 Zihan Xu
Zihan Xu Wenjun Ding
Wenjun Ding Xiaobei Deng
Xiaobei Deng- 1Faculty of Public Health, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2Laboratory of Environment and Health, College of Life Sciences, University of Chinese Academy of Sciences, Beijing, China
Epithelial-mesenchymal transition (EMT) refers to the conversion of epithelial cells to mesenchymal phenotype, which endows the epithelial cells with enhanced migration, invasion, and extracellular matrix production abilities. These characteristics link EMT with the pathogenesis of organ fibrosis and cancer progression. Recent studies have preliminarily established that fine particulate matter with an aerodynamic diameter of less than 2.5 μm (PM2.5) is correlated with EMT initiation. In this pathological process, PM2.5 particles, excessive reactive oxygen species (ROS) derived from PM2.5, and certain components in PM2.5, such as ions and polyaromatic hydrocarbons (PAHs), have been implicated as potential EMT mediators that are linked to the activation of transforming growth factor β (TGF-β)/SMADs, NF-κB, growth factor (GF)/extracellular signal-regulated protein kinase (ERK), GF/phosphatidylinositol 3-kinase (PI3K)/Akt, wingless/integrated (Wnt)/β-catenin, Notch, Hedgehog, high mobility group box B1 (HMGB1)-receptor for advanced glycation end-products (RAGE), and aryl hydrocarbon receptor (AHR) signaling cascades and to cytoskeleton rearrangement. These pathways directly and indirectly transduce pro-EMT signals that regulate EMT-related gene expression in epithelial cells, finally inducing the characteristic alterations in morphology and functions of epithelia. In addition, novel associations between autophagy, ATP citrate lyase (ACLY), and exosomes with PM2.5-induced EMT have also been summarized. However, some debates and paradoxes remain to be consolidated. This review discusses the potential molecular mechanisms underlying PM2.5-induced EMT, which might account for the latent role of PM2.5 in cancer progression and fibrogenesis.
Introduction
Air pollution, especially that of developing countries, has become increasingly severe with the process of industrialization. Even before atmospheric brown cloud (ABC) was first observed in Asia, ambient particulate matter (PM) has been widely regarded as one of the major airborne pollutants. PM is composed of solids and/or liquids suspended in the atmosphere with a large variation in size. PM can be classified into four different categories according to aerodynamic diameters: total suspended particulates (TSPs); inhalable particles with an aerodynamic diameter less than 10 μm (PM10); fine PM with an aerodynamic diameter less than 2.5 μm (PM2.5); and ultrafine particles (PM0.1). Owing to the relatively high dispersity and small diameter, PM2.5 can be inhaled and deposited in airways and alveoli and can even penetrate into the bloodstream, systemically spreading and inducing damage to multiple systems and organs (Wang et al., 2013; Falcon-Rodriguez et al., 2016; Liu et al., 2019). PM2.5 is derived from industrial and vehicle emissions, mineral dust, biomass incineration, and other diverse sources; thus, the composition of PM2.5 is extremely complex (Belis et al., 2013; Falcon-Rodriguez et al., 2016). Transitional metals, soluble salts, organic/elemental carbon, and microbes that adhere to PM2.5 particles contribute to the toxic effects of PM2.5 including inflammation, DNA damage, mutations, reactive oxygen species (ROS) accumulation, and mitochondria dysfunction. All these abnormal intracellular events are highly correlated with the pathogenesis and progression of a series of human diseases (Ghio et al., 2012; Valavanidis et al., 2013; Feng et al., 2016; Qi et al., 2019). Outdoor PM2.5 exposure has been listed as the fifth most influential risk factor for global death, which caused 4.2 million deaths in 2015 (Schraufnagel et al., 2018). Besides, epidemiological studies have shown that PM2.5 exposure was highly correlated with the morbidity and mortality of respiratory and cardiovascular diseases. Maternal exposure to PM2.5 is also linked to an increased risk of airway disease development in offspring (Saha et al., 2018; Sanyal et al., 2018).
Epithelial-mesenchymal transition (EMT) is a process through which epithelial cells lose their apical-basal polarity and acquire the mesenchymal phenotype (Katsuno et al., 2013; Chaffer et al., 2016). A series of underlying molecular events, especially the repression of E-cadherin expression, the upregulation of N-cadherin and vimentin, and the disruption of adherin junctions, have been uncovered (Kourtidis et al., 2017; Miyazono et al., 2018). Other significant molecular features underlying EMT include cytoskeleton remodeling, such as actin stress fiber formation, and induction of mesenchymal biomarkers, including α-smooth muscle actin (α-SMA) and fibronectin (Kalluri and Weinberg, 2009). As EMT endows the epithelia with the potential of myofibroblast differentiation, increased motility, migration and invasion ability, this cellular event, if abnormally activated, will inevitably mediate the progression of organ fibrosis and cancer metastasis by facilitating the secretion of the extracellular matrix components or the dissociation of neoplastic cells from primary tumors and the subsequent intravasation (Stone et al., 2016; Yan et al., 2018; Lu and Kang, 2019). Furthermore, the enhanced anti-apoptosis and drug detoxification characteristics of cancer cells that have undergone EMT also result in drug resistance in cancer chemotherapy (Santamaria et al., 2019).
To date, three EMT subtypes have been determined based on their biological functions in tissue development and pathogenesis. Type 1 EMT is associated with the formation and implantation of embryo together with organ development. Type 2 EMT functions in wound healing by contributing to fibroblast generation and extracellular matrix production. Type 3 EMT refers to the phenotypic transformation in neoplastic epithelial cells, which enhances anti-apoptosis and invasion abilities (Kalluri, 2009; Kalluri and Weinberg, 2009; Zou et al., 2017).
Although massive statistical analyses have established the causal link between environmental PM2.5 exposure and morbidity/mortality of multiple cancers as well as fibrotic diseases, and “outdoor air pollution, particulate matter in” has been indexed as one of 120 primary carcinogens, the exact in-depth mechanisms at the cellular or microscopic level are still largely unknown (Kim et al., 2018; Sese et al., 2018; Cheng et al., 2019; International Agency for Research on Cancer, 2019). As these diseases are currently largely irreversible and lethal, targeted approaches to prevent their onset should be developed. Current research has demonstrated that acute or chronic PM2.5 exposure elicited the expression of characteristic EMT markers and EMT-related transcription factors (TFs) in the epithelia (Wei et al., 2017; Chi et al., 2018; Xu et al., 2019a). Similar altered expression patterns have also been detected in the lungs after mice were exposed to PM2.5 (Fu et al., 2019). Hence, it is possible that PM2.5 might serve as an exogenous promoter of fibrosis and cancer by initiating abnormal EMT in corresponding target epithelial tissues and cells. Although the molecular toxicology of PM2.5-induced EMT has not been well studied, on the basis of existing publications, this review aims to describe validated or potential molecular processes, especially the signaling pathways, participating in the action of PM2.5 on pro-fibrotic or neoplastic EMT.
Overview of the Molecular Players in Epithelial-Mesenchymal Transition
Key Transcription Factors
The loss of E-cadherin is a typical EMT hallmark. The TFs that control the expression of E-cadherin include the Zn-finger proteins Snail and Slug, the two-handed Zn-finger proteins ZEB1 and ZEB2, and the basic helix-loop-helix family protein TCF and Twist have also been identified (Guaita et al., 2002; Burger et al., 2017). These TFs directly bind to 5′ E-boxes of the human E-cadherin promoter and further silence the transcription of CDH1 (Batlle et al., 2000; Guaita et al., 2002; Vesuna et al., 2008). Lymphoid enhancer-binding factor-1 (LEF-1), a member of TCF/LEF family, also regulates EMT by recruiting the coactivator β-catenin to enhancers (Gilles et al., 2003; Santiago et al., 2017). Interactions and crosstalk between TFs have also been extensively recognized. Snail can induce the expression of ZEB1/2, Twist, Slug, and LEF-1 (Guaita et al., 2002; Dave et al., 2011; Cevenini et al., 2018). Twist can serve as the transcriptional initiator of Slug and ZEB1, whereas TCF-4 functions indirectly via ZEB1 (Taube et al., 2010; Sanchez-Tillo et al., 2011). LEF-1 can also exert its pro-EMT ability indirectly by upregulating Slug expression (Lambertini et al., 2010). In addition, a member of the high-mobility group (HMG) protein family, the transforming growth factor β (TGF-β)/SMAD2/3-dependent TF HMGA2, induces the expression of Snail, Slug, and Twist and mediates EMT (Thuault et al., 2006, 2008). NF-κB, a versatile TF involved in the regulation of proliferation, adhesion, and inflammation, executes its pro-EMT ability via Snail, Slug, and ZEB1/2 (Min et al., 2008; Sarkar et al., 2008; Techasen et al., 2012). Key TFs related to EMT and their transcriptional interactions are roughly summarized in Table 1.
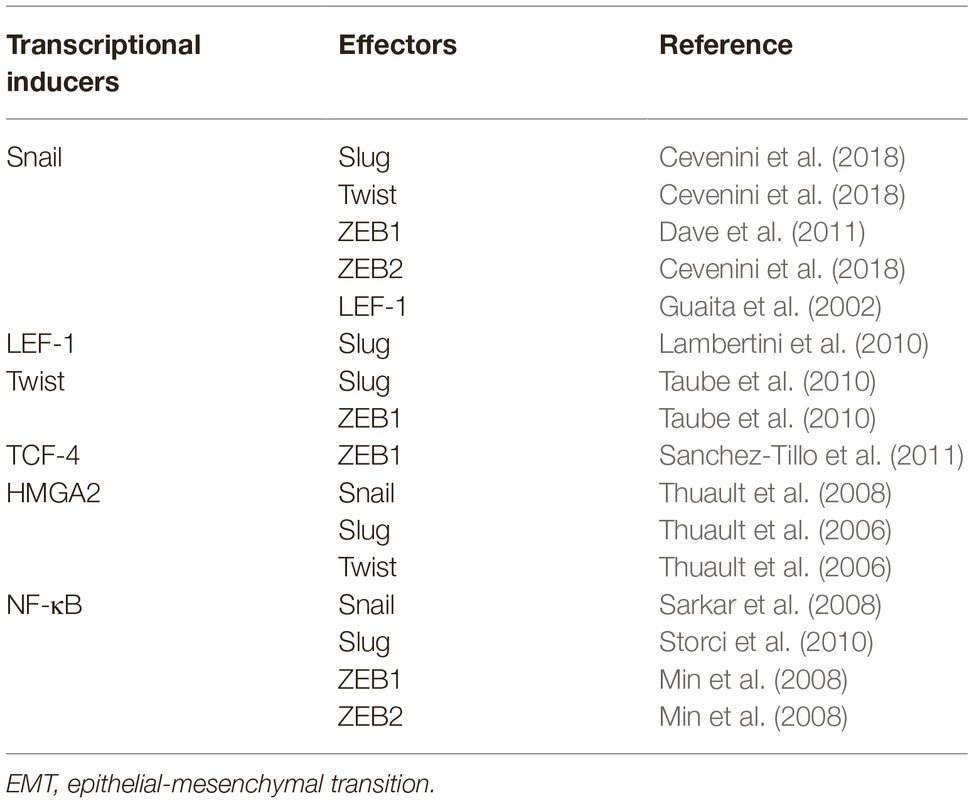
Table 1. Positive transcriptional regulation among pro-EMT transcription factors.
Pro-Epithelial-Mesenchymal Transition Signaling Cascades
TGF-β is considered as the most powerful initiator of EMT. TGF-β can directly and positively regulate mesenchymal markers such as α-SMA and type I collagen (COL1) and is also a potent regulator of cytoskeleton remodeling (Zavadil and Bottinger, 2005; Fernandez and Eickelberg, 2012). The type II TGF-β receptor (TβRII) is initially recruited by activated TGF-β and forms a hetero-tetrameric complex with the type I TGF-β receptor (TβRI) by phosphorylating the GS domain of TβRI (Miyazawa et al., 2002; Huang and Chen, 2012; Vander Ark et al., 2018). SMAD2 and SMAD3, receptor-activated SMAD proteins (R-SMADs), are cytoplasmic mediators of TGF-β signaling whose recruitment and phosphorylation by TβRI result in their translocation to the nucleus in assistance of SMAD4 (Fabregat and Caballero-Diaz, 2018). SMAD2/3/4 together with other coactivators activates HMGA2, Snail, Slug, and ZEB1/2 expression, which further initiates the phenotypic transformation of EMT (Thuault et al., 2006; Katsuno et al., 2013). Another set of SMAD signaling cascades consisting of bone morphogenetic protein receptors (BMPRs) and the SMAD 1/5/8 also enhances Slug and Twist expression (McCormack et al., 2013; Li et al., 2014; Ahn et al., 2016; Wang et al., 2016). SMAD6 and SMAD7, two inhibitory SMADs (I-SMADs), participate in the inhibition of signal transduction via competitive binding with TβRI and BMPRI (Itoh and ten Dijke, 2007; Gonzalez-Nunez et al., 2013; Koganti et al., 2018). There are also non-canonical TGF-β cascades linked with EMT, in which TβRII phosphorylates Par6 and further modulates activity of RhoA via ubiquitination mediated by Smurf1, resulting in the loss of tight junction and EMT (Choi et al., 2017; Simeone et al., 2018).
β-Catenin is the main effector of Wnt pathway. The cytoplasmic accumulation of β-catenin after activation of Wnt signaling, or the release of β-catenin due to the loss of E-cadherin, which allow the interactions between β-catenin and LEF-1, can induce the upregulation of Snail, Slug, Twist and mesenchymal markers such as fibronectin (Guo et al., 2012; Ghahhari and Babashah, 2015). When the Wnt ligands bind to Frizzle and LDL receptor-related protein-5/6 (LRP5/6) receptors, extracellular signals are transduced by LRP to phosphorylated Disheveled (Dsh), which in turn block the activity of glycogen synthase kinase-3β (GSK-3β), a key EMT suppressor that mediates the ubiquitination of Snail and the degradative phosphorylation of β-catenin (Doble and Woodgett, 2007; Heuberger and Birchmeier, 2010; Lam and Gottardi, 2011; Gonzalez and Medici, 2014).
The receptor tyrosine kinases (RTKs) and their downstream effectors are also pivotal in EMT. There are numerous growth factors (GFs), including epidermal GF (EGF), fibroblast GF (FGF), platelet-derived GF (PDGF), and insulin-like GF (IGF), which serve as RTK ligands (Jechlinger et al., 2006; Cevenini et al., 2018; Shu et al., 2018; Yamaoka et al., 2018). Binding of GFs with RTKs and the ensuing tyrosine autophosphorylation of RTKs can result in binding of GTP to Ras and the recruitment of Raf, activating mitogen-activated protein kinase (MAPK)/extracellular signal-regulated protein kinase (ERK) kinase (MEK), which further phosphorylates ERKs. Ligand-bound RTKs also activate phosphoinositide 3-kinase (PI3K), which activates Akt via PIP3 converted from PIP2. Activation of Akt and ERK leads to the inactivating phosphorylation of GSK-3β, which promotes EMT (Oloumi et al., 2004; Zhou et al., 2004; Ding et al., 2005; Doble and Woodgett, 2007; Lemmon and Schlessinger, 2010; Tripathi and Garg, 2018). Akt activation also contributes to signal transduction through the NF-κB/Snail pathway (Julien et al., 2007).
