 Paola Bellosta
Paola Bellosta Alessia Soldano
Alessia Soldano- 1Laboratory of Metabolism of Cell Growth and Neuronal Survival, Department of Cellular, Computational and Integrative Biology (CIBio), University of Trento, Trento, Italy
- 2Department of Medicine, New York University Langone Medical Center, New York, NY, United States
- 3Laboratory of Translational Genomics, Department of Cellular, Computational and Integrative Biology (CIBio), University of Trento, Trento, Italy
Autism Spectrum Disorder (ASD) is a complex group of multi-factorial developmental disorders that leads to communication and behavioral defects. Genetic alterations have been identified in around 20% of ASD patients and the use of genetic models, such as Drosophila melanogaster, has been of paramount importance in deciphering the significance of these alterations. In fact, many of the ASD associated genes, such as FMR1, Neurexin, Neuroligins and SHANK encode for proteins that have conserved functions in neurons and during synapse development, both in humans and in the fruit fly. Drosophila is a prominent model in neuroscience due to the conserved genetic networks that control neurodevelopmental processes and to the ease of manipulating its genetics. In the present review we will describe recent advances in the field of ASD with a particular focus on the characterization of genes where the use of Drosophila has been fundamental to better understand their function.
Autism
Autism Spectrum Disorder (ASD) is a complex developmental neurological disease characterized by persistent deficits in social behaviors (communication, interaction), presence of repetitive and restrictive comportments and is often associated with motor deficits and sleep abnormalities, among others. Among individuals suffering from ASD, there is a high frequency of intellectual disability and mental retardation, although the described frequency is variable due to the difficulty in assessing cognitive performance in certain groups of ASD patients (O’Brien and Pearson, 2004; Chakrabarti and Fombonne, 2005). Autism is not considered a single gene disorder because it is caused by both genetic and non-genetic risk factors that induce a complex range of different symptoms for which the precise causes are unknown (Park et al., 2016). Genetic disorders, such as Fragile X syndrome (FXS), Down syndrome, and, more recently, Asperger’s and Rett syndrome, have been associated with ASD. In less than 20% of patients has a clear monogenic cause for ASD been identified and most of these studies highlighted mutations in genes involved in several aspects of synapse biology, such as synaptogenesis/synaptic plasticity/morphology/function and axon motility (De Rubeis et al., 2014; Iossifov et al., 2014; Luo et al., 2018).
ASD Associated Defects in Synaptogenesis and Synaptic Plasticity
The identification of ASD susceptibility genes involved in various aspects of synapse biology, lead to the hypothesis that aberrant synaptogenesis/synaptic function might be a central process in ASD (Peca and Feng, 2012; Zoghbi and Bear, 2012). Multiple studies in animal models converge on the concept that reproducing alterations in ASD genes leads to aberrant synaptic morphology and function (Peca and Feng, 2012; Zoghbi and Bear, 2012). Interestingly, observation of post-mortem ASD patients’ tissues indicate that dendritic spines, postsynaptic sites in the mammalian brain, are present at a higher density in ASD patients and this condition is most commonly found in ASD subjects with lower levels of cognitive performance (Hutsler and Zhang, 2010). Moreover, ASD patients have an increased density of dendritic spines in layer V pyramidal neurons and reduced developmental spine pruning, a process needed to achieve correct neuronal communication (Tang et al., 2014). This is of particular interest since it has been postulated that ASD might be caused by an altered balance between excitatory and inhibitory synapses, probably due to defects in synapse elimination/formation (Ramocki and Zoghbi, 2008; Gatto and Broadie, 2010).
Drosophila as a Model to Study ASD
Drosophila is an excellent model to study ASD to understand the consequences of genetic alterations found in ASD patients and to identify the molecular mechanisms underlying the role of ASD related genes in synaptic function and plasticity (Doi et al., 2016; Tian et al., 2017). Moreover, 75% of the human disease genes have orthologs in Drosophila (Bier, 2005), rendering the fruit fly a highly tractable genetic model organism to understand the molecular bases of ASDs. In the past decade the panel of genetic tools that can be used to study human disease genes has expanded massively (Table 1; Chow and Reiter, 2017). Drosophila has been used for classical unbiased screens, using either mutagens to induce random mutations in the genome or genome-wide RNAi/CRISPR screens, to identify genes that lead to ASDs-like phenotypes. On the other hand, known ASDs genes have been perturbed to mimic the patient’s condition and to study the biological consequences of these alterations.

Table 1. Summary of the genetic tools that can be used to study the physiological role of ASD genes and to understand their contribution, alone and in combination with others, to ASD development.
In the present review we describe the latest studies that use Drosophila to clarify the function of the most representative genes associated with ASD (Figure 1).
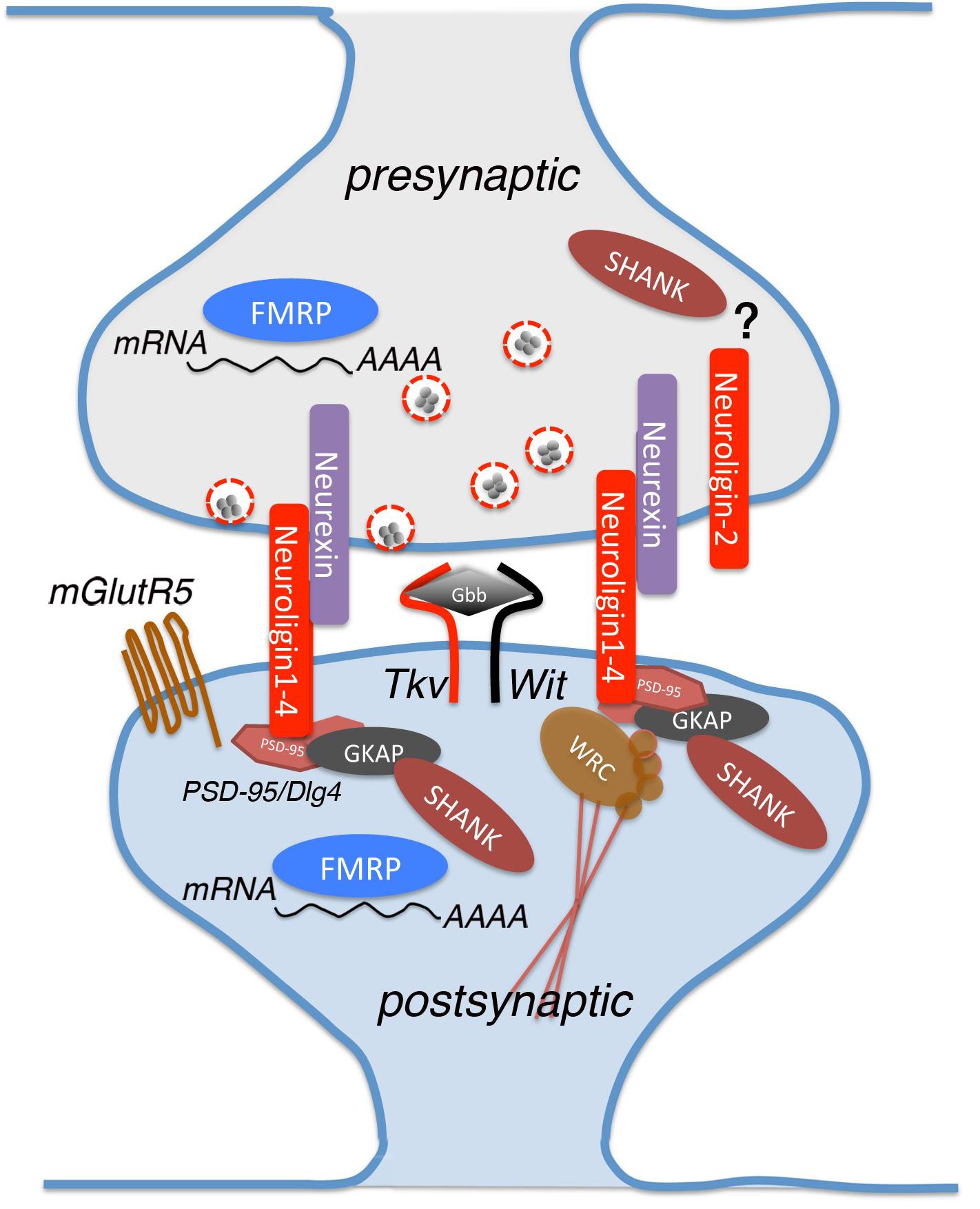
