 Matthew Mikhael
Matthew Mikhael Christian Makar
Christian Makar Amir Wissa
Amir Wissa Trixie Le
Trixie Le Mansoureh Eghbali
Mansoureh Eghbali Soban Umar
Soban Umar- Division of Molecular Medicine, Department of Anesthesiology and Perioperative Medicine, David Geffen School of Medicine, UCLA, Los Angeles, CA, United States
Pulmonary hypertension (PH) is a pulmonary vascular disease characterized by increased pulmonary artery pressures. Long standing pulmonary arterial pressure overload leads to right ventricular (RV) hypertrophy, RV failure, and death. RV failure is a major determinant of survival in PH. Oxidative stress has been associated with the development of RV failure secondary to PH. Here we summarize the structural and functional changes in the RV in response to sustained pulmonary arterial pressure overload. Furthermore, we review the pre-clinical and clinical studies highlighting the association of oxidative stress with pulmonary vasculature and RV remodeling in chronic PH. Targeting oxidative stress promises to be an effective therapeutic strategy for the treatment of RV failure.
Pulmonary Hypertension and Its Effects on the Right Ventricle
Pulmonary hypertension (PH) is characterized by an average mean pulmonary artery pressure of 25 mmHg or greater at rest. Long standing right ventricular (RV) pressure overload in PH leads to RV hypertrophy and eventually RV failure and death (Grosse et al., 2017). Early detection of PH can lead to a better prognosis and increase in the average survival rate (McLaughlin, 2013).
Considering healthy pulmonary circulation maintains low pressure compared to the systemic circulation, the RV walls are thinner and less resilient than the left ventricular (LV) walls (Badano et al., 2010). In response to chronic pressure overload due to PH, the RV undergoes adaptations including myocardial hypertrophy, followed by contractile impairment, and activation of myocardial renin-angiotensin-aldosterone system. Despite all these adaptations, RV may still exhibit dysfunction due to afterload mismatch. This RV dysfunction can be attributed to a reduced cardiac output, and functional tricuspid regurgitation. As hypertrophy and dilation continue to affect the RV, the RV becomes more spherical and its cross-sectional area enlarges, and the interventricular septum flattens; ultimately causing LV dysfunction (Badano et al., 2010; Figure 1).
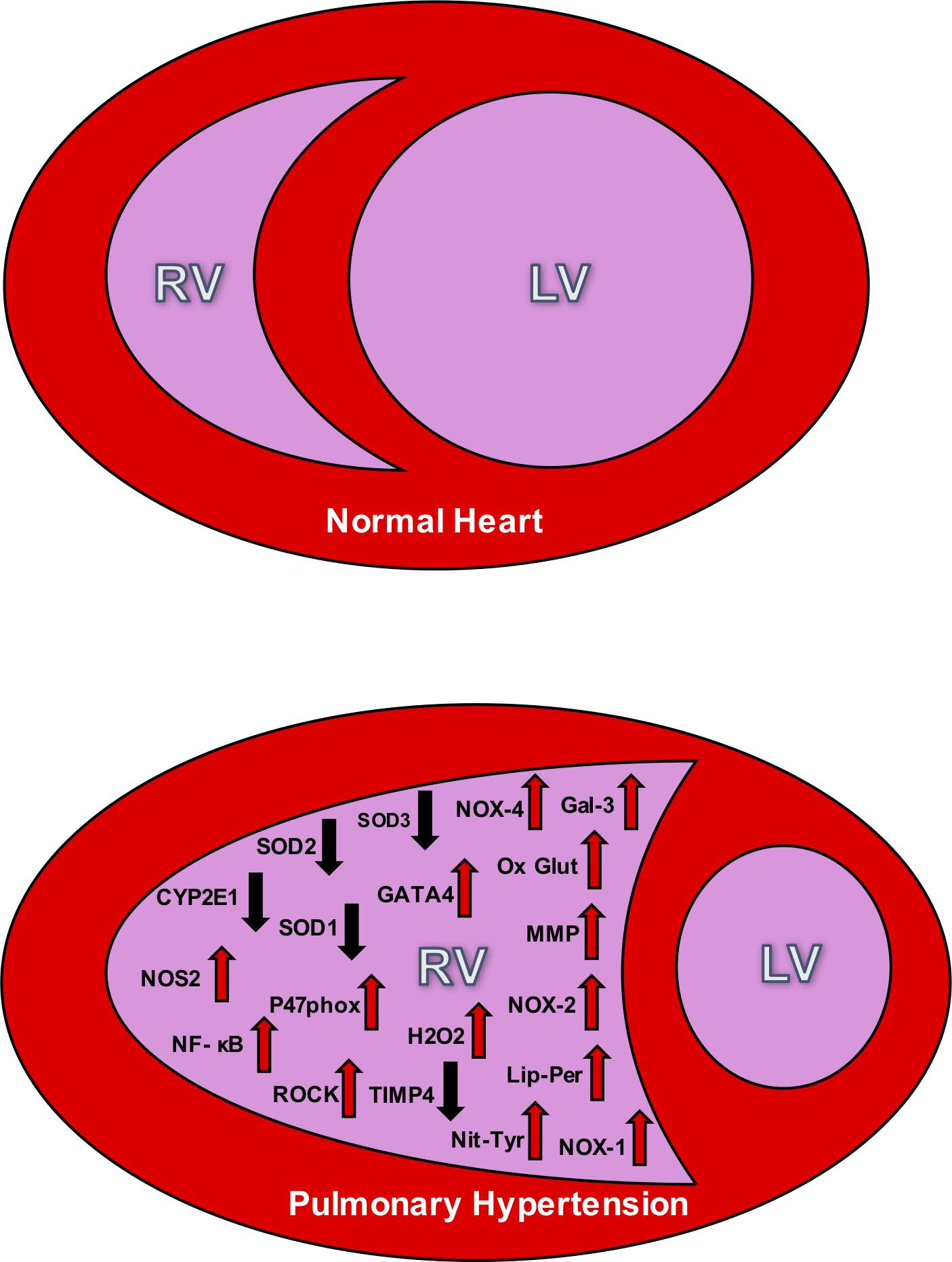
Figure 1. The top panel depicts the relative size difference between the right ventricle (RV) and the left ventricle (LV) in a normal heart. The bottom panel shows the effect of RV hypertrophy and the change in structure that accompanies PH. This bottom figure also illustrates the oxidative stress markers seen in the RV during PH. The up arrows signify increases in the markers during PH, and down arrows signify decreases. NOX, NADPH oxidase; SOD, superoxide dismutase; NOS, nitric oxide synthase; Ox Glut, oxidized glutathiones; TIMP, tissue inhibitor of metalloproteinase; MMP, matrix metalloproteinase; ROCK, Rho kinase; H2O2, peroxide; Lip-Per, lipid peroxidation; Nit-Tyr, nitrotyrosine. Sources: Gal 3 (He), NOX-4 (He, Cowley, Li), SOD3 (Zelko), SOD2 (Maron), SOD 1 (Cowley), GATA4 (Park and Suzuki), CYP2El (Potus), NOS2 (Dunlop), NF- κB (Boehm), oxidized glutathiones (Wang, Türck), nitrotyrosine (Qipshidze, Demarco), TIMP4 (Qipshidze), MMP (Qipshidze), NOX-2 (Ikeda,Li), p47phox (Ikeda), Rho kinase (Ikeda, Alzoubi), NOX-1 (Galhotra, Meghwani, Türck), hydrogen peroxide (Brandes), and lipid peroxidation (Puukila).
Pulmonary Hypertension-Induced Right Ventricular Hypertrophy
Right ventricular hypertrophy (RVH) is an adaptive transformation in the RV tissue that is usually triggered by chronic pressure overload. This pressure overload can result from stenotic valvular heart diseases or more commonly, pulmonary vascular disease associated with PH (Ikeda et al., 2014; Avazmohammadi et al., 2017). Pressure overload induces different responses in the LV and the RV due to their differing embryonic formations. Both chambers typically start developing at a 3.5 mm thickness prenatally but will grow at different rates after birth. The RV will grow to a 4 mm thickness while the LV quadruples in size to about 11 mm (Ryan and Archer, 2014). Thus in order to accommodate for the higher chronic pulmonary arterial pressures that accompany the different types of PH, these two relative thicknesses will change.
