
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 05 June 2019
Sec. Cardiac Electrophysiology
Volume 10 - 2019 | https://doi.org/10.3389/fphys.2019.00700
 Eyal Nof1,2Leonid Vysochek1Eshcar Meisel1,2Elena Burashnikov3Charles Antzelevitch3,4,5Jerome Clatot3Roy Beinart1,2David Luria1†Michael Glikson1,2†
Eyal Nof1,2Leonid Vysochek1Eshcar Meisel1,2Elena Burashnikov3Charles Antzelevitch3,4,5Jerome Clatot3Roy Beinart1,2David Luria1†Michael Glikson1,2† Shimrit Oz1*
Shimrit Oz1*Mutations in the SCN5A gene, encoding the cardiac voltage-gated sodium channel NaV1.5, are associated with inherited cardiac arrhythmia and conduction disease. Ca2+-dependent mechanisms and the involvement of β-subunit (NaVβ) in NaV1.5 regulation are not fully understood. A patient with severe sinus-bradycardia and cardiac conduction-disease was genetically evaluated and compound heterozygosity in the SCN5A gene was found. Mutations were identified in the cytoplasmic DIII-IV linker (K1493del) and the C-terminus (A1924T) of NaV1.5, both are putative CaM-binding domains. These mutants were functionally studied in human embryonic kidney (HEK) cells and HL-1 cells using whole-cell patch clamp technique. Calmodulin (CaM) interaction and cell-surface expression of heterologously expressed NaV1.5 mutants were studied by pull-down and biotinylation assays. The mutation K1493del rendered NaV1.5 non-conductive. NaV1.5K1493del altered the gating properties of co-expressed functional NaV1.5, in a Ca2+ and NaVβ1-dependent manner. NaV1.5A1924T impaired NaVβ1-dependent gating regulation. Ca2+-dependent CaM-interaction with NaV1.5 was blunted in NaV1.5K1493del. Electrical charge substitution at position 1493 did not affect CaM-interaction and channel functionality. Arrhythmia and conduction-disease -associated mutations revealed Ca2+-dependent gating regulation of NaV1.5 channels. Our results highlight the role of NaV1.5 DIII-IV linker in the CaM-binding complex and channel function, and suggest that the Ca2+-sensing machinery of NaV1.5 involves NaVβ1.
Sodium current (INa) upstroke is a hallmark of the action-potential in excitable cells. The SCN5A gene encodes the pore-forming α-subunit of the cardiac sodium channel NaV1.5. NaV1.5 channels are expressed in cardiomyocytes as well as the cardiac His-Purkinje system. Accordingly, NaV1.5 loss-of-function mutations are associated with cardiac arrhythmia and conduction defects (Schott et al., 1999; Tan et al., 2001; Holst et al., 2010; Zumhagen et al., 2013). Although INa does not contribute to the action potential of pacemaker cells, its presence in the periphery of the sinoatrial node can modulate impulse conduction and heart rate (Kodama et al., 1997). Hence, loss-of-function mutations in SCN5A can result in a nodal dysfunction (Benson et al., 2003; Lei et al., 2008; Gui et al., 2010; Ziyadeh-Isleem et al., 2014; Chiang et al., 2015; Milanesi et al., 2015).
The NaV1.5 α-subunit is composed of four homologous domains (DI-DIV), each containing six transmembrane repeats. The domains are joined by three cytosolic linkers and flanked by cytosolic C- and N-termini (see Figure 1C). The α-subunit forms a functional monomer; however, multi-channel assembly and functional coupling among monomeric channels have been reported (Keller et al., 2005; Poelzing et al., 2006; Clatot et al., 2017). Moreover, the α-subunit is the core of a macromolecular complex that interacts with auxiliary proteins that modulate expression, trafficking, localization and gating of NaV1.5; among them the regulatory β-subunits (NaVβ) and the Ca2+-sensor calmodulin (CaM) (Abriel, 2010).