Figure 1. A schematic view of a glutamatergic synapse, showing the proteins analyzed in this review and implicated in Autism Spectrum Disorder (ASD). Neurexin and neuroligin are located in pre- and post-synaptic sites where they interact with multiple partners for mediating synapse development and maturation. Neuroligins binds to PSD-95, which interacts indirectly with Shank by intracellular complexes containing also Guanylate-kinase-associated protein (GKAP). dNlg1 and 3 act mostly in pre-synaptic terminals while dNlg2 functions is both pre- and post-synaptic ends. Other proteins involved in the synaptic establishment are FMRP RNA-binding and Thickveins (Tkv), that are expressed in both synaptic sites, mGlutR5 and Wishful thinking (Wit) that are found specifically on the post-synaptic membrane. Glass Bottom Boat (Gbb) is the ligand of both Tkv and Wit, homologous to vertebrate bone morphogenic protein (BMP).
dfmr1
Fragile X syndrome (FXS) is a neuro-developmental disease that leads to intellectual disability and is the most common form of autism of monogenic origin (Mila et al., 2018). FXS is caused by a variable expansion of a trinucleotide (CGG) repeat in the 5′ UTR of the fragile X mental retardation-1 gene (FMR1), or less frequently, by point mutations in FMR1 (Collins et al., 2010; Handt et al., 2014), that leads to loss of FMR1 protein (Pieretti et al., 1991). FMR1 encodes for an RNA-binding protein, FMRP, that mainly inhibits translation by binding to specific sequences on mRNAs (Darnell et al., 2011; Ascano et al., 2012).
Drosophila harbors only one FMRP ortholog, dfmr1, that shares high homology with its mammalian counterpart (Wan et al., 2000). A recent study using fruit flies suggested that the molecular function of dfmr1 might not only be translation repression. Ribosome-profiling of oocytes upon dfmr1 knockdown shows that dfmr1 RNAi leads to both enhanced and reduced mRNA translation in proportion to protein size, with dmfr1 predominantly up-regulating bigger proteins (Greenblatt and Spradling, 2018). Interestingly, many of the down-regulated genes are orthologs of genes implicated in ASD, such as the E2 Ubiquitin-conjugating enzyme BIRC6, or the Vacuolar H + ATPase DMXL2, both of which are associated with intellectual disabilities and neurodevelopmental disorders in humans, a result that outlines the relevance of using Drosophila genetics to gain insights into these human pathologies.
dfmr1 plays a central role in synaptic plasticity, indeed loss-of-function mutants of dfmr1 show synaptic overgrowth, increased number and enlargement of synaptic boutons, and excessive branching at the Neuromuscular Junctions (NMJ). Mutations in dfmr1 affect synaptic transmission at histaminergic photoreceptor synapses (central) and glutamatergic NMJ synapses (peripheral) (Zhang et al., 2001).
dfmr1 controls brain development and neural circuit assembly (Morales et al., 2002). Loss of dfmr1 causes axon extension defects of Dorsal Cluster neurons (DC) and lateral neurons (LNvs), and neurite-branching abnormalities in DC neurons. Interestingly, loss and gain of function of dfmr1 lead to similar phenotypic defects, indicating that the levels of dfmr1 are critical for brain development (Morales et al., 2002). The role of dfmr1 in regulating axon morphology has also been demonstrated in the Mushroom Body neurons (MB), a higher hierarchy circuit involved in olfactory learning and memory (Lee et al., 1999; Akalal et al., 2006). Loss of dfmr1 in all MB neuronal classes increases structural complexity and induces growth of additional processes from neuronal soma, supporting the overbranching and overgrowth phenotype visible in dendrites and axons (Michel et al., 2004; Pan et al., 2004; Tessier and Broadie, 2008). dfmr1 also controls remodeling of two classes of MB body extrinsic input and output neurons, namely GABA-ergic MVP2 (MBON-γ1pedc > α/β) and projection neuron (PN). In fact, the dendritic arborizations of these neurons are enlarged in dfmr1 null animals. MVP2 and PN neurons respond in the opposite way to this activity by remodeling their dendritic arbor and dfmr1 is required for this function (Doll and Broadie, 2015).
Loss of dfmr1 causes several behavioral defects including deficits in memory (Coffee et al., 2010; Gatto et al., 2014), associative learning defects (Choi et al., 2010; Santos et al., 2014; Doll and Broadie, 2016) and alteration of circadian rhythm (Sofola et al., 2008; Gatto and Broadie, 2009), which are all known to be linked to defects in LNvs morphology (Dockendorff et al., 2002; Sofola et al., 2008). Gene expression analysis at different times during the day (Circadian-time points CT) highlighted a subset of mRNAs and miRNAs that, in dfmr1 mutant flies, were altered at a specific time point only. This pattern of gene expression alteration reflects a circadian rhythm-dependent alteration (Xu et al., 2012). dfmr1 mutants also exhibit sleep defects: showing a prolonged “sleep phase”, which is reduced by overexpression of dfmr1 in the MB (Bushey et al., 2009), and a deeper sleep (night-like) phenotype at daylight (van Alphen et al., 2013). Similarly, patients with FXS suffer from sleep disorders suggesting a conserved function of FMR1 in controlling components of the circadian rhythm.
dfmr1 modulates grooming behavior, recapitulating the repetitive behavior observed in ASD patients, and is therefore of great relevance for translational studies. dfmr1 mutants groom more than control flies; this phenotype worsens with age and can be suppressed by treatment with reserpine, which blocks the Drosophila vesicular monoamine transporter (dVMAT) (Tauber et al., 2011).
dfmr1 mutants show also impairment in odor-induced attraction and aversion, due to reduced lateral interactions across the olfactory glomeruli and impairment of the lateral inhibition in the antennal lobe caused by weaker inhibition from GABAergic interneurons (Franco et al., 2017). These results are of great interest in comparative studies in humans given that alterations in GABAergic transmission and lack of inhibition might be central components of the neuropathology of FXS.