Right ventricular cardiac output depends on the synchronization between the force of contractile myocytes and the resistance to the blood flow through the pulmonary system. Normally, the RV can accommodate large but acute changes in volume loading during high levels of physical exercise, for example. However, there is a limited contractile reserve to keep the ejection fraction at homeostatic levels. In clinical cases and pre-clinical studies of animal models, the dilation of the RV is proportional to the severity of the adjacent vascular pressures (Watts et al., 2010). Hence, as afterload increases, the RV must significantly increase in size in order to maintain a suitable stroke volume. As a result, the naturally low pressure and elastic RV becomes large and inelastic, pushing on the interventricular septum to subsequently decrease the size of the LV (see Figure 1).
Some patients can live for years with adaptive RV hypertrophy by overcoming the high pressures, but when the RV becomes maladaptive, the complications follow soon afterward. Despite numerous studies highlighting the structural and functional changes of the RV secondary to PH, the transcriptomic profile, and molecular pathways that regulate these changes in the RV are not well studied (Lahm et al., 2018). For example, regulators such as extracellular ubiquitin, STAT3, Nrf2, and Ang-2/Tie-2 have all been identified as angiogenic regulators that contribute to LV morphological change, but it is not known whether these same pathways are regulating the RV as well (Lahm et al., 2018).
Right Ventricular Failure Secondary to Chronic Pulmonary Hypertension
Right ventricular function is the leading prognostic determinant of survival in PH patients. For example, 5-year survival in patients with pulmonary arterial hypertension (PAH) is strongly correlated with RV ejection fraction (RVEF) compared to pulmonary artery pressures or pulmonary vascular resistance (van de Veerdonk et al., 2011). However, there is no true definition for right ventricular failure (RVF) as it clinically varies from case to case and the therapeutic options for RVF are very limited. Recently, the American Thoracic Society defined RVF as “a complex clinical syndrome characterized by insufficient delivery of blood from the RV in the setting of elevated systemic venous pressure at rest or exercise.” This widespread phenomenon (affecting nearly 70 million in the United States alone) is a direct result of RV hypertrophy as the overworked myocytes experience increased afterload and lose their contractility (van de Veerdonk et al., 2011). While RV hypertrophy is just the initial compensatory mechanism for high pulmonary vascular pressures, RVF takes effect once the hypertrophy becomes maladaptive.
Role of Oxidative Stress in the Lungs and Pulmonary Vasculature in Pulmonary Hypertension
Oxidative stress occurs when the number of reactive oxygen species (ROS) exceeds that which the biological system can combat and neutralize. ROS are oxygen containing molecules with an odd number of electrons, termed free radicals. These free radicals can oxidize molecules, such as lipids and DNA, causing a multitude of harmful effects. The pulmonary vasculature undergoes morphological changes in PH that can be mediated by oxidative stress. Past research has shown the expression of various oxidative stress markers changes in the lungs and pulmonary vasculature of animals and humans with PH.
In animal studies, Xu et al. (2011) found that increased oxidative stress contributes to the PH development in rodents. In normal conditions, extracellular superoxide dismutase (SOD3) is expressed in high concentrations in the lungs and is responsible for removing extracellular superoxide anions, a type of free radical contributing to oxidative stress. The absence of SOD3 resulted in significantly worse PH in hypoxic SOD3 knockout mice and MCT rats with a SOD3 loss-of-function gene mutation (Xu et al., 2011). In another study, Wang L. et al. (2017) examined the effects of 17β-estradiol and 2-methoxyestradiol on the oxidative stress-hypoxia inducible factor-1 (OS-HIF-1) pathway in rats with hypoxia-induced PH. Hypoxic rats had a significant increase in oxidative stress levels as indicated by increased serum ROS levels, decreased serum SOD, and decreased manganese SOD (MnSOD) levels. Furthermore, MnSOD mRNA and protein levels were decreased in the lung tissue (Wang L. et al., 2017).
Bertoli et al. (2018) investigated the effects of chronic iron-overload on the resistance pulmonary arteries in rats. They found that rats treated with a high dose of iron dextran demonstrated increased vasoconstriction and vascular hyper-reactivity and reduced NO which were reversed by antioxidant therapy (Bertoli et al., 2018). Hoshikawa et al. (2001) found that the free radical phosphatidylcholine hydroperoxide (PCOOH) increased within 7 days following hypoxic exposure in rat lungs, and treating the rats with antioxidant N-acetylcysteine resulted in suppression of lung PCOOH levels. The activity of xanthine oxidase (XO), an enzyme that generates ROS, was also increased in rat lung tissue over time from day 1 through day 21 of hypoxia. Treating rats with the XO inhibitor allopurinol significantly inhibited the hypoxia-induced PH and resulted in decreased lung PCOOH levels (Hoshikawa et al., 2001).
Shi et al. (2018) used Baicalein, a flavonoid with known anti-proliferative and anti-inflammatory effects, to reduce oxidative stress in the lungs in an attempt to ameliorate hemodynamic and pulmonary vascular changes associated with MCT-induced PH in rats. Baicalein treatment inhibited inflammatory biomarkers (such as IL-6, TNF-α, and IL-1β) and reduced the Bax/Bcl-2 ratio and levels of cleaved caspase-3 which led to an increase in SOD production resulting in lowered ROS and inhibition of the NF-κB pathway (Shi et al., 2018).
On review of human studies, Bowers et al. (2004) demonstrated that histological lung sections from patients with severe PH exhibited increased oxidative stress than healthy controls, and chronic prostacyclin infusion, a commonly prescribed therapy for PH, exerted an anti-inflammatory effect.
Preterm infants with persistent pulmonary hypertension of the newborn (PPHN) present distinct challenges due to their lack of antioxidant defense mechanisms. Wedgwood et al. (2019) argued that oxidant stress in PPHN results from free radical generation from underlying lung disease or from free radicals generated by supplemental oxygen. These free radicals in turn act on the NO pathway resulting in decreased cGMP levels and pulmonary vasoconstriction (Wedgwood et al., 2019).
Kikuchi et al. (2018) recently demonstrated that Selenoprotein P (SeP), an extracellular protein responsible for maintaining cellular metabolism, exhibits a 32-fold increase in human PAH-PASMCs compared with control PASMCs. They reported that SeP promoted PASMC proliferation and resistance to apoptosis through increased oxidative stress and mitochondrial dysfunction associated with dysregulated glutathione metabolism. In addition, SeP-knockout mice (SeP–/–) exposed to chronic hypoxia demonstrated significantly reduced PH and pulmonary vascular remodeling (Kikuchi et al., 2018).
Krüppel-like factor 4 (KLF4) plays an important role in the protection of endothelial cells (ECs) by regulating vasodilation, inflammation, coagulation, and oxidative stress. In a recent study, Ban et al. (2019) demonstrated that KLF4 undergoes S-nitrosation in response to nitrosative stress in the human umbilical vein ECs, which is mediated by endothelin-1 and is inhibited by endothelin receptor antagonist Bosentan. Furthermore, they demonstrated that S-nitrosated KLF4 was significantly increased in lung tissues, along with decreased nuclear localization of KLF4 in rats with hypoxia-induced PH (Ban et al., 2019). In another study, Masri et al. (2008) hypothesized that ROS consumption of NO may contribute to low NO levels and development of PH in human lungs. They found that antioxidants, glutathione peroxidase and SOD activities were decreased in IPAH lungs compared to controls, while catalase and glutathione activities were similar among the groups (Masri et al., 2008).