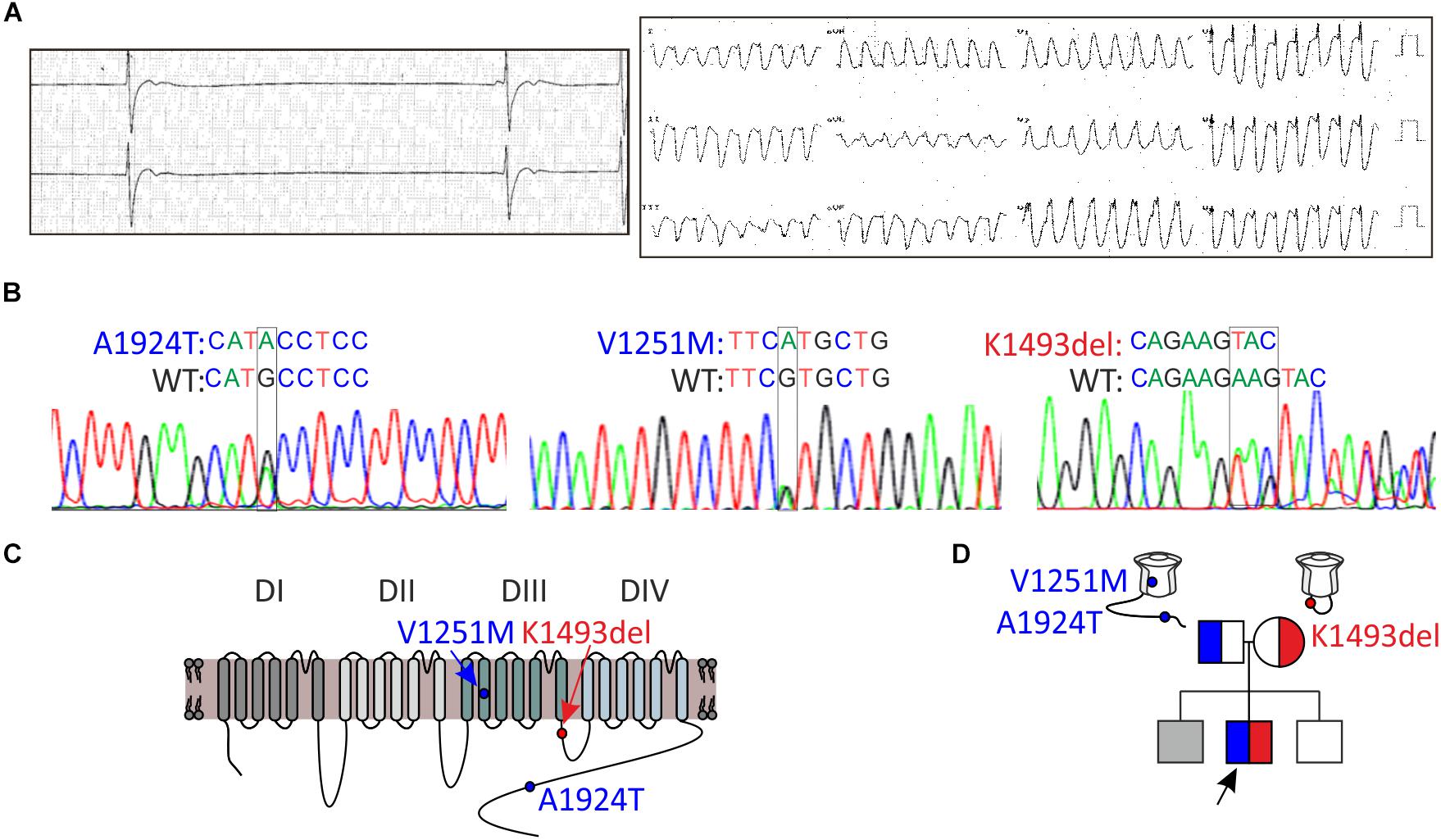
Figure 1. Clinical and genetic characteristics of the proband. (A) Left, Holter recording at rest. Right, 12-lead ECG during exercise. (B) Sequence chromatogram of SCN5A variants. (C) Schematic illustration of NaV1.5 protein and the location of the variants found. (D) Pedigree tree of the family. Circle indicates female, squares indicate male. The arrow points to the proband. In gray, suspected affected sibling. Red for the maternal allele variant, blue for the paternal allele variant.