The Janus kinase/signal transducers and activators of transcription-3 (JAK/STAT3) pathway, which were initially defined as the inflammation response-related pathway, also actively participate in EMT. Interleukin-6 (IL-6) activates JAK after binding to IL-6 receptor, which subsequently facilitates the phosphorylation and the following dimerization of STAT3. STAT3 homodimer then translocates into the nucleus and regulate the transcription (Yan et al., 2018). The IL-6/JAK/STAT3 pathway can effectively upregulate pro-EMT TFs, including Snail, Slug, and Twist and modify the expression of EMT-related genes (Colomiere et al., 2009; Sullivan et al., 2009; Yadav et al., 2011; Chen et al., 2015). In addition, interleukin-8 (IL-8) can also induce similar JAK/STAT3-dependent responses through interactions with IL-8 receptor (Fernando et al., 2011; Fu et al., 2015).
The sonic hedgehog (Shh) signaling pathway consisting of Shh, protein patch homolog (PTCH), smoothend (SMO), and glima-associated oncogene-1/2/3 (Gli1/2/3) mediates embryonic development. Recently, the importance of this pathway in cancer and EMT has also been revealed (McCubrey et al., 2016; Zou et al., 2017). Upon binding to the transmembrane G protein-coupled receptor PTCH, secretive Shh releases the repressed SMO-bound PTCH, which further induces the transcriptional activity of Glis, promoting the transcription of Snail, Slug, and other EMT-related gene targets (Li et al., 2006; Pain et al., 2014; Flemban and Qualtrough, 2015; Zhang et al., 2016a).
Novel studies have described the participation of Notch signaling, an original paracrine pathway, in angiogenesis, tumor progression, cell proliferation, and EMT induction. The initiation of Notch relies on the trans-interactions between Notch ligands (Jag1, Jag2, Delta1, Delta3, and Delta4) at cell surface of signaling-sending cells and extracellular domains of noncovalent heterodimeric transmembrane Notch receptors (Notch1, Notch2, Notch3, and Notch4) on the surface of signaling-receiving cells. An initial cleavage at the extracellular site of the notch receptor mediated by a disintegrin and metalloproteinase (ADAM) follows the activation of Notch receptor (Meurette and Mehlen, 2018; Chatterjee and Sil, 2019). After the second intracellular cleavage mediated by γ-secretase, Notch intracellular domain (NICD) with activated transcriptional activity is released and then translocates into the nucleus, mediating the transcriptional upregulation of hairy and enhancer of split (HES) family genes and the HES related with YRPW motif (HEY) family genes in cooperation with co-regulators. These effectors further induce the corresponding downstream transcriptional modification, such as the induction of Snail and Slug (Thiery et al., 2009; Vinson et al., 2016; Leonetti et al., 2019).
A specific consequence of the multiple signal transduction networks reviewed above is the increase of cell motility. To drive the process of migration and invasion, the subtle remodeling of cytoskeleton dynamics accompanied by the tuning of epithelial adhesion, which is directly regulated by focal adhesion kinase (FAK), Rho family GTPase, and Rho-associated protein kinase (ROCK) signaling, is highly activated, leading to actin polymerization, stress fiber assembly, lamellipodia and filopodia formation, and the creeping-like motions of the cells (Cavallaro and Christofori, 2001; Yilmaz and Christofori, 2009; Jiang et al., 2017; Jansen et al., 2018). A schematic overview of the pro-EMT pathways and key messengers is shown in Figure 1.
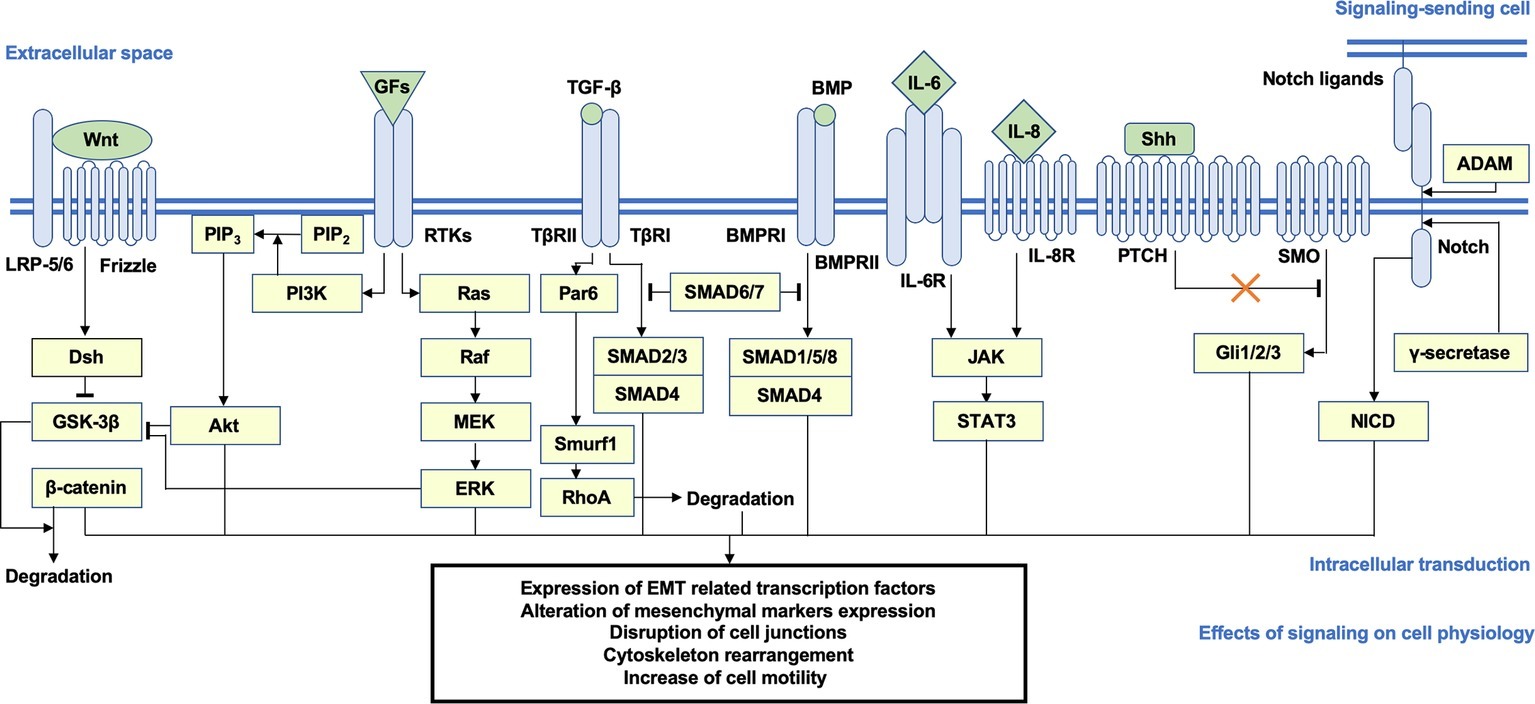
Figure 1. Brief schema of the putative signaling transduction mechanisms underlying EMT. Activation of the Wnt/β-catenin, PI3K/Akt, Ras/ERK, TGF-β/SMAD2/3, BMP/SMAD1/5/8, JAK/STAT3, Shh, and Notch pathways is highly correlated with EMT. After ligand-receptor binding, intracellular secondary messengers are activated and initiated downstream transduction, which generally induce the nuclear translocation of signaling-specific TFs and the transcriptional regulation of EMT-related genes, such as CDH1 and CDH2, EMT TFs, and mesenchymal markers, accompanied by a series of alterations on cellular physiological or pathological activities (e.g., disjunction of adherin junctions, cytoskeleton remodeling, and increase of cellular motility). Arrows represent the molecular interactions in which downstream messengers are activated; T shape arrows represent inhibitive molecular interactions. EMT, epithelial-mesenchymal transition; PI3K, phosphoinositide 3-kinase; ERK, extracellular signal-regulated protein kinase; TGF-β, transforming growth factor β; JAK, Janus kinase; Shh, sonic hedgehog; TFs, transcription factors.
Potential Mechanisms of PM2.5-Induced Epithelial-Mesenchymal Transition
Potential Reactive Oxygen Species-Dependent Mechanisms in PM2.5-Induced Epithelial-Mesenchymal Transition
ROS are physiologically produced as byproducts of biological reactions in the cytoplasm, mitochondria, and peroxisomes catalyzed by cytochrome P450 and NADPH oxidase (NOX). ROS have primary functions to actively defend against invading pathogens (Bardaweel et al., 2018; Lee et al., 2018). However, excessive cytoplasmic retention of ROS generally results in cellular oxidative stress, which can alter intracellular signaling, DNA damage, and apoptosis (Danielsen et al., 2009; Deng et al., 2014; Vattanasit et al., 2014; Cho et al., 2018). Intracellular ROS can be generated by cellular exposure to external contaminants, including the main components of PM2.5, such as black carbon, redox-active metals (e.g., Fe, Cu, Ni, Zn, Cr, As, and Mn), and PAHs (e.g., naphthalene, benzo-a-pyrene, and anthracene) (Shuster-Meiseles et al., 2016; Niranjan and Thakur, 2017; Sun et al., 2018). PM2.5 particles have also been characterized as strong inducers of ROS, as they directly release ROS, catalyze redox reactions, or participate in redox cycles as substrates (Squadrito et al., 2001; Knaapen et al., 2004; Risom et al., 2005; Valavanidis et al., 2013; Cho et al., 2018; Jin et al., 2018).
As ROS has been well characterized as one of the fundamental intermediaries of the PM2.5-induced response and injury, and evidence verifying PM2.5-induced EMT via ROS in vivo and in vitro has been emerging, this section will be mainly focused on the potential pathogenic role of PM2.5-derived ROS in EMT (Thevenot et al., 2013; Yan et al., 2016; Yi et al., 2017; Palleschi et al., 2018; Li et al., 2018b). Some essential information including the sources and the concentrations of PM2.5 utilized, as well as the antioxidant used for treatment and the effective concentrations, is summarized in Table 2.
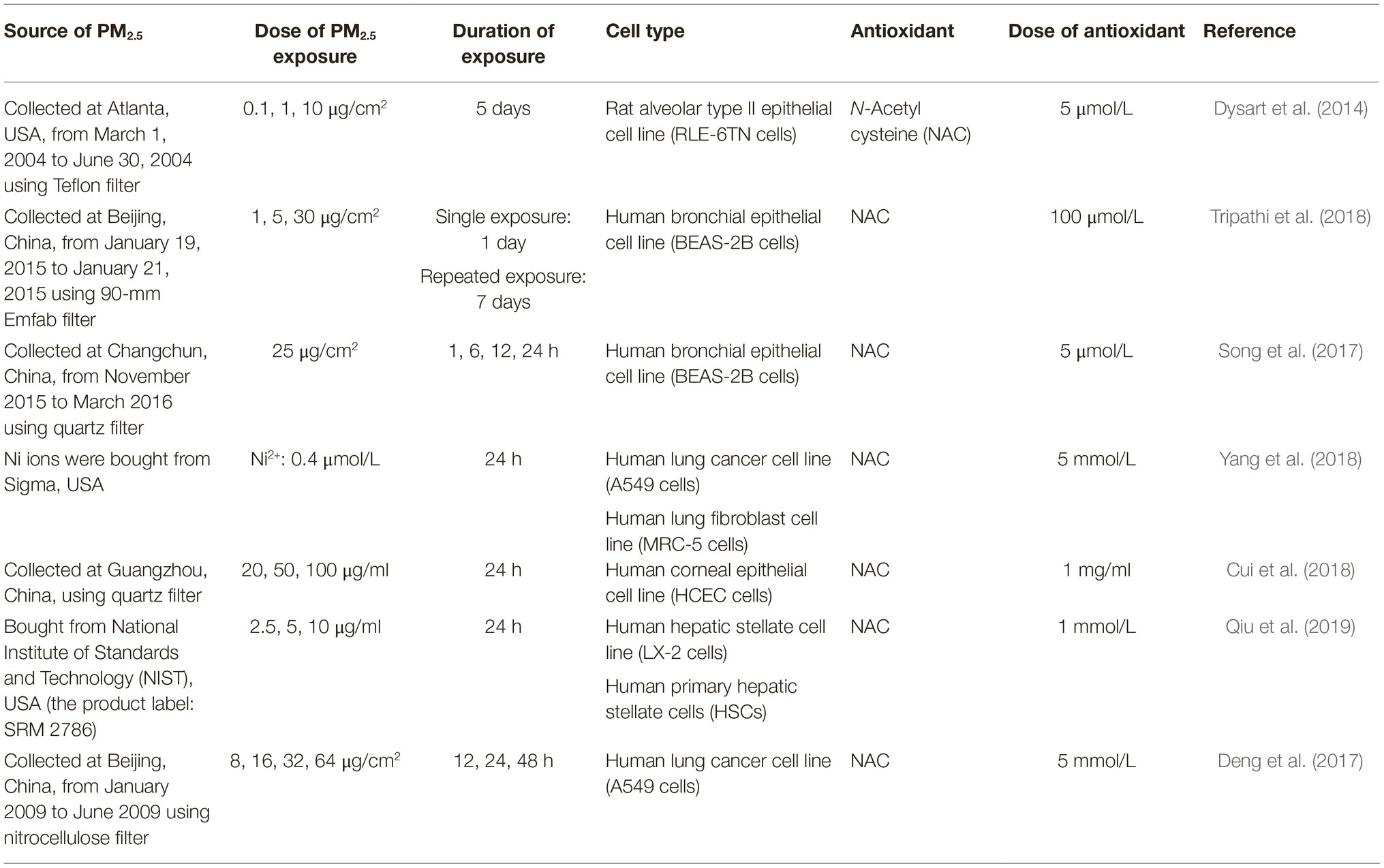
Table 2. Summary table about exposure assays of some studies reviewed in section “Potential Reactive Oxygen Species-Dependent Mechanisms in PM2.5-Induced Epithelial-Mesenchymal Transition.”
PM2.5-Reactive Oxygen Species Participates in SMAD-Dependent Epithelial-Mesenchymal Transition
TGF-β has been shown to involve ROS formation via NOX4, whereas ROS also stimulate the epithelial excretion of TGF-β (Latella, 2018). ROS can also convert latent TGF-β to its active form by oxidizing the latency-associated peptide (LAP) (Liu and Gaston Pravia, 2010; Montorfano et al., 2014; Jaffer et al., 2015). Excessive ROS derived from protein oxidation can induce TβRI/II expression and further initiate TGF-β/SMAD signaling, which induces EMT in hepatocytes in vivo and in vitro (Sun et al., 2017). These studies indicate that ROS can activate TGF-β/SMAD signaling at different points along the pathway, including ligand-receptor interactions and intracellular signaling.
An in vivo study demonstrated that long-term PM2.5 exposure induced increased TGF-β1 expression, SMAD2/3 phosphorylation, and collagen accumulation in mice lungs, indicating potential links among PM2.5 exposure, the TGF-β1/SMAD2/3 pathway, and pro-fibrotic EMT (Gu et al., 2017). Another study supported this pathogenesis in mice model: Maternal exposure to PM2.5 could also result in significant oxidative stress, activation of TGF-β/SMAD3 signaling, and EMT in fetal mice lung, which contributed to postnatal pulmonary dysfunction (Tang et al., 2017). In addition, exposure of mice to PM2.5 for 4 weeks not only elicited SMAD3 phosphorylation and cardiac fibrosis but also induced excessive ROS accumulation and NOX4 expression in heart tissues, indicating that the potential occurrence of PM2.5-induced EMT or endothelial-mesenchymal transition (EndMT) might be ROS dependent (Qin et al., 2018). Our previous in vitro study validated this mechanism: The release of TGF-β1, the activation of the TGF-β1/SMAD3 pathway, and the altered expression of hallmark EMT proteins in human bronchial epithelial cells were observed after 30-passage low-dose PM2.5 exposure (Xu et al., 2019a). In another study, the exposure of alveolar epithelial cells to PM2.5 not only induced the activation of latent TGF-β but also increased cell contractility and elongated cellular morphology in a dose-dependent manner. These effects could be reversed by NAC treatment, demonstrating the role of ROS in this pathogenic event (Dysart et al., 2014). In addition, increased TGF-β1 mRNA expression was observed in bronchial epithelial cells after 7 days’ consecutive exposure to PM2.5, and treatment with the antioxidant NAC could also effectively offset the toxicity of PM2.5 exposure (Tripathi et al., 2018). However, the detailed EMT transcription patterns induced by PM2.5-induced ROS through the SMAD2/3-dependent pathway remain unknown.