FXS patients, together with most individuals with ASD, suffer from dysfunctions in sensory processing (SPD), meaning they respond to a certain behavioral stimulus differently than individuals in the average population (Sinclair et al., 2017). This dysfunction has been investigated in flies by studying the sensory processing of the Drosophila stress odorant (dSO) (Androschuk et al., 2018). dfmr1 null animals have lost dSO avoidance-behavior and dfmr1 is required in the MB and glia to mediate the dSO sensory response. This behavioral defect can be pharmacologically rescued by feeding adults with molecules that target cAMP/cGMP signaling pathways, such as the cAMP-increasing agent IBMX (3-isobutyl-1-methylxanthine), and the cAMP-dependent PKA activator and the cGMP dependent phosphodiesterase inhibitor 8-CPT (8-(4-Chlorophenylthio)adenosine 3′,5′-cyclic monophosphate), suggesting a potential use of these drugs In ASD treatments (Franco et al., 2017).
Up to now only few potentially pathogenic mutations have been identified in FXS patients. The most studied is an isoleucine to asparagine substitution (I304N) within the second K-homologous (KH) domain of the human FMRP, which is associated with very severe FXS (De Boulle et al., 1993). However, mutations in the highly conserved isoleucine residues I244N and I307N of the KH domain in Drosophila resulted in dfmr1 null-like, MB β-lobe midline crossing phenotype, though at a lower frequencies than in dfmr1 mutants. These KH mutants also fail to retain rhythmic locomotion activity in constant darkness, but with a milder phenotype than in dfmr1 null animals (Banerjee et al., 2007). More recently, Okray et al. (2015) characterized in Drosophila a new FMR1 frameshift mutation (Guanine insertion in exon-15) found in a patient with FXS. This mutation generates a novel peptide sequence with a premature stop codon, resulting in the truncation of the FMRP protein at the C-terminus and loss of the arginine-glycine-rich motif (RGG box), which is one of the FMRP RNA-binding domains. Overexpression of a mutant form of dfmr1 (dfmr1-ΔC+NLS allele), which closely mimics the human variant, in LNvs, results in axons that fail to extend medially, leads to aberrant bifurcations of axonal bundle and to the formation of axonal “tangles.”
Neurexin and Neuroligins
Neurexin (Nrx) and Neuroligins (Nlgs) are adhesion molecules that function as trans-synaptic binding partners involved in synaptogenesis (Knight et al., 2011). Several genetic alterations including point mutations, deletions and translocation events have been identified in NRXN1, NLGN3 and NLGN4 in ASD patients (Laumonnier et al., 2004; Kim et al., 2008; Yan et al., 2008).
In Drosophila, loss of dNlgs (Drosophila harbors 4 dNlgs) and dNrx results in developmental defects at the NMJ such as an altered number of boutons, aberrant presynaptic/postsynaptic structure, and impaired synaptic transmission (Sun et al., 2011; Chen et al., 2012; Hahn et al., 2013; Xing et al., 2014). In particular, dNlg1, 2, and 4 have a positive effect on synaptic growth at the NMJ and their loss leads to a reduction of synaptic boutons (Banovic et al., 2010; Sun et al., 2011), while loss of dNlg3 leads to the opposite phenotype (Xing et al., 2014). dNlg1 and 3 act mostly in pre-synaptic terminals while dNlg2 functions in both pre- and post-synaptic ends (Chen et al., 2012; Xing et al., 2014). dNlgs and dNrx work together to coordinate these functions.
Recent studies dissected the molecular mechanisms underlying these functions: Zhang and colleagues (Zhang et al., 2017) demonstrated that dNlg4 modulates BMP signaling by maintaining the protein levels of the type-I BMP receptor Thick Veins (Tkv) at the presynaptic sites. BMP signaling seems to be a target of several dNlgs/dNrx complexes; in fact, it has been demonstrated that Tkv levels are also reduced in dNlg1 and dNrx mutants (Banerjee et al., 2017). Interestingly, mutants of the type-II BMP receptor Wishful Thinking (Wit) show phenotypic similarities to dNlg1 and dNrx mutants (Banerjee and Riordan, 2018). dNrx, dNlg1 and Wit seem to form a complex at the NMJ, where dNrx and dNlg1 are required for both localization and stability of Wit. dNrx is found in a complex with Wit and its ligand Gbb, the ortholog of vertebrate BMP, and other downstream effectors to allow proper axonal transport and microtubule organization (Banerjee and Riordan, 2018).
dNlg1 also directly affects the actin cytoskeleton via interaction with the WAVE regulatory complex (WRC), one of the key players in F-actin assembly (Xing et al., 2018). In particular, dNlg1 mediates the effect of dNrx on actin at post-synaptic terminals by binding to the WRC and recruiting it to the post-synaptic membrane. dNlg1-WRC interaction mediates post-synaptic F-actin assembly, which is required for normal NMJ assembly and boutons growth, while dNrx and dNlg4 control axonal branching (Liu et al., 2017). dNrx is also expressed in the axon terminals and interstitial branches of L4 lamina neurons that project into the medulla neuropil, and is required for L4 columnar restriction. In particular, dNlg4/dNrx interaction promotes dNrx clustering on the membrane which results in dNrx/Ephrin interaction and subsequent Ephrin clustering (Neriec and Desplan, 2016).
In mammals, Neurexins and Neuroligins are also central for the establishment of functional synaptic networks (Sudhof, 2017). The findings described in Drosophila strongly support that dNlgs and dNrx have a primary role in synapse formation/maintenance and outline how these signaling pathways might be further assessed as pharmacological targets.
Shank
The family of SH3 and multiple ankyrin repeat domains proteins (SHANKs) is composed of three members: SHANK1, 2 and 3. These proteins are scaffolding proteins present at the post-synaptic density in glutamatergic synapses. SHANK3 deletions, duplications, and mutations have been frequently reported in patients with ASD (Durand et al., 2007; Boccuto et al., 2013; Leblond et al., 2014). SHANK3 mutations are one of the most prevalent monogenic causes of ASD, accounting for at least 0.69% of all cases, and patients harboring SHANK3 truncating mutations display autism combined with moderate to severe intellectual disabilities. Moreover, 22q13.3 deletion syndrome, also known as Phelan–McDermid syndrome, which is characterized by ASD or ASD-traits, is caused by deletions and mutations that lead to the loss of a functional copy of SHANK3 (Soorya et al., 2013). Recent META-analysis of SHANK family mutations in ASD identified deletions disrupting SHANK1 and SHANK2 genes in patients, but not duplication of either (Leblond et al., 2014). This study also suggested the existence of a gradient of severity in cognitive impairment depending on the SHANK gene mutated. So far, the molecular mechanisms underlying SHANK functions remain partially unclear and studies using Drosophila have contributed significantly in addressing this question.
