). Structural and functional impact of troponin C-mediated Ca2+ sensitization on myofilament lattice spacing and cross-bridge mechanics in mouse cardiac muscle. J. Mol. Cell Cardiol. 123, 26–37. doi: 10.1016/j.yjmcc.2018.08.015
Hancock, W. O., Huntsman, L. L., and Gordon, A. M. (1997). Models of calcium activation account for differences between skeletal and cardiac force redevelopment kinetics. J. Muscle Res. Cell. Motil. 18, 671–681.
Hershberger, R. E., Hedges, D. J., and Morales, A. (2013). Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 10, 531–547. doi: 10.1038/nrcardio.2013.105
Hershberger, R. E., Norton, N., Morales, A., Li, D., Siegfried, J. D., and Gonzalez-Quintana, J. (2010). Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ. Cardiovasc. Genet. 3, 155–161. doi: 10.1161/CIRCGENETICS.109.912345
Ho, Y. L., Lin, Y. H., Tsai, W. Y., Hsieh, F. J., and Tsai, H. J. (2009). Conditional antisense-knockdown of zebrafish cardiac troponin C as a new animal model for dilated cardiomyopathy. Circ. J. 73, 1691–1697. doi: 10.1253/circj.cj-09-0210
Holroyde, M. J., Robertson, S. P., Johnson, J. D., Solaro, R. J., and Potter, J. D. (1980). The calcium and magnesium binding sites on cardiac troponin and their role in the regulation of myofibrillar adenosine triphosphatase. J. Biol. Chem. 255, 11688–11693.
Johnston, J. R., Chase, P. B., and Pinto, J. R. (2018). Troponin through the looking-glass: emerging roles beyond regulation of striated muscle contraction. Oncotarget 9, 1461–1482. doi: 10.18632/oncotarget.22879
Landesberg, A., and Sideman, S. (1994). Coupling calcium binding to troponin C and cross-bridge cycling in skinned cardiac cells. Am J Physiol 266(3 Pt 2), H1260–H1271.
Landstrom, A. P., Parvatiyar, M. S., Pinto, J. R., Marquardt, M. L., Bos, J. M., Tester, D. J., et al. (2008). Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J. Mol. Cell Cardiol. 45, 281–288. doi: 10.1016/j.yjmcc.2008.05.003
Lee, T. M., Hsu, D. T., Kantor, P., Towbin, J. A., Ware, S. M., Colan, S. D., et al. (2017). Pediatric cardiomyopathies. Circ. Res. 121, 855–873. doi: 10.1161/CIRCRESAHA.116.309386
Li, M. X., and Hwang, P. M. (2015). Structure and function of cardiac troponin C (TNNC1): implications for heart failure, cardiomyopathies, and troponin modulating drugs. Gene 571, 153–166. doi: 10.1016/j.gene.2015.07.074
Li, Y., Charles, P. Y., Nan, C., Pinto, J. R., Wang, Y., Liang, J., et al. (2010). Correcting diastolic dysfunction by Ca2+ desensitizing troponin in a transgenic mouse model of restrictive cardiomyopathy. J. Mol. Cell. Cardiol. 49, 402–411. doi: 10.1016/j.yjmcc.2010.04.017
Lindhout, D. A., and Sykes, B. D. (2003). Structure and dynamics of the C-domain of human cardiac troponin C in complex with the inhibitory region of human cardiac troponin I. J. Biol. Chem. 278, 27024–27034. doi: 10.1074/jbc.M302497200
Lipshultz, S. E., Sleeper, L. A., Towbin, J. A., Lowe, A. M., Orav, E. J., Cox, G. F., et al. (2003). The incidence of pediatric cardiomyopathy in two regions of the United States. N. Engl. J. Med. 348, 1647–1655. doi: 10.1056/NEJMoa021715
Loong, C. K., Takeda, A. K., Badr, M. A., Rogers, J. S., and Chase, P. B. (2013). Slowed dynamics of thin filament regulatory units reduces Ca2+-sensitivity of cardiac biomechanical function. Cell Mol. Bioeng. 6, 183–198. doi: 10.1007/s12195-013-0269-8
