 Marissa Sumathipala
Marissa Sumathipala Enrico Maiorino
Enrico Maiorino Scott T. Weiss
Scott T. Weiss Amitabh Sharma
Amitabh Sharma
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 16 July 2019
Sec. Systems Biology Archive
Volume 10 - 2019 | https://doi.org/10.3389/fphys.2019.00888
This article is part of the Research Topic Integrated Omics for Defining Interactomes View all 9 articles
Recently, long-non-coding RNAs (lncRNAs) have attracted attention because of their emerging role in many important biological mechanisms. The accumulating evidence indicates that the dysregulation of lncRNAs is associated with complex diseases. However, only a few lncRNA-disease associations have been experimentally validated and therefore, predicting potential lncRNAs that are associated with diseases become an important task. Current computational approaches often use known lncRNA-disease associations to predict potential lncRNA-disease links. In this work, we exploited the topology of multi-level networks to propose the LncRNA rankIng by NetwOrk DiffusioN (LION) approach to identify lncRNA-disease associations. The multi-level complex network consisted of lncRNA-protein, protein–protein interactions, and protein-disease associations. We applied the network diffusion algorithm of LION to predict the lncRNA-disease associations within the multi-level network. LION achieved an AUC value of 96.8% for cardiovascular diseases, 91.9% for cancer, and 90.2% for neurological diseases by using experimentally verified lncRNAs associated with diseases. Furthermore, compared to a similar approach (TPGLDA), LION performed better for cardiovascular diseases and cancer. Given the versatile role played by lncRNAs in different biological mechanisms that are perturbed in diseases, LION’s accurate prediction of lncRNA-disease associations helps in ranking lncRNAs that could function as potential biomarkers and potential drug targets.
Non-coding RNAs can be classified broadly in two types: small non-coding RNAs and long non-coding (lnc) RNAs that are more than 200 nucleotides (Kapranov et al., 2007; Kung et al., 2013). LncRNAs are discrete transcription units located in sequence space, which do not overlap protein coding genes (Kung et al., 2013). Recently, lncRNAs have received widespread attention due to their diverse roles in biological regulation, developmental processes, and diseases (Mercer et al., 2009; Orom et al., 2010; Moran et al., 2012; Sun and Kraus, 2015; Ulitsky, 2016). With a wide array of regulatory functions in epigenetic, transcriptional, post-transcriptional regulation including histone modification, DNA methylation, and transcriptional co-regulation, it is not surprising that the dysregulation of lncRNAs have been reported in many diseases (Liu et al., 2013; Shi et al., 2013; Chakravarty et al., 2014; Kataoka and Wang, 2014; Ma et al., 2016). Furthermore, increasing evidence suggests that the regulatory role of lncRNAs in biological processes often involves interactions with proteins (Ferre et al., 2016; Xiao et al., 2017). The impact of each lncRNA may be determined by its ability to perform numerous tasks in the cell by interacting with proteins, DNA and RNA molecules.
To assist in understanding the pathogenesis of complex diseases, there have been efforts to infer potential associations between lncRNAs and diseases using lncRNA-protein interaction data (Chen and Yan, 2013; Li et al., 2014; Yang et al., 2014; Liu et al., 2017). Computational approaches have been developed to predict lncRNA-protein interactions, such as lncPro and RPI-Pred (Suresh et al., 2015; Shi et al., 2017; Xiao et al., 2017; Zheng et al., 2017). Several computation methods like LncRNADisease (Wang et al., 2016), GrwLDA (Gu et al., 2017), TPGLDA (Ding et al., 2018), and KATZLDA (Chen, 2015) uncover potential lncRNA-disease associations by integrating lncRNA functional similarities, lncRNA expression profiles, known lncRNA-disease associations, disease semantic similarities, and gene-disease associations. Studies seeking to predict non-coding RNAs in disease by constructing heterogenous networks with multiple types of biological interactions include ncPred, which uses resource transfer on a tripartite network (Alaimo et al., 2014); ComiRNet, which applies clustering to miRNA-gene regulatory networks (Pio et al., 2015); and LP-HCLUS, which integrates across interactions between lncRNAs, miRNAs, diseases, and genes (Barracchia et al., 2018). Most of these approaches use experimentally known lncRNA-disease associations as part of their input data and infer new lncRNA-disease associations.
An emergent concept postulates that a disease reflects the interplay of multiple biomolecules, and is rarely a straightforward consequence of an abnormality in a single gene encoding protein (Barabasi et al., 2011; Vidal et al., 2011; Sharma et al., 2015; Tasan et al., 2015; Sonawane et al., 2019). Given that each lncRNA may regulate multiple protein targets, and each protein may interact with multiple lncRNAs and with other proteins, it is crucial to integrate lncRNA-protein and protein–protein interactions in a heterogeneous network model to fully understand their dynamics at a molecular level. As the prediction of lncRNA disease associations are at a very early stage, known lncRNA-disease associations are limited. Here, we use the information flow-based method that exploits the connectivity structure among proteins and lncRNAs to predict novel lncRNA-disease associations. Diffusion-based methods are based on the notion that products of genes associated with diseases have a strong tendency to interact with each other in terms of the cumulative strength of paths that connect the corresponding proteins. These methods estimate the most redundant paths on the network, identifying the destination nodes (lncRNAs) that are most likely to be reached when starting from the seeds (disease proteins). When a node has a high score it means that the paths leading to it are highly redundant, which in turn implies that even if a portion of the network edges were missing due to incompleteness of data the results would be similar. This is in contrast with shortest-path-based methods that can instead be very sensitive to the removal of some critical links. We and others have previously proposed network diffusion approaches that model the information flow in molecular networks to localize the disease network neighborhood (Sharma et al., 2018), identify biomarkers in genome-wide studies (Qian et al., 2014), find significantly mutated pathways in cancer (Vandin et al., 2011), and prioritize disease genes (Navlakha and Kingsford, 2010). These studies successfully exploit the topology or structure of molecular interactions, called the interactome, even in incomplete space. With the growing availability of lncRNA-related interactome data, generalizing the guilt-by-association principle to predict lncRNA candidates might help in revealing the role of lncRNA in complex, interconnected disease mechanisms.
In this work, we propose LncRNA rankIng by NetwOrk DiffusioN (LION). LION is a network-diffusion method that integrates lncRNA-protein, protein–protein, and disease-protein networks to prioritize important lncRNAs for diseases. First, we construct a multi-level complex network (tripartite network) consisting of lncRNA-protein, protein–protein, and protein-disease associations. Next, we apply a random walk network diffusion algorithm. The random walk method exploits the local network neighborhood of diseases to measure the proximity of lncRNAs to the disease genes based on the probabilities of the connecting edges. It is possible to identify which lncRNA is connected to a given disease on the basis of the probability of reachability in the heterogeneous network. To evaluate LION, we utilize the available experimentally verified lncRNA disease associations (Chen et al., 2013) to demonstrate the performance of our method and compare with a similar method (TPGLDA) to demonstrate the performance advantages of our approach.
The majority of current methods (Chen and Yan, 2013; Liu et al., 2014; Sun et al., 2014; Yang et al., 2014; Lu C. et al., 2018) use the known lncRNA-disease interactions to compute the novel associations. Here, we predicted the lncRNA-disease network without a priori lncRNA-disease information. We first constructed a tripartite network from 28,488 protein-disease associations compiled from OMIM and GWAS databases, 141,296 protein–protein interactions, and 3,998 lncRNA-protein interactions (Figure 1). Next, we applied LION to prioritize lncRNAs, computing the probability of a random walker moving from a disease protein to a lncRNA. In the end, a final bipartite lncRNA-disease network was constructed from the predictions of 747 diseases. This lncRNA-disease network consisted of 304,868 weighted lncRNA-disease edges, where each link represents a predicted association between a disease and lncRNA that is proximal to its corresponding disease genes.