NaV1.5 is regulated by Ca2+, but the role and mechanism of this regulation is still debated (Van Petegem et al., 2012; Ben-Johny et al., 2014; Gabelli et al., 2016; Pitt and Lee, 2016; Johnson, 2019). CaM interacts with NaV1.5 C-terminus (CT) (Gabelli et al., 2014; Wang et al., 2014), and the linker between domains DIII-IV (DIII-IV linker) (Sarhan et al., 2012; Johnson et al., 2018). However, the contribution of each CaM-interaction domain to the Ca2+-sensing machinery of NaV1.5 is not fully established. NaVβ1 interacts non-covalently with NaV1.5 and can modulate INa gating, as well as transcription and cell-adhesion (Calhoun and Isom, 2014). The involvement of NaVβ1 in Ca2+-dependent gating regulation is not clear.
We report a novel combination of SCN5A variants, K1493del and A1924T, in a patient with sinus-bradycardia and cardiac conduction-disease. Heterozygous A1924T has been previously associated with Brugada syndrome (BrS) (Rook et al., 1999), and homozygous A1924T with sinus-bradycardia with conduction delay (Lopez et al., 2011). Heterozygous K1493del has been associated with isolated conduction disease (Zumhagen et al., 2013). Here, we studied the molecular basis of Ca2+- and CaM-dependent NaV1.5 modulation, using the disease-associated mutations.
The proband and his parents gave written informed consents for both the clinical and genetic studies, which were approved by the Institutional Ethics-Committee of the Sheba Medical Center, Tel-Hashomer (approval 2853/03). Evaluation included resting electrocardiogram (ECG), 24-h Holter monitoring (DELMAR® systems; Impresario 3.04.0089), two-dimensional echocardiography, treadmill exercise test, cardiac MRI, ajmaline test and an electrophysiological-study.
Heparinized blood was drawn from the proband and his parents; DNA was extracted using a commercial kit (Gentra System Inc., Minneapolis, MN, United States) and the exons and exon-intron boundaries of the following genes: HCN4, KCNJ2, KCNJ12, and SCN5A were amplified by PCR (Verities PCR, Applied Biosystems, Austin, TX, United States). The PCR products were purified (Exosap-IT, USB, Isogen Life-Science, Netherlands) and sequenced in both directions (BigDye Terminator v3.1 cycle sequencing Kit and 3130xL Genetic Analyzer, Applied Biosystems).
A DNA construct of the paternal NaV1.5 variant was prepared with two variants, A1924T and V1251M, on the same DNA construct and denoted as NaV1.5A1924T^*. In the maternal channel, NaV1.5K1493del, one lysine of the doublet in positions 1492-3 was deleted. N-terminally tagged green-fluorescent protein (GFP)-NaV1.5 was used (Zimmer et al., 2002). In the constructs K1493A/E/R the second lysine of the lysine-doublet was substituted with the amino-acids indicated. Point-mutations were prepared by site-directed mutagenesis using a standard PCR (Roche, IN, United States), and followed by sequencing of the entire coding sequence. Rat and human -NaVβ1 (rNaVβ1 and hNaVβ1, respectively) were cloned into pcDNA3 vector using HindIII and EcoRI sites, and followed by an internal ribosome entry site (IRES) and the red fluorescent-protein (RFP) sequence between EcoRI and NotI sites (rNaVβ1/RFP and hNaVβ1/RFP, respectively). NaV1.5 (NM_198056.2), calmodulin (M19312), rNaVβ1 (M91808) hNaVβ1 (NP_001028), and GFP were all in a pcDNA3 vector.
HEK293 cells were maintained in Dulbecco’s Modified Eagle’s Medium supplemented with 10% Fetal Bovine Serum, 100 U/ml penicillin, 10 mg/ml streptomycin and 2 mM L-Glutamine (Biological Industries, Kibbutz Beit-Haemek, Israel) at 37°C with 5% CO2. For electrophysiological experiments, transfections were performed in 35 mm dish using Trans-ITx2 (Mirus, Madison, WI, United States) according to the manufacturer’s instructions. 1 μg of each construct was used for transfection (NaV1.5, NaVβ1/RFP, and CaM), except in Figure 2A, where 3 μg of GFP tagged- K1493del mutated -NaV1.5 (GFP-NaV1.5K1493del) and NaVβ1/RFP (μg DNA ratio 1:1) were used. When indicated, 0.5 μg GFP was added as a transfection marker. On the following day, cells were plated on coverslips. For biochemical experiments, cells in 10-cm dishes were transfected using the Calcium-Phosphate method. 5–15 μg DNA of NaV1.5 α-subunit were used, rNaVβ1 was added in a 0.6 β:α molar ratio unless otherwise indicated. In Figure 5B, CaM was added in a 2 CaM:α molar ratio. Experiments were performed 48–72 h after transfection.