Drosophila harbors only one ortholog of the SHANK family called Prosap/Shank (Liebl and Featherstone, 2008). Harris et al. (2016) described that Shank localizes to the post-synaptic membrane at the NMJ where it is involved in the regulation of synapse morphology and maturation. The levels of Shank at synapses are critical; Shank mutants exhibit a 24% reduction in synaptic boutons and an excessively high number of immature synaptic structures. On the other hand, Shank heterozygous animals show an intermediate phenotype, with a 15% reduction in boutons numbers but no increase in immature synaptic structures. Interestingly, post-synaptic Shank overexpression leads to phenotypes similar to those observed in Shank mutants, confirming that the levels of Shank are critical to achieve normal synaptic development. Shank defects have been associated with the modulation of Wnt/FNI (Frizzled Nuclear Import) pathway at the post-synaptic terminal. Shank affects the internalization of the Frizzled-2 (Fz2) receptor, most likely by organizing molecules associated with its internalization and trafficking to the nucleus (Harris et al., 2016). A more recent study from Wu et al. (2017) described Shank expression in axons and at the presynaptic terminal, but not at the post-synaptic sites of the NMJs. Moreover, they generated new Shank mutant alleles that show normal morphology at the NMJ and at the post-synaptic density. The authors focused on the role of Shank in the CNS since the protein, like its mammalian counterpart, is expressed in the brain and enriched in the neuropil region. Loss of Shank leads to developmental defects of the synapses in the larval MB Calyx, where the protein exerts its function at both pre- and post-synaptic sites. Synapse defects are visible also in the adult MB Calyx, that presents altered microglomeruli and abnormal localization of the α7 subunit of nicotinic acetylcholine receptor (AChR Dα7) and Choline acetyltransferase (ChAT). These abnormalities result in significant impairment of the olfactory learning in Shank mutants.
mGluR
A genome-wide association study (GWAS) of copy-number variation (CNVs) in patients with autism that lead to defective gene family interaction networks (GFINs) (Hadley et al., 2014) identified CNVs in the metabotropic glutamate receptor (mGluR) signaling pathway in 5.8% of patients with ASD.
The involvement of mGluR in autism has been highlighted in its involvement in FXS. The “mGluR theory” states that loss of FMRP in FXS results in increased glutamatergic signaling via mGluR5, leading to uncontrolled increases in local mRNA translation (Pop et al., 2014). In fact, mGluR activation normally stimulates synthesis of proteins involved in stabilization of long-term depression (LTD) (Weiler et al., 1997). In FXR patients, this translation stimulation is not balanced by the presence of FMRP and leads to increased AMPA receptor internalization and destabilization of the synapses.
As described in the previous section, loss of dfmr1 activity in Drosophila mimics classic FXS symptoms and the impact of mGluR inhibition on these phenotypes has been studied by several groups. McBride et al. (2005) demonstrated that treatment with mGluR antagonists or Lithium Chloride (LiCl), during development and adulthood, restores the naive courtship levels of the dfmr1 mutants. Similar treatments also rescue dfmr1 defects in immediate recall-memory and the lack of short-term memory. Moreover, the treatment with mGlur antagonists greatly reduces axon growth defects (β lobe overgrowth) observed in the MB of dfmr1 mutants. Interestingly, the free running rest-activity rhythm defects of dfmr1 mutant flies are not rescued by these treatments, suggesting that not all the phenotypes observed in dfmr1 null flies are due to upregulation of mGluR signaling (McBride et al., 2005). Recently, the study of the relationship between mGluR and dfmr1 was extended by investigating the effect of aging on dfmr1 mutants. In particular, Choi et al. (2010) demonstrated that dfmr1 mutants show an age-dependent loss of learning that was rescued by the administration of mGluR antagonists and LiCl. Interestingly, treatment during development rescued the learning defect but not the courtship phenotype, indicating that the rescue obtained by treatment during development alone is not permanent. In fact, when aged flies were treated during development and adulthood or during adulthood alone, the naive courtship was restored (Choi et al., 2010). The interconnection between dfmr1 and mGluR has been demonstrated also through genetic interaction, where loss of dfmr1 was shown to partially alleviate the phenotypes at the NMJ resulting from loss of mGluR, possibly via reduction of translational inhibition. Similarly, loss of mGluR partially rescues the defects caused by loss of dfmr1 and the consequent impairment of translation regulation (Repicky and Broadie, 2009).
Dopamine Network
The dopamine (DA) network has been widely associated with ASD, where mutations in genes of the DA signaling, such as the Dopamine transporter (DAT), Synataxin 1 (STX1), the DA-receptors, and enzymes involved in DA metabolism, have been associated with autism. Work from several groups suggested that dopamine imbalances in specific circuits of the brain could lead to ASD related behavior (Gadow et al., 2010; Nakamura et al., 2010; Paval, 2017). Moreover, increased size of DA-enriched brain regions, such as the striatum, has been associated with the severity of the disorder (Langen et al., 2014).
Several years ago, a new missense mutation in the human DAT gene (hDAT-T356M) was identified. This mutation results in reduced ability to accumulate intracellular DA, due to an increased dopamine efflux (Hamilton et al., 2013). The functional consequences of this mutation have been studied in Drosophila by expressing the hDAT-T356M in DAT null mutant flies. These animals show hyperactivity as compared to flies expressing the wt hDAT gene due to increased extracellular levels of DA and abnormal dopamine efflux (Hamilton et al., 2013).
Exome sequencing studies in ASD patients led to the identification of missense variants in the hDAT (hDAT-R51W) and in STX1A (STX1A-R26Q) genes. The analysis of these mutations showed defects in the reverse transport of DA that leads to behavioral abnormalities (De Rubeis et al., 2014; Iossifov et al., 2014; Cartier et al., 2015). Mechanistically, the STX1A-R26Q variant is less phosphorylated by Caseine Kinase-2 (CK2), a modification that supports the reverse transport of DA and leads to a reduction in DA efflux. Similarly, the hDAT-R51W variant shows a reduced interaction with STX1 and reduced DA efflux. The effects of these mutations have been characterized in vivo in Drosophila by assessing locomotion. In fact, Amphetamine (AMPH) feeding stimulates Drosophila locomotion but only in the presence of a fully functional DA network. Moreover, expression of a dominant negative form of CK2, mimicking the STX1A-R26Q variant, in DA neurons renders flies insensitive to AMPH. On the other hand, flies harboring the hDAT-R51W mutation increased their locomotion upon AMPH significantly less than wt hDAT expressing flies, confirming the reduced ability of AMPH to cause DA efflux in hDAT R/W mutants (Cartier et al., 2015).