In summary, there is growing evidence implicating oxidative stress in the pathogenesis of PH in pre-clinical and clinical studies. It is important to identify the expression and function of oxidative stress markers in specific cell types in the lungs and pulmonary vasculature in order to help devise novel targeted therapies.
Contribution of Oxidative Stress to RV Remodeling in PH
Oxidative stress plays a key role in pulmonary vascular remodeling, which in turn increases the RV after-load leading to RV hypertrophy and eventual RV failure (Bello-Klein et al., 2018). Additionally, it has been shown that the RV is more vulnerable to oxidative stress than the LV (Reddy and Bernstein, 2015), potentially because the RV cannot upregulate manganese superoxide dismutase expression, an enzyme that combats ROS (Shults et al., 2018).
Formation of ROS in many animal models of PH are derived from NADP oxidase activity, xanthine oxidase, and endothelial NOS. As PH progresses, circulating monocytes accumulate in the pulmonary arterioles and start to generate ROS to induce cell proliferation and fibrosis in the RV and small pulmonary arteries (Demarco et al., 2010). In an experimental model of PH induced by MCT in rats, high fat diet resulted in high levels of free radical formation in the dilated RV (Khoo et al., 2012). In Sugen-hypoxia model of PH in rats, a significant increase in oxidative stress caused cardiomyocyte deterioration. Furthermore, oxidative stress promoted perivascular fibrosis, leading to RV remodeling (Woo et al., 2017). In a rat model of angio-proliferative PH, although RV pressures were similar between males and females, males had worse RV hypertrophy, fibrosis, dysfunction, and survival compared to females. The reduced RV fibrosis in females was likely due to protection against oxidative stress as it correlated with increased caveolin-1 and decreased endothelial nitric oxide (NO) synthase-derived superoxide (Rafikova et al., 2015).
A decrease in SOD3 causes an imbalance between oxidants and antioxidants, and may therefore lead to vascular remodeling and PH. In SOD3 knockout mouse model with silica-induced PH, RV pressures were significantly higher compared to the wild type control mice (Zelko et al., 2018). The isoform SOD2 stabilizes the superoxide anion to hydrogen peroxide, which is less likely to damage the RV vasculature (Maron and Abman, 2017). Hence, the commonly used animal models of PH demonstrate association of oxidative stress markers with the changes accompanying RV remodeling.
Molecular Pathways Associated With RV Oxidative Stress in Pulmonary Hypertension
There are several proposed pathways leading to RV oxidative stress in PH. PH creates ROS, which in turn causes carbonylation of annexin A1 protein that results in its degradation by proteasomes in the RV. Annexin A1 is an inhibitor of CBF/NF-Y, a transcription factor that functions to activate the GATA4 gene. The GATA4 gene is a regulator of hypertrophy in the RV. In animal models of PH, increased GATA4 expression in the RV has been observed (Park et al., 2010; Suzuki and Shults, 2017). In an RV transcriptomic analysis of MCT-induced PH in Sprague-Dawley rats, Annexin A1 was identified to be dysregulated. Furthermore, the transcriptomic analysis of the RV showed that CYP2El, a gene that combats oxidative stress, is downregulated in PH (Potus et al., 2018).
When the RV experiences pressure overload, it uses NO to decrease the afterload by promoting pulmonary vasodilation. NO synthase 2 (NOS2) is induced in cardiac fibroblasts under RV pressure overload. Induction of NOS2 has been associated with collagen formation, which causes fibrosis in the RV and impaired function. Oxidative stress and cytokines which are elevated in PH, result in increased NF-κB expression. The NOS2 promotor has a NF-κB binding site that promotes transcription of NOS2. This creates oxidative stress, ROS formation, and eventual collagen deposition, which leads to fibrotic formation in the RV (Boehm et al., 2018).
Galectin-3 (Gal-3), a biomarker for LV remodeling, has been shown to interact with NOX4-derived oxidative stress promoting cardiac fibrosis in the RV in PH patients (He et al., 2017). Budas et al. (2018) studied the effects of inhibition of redox-sensitive apical MAPK and apoptosis signal-regulating kinase 1 (ASK1) on RV remodeling in rats with PH. Inhibiting ASK1 reduced remodeling of the pulmonary vasculature and the RV. ASK1 inhibitor, GS-444217 was shown to improve cardiac function and reduce fibrosis. GS-444217 decreased phosphorylation of p38 and JNK (c-Jun N-terminal kinase), and reduced cardiac fibroblast activation. Fibrosis formation causes a further increase in the severity of symptoms in PH patients. In a study by Wang X. et al. (2017) rats with Sugen-induced PH developed RV hypertrophy with increased levels of oxidized glutathiones, xanthine, and uric acid in the RV, which suggested higher production of ROS by xanthine oxidase. Also, there was a 30-fold lower level of antioxidant α-tocopherol nicotinate in the RV (Wang X. et al., 2017).
Glucose-6-phosphate (G6P) dehydrogenase is a major source of NADP which is a substrate for NADP oxidase in the RV. If G6P is inhibited, it reduces hypertrophy in the RV. One possible inhibitor of G6P is a 17-ketosteroid called Dehydroepiandrosterone, which was also shown to improve LV diastolic function (Rawat et al., 2014).
Oxidative stress generates nitrotyrosine residues in tissue inhibitor of metalloproteinase (TIMPs) which frees active martix metalloproteinase (MMP). It was hypothesized that the imbalance in MMP to TIMP causes fibrosis and ultimately RVF. It was found that folic acid did increase TIMP-4 and decreased different isoforms of MMPs. In pulmonary arterial constriction models, this was the opposite where TIMP-4 was decreased and MMP was increased. As a result, folic acid decreased levels of ROS in the RV wall, interstitial fibrosis, and RV pressures affirming the correlation between the oxidative stress and the RVF through the MMP to TIMP ratio (Qipshidze et al., 2012).
Ikeda et al. (2014) measured expression levels of oxidative stress related genes in the RV 24 h after pulmonary arterial constriction. Oxidative stress genes, such as p47phox and NOX2, were upregulated in the RV. Rho kinase (ROCK2) was rapidly induced after pulmonary arterial constriction in the RV free wall. In dominant negative Rho kinase mice, there was no presence of oxidative species. The timing between these steps are significant because it illustrates a correlation between Rho kinase, high pressures in the RV, and oxidative species (Ikeda et al., 2014). A possible treatment for Rho kinase overexpression is dehydroepiandrosterone (DHEA), a steroid, which has been shown to decrease NADPH levels in the RV. DHEA reduced the activity of Rho kinase, which in turn resulted in reduction of active forms of STAT3 and NFATc3 leading to rescue of RV remodeling. Overall the antioxidant activity of DHEA preserved contractility and slightly reduced high RVSP resulting from PH (Alzoubi et al., 2013).
A study by Dunlop et al. (2014) previously found that extended exposure to inhaled NO and chronic hypoxia causes a complete RV systolic dysfunction. A follow up study was done using therapeutic hypercapnia (10% CO2) in order to block the Interleukin (IL)-1α pathway which in turn inhibits NOS-2. This causes a complete normalization of RV hypertrophy (Dunlop et al., 2014). Chovanec et al. (2009) theorized another mechanism of hypercapnia, in which the release of oxygen radicals and NO are directly inhibited while in a hypoxic environment. In summary, there are multiple pathways leading to generation of oxidative stress in the RV secondary to pressure overload (see Table 1 for summary of pathways).