Marques, M. A., Pinto, J. R., Moraes, A. H., Iqbal, A., de Magalhaes, M. T., Monteiro, J., et al. (2017). Allosteric transmission along a loosely structured backbone allows a cardiac troponin C mutant to function with only One Ca2+ Ion. J. Biol. Chem. 292, 2379–2394. doi: 10.1074/jbc.M116.765362
Memo, M., Leung, M. C., Ward, D. G., Dos Remedios, C., Morimoto, S., Zhang, L., et al. (2013). Familial dilated cardiomyopathy mutations uncouple troponin I phosphorylation from changes in myofibrillar Ca2+ sensitivity. Cardiovasc. Res. 99, 65–73. doi: 10.1093/cvr/cvt071
Messer, A. E., and Marston, S. B. (2014). Investigating the role of uncoupling of troponin I phosphorylation from changes in myofibrillar Ca2+-sensitivity in the pathogenesis of cardiomyopathy. Front. Physiol. 5:315. doi: 10.3389/fphys.2014.00315
Mogensen, J., Murphy, R. T., Shaw, T., Bahl, A., Redwood, C., Watkins, H., et al. (2004). Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 44, 2033–2040. doi: 10.1016/j.jacc.2004.08.027
Nanni, L., Pieroni, M., Chimenti, C., Simionati, B., Zimbello, R., Maseri, A., et al. (2003). Hypertrophic cardiomyopathy: two homozygous cases with “typical” hypertrophic cardiomyopathy and three new mutations in cases with progression to dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 309, 391–398. doi: 10.1016/j.bbrc.2003.08.014
Parvatiyar, M. S., Landstrom, A. P., Figueiredo-Freitas, C., Potter, J. D., Ackerman, M. J., and Pinto, J. R. (2012). A mutation in TNNC1-encoded cardiac troponin C, TNNC1-A31S, predisposes to hypertrophic cardiomyopathy and ventricular fibrillation. J. Biol. Chem. 287, 31845–31855. doi: 10.1074/jbc.M112.377713
Pinto, J. R., Muller-Delp, J., and Chase, P. B. (2017). Will you still need me (Ca2+, TnT, and DHPR), will you still cleave me (calpain), when I’m 64? Aging Cell 16, 202–204. doi: 10.1111/acel.12560
Pinto, J. R., Parvatiyar, M. S., Jones, M. A., Liang, J., Ackerman, M. J., and Potter, J. D. (2009). A functional and structural study of troponin C mutations related to hypertrophic cardiomyopathy. J. Biol. Chem. 284, 19090–19100. doi: 10.1074/jbc.M109.007021
Pinto, J. R., Reynaldo, D. P., Parvatiyar, M. S., Dweck, D., Liang, J., Jones, M. A., et al. (2011a). Strong cross-bridges potentiate the Ca2+ affinity changes produced by hypertrophic cardiomyopathy cardiac troponin C mutants in myofilaments: a fast kinetic approach. J. Biol. Chem. 286, 1005–1013. doi: 10.1074/jbc.M110.168583
Pinto, J. R., Siegfried, J. D., Parvatiyar, M. S., Li, D., Norton, N., Jones, M. A., et al. (2011b). Functional characterization of TNNC1 rare variants identified in dilated cardiomyopathy. J. Biol. Chem. 286, 34404–34412. doi: 10.1074/jbc.M111.267211
Ploski, R., Rydzanicz, M., Ksiazczyk, T. M., Franaszczyk, M., Pollak, A., Kosinska, J., et al. (2016). Evidence for troponin C (TNNC1) as a gene for autosomal recessive restrictive cardiomyopathy with fatal outcome in infancy. Am. J. Med. Genet. A 170, 3241–3248. doi: 10.1002/ajmg.a.37860
Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., and Zhang, F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308. doi: 10.1038/nprot.2013.143
Regnier, M., Rivera, A. J., Chase, P. B., Smillie, L. B., and Sorenson, M. M. (1999). Regulation of skeletal muscle tension redevelopment by troponin C constructs with different Ca2+ affinities. Biophys. J. 76, 2664–2672. doi: 10.1016/s0006-3495(99)77418-7