Concluding Remarks
Drosophila melanogaster is an extremely useful model to understand the molecular mechanisms underlying the function of ASD associated genes in brain development and function (Figure 2).
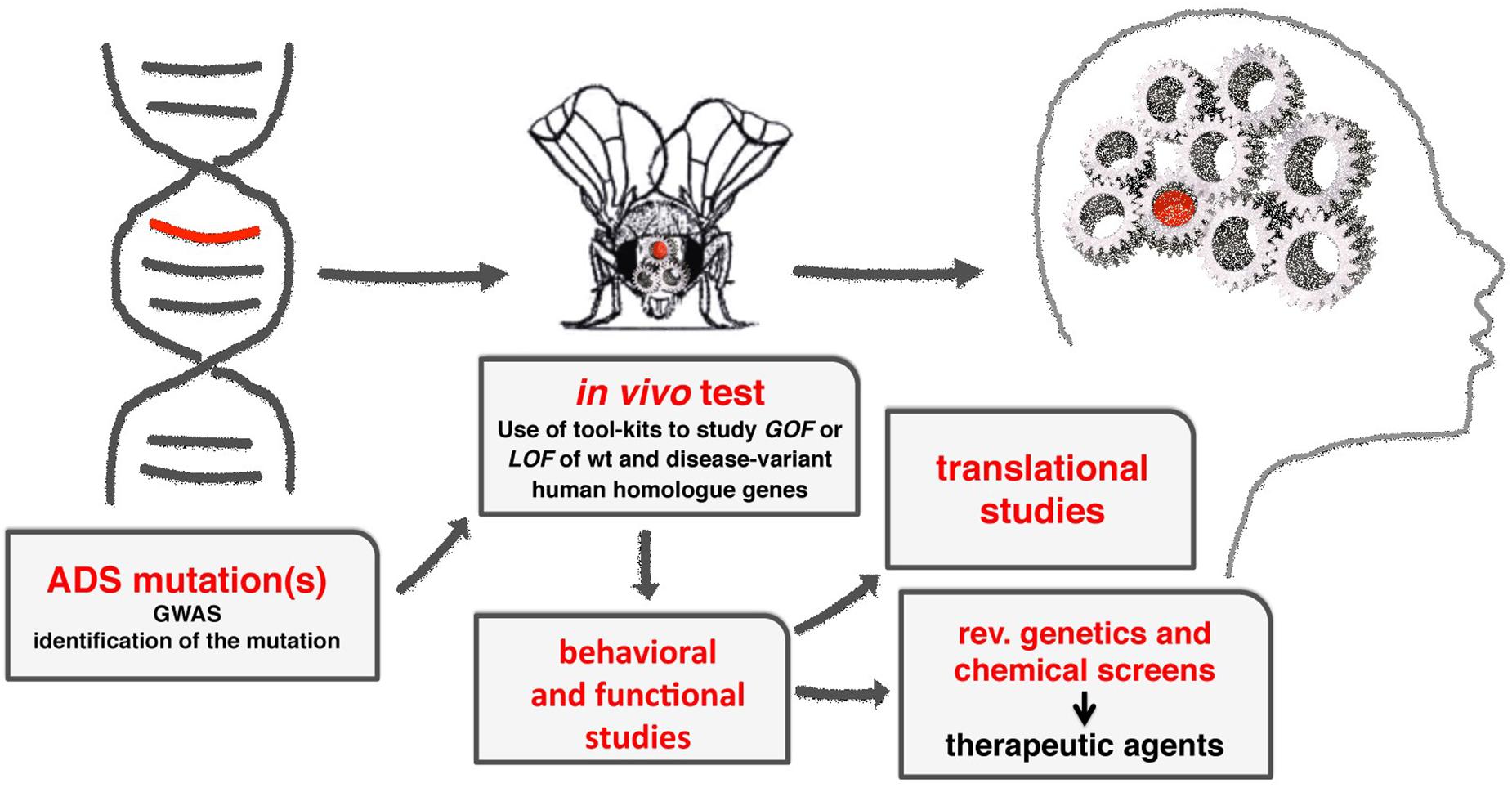
Figure 2. A diagram showing the flow-through to study the molecular mechanisms of ADS using Drosophila. From the initial identification of human genes associated to ADS, to the characterization of their functions using GOF and LOF experiments, applying the milieu of genetic tools available (see Table 1). Using transgenic animals, functional and behavioral studies are used to provide the translational benefits necessary to identify or clarify the function of the gene in humans. Moreover, the fruit fly can be easily adapted to perform reverse genetics or chemical screens to identify novel genes or therapeutic drugs in ADSs.
Moreover, the fast growing body of GWAS provides detailed information on the presence of genomic alterations in patients, for which the functional consequences and their relevance in ASD are difficult to interpret (i.e., gene redundancy, complex networks etc.). The fruit fly allows for the analysis of the effects of multiple genetic modifications in different subsets of cells, allowing for the discrimination of the contributions of combinations of genetic alterations co-occurring in ASD patients.
Therefore, the combination of genomic analysis of ASD patients together with the use of an easy to manipulate in vivo model with a robust and comparable neuronal development, will be essential to gain insight into the pathogenesis of these disorders.
Author Contributions
Both authors listed contributed to writing and reviewing the manuscript.
Funding
Funding from Cariplo Foundation 2014703 and EHDN 689 for PB. AS is supported by funding from the European Union’s Horizon 2020 Research and Innovation programme under the Marie Skłodowska Curie grant agreement No. 752621.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Sheri Zola for critically reading and correcting the manuscript.