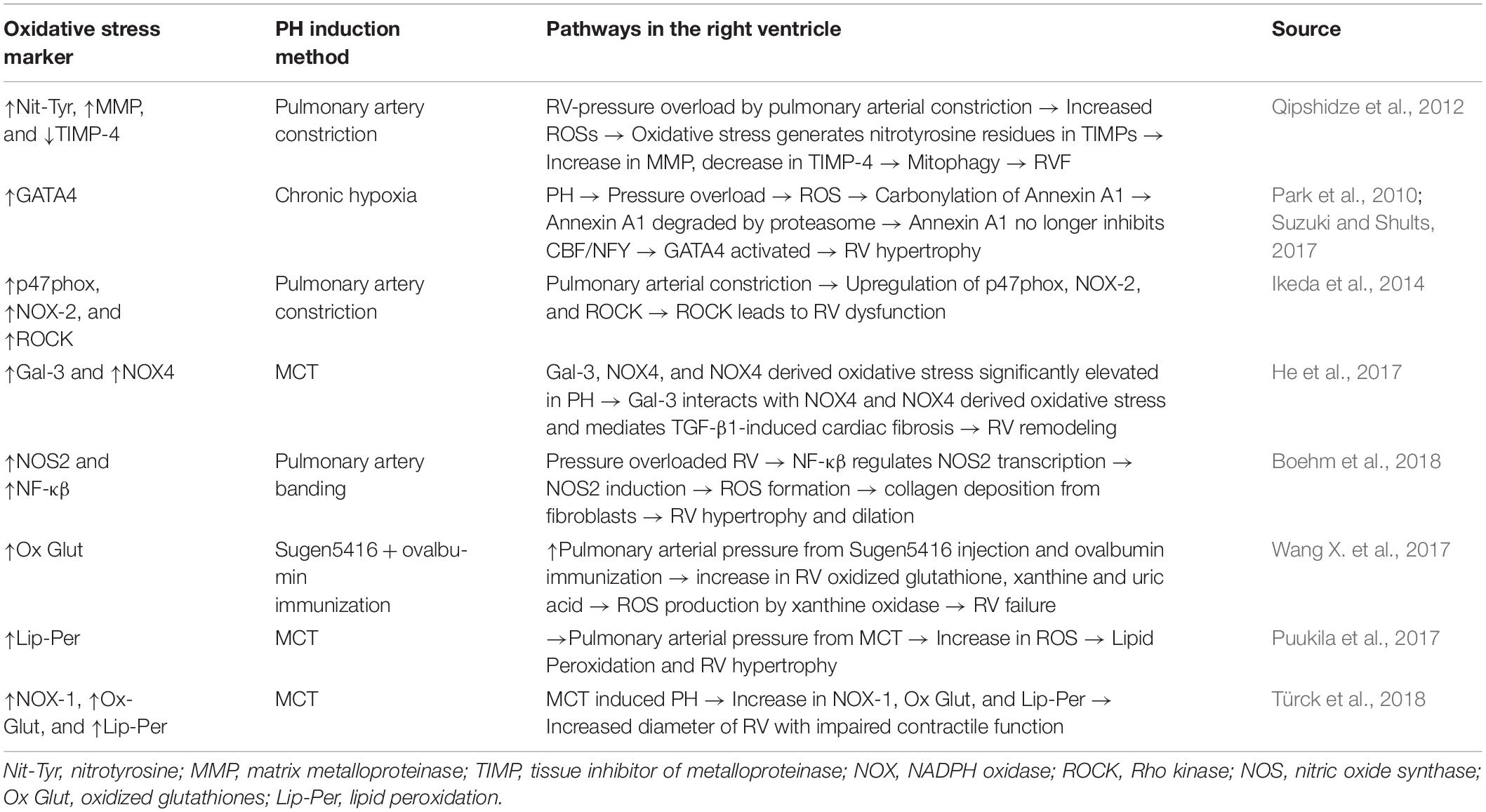
Table 1. Mechanistic pathways for the regulatory oxidative markers found in the right ventricle (RV) of pulmonary hypertension (PH) models.
Targeting Oxidative Stress in RV Remodeling Associated With Pulmonary Hypertension
Pharmacological Treatments
A number of therapeutic strategies have been tested to alleviate the effects of oxidative stress in PH. Antioxidants are the primary agents that inhibit oxidation of biomolecules by ROS. However, oxidative stress regulation in the RV can be controlled by epigenetics, and a diverse group of protein interactions. Therefore solely administering antioxidants in bulk amounts is unlikely to be effective (Suzuki and Shults, 2017).
Many new therapies have been tested that simultaneously ameliorate the effects of both oxidative stress and PH. Dihydroartemisinin (DHA), an agent that has anti-inflammatory, anti-malaria, and anti-tumor effects has been shown to reduce the symptoms of MCT-induced PH. DHA inhibits pulmonary arterial EC proliferation and reduces oxidative stress by increasing SOD expression and reducing ROS (Yu et al., 2018). PPARα agonist fenofibrate (FF) is another discovered therapy against oxidative stress and PH. In MCT-induced PH rats, FF was orally administered to the rats 3-days post MCT. FF reduced RV hypertrophy, oxidative stress, ROS, NADPH Oxidase (NOX-1) expression, and increased the Bcl2/Bax ratio in the RV that was caused by PH (Galhotra et al., 2018). Trapidil, a vasodilator, has also been shown to mitigate the effects of oxidative stress and PH. Increased lipid and glutathione peroxidation as well as an increase in RV diameter has been reported in MCT model of PH in Wistar rats. Trapidil reduced NOX-1 activity, reduced glutathione oxidation in the RV, and reduced the diameter of the RV (Türck et al., 2018). Rats treated with high doses of iron caused pathological remodeling in the RV. The oxidative stress that causes the hypertrophy was mediated by Angiotensin II Type 1 receptor (AT1). An AT1 antagonist was administered orally and was shown to restore vascular function (Bertoli et al., 2018). It has been shown that α1-adrenergic receptor (α1-AR) antagonist A61603 improves left ventricular failure. In the bleomycin model of RV failure, A61603 increased cellular SOD1 and decreased NOX4, myocyte necrosis, and fibrosis in the RV (Cowley et al., 2017). Pterostilbene (PTS), a phytophenol, paired with hydroxypropyl-β-cyclodextrin (HPβCD) at high doses completely prevents RV failure and hypertrophy caused by PH, while protecting systolic function by reducing NADPH oxidase-dependent superoxide anions (Dos Santos Lacerda et al., 2017).
According to a study by Zimmer et al. (2017) it was initially believed that aerobic exercise would increase RV function in MCT-induced PH rats. However, they found no change in oxidative stress due to stable levels of eNOS activity (Zimmer et al., 2017). Contrary to the findings from Zimmer et al. (2017) aerobic exercise has been shown to promote hydrogen peroxide and vascular endothelial growth factor which ultimately improve RV function, in spite of ROS created by hydrogen peroxide. However, it is not specified if other antioxidant markers are associated with aerobic exercise (Colombo et al., 2016). In addition, aerobic exercise at early stages of PH decreases mitochondrial oxidative stress, demonstrating cardioprotective effects (Moreira-Gonçalves et al., 2015). Each study focused on a different oxidative stress marker, which led them to different conclusions on the effect of exercise on oxidative damage. However, all three studies have shown that RV hypertrophy is unaffected by aerobic exercise.
EUK-134, a known antioxidant, reduced cardiomyocyte hypertrophy and decreased end systolic volume in MCT-induced PH rats (Redout et al., 2010). Thus, this was one of the first studies that demonstrated the importance of ROS in contractile dysfunction due to PH and how antioxidants such as EUK-134 can be used for PH treatment.
Herbal Treatments
Many of the novel therapies used to target oxidative stress and PH simultaneously, are derived from natural herbs and plants. One such therapy that showed similar effects to FF is ocimum sanctum (OS), a plant predominantly found in India. Similar to FF treatment, OS has shown a decrease in both RV hypertrophy and NOX-1 expression and an increase in Bcl2/Bax ratio in the RV tissue (Meghwani et al., 2018). The stem bark of a plant called Terminalia arjuna and Carvacrol, an oil in oregano and thyme, was also used to increase the Bcl2/Bax ratio in the pulmonary artery SMC (Zhang et al., 2016; Meghwani et al., 2017). Similar to the previously mentioned therapies, Copaiba oil has also been shown to decrease RV hypertrophy, and reduce oxidation of proteins and lipids in the RV of the MCT group (Campos-Carraro et al., 2018). In hypoxia-induced PH rats, Trimethoxystilbene (a novel polypehnol found in grapes and red wine with known anti-inflammatory effects) treatment prevented RVH by inhibiting the NOX/VPO1 pathway that mediates oxidative stress (Liu et al., 2014). NOX and VPO1 work together to cause higher levels of peroxide, which leads to smooth muscle cell proliferation (Brandes, 2011).