In summary, these data demonstrate that PM2.5 might induce EMT via ROS in a TGF-β/SMAD2/3 signaling-dependent manner. However, additional research is needed to further establish a firm causal link.
PM2.5-Reactive Oxygen Species Might Induce Epithelial-Mesenchymal Transition via NF-κB Cascades
The activation of NF-κB can be evoked by ROS. NF-κB is activated as the response of cellular oxidative stress, responsible for inflammation, apoptosis, and differentiation. Moreover, the activation of NF-κB by ROS is also correlated with Snail upregulation and EMT phenotype transformation in epithelial cells (Valavanidis et al., 2013; Cichon and Radisky, 2014). Different antioxidant treatments targeting ROS in human non-small cell lung carcinoma cells, pancreatic cancer cells, and hepatocellular carcinomatous cells all effectively inhibit NF-κB activation, which further represses matrix metalloproteinase (MMP) expression together with cellular migration and invasion (Lee and Lim, 2011; Chao et al., 2017; Huang and Xin, 2018). In addition, IL-6 and IL-8, which are transcriptionally upregulated by ROS-activated NF-κB, also exert pro-EMT abilities via downstream JAK/STAT3 signaling (Gupta et al., 2012). In gastric cancer cells, even the regulatory effects of IL-6/JAK/STAT3 on expression of pro-EMT TFs and EMT markers relied on NOX4, indicating the potential comprehensive role of ROS in the transduction of NF-κB/IL-6 signaling (Gao et al., 2017). NF-κB is also able to induce TNF-α expression in response to ROS stimulation, and activated TNF-α receptors in turn upregulate NF-κB transcription with further Snail expression, implying positive feedback loops underlying ROS-induced EMT via NF-κB (Dong et al., 2007).
A novel publication confirmed the positive role of NF-κB in PM2.5-induced EMT. Organic extracts of PM2.5 enhanced the binding of NF-κB with the promoter of long non-coding RNA (lncRNA) MALAT1, and MALAT1 relieved the expression silencing of ZEB1 by sponging miR-204 targeting ZEB1 mRNA, resulting in the mesenchymal phenotype transition of pulmonary epithelial cells (Luo et al., 2019). Another novel study showed the correlation between PM2.5, ROS, NF-κB, and pro-fibrotic EMT in vivo. The oxidative stress within the rat lungs exacerbated during PM2.5 exposure but disappeared after the termination of exposure, and the upregulation of RelA/p65, deteriorated pulmonary fibrosis, and EMT in rat lungs were observed in the post-exposure phase, indicating that PM2.5 might lead to pulmonary fibrosis by oxidative stress-initiated NF-κB/inflammation/EMT pathway (Sun et al., 2019). Moreover, a previous study demonstrated that NF-κB activation following PM2.5 exposure can be ROS dependent: The inhibition of ROS effectively abrogated the sustained NF-κB activation in BEAS-2B bronchial epithelial cells induced by PM2.5 exposure (Song et al., 2017). Mice exposed to ultrafine carbon black particles showed oxidant-dependent NF-κB activation and increased expression of NF-κB responsive genes including TNF-α and IL-6 in lung tissues (Shukla et al., 2000). Silica particles, a kind of well-described pro-fibrotic agent, were able to induce a similar pathological response in silicosis murine models, whereas non-fibrogenic particles failed to induce this effect. These results implied that ROS-derived NF-κB and its downstream inflammatory mediators actively participate in fibrogenesis, in which EMT might act as a major participant (Hubbard et al., 2002). Additionally, an in vitro study showed increased IL-6 level and downstream JAK2/STAT3 signaling activation concomitant with the emergence of oxidative stress in PM2.5-treated alveolar cancer cells, whereas a similar pattern was also observed in another in vitro study, in which ROS accumulation, IL-6/IL-8 release, and JAK2/STAT3 signaling pathway activation were observed to be mediated by cytochrome P450, family 1, member A1/B1 (CYP1A1/B1) after PM2.5 exposure to human bronchial epithelial cells (Yuan et al., 2019). This evidence partly substantiated the hypothesis that PM2.5 might induce EMT via ROS/NF-κB/IL-6/8/JAK/STAT3 axis (Xu et al., 2015). Although the NF-κB activation and following release of IL-6, IL-8, TNF-α, or mitochondrial dysfunction in epithelial tissues induced by PM2.5-derived ROS have been validated to be significant in lung inflammation and cell death, the in-depth regulatory role of this shared pathway in EMT is largely unknown (Bourgeois and Owens, 2014; Leclercq et al., 2018; Liu et al., 2018; Wang et al., 2018; Xin et al., 2018). Currently, the link between cigarette smoke extract (CSE) and pulmonary inflammation together with EMT in airway epithelia has been preliminarily verified. CSE-induced inflammatory responses in human bronchial cells include NF-κB activation and further increased IL-6 transcription, which promote phenotype transformation via downregulation of miR-200-c, a proven anti-EMT micro-RNA (miRNA) family member. Hence, ROS in conjunction with NF-κB might lie at the intersection of PM2.5-derived inflammation and EMT, although the exact mechanisms remain to be elucidated (Zhao et al., 2013). Controversy on this issue also existed. Fe3O4, one of the major anthropogenic components in PM, could result in a delayed NF-κB response in A549 neoplastic pulmonary epithelial cells as a result of the decrease in IκB degradation induced by excessive ROS (Konczol et al., 2011).
In summary, whether PM2.5-intracellular ROS induced NF-κB phosphorylation could initiate EMT is still obscure, as the small amount of peripheral proof is too weak to conclusively establish this stand.
PM2.5, Reactive Oxygen Species, and Receptor Tyrosine Kinase Signaling
ROS are strong initiators of pro-EMT PI3K/Akt and Ras/ERK signaling. ROS have been shown to directly induce the phosphorylation of EGF and PDGF receptors in the absence of ligand and to enhance VEGF-induced receptor phosphorylation via oxidative repression of the intracellular inhibitors protein tyrosine phosphatase-1B (PTP1B) and density-enhanced phosphatase-1 (DEP1) (Lei and Kazlauskas, 2009; Oshikawa et al., 2010; Leon-Buitimea et al., 2012). The ROS-induced conversion of PIP2 to PIP3 also initiates Akt signaling events (Zhang et al., 2016b). Additionally, glutathione peroxidase inactivation-induced ROS accumulation resulted in the acquisition of mesenchymal characteristics of pancreatic cancer cells by the Akt/GSK-3β/Snail regulation (Meng et al., 2018). Likewise, the inhibition of GSK-3β, which increases Snail transcriptional activity and an EMT-like phenotype following ROS-stimulated ERK activation instead of Akt, was observed in Helicobacter pylori-treated gastric cancer cells (Ngo et al., 2016). Besides, the definitive role of ROS in activating ERK to promote the invasion of vascular smooth muscle cells and keratinocytes has also been unfolded. ROS-mediated ERK phosphorylation is also a significant alternative pathway for TGF-β-induced EMT in renal tubular epithelial cells (Rhyu et al., 2005; Wu, 2006).
PM2.5 has been extensively regarded to stimulate the ERK or Akt pathway. PM2.5 could induce MMP-13 expression, invasion, and migration of hepatocellular carcinoma cells via Akt (Zhang et al., 2017a). The increased secretion of EGF by human alveolar epithelial cells after PM2.5 exposure and the further initiation of intracellular EGFR/ERK/NF-κB have also been observed (Jeong et al., 2017). In addition, a microfluidic system-based study on PM2.5-treated bronchial epithelial cells showed significant activation of PI3K/Akt pathway, FGF/FGFR/MAPK/VEGF signaling, and the JAK/STAT pathway, which led to cell proliferation and apoptosis evasion. And increased intracellular and mitochondrial ROS levels implicated that PM2.5-derived ROS could be correlated with the activation of these signaling series (Zheng et al., 2017).
In addition to ERK, other members of the MAPK family have been shown to participate in PM2.5 responses. The coordination of p38 MAPK with proliferating cell nuclear antigen (PCNA) was responsible for PM2.5-induced vascular smooth muscle cell proliferation, and the positive role of ROS was also implied by upregulation of superoxide dismutase (SOD), a cellular oxidative stress biomarker (Wan et al., 2018). Nevertheless, current research on the toxicology of PM2.5-ROS also primarily focuses on the linkage between Akt or MAPK activation and inflammation or apoptosis, suggesting a double-edged role of PM2.5-ROS that could either induce cell death or enhance cell viability (An et al., 2018; Li et al., 2018a). Abundant in PM2.5, Ni2+ was shown to be responsible for cellular oxidative stress and the phenotype transformation of alveolar epithelial cells, and ROS/Akt-dependent MMP-9 and COL1 induction. Similarly, Ni2+/ROS/Akt also led to the inflammation of pulmonary epithelia and mice lung tissues (Yang et al., 2018). Hence, there is also crosstalk between PM2.5-ROS-induced inflammation and EMT via RTK-related cascades. However, further research is needed to clarify the interplay of PM2.5, ROS, Akt, and ERK with EMT.
PM2.5-Derived Reactive Oxygen Species: The Double-Edged Sword on Cytoskeleton Remodeling
Increase of cell invasion/migration is the characteristic of EMT regulated by subtle cytoskeleton remodeling and shifting in adhesion. TGF-β-induced intracellular ROS via NOX4 accelerates the polymerization of globular actin (G-actin) to fibrous actin (F-actin) in human umbilical vein endothelial cells, which is essential for lamellipodia and filopodia formation (Hu et al., 2005; Lamouille et al., 2014). ROS generated by NOX1 was also able to induce the spindle-like cellular morphology, EMT, and differentiation of human gingival epithelial cells via the post-transcriptional accumulation of CK18, a kind of pro-EMT intermediate filament (Sattayakhom et al., 2014). And ROS/ErbB2 signaling was also reported to be responsible for the aggressiveness of breast epithelial cells by phosphorylating FAK and activating RhoC (Wang et al., 2017). In another study, although mere H2O2 treatment was not sufficient to induce complete EMT in retinal pigment epithelial cells due to the transiency of the stimulation, ROS could also initiate Rho-GTP binding, resulting in stress fiber formation (Inumaru et al., 2009).
PM2.5 has been verified as the modulator of cytoskeleton inducing incomplete contact between adjacent human bronchial epithelial cells, in which strong oxidative stress was also observed (Longhin et al., 2016). Besides, long-term chronic PM2.5 exposure to human bronchial epithelial cells could enhance RhoA-GTP binding and stress fiber formation, although no increase of cellular proliferation was observed (Dornhof et al., 2017). In another research, however, PM2.5 that stimulated ROS generation in corneal epithelial cells failed to promote EMT via cytoskeletal regulation. On the contrary, PM2.5 inhibited FAK and paxillin phosphorylation, which further repressed RhoA activity and epithelial cell migration. Now that these inhibitory processes could be reversed with NAC treatment, the discrepant roles of PM2.5-derived ROS in cytoskeleton regulation have been identified (Cui et al., 2018). In summary, the lack of comprehensive data leaves the role of PM2.5 in cytoskeleton modulation shadowy.
Something New: PM2.5-Reactive Oxygen Species-Induced Autophagy Participates in Epithelial-Mesenchymal Transition
Autophagy is a cellular degradation process that is activated in response to environmental stressors such as GF depletion and hypoxia (Yang et al., 2015; Marcucci et al., 2017). Recent evidence has linked autophagy and EMT through shared signaling pathways and functional causality in cancer progression and metastasis (Lv et al., 2012; Li et al., 2013; Catalano et al., 2015; Gugnoni et al., 2016; Zhang et al., 2017b). A recent study verified that PM2.5 participated in liver fibrosis via ROS-mediated mitophagy. Hepatic stellate cells, a principal liver cell type with EMT plasticity, displayed enhanced α-SMA and COL1 expression after PM2.5 treatment. These processes were mediated by ROS-induced PTEN-induced kinase 1 (PINK1) and Parkin, which signified mitophagy (Choi et al., 2009; Qiu et al., 2019). In lung cancer cells, PM2.5-induced cell migration and invasion depended on autophagy, which was initially induced by increased intracellular ROS and its downstream effector lncRNA, loc146880 (Deng et al., 2017). Another study revealed an alternative mechanism underlying PM2.5-autophagy-EMT in lung cancer cells, in which upregulated lncRNA LCPAT1 served as a messenger that mediated autophagy and cell migration by interacting with RCC2 after PM2.5 exposure (Lin et al., 2018). This evidence not only validates the correlation between autophagy and PM2.5-induced epithelia phenotype transformation but also provides new insight into the epigenetic toxicology of PM2.5.
Other Molecular Events Underlying PM2.5-Induced Epithelial-Mesenchymal Transition
Information about exposure assays in this section is selectively summarized in Table 3.