References
Akalal, D.-B. G., Wilson, C. F., Zong, L., Tanaka, N. K., Ito, K., and Davis, R. L. (2006). Roles for Drosophila mushroom body neurons in olfactory learning and memory. Learn. Mem. 13, 659–668. doi: 10.1101/lm.221206
Androschuk, A., He, R. X., Weber, S., Rosenfelt, C., and Bolduc, F. V. (2018). Stress odorant sensory response dysfunction in Drosophila fragile X syndrome mutants. Front. Mol. Neurosci. 11:242. doi: 10.3389/fnmol.2018.00242
Ascano, M. Jr., Mukherjee, N., Bandaru, P., Miller, J. B., Nusbaum, J. D., Corcoran, D. L., et al. (2012). FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature 492, 382–386. doi: 10.1038/nature11737
Banerjee, P., Nayar, S., Hebbar, S., Fox, C. F., Jacobs, M. C., Park, J. H., et al. (2007). Substitution of critical isoleucines in the KH domains of Drosophila fragile X protein results in partial loss-of-function phenotypes. Genetics 175, 1241–1250. doi: 10.1534/genetics.106.068908
Banerjee, S., and Riordan, M. (2018). Coordinated regulation of axonal microtubule organization and transport by Drosophila neurexin and BMP pathway. Sci. Rep. 8:17337. doi: 10.1038/s41598-018-35618-7
Banerjee, S., Venkatesan, A., and Bhat, M. A. (2017). Neurexin, neuroligin and wishful thinking coordinate synaptic cytoarchitecture and growth at neuromuscular junctions. Mol. Cell. Neurosci. 78, 9–24. doi: 10.1016/j.mcn.2016.11.004
Banovic, D., Khorramshahi, O., Owald, D., Wichmann, C., Riedt, T., Fouquet, W., et al. (2010). Drosophila neuroligin 1 promotes growth and postsynaptic differentiation at glutamatergic neuromuscular junctions. Neuron 66, 724–738. doi: 10.1016/j.neuron.2010.05.020
Bier, E. (2005). Drosophila, the golden bug, emerges as a tool for human genetics. Nat. Rev. Genet. 6, 9–23. doi: 10.1038/nrg1503
Boccuto, L., Lauri, M., Sarasua, S. M., Skinner, C. D., Buccella, D., Dwivedi, A., et al. (2013). Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur. J. Hum. Genet. 21, 310–316. doi: 10.1038/ejhg.2012.175
Bushey, D., Tononi, G., and Cirelli, C. (2009). The Drosophila fragile X mental retardation gene regulates sleep need. J. Neurosci. 29, 1948–1961. doi: 10.1523/JNEUROSCI.4830-08.2009
Cartier, E., Hamilton, P. J., Belovich, A. N., Shekar, A., Campbell, N. G., Saunders, C., et al. (2015). Rare autism-associated variants implicate syntaxin 1 (STX1 R26Q) phosphorylation and the dopamine transporter (hDAT R51W) in dopamine neurotransmission and behaviors. EBioMedicine 2, 135–146. doi: 10.1016/j.ebiom.2015.01.007
Chakrabarti, S., and Fombonne, E. (2005). Pervasive developmental disorders in preschool children: confirmation of high prevalence. Am. J. Psychiatry 162, 1133–1141. doi: 10.1176/appi.ajp.162.6.1133
Chen, Y. C., Lin, Y. Q., Banerjee, S., Venken, K., Li, J., Ismat, A., et al. (2012). Drosophila neuroligin 2 is required presynaptically and postsynaptically for proper synaptic differentiation and synaptic transmission. J. Neurosci. 32, 16018–16030. doi: 10.1523/JNEUROSCI.1685-12.2012
Choi, C. H., McBride, S. M., Schoenfeld, B. P., Liebelt, D. A., Ferreiro, D., Ferrick, N. J., et al. (2010). Age-dependent cognitive impairment in a Drosophila fragile X model and its pharmacological rescue. Biogerontology 11, 347–362. doi: 10.1007/s10522-009-9259-6
Chow, C. Y., and Reiter, L. T. (2017). Etiology of human genetic disease on the fly. Trends Genet. 33, 391–398. doi: 10.1016/j.tig.2017.03.007
Coffee, R. L., Tessier, C. R., Woodruff, E. A., and Broadie, K. (2010). Fragile X mental retardation protein has a unique, evolutionarily conserved neuronal function not shared with FXR1P or FXR2P. Dis. Models Mech. 3, 471–485. doi: 10.1242/dmm.004598
Collins, S. C., Bray, S. M., Suhl, J. A., Cutler, D. J., Coffee, B., Zwick, M. E., et al. (2010). Identification of novel FMR1 variants by massively parallel sequencing in developmentally delayed males. Am. J. Med. Genet. A 152A, 2512–2520. doi: 10.1002/ajmg.a.33626
Darnell, J. C., Van Driesche, S. J., Zhang, C., Hung, K. Y., Mele, A., Fraser, C. E., et al. (2011). FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261. doi: 10.1016/j.cell.2011.06.013
De Boulle, K., Verkerk, A. J., Reyniers, E., Vits, L., Hendrickx, J., Van Roy, B., et al. (1993). A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat. Genet. 3, 31–35. doi: 10.1038/ng0193-31
De Rubeis, S., He, X., Goldberg, A. P., Poultney, C. S., Samocha, K., Cicek, A. E., et al. (2014). Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215. doi: 10.1038/nature13772
Dockendorff, T. C., Su, H. S., McBride, S. M., Yang, Z., Choi, C. H., Siwicki, K. K., et al. (2002). Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron 34, 973–984. doi: 10.1016/s0896-6273(02)00724-9
Doi, A., Takagi, M., Fujimoto, K., Kakihara, J., Hayashi, Y., Tatsumi, H., et al. (2016). Long rp’ tachycardia with unusual entrainment responses: what is the mechanism? J. Cardiovasc. Electrophysiol. 27, 1242–1244. doi: 10.1111/jce.12987
Doll, C. A., and Broadie, K. (2015). Activity-dependent FMRP requirements in development of the neural circuitry of learning and memory. Development 142, 1346–1356. doi: 10.1242/dev.117127
Doll, C. A., and Broadie, K. (2016). Neuron class-specific requirements for fragile X mental retardation protein in critical period development of calcium signaling in learning and memory circuitry. Neurobiol. Dis. 89, 76–87. doi: 10.1016/j.nbd.2016.02.006
Durand, C. M., Betancur, C., Boeckers, T. M., Bockmann, J., Chaste, P., Fauchereau, F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27. doi: 10.1038/ng1933
Franco, L. M., Okray, Z., Linneweber, G. A., Hassan, B. A., and Yaksi, E. (2017). Reduced lateral inhibition impairs olfactory computations and behaviors in a drosophila model of fragile X syndrome. Curr. Biol. 27, 1111–1123. doi: 10.1016/j.cub.2017.02.065
Gadow, K. D., Devincent, C. J., Olvet, D. M., Pisarevskaya, V., and Hatchwell, E. (2010). Association of DRD4 polymorphism with severity of oppositional defiant disorder, separation anxiety disorder and repetitive behaviors in children with autism spectrum disorder. Eur. J. Neurosci. 32, 1058–1065. doi: 10.1111/j.1460-9568.2010.07382.x