Grape seed procyanidin extract (GSPE) has been shown to prevent hypoxia-induced PH through antioxidant properties resulting in decreased RVSP and RV dilation. GSPE increased SOD and decreased NOX4 mRNA levels simultaneously, which decreased ROS production in pulmonary arterial SMC (Jin et al., 2016). Sesamin, an extract from sesame seeds, was used to inhibit NOX2 and NOX4 in the pulmonary vessels. This consequentially lowered RVSP and RV hypertrophy index (Li et al., 2015). The decrease of NOX4 and increase of SOD1 balances the antioxidant imbalance caused by PH. Secoisolariciresinol diglucoside (SDG), derived from flaxseed, was given to MCT rats with RV hypertrophy and increased lipid peroxidation. It did not decrease RV hypertrophy, however, it decreased ROS levels and SOD activity in the RV. When rats were pretreated with SDG, it was more effective in decreasing RV dysfunction (Puukila et al., 2017). When MCT rats were treated with Withania somnifera, a clinical antioxidant, oxidative markers such as levels of ROS were decreased in the lungs, which led to decreased RVSP (Kaur et al., 2015). The therapeutic effects of protandim, which enables Nrf2 to upregulate the expression of genes that code for antioxidants, were examined in Su/Hx model in rats. Protandim was found only to protect against RV failure, without affecting other PH symptoms like angio-obliteration caused by PH (Voelkel et al., 2013). This is important because a well-functioning RV is an important prognostic indicator of survival in PH patients.
In summary, there is a growing body of evidence demonstrating the association of oxidative stress to the development of RV remodeling in PH. Investigators have tested novel therapies targeting oxidative stress markers and molecular pathways to treat experimental RV failure. Further pre-clinical and clinical research is needed to validate the efficacy of these experimental therapies for targeting RV oxidative stress in PH.
Past Failures of Antioxidant PH Therapies in the RV and Lungs
A variety of antioxidant therapies have been tested for the treatment of pre-clinical PH. Some studies have focused on targeting oxidative stress in the lungs, whereas others have focused more on the effects of antioxidant therapies in the RV. RV failure is the primary cause of death in PH patients, and although treatment of patients with vasodilator antioxidants may reduce pulmonary vascular resistance, this effect may not be accompanied by improvements in the RV (Drake et al., 2013).
Antioxidant therapies targeting the lungs and RV in PH have shown prior failures. NO can modulate cellular and physiological processes to limit oxidative stress. Saugstad (2000) argued that NO can act as an antioxidant and pro-oxidant depending on the dose of NO administered and the presence of other oxidants. Previous rat studies found that NO in high concentrations induces free radical mediated injury in rat lungs. Oxidative stress was only partially treated in the hyperoxic condition, which in itself, helped exacerbate the disease (Saugstad, 2000). L-arginine, a substrate needed to produce endogenous NO was studied to see whether it prevents RV and pulmonary vascular hypertrophy in hypoxic rats. Long term oral L-arginine failed to prevent RV and pulmonary vascular hypertrophy in the rats with hypoxia-induced PH (Laursen et al., 2008). Horstman et al. (1998) used a direct administration of low concentrations of inhaled NO and found that inhaled NO attenuates RV hypertrophy and vascular remodeling secondary to hypoxia, but has no such effects in MCT-induced PH. This shows that the effectiveness of inhaled NO in attenuating remodeling in PH is dependent on the mechanism by which PH was induced. Another group also found mechanism-dependent results when they looked to make a determination between vasodilator doses of inhaled NO and the prevention of progression of PH in MCT rats. In MCT-induced PH, there was a decrease in pulmonary artery pressure. However, in chronic hypoxic conditions inhaled NO did not prevent development of PH with no changes in the RV (Maruyama et al., 1997).
The use of continuous inhalation of NO as a treatment was also used in a study of rats exposed to chronic hypoxia followed by normoxia, but there was no effect on alleviating pulmonary vascular remodeling in the PH lungs (Jiang et al., 2004). Prior studies on the umbilical vein endothelial cells led to the belief that NO can decrease malignant ET-1 overexpression which in theory should minimize the effects of oxidative stress. However, in a group of rats with hypoxia induced PH, investigators found that NO had no impact on the increased ET-1 expression in rat lungs with PH induced by hypoxic conditions (Earley et al., 2002).
A novel attempt at using the administration of chronic oxygen, instead of NO, was used as a possible antioxidant therapy for PH. However, results showed that the high influx of oxygen resulted in increased RV inflammation, pulmonary arterial hypertrophy, and SOD activity. Rats that received MCT and oxygen therapy recruited leukocytes that promoted RV inflammation and failure. And despite increases in SOD activity, antioxidants were not able to sufficiently counteract the oxidative stress caused by the combination of MCT and chronic oxygen therapy (Fujita et al., 2018).
As discussed earlier in this review, protandim which upregulates the expression of genes encoding antioxidants such as SOD and heme oxygenase-1 (HO-1), was found only to protect against RV failure, without affecting pulmonary vascular angioobliterative lesions caused by PH. The authors argued that antioxidants may affect pulmonary vascular remodeling when administered early during the development of the pulmonary vascular lesions while antioxidants may impact RV failure even late in the disease process (Voelkel et al., 2013).
Several studies have demonstrated the efficacy of statins in experimental models of PH, by reducing oxidative stress and inflammation, inhibiting PASMC proliferation, and inducing PASMC/PAEC apoptosis and vasodilatation (Nishimura et al., 2002, 2003; Taraseviciene-Stewart et al., 2006; Girgis et al., 2007). These results were also supported by observational studies in PH patients (Kao, 2005; King and Day, 2011). However, a phase-II randomized clinical trial of aspirin and simvastatin for PAH (ASA-STAT) failed to demonstrate a significant effect on the six-minute walk distance between simvastatin and placebo groups (Kawut et al., 2011).
To summarize, although antioxidants have shown promise in the treatment of experimental PH and RV failure, there have been reports of failure of some of these therapies. Understanding the precise role of oxidative stress in the pathogenesis and progression of PH and RV failure will help in developing targeted and effective antioxidant therapies.
Conclusion
Long standing PH leads to RV hypertrophy and RV failure. Accumulating evidence suggests involvement of oxidative stress in the development and progression of RV remodeling associated with PH. Targeting RV oxidative stress associated with PH may lead to the development of novel therapies for RV failure secondary to PH.
Author Contributions
MM, CM, AW, TL, and ME contributed in drafting and revising the manuscript. SU contributed in drafting and revising the manuscript, and provided the supervision.