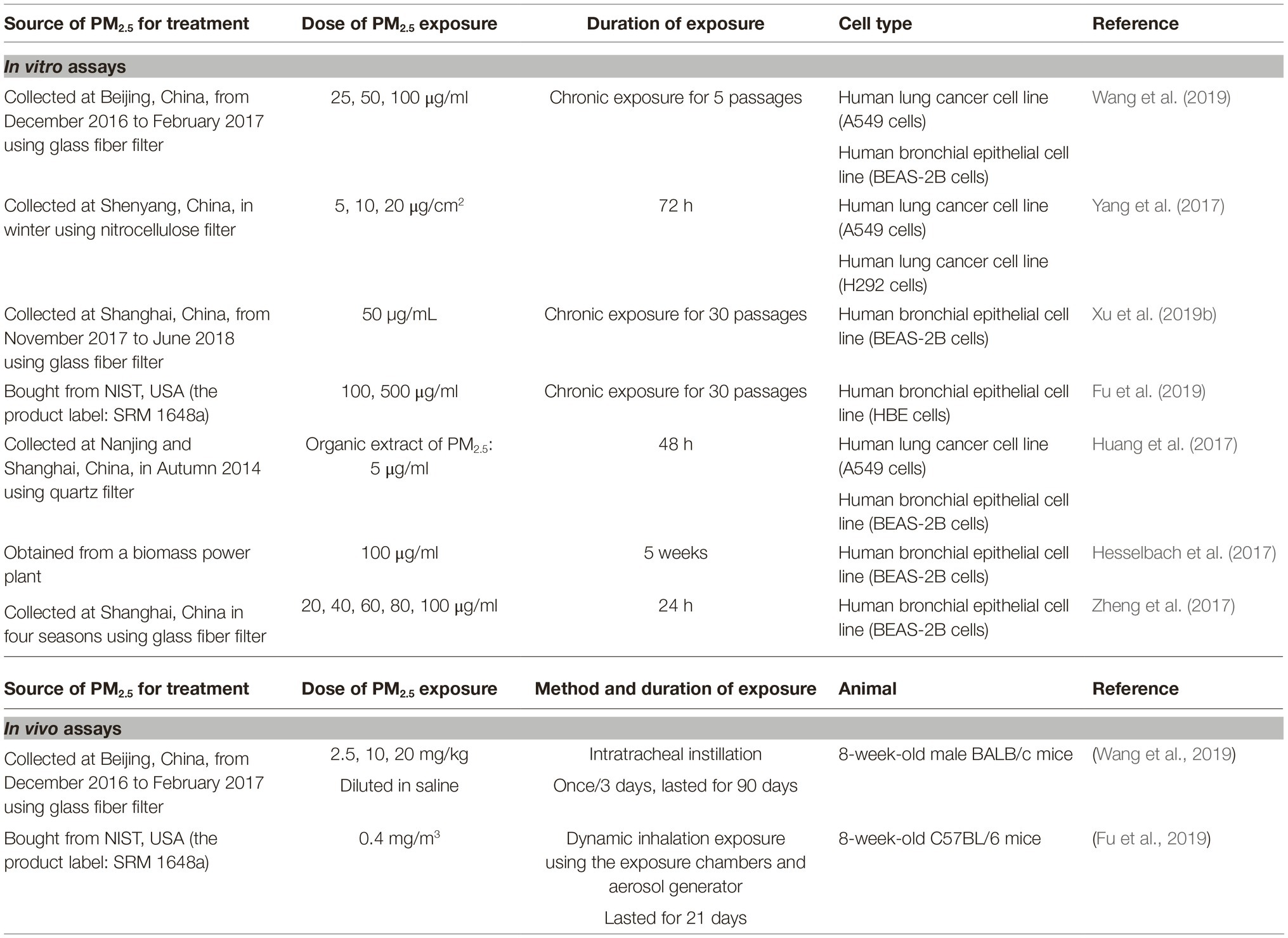
Table 3. Summary table about exposure assays of some studies reviewed in section “Other Molecular Events Underlying PM2.5-Induced Epithelial-Mesenchymal Transition.”
Participation of Novel Signaling and Molecules in PM2.5-Induced Epithelial-Mesenchymal Transition
PM2.5 can also activate EMT via diverse signaling pathways, including HMGB1-receptor for advanced glycation end-products (RAGE), Shh, Wnt/β-catenin, and Notch pathway together with SMAD1 signaling, in which the role of ROS has not been elucidated. HMGB1 and its cognate receptor RAGE were both induced in human bronchial epithelial cells and in the murine airway in response to PM2.5 exposure. HMGB1 binding further activated TGF-β1 and PDGF release, providing strong evidence that HMGB1 is a mediator of PM2.5-induced EMT (Zou et al., 2018). Increased Shh expression activated downstream Shh/Gli1 signal transduction and further enhanced Snail expression as well as cell migration after low dose PM2.5 exposure to human bronchial smooth muscle cells (Ye et al., 2018). Moreover, the enhanced expression of Notch1 together with downstream TFs Hes1, Snail, Slug and biomarkers of EMT was detected in lungs of mice after intratracheal instillation of PM2.5 with varied concentrations. The induction of cancer stem cells markers including OCT4 and SOX2 in A549 neoplastic pulmonary epithelial cells following the activation of PM2.5/Notch1 pathway further validated the pro-EMT property of PM2.5 (Wang et al., 2019). Furthermore, unlike the SMADs-dependent EMT mechanisms stated above, SMAD1 activity could also be indirectly induced by repressing expression of SMAD6/7 after PM2.5 treatment on neoplastic pulmonary epithelial cells, resulting in the inhibition of SMAD1 degradation and induction of the spindle-like cellular morphology with cadherin switch (Yang et al., 2017). Interestingly, rather than directly responding to PM2.5 exposure, Wnt/β-catenin signaling promoted the proliferation of human alveolar cancer cells and xenograft tumor growth via PM2.5-induced Wnt3a-enriched exosomes (Xu et al., 2018). Our recent research was focused on exosomal miRNAs of EMT-like pulmonary epithelia after PM2.5 exposure. It predicted the abnormal regulation of multiple cancer-related pathways by exosomal miRNAs in a systematic perspective, further emphasizing the significance of diverse interactions between exosomes and signaling pathways in the process of PM2.5-induced EMT (Xu et al., 2019b). ATP citrate lyase (ACLY) linking glycolytic and lipidic metabolism has been considered as a novel anticancer target, and recently, it has also been found responsible for PM2.5-induced EMT (Granchi, 2018). Increase of intracellular ACLY expression and citrate acid levels synchronized with EMT phenotype transformation of human bronchial epithelial cells after long-term PM2.5 exposure, and the knock down of ACLY significantly abrogated the in vivo metastasis ability of the cells (Fu et al., 2019).
Polyaromatic Hydrocarbons Might Function in PM2.5-Induced Epithelial-Mesenchymal Transition Through Aryl Hydrocarbon Receptor
Showing high affinity for a variety of PAHs, ligand-bound aryl hydrocarbon receptor (AHR) is able to interact with Ah receptor nuclear translocator protein (ARNT) and further mediate the toxicities of PAHs by stimulating the expression of target genes such as CYP1A1, which oxidizes PAHs and generally enhances their toxicities (Safe et al., 2013; Noakes, 2015). The AHR-ARNT complex can also promote cell proliferation by binding to the Slug promoter, activating ERK in the absence of external GFs, and enhancing FGF, PDGFA, and PCNA production (Shimba et al., 2002; Diry et al., 2006; Randi et al., 2008; Wang et al., 2009; Safe et al., 2013; John et al., 2014). Because PAHs account for a large portion of the organic constituents in PM2.5, their contribution to PM2.5-induced EMT might be nonnegligible. The significant cyp1a1 upregulation following the exposure of HBE cells to diesel exhaust particles (DEPs) or the exposure of BEAS-2B and A549 cells to PM2.5 proved the ability of PM to activate AHR signaling (Gualtieri et al., 2011, 2012; Rynning et al., 2018). Likewise, inhibiting AHR or CYP1A1 in BEAS-2B and A549 epithelial cells before PM2.5 exposure could effectively rescue PM2.5-induced E-cadherin downregulation and N-cadherin upregulation. In this study, lncRNA MALAT1 showed synchronized expression tendency with N-cadherin, indicating that it functions as an effector regulating EMT at downstream of PM2.5/AHR signaling, in addition to its role in NF-κB pathway reviewed above (Huang et al., 2017). These studies indicated that PM2.5/PAHs/AHR/CYP1A1 signaling significantly participates in EMT initiation, although more exploration is urgently needed.
PM2.5 Induces Pro-Epithelial-Mesenchymal Transition Epigenetic Regulations
Epigenetic regulations through lncRNAs and miRNAs underlying PM2.5-induced EMT have been partly reviewed above. Besides that, miR-16-1-3p directly targeting Twist mRNA could be inhibited by PM2.5 exposure, participating in PM2.5 exposure-induced EMT of hepatocellular carcinoma cells (Zhang and Li, 2019). Moreover, PM2.5 exposure also participates in initiation of EMT of bronchial epithelial cells by altering DNA methylation (Li et al., 2017). After 5 weeks of PM2.5 exposure, hypomethylated CpG islands (CpGs) in the genome of bronchial epithelial cells were significantly enriched in genes associated with GTPase activity, extracellular matrix organization, and GF stimuli, whereas hypermethylated CpGs were clustered in genes responsible for cell adhesion and ion transportation (Hesselbach et al., 2017). Another set of DNA methylation analyses on bronchial epithelial cells after PM2.5 exposure for 24 h showed comparable results, as Gene Ontology (GO) terms related with regulation on actin filament and adherin junction were significantly enriched in hypomethylated gene list (Shi et al., 2019). Because these annotated genes play dominant roles in EMT, these in silico analyses strongly indicated an association between PM2.5 exposure and EMT from a systematic perspective.
Calcium Might Act as an Initiator of Epithelial-Mesenchymal Transition in PM2.5?
Calcium signaling is highly correlated with multiple malignant cancers. Alterations in the expression of Ca2+ channels or transporters have been identified in tumor cells, indicating that ectopic Ca2+ signal transduction might play a role in EMT (Cui et al., 2017). For example, the activation of transient receptor potential cation channel 7 (TRPM7) and Ca2+ influx through TRPM7 indicates the initiation of EMT. Furthermore, EGF-induced TRPM7-Ca2+-dependent STAT3 activation significantly upregulated Twist, Snail, and EMT marker expression in breast cancer cells (Davis et al., 2014). Ca2+ is also able to elicit the expression of the ATP-binding cassette subfamily C member 3 (ABCC3), a promoter of Twist and EMT in breast cancer cells (Stewart et al., 2015). As Ca2+ has been well characterized as one of the most plentiful cations in PM2.5, and PM2.5 is able to penetrate into the bloodstream or be taken by cells, PM2.5 exposure might result in increased Ca2+ concentration in microenvironment around the epithelia or cytoplasm, which could further regulate the activity of pro-EMT Ca2+ signaling (Huang et al., 2018; Qin et al., 2018; Xie and Zhao, 2018). In our recent study, pulmonary epithelial cells showed EMT-like phenotype after long-term low dose PM2.5 exposure, and a subset of the intracellular differentially expressed genes was significantly related to the cellular response to calcium ion (Xu et al., 2019b). However, although limited in vitro studies have reported that PM2.5 exposure could raise the intracellular Ca2+ concentration of BEAS-2B cells and another non-epithelial cell line, the biological correlation of this event with EMT was yet to be clearly figured out (Zheng et al., 2017; Zhao et al., 2019).
Conclusions
Accumulating evidence has shown that PM2.5 can exert its toxic effect on somatic cells resulting in disorders including carcinogenesis and fibrosis. As EMT endows the epithelia with the characteristics of mesenchymal cells, it has been recognized as one of the major pathogenic mechanisms that promote malignant tumor metastasis and fibrogenesis. We and other researchers have originally identified PM2.5 as an initiator of EMT. On the basis of studies investigating PM2.5 toxicology, in this review, we describe that PM2.5 might induce EMT by intermediary ROS, which induce the secretion of TGF-β, IL-6, IL-8, and TNF-α, as well as the activation of the SMADs, NF-κB, JAK/STAT3, ERK, Akt, and Rho GTPase-dependent cascades. However, only the role of PM2.5-derived ROS in EMT induction mediated by TGF-β/SMAD2/3 pathway has been relatively clearly uncovered, whereas other mechanisms are revealed by limited research. Because PM2.5-induced inflammation and apoptosis largely share collective key messengers with putative EMT molecular processes, the underlying subtle mechanisms that regulate these cellular events remain to be elucidated. In addition, preliminary evidence has shown a correlation between PM2.5-ROS-induced autophagy and EMT, although the interactions at the molecular level remain to be explored. Furthermore, PM2.5-induced EMT is also dependent upon HMGB1-RAGE, Shh, Wnt/β-catenin, Notch, SMAD1 signaling, and ACLY enzyme, in which the involvement of ROS remains unclear. It is worth mentioning that epigenetic regulations involving miRNAs, lncRNAs, and DNA methylation might act as transcriptional or posttranscriptional regulators in PM2.5-induced EMT. PAHs and Ca2+, the primary constituents of PM2.5, might specifically induce EMT by AHR and Ca2+ signaling, respectively Figure 2.

Figure 2. Potential regulatory routes of PM2.5 in EMT. PM2.5 might function as an EMT initiator by activating TGF-β/SMAD2/3, NF-κB, ERK, Akt, and mediating cytoskeleton rearrangement via ROS. In addition to epigenetic mechanisms such as lncRNAs, miRNAs, and DNA methylation regulation, HMGB1-RAGE, Shh, SMAD1, Wnt3a/β-catenin, and Notch pathways have also been originally validated as effector signaling of PM2.5 in the process of EMT. ACLY might act as a key metabolic modulator in PM2.5-induced EMT. Autophagy following PM2.5-ROS can also promote EMT. Specific components within PM2.5 might be EMT promoters, as PAHs can initiate EMT via AHR/CYP1A1 signaling, and Ca2+ ions might function by activating Ca2+ signaling. EMT, epithelial-mesenchymal transition; TGF-β, transforming growth factor β; ERK, extracellular signal-regulated protein kinase; ROS, reactive oxygen species; RAGE, receptor for advanced glycation end-products; Shh, sonic hedgehog; ACLY, ATP citrate lyase; PAHs, polyaromatic hydrocarbons; AHR, aryl hydrocarbon receptor.
However, our present understanding of the toxicology of PM2.5 is still highly limited, as studies on PM2.5 toxicology are confined to a series of factors that might result in ineluctable systematic errors. It is clear that different sampling sites inevitably bring about variance of PM2.5 composition, as climate characteristics and social, economic, and cultural features are extremely diverse around the world. This natural diversity might make the results of different studies incomparable. Nevertheless, detailed studies investigating the toxicology of the exact components of PM2.5 could be difficult to perform. Second, although sampling protocols have been preliminarily standardized, inconsistencies between the actual operations of researchers uncontrollably exist, which might lead to a reduction in internal authenticity. Besides, the methods of PM2.5 treatment, including the selected vehicle solvent and the optimal concentrations to imitate the actual exposure state of humans, also significantly affect the physiology and pathological responses of cultured cells and murine models. According to existing studies, NAC is the widely used antioxidant against PM2.5 exposure, which shows strong efficacy of ROS elimination in vitro. However, the opposite effect of NAC on lung cancer cells, that is, growth promotion effect by reducing ROS, has been observed (Sayin et al., 2014). Besides, the failure of PANTHER combined therapy consisting of prednisone, azathioprine, and NAC designed for idiopathic pulmonary fibrosis also indicates that precise drug prevention is still far from mature (Idiopathic Pulmonary Fibrosis Clinical Research Network et al., 2012). Therefore, additional in-depth research and consensus are urgently needed. Owing to the relatively limited understanding of the underlying mechanisms of PM2.5-induced EMT, we comprehensively reviewed potential molecular events participating in PM2.5-induced EMT in various epithelial cell types that might be targeted by PM2.5 exposure. As tissue/cell type-specific responses to PM2.5 inevitably exist, the definitive role of PM2.5 in EMT should be further explored and confirmed.
Author Contributions
ZX was responsible for literature searching, information aggregation, and manuscript preparation. WD participated in the review and language revision of this manuscript. XD contributed to the concept of this review and participated in the manuscript revision. XD and WD obtained the funding and financially supported the publication of this review. All authors have read and approved the final version of this manuscript.