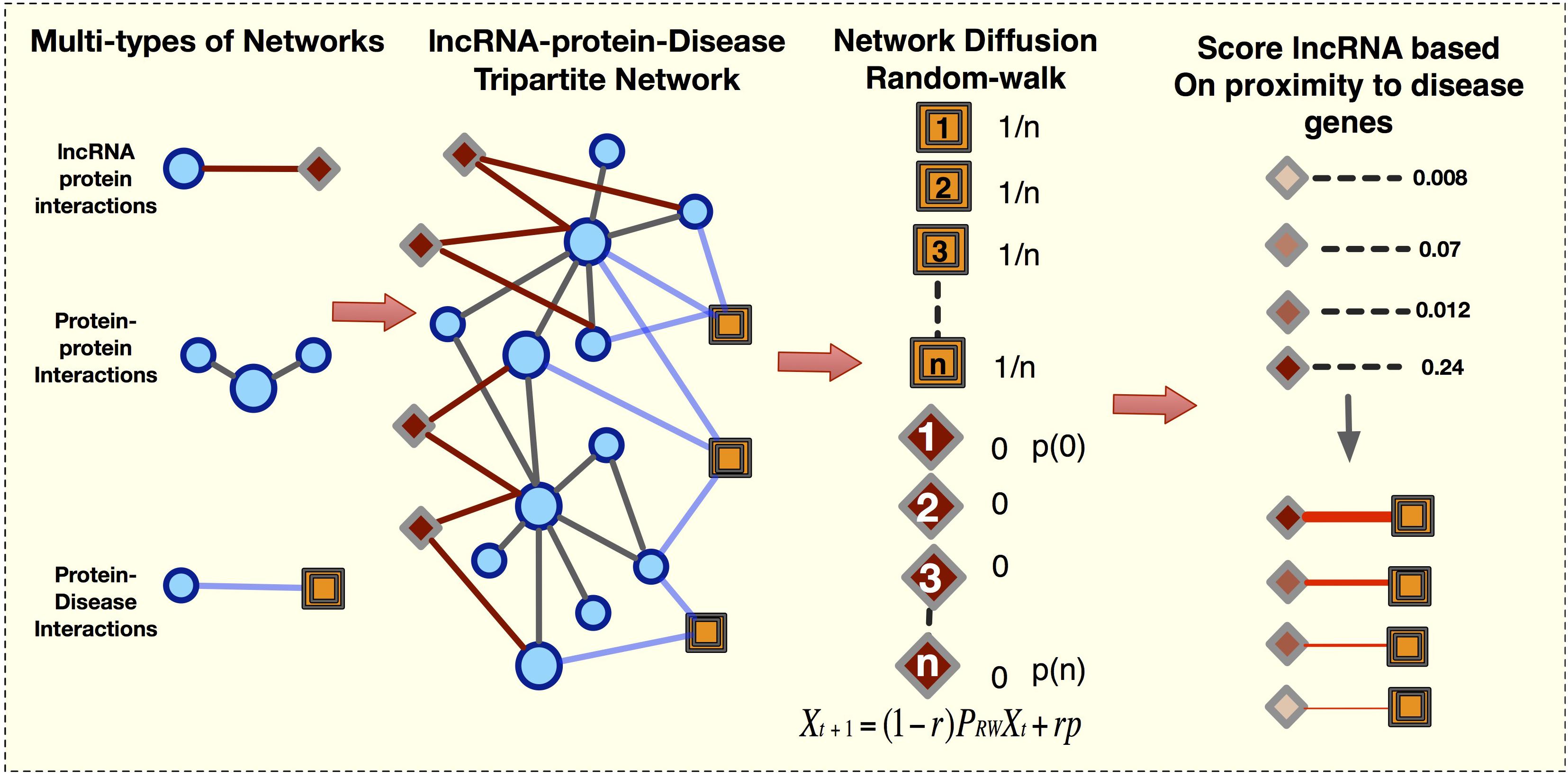
Figure 1. Framework to create the lncRNA-Disease-Network (LDN). We first construct the lncRNA-gene-disease tripartite network and next apply network diffusion method to rank lncRNA disease associations.
We used the LncRNADisease experimental lncRNA-disease dataset to assess the predictive ability of LION, described in materials and methods. The dataset contains 372 lncRNAs, 245 diseases, and 1,101 lncRNA-disease associations. We calculated area under the receiver operating characteristic (ROC) curve to evaluate the predictive performance of our method. For the receiver operating characteristic curve, we plotted sensitivity and specificity at different thresholds, using the predicted lncRNA-disease edge weights as the thresholds. A predictor making random guesses would have an AUC of 0.5 and a predictor with perfect performance would have an AUC of 1. Typically, an AUC above 0.85 is considered good performance and an AUC above 0.95 is considered excellent performance.
We assessed the relative performance of LION by computing and analyzing three ROC curves: (1) LION method, (2) a current state-of-the-art method called TPGLDA, (3) a randomized network model as a negative control. TPGLDA uses known lncRNA-disease associations and known gene-disease associations, and makes predictions using a resource allocation algorithm that creates interaction profiles at each lncRNA. We generated the random network by starting with the lncRNA-disease network predicted with LION, and randomly shuffling node labels to create a random graph null model with the same connectivity structure, enabling comparison with LION’s predictions as a control.
To assess the overall performance of LION in predicting lncRNA-disease associations, we first applied LION to predict lncRNA-disease associations for three broad categories of diseases: cardiovascular diseases, cancers, and neurological diseases (Figure 2). LION yielded high performance for all three with AUCs all above 0.9. In contrast, the randomized network had a low AUC of approximately 0.5, which corresponds to a predictor making random guesses when the lncRNA-disease associations themselves are randomly assigned. The high AUCs above 0.9 with LION indicates our method is accurately predicting biologically relevant lncRNA-disease associations by inferring them from interactome and lncRNAome data. The AUC performance of TPGLDA was 0.809, 0.790, and 0.933 for cardiovascular disease, cancers, and neurological disease, respectively. Compared to TPGLDA, LION has improved performance in cardiovascular diseases and cancers, and comparable performance in neurological diseases. This confirms the ability of LION to make equivalent or higher accuracy predictions with respect to TPGLDA; in particular, LION does so without experimental lncRNA-disease data as an input.
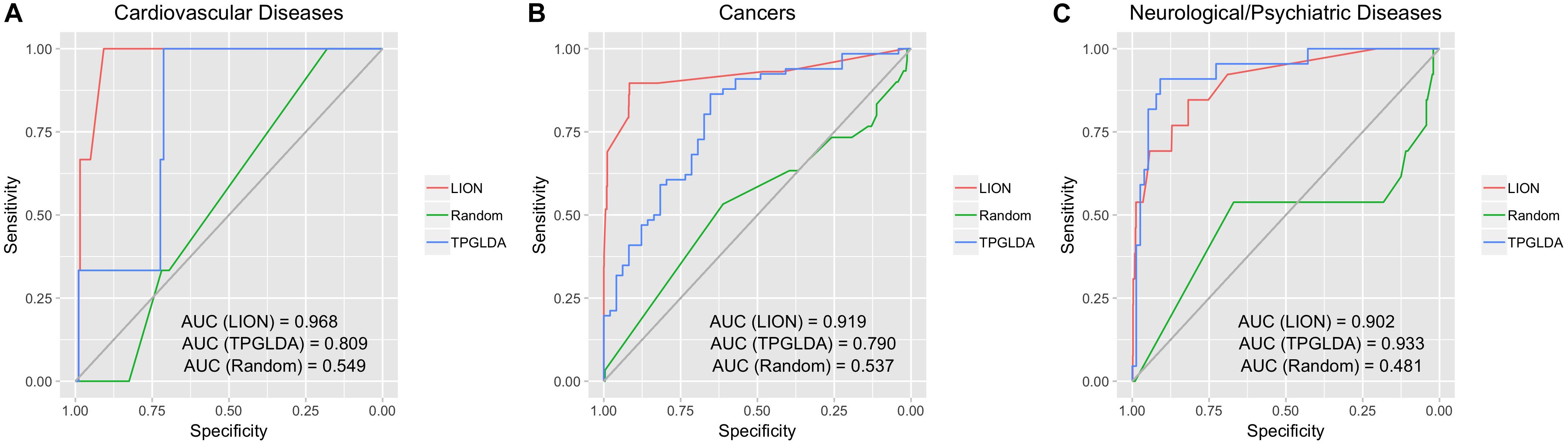
Figure 2. LION’s performance in predicting lncRNA-disease associations for three broad groups of diseases. For each, three receiver operating characteristic (ROC) curves are shown: (1) LION, (2) TPGLDA, a current state-of-the-art method for lncRNA-disease association prediction, (3) randomized network generated with node label shuffling as a negative control. Area under the ROC curve (AUC) values are listed for each ROC curve. (A) ROC plot for cardiovascular disease. (B) ROC plot for cancers. (C) ROC plot for neurological and psychiatric diseases.