Funding
SU was supported by a Mentored Research Training Grant from the Foundation of Anesthesia Education and Research (FAER) and a K08 Grant (1K08 HL141995 01A1) from the NIH (NHLBI).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alzoubi, A., Toba, M., Abe, K., O’Neill, K. D., Rocic, P., Fagan, K. A., et al. (2013). Dehydroepiandrosterone restores right ventricular structure and function in rats with severe pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 304, H1708–H1718. doi: 10.1152/ajpheart.00746.2012
Avazmohammadi, R., Hill, M., Simon, M., and Sacks, M. (2017). Transmural remodeling of right ventricular myocardium in response to pulmonary arterial hypertension. APL Bioeng. 1:016105. doi: 10.1063/1.5011639
Badano, L. P., Ginghina, C., Easaw, J., Muraru, D., Grillo, M. T., Lancellotti, P., et al. (2010). Right ventricle in pulmonary arterial hypertension: haemodynamics, structural changes, imaging, and proposal of a study protocol aimed to assess remodelling and treatment effects. Eur. J. Echocardiogr. 11, 27–37. doi: 10.1093/ejechocard/jep152
Ban, Y., Liu, Y., Li, Y., Zhang, Y., Xiao, L., Gu, Y., et al. (2019). S-nitrosation impairs KLF4 activity and instigates endothelial dysfunction in pulmonary arterial hypertension. Redox Biol. 21:101099. doi: 10.1016/j.redox.2019.101099
Bello-Klein, A., Mancardi, D., Araujo, A. S., Schenkel, P. C., Turck, P., and de Lima Seolin, B. G. (2018). Role of redox homeostasis and inflammation in the pathogenesis of pulmonary arterial hypertension. Curr. Med. Chem. 25, 1340–1351. doi: 10.2174/0929867325666171226114838
Bertoli, S. R., Marques, V. B., Rossi, E. M., Krause, M., Carneiro, M. T. W. D., Simões, M. R., et al. (2018). Chronic iron overload induces vascular dysfunction in resistance pulmonary arteries associated with right ventricular remodeling in rats. Toxicol. Lett. 295, 296–306. doi: 10.1016/j.toxlet.2018.07.010
Boehm, M., Novoyatleva, T., Kojonazarov, B., Veit, F., Weissmann, N., Ghofrani, H. A., et al. (2018). Nitric oxide synthase 2 induction promotes right ventricular fibrosis. Am. J. Respir. Cell Mol. Biol. 60, 346–356. doi: 10.1165/rcmb.2018-0069OC
Bowers, R., Cool, C., Murphy, R. C., Tuder, R. M., Hopken, M. W., Flores, S. C., et al. (2004). Oxidative stress in severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 169, 764–769. doi: 10.1164/rccm.200301-147OC
Brandes, R. P. (2011). Vascular peroxidase 1/peroxidasin: a complex protein with a simple function? Cardiovasc. Res. 91, 1–2. doi: 10.1093/cvr/cvr120
Budas, G. R., Boehm, M., Kojonazarov, B., Viswanathan, G., Tian, X., Veeroju, S., et al. (2018). ASK1 inhibition halts disease progression in preclinical models of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 197, 373–385. doi: 10.1164/rccm.201703-0502OC
Campos-Carraro, C., Turck, P., de Lima-Seolin, B. G., Tavares, A. M. V., Dos Santos Lacerda, D., Corssac, G. B., et al. (2018). Copaiba oil attenuates right ventricular remodeling by decreasing myocardial apoptotic signaling in monocrotaline-induced rats. J. Cardiovasc. Pharmacol. 72, 214–221.
Chovanec, M., Novotná, J., Wilhelm, J., Hampl, V., Vízek, M., and Herget, J. (2009). Hypercapnia attenuates hypoxic pulmonary hypertension by inhibiting lung radical injury. Physiol. Res. 58(Suppl. 2), S79–S85.
Colombo, R., Siqueira, R., Conzatti, A., de Lima Seolin, B. G., Fernandes, T. R. G., Godoy, A. E. G., et al. (2016). Exercise training contributes to H2O2/VEGF signaling in the lung of rats with monocrotaline-induced pulmonary hypertension. Vascul. Pharmacol. 87, 49–59. doi: 10.1016/j.vph.2016.06.006
Cowley, P. M., Wang, G., Joshi, S., Swigart, P. M., Lovett, D. H., Simpson, P. C., et al. (2017). α1A-Subtype adrenergic agonist therapy for the failing right ventricle. Am. J. Physiol. Heart Circ. Physiol. 313, H1109–H1118. doi: 10.1152/ajpheart.00153.2017
Demarco, V. G., Whaley-Connell, A. T., Sowers, J. R., Habibi, J., and Dellsperger, K. C. (2010). Contribution of oxidative stress to pulmonary arterial hypertension. World J. Cardiol. 2, 316–324. doi: 10.4330/wjc.v2.i10.316
Dos Santos Lacerda, D., Türck, P., Gazzi de Lima-Seolin, B., Colombo, R., Duarte Ortiz, V., Poletto Bonetto, J. H., et al. (2017). Pterostilbene reduces oxidative stress, prevents hypertrophy and preserves systolic function of right ventricle in cor pulmonale model. Br. J. Pharmacol. 174, 3302–3314. doi: 10.1111/bph.13948
Drake, J. I., Gomez-Arroyo, J., Dumur, C. I., Kraskauskas, D., Natarajan, R., Bogaard, H. J., et al. (2013). Chronic carvedilol treatment partially reverses the right ventricular failure transcriptional profile in experimental pulmonary hypertension. Physiol. Genomics 45, 449–461. doi: 10.1152/physiolgenomics.00166.2012
Dunlop, K., Gosal, K., Kantores, C., Ivanovska, J., Dhaliwal, R., Desjardins, J.-F., et al. (2014). Therapeutic hypercapnia prevents inhaled nitric oxide-induced right-ventricular systolic dysfunction in juvenile rats. Free Radic. Biol. Med. 69, 35–49. doi: 10.1016/j.freeradbiomed.2014.01.008
Earley, S., Nelin, L. D., Chicoine, L. G., and Walker, B. R. (2002). Hypoxia-induced pulmonary endothelin-1 expression is unaltered by nitric oxide. J. Appl. Physiol. 92, 1152–1158. doi: 10.1152/japplphysiol.00829.2001
Fujita, N., Yamasaki, N., Eto, K., Asaeda, M., Kuwahara, W., and Imagita, H. (2018). Oxygen therapy may worsen the survival rate in rats with monocrotaline-induced pulmonary arterial hypertension. PLoS One 13:e0204254. doi: 10.1371/journal.pone.0204254
Galhotra, P., Prabhakar, P., Meghwani, H., Mohammed, S. A., Banerjee, S. K., Seth, S., et al. (2018). Beneficial effects of fenofibrate in pulmonary hypertension in rats. Mol. Cell. Biochem. 449, 185–194. doi: 10.1007/s11010-018-3355-3
Girgis, R. E., Mozammel, S., Champion, H. C., Li, D., Peng, X., Shimoda, L., et al. (2007). Regression of chronic hypoxic pulmonary hypertension by simvastatin. Am. J. Physiol. Lung. Cell Mol. Physiol. 292, L1105–L1110.