Funding
This study was financially supported by the National Natural Science Foundation of China (grant nos. 21777099, XD; 91643206 and U1432245, WD), Shanghai Jiao Tong University “New Young Teachers Startup Plan” (grant no. 15x100040063, XD), Shanghai College Student Innovation and Entrepreneurship Training Project (grant no. IAP11151, ZX), and the CAS/SAFEA International Partnership Program for Creative Research Teams.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
EMT, Epithelial mesenchymal transition; PM2.5, Fine particulate matter with an aerodynamic diameter less than 2.5 μm; ROS, Reactive oxygen species; PAHs, Polyaromatic hydrocarbons; ABC, Atmospheric brown cloud; TSP, Total suspended particulates; PM10, Inhalable particles with an aerodynamic diameter less than 10 μm; PM0.1, Ultrafine particles; α-SMA, α-Smooth muscle actin; TFs, Transcription factors; LEF-1, Lymphoid enhancer-binding factor-1; HMG, High-mobility group protein family; TGF-β, Transforming growth factor β; COL1, Type I collagen; TβRII, The type II TGF-β receptor; TβRI, The type I TGF-β receptor; R-SMADs, Receptor-activated SMAD proteins; BMPR, Bone morphogenetic protein receptor; I-SMADs, Inhibitory SMADs; Wnt, Wingless/Integrated; LRP5/6, LDL receptor-related protein-5/6; Dsh, Disheveled; GSK-3β, Glycogen synthase kinase-3β; RTKs, Receptor tyrosine kinases; GFs, Growth factors; EGF, Epidermal growth factor; FGF, Fibroblast growth factor; PDGF, Platelet-derived growth factor; IGF, Insulin-like growth factor; MEK, MAPK/ERK kinase; ERK, Extracellular signal-regulated protein kinases; PI3K, Phosphoinositide 3-kinase; PKB/Akt, Protein kinase B; JAK, The Janus kinase; STAT3, Signal transducers and activators of transcription-3; IL-6, Interleukin-6; IL-8, Interleukin-8; Shh, The sonic hedgehog; PTCH, Protein patch homolog; SMO, Smoothend; Gli1/2/3, Glima-associated oncogene-1/2/3; ADAM, A disintegrin and metalloproteinase; NICD, Notch intracellular domain; HES, Hairy and enhancer of split family; HEY, The HES related with YRPW motif family; FAK, Focal adhesion kinase; ROCK, Rho-associated protein kinase; NOX, NADPH oxidase; LAP, Latency-associated peptide; EndMT, Endothelial-mesenchymal transition; NAC, N-acetyl cysteine; MMP, Matrix metalloproteinase; CYP1A1/B1, Cytochrome P450, family 1, member A1/B1; LncRNA, Long non-coding RNA; CSE, Cigarette smoke extract; MiRNA, Micro-RNA; PTP1B, Protein tyrosine phosphatase-1B; DEP1, Density-enhanced phosphatase-1; MAPK, Mitogen-activated protein kinase; PCNA, Proliferating cell nuclear antigen; SOD, Superoxide dismutase; F-actin, Fibrous actin; G-actin, Globular actin; PINK1, PTEN-induced kinase 1; RAGE, Receptor for advanced glycation end-products; ACLY, ATP citrate lyase; AHR, Aryl hydrocarbon receptor; ARNT, Ah receptor nuclear translocator protein; DEPs, Diesel exhaust particles; CpGs, CpG islands; GO, Gene Ontology; TRPM7, Transient receptor potential cation channel 7; ABCC3, ATP-binding cassette subfamily C member 3.
References
Ahn, B. N., Karadeniz, F., Kong, C. S., Nam, K. H., Jang, M. S., Seo, Y., et al. (2016). Dioxinodehydroeckol enhances the differentiation of osteoblasts by regulating the expression of Phospho-Smad1/5/8. Mar. Drugs 14:E168. doi: 10.3390/md14090168
An, J., Zhou, Q., Wu, M., Wang, L., Zhong, Y., Feng, J., et al. (2018). Interactions between oxidative stress, autophagy and apoptosis in A549 cells treated with aged black carbon. Toxicol. In Vitro 54, 67–74. doi: 10.1016/j.tiv.2018.09.008
Bardaweel, S. K., Gul, M., Alzweiri, M., Ishaqat, A., HA, A. L., and Bashatwah, R. M. (2018). Reactive oxygen species: the dual role in physiological and pathological conditions of the human body. Eurasian J. Med. 50, 193–201. doi: 10.5152/eurasianjmed.2018.17397
Batlle, E., Sancho, E., Franci, C., Dominguez, D., Monfar, M., Baulida, J., et al. (2000). The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2, 84–89. doi: 10.1038/35000034
Belis, C. A., Karagulian, F., Larsen, B. R., and Hopke, P. K. (2013). Critical review and meta-analysis of ambient particulate matter source apportionment using receptor models in Europe. Atmos. Environ. 69, 94–108. doi: 10.1016/j.atmosenv.2012.11.009
Bourgeois, B., and Owens, J. W. (2014). The influence of hurricanes Katrina and Rita on the inflammatory cytokine response and protein expression in A549 cells exposed to PM2.5 collected in the Baton Rouge-Port Allen industrial corridor of southeastern Louisiana in 2005. Toxicol. Mech. Methods 24, 220–242. doi: 10.3109/15376516.2014.881945
Burger, G. A., Danen, E. H. J., and Beltman, J. B. (2017). Deciphering epithelial-mesenchymal transition regulatory networks in cancer through computational approaches. Front. Oncol. 7:162. doi: 10.3389/fonc.2017.00162
Catalano, M., D’Alessandro, G., Lepore, F., Corazzari, M., Caldarola, S., Valacca, C., et al. (2015). Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol. Oncol. 9, 1612–1625. doi: 10.1016/j.molonc.2015.04.016
Cavallaro, U., and Christofori, G. (2001). Cell adhesion in tumor invasion and metastasis: loss of the glue is not enough. Biochim. Biophys. Acta 1552, 39–45. doi: 10.1016/s0304-419x(01)00038-5
Cevenini, A., Orru, S., Mancini, A., Alfieri, A., Buono, P., and Imperlini, E. (2018). Molecular signatures of the insulin-like growth factor 1-mediated epithelial-mesenchymal transition in breast, lung and gastric cancers. Int. J. Mol. Sci. 19:2411. doi: 10.3390/ijms19082411
Chaffer, C. L., San Juan, B. P., Lim, E., and Weinberg, R. A. (2016). EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 35, 645–654. doi: 10.1007/s10555-016-9648-7
Chao, W., Deng, J. S., Li, P. Y., Liang, Y. C., and Huang, G. J. (2017). 3,4-Dihydroxybenzalactone suppresses human non-small cell lung carcinoma cells metastasis via suppression of epithelial to mesenchymal transition, ROS-mediated PI3K/AKT/MAPK/MMP and NFkappaB signaling pathways. Molecules 22:537. doi: 10.3390/molecules22040537
Chatterjee, S., and Sil, P. C. (2019). Targeting the crosstalks of Wnt pathway with hedgehog and notch for cancer therapy. Pharmacol. Res. 142, 251–261. doi: 10.1016/j.phrs.2019.02.027
Chen, W., Gao, Q., Han, S., Pan, F., and Fan, W. (2015). The CCL2/CCR2 axis enhances IL-6-induced epithelial-mesenchymal transition by cooperatively activating STAT3-twist signaling. Tumour Biol. 36, 973–981. doi: 10.1007/s13277-014-2717-z
Cheng, I., Tseng, C., Wu, J., Yang, J., Conroy, S. M., Shariff-Marco, S., et al. (2019). Association between ambient air pollution and breast cancer risk: the multiethnic cohort study. Int. J. Cancer 1–13. doi: 10.1002/ijc.32308
Chi, Y., Huang, Q., Lin, Y., Ye, G., Zhu, H., and Dong, S. (2018). Epithelial-mesenchymal transition effect of fine particulate matter from the Yangtze River Delta region in China on human bronchial epithelial cells. J. Environ. Sci. 66, 155–164. doi: 10.1016/j.jes.2017.05.002
Cho, C. C., Hsieh, W. Y., Tsai, C. H., Chen, C. Y., Chang, H. F., and Lin, C. S. (2018). In vitro and in vivo experimental studies of PM2.5 on disease progression. Int. J. Environ. Res. Public Health 15:E1380. doi: 10.3390/ijerph15071380
Choi, E. K., Kim, J. G., Kim, H. J., Cho, J. Y., Jeong, H., Park, Y., et al. (2017). Regulation of RhoA GTPase and novel target proteins for ROCK. Small GTPases 3, 1–8. doi: 10.1080/21541248.2017.1364831
Choi, S. S., Omenetti, A., Witek, R. P., Moylan, C. A., Syn, W. K., Jung, Y., et al. (2009). Hedgehog pathway activation and epithelial-to-mesenchymal transitions during myofibroblastic transformation of rat hepatic cells in culture and cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 297, G1093–G1106. doi: 10.1152/ajpgi.00292.2009
Cichon, M. A., and Radisky, D. C. (2014). ROS-induced epithelial-mesenchymal transition in mammary epithelial cells is mediated by NF-kB-dependent activation of snail. Oncotarget 5, 2827–2838. doi: 10.18632/oncotarget.1940
Colomiere, M., Ward, A. C., Riley, C., Trenerry, M. K., Cameron-Smith, D., Findlay, J., et al. (2009). Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial-mesenchymal transition in ovarian carcinomas. Br. J. Cancer 100, 134–144. doi: 10.1038/sj.bjc.6604794
Cui, Y. H., Hu, Z. X., Gao, Z. X., Song, X. L., Feng, Q. Y., Yang, G., et al. (2018). Airborne particulate matter impairs corneal epithelial cells migration via disturbing FAK/RhoA signaling pathway and cytoskeleton organization. Nanotoxicology 12, 312–324. doi: 10.1080/17435390.2018.1440651
Cui, C., Merritt, R., Fu, L., and Pan, Z. (2017). Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 7, 3–17. doi: 10.1016/j.apsb.2016.11.001
Danielsen, P. H., Loft, S., Kocbach, A., Schwarze, P. E., and Moller, P. (2009). Oxidative damage to DNA and repair induced by Norwegian wood smoke particles in human A549 and THP-1 cell lines. Mutat. Res. 674, 116–122. doi: 10.1016/j.mrgentox.2008.10.014
Dave, N., Guaita-Esteruelas, S., Gutarra, S., Frias, A., Beltran, M., Peiro, S., et al. (2011). Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J. Biol. Chem. 286, 12024–12032. doi: 10.1074/jbc.M110.168625
Davis, F. M., Azimi, I., Faville, R. A., Peters, A. A., Jalink, K., Putney, J. W. Jr., et al. (2014). Induction of epithelial-mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene 33, 2307–2316. doi: 10.1038/onc.2013.187
Deng, X., Feng, N., Zheng, M., Ye, X., Lin, H., Yu, X., et al. (2017). PM2.5 exposure-induced autophagy is mediated by lncRNA loc146880 which also promotes the migration and invasion of lung cancer cells. Biochim. Biophys. Acta Gen. Subj. 1861, 112–125. doi: 10.1016/j.bbagen.2016.11.009
Deng, X., Zhang, F., Wang, L., Rui, W., Long, F., Zhao, Y., et al. (2014). Airborne fine particulate matter induces multiple cell death pathways in human lung epithelial cells. Apoptosis 19, 1099–1112. doi: 10.1007/s10495-014-0980-5
Ding, Q., Xia, W., Liu, J. C., Yang, J. Y., Lee, D. F., Xia, J., et al. (2005). Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol. Cell 19, 159–170. doi: 10.1016/j.molcel.2005.06.009
Diry, M., Tomkiewicz, C., Koehle, C., Coumoul, X., Bock, K. W., Barouki, R., et al. (2006). Activation of the dioxin/aryl hydrocarbon receptor (AhR) modulates cell plasticity through a JNK-dependent mechanism. Oncogene 25, 5570–5574. doi: 10.1038/sj.onc.1209553
Doble, B. W., and Woodgett, J. R. (2007). Role of glycogen synthase kinase-3 in cell fate and epithelial-mesenchymal transitions. Cells Tissues Organs 185, 73–84. doi: 10.1159/000101306
Dong, R., Wang, Q., He, X. L., Chu, Y. K., Lu, J. G., and Ma, Q. J. (2007). Role of nuclear factor kappa B and reactive oxygen species in the tumor necrosis factor-alpha-induced epithelial-mesenchymal transition of MCF-7 cells. Braz. J. Med. Biol. Res. 40, 1071–1078. doi: 10.1590/S0100-879X2007000800007
Dornhof, R., Maschowski, C., Osipova, A., Giere, R., Seidl, M., Merfort, I., et al. (2017). Stress fibers, autophagy and necrosis by persistent exposure to PM2.5 from biomass combustion. PLoS One 12:e0180291. doi: 10.1371/journal.pone.0180291
Dysart, M. M., Galvis, B. R., Russell, A. G., and Barker, T. H. (2014). Environmental particulate (PM2.5) augments stiffness-induced alveolar epithelial cell mechanoactivation of transforming growth factor beta. PLoS One 9:e106821. doi: 10.1371/journal.pone.0106821
Fabregat, I., and Caballero-Diaz, D. (2018). Transforming growth factor-beta-induced cell plasticity in liver fibrosis and hepatocarcinogenesis. Front. Oncol. 8:357. doi: 10.3389/fonc.2018.00357
Falcon-Rodriguez, C. I., Osornio-Vargas, A. R., Sada-Ovalle, I., and Segura-Medina, P. (2016). Aeroparticles, composition, and lung diseases. Front. Immunol. 7:3. doi: 10.3389/fimmu.2016.00003
Feng, S., Gao, D., Liao, F., Zhou, F., and Wang, X. (2016). The health effects of ambient PM2.5 and potential mechanisms. Ecotoxicol. Environ. Saf. 128, 67–74. doi: 10.1016/j.ecoenv.2016.01.030
Fernandez, I. E., and Eickelberg, O. (2012). The impact of TGF-beta on lung fibrosis: from targeting to biomarkers. Proc. Am. Thorac. Soc. 9, 111–116. doi: 10.1513/pats.201203-023AW
Fernando, R. I., Castillo, M. D., Litzinger, M., Hamilton, D. H., and Palena, C. (2011). IL-8 signaling plays a critical role in the epithelial-mesenchymal transition of human carcinoma cells. Cancer Res. 71, 5296–5306. doi: 10.1158/0008-5472.CAN-11-0156
Flemban, A., and Qualtrough, D. (2015). The potential role of hedgehog signaling in the luminal/basal phenotype of breast epithelia and in breast cancer invasion and metastasis. Cancer 7, 1863–1884. doi: 10.3390/cancers7030866
Fu, X. T., Dai, Z., Song, K., Zhang, Z. J., Zhou, Z. J., Zhou, S. L., et al. (2015). Macrophage-secreted IL-8 induces epithelial-mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/snail pathway. Int. J. Oncol. 46, 587–596. doi: 10.3892/ijo.2014.2761
Fu, Y., Lu, R., Cui, J., Sun, H., Yang, H., Meng, Q., et al. (2019). Inhibition of ATP citrate lyase (ACLY) protects airway epithelia from PM2.5-induced epithelial-mesenchymal transition. Ecotoxicol. Environ. Saf. 167, 309–316. doi: 10.1016/j.ecoenv.2018.10.033
Gao, X., Sun, J., Huang, C., Hu, X., Jiang, N., and Lu, C. (2017). RNAi-mediated silencing of NOX4 inhibited the invasion of gastric cancer cells through JAK2/STAT3 signaling. Am. J. Transl. Res. 9, 4440–4449.
Ghahhari, N. M., and Babashah, S. (2015). Interplay between microRNAs and WNT/beta-catenin signalling pathway regulates epithelial-mesenchymal transition in cancer. Eur. J. Cancer 51, 1638–1649. doi: 10.1016/j.ejca.2015.04.021
Ghio, A. J., Carraway, M. S., and Madden, M. C. (2012). Composition of air pollution particles and oxidative stress in cells, tissues, and living systems. J. Toxicol. Environ. Health B Crit. Rev. 15, 1–21. doi: 10.1080/10937404.2012.632359
Gilles, C., Polette, M., Mestdagt, M., Nawrocki-Raby, B., Ruggeri, P., Birembaut, P., et al. (2003). Transactivation of vimentin by beta-catenin in human breast cancer cells. Cancer Res. 63, 2658–2664.