Having demonstrated high performance for three broad disease groups, we next evaluated LION on four individual cancers (Figure 3). The computed AUCs for LION were 0.957, 0.971, 0.954, and 0.967 for breast cancer, blood cancers, ovarian cancer, and bladder cancer, respectively. With AUCs exceeding 0.95, LION demonstrated excellent performance in predicting lncRNAs for individual cancers. Similar to the broad disease groups of Figure 2, the random network ROC curve for the cancers had much lower AUCs of around 0.5. When compared against the TPGLDA method, we see that LION has an improved or roughly equal performance to TPGLDA; AUCs for TPGLDA were 0.959, 0.899, 0.812, and 0.714 for breast cancer, blood cancers, ovarian cancers, and bladder cancers, respectively. For breast cancer and blood cancers, performance was roughly equal in both methods, while in the case of ovarian and bladder cancer, LION outperformed TPGLDA. These results further confirm the promise of LION to make accurate predictions from interactome and lncRNA-protein data, without prior information of lncRNA-disease associations.
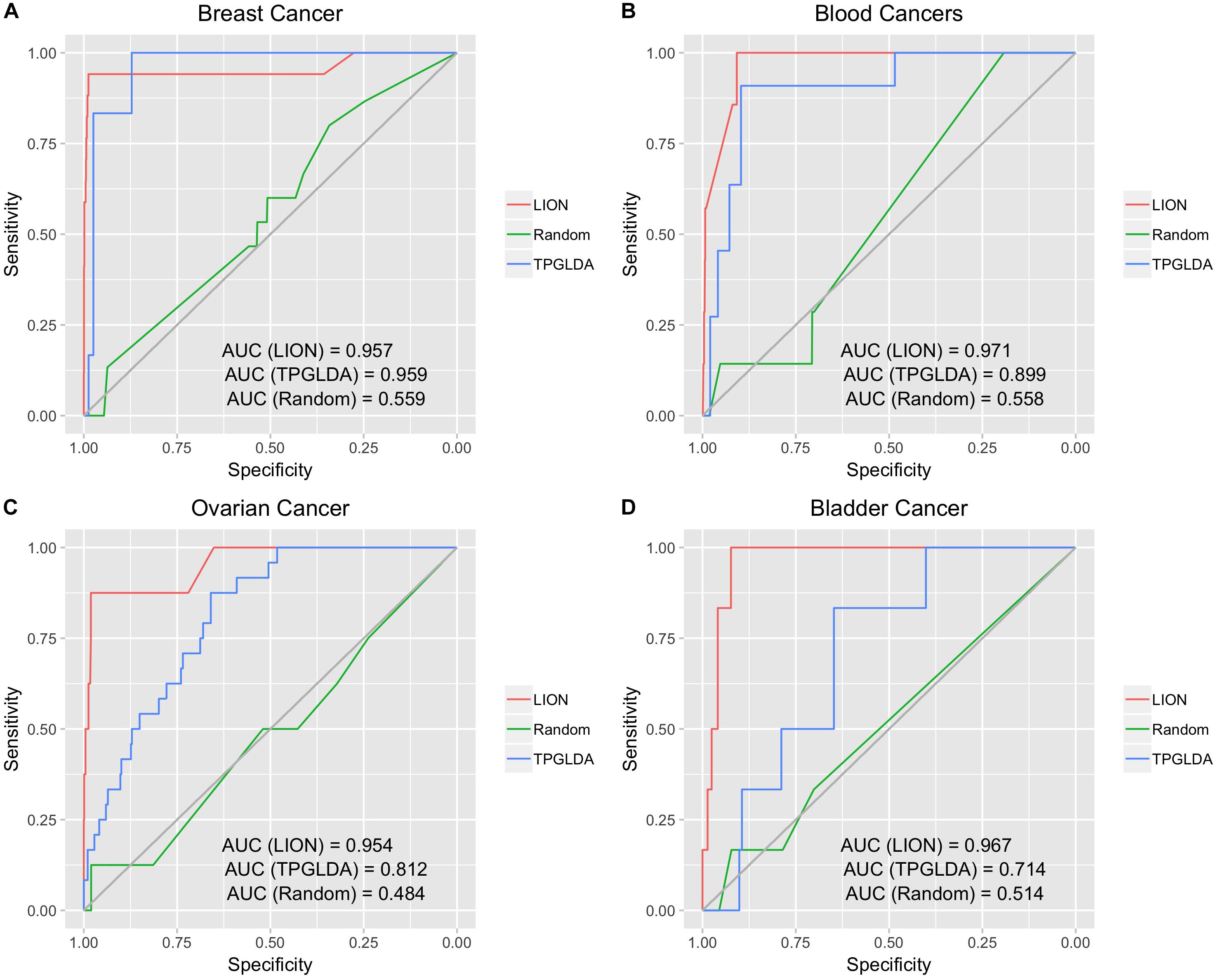
Figure 3. Performance in predicting lncRNA-disease associations for four individual cancers. LION outperformed the random network on all four cancers, and had higher or comparable performance than TPGLDA for all four. (A) ROC plot for breast cancer. (B) ROC plot for blood cancers. (C) ROC plot for ovarian cancer. (D) ROC plot for bladder cancer.
We tested the statistical significance of differences in predictions made by LION, the randomized network, and TPGLDA methods using the Wilcox rank sum test. For each of the 7 diseases in Figures 2, 3, we ran two Wilcox tests: (1) comparing edge weights between LION and the randomized network, (2) comparing edge weights between LION and TPGLDA. We found statistically significant differences between LION’s predictions and the randomized predictions (p < 0.01), and between LION and TGPLDA (p < 0.01). The results of the Wilcox test confirm that LION’s better performance, seen in Figures 2, 3, arise from an improved ranking of disease-associated lncRNAs that are significantly different from either the TGPLDA rankings or from the randomized rankings.
We next conducted a qualitative evaluation of the top 50 predicted lncRNAs for four individual cancers to gain insight into their biological relevance to the disease. We identified biological roles of lncRNAs using the LncRNADisease database and experimental studies in the literature. For breast cancer, the top 50 predictions contained eight of the nine experimentally validated lncRNAs for breast cancer reported in LncRNADisease. The eight lncRNAs were MALAT1, XIST, HOTAIR, MEG3, GAS5, H19, CDKN2B-AS1, and PVT1. Each of these lncRNAs been validated by genetic profiling studies or in vivo and in vitro experimental studies, reported in the LncRNADisease database. For example, MALAT1 is a regulator of alternative splicing in breast cancer (Moran et al., 2012; Qi and Du, 2013; Jiang and Bikle, 2014); HOTAIR is overexpressed in a quarter of breast cancers (Gupta et al., 2010; Hung and Chang, 2010); the role of XIST in X chromosome inactivation is linked to BRCA1 tumors (Vincent-Salomon et al., 2007); MEG3 suppresses breast cancer through AKT (Zhang et al., 2017); GAS5 is downregulated in breast cancer and can induce apoptosis (Mourtada-Maarabouni et al., 2009). H19 is the direct target of a critical breast oncogene that triggers tumorigenesis (Barsyte-Lovejoy et al., 2006). CDKN2B-AS1 was identified through a GWAS study of breast cancer susceptibility loci (Turnbull et al., 2010). PVT1 was mechanistically linked to a common loci of breast cancer risk through its role as an apoptotic inhibitor (Guan et al., 2007). Apart from these eight, several additional predictions in the top 50 have been recently validated and reported in the literature, but not yet added to the LncRNADisease database. These lncRNAs include DANCR, NEAT1, RMST, and KCNQ1OT1 (Ke et al., 2016; Sha et al., 2017; Feng et al., 2018; Wang et al., 2018). One of the top ranked lncRNAs, HULC, has been linked to other forms of cancer but not to breast cancer. HULC decreases miRNA 15a, which in turn increases expression of p62, a critical cancer signaling protein; both miR15a and p62 have been linked to breast cancer (Patel et al., 2016; Xu et al., 2017). Given the biological roles of HULC’s two known targets in breast cancer pathways, HULC may be a potentially important lncRNA for breast cancer.
For bladder cancer, we found six experimentally validated lncRNAs in the LncRNADisease database, all of which were in the top 50 LION predictions. Of these, XIST, H19, MALAT1, MEG3 play a role in tumor proliferation, suppression, and metastasis of bladder cancer (Ariel et al., 2000; Ying et al., 2013; Martens-Uzunova et al., 2014; Wu et al., 2014). UCA and TUG1 are both known to promote cell proliferation and tumorigenesis in bladder carcinomas (Wang et al., 2012; Han et al., 2013). A top ranked lncRNA prediction, DBH-AS1 has not been linked to bladder cancer experimentally, but it regulates the miRNA-138-5p, a key inducer of bladder cancer carcinogenesis (Yang et al., 2016; Bao et al., 2018). Thus, DBH-AS1 may be a potential novel lncRNA target for bladder cancer.