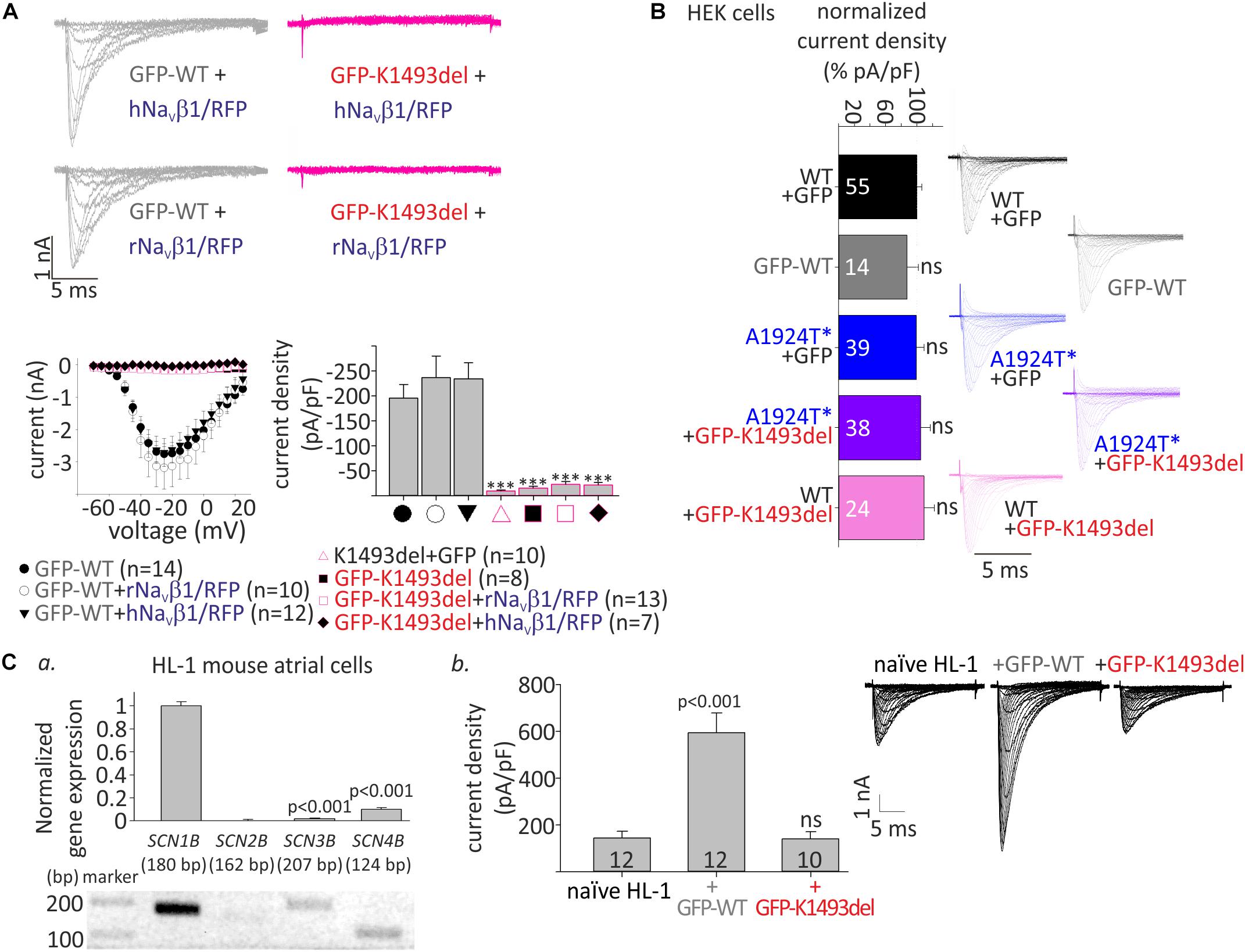
Figure 2. Whole-cell INa of WT and mutant NaV1.5 in HEK and HL-1 cells. (A) Top, representative currents elicited from resting potential by depolarizing steps (–70 until –20 mV in 5 mV interval) in HEK cells. Bottom, voltage-dependent amplitudes and maximal current densities, in cells expressing GFP-NaV1.5WT, GFP-NaV1.5K1493del, and NaV1.5K1493del, with and without r/hNaVβ1 co-expressing RFP in the same vector. All groups were compared to GFP-WT for statistical significance. (B) WT and mutant NaV1.5 expressed in HEK cells (without NaVβ subunit). Current densities were normalized to the mean current density of NaV1.5WT in the same experiment and summarized from 9 experiments. Currents in A and the representative currents in B were recorded in [10 mM EGTA]in. (Ca), Gene expression levels of SCNB1-4 in HL-1 cells. Top, gene levels were quantified by real-time PCR and normalized to GAPDH. Bottom, semi-quantitative PCR. (b), INa measured in naïve and transfected HL-1 cells, in [10 mM EGTA]in. Mean current densities and representative cells are shown. Statistical significance was determined by one-way ANOVA, followed by Holm–Sidak test. ns, not significant.