Gonzalez, D. M., and Medici, D. (2014). Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 7:re8. doi: 10.1126/scisignal.2005189
Gonzalez-Nunez, M., Munoz-Felix, J. M., and Lopez-Novoa, J. M. (2013). The ALK-1/Smad1 pathway in cardiovascular physiopathology. A new target for therapy? Biochim. Biophys. Acta 1832, 1492–1510. doi: 10.1016/j.bbadis.2013.05.016
Granchi, C. (2018). ATP citrate lyase (ACLY) inhibitors: an anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur. J. Med. Chem. 157, 1276–1291. doi: 10.1016/j.ejmech.2018.09.001
Gu, L. Z., Sun, H., and Chen, J. H. (2017). Histone deacetylases 3 deletion restrains PM2.5-induced mice lung injury by regulating NF-kappaB and TGF-beta/Smad2/3 signaling pathways. Biomed. Pharmacother. 85, 756–762. doi: 10.1016/j.biopha.2016.11.094
Guaita, S., Puig, I., Franci, C., Garrido, M., Dominguez, D., Batlle, E., et al. (2002). Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J. Biol. Chem. 277, 39209–39216. doi: 10.1074/jbc.M206400200
Gualtieri, M., Longhin, E., Mattioli, M., Mantecca, P., Tinaglia, V., Mangano, E., et al. (2012). Gene expression profiling of A549 cells exposed to Milan PM2.5. Toxicol. Lett. 209, 136–145. doi: 10.1016/j.toxlet.2011.11.015
Gualtieri, M., Ovrevik, J., Mollerup, S., Asare, N., Longhin, E., Dahlman, H. J., et al. (2011). Airborne urban particles (Milan winter-PM2.5) cause mitotic arrest and cell death: effects on DNA, mitochondria, AhR binding and spindle organization. Mutat. Res. 713, 18–31. doi: 10.1016/j.mrfmmm.2011.05.011
Gugnoni, M., Sancisi, V., Manzotti, G., Gandolfi, G., and Ciarrocchi, A. (2016). Autophagy and epithelial-mesenchymal transition: an intricate interplay in cancer. Cell Death Dis. 7:e2520. doi: 10.1038/cddis.2016.415
Guo, Y., Xiao, L., Sun, L., and Liu, F. (2012). Wnt/beta-catenin signaling: a promising new target for fibrosis diseases. Physiol. Res. 61, 337–346.
Gupta, S. C., Hevia, D., Patchva, S., Park, B., Koh, W., and Aggarwal, B. B. (2012). Upsides and downsides of reactive oxygen species for cancer: the roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid. Redox Signal. 16, 1295–1322. doi: 10.1089/ars.2011.4414
Hesselbach, K., Kim, G. J., Flemming, S., Haupl, T., Bonin, M., Dornhof, R., et al. (2017). Disease relevant modifications of the methylome and transcriptome by particulate matter (PM2.5) from biomass combustion. Epigenetics 12, 779–792. doi: 10.1080/15592294.2017.1356555
Heuberger, J., and Birchmeier, W. (2010). Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2:a002915. doi: 10.1101/cshperspect.a002915
Hu, T., Ramachandrarao, S. P., Siva, S., Valancius, C., Zhu, Y., Mahadev, K., et al. (2005). Reactive oxygen species production via NADPH oxidase mediates TGF-beta-induced cytoskeletal alterations in endothelial cells. Am. J. Physiol. Ren. Physiol. 289, F816–F825. doi: 10.1152/ajprenal.00024.2005
Huang, F., and Chen, Y. G. (2012). Regulation of TGF-beta receptor activity. Cell Biosci. 2:9. doi: 10.1186/2045-3701-2-9
Huang, Q., Chi, Y., Deng, J., Liu, Y., Lu, Y., Chen, J., et al. (2017). Fine particulate matter 2.5 exerted its toxicological effect by regulating a new layer, long non-coding RNA. Sci. Rep. 7:9392. doi: 10.1038/s41598-017-09818-6
Huang, M., and Xin, W. (2018). Matrine inhibiting pancreatic cells epithelial-mesenchymal transition and invasion through ROS/NF-kappaB/MMPs pathway. Life Sci. 192, 55–61. doi: 10.1016/j.lfs.2017.11.024
Huang, X., Zhang, J., Luo, B., Wang, L., Tang, G., Liu, Z., et al. (2018). Water-soluble ions in PM2.5 during spring haze and dust periods in Chengdu, China: variations, nitrate formation and potential source areas. Environ. Pollut. 243, 1740–1749. doi: 10.1016/j.envpol.2018.09.126
Hubbard, A. K., Timblin, C. R., Shukla, A., Rincon, M., and Mossman, B. T. (2002). Activation of NF-kappaB-dependent gene expression by silica in lungs of luciferase reporter mice. Am. J. Phys. Lung Cell. Mol. Phys. 282, L968–L975. doi: 10.1152/ajplung.00327.2001
International Agency for Research on Cancer (IARC) (2019). Agents classified by the IARC monographs. Available from: https://monographs.iarc.fr/agents-classified-by-the-iarc/
Idiopathic Pulmonary Fibrosis Clinical Research NetworkRaghu, G., Anstrom, K. J., King, T. E. Jr., Lasky, J. A., and Martinez, F. J. (2012). Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 366, 1968–1977. doi: 10.1056/NEJMoa1113354
Inumaru, J., Nagano, O., Takahashi, E., Ishimoto, T., Nakamura, S., Suzuki, Y., et al. (2009). Molecular mechanisms regulating dissociation of cell-cell junction of epithelial cells by oxidative stress. Genes Cells 14, 703–716. doi: 10.1111/j.1365-2443.2009.01303.x
Itoh, S., and ten Dijke, P. (2007). Negative regulation of TGF-beta receptor/Smad signal transduction. Curr. Opin. Cell Biol. 19, 176–184. doi: 10.1016/j.ceb.2007.02.015
Jaffer, O. A., Carter, A. B., Sanders, P. N., Dibbern, M. E., Winters, C. J., Murthy, S., et al. (2015). Mitochondrial-targeted antioxidant therapy decreases transforming growth factor-beta-mediated collagen production in a murine asthma model. Am. J. Respir. Cell Mol. Biol. 52, 106–115. doi: 10.1165/rcmb.2013-0519OC
Jansen, S., Gosens, R., Wieland, T., and Schmidt, M. (2018). Paving the rho in cancer metastasis: rho GTPases and beyond. Pharmacol. Ther. 183, 1–21. doi: 10.1016/j.pharmthera.2017.09.002
Jechlinger, M., Sommer, A., Moriggl, R., Seither, P., Kraut, N., Capodiecci, P., et al. (2006). Autocrine PDGFR signaling promotes mammary cancer metastasis. J. Clin. Invest. 116, 1561–1570. doi: 10.1172/JCI24652
Jeong, S. C., Cho, Y., Song, M. K., Lee, E., and Ryu, J. C. (2017). Epidermal growth factor receptor (EGFR)-MAPK-nuclear factor(NF)-kappaB-IL8: a possible mechanism of particulate matter(PM) 2.5-induced lung toxicity. Environ. Toxicol. 32, 1628–1636. doi: 10.1002/tox.22390
Jiang, J., Wang, K., Chen, Y., Chen, H., Nice, E. C., and Huang, C. (2017). Redox regulation in tumor cell epithelial-mesenchymal transition: molecular basis and therapeutic strategy. Signal Transduct. Target. Ther. 2:17036. doi: 10.1038/sigtrans.2017.36
Jin, X., Xue, B., Zhou, Q., Su, R., and Li, Z. (2018). Mitochondrial damage mediated by ROS incurs bronchial epithelial cell apoptosis upon ambient PM2.5 exposure. J. Toxicol. Sci. 43, 101–111. doi: 10.2131/jts.43.101
John, K., Lahoti, T. S., Wagner, K., Hughes, J. M., and Perdew, G. H. (2014). The ah receptor regulates growth factor expression in head and neck squamous cell carcinoma cell lines. Mol. Carcinog. 53, 765–776. doi: 10.1002/mc.22032
Julien, S., Puig, I., Caretti, E., Bonaventure, J., Nelles, L., van Roy, F., et al. (2007). Activation of NF-kappaB by Akt upregulates snail expression and induces epithelium mesenchyme transition. Oncogene 26, 7445–7456. doi: 10.1038/sj.onc.1210546
Kalluri, R. (2009). EMT: when epithelial cells decide to become mesenchymal-like cells. J. Clin. Invest. 119, 1417–1419. doi: 10.1172/JCI39675
Kalluri, R., and Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428. doi: 10.1172/JCI39104
Katsuno, Y., Lamouille, S., and Derynck, R. (2013). TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 25, 76–84. doi: 10.1097/CCO.0b013e32835b6371
Kim, H. B., Shim, J. Y., Park, B., and Lee, Y. J. (2018). Long-term exposure to air pollutants and cancer mortality: a meta-analysis of cohort studies. Int. J. Environ. Res. Public Health 15, 1–15. doi: 10.3390/ijerph15112608
Knaapen, A. M., Borm, P. J., Albrecht, C., and Schins, R. P. (2004). Inhaled particles and lung cancer. Part A: mechanisms. Int. J. Cancer 109, 799–809. doi: 10.1002/ijc.11708
Koganti, P., Levy-Cohen, G., and Blank, M. (2018). Smurfs in protein homeostasis, signaling, and cancer. Front. Oncol. 8:295. doi: 10.3389/fonc.2018.00295
Konczol, M., Ebeling, S., Goldenberg, E., Treude, F., Gminski, R., Giere, R., et al. (2011). Cytotoxicity and genotoxicity of size-fractionated iron oxide (magnetite) in A549 human lung epithelial cells: role of ROS, JNK, and NF-kappaB. Chem. Res. Toxicol. 24, 1460–1475. doi: 10.1021/tx200051s
Kourtidis, A., Lu, R., Pence, L. J., and Anastasiadis, P. Z. (2017). A central role for cadherin signaling in cancer. Exp. Cell Res. 358, 78–85. doi: 10.1016/j.yexcr.2017.04.006
Lam, A. P., and Gottardi, C. J. (2011). Beta-catenin signaling: a novel mediator of fibrosis and potential therapeutic target. Curr. Opin. Rheumatol. 23, 562–567. doi: 10.1097/BOR.0b013e32834b3309
Lambertini, E., Franceschetti, T., Torreggiani, E., Penolazzi, L., Pastore, A., Pelucchi, S., et al. (2010). SLUG: a new target of lymphoid enhancer factor-1 in human osteoblasts. BMC Mol. Biol. 11:13. doi: 10.1186/1471-2199-11-13
Lamouille, S., Xu, J., and Derynck, R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196. doi: 10.1038/nrm3758
Latella, G. (2018). Redox imbalance in intestinal fibrosis: beware of the TGFbeta-1, ROS, and Nrf2 connection. Dig. Dis. Sci. 63, 312–320. doi: 10.1007/s10620-017-4887-1
Leclercq, B., Kluza, J., Antherieu, S., Sotty, J., Alleman, L. Y., Perdrix, E., et al. (2018). Air pollution-derived PM2.5 impairs mitochondrial function in healthy and chronic obstructive pulmonary diseased human bronchial epithelial cells. Environ. Pollut. 243, 1434–1449. doi: 10.1016/j.envpol.2018.09.062
Lee, B. W. L., Ghode, P., and Ong, D. S. T. (2018). Redox regulation of cell state and fate. Redox Biol. doi: 10.1016/j.redox.2018.11.014
Lee, J., and Lim, K. T. (2011). Inhibitory effect of phytoglycoprotein (38 kDa) on expression of matrix metalloproteinase-9 in 12-O-tetradecanoylphorbol-13-acetate-treated HepG2cells. Naunyn Schmiedeberg's Arch. Pharmacol. 384, 185–196. doi: 10.1007/s00210-011-0663-5
Lei, H., and Kazlauskas, A. (2009). Growth factors outside of the platelet-derived growth factor (PDGF) family employ reactive oxygen species/Src family kinases to activate PDGF receptor alpha and thereby promote proliferation and survival of cells. J. Biol. Chem. 284, 6329–6336. doi: 10.1074/jbc.M808426200
Lemmon, M. A., and Schlessinger, J. (2010). Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134. doi: 10.1016/j.cell.2010.06.011
Leon-Buitimea, A., Rodriguez-Fragoso, L., Lauer, F. T., Bowles, H., Thompson, T. A., and Burchiel, S. W. (2012). Ethanol-induced oxidative stress is associated with EGF receptor phosphorylation in MCF-10A cells overexpressing CYP2E1. Toxicol. Lett. 209, 161–165. doi: 10.1016/j.toxlet.2011.12.009
Leonetti, A., Facchinetti, F., Minari, R., Cortellini, A., Rolfo, C. D., Giovannetti, E., et al. (2019). Notch pathway in small-cell lung cancer: from preclinical evidence to therapeutic challenges. Cell. Oncol. 42, 261–273. doi: 10.1007/s13402-019-00441-3
Li, X., Deng, W., Nail, C. D., Bailey, S. K., Kraus, M. H., Ruppert, J. M., et al. (2006). Snail induction is an early response to Gli1 that determines the efficiency of epithelial transformation. Oncogene 25, 609–621. doi: 10.1038/sj.onc.1209077
Li, J. H., Li, W. X., Bai, C. X., and Song, Y. L. (2017). Particulate matter-induced epigenetic changes and lung cancer. Clin. Respir. J. 11, 539–546. doi: 10.1111/crj.12389
Li, A., Xia, X., Yeh, J., Kua, H., Liu, H., Mishina, Y., et al. (2014). PDGF-AA promotes osteogenic differentiation and migration of mesenchymal stem cell by down-regulating PDGFRalpha and derepressing BMP-Smad1/5/8 signaling. PLoS One 9:e113785. doi: 10.1371/journal.pone.0113785
Li, J., Yang, B., Zhou, Q., Wu, Y., Shang, D., Guo, Y., et al. (2013). Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis 34, 1343–1351. doi: 10.1093/carcin/bgt063
Li, J., Zhou, Q., Yang, T., Li, Y., Zhang, Y., Wang, J., et al. (2018a). SGK1 inhibits PM2.5-induced apoptosis and oxidative stress in human lung alveolar epithelial A549cells. Biochem. Biophys. Res. Commun. 496, 1291–1295. doi: 10.1016/j.bbrc.2018.02.002
Li, R., Zhou, R., and Zhang, J. (2018b). Function of PM2.5 in the pathogenesis of lung cancer and chronic airway inflammatory diseases. Oncol. Lett. 15, 7506–7514. doi: 10.3892/ol.2018.8355
Lin, H., Zhang, X., Feng, N., Wang, R., Zhang, W., Deng, X., et al. (2018). LncRNA LCPAT1 mediates smoking/ particulate matter 2.5-induced cell autophagy and epithelial-mesenchymal transition in lung cancer cells via RCC2. Cell. Physiol. Biochem. 47, 1244–1258. doi: 10.1159/000490220
Liu, R. M., and Gaston Pravia, K. A. (2010). Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radic. Biol. Med. 48, 1–15. doi: 10.1016/j.freeradbiomed.2009.09.026
Liu, L., Wan, C., Zhang, W., Guan, L., Tian, G., Zhang, F., et al. (2018). MiR-146a regulates PM1 -induced inflammation via NF-kappaB signaling pathway in BEAS-2B cells. Environ. Toxicol. 33, 743–751. doi: 10.1002/tox.22561
Liu, B., Wu, S. D., Shen, L. J., Zhao, T. X., Wei, Y., Tang, X. L., et al. (2019). Spermatogenesis dysfunction induced by PM2.5 from automobile exhaust via the ROS-mediated MAPK signaling pathway. Ecotoxicol. Environ. Saf. 167, 161–168. doi: 10.1016/j.ecoenv.2018.09.118