Grosse, A., Grosse, C., and Lang, I. M. (2017). Distinguishing chronic thromboembolic pulmonary hypertension from other causes of pulmonary hypertension using CT. Am. J. Roentgenol. 209, 1228–1238. doi: 10.2214/AJR.17.17871
He, J., Li, X., Luo, H., Li, T., Zhao, L., Qi, Q., et al. (2017). Galectin-3 mediates the pulmonary arterial hypertension-induced right ventricular remodeling through interacting with NADPH oxidase 4. J. Am. Soc. Hypertens. 11, 275–289.e2. doi: 10.1016/j.jash.2017.03.008
Horstman, D. J., Frank, D. U., and Rich, G. F. (1998). Prolonged inhaled NO attenuates hypoxic, but not monocrotaline-induced, pulmonary vascular remodeling in rats. Anesth. Analg. 86, 74–81. doi: 10.1097/00000539-199801000-00015
Hoshikawa, Y., Ono, S., Suzuki, S., Tanita, T., Chida, M., Song, C., et al. (2001). Generation of oxidative stress contributes to the development of pulmonary hypertension induced by hypoxia. J. Appl. Physiol. 90, 1299–1306. doi: 10.1152/jappl.2001.90.4.1299
Ikeda, S., Satoh, K., Kikuchi, N., Miyata, S., Suzuki, K., Omura, J., et al. (2014). Crucial role of rho-kinase in pressure overload-induced right ventricular hypertrophy and dysfunction in mice. Arterioscler. Thromb. Vasc. Biol. 34, 1260–1271. doi: 10.1161/ATVBAHA.114.303320
Jiang, B. H., Maruyama, J., Yokochi, A., Iwasaki, M., Amano, H., Mitani, Y., et al. (2004). Prolonged nitric oxide inhalation fails to regress hypoxic vascular remodeling in rat lung. Chest 125, 2247–2252. doi: 10.1378/chest.125.6.2247
Jin, H., Liu, M., Zhang, X., Pan, J., Han, J., Wang, Y., et al. (2016). Grape seed procyanidin extract attenuates hypoxic pulmonary hypertension by inhibiting oxidative stress and pulmonary arterial smooth muscle cells proliferation. J. Nutr. Biochem. 36, 81–88. doi: 10.1016/j.jnutbio.2016.07.006
Kao, P. N. (2005). Simvastatin treatment of pulmonary hypertension: an observational case series. Chest 127, 1446–1452. doi: 10.1016/s0012-3692(15)34501-3
Kaur, G., Singh, N., Samuel, S. S., Bora, H. K., Sharma, S., Pachauri, S. D., et al. (2015). Withania somnifera shows a protective effect in monocrotaline-induced pulmonary hypertension. Pharm. Biol. 53, 147–157. doi: 10.3109/13880209.2014.912240
Kawut, S. M., Bagiella, E., Lederer, D. J., Shimbo, D., Horn, E. M., Roberts, K. E., et al. (2011). Randomized clinical trial of aspirin and simvastatin for pulmonary arterial hypertension: ASA-STAT. Circulation 123, 2985–2993. doi: 10.1161/CIRCULATIONAHA.110.015693
Khoo, N. K. H., Cantu-Medellin, N., Devlin, J. E., St Croix, C. M., Watkins, S. C., Fleming, A. M., et al. (2012). Obesity-induced tissue free radical generation: an in vivo immuno-spin trapping study. Free Radic. Biol. Med. 52, 2312–2319. doi: 10.1016/j.freeradbiomed.2012.04.011
Kikuchi, N., Satoh, K., Kurosawa, R., Yaoita, N., Elias-Al-Mamun, M., Siddique, M. A. H., et al. (2018). Selenoprotein P promotes the development of pulmonary arterial hypertension. Circulation 138, 600–623. doi: 10.1161/CIRCULATIONAHA.117.033113
King, W. T., and Day, R. W. (2011). Treatment of pediatric pulmonary hypertension with simvastatin: an observational study. Pediatr. Pulmonol. 46, 261–265. doi: 10.1002/ppul.21361
Lahm, T., Douglas, I. S., Archer, S. L., Bogaard, H. J., Chesler, N. C., Haddad, F., et al. (2018). Assessment of right ventricular function in the research setting: knowledge gaps and pathways forward. An official American thoracic society research statement. Am. J. Respir. Crit. Care Med. 198, e15–e43. doi: 10.1164/rccm.201806-1160ST
Laursen, B. E., Dam, M. Y., Mulvany, M. J., and Simonsen, U. (2008). Hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy is unaltered by long-term oral L-arginine administration. Vascul. Pharmacol. 49, 71–76. doi: 10.1016/j.vph.2008.03.001
Li, X., Gao, Y., Li, S., and Yang, J. (2015). [Effect of sesamin on pulmonary vascular remodeling in rats with monocrotaline-induced pulmonary hypertension]. China J. Chin. Mater. Medica 40, 1355–1361.
Liu, B., Luo, X.-J., Yang, Z.-B., Zhang, J.-J., Li, T.-B., Zhang, X.-J., et al. (2014). Inhibition of NOX/VPO1 pathway and inflammatory reaction by trimethoxystilbene in prevention of cardiovascular remodeling in hypoxia-induced pulmonary hypertensive rats. J. Cardiovasc. Pharmacol. 63, 567–576. doi: 10.1097/FJC.0000000000000082
Maron, B. A., and Abman, S. H. (2017). Translational advances in the field of pulmonary hypertension. Focusing on developmental origins and disease inception for the prevention of pulmonary hypertension. Am. J. Respir. Crit. Care Med. 195, 292–301. doi: 10.1164/rccm.201604-0882PP
Maruyama, J., Maruyama, K., Mitani, Y., Kitabatake, M., Yamauchi, T., and Miyasaka, K. (1997). Continuous low-dose NO inhalation does not prevent monocrotaline-induced pulmonary hypertension in rats. Am. J. Physiol. 272, H517–H524. doi: 10.1152/ajpheart.1997.272.1.H517
Masri, F. A., Comhair, S. A., Dostanic-Larson, I., Kaneko, F. T., Dweik, R. A., Arroliga, A. C., et al. (2008). Deficiency of lung antioxidants in idiopathic pulmonary arterial hypertension. Clin. Transl. Sci. 1, 99–106.
McLaughlin, V. (2013). Managing pulmonary arterial hypertension and optimizing treatment options: prognosis of pulmonary artery hypertension. Am. J. Cardiol. 111, 10C–15C. doi: 10.1016/j.amjcard.2013.01.319
Meghwani, H., Prabhakar, P., Mohammed, S. A., Dua, P., Seth, S., Hote, M. P., et al. (2018). Beneficial effect of Ocimum sanctum (Linn) against monocrotaline-induced pulmonary hypertension in rats. Medicines 5:E34. doi: 10.3390/medicines5020034
Meghwani, H., Prabhakar, P., Mohammed, S. A., Seth, S., Hote, M. P., Banerjee, S. K., et al. (2017). Beneficial effects of aqueous extract of stem bark of Terminalia arjuna (Roxb.), An ayurvedic drug in experimental pulmonary hypertension. J. Ethnopharmacol. 197, 184–194. doi: 10.1016/j.jep.2016.07.029
Moreira-Gonçalves, D., Ferreira, R., Fonseca, H., Padrão, A. I., Moreno, N., Silva, A. F., et al. (2015). Cardioprotective effects of early and late aerobic exercise training in experimental pulmonary arterial hypertension. Basic Res. Cardiol. 110:57. doi: 10.1007/s00395-015-0514-5
Nishimura, T., Faul, J. L., Berry, G. J., Vaszar, L. T., Qiu, D., Pearl, R. G., et al. (2002). Simvastatin attenuates smooth muscle neointimal proliferation and pulmonary hypertension in rats. Am. J. Respir. Crit. Care Med. 166, 1403–1408. doi: 10.1164/rccm.200203-268oc
Nishimura, T., Vaszar, L. T., Faul, J. L., Zhao, G., Berry, G. J., Shi, L., et al. (2003). Simvastatin rescues rats from fatal pulmonary hypertension by inducing apoptosis of neointimal smooth muscle cells. Circulation 108, 1640–1645. doi: 10.1161/01.cir.0000087592.47401.37
Park, A.-M., Wong, C.-M., Jelinkova, L., Liu, L., Nagase, H., and Suzuki, Y. J. (2010). Pulmonary hypertension-induced GATA4 activation in the right ventricle. Hypertens 56, 1145–1151. doi: 10.1161/HYPERTENSIONAHA.110.160515
Potus, F., Hindmarch, C. C. T., Dunham-Snary, K. J., Stafford, J., and Archer, S. L. (2018). Transcriptomic signature of right ventricular failure in experimental pulmonary arterial hypertension: deep sequencing demonstrates mitochondrial, fibrotic, inflammatory and angiogenic abnormalities. Int. J. Mol. Sci. 19:E2730. doi: 10.3390/ijms19092730