HL-1 cells were plated in gelatin/fibronectin-coated dishes, in Claycomb media supplemented with 10% Fetal Bovine Serum, 100 μM norepinephrine (Sigma, St. Louis, Mo, United States), 100 U/ml penicillin, 10 mg/ml streptomycin and 2 mM L-Glutamine (Biological Industries). Transfections using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, United States) were performed in 35 mm dishes. On the following day, cells were plated on gelatin/fibronectin-coated coverslips. Experiments were performed 48 h after transfection.
Total RNA was extracted using RNeasy Plus mini kit (Qiagen, Hilden, Germany). Reverse transcription with random primers was performed using the high capacity cDNA reverse transcription kit (Applied Biosystems, CA, United States). cDNA was amplified using KAPA HiFi HotStart ReadyMix (Roche). Quantitative real-time PCR was performed using ABI Step-one plus sequence detection system (Applied Biosystems) with Fast SYBR Green Master Mix reagent (Applied Biosystems). The primers for mouse genes used were, for NaVβ1 (NM_011322.2): SCN1B Fw (CGAGGCTGTGTATGGGATGAC)/SCN1B Rv (CCCTCAAAGCGCTCATCTTC), for NaVβ2 (NM_001014761.2): SCN2B Fw (GTGAACCACAAGCAGT TCTCT)/SCN2B Rv (TGACACGTCGTACTTACTGGG), for NaVβ3 (NM_001286614.1): SCN3B Fw (TGTAATGTG TCCAGGGAGTTTG)/SCN3B Rv (TTCGGCCTTAGAGACCT TTCTG) and for NaVβ4 (NM_001013390.3): SCN4B Fw (GGAACCGAGGCAATACTCAGG)/SCN4B Rv (CCGTTAA TAGCGTAGATGGTGGT). Gene expression was quantified using the 2^−ΔΔCt method by normalization to the housekeeping gene GAPDH (Fw: TCGTCCCGTAG ACAAAATGG/Rv: TTGAGGTCAATGAAGGGGTC).
Currents were recorded using the whole-cell configuration of the patch-clamp technique at 23°C. Red- and/or green- fluorescent cells were selected for recording. Signals were amplified using an Axopatch 200B patch-clamp amplifier (Axon Instruments, Foster City, CA, United States), sampled at 100 kHz and filtered with a low-pass Bessel filter at 10 kHz. Data was acquired with DigiData1440A and analyzed using pCLAMP 10 software (Axon Instruments). Patch pipettes (Harvard apparatus) resistance was 1.5–4 MΩ. Cells with access resistance over 5 MΩ were discarded. Series resistance was compensated by 85%. Leak currents were subtracted using a P/3 protocol. External solution contained (in mM) 137 NaCl, 4 KCl, 10 Hepes, 10 Glucose, 1.8 CaCl2, and 1 MgCl2 titrated with NaOH to pH 7.4, and adjusted to 310 mOsm. Internal solution contained (in mM) 100 CsF, 25 NaF, 10 Hepes, 10 NaCl, 2 Mg2+-ATP and 10 ethylene glycol-bis (β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), titrated with CsOH to pH 7.2 and adjusted to 295 mOsm, this pipette solution is (10 mM EGTA)in. For fast Ca2+ chelation, EGTA was replaced with 10 mM 1,2-BIS (2-AMINOPHENOXY)-ETHANE-N,N,N′N′-TETRAACETIC ACID [(10 mM BAPTA)in]. For pipette solution with 10 μM Ca2+, EGTA was replaced with 1 mM BAPTA and 1 mM CaCl2 [(10 μM Ca2+)in]. EGTA was replaced with 5 mM N-(2-Hydroxyethyl)ethylenediamine-N,N′,N′-triacetic acid (HEDTA) and 2.5 mM CaCl2 for [10 μM Ca2+]in/HEDTA (calculated with WebMaxC extended program1). Pronase (Roche) was dissolved in [10 mM EGTA]in solution.