Longhin, E., Capasso, L., Battaglia, C., Proverbio, M. C., Cosentino, C., Cifola, I., et al. (2016). Integrative transcriptomic and protein analysis of human bronchial BEAS-2B exposed to seasonal urban particulate matter. Environ. Pollut. 209, 87–98. doi: 10.1016/j.envpol.2015.11.013
Lu, W., and Kang, Y. (2019). Epithelial-mesenchymal plasticity in cancer progression and metastasis. Dev. Cell 49, 361–374. doi: 10.1016/j.devcel.2019.04.010
Luo, F., Wei, H., Guo, H., Li, Y., Feng, Y., Bian, Q., et al. (2019). LncRNA MALAT1, a lncRNA acting via the miR-204/ZEB1 pathway, mediates the EMT induced by organic extract of PM2.5 in lung bronchial epithelial cells. Am. J. Phys. Lung Cell. Mol. Phys. 317, L87–L98. doi: 10.1152/ajplung.00073.2019
Lv, Q., Wang, W., Xue, J., Hua, F., Mu, R., Lin, H., et al. (2012). DEDD interacts with PI3KC3 to activate autophagy and attenuate epithelial-mesenchymal transition in human breast cancer. Cancer Res. 72, 3238–3250. doi: 10.1158/0008-5472.CAN-11-3832
Marcucci, F., Ghezzi, P., and Rumio, C. (2017). The role of autophagy in the cross-talk between epithelial-mesenchymal transitioned tumor cells and cancer stem-like cells. Mol. Cancer 16:3. doi: 10.1186/s12943-016-0573-8
McCormack, N., Molloy, E. L., and O'Dea, S. (2013). Bone morphogenetic proteins enhance an epithelial-mesenchymal transition in normal airway epithelial cells during restitution of a disrupted epithelium. Respir. Res. 14:36. doi: 10.1186/1465-9921-14-36
McCubrey, J. A., Rakus, D., Gizak, A., Steelman, L. S., Abrams, S. L., Lertpiriyapong, K., et al. (2016). Effects of mutations in Wnt/beta-catenin, hedgehog, notch and PI3K pathways on GSK-3 activity-diverse effects on cell growth, metabolism and cancer. Biochim. Biophys. Acta 1863, 2942–2976. doi: 10.1016/j.bbamcr.2016.09.004
Meng, Q., Shi, S., Liang, C., Liang, D., Hua, J., Zhang, B., et al. (2018). Abrogation of glutathione peroxidase-1 drives EMT and chemoresistance in pancreatic cancer by activating ROS-mediated Akt/GSK3beta/snail signaling. Oncogene 37, 5843–5857. doi: 10.1038/s41388-018-0392-z
Meurette, O., and Mehlen, P. (2018). Notch signaling in the tumor microenvironment. Cancer Cell 34, 536–548. doi: 10.1016/j.ccell.2018.07.009
Min, C., Eddy, S. F., Sherr, D. H., and Sonenshein, G. E. (2008). NF-kappaB and epithelial to mesenchymal transition of cancer. J. Cell. Biochem. 104, 733–744. doi: 10.1002/jcb.21695
Miyazawa, K., Shinozaki, M., Hara, T., Furuya, T., and Miyazono, K. (2002). Two major Smad pathways in TGF-beta superfamily signalling. Genes Cells 7, 1191–1204. doi: 10.1046/j.1365-2443.2002.00599.x
Miyazono, K., Katsuno, Y., Koinuma, D., Ehata, S., and Morikawa, M. (2018). Intracellular and extracellular TGF-beta signaling in cancer: some recent topics. Front. Med. 12, 387–411. doi: 10.1007/s11684-018-0646-8
Montorfano, I., Becerra, A., Cerro, R., Echeverria, C., Saez, E., Morales, M. G., et al. (2014). Oxidative stress mediates the conversion of endothelial cells into myofibroblasts via a TGF-beta1 and TGF-beta2-dependent pathway. Lab. Investig. 94, 1068–1082. doi: 10.1038/labinvest.2014.100
Ngo, H. K., Lee, H. G., Piao, J. Y., Zhong, X., Lee, H. N., Han, H. J., et al. (2016). Helicobacter pylori induces snail expression through ROS-mediated activation of Erk and inactivation of GSK-3beta in human gastric cancer cells. Mol. Carcinog. 55, 2236–2246. doi: 10.1002/mc.22464
Niranjan, R., and Thakur, A. K. (2017). The toxicological mechanisms of environmental soot (black carbon) and carbon black: focus on oxidative stress and inflammatory pathways. Front. Immunol. 8:763. doi: 10.3389/fimmu.2017.00763
Noakes, R. (2015). The aryl hydrocarbon receptor: a review of its role in the physiology and pathology of the integument and its relationship to the tryptophan metabolism. Int. J. Tryptophan Res. 8, 7–18. doi: 10.4137/IJTR.S19985
Oloumi, A., McPhee, T., and Dedhar, S. (2004). Regulation of E-cadherin expression and beta-catenin/Tcf transcriptional activity by the integrin-linked kinase. Biochim. Biophys. Acta 1691, 1–15. doi: 10.1016/j.bbamcr.2003.12.002
Oshikawa, J., Urao, N., Kim, H. W., Kaplan, N., Razvi, M., McKinney, R., et al. (2010). Extracellular SOD-derived H2O2 promotes VEGF signaling in caveolae/lipid rafts and post-ischemic angiogenesis in mice. PLoS One 5:e10189. doi: 10.1371/journal.pone.0010189
Pain, M., Bermudez, O., Lacoste, P., Royer, P. J., Botturi, K., Tissot, A., et al. (2014). Tissue remodelling in chronic bronchial diseases: from the epithelial to mesenchymal phenotype. Eur. Respir. Rev. 23, 118–130. doi: 10.1183/09059180.00004413
Palleschi, S., Rossi, B., Armiento, G., Montereali, M. R., Nardi, E., Mazziotti Tagliani, S., et al. (2018). Toxicity of the readily leachable fraction of urban PM2.5 to human lung epithelial cells: role of soluble metals. Chemosphere 196, 35–44. doi: 10.1016/j.chemosphere.2017.12.147
Qi, Z., Song, Y., Ding, Q., Liao, X., Li, R., Liu, G., et al. (2019). Water soluble and insoluble components of PM2.5 and their functional cardiotoxicities on neonatal rat cardiomyocytes in vitro. Ecotoxicol. Environ. Saf. 168, 378–387. doi: 10.1016/j.ecoenv.2018.10.107
Qin, G., Xia, J., Zhang, Y., Guo, L., Chen, R., and Sang, N. (2018). Ambient fine particulate matter exposure induces reversible cardiac dysfunction and fibrosis in juvenile and older female mice. Part. Fibre Toxicol. 15:27. doi: 10.1186/s12989-018-0264-2
Qiu, Y. N., Wang, G. H., Zhou, F., Hao, J. J., Tian, L., Guan, L. F., et al. (2019). PM2.5 induces liver fibrosis via triggering ROS-mediated mitophagy. Ecotoxicol. Environ. Saf. 167, 178–187. doi: 10.1016/j.ecoenv.2018.08.050
Randi, A. S., Sanchez, M. S., Alvarez, L., Cardozo, J., Pontillo, C., and Kleiman de Pisarev, D. L. (2008). Hexachlorobenzene triggers AhR translocation to the nucleus, c-Src activation and EGFR transactivation in rat liver. Toxicol. Lett. 177, 116–122. doi: 10.1016/j.toxlet.2008.01.003
Rhyu, D. Y., Yang, Y., Ha, H., Lee, G. T., Song, J. S., Uh, S. T., et al. (2005). Role of reactive oxygen species in TGF-beta1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J. Am. Soc. Nephrol. 16, 667–675. doi: 10.1681/ASN.2004050425
Risom, L., Moller, P., and Loft, S. (2005). Oxidative stress-induced DNA damage by particulate air pollution. Mutat. Res. 592, 119–137. doi: 10.1016/j.mrfmmm.2005.06.012
Rynning, I., Neca, J., Vrbova, K., Libalova, H., Rossner, P. Jr., Holme, J. A., et al. (2018). In vitro transformation of human bronchial epithelial cells by diesel exhaust particles: gene expression profiling and early toxic responses. Toxicol. Sci. 166, 51–64. doi: 10.1093/toxsci/kfy183
Safe, S., Lee, S. O., and Jin, U. H. (2013). Role of the aryl hydrocarbon receptor in carcinogenesis and potential as a drug target. Toxicol. Sci. 135, 1–16. doi: 10.1093/toxsci/kft128
Saha, P., Johny, E., Dangi, A., Shinde, S., Brake, S., Eapen, M. S., et al. (2018). Impact of maternal air pollution exposure on children's lung health: an indian perspective. Toxics 6:68. doi: 10.3390/toxics6040068
Sanchez-Tillo, E., de Barrios, O., Siles, L., Cuatrecasas, M., Castells, A., and Postigo, A. (2011). Beta-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc. Natl. Acad. Sci. USA 108, 19204–19209. doi: 10.1073/pnas.1108977108
Santamaria, P. G., Moreno-Bueno, G., and Cano, A. (2019). Contribution of epithelial plasticity to therapy resistance. J. Clin. Med. 8:676. doi: 10.3390/jcm8050676
Santiago, L., Daniels, G., Wang, D., Deng, F. M., and Lee, P. (2017). Wnt signaling pathway protein LEF1 in cancer, as a biomarker for prognosis and a target for treatment. Am. J. Cancer Res. 7, 1389–1406.
Sanyal, S., Rochereau, T., Maesano, C. N., Com-Ruelle, L., and Annesi-Maesano, I. (2018). Long-term effect of outdoor air pollution on mortality and morbidity: a 12-year follow-up study for metropolitan France. Int. J. Environ. Res. Public Health 15, 1–8. doi: 10.3390/ijerph15112487
Sarkar, F. H., Li, Y., Wang, Z., and Kong, D. (2008). NF-kappaB signaling pathway and its therapeutic implications in human diseases. Int. Rev. Immunol. 27, 293–319. doi: 10.1080/08830180802276179
Sattayakhom, A., Chunglok, W., Ittarat, W., and Chamulitrat, W. (2014). Study designs to investigate Nox1 acceleration of neoplastic progression in immortalized human epithelial cells by selection of differentiation resistant cells. Redox Biol. 2, 140–147. doi: 10.1016/j.redox.2013.12.010
Sayin, V. I., Ibrahim, M. X., Larsson, E., Nilsson, J. A., Lindahl, P., and Bergo, M. O. (2014). Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 6:221ra215. doi: 10.1126/scitranslmed.3007653
Schraufnagel, D. E., Balmes, J., Cowl, C. T., De Matteis, S., Jung, S. H., Mortimer, K., et al. (2018). Air pollution and non-communicable diseases: a review by the forum of international respiratory societies' environmental committee, part 1: the damaging effects of air pollution. Chest 155, 409–416. doi: 10.1016/j.chest.2018.10.042
Sese, L., Nunes, H., Cottin, V., Sanyal, S., Didier, M., Carton, Z., et al. (2018). Role of atmospheric pollution on the natural history of idiopathic pulmonary fibrosis. Thorax 73, 145–150. doi: 10.1136/thoraxjnl-2017-209967
Shi, Y., Zhao, T., Yang, X., Sun, B., Li, Y., Duan, J., et al. (2019). PM2.5-induced alteration of DNA methylation and RNA-transcription are associated with inflammatory response and lung injury. Sci. Total Environ. 650, 908–921. doi: 10.1016/j.scitotenv.2018.09.085
Shimba, S., Komiyama, K., Moro, I., and Tezuka, M. (2002). Overexpression of the aryl hydrocarbon receptor (AhR) accelerates the cell proliferation of A549 cells. J. Biochem. 132, 795–802. doi: 10.1093/oxfordjournals.jbchem.a003289
Shu, D. Y., Wojciechowski, M., and Lovicu, F. J. (2018). ERK1/2-mediated EGFR-signaling is required for TGFbeta-induced lens epithelial-mesenchymal transition. Exp. Eye Res. 178, 108–121. doi: 10.1016/j.exer.2018.09.021
Shukla, A., Timblin, C., BeruBe, K., Gordon, T., McKinney, W., Driscoll, K., et al. (2000). Inhaled particulate matter causes expression of nuclear factor (NF)-kappaB-related genes and oxidant-dependent NF-kappaB activation in vitro. Am. J. Respir. Cell Mol. Biol. 23, 182–187. doi: 10.1165/ajrcmb.23.2.4035
Shuster-Meiseles, T., Shafer, M. M., Heo, J., Pardo, M., Antkiewicz, D. S., Schauer, J. J., et al. (2016). ROS-generating/ARE-activating capacity of metals in roadway particulate matter deposited in urban environment. Environ. Res. 146, 252–262. doi: 10.1016/j.envres.2016.01.009
Simeone, P., Trerotola, M., Franck, J., Cardon, T., Marchisio, M., Fournier, I., et al. (2018). The multiverse nature of epithelial to mesenchymal transition. Semin. Cancer Biol. 58, 1–10. doi: 10.1016/j.semcancer.2018.11.004
Song, L., Li, D., Li, X., Ma, L., Bai, X., Wen, Z., et al. (2017). Exposure to PM2.5 induces aberrant activation of NF-kappaB in human airway epithelial cells by downregulating miR-331 expression. Environ. Toxicol. Pharmacol. 50, 192–199. doi: 10.1016/j.etap.2017.02.011
Squadrito, G. L., Cueto, R., Dellinger, B., and Pryor, W. A. (2001). Quinoid redox cycling as a mechanism for sustained free radical generation by inhaled airborne particulate matter. Free Radic. Biol. Med. 31, 1132–1138. doi: 10.1016/S0891-5849(01)00703-1
Stewart, T. A., Azimi, I., Thompson, E. W., Roberts-Thomson, S. J., and Monteith, G. R. (2015). A role for calcium in the regulation of ATP-binding cassette, sub-family C, member 3 (ABCC3) gene expression in a model of epidermal growth factor-mediated breast cancer epithelial-mesenchymal transition. Biochem. Biophys. Res. Commun. 458, 509–514. doi: 10.1016/j.bbrc.2015.01.141
Stone, R. C., Pastar, I., Ojeh, N., Chen, V., Liu, S., Garzon, K. I., et al. (2016). Epithelial-mesenchymal transition in tissue repair and fibrosis. Cell Tissue Res. 365, 495–506. doi: 10.1007/s00441-016-2464-0
Storci, G., Sansone, P., Mari, S., D'Uva, G., Tavolari, S., Guarnieri, T., et al. (2010). TNFalpha up-regulates SLUG via the NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype. J. Cell. Physiol. 225, 682–691. doi: 10.1002/jcp.22264
Sullivan, N. J., Sasser, A. K., Axel, A. E., Vesuna, F., Raman, V., Ramirez, N., et al. (2009). Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 28, 2940–2947. doi: 10.1038/onc.2009.180
Sun, J., Shen, Z., Zeng, Y., Niu, X., Wang, J., Cao, J., et al. (2018). Characterization and cytotoxicity of PAHs in PM2.5 emitted from residential solid fuel burning in the Guanzhong plain, China. Environ. Pollut. 241, 359–368. doi: 10.1016/j.envpol.2018.05.076
Sun, B., Shi, Y., Li, Y., Jiang, J., Liang, S., Duan, J., et al. (2019). Short-term PM2.5 exposure induces sustained pulmonary fibrosis development during post-exposure period in rats. J. Hazard Mater. 1–11. doi: 10.1016/j.jhazmat.2019.121566 (in press).