A qualitative evaluation of ovarian cancer revealed the top 20 predictions by LION contained all four experimentally validated lncRNAs: MALAT1, H19, HOTAIR, and PVT1. Via the MAPK pathway, MALAT1 promotes ovarian cancer cell proliferation and migration (Zou et al., 2016). H19 has been investigated as a targeted therapy strategy for ovarian cancer and its inhibition suppresses tumor growth (Mizrahi et al., 2009). HOTAIR is a predictor of patient prognosis, including features such as tumor grade and survival (Qiu et al., 2014). Inhibition of PVT1 is linked to induction of an apoptotic response and proliferation inhibition of ovarian cancer cell lines [17908964]. The lncRNA TERC was highly ranked, but our literature search did not uncover a study linking TERC and ovarian cancer. A component of the telomerase enzyme complex, TERC is implicated in maintaining telomere length and therefore genetic susceptibility to aging related diseases, such as ovarian cancer (Grammatikakis et al., 2014). Our ranking and literature search on the biological role of TERC suggests it may be a new lncRNA that could be related to ovarian cancer.
The top 50 ranked lncRNAs for blood cancer contained 4 of the 11 experimentally validated lncRNAs. Polymorphisms in the gene encoding lncRNA CDKN2B-AS1 are associated with lymphoblastic leukemia (Iacobucci et al., 2011). A chromosomal translocation mutation implicated in B-cell lymphoma was linked to the GAS5 lncRNA; the mutation causes fusion of the GAS5 transcript to the BCL6 gene (Nakamura et al., 2008). Similarly, a common chromosome eight breakpoint mutation in Burkitt’s lymphoma was linked to the PVT1 locus in a mouse model, implicating PVT1 in disease tumorigenesis (Graham and Adams, 1986). In a genetic profiling study of acute myeloid leukemia patients, hypermethylation of the imprinted gene MEG3 was linked to significantly reduced survival (Benetatos et al., 2010). Similar to breast cancer, our literature search uncovered several high ranked lncRNAs that are linked to blood cancers but yet not been added to the LncRNADisease database, such as MIAT, MALAT1, XIST, CRNDE (Ellis et al., 2012; Yildirim et al., 2013; Sattari et al., 2016; Ahmadi et al., 2018). One of the top ranked lncRNAs, SNHG15, has not been experimentally linked to blood cancer; however, its regulation of the ubiquitin-proteasome system and its established oncogenic role in osteosarcoma suggest it may be a promising lncRNA for blood cancers (Ireland, 1986; Jiang et al., 2018).
As the role of lncRNAs in myocardial infarction has been well studied, we examined predictions for myocardial infarction as a case study to further validate our lncRNA-disease predictions. Myocardial infarction is one of the world’s leading causes of death (Benjamin et al., 2017). As shown in Table 1, all top five lncRNAs are not only validated in the experimental lncRNA-disease dataset, but also by experimental studies in the literature, providing further validation for our method. For example, clinical studies have found HOTAIR is downregulated in serum of patients and in vivo experiments revealed HOTAIR has a cardioprotective role (Gao et al., 2017; Lu W. et al., 2018). This indicates that HOTAIR is a promising candidate as a clinical biomarker for non-invasive diagnosis and potential therapeutic target for myocardial infarction.
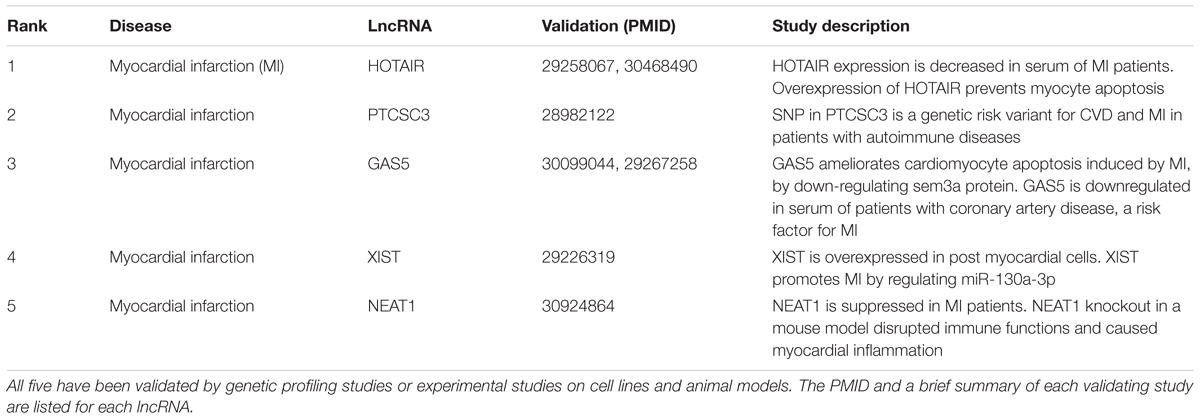
Table 1. Top five lncRNA predictions by LION for Myocardial Infarction.
Since LION does not rely on known, experimentally verified lncRNA-disease associations to make predictions, it is not restricted to only diseases for which experimental data is available. We applied LION to predict potential novel lncRNAs for respiratory diseases, where the role of lncRNAs is least explored. Respiratory diseases are a class of genetically complex diseases where the molecular and regulatory genomic underpinnings, and particularly the role of lncRNAs, are not well understood. In particular, we focused on respiratory tract infections (RTIs) and chronic obstructive pulmonary disease (COPD).
Lower RTIs are the most common infection and one of the leading causes of death in the United States by infection (File, 2000). COPD is the third leading cause of death in the United States, with about 15 million cases per year in the United States (Doney et al., 2014). Tables 2, 3 show the top five predictions for RTIs and COPD, respectively. None of the top predicted lncRNAs have been linked to RTIs. Of the lncRNAs predicted for COPD, only MEG3 has been associated with COPD in the literature. The unconfirmed lncRNAs present novel potential lncRNAs that could be further studied as regulatory drivers of the disease, clinical biomarkers, and therapeutic targets. MEG3, a top lncRNA for both COPD and RTIs, is differentially expressed in pulmonary fibrosis and in COPD (Tang et al., 2016; Gokey et al., 2018), indicating it may play a key role in the pathology of multiple respiratory diseases. IFNG-AS1, a top lncRNA for only RTIs but not COPD, has been linked to T helper cell responses (Peng et al., 2015), suggesting it may play a role in the immune response component of RTIs. RTIs, COPD, and myocardial infarctions all have HOTAIR in the top five predictions. This suggests HOTAIR’s roles in gene methylation and epigenetic differentiation may contribute to it being strongly implicated in many diseases caused by a combination of environmental and genetic factors.

Table 2. Top five lncRNA predictions by LION for respiratory tract infections.
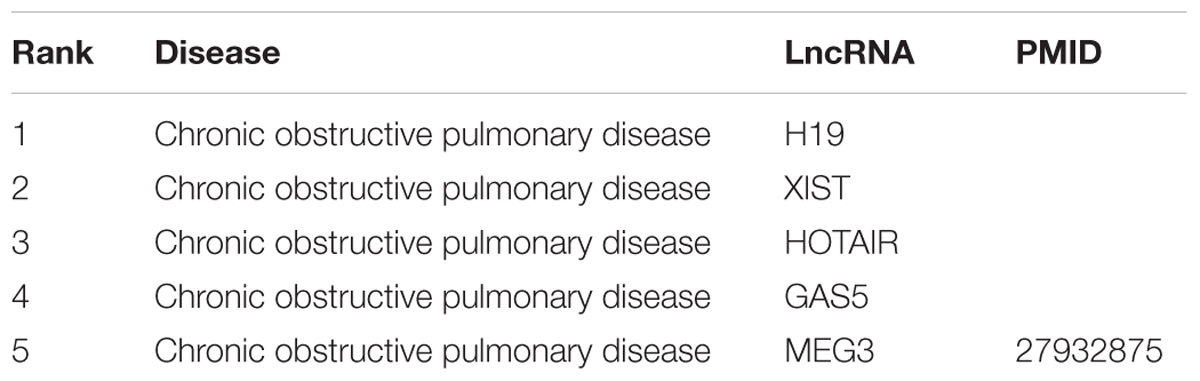
Table 3. Top five lncRNA predictions by LION for Chronic obstructive pulmonary disease (COPD).