Activation protocol was initiated after 2 min, steady-state inactivation (SSI) protocol after 3.25 min and recovery from inactivation after 8 min from the rupture of the cell membrane.
Holding potential was −120 mV. Current-densities were obtained by dividing the peak current by the cell capacitance. Normalized current-densities were calculated by dividing the current density of each cell with the mean NaV1.5WT current density measured on the same day of the experiment, using the same pipette solution composition. The voltage-dependent activation was calculated by fitting currents, generated by steps from −80 to 40 mV in 5 mV increments for 20 ms, with a modified Boltzmann equation: I = [Gmax*(V-Vrev)]/[1+exp(Va-V)/k], where I is the peak current for the test potential V, Gmax is maximum conductance, Vrev is the reversal potential, Va is the potential for half-activation or half-availability, and k is the slope factor. The normalized conductance was determined by modified Ohm’s law G/Gmax = I/Gmax (V-Vrev).
Steady-state inactivation was assayed by a 20 ms test pulse to −20 mV after a 500 ms pre-pulse to varying voltages (from −140 to −45 mV in 5 mV steps). SSI curves were fitted with Boltzmann equation: I = 1/[1 + exp(Va − V)/k],
Recovery from inactivation curve was obtained by a 1 s conditioning pulse (I1) to -20 mV followed by a test pulse (I2) to -20 mV after a varying time (1–24 ms, with 1 ms steps) at -120 mV. Fractional recovery was calculated as I2/I1. The time constant (τ) and amplitude (A) of recovery from inactivation were obtained by fitting the data with the function y = A(1−e–t/τ).
Waveform’s current decay was fitted to one -exponent fit using Levenberg-Marquardt algorithm, in the form f(t) = Ae–t/τ +C.
For biotinylation experiments, cells were incubated with 0.5 mg/ml EZ-Link Sulfo-NHS-SS-Biotin (Pierce, Rockford, IL, United States) in phosphate buffer saline (pH 8) containing 1 mM Ca2+ and 0.5 mM Mg2+ (PBS-CM), for 30 min at 4°C. The reaction was terminated by incubation in 50 mM glycine in PBS-CM, for 10 min at 4°C. Cells were scraped and washed with PBS-CM supplemented with 0.5 mM phenylmethylsulfonyl fluoride (PMSF) at 4°C. Cells were lysed in TNE buffer (in mM): 20 Tris–HCl pH 8, 150 NaCl, 1 EGTA, and 1% NP40 supplemented with 0.5 PMSF, protease inhibitor mixture (Roche), 25 β-glycerol phosphate and 1 Na3VO4, for 30 min on ice. A 25 μg sample was taken for input. An equal amount of total protein from each group was incubated with streptavidin-agarose beads (Pierce) for 2 h at 4°C in a rotating device. Samples were washed with TNE buffer supplemented with 0.5 mM PMSF, and proteins were eluted by incubation with sample buffer and freshly added 100 mM DTT, for 1 h at room temperature, and then for 5 min at 65°C. NaV1.5WT and NaV1.5K1493del intensities were compared in groups transfected with an equal amount of DNA.
For CaM pull-down experiments, cells were collected in PBS supplemented with 0.5 mM PMSF, and lysed with (in mM) 20 Tris–HCl pH 7.5, 150 NaCl, and 1% Triton x100, supplemented with 0.5 PMSF, protease inhibitor mixture (Roche), 25 β-glycerol phosphate and 1 Na3VO4, for 30 min on ice. A 25 μg sample was taken for input. CaM agarose beads (A6112, Sigma) were washed with (in mM) 20 Tris–HCl pH 7.5, 150 NaCl added with either 2 CaCl2, or 10 EGTA, then were incubated with an equal amount of total protein from each group, supplemented with either 2 CaCl2 or 10 EGTA, for 2 h at 4°C in a rotating device. Samples were washed with (in mM) 20 Tris–HCl pH 7.5, 150 NaCl and 1% Triton x100 supplemented with 0.5 PMSF, and proteins were eluted with sample buffer, for 5 min at 65°C.