Sun, S., Xie, F., Zhang, Q., Cui, Z., Cheng, X., Zhong, F., et al. (2017). Advanced oxidation protein products induce hepatocyte epithelial-mesenchymal transition via a ROS-dependent, TGF-beta/Smad signaling pathway. Cell Biol. Int. 41, 842–853. doi: 10.1002/cbin.10792
Tang, W., Du, L., Sun, W., Yu, Z., He, F., Chen, J., et al. (2017). Maternal exposure to fine particulate air pollution induces epithelial-to-mesenchymal transition resulting in postnatal pulmonary dysfunction mediated by transforming growth factor-beta/Smad3 signaling. Toxicol. Lett. 267, 11–20. doi: 10.1016/j.toxlet.2016.12.016
Taube, J. H., Herschkowitz, J. I., Komurov, K., Zhou, A. Y., Gupta, S., Yang, J., et al. (2010). Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA 107, 15449–15454. doi: 10.1073/pnas.1004900107
Techasen, A., Namwat, N., Loilome, W., Bungkanjana, P., Khuntikeo, N., Puapairoj, A., et al. (2012). Tumor necrosis factor-alpha (TNF-alpha) stimulates the epithelial-mesenchymal transition regulator snail in cholangiocarcinoma. Med. Oncol. 29, 3083–3091. doi: 10.1007/s12032-012-0305-x
Thevenot, P. T., Saravia, J., Jin, N., Giaimo, J. D., Chustz, R. E., Mahne, S., et al. (2013). Radical-containing ultrafine particulate matter initiates epithelial-to-mesenchymal transitions in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 48, 188–197. doi: 10.1165/rcmb.2012-0052OC
Thiery, J. P., Acloque, H., Huang, R. Y., and Nieto, M. A. (2009). Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890. doi: 10.1016/j.cell.2009.11.007
Thuault, S., Tan, E. J., Peinado, H., Cano, A., Heldin, C. H., and Moustakas, A. (2008). HMGA2 and Smads co-regulate SNAIL1 expression during induction of epithelial-to-mesenchymal transition. J. Biol. Chem. 283, 33437–33446. doi: 10.1074/jbc.M802016200
Thuault, S., Valcourt, U., Petersen, M., Manfioletti, G., Heldin, C. H., and Moustakas, A. (2006). Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J. Cell Biol. 174, 175–183. doi: 10.1083/jcb.200512110
Tripathi, P., Deng, F., Scruggs, A. M., Chen, Y., and Huang, S. K. (2018). Variation in doses and duration of particulate matter exposure in bronchial epithelial cells results in upregulation of different genes associated with airway disorders. Toxicol. In Vitro 51, 95–105. doi: 10.1016/j.tiv.2018.05.004
Tripathi, K., and Garg, M. (2018). Mechanistic regulation of epithelial-to-mesenchymal transition through RAS signaling pathway and therapeutic implications in human cancer. J. Cell Commun. Signal. 12, 513–527. doi: 10.1007/s12079-017-0441-3
Valavanidis, A., Vlachogianni, T., Fiotakis, K., and Loridas, S. (2013). Pulmonary oxidative stress, inflammation and cancer: respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int. J. Environ. Res. Public Health 10, 3886–3907. doi: 10.3390/ijerph10093886
Vander Ark, A., Cao, J., and Li, X. (2018). TGF-beta receptors: in and beyond TGF-beta signaling. Cell. Signal. 52, 112–120. doi: 10.1016/j.cellsig.2018.09.002
Vattanasit, U., Navasumrit, P., Khadka, M. B., Kanitwithayanun, J., Promvijit, J., Autrup, H., et al. (2014). Oxidative DNA damage and inflammatory responses in cultured human cells and in humans exposed to traffic-related particles. Int. J. Hyg. Environ. Health 217, 23–33. doi: 10.1016/j.ijheh.2013.03.002
Vesuna, F., van Diest, P., Chen, J. H., and Raman, V. (2008). Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem. Biophys. Res. Commun. 367, 235–241. doi: 10.1016/j.bbrc.2007.11.151
Vinson, K. E., George, D. C., Fender, A. W., Bertrand, F. E., and Sigounas, G. (2016). The notch pathway in colorectal cancer. Int. J. Cancer 138, 1835–1842. doi: 10.1002/ijc.29800
Wan, Q., Liu, Z., and Yang, Y. (2018). Puerarin inhibits vascular smooth muscle cells proliferation induced by fine particulate matter via suppressing of the p38 MAPK signaling pathway. BMC Complement. Altern. Med. 18:146. doi: 10.1186/s12906-018-2206-9
Wang, C. K., Chang, H., Chen, P. H., Chang, J. T., Kuo, Y. C., Ko, J. L., et al. (2009). Aryl hydrocarbon receptor activation and overexpression upregulated fibroblast growth factor-9 in human lung adenocarcinomas. Int. J. Cancer 125, 807–815. doi: 10.1002/ijc.24348
Wang, Y., Sun, B., Zhao, X., Zhao, N., Sun, R., Zhu, D., et al. (2016). Twist1-related miR-26b-5p suppresses epithelial-mesenchymal transition, migration and invasion by targeting SMAD1 in hepatocellular carcinoma. Oncotarget 7, 24383–24401. doi: 10.18632/oncotarget.8328
Wang, Y., Xu, M., Ke, Z. J., and Luo, J. (2017). Cellular and molecular mechanisms underlying alcohol-induced aggressiveness of breast cancer. Pharmacol. Res. 115, 299–308. doi: 10.1016/j.phrs.2016.12.005
Wang, J., Zhang, W. J., Xiong, W., Lu, W. H., Zheng, H. Y., Zhou, X., et al. (2018). PM2.5 stimulated the release of cytokines from BEAS-2B cells through activation of IKK/NF-kappaB pathway. Hum. Exp. Toxicol. 38, 311–320. doi: 10.1177/0960327118802628
Wang, G., Zhen, L., Lu, P., Jiang, R., and Song, W. (2013). Effects of ozone and fine particulate matter (PM2.5) on rat cardiac autonomic nervous system and systemic inflammation. Wei Sheng Yan Jiu 42, 554–560.
Wang, Y., Zhong, Y., Hou, T., Liao, J., Zhang, C., Sun, C., et al. (2019). PM2.5 induces EMT and promotes CSC properties by activating notch pathway in vivo and vitro. Ecotoxicol. Environ. Saf. 178, 159–167. doi: 10.1016/j.ecoenv.2019.03.086
Wei, H., Liang, F., Cheng, W., Zhou, R., Wu, X., Feng, Y., et al. (2017). The mechanisms for lung cancer risk of PM2.5: induction of epithelial-mesenchymal transition and cancer stem cell properties in human non-small cell lung cancer cells. Environ. Toxicol. 32, 2341–2351. doi: 10.1002/tox.22437
Wu, W. S. (2006). The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev. 25, 695–705. doi: 10.1007/s10555-006-9037-8
Xie, Y., and Zhao, B. (2018). Chemical composition of outdoor and indoor PM2.5 collected during haze events: transformations and modified source contributions resulting from outdoor-to-indoor transport. Indoor Air 28, 828–839. doi: 10.1111/ina.12503
Xin, L., Che, B., Zhai, B., Luo, Q., Zhang, C., Wang, J., et al. (2018). 1,25-Dihydroxy vitamin D3 attenuates the oxidative stress-mediated inflammation induced by PM2.5 via the p38/NF-kappaB/NLRP3 pathway. Inflammation 42, 702–713. doi: 10.1007/s10753-018-0928-y
Xu, H., Jiao, X., Wu, Y., Li, S., Cao, L., and Dong, L. (2018). Exosomes derived from PM2.5 treated lung cancer cells promote the growth of lung cancer via the Wnt3a/betacatenin pathway. Oncol. Rep. 41, 1180–1188. doi: 10.3892/or.2018.6862
Xu, Z., Li, Z., Liao, Z., Gao, S., Hua, L., Ye, X., et al. (2019a). PM2.5 induced pulmonary fibrosis in vivo and in vitro. Ecotoxicol. Environ. Saf. 171, 112–121. doi: 10.1016/j.ecoenv.2018.12.061
Xu, Z., Wang, N., Xu, Y., Hua, L., Zhou, D., Zheng, M., et al. (2019b). Effects of chronic PM2.5 exposure on pulmonary epithelia: transcriptome analysis of mRNA-exosomal miRNA interactions. Toxicol. Lett. 316, 49–59. doi: 10.1016/j.toxlet.2019.09.010
Xu, Z., Zhang, Z., Ma, X., Ping, F., and Zheng, X. (2015). Effect of PM2.5 on oxidative stress-JAK/STAT signaling pathway of human bronchial epithelial cells. Wei Sheng Yan Jiu 44, 451–455.
Yadav, A., Kumar, B., Datta, J., Teknos, T. N., and Kumar, P. (2011). IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol. Cancer Res. 9, 1658–1667. doi: 10.1158/1541-7786.MCR-11-0271
Yamaoka, T., Kusumoto, S., Ando, K., Ohba, M., and Ohmori, T. (2018). Receptor tyrosine kinase-targeted cancer therapy. Int. J. Mol. Sci. 19:3491. doi: 10.3390/ijms19113491
Yan, Z., Jin, Y., An, Z., Liu, Y., Samet, J. M., and Wu, W. (2016). Inflammatory cell signaling following exposures to particulate matter and ozone. Biochim. Biophys. Acta 1860, 2826–2834. doi: 10.1016/j.bbagen.2016.03.030
Yan, L., Xu, F., and Dai, C. L. (2018). Relationship between epithelial-to-mesenchymal transition and the inflammatory microenvironment of hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 37:203. doi: 10.1186/s13046-018-0887-z
Yang, L., Lin, Z., Wang, Y., Li, C., Xu, W., Li, Q., et al. (2018). Nickle(II) ions exacerbate bleomycin-induced pulmonary inflammation and fibrosis by activating the ROS/Akt signaling pathway. Environ. Sci. Pollut. Res. Int. 25, 4406–4418. doi: 10.1007/s11356-017-0525-x
Yang, D., Ma, M., Zhou, W., Yang, B., and Xiao, C. (2017). Inhibition of miR-32 activity promoted EMT induced by PM2.5 exposure through the modulation of the Smad1-mediated signaling pathways in lung cancer cells. Chemosphere 184, 289–298. doi: 10.1016/j.chemosphere.2017.05.152
Yang, M. C., Wang, H. C., Hou, Y. C., Tung, H. L., Chiu, T. J., and Shan, Y. S. (2015). Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol. Cancer 14:179. doi: 10.1186/s12943-015-0449-3
Ye, X., Hong, W., Hao, B., Peng, G., Huang, L., Zhao, Z., et al. (2018). PM2.5 promotes human bronchial smooth muscle cell migration via the sonic hedgehog signaling pathway. Respir. Res. 19:37. doi: 10.1186/s12931-017-0702-y
Yi, L., Wei, C., and Fan, W. (2017). Fine-particulate matter (PM2.5), a risk factor for rat gestational diabetes with altered blood glucose and pancreatic GLUT2 expression. Gynecol. Endocrinol. 33, 611–616. doi: 10.1080/09513590.2017.1301923
Yilmaz, M., and Christofori, G. (2009). EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 28, 15–33. doi: 10.1007/s10555-008-9169-0
Yuan, Q., Chen, Y., Li, X., Zhang, Z., and Chu, H. (2019). Ambient fine particulate matter (PM2.5) induces oxidative stress and pro-inflammatory response via up-regulating the expression of CYP1A1/1B1 in human bronchial epithelial cells in vitro. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 839, 40–48. doi: 10.1016/j.mrgentox.2018.12.005
Zavadil, J., and Bottinger, E. P. (2005). TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 24, 5764–5774. doi: 10.1038/sj.onc.1208927
Zhang, X., Cheng, Q., Yin, H., and Yang, G. (2017b). Regulation of autophagy and EMT by the interplay between p53 and RAS during cancer progression (review). Int. J. Oncol. 51, 18–24. doi: 10.3892/ijo.2017.4025
Zhang, H., and Li, Z. (2019). microRNA-16 via Twist1 inhibits EMT induced by PM2.5 exposure in human hepatocellular carcinoma. Open Med. 14, 673–682. doi: 10.1515/med-2019-0078
Zhang, Q., Luo, Q., Yuan, X., Chai, L., Li, D., Liu, J., et al. (2017a). Atmospheric particulate matter 2.5 promotes the migration and invasion of hepatocellular carcinoma cells. Oncol. Lett. 13, 3445–3450. doi: 10.3892/ol.2017.5947
Zhang, J., Tian, X. J., and Xing, J. (2016a). Signal transduction pathways of EMT induced by TGF-beta, SHH, and WNT and their crosstalks. J. Clin. Med. 5, 1–18. doi: 10.3390/jcm5040041
Zhang, J., Wang, X., Vikash, V., Ye, Q., Wu, D., Liu, Y., et al. (2016b). ROS and ROS-mediated cellular signaling. Oxidative Med. Cell. Longev. 2016:4350965. doi: 10.1155/2016/4350965
Zhao, H., Tong, G., Liu, J., Wang, J., Zhang, H., Bai, J., et al. (2019). IP3R and RyR channels are involved in traffic-related PM2.5-induced disorders of calcium homeostasis. Toxicol. Ind. Health 35, 339–348. doi: 10.1177/0748233719843763
Zhao, Y., Xu, Y., Li, Y., Xu, W., Luo, F., Wang, B., et al. (2013). NF-kappaB-mediated inflammation leading to EMT via miR-200c is involved in cell transformation induced by cigarette smoke extract. Toxicol. Sci. 135, 265–276. doi: 10.1093/toxsci/kft150
Zheng, L., Liu, S., Zhuang, G., Xu, J., Liu, Q., Zhang, X., et al. (2017). Signal transductions of BEAS-2B cells in response to carcinogenic PM2.5 exposure based on a microfluidic system. Anal. Chem. 89, 5413–5421. doi: 10.1021/acs.analchem.7b00218
Zhou, B. P., Deng, J., Xia, W., Xu, J., Li, Y. M., Gunduz, M., et al. (2004). Dual regulation of snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 6, 931–940. doi: 10.1038/ncb1173
Zou, W., He, F., Liu, S., Pu, J., Hu, J., Sheng, Q., et al. (2018). PM2.5 induced the expression of fibrogenic mediators via HMGB1-RAGE signaling in human airway epithelial cells. Can. Respir. J. 2018:1817398. doi: 10.1155/2018/1817398
Keywords: PM2.5, epithelial-mesenchymal transition, cancer, fibrosis, signaling pathway, molecular toxicology, air pollution, environment and human health
Citation: Xu Z, Ding W and Deng X (2019) PM2.5, Fine Particulate Matter: A Novel Player in the Epithelial-Mesenchymal Transition? Front. Physiol. 10:1404. doi: 10.3389/fphys.2019.01404
Edited by:
Rafael Kramann, University Hospital RWTH Aachen, GermanyReviewed by:
Limin Lu, Fudan University, ChinaXiao-Ming Meng, Anhui Medical University, China
Patrick Tang, The Chinese University of Hong Kong, China
Copyright © 2019 Xu, Ding and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaobei Deng, dengxiaobei@sjtu.edu.cn; Wenjun Ding, dingwj@ucas.ac.cn