Motivated by the success of network based methods in extrapolating information from the interactome (Navlakha and Kingsford, 2010; Vidal et al., 2011; Menche et al., 2015; Sharma et al., 2018), we develop a lncRNA-gene-disease tripartite graph and collapse it into a weighted lncRNA-disease bipartite graph using a random walk. The powerful predictive ability and applicability of LION for virtually any disease arises from its unique method of multi-layer network construction and ranking with a network diffusion algorithm. Accumulating evidence suggests the importance of lncRNA’s role in various biological processes and in predicting novel lncRNAs for diseases has significant medical and biological implications. Most of the current methods infer potential lncRNA disease associations based on existing knowledge of lncRNA-disease relationships (Chen and Yan, 2013; Chen, 2015; Gu et al., 2017; Ding et al., 2018). In contrast to current state-of-the-art methods (Chen et al., 2017), we predicted lncRNA-disease associations by using the topology of a heterogeneous network comprising lncRNA-proteins, protein–protein, and protein-disease interactions. Moreover, LION is not restricted to predicting lncRNAs for diseases with known lncRNAs, it can make predictions for any disease by using their known disease proteins. To our knowledge, this approach is the first of its kind to make accurate predictions from protein interactome and lncRNA-protein data, without requiring known lncRNA-disease associations.
Making lncRNA-disease predictions without a priori information does not impact the performance of LION. When compared against experimental lncRNA-disease associations, LION accurately predicted lncRNAs for both broad disease categories (cancers, cardiovascular diseases, and neurological/psychiatric diseases) and individual cancers (breast, blood, ovarian, and bladder), with AUCs all over 0.85. In contrast, the negative control, a lncRNA-disease network randomized with node label shuffling, had AUCs of only 0.5. In comparison to the current state-of-the-art method for lncRNA-disease prediction, TPGLDA, LION had improved or equal performance. LION had improved performance for cardiovascular diseases, cancers, ovarian cancer, and bladder cancer. LION performed equally well as TPGLDA on neurological/psychiatric diseases, breast cancer, and blood cancers. LION has potential value for predicting novel lncRNAs, demonstrated through analysis of case studies in COPD and respiratory tract infections. LION has potential applications in biomedical research to shed light on molecular underpinnings of disease, prioritize putative therapeutic targets and biomarkers, and elucidate disease-disease relationships.
Despite its promise, the limitations that influence the prediction of LION include the literature biases in the protein-disease, lncRNA-proteins, and protein–protein interactions datasets. In particular, as lncRNA-protein associations are likely quite incomplete owing to the recent discovery of lncRNAs, the lncRNA-protein dataset may be skewed toward a few well studied lncRNAs. Furthermore, the missing links could bias the predictions toward the well-studied lncRNAs, such as HOTAIR, which we found in the top five predictions for myocardial infarction, COPD, and respiratory tract infections. A second limitation is the unweighted and incomplete GWAS and OMIM gene-disease datasets used to build the tripartite network; LION would be improved by weights that enable distinguishing crucial disease genes, such as through using differential gene expression data. We plan to include more complete interaction datasets to improve the accuracy of prediction in the future.
To obtain genome-wide lncRNA and protein-coding gene associations, we combine three sources:
We downloaded known lncRNA-protein interaction datasets from the following databases. (i) lncRInter, a reliable and high quality lncRNA interaction database containing experimentally validated data whose lncRNA interaction datasets are extracted from peer-reviewed publications (Liu et al., 2017). (ii) NPInter v3.0 contains experimentally verified interactions between non-coding RNAs, especially lncRNAs, and other molecules (proteins, mRNAs, genomic DNAs) (Hao et al., 2016). (iii) EVLncRNAs, a high-quality and integrated database that manually curates all types of experimentally validated lncRNAs (Supplementary Table S1; Zhou et al., 2018).
By analyzing millions of RBP binding sites from 117 CLIP-Seq datasets generated by 50 independent studies, starBase V2.0 has identified 22,735 RBP–lncRNA regulatory relationships (Supplementary Table S1; Li et al., 2014).
We combine several sources of protein interactions: (i) regulatory interactions derived from transcription factors binding to regulatory elements; (ii) binary interactions from several yeast two-hybrid high-throughput and literature-curated datasets; (iii) literature-curated interactions derived mostly from low-throughput experiments; (iv) metabolic enzyme-coupled interactions; (v) protein complexes; (vi) kinase-substrate pairs; and (vii) signaling interactions. The union of all interactions from (i) to (vii) yields a network of 15,949 proteins that are interconnected by 217,140 interactions (Supplementary Table S2; Menche et al., 2015).
A total of 28,488 associations between protein-coding genes and diseases in the OMIM and GWAS were downloaded from DisGeNET (Supplementary Table S3; Pinero et al., 2017).
To predict and rank disease associated lncRNAs, we first constructed a tripartite lncRNA-protein-disease network from the datasets described in section “Data Sources and Construction of Multi-Level Complex Network” (Figure 1).
Next, we utilized a random walk with restart to rank lncRNAs for each disease using network based proximity between a lncRNA and a disease’s known proteins (Kohler et al., 2008). For this, we constructed a subnetwork of the tripartite network, comprised of all the disease genes and their nearest neighbors, as well as the lncRNAs regulating the proteins. We performed this step to localize the lncRNA predictions to each disease network neighborhood in the interactome. Indeed, based on the local impact hypothesis, molecular entities involved in similar diseases have an increased tendency to interact with each other and to localize in a specific network neighborhood (Sharma et al., 2015).
For example, with myocardial infarction (MI), we created a subnetwork with (1) 1228 protein–protein interactions between the 27 known MI genes and their nearest neighbors, and (2) 2,175 lncRNA-protein interactions involving 1235 lncRNAs. The MI seed genes are included in Supplementary Table S4 and the adjacency matrix representing the MI subnetwork is in Supplementary Table S5.
We then executed a random walk process to predict the lncRNA associated to the 27 known MI genes. The basic idea of LION is a walker starting from a single or group of disease genes and visiting other genes and lncRNAs (nodes) in the multi-level network by taking a series of random walking steps. On every moving step, the walker moves from its current node to the neighboring nodes and therefore a distribution value is calculated for every node in the network, which denotes the probability that a walker is at a given node at the current step. At each step, the walker has a probability r = 0.5 to be relocated on the starting genes. The probability distribution at step k + 1 is described by the iterative form:
where, pk is a vector where the i-th element is the probability of being at node i at step k. p0 is the uniform distribution over all starting disease genes. W is the column normalized graph adjacency matrix. By iterating the process until convergence (|| pk+1 − pk || L1 < 10-6) we obtained the steady state probability p∞, and 1235 lncRNAs were ranked according to their values in p∞. Pseudocode for the network diffusion algorithm is included in the Supplementary Information.
This process was repeated for all of the 747 diseases for which a non-bipartite and connected subnetwork could be constructed. These conditions are required for the random walk probabilities to converge to a unique limiting probability distribution.
The final predicted lncRNA-disease network contains 304,868 weighted lncRNA-disease associations between 747 diseases and 1,346 lncRNAs. Each association represents a predicted link between a disease and a lncRNA proximal to the disease’s genes. The number of lncRNAs ranked for each disease has a median of 156 and the number of diseases targeted by each lncRNA has a median of 195. Supplementary Table S6 contains the predicted lncRNA-disease associations for the 4 types of cancer (breast, bladder, blood, and ovarian) and 3 cardiovascular diseases (myocardial infarction, respiratory tract infections, and chronic obstructive pulmonary disease), analyzed in the section “Results.”
To validate the lncRNA-disease predictions by LION, we use the LncRNADisease dataset (Chen et al., 2013), a manually curated database of 1,101 experimentally validated lncRNA-disease associations between 245 diseases and 372 lncRNAs. Using the LncRNADisease experimental dataset as ground truths and predicted lncRNA-disease edge weights from LION, we create receiver operating characteristic (ROC) curves and compute area under the ROC curve (AUC). We created ROC curves first for broad disease categories – cancers, cardiovascular diseases, and neurological/psychiatric diseases – and next for specific diseases – breast, bladder, ovarian, and blood cancers. As a negative control, we created a random graph null model by shuffling node labels on the bipartite lncRNA-disease network. To assess if LION is performing as well as current methods, we compare with the TPGLDA method (Ding et al., 2018). Using the same experimental LncRNADisease dataset, we also create ROC curves for both TPGLDA and the randomized network.
AS and MS conceived the idea of the study. SW co-supervised the analyses. MS and EM performed the computational and statistical analyses. All authors contributed to the interpretation of the results and in writing the manuscript, read, and approved the final manuscript.
We acknowledge the support by the National Institutes of Health (NIH) grants R01 HL118455-04-1, R01 HL149302-01, and P01 HL13285. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Marc Santolini for his help and discussion.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2019.00888/full#supplementary-material
TABLE S1 | Dataset of lncRNA-protein interactions. Contains 391,791 interactions from lncRNAInter, NPInter, EVLncRNAs and starBas between 1,779 lncRNAs and 14,951 proteins.