Gatto, C. L., and Broadie, K. (2009). Temporal requirements of the fragile x mental retardation protein in modulating circadian clock circuit synaptic architecture. Front. Neural Circuits 3:8. doi: 10.3389/neuro.04.008.2009
Gatto, C. L., and Broadie, K. (2010). Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental disorder models. Front. synaptic Neurosci. 2:4. doi: 10.3389/fnsyn.2010.00004
Gatto, C. L., Pereira, D., and Broadie, K. (2014). GABAergic circuit dysfunction in the Drosophila fragile X syndrome model. Neurobiol. Dis. 65, 142–159. doi: 10.1016/j.nbd.2014.01.008
Greenblatt, E. J., and Spradling, A. C. (2018). Fragile X mental retardation 1 gene enhances the translation of large autism-related proteins. Science 361, 709–712. doi: 10.1126/science.aas9963
Hadley, D., Wu, Z. L., Kao, C., Kini, A., Mohamed-Hadley, A., Thomas, K., et al. (2014). The impact of the metabotropic glutamate receptor and other gene family interaction networks on autism. Nat. Commun. 5:4074. doi: 10.1038/ncomms5074
Hahn, N., Geurten, B., Gurvich, A., Piepenbrock, D., Kastner, A., Zanini, D., et al. (2013). Monogenic heritable autism gene neuroligin impacts Drosophila social behaviour. Behav. Brain Res. 252, 450–457. doi: 10.1016/j.bbr.2013.06.020
Hamilton, P. J., Campbell, N. G., Sharma, S., Erreger, K., Herborg Hansen, F., Saunders, C., et al. (2013). De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Mol. Psychiatry 18, 1315–1323. doi: 10.1038/mp.2013.102
Handt, M., Epplen, A., Hoffjan, S., Mese, K., Epplen, J. T., and Dekomien, G. (2014). Point mutation frequency in the FMR1 gene as revealed by fragile X syndrome screening. Mol. Cell. Probes 28, 279–283. doi: 10.1016/j.mcp.2014.08.003
Harris, K. P., Akbergenova, Y., Cho, R. W., Baas-Thomas, M. S., and Littleton, J. T. (2016). Shank modulates postsynaptic wnt signaling to regulate synaptic development. J. Neurosci. 36, 5820–5832. doi: 10.1523/JNEUROSCI.4279-15.2016
Hutsler, J. J., and Zhang, H. (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309, 83–94. doi: 10.1016/j.brainres.2009.09.120
Iossifov, I., O’Roak, B. J., Sanders, S. J., Ronemus, M., Krumm, N., Levy, D., et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. doi: 10.1038/nature13908
Kim, H. G., Kishikawa, S., Higgins, A. W., Seong, I. S., Donovan, D. J., Shen, Y., et al. (2008). Disruption of neurexin 1 associated with autism spectrum disorder. Am. J. Hum. Genet. 82, 199–207. doi: 10.1016/j.ajhg.2007.09.011
Knight, D., Xie, W., and Boulianne, G. L. (2011). Neurexins and neuroligins: recent insights from invertebrates. Mol. Neurobiol. 44, 426–440. doi: 10.1007/s12035-011-8213-1
Langen, M., Bos, D., Noordermeer, S. D., Nederveen, H., van Engeland, H., and Durston, S. (2014). Changes in the development of striatum are involved in repetitive behavior in autism. Biol. Psychiatry 76, 405–411. doi: 10.1016/j.biopsych.2013.08.013
Laumonnier, F., Bonnet-Brilhault, F., Gomot, M., Blanc, R., David, A., Moizard, M. P., et al. (2004). X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 74, 552–557. doi: 10.1086/382137
Leblond, C. S., Nava, C., Polge, A., Gauthier, J., Huguet, G., Lumbroso, S., et al. (2014). Meta-analysis of SHANK mutations in autism spectrum disorders: a gradient of severity in cognitive impairments. PLoS Genet. 10:e1004580. doi: 10.1371/journal.pgen.1004580
Lee, T., Lee, A., and Luo, L. (1999). Development of the Drosophila mushroom bodies: sequential generation of three distinct types of neurons from a neuroblast. Development 126, 4065–4076.
Liebl, F. L., and Featherstone, D. E. (2008). Identification and investigation of Drosophila postsynaptic density homologs. Bioinform. Biol. Insights 2, 369–381.
Liu, L., Tian, Y., Zhang, X. Y., Zhang, X., Li, T., Xie, W., et al. (2017). Neurexin restricts axonal branching in columns by promoting ephrin clustering. Dev. Cell 41, 94.e4–106.e4. doi: 10.1016/j.devcel.2017.03.004
Luo, W., Zhang, C., Jiang, Y. H., and Brouwer, C. R. (2018). Systematic reconstruction of autism biology from massive genetic mutation profiles. Sci. Adv. 4:e1701799. doi: 10.1126/sciadv.1701799
McBride, S. M., Choi, C. H., Wang, Y., Liebelt, D., Braunstein, E., Ferreiro, D., et al. (2005). Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron 45, 753–764. doi: 10.1016/j.neuron.2005.01.038
Michel, C. I., Kraft, R., and Restifo, L. L. (2004). Defective neuronal development in the mushroom bodies of Drosophila fragile X mental retardation 1 mutants. J. Neurosci. 24, 5798–5809. doi: 10.1523/jneurosci.1102-04.2004
Mila, M., Alvarez-Mora, M. I., Madrigal, I., and Rodriguez-Revenga, L. (2018). Fragile X syndrome: an overview and update of the FMR1 gene. Clin. Genet. 93, 197–205. doi: 10.1111/cge.13075
Morales, J., Hiesinger, P. R., Schroeder, A. J., Kume, K., Verstreken, P., Jackson, F. R., et al. (2002). Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron 34, 961–972. doi: 10.1016/s0896-6273(02)00731-6
Nakamura, K., Sekine, Y., Ouchi, Y., Tsujii, M., Yoshikawa, E., Futatsubashi, M., et al. (2010). Brain serotonin and dopamine transporter bindings in adults with high-functioning autism. Arch. Gen. Psychiatry 67, 59–68. doi: 10.1001/archgenpsychiatry.2009.137
Neriec, N., and Desplan, C. (2016). From the eye to the brain: development of the Drosophila visual system. Curr. Top. Dev. Biol. 116, 247–271. doi: 10.1016/bs.ctdb.2015.11.032
O’Brien, G., and Pearson, J. (2004). Autism and learning disability. Autism 8, 125–140. doi: 10.1177/1362361304042718
Okray, Z., de Esch, C. E., Van Esch, H., Devriendt, K., Claeys, A., Yan, J., et al. (2015). A novel fragile X syndrome mutation reveals a conserved role for the carboxy-terminus in FMRP localization and function. EMBO Mol. Med. 7, 423–437. doi: 10.15252/emmm.201404576
Pan, L., Zhang, Y. Q., Woodruff, E., and Broadie, K. (2004). The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr. Biol. 14, 1863–1870. doi: 10.1016/j.cub.2004.09.085
Park, H. R., Lee, J. M., Moon, H. E., Lee, D. S., Kim, B. N., Kim, J., et al. (2016). A short review on the current understanding of autism spectrum disorders. Exp. Neurobiol. 25, 1–13. doi: 10.5607/en.2016.25.1.1
Paval, D. (2017). A dopamine hypothesis of autism spectrum disorder. Dev. Neurosci. 39, 355–360. doi: 10.1159/000478725