Puukila, S., Fernandes, R. O., Türck, P., Carraro, C. C., Bonetto, J. H. P., de Lima-Seolin, B. G., et al. (2017). Secoisolariciresinol diglucoside attenuates cardiac hypertrophy and oxidative stress in monocrotaline-induced right heart dysfunction. Mol. Cell. Biochem. 432, 33–39. doi: 10.1007/s11010-017-2995-z
Qipshidze, N., Tyagi, N., Metreveli, N., Lominadze, D., and Tyagi, S. C. (2012). Autophagy mechanism of right ventricular remodeling in murine model of pulmonary artery constriction. Am. J. Physiol. Heart Circ. Physiol. 302, H688–H696. doi: 10.1152/ajpheart.00777.2011
Rafikova, O., Rafikov, R., Meadows, M. L., Kangath, A., Jonigk, D., and Black, S. M. (2015). The sexual dimorphism associated with pulmonary hypertension corresponds to a fibrotic phenotype. Pulm. Circ. 5, 184–197. doi: 10.1086/679724
Rawat, D. K., Alzoubi, A., Gupte, R., Chettimada, S., Watanabe, M., Kahn, A. G., et al. (2014). Increased reactive oxygen species, metabolic maladaptation, and autophagy contribute to pulmonary arterial hypertension-induced ventricular hypertrophy and diastolic heart failure. Hypertens 64, 1266–1274. doi: 10.1161/HYPERTENSIONAHA.114.03261
Reddy, S., and Bernstein, D. (2015). Molecular mechanisms of right ventricular failure. Circulation 132, 1734–1742. doi: 10.1161/CIRCULATIONAHA.114.012975
Redout, E. M., van der Toorn, A., Zuidwijk, M. J., van de Kolk, C. W. A., van Echteld, C. J. A., Musters, R. J. P., et al. (2010). Antioxidant treatment attenuates pulmonary arterial hypertension-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 298, H1038–H1047. doi: 10.1152/ajpheart.00097.2009
Ryan, J. J., and Archer, S. L. (2014). The right ventricle in pulmonary arterial hypertension: disorders of metabolism, angiogenesis and adrenergic signaling in right ventricular failure. Circ. Res. 115, 176–188. doi: 10.1161/CIRCRESAHA.113.301129
Saugstad, O. D. (2000). Does nitric oxide prevent oxidative mediated lung injury? Acta Paediatr. Oslo Nor. 1992, 905–907. doi: 10.1111/j.1651-2227.2000.tb00405.x
Shi, R., Wei, Z., Zhu, D., Fu, N., Wang, C., Yin, S., et al. (2018). Baicalein attenuates monocrotaline-induced pulmonary arterial hypertension by inhibiting vascular remodeling in rats. Pulm. Pharmacol. Ther. 48, 124–135. doi: 10.1016/j.pupt.2017.11.003
Shults, N. V., Melnyk, O., Suzuki, D. I., and Suzuki, Y. J. (2018). Redox biology of right-sided heart failure. Antioxidants 7:106. doi: 10.3390/antiox7080106
Suzuki, Y. J., and Shults, N. V. (2017). Redox signaling in the right ventricle. Adv. Exp. Med. Biol. 967, 315–323. doi: 10.1007/978-3-319-63245-2_19
Taraseviciene-Stewart, L., Scerbavicius, R., Choe, K. H., Cool, C., Wood, K., Tuder, R. M., et al. (2006). Simvastatin causes endothelial cell apoptosis and attenuates severe pulmonary hypertension. Am. J. Physiol. Lung. Cell. Mol. Physiol. 291, L668–L676.
Türck, P., Lacerda, D. S., Carraro, C. C., de Lima-Seolin, B. G., Teixeira, R. B., Poletto Bonetto, J. H., et al. (2018). Trapidil improves hemodynamic, echocardiographic and redox state parameters of right ventricle in monocrotaline-induced pulmonary arterial hypertension model. Biomed. Pharmacother. 103, 182–190. doi: 10.1016/j.biopha.2018.04.001
van de Veerdonk, M. C., Kind, T., Marcus, J. T., Mauritz, G.-J., Heymans, M. W., Bogaard, H.-J., et al. (2011). Progressive right ventricular dysfunction in patients with pulmonary arterial hypertension responding to therapy. J. Am. Coll. Cardiol. 58, 2511–2519. doi: 10.1016/j.jacc.2011.06.068
Voelkel, N. F., Bogaard, H. J., Al Husseini, A., Farkas, L., Gomez-Arroyo, J., and Natarajan, R. (2013). Antioxidants for the treatment of patients with severe angioproliferative pulmonary hypertension? Antioxid. Redox Signal. 18, 1810–1817. doi: 10.1089/ars.2012.4828
Wang, L., Zheng, Q., Yuan, Y., Li, Y., and Gong, X. (2017). Effects of 17β-estradiol and 2-methoxyestradiol on the oxidative stress-hypoxia inducible factor-1 pathway in hypoxic pulmonary hypertensive rats. Exp. Ther. Med. 13, 2537–2543. doi: 10.3892/etm.2017.4243
Wang, X., Shults, N. V., and Suzuki, Y. J. (2017). Oxidative profiling of the failing right heart in rats with pulmonary hypertension. PLoS One 12:e0176887. doi: 10.1371/journal.pone.0176887
Watts, J. A., Marchick, M. R., and Kline, J. A. (2010). Right ventricular heart failure from pulmonary embolism: key distinctions from chronic pulmonary hypertension. J. Card. Fail. 16, 250–259. doi: 10.1016/j.cardfail.2009.11.008
Wedgwood, S., Steinhorn, R. H., and Lakshminrusimha, S. (2019). Optimal oxygenation and role of free radicals in PPHN. Free Radic. Biol. Med. doi: 10.1016/j.freeradbiomed.2019.04.001 [Epub ahead of print].
Woo, E., Kato, R., Imano, H., Fujiwara, Y., Ijiri, Y., Okada, Y., et al. (2017). Capillary degeneration and right ventricular remodeling due to hypoxic stress with Sugen5416. Curr. Vasc. Pharmacol. 15, 589–598. doi: 10.2174/1570161115666170427154217
Xu, D., Guo, H., Xu, X., Lu, Z., Fassett, J., Hu, X., et al. (2011). Exacerbated pulmonary arterial hypertension and right ventricular hypertrophy in animals with loss of function of extracellular superoxide dismutase. Hypertens 58, 303–309. doi: 10.1161/HYPERTENSIONAHA.110.166819
Yu, H., Liu, J., Dong, Y., Xu, M., Xu, L., Guan, H., et al. (2018). Anti-hypoxic effect of dihydroartemisinin on pulmonary artery endothelial cells. Biochem. Biophys. Res. Commun. 506, 840–846. doi: 10.1016/j.bbrc.2018.10.176
Zelko, I. N., Zhu, J., and Roman, J. (2018). Role of SOD3 in silica-related lung fibrosis and pulmonary vascular remodeling. Respir. Res. 19:221. doi: 10.1186/s12931-018-0933-6
Zhang, Q., Fan, K., Wang, P., Yu, J., Liu, R., Qi, H., et al. (2016). Carvacrol induces the apoptosis of pulmonary artery smooth muscle cells under hypoxia. Eur. J. Pharmacol. 770, 134–146. doi: 10.1016/j.ejphar.2015.11.037
Zimmer, A., Teixeira, R. B., Bonetto, J. H. P., Siqueira, R., Carraro, C. C., Donatti, L. M., et al. (2017). Effects of aerobic exercise training on metabolism of nitric oxide and endothelin-1 in lung parenchyma of rats with pulmonary arterial hypertension. Mol. Cell. Biochem. 429, 73–89. doi: 10.1007/s11010-016-2937-1
Keywords: oxidative stress, right ventricle, pulmonary hypertension, remodeling, RV failure
Citation: Mikhael M, Makar C, Wissa A, Le T, Eghbali M and Umar S (2019) Oxidative Stress and Its Implications in the Right Ventricular Remodeling Secondary to Pulmonary Hypertension. Front. Physiol. 10:1233. doi: 10.3389/fphys.2019.01233
Received: 28 February 2019; Accepted: 09 September 2019;
Published: 24 September 2019.
Edited by:
Feng Chen, Augusta University, United StatesReviewed by:
Saurabh Aggarwal, The University of Alabama at Birmingham, United StatesDeepesh Pandey, Johns Hopkins University, United States
Copyright © 2019 Mikhael, Makar, Wissa, Le, Eghbali and Umar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Soban Umar, c3VtYXJAbWVkbmV0LnVjbGEuZWR1
†These authors have contributed equally to this work as co-first authors