TABLE S2 | Dataset of protein-protein interactions. Contains 217,160 interactions between 15,961 proteins.
TABLE S3 | Complete dataset of disease-gene associations downloaded from DisGeNET. Contains 28,488 disease-gene associations between 4,454 diseases and 6,464 genes.
TABLE S4 | Set of 27 known disease genes for myocardial infarction for use in seeding a random walk.
TABLE S5 | Adjacency matrix representing an example subnetwork for myocardial infarction.
TABLE S6 | Predicted lncRNA-disease associations for breast cancer, blood cancer, bladder cancer, ovarian cancer, myocardial infarction, respiratory tract infections, and chronic obstructive pulmonary disease.
DATA SHEET S1 | Pseudocode for LION algorithm.
Ahmadi, A., Kaviani, S., Yaghmaie, M., Pashaiefar, H., Ahmadvand, M., Jalili, M., et al. (2018). Altered expression of MALAT1 lncRNA in chronic lymphocytic leukemia patients, correlation with cytogenetic findings. Blood Res. 53, 320–324. doi: 10.5045/br.2018.53.4.320
Alaimo, S., Giugno, R., and Pulvirenti, A. (2014). ncPred: ncRNA-disease association prediction through tripartite network-based inference. Front. Bioeng. Biotechnol. 2:71. doi: 10.3389/fbioe.2014.00071
Ariel, I., Sughayer, M., Fellig, Y., Pizov, G., Ayesh, S., Podeh, D., et al. (2000). The imprinted H19 gene is a marker of early recurrence in human bladder carcinoma. Mol. Pathol. 53, 320–323. doi: 10.1136/mp.53.6.320
Bao, J., Chen, X., Hou, Y., Kang, G., Li, Q., and Xu, Y. (2018). LncRNA DBH-AS1 facilitates the tumorigenesis of hepatocellular carcinoma by targeting miR-138 via FAK/Src/ERK pathway. Biomed. Pharmacother. 107, 824–833. doi: 10.1016/j.biopha.2018.08.079
Barabasi, A. L., Gulbahce, N., and Loscalzo, J. (2011). Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 12, 56–68. doi: 10.1038/nrg2918
Barracchia, E. P., Pio, G., Malerba, D., and Ceci, M. (2018). Identifying lncRNA-disease relationships via heterogeneous clustering. New Front. Mining Compl. Patt. 10785, 35–48. doi: 10.1007/978-3-319-78680-3_3
Barsyte-Lovejoy, D., Lau, S. K., Boutros, P. C., Khosravi, F., Jurisica, I., Andrulis, I. L., et al. (2006). The c-Myc oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Res. 66, 5330–5337. doi: 10.1158/0008-5472.can-06-0037
Benetatos, L., Hatzimichael, E., Dasoula, A., Dranitsaris, G., Tsiara, S., Syrrou, M., et al. (2010). CpG methylation analysis of the MEG3 and SNRPN imprinted genes in acute myeloid leukemia and myelodysplastic syndromes. Leuk. Res. 34, 148–153. doi: 10.1016/j.leukres.2009.06.019
Benjamin, E. J., Blaha, M. J., Chiuve, S. E., Cushman, M., Das, S. R., Deo, R., et al. (2017). Heart disease and stroke statistics-2017 update: a report from the american heart association. Circulation 135, e146–e603.
Chakravarty, D., Sboner, A., Nair, S. S., Giannopoulou, E., Li, R., Hennig, S., et al. (2014). The oestrogen receptor alpha-regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat. Commun. 5:5383. doi: 10.1038/ncomms6383
Chen, G., Wang, Z., Wang, D., Qiu, C., Liu, M., Chen, X., et al. (2013). LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 41, D983–D986. doi: 10.1093/nar/gks1099
Chen, X. (2015). KATZLDA: KATZ measure for the lncRNA-disease association prediction. Sci. Rep. 5:16840. doi: 10.1038/srep16840
Chen, X., Yan, C. C., Zhang, X., and You, Z. H. (2017). Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief. Bioinform. 18, 558–576. doi: 10.1093/bib/bbw060
Chen, X., and Yan, G. Y. (2013). Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics 29, 2617–2624. doi: 10.1093/bioinformatics/btt426
Ding, L., Wang, M., Sun, D., and Li, A. (2018). TPGLDA: Novel prediction of associations between lncRNAs and diseases via lncRNA-disease-gene tripartite graph. Sci. Rep. 8:1065. doi: 10.1038/s41598-018-19357-3
Doney, B., Hnizdo, E., Syamlal, G., Kullman, G., Burchfiel, C., Martin, C. J., et al. (2014). Prevalence of chronic obstructive pulmonary disease among US working adults aged 40 to 70 years. National Health Interview Survey data 2004 to 2011. J. Occup. Environ. Med. 56, 1088–1093. doi: 10.1097/JOM.0000000000000232
Ellis, B. C., Molloy, P. L., and Graham, L. D. (2012). CRNDE: a long non-coding rna involved in cancer, neurobiology, and development. Front. Genet. 3:270. doi: 10.3389/fgene.2012.00270
Feng, W., Wang, C., Liang, C., Yang, H., Chen, D., Yu, X., et al. (2018). The dysregulated expression of KCNQ1OT1 and its interaction with downstream factors miR-145/CCNE2 in breast cancer cells. Cell Physiol. Biochem. 49, 432–446. doi: 10.1159/000492978
Ferre, F., Colantoni, A., and Helmer-Citterich, M. (2016). Revealing protein-lncRNA interaction. Brief. Bioinform. 17, 106–116. doi: 10.1093/bib/bbv031
File, T. M. (2000). The epidemiology of respiratory tract infections. Semin. Respir. Infect. 15, 184–194. doi: 10.1053/srin.2000.18059
Gao, L., Liu, Y., Guo, S., Yao, R., Wu, L., Xiao, L., et al. (2017). Circulating long noncoding RNA HOTAIR is an essential mediator of acute myocardial infarction. Cell Physiol. Biochem. 44, 1497–1508. doi: 10.1159/000485588
Gokey, J. J., Snowball, J., Sridharan, A., Speth, J. P., Black, K. E., Hariri, L. P., et al. (2018). MEG3 is increased in idiopathic pulmonary fibrosis and regulates epithelial cell differentiation. JCI Insight 3:122490. doi: 10.1172/jci.insight.122490
Graham, M., and Adams, J. M. (1986). Chromosome 8 breakpoint far 3’ of the c-myc oncogene in a Burkitt’s lymphoma 2;8 variant translocation is equivalent to the murine pvt-1 locus. EMBO J. 5, 2845–2851. doi: 10.1002/j.1460-2075.1986.tb04578.x
Grammatikakis, I., Panda, A. C., Abdelmohsen, K., and Gorospe, M. (2014). Long noncoding RNAs(lncRNAs) and the molecular hallmarks of aging. Aging 6, 992–1009. doi: 10.18632/aging.100710
Gu, C., Liao, B., Li, X., Cai, L., Li, Z., Li, K., et al. (2017). Global network random walk for predicting potential human lncRNA-disease associations. Sci. Rep. 7:12442. doi: 10.1038/s41598-017-12763-z
Guan, Y., Kuo, W. L., Stilwell, J. L., Takano, H., Lapuk, A. V., Fridlyand, J., et al. (2007). Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin. Cancer Res. 13, 5745–5755. doi: 10.1158/1078-0432.ccr-06-2882
Gupta, R. A., Shah, N., Wang, K. C., Kim, J., Horlings, H. M., Wong, D. J., et al. (2010). Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076. doi: 10.1038/nature08975
Han, Y., Liu, Y., Gui, Y., and Cai, Z. (2013). Long intergenic non-coding RNA TUG1 is overexpressed in urothelial carcinoma of the bladder. J Surg. Oncol. 107, 555–559. doi: 10.1002/jso.23264
Hao, Y., Wu, W., Li, H., Yuan, J., Luo, J., Zhao, Y., et al. (2016). NPInter v3.0: an upgraded database of noncoding RNA-associated interactions. Database 2016:baw057. doi: 10.1093/database/baw057
Hung, T., and Chang, H. Y. (2010). Long noncoding RNA in genome regulation: prospects and mechanisms. RNA Biol. 7, 582–585. doi: 10.4161/rna.7.5.13216
Iacobucci, I., Sazzini, M., Garagnani, P., Ferrari, A., Boattini, A., Lonetti, A., et al. (2011). A polymorphism in the chromosome 9p21 ANRIL locus is associated to Philadelphia positive acute lymphoblastic leukemia. Leuk. Res. 35, 1052–1059. doi: 10.1016/j.leukres.2011.02.020