Robinson, P., Griffiths, P. J., Watkins, H., and Redwood, C. S. (2007). Dilated and hypertrophic cardiomyopathy mutations in troponin and α-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ. Res. 101, 1266–1273. doi: 10.1161/CIRCRESAHA.107.156380
Takeda, S., Yamashita, A., Maeda, K., and Maeda, Y. (2003). Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature 424, 35–41. doi: 10.1038/nature01780
van der Velden, J., and Stienen, G. J. M. (2019). Cardiac disorders and pathophysiology of sarcomeric proteins. Physiol. Rev. 99, 381–426. doi: 10.1152/physrev.00040.2017
Veltri, T., de Oliveira, G. A., Bienkiewicz, E. A., Palhano, F. L., Marques, M. A., Moraes, A. H., et al. (2017a). Amide hydrogens reveal a temperature-dependent structural transition that enhances site-II Ca2+-binding affinity in a C-domain mutant of cardiac troponin C. Sci. Rep. 7:691. doi: 10.1038/s41598-017-00777-6
Veltri, T., Landim-Vieira, M., Parvatiyar, M. S., Gonzalez-Martinez, D., Dieseldorff Jones, K. M., Michell, C. A., et al. (2017b). Hypertrophic cardiomyopathy cardiac troponin c mutations differentially affect slow skeletal and cardiac muscle regulation. Front. Physiol. 8:221. doi: 10.3389/fphys.2017.00221
Wang, Y., Pinto, J. R., Solis, R. S., Dweck, D., Liang, J., Diaz-Perez, Z., et al. (2012). Generation and functional characterization of knock-in mice harboring the cardiac troponin I-R21C mutation associated with hypertrophic cardiomyopathy. J. Biol. Chem. 287, 2156–2167. doi: 10.1074/jbc.M111.294306
Willott, R. H., Gomes, A. V., Chang, A. N., Parvatiyar, M. S., Pinto, J. R., and Potter, J. D. (2010). Mutations in Troponin that cause HCM, DCM AND RCM: what can we learn about thin filament function? J. Mol. Cell. Cardiol. 48, 882–892. doi: 10.1016/j.yjmcc.2009.10.031
Wu, H., Lee, J., Vincent, L. G., Wang, Q., Gu, M., Lan, F., et al. (2015). Epigenetic regulation of phosphodiesterases 2A and 3A underlies compromised β-adrenergic signaling in an iPSC model of dilated cardiomyopathy. Cell Stem Cell 17, 89–100. doi: 10.1016/j.stem.2015.04.020
Keywords: genetic analysis, cardiac troponin C, missense variant, dilated cardiomyopathy, TNNC1
Citation: Landim-Vieira M, Johnston JR, Ji W, Mis EK, Tijerino J, Spencer-Manzon M, Jeffries L, Hall EK, Panisello-Manterola D, Khokha MK, Deniz E, Chase PB, Lakhani SA and Pinto JR (2020) Familial Dilated Cardiomyopathy Associated With a Novel Combination of Compound Heterozygous TNNC1 Variants. Front. Physiol. 10:1612. doi: 10.3389/fphys.2019.01612
Received: 19 July 2019; Accepted: 23 December 2019;
Published: 22 January 2020.
Edited by:
Julian Stelzer, Case Western Reserve University, United StatesReviewed by:
Jonathan P. Davis, The Ohio State University, United StatesCharles Redwood, University of Oxford, United Kingdom
Beata M. Wolska, The University of Illinois at Chicago, United States
Copyright © 2020 Landim-Vieira, Johnston, Ji, Mis, Tijerino, Spencer-Manzon, Jeffries, Hall, Panisello-Manterola, Khokha, Deniz, Chase, Lakhani and Pinto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saquib A. Lakhani, c2FxdWliLmxha2hhbmlAeWFsZS5lZHU=; Jose Renato Pinto, am9zZS5waW50b0BtZWQuZnN1LmVkdQ==
†These authors have contributed equally to this work