Peca, J., and Feng, G. (2012). Cellular and synaptic network defects in autism. Curr. Opin. Neurobiol. 22, 866–872. doi: 10.1016/j.conb.2012.02.015
Pieretti, M., Zhang, F. P., Fu, Y. H., Warren, S. T., Oostra, B. A., Caskey, C. T., et al. (1991). Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66, 817–822. doi: 10.1016/0092-8674(91)90125-i
Pop, A. S., Gomez-Mancilla, B., Neri, G., Willemsen, R., and Gasparini, F. (2014). Fragile X syndrome: a preclinical review on metabotropic glutamate receptor 5 (mGluR5) antagonists and drug development. Psychopharmacology 231, 1217–1226. doi: 10.1007/s00213-013-3330-3
Ramocki, M. B., and Zoghbi, H. Y. (2008). Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature 455, 912–918. doi: 10.1038/nature07457
Repicky, S., and Broadie, K. (2009). Metabotropic glutamate receptor-mediated use-dependent down-regulation of synaptic excitability involves the fragile X mental retardation protein. J. Neurophysiol. 101, 672–687. doi: 10.1152/jn.90953.2008
Santos, A. R., Kanellopoulos, A. K., and Bagni, C. (2014). Learning and behavioral deficits associated with the absence of the fragile X mental retardation protein: what a fly and mouse model can teach us. Learn. Mem. 21, 543–555. doi: 10.1101/lm.035956.114
Sinclair, D., Oranje, B., Razak, K. A., Siegel, S. J., and Schmid, S. (2017). Sensory processing in autism spectrum disorders and Fragile X syndrome-From the clinic to animal models. Neurosci. Biobehav. Rev. 76(Pt B), 235–253. doi: 10.1016/j.neubiorev.2016.05.029
Sofola, O., Sundram, V., Ng, F., Kleyner, Y., Morales, J., Botas, J., et al. (2008). The Drosophila FMRP and LARK RNA-binding proteins function together to regulate eye development and circadian behavior. J. Neurosci. 28, 10200–10205. doi: 10.1523/JNEUROSCI.2786-08.2008
Soorya, L., Kolevzon, A., Zweifach, J., Lim, T., Dobry, Y., Schwartz, L., et al. (2013). Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol. Autism 4:18. doi: 10.1186/2040-2392-4-18
Sudhof, T. C. (2017). Synaptic neurexin complexes: a molecular code for the logic of neural circuits. Cell 171, 745–769. doi: 10.1016/j.cell.2017.10.024
Sun, M., Xing, G., Yuan, L., Gan, G., Knight, D., With, S. I., et al. (2011). Neuroligin 2 is required for synapse development and function at the Drosophila neuromuscular junction. J. Neurosci. 31, 687–699. doi: 10.1523/jneurosci.3854-10.2011
Tang, G., Gudsnuk, K., Kuo, S. H., Cotrina, M. L., Rosoklija, G., Sosunov, A., et al. (2014). Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143. doi: 10.1016/j.neuron.2014.07.040
Tauber, J. M., Vanlandingham, P. A., and Zhang, B. (2011). Elevated levels of the vesicular monoamine transporter and a novel repetitive behavior in the Drosophila model of fragile X syndrome. PLoS One 6:e27100. doi: 10.1371/journal.pone.0027100
Tessier, C. R., and Broadie, K. (2008). Drosophila fragile X mental retardation protein developmentally regulates activity-dependent axon pruning. Development 135, 1547–1557. doi: 10.1242/dev.015867
Tian, Y., Zhang, Z. C., and Han, J. (2017). Drosophila studies on autism spectrum disorders. Neurosci. Bull. 33, 737–746. doi: 10.1007/s12264-017-0166-6
van Alphen, B., Yap, M. H., Kirszenblat, L., Kottler, B., and van Swinderen, B. (2013). A dynamic deep sleep stage in Drosophila. J. Neurosci. 33, 6917–6927. doi: 10.1523/JNEUROSCI.0061-13.2013
Wan, L., Dockendorff, T. C., Jongens, T. A., and Dreyfuss, G. (2000). Characterization of dFMR1, a Drosophila melanogaster homolog of the fragile X mental retardation protein. Mol. Cell. Biol. 20, 8536–8547. doi: 10.1128/mcb.20.22.8536-8547.2000
Weiler, I. J., Irwin, S. A., Klintsova, A. Y., Spencer, C. M., Brazelton, A. D., Miyashiro, K., et al. (1997). Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc. Natl. Acad. Sci. U.S.A. 94, 5395–5400. doi: 10.1073/pnas.94.10.5395
Wu, S., Gan, G., Zhang, Z., Sun, J., Wang, Q., Gao, Z., et al. (2017). A presynaptic function of shank protein in Drosophila. J. Neurosci. 37, 11592–11604. doi: 10.1523/JNEUROSCI.0893-17.2017
Xing, G., Gan, G., Chen, D., Sun, M., Yi, J., Lv, H., et al. (2014). Drosophila neuroligin3 regulates neuromuscular junction development and synaptic differentiation. J. Biol. Chem. 289, 31867–31877. doi: 10.1074/jbc.M114.574897
Xing, G., Li, M., Sun, Y., Rui, M., Zhuang, Y., Lv, H., et al. (2018). Neurexin-Neuroligin 1 regulates synaptic morphology and functions via the WAVE regulatory complex in Drosophila neuromuscular junction. eLife 7:e30457. doi: 10.7554/eLife.30457
Xu, S., Poidevin, M., Han, E., Bi, J., and Jin, P. (2012). Circadian rhythm-dependent alterations of gene expression in Drosophila brain lacking fragile X mental retardation protein. PLoS One 7:e37937. doi: 10.1371/journal.pone.0037937
Yan, J., Noltner, K., Feng, J., Li, W., Schroer, R., Skinner, C., et al. (2008). Neurexin 1alpha structural variants associated with autism. Neurosci. Lett. 438, 368–370. doi: 10.1016/j.neulet.2008.04.074
Zhang, X., Rui, M., Gan, G., Huang, C., Yi, J., Lv, H., et al. (2017). Neuroligin 4 regulates synaptic growth via the bone morphogenetic protein (BMP) signaling pathway at the Drosophila neuromuscular junction. J. Biol. Chem. 292, 17991–18005. doi: 10.1074/jbc.M117.810242
Zhang, Y. Q., Bailey, A. M., Matthies, H. J., Renden, R. B., Smith, M. A., Speese, S. D., et al. (2001). Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 107, 591–603. doi: 10.1016/s0092-8674(01)00589-x
Keywords: autism (ASD), shank, FMR1, neurexin, neuroligins, mGlu receptor 5, Drosophila, dopamine
Citation: Bellosta P and Soldano A (2019) Dissecting the Genetics of Autism Spectrum Disorders: A Drosophila Perspective. Front. Physiol. 10:987. doi: 10.3389/fphys.2019.00987
Received: 28 April 2019; Accepted: 18 July 2019;
Published: 07 August 2019.
Edited by:
Gabriella Mazzotta, University of Padua, ItalyReviewed by:
Lawrence Todd Reiter, The University of Tennessee Health Science Center, United StatesJae Park, The University of Tennessee, Knoxville, United States
Copyright © 2019 Bellosta and Soldano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Bellosta, cGFvbGEuYmVsbG9zdGFAdW5pdG4uaXQ=; Alessia Soldano, YWxlc3NpYS5zb2xkYW5vQHVuaXRuLml0