Ireland, M. P. (1986). Studies on the effects of dietary beryllium at two different calcium concentrations in Achatina fulica (Pulmonata). Comp. Biochem. Physiol. C 83, 435–438. doi: 10.1016/0742-8413(86)90149-0
Jiang, H., Li, T., Qu, Y., Wang, X., Li, B., Song, J., et al. (2018). Long non-coding RNA SNHG15 interacts with and stabilizes transcription factor Slug and promotes colon cancer progression. Cancer Lett. 425, 78–87. doi: 10.1016/j.canlet.2018.03.038
Jiang, Y. J., and Bikle, D. D. (2014). LncRNA: a new player in 1alpha, 25(OH)(2) vitamin D(3) /VDR protection against skin cancer formation. Exp. Dermatol. 23, 147–150. doi: 10.1111/exd.12341
Kapranov, P., Cheng, J., Dike, S., Nix, D. A., Duttagupta, R., Willingham, A. T., et al. (2007). RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316, 1484–1488. doi: 10.1126/science.1138341
Kataoka, M., and Wang, D. Z. (2014). Non-coding RNAs including miRNAs and lncRNAs in cardiovascular biology and disease. Cells 3, 883–898. doi: 10.3390/cells3030883
Ke, H., Zhao, L., Feng, X., Xu, H., Zou, L., Yang, Q., et al. (2016). NEAT1 is required for survival of breast cancer cells through FUS and miR-548. Gene Regul. Syst. Bio. 10, 11–17. doi: 10.4137/GRSB.S29414
Kohler, S., Bauer, S., Horn, D., and Robinson, P. N. (2008). Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet. 82, 949–958. doi: 10.1016/j.ajhg.2008.02.013
Kung, J. T., Colognori, D., and Lee, J. T. (2013). Long noncoding RNAs: past, present, and future. Genetics 193, 651–669. doi: 10.1534/genetics.112.146704
Li, J. H., Liu, S., Zheng, L. L., Wu, J., Sun, W. J., Wang, Z. L., et al. (2014). Discovery of protein-lncRNA interactions by integrating large-scale CLIP-Seq and RNA-Seq datasets. Front. Bioeng. Biotechnol. 2:88. doi: 10.3389/fbioe.2014.00088
Liu, C. J., Gao, C., Ma, Z., Cong, R., Zhang, Q., and Guo, A. Y. (2017). lncRInter: a database of experimentally validated long non-coding RNA interaction. J. Genet. Genomics 44, 265–268. doi: 10.1016/j.jgg.2017.01.004
Liu, M. X., Chen, X., Chen, G., Cui, Q. H., and Yan, G. Y. (2014). A computational framework to infer human disease-associated long noncoding RNAs. PLoS One 9:e84408. doi: 10.1371/journal.pone.0084408
Liu, Q., Huang, J., Zhou, N., Zhang, Z., Zhang, A., Lu, Z., et al. (2013). LncRNA loc285194 is a p53-regulated tumor suppressor. Nucleic Acids Res. 41, 4976–4987. doi: 10.1093/nar/gkt182
Lu, C., Yang, M., Luo, F., Wu, F. X., Li, M., Pan, Y., et al. (2018). Prediction of lncRNA-disease associations based on inductive matrix completion. Bioinformatics 34, 3357–3364. doi: 10.1093/bioinformatics/bty327
Lu, W., Zhu, L., Ruan, Z. B., Wang, M. X., Ren, Y., and Li, W. (2018). HOTAIR promotes inflammatory response after acute myocardium infarction by upregulating RAGE. Eur. Rev. Med. Pharmacol. Sci. 22, 7423–7430. doi: 10.26355/eurrev_201811_16282
Ma, C., Shi, X., Zhu, Q., Li, Q., Liu, Y., Yao, Y., et al. (2016). The growth arrest-specific transcript 5 (GAS5): a pivotal tumor suppressor long noncoding RNA in human cancers. Tumour Biol. 37, 1437–1444. doi: 10.1007/s13277-015-4521-9
Martens-Uzunova, E. S., Bottcher, R., Croce, C. M., Jenster, G., Visakorpi, T., and Calin, G. A. (2014). Long noncoding RNA in prostate, bladder, and kidney cancer. Eur. Urol. 65, 1140–1151. doi: 10.1016/j.eururo.2013.12.003
Menche, J., Sharma, A., Kitsak, M., Ghiassian, S. D., Vidal, M., Loscalzo, J., et al. (2015). Disease networks. Uncovering disease-disease relationships through the incomplete interactome. Science 347:1257601. doi: 10.1126/science.1257601
Mercer, T. R., Dinger, M. E., and Mattick, J. S. (2009). Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10, 155–159. doi: 10.1038/nrg2521
Mizrahi, A., Czerniak, A., Levy, T., Amiur, S., Gallula, J., Matouk, I., et al. (2009). Development of targeted therapy for ovarian cancer mediated by a plasmid expressing diphtheria toxin under the control of H19 regulatory sequences. J. Transl. Med. 7:69. doi: 10.1186/1479-5876-7-69
Moran, V. A., Perera, R. J., and Khalil, A. M. (2012). Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res. 40, 6391–6400. doi: 10.1093/nar/gks296
Mourtada-Maarabouni, M., Pickard, M. R., Hedge, V. L., Farzaneh, F., and Williams, G. T. (2009). GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene 28, 195–208. doi: 10.1038/onc.2008.373
Nakamura, Y., Takahashi, N., Kakegawa, E., Yoshida, K., Ito, Y., Kayano, H., et al. (2008). The GAS5 (growth arrest-specific transcript 5) gene fuses to BCL6 as a result of t(1;3)(q25;q27) in a patient with B-cell lymphoma. Cancer Genet. Cytogenet. 182, 144–149. doi: 10.1016/j.cancergencyto.2008.01.013
Navlakha, S., and Kingsford, C. (2010). The power of protein interaction networks for associating genes with diseases. Bioinformatics 26, 1057–1063. doi: 10.1093/bioinformatics/btq076
Orom, U. A., Derrien, T., Beringer, M., Gumireddy, K., Gardini, A., Bussotti, G., et al. (2010). Long noncoding RNAs with enhancer-like function in human cells. Cell 143, 46–58. doi: 10.1016/j.cell.2010.09.001
Patel, N., Garikapati, K. R., Ramaiah, M. J., Polavarapu, K. K., Bhadra, U., and Bhadra, M. P. (2016). miR-15a/miR-16 induces mitochondrial dependent apoptosis in breast cancer cells by suppressing oncogene BMI1. Life Sci. 164, 60–70. doi: 10.1016/j.lfs.2016.08.028
Peng, H., Liu, Y., Tian, J., Ma, J., Tang, X., Rui, K., et al. (2015). The long noncoding RNA IFNG-AS1 promotes T helper type 1 cells response in patients with hashimoto’s thyroiditis. Sci. Rep. 5:17702. doi: 10.1038/srep17702
Pinero, J., Bravo, A., Queralt-Rosinach, N., Gutierrez-Sacristan, A., Deu-Pons, J., Centeno, E., et al. (2017). DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 45, D833–D839. doi: 10.1093/nar/gkw943
Pio, G., Ceci, M., Malerba, D., and D’Elia, D. (2015). ComiRNet: a web-based system for the analysis of miRNA-gene regulatory networks. BMC Bioinform. 16(Suppl. 9):S7. doi: 10.1186/1471-2105-16-S9-S7
Qi, P., and Du, X. (2013). The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Mod. Pathol. 26, 155–165. doi: 10.1038/modpathol.2012.160
Qian, Y., Besenbacher, S., Mailund, T., and Schierup, M. H. (2014). Identifying disease associated genes by network propagation. BMC Syst. Biol. 8(Suppl. 1):S6. doi: 10.1186/1752-0509-8-S1-S6
Qiu, J. J., Lin, Y. Y., Ye, L. C., Ding, J. X., Feng, W. W., Jin, H. Y., et al. (2014). Overexpression of long non-coding RNA HOTAIR predicts poor patient prognosis and promotes tumor metastasis in epithelial ovarian cancer. Gynecol. Oncol. 134, 121–128. doi: 10.1016/j.ygyno.2014.03.556
Sattari, A., Siddiqui, H., Moshiri, F., Ngankeu, A., Nakamura, T., Kipps, T. J., et al. (2016). Upregulation of long noncoding RNA MIAT in aggressive form of chronic lymphocytic leukemias. Oncotarget 7, 54174–54182. doi: 10.18632/oncotarget.11099
Sha, S., Yuan, D., Liu, Y., Han, B., and Zhong, N. (2017). Targeting long non-coding RNA DANCR inhibits triple negative breast cancer progression. Biol. Open 6, 1310–1316. doi: 10.1242/bio.023135
Sharma, A., Kitsak, M., Cho, M. H., Ameli, A., Zhou, X., Jiang, Z., et al. (2018). Integration of molecular interactome and targeted interaction analysis to identify a COPD disease network module. Sci. Rep. 8:14439. doi: 10.1038/s41598-018-32173-z
Sharma, A., Menche, J., Huang, C. C., Ort, T., Zhou, X., Kitsak, M., et al. (2015). A disease module in the interactome explains disease heterogeneity, drug response and captures novel pathways and genes in asthma. Hum. Mol. Genet. 24, 3005–3020. doi: 10.1093/hmg/ddv001
Shi, J. Y., Huang, H., Zhang, Y. N., Long, Y. X., and Yiu, S. M. (2017). Predicting binary, discrete and continued lncRNA-disease associations via a unified framework based on graph regression. BMC Med. Genomics 10:65. doi: 10.1186/s12920-017-0305-y
Shi, X., Sun, M., Liu, H., Yao, Y., and Song, Y. (2013). Long non-coding RNAs: a new frontier in the study of human diseases. Cancer Lett. 339, 159–166. doi: 10.1016/j.canlet.2013.06.013
Sonawane, A. R., Weiss, S. T., Glass, K., and Sharma, A. (2019). Network medicine in the age of biomedical big data. Front. Genet. 10:294. doi: 10.3389/fgene.2019.00294
Sun, J., Shi, H., Wang, Z., Zhang, C., Liu, L., Wang, L., et al. (2014). Inferring novel lncRNA-disease associations based on a random walk model of a lncRNA functional similarity network. Mol. Biosyst. 10, 2074–2081. doi: 10.1039/c3mb70608g
Sun, M., and Kraus, W. L. (2015). From discovery to function: the expanding roles of long noncoding RNAs in physiology and disease. Endocr. Rev. 36, 25–64. doi: 10.1210/er.2014-1034
Suresh, V., Liu, L., Adjeroh, D., and Zhou, X. (2015). RPI-Pred: predicting ncRNA-protein interaction using sequence and structural information. Nucleic Acids Res. 43, 1370–1379. doi: 10.1093/nar/gkv020
Tang, W., Shen, Z., Guo, J., and Sun, S. (2016). Screening of long non-coding RNA and TUG1 inhibits proliferation with TGF-beta induction in patients with COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 11, 2951–2964. doi: 10.2147/copd.s109570
Tasan, M., Musso, G., Hao, T., Vidal, M., MacRae, C. A., and Roth, F. P. (2015). Selecting causal genes from genome-wide association studies via functionally coherent subnetworks. Nat. Methods 12, 154–159. doi: 10.1038/nmeth.3215
Turnbull, C., Ahmed, S., Morrison, J., Pernet, D., Renwick, A., Maranian, M., et al. (2010). Genome-wide association study identifies five new breast cancer susceptibility loci. Nat. Genet. 42, 504–507. doi: 10.1038/ng.586
Ulitsky, I. (2016). Evolution to the rescue: using comparative genomics to understand long non-coding RNAs. Nat. Rev. Genet. 17, 601–614. doi: 10.1038/nrg.2016.85
Vandin, F., Upfal, E., and Raphael, B. J. (2011). Algorithms for detecting significantly mutated pathways in cancer. J. Comput. Biol. 18, 507–522. doi: 10.1089/cmb.2010.0265
Vidal, M., Cusick, M. E., and Barabasi, A. L. (2011). Interactome networks and human disease. Cell 144, 986–998. doi: 10.1016/j.cell.2011.02.016
Vincent-Salomon, A., Ganem-Elbaz, C., Manie, E., Raynal, V., Sastre-Garau, X., Stoppa-Lyonnet, D., et al. (2007). X inactive-specific transcript RNA coating and genetic instability of the X chromosome in BRCA1 breast tumors. Cancer Res. 67, 5134–5140. doi: 10.1158/0008-5472.can-07-0465
Wang, J., Ma, R., Ma, W., Chen, J., Yang, J., Xi, Y., et al. (2016). LncDisease: a sequence based bioinformatics tool for predicting lncRNA-disease associations. Nucleic Acids Res. 44:e90. doi: 10.1093/nar/gkw093
Wang, L., Liu, D., Wu, X., Zeng, Y., Li, L., Hou, Y., et al. (2018). Long non-coding RNA (LncRNA) RMST in triple-negative breast cancer (TNBC): Expression analysis and biological roles research. J. Cell Physiol. 233, 6603–6612. doi: 10.1002/jcp.26311
Wang, Y., Chen, W., Yang, C., Wu, W., Wu, S., Qin, X., et al. (2012). Long non-coding RNA UCA1a(CUDR) promotes proliferation and tumorigenesis of bladder cancer. Int. J. Oncol. 41, 276–284. doi: 10.3892/ijo.2012.1443
Wu, X. S., Wang, X. A., Wu, W. G., Hu, Y. P., Li, M. L., Ding, Q., et al. (2014). MALAT1 promotes the proliferation and metastasis of gallbladder cancer cells by activating the ERK/MAPK pathway. Cancer Biol. Ther. 15, 806–814. doi: 10.4161/cbt.28584
Xiao, Y., Zhang, J., and Deng, L. (2017). Prediction of lncRNA-protein interactions using HeteSim scores based on heterogeneous networks. Sci. Rep. 7:3664. doi: 10.1038/s41598-017-03986-1
Xu, L. Z., Li, S. S., Zhou, W., Kang, Z. J., Zhang, Q. X., Kamran, M., et al. (2017). p62/SQSTM1 enhances breast cancer stem-like properties by stabilizing MYC mRNA. Oncogene 36, 304–317. doi: 10.1038/onc.2016.202
Yang, R., Liu, M., Liang, H., Guo, S., Guo, X., Yuan, M., et al. (2016). miR-138-5p contributes to cell proliferation and invasion by targeting Survivin in bladder cancer cells. Mol. Cancer 15:82.
Yang, X., Gao, L., Guo, X., Shi, X., Wu, H., Song, F., et al. (2014). A network based method for analysis of lncRNA-disease associations and prediction of lncRNAs implicated in diseases. PLoS One 9:e87797. doi: 10.1371/journal.pone.0087797
Yildirim, E., Kirby, J. E., Brown, D. E., Mercier, F. E., Sadreyev, R. I., Scadden, D. T., et al. (2013). Xist RNA is a potent suppressor of hematologic cancer in mice. Cell 152, 727–742. doi: 10.1016/j.cell.2013.01.034
Ying, L., Huang, Y., Chen, H., Wang, Y., Xia, L., Chen, Y., et al. (2013). Downregulated MEG3 activates autophagy and increases cell proliferation in bladder cancer. Mol. Biosyst. 9, 407–411. doi: 10.1039/c2mb25386k
Zhang, C. Y., Yu, M. S., Li, X., Zhang, Z., Han, C. R., and Yan, B. (2017). Overexpression of long non-coding RNA MEG3 suppresses breast cancer cell proliferation, invasion, and angiogenesis through AKT pathway. Tumour Biol. 39:1010428317701311. doi: 10.1177/1010428317701311
Zheng, X., Wang, Y., Tian, K., Zhou, J., Guan, J., Luo, L., et al. (2017). Fusing multiple protein-protein similarity networks to effectively predict lncRNA-protein interactions. BMC Bioinform. 18:420. doi: 10.1186/s12859-017-1819-1
Zhou, B., Zhao, H., Yu, J., Guo, C., Dou, X., Song, F., et al. (2018). EVLncRNAs: a manually curated database for long non-coding RNAs validated by low-throughput experiments. Nucleic Acids Res. 46, D100–D105. doi: 10.1093/nar/gkx677
Keywords: lncRNA, network medicine, interactome, network diffusion, disease, protein–protein interactions, disease network
Citation: Sumathipala M, Maiorino E, Weiss ST and Sharma A (2019) Network Diffusion Approach to Predict LncRNA Disease Associations Using Multi-Type Biological Networks: LION. Front. Physiol. 10:888. doi: 10.3389/fphys.2019.00888
Received: 01 January 2019; Accepted: 26 June 2019;
Published: 16 July 2019.
Edited by:
Sudipto Saha, Bose Institute, IndiaReviewed by:
Paolo Tieri, Italian National Research Council (CNR), ItalyCopyright © 2019 Sumathipala, Maiorino, Weiss and Sharma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amitabh Sharma, cmVhc2hAY2hhbm5pbmcuaGFydmFyZC5lZHU=; YW1pdGFiaHNoYXJtYWFAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.