Normalized NaV1.5 intensity was calculated by dividing NaV1.5 intensity by a normalizer, γ-tubulin for inputs (NaV1.5-inputNORM = NaV1.5/γ-tubulin) and Na-K ATPase for plasma membrane-bound fraction (NaV1.5-PMNORM = NaV1.5/Na-K ATPase). Percent of total expression was calculated by dividing intensities of NaV1.5-inputNORM with a control group from the same experiment. Trafficking was calculated by dividing NaV1.5-PMNORM by NaV1.5-inputNORM, and presented as % trafficking from the control group in the same experiment. CaM-beads binding level was quantified by dividing bound by input fractions and normalized to control group in the same experiment.
The antibodies that were used are hNaV1.5 (ASC-013, Alomone, Jerusalem, Israel), Na-K ATPase (ANP-001, Alomone), γ-tubulin (T5192, Sigma), and CaM (05-173, Millipore Corp., Temecula, CA, United States).
Densitometry of Western blot bands was analyzed using ImageJ (NIH, United States). Figures were prepared using CorelDrawX8 (Corel Corp., Ottawa, Canada). Statistical analyses were performed using SigmaPlot 13 (Systat Software, CA, United States). Data are presented as mean ± SEM. One-way ANOVA followed by the multiple comparison Holm–Sidak post hoc test was used to compare several groups. Two-tailed Student’s t-test was used to compare two groups.
A 16-year-old male presented with a syncope during exercise. Physical examination revealed a healthy looking young man without any abnormalities. He had bradycardia for many years but was not previously symptomatic. A 24-h Holter recording showed an average heart rate of 50 (range: 24–110) due to sinus-bradycardia and occasional junctional rhythm with sinus-pauses up to 5.8 s (Figure 1A, left). A wide complex tachycardia (WCT) of 193 beat-per-minute was recorded during exercise testing (Figure 1A, right). Echocardiography and cardiac-MRI did not reveal any structural abnormalities or areas of late gadolinium enhancement. Ajmaline provocation test ruled out BrS. Electrophysiological-study demonstrated a prolonged A-H interval of 260 ms and H–V interval of 75 ms. During rapid pacing (420 CL), the H–V interval increased to over 100 ms. Atrial-flutter was inducible with 1:1 conduction, leading to “clinical” WCT. The proband developed symptomatic bradycardia and a permanent pacemaker was eventually implanted.
DNA screening of the proband revealed compound heterozygosity in the SCN5A gene. The paternal allele had two missense variants, leading to amino-acid substitutions of alanine by threonine in position 1924 (A1924T) and valine by methionine in position 1251 (V1251M). The variant V1251M is a benign polymorphism (Kapplinger et al., 2015). The maternal allele had an in-frame, single codon deletion, resulting in a removal of one out of two adjacent lysines, in positions 1492-1493 (K1493del) (Figures 1B,C). We cannot rule out the presence of mutation(s) in additional gene(s) that were not identified in the present gene screen.
Holter-testing of both parents did not reveal bradycardia, but the father displayed premature ventricular contractions (PVCs) with Right Bundle Branch Block (RBBB) pattern in V1 on 12-lead ECG. One brother did not have bradycardia. Another brother had bradycardia but he declined any clinical or genetic evaluation (Figure 1D).
Although K1493del and A1924T heterozygotes were previously reported with bradyarrhythmias (Rook et al., 1999; Zumhagen et al., 2013), the heterozygotes in this report were asymptomatic, probably due to reduced disease penetrance in these individuals. The extent and reasons for SCN5A mutation expression variability are not entirely clear. Reasons for the variability may include epigenetic gene silencing, age, gender, environmental factors and other genetic modifiers (Liu et al., 2016; Verkerk et al., 2018). We suggest that the accumulated effects in the compound-heterozygote may have increased the penetrance and severity of the arrhythmic phenotype compared with the heterozygotes in the reported family.
To examine the functional consequences of the variants, we expressed NaV1.5, WT and mutants, in HEK cells and measured whole-cell INa. We used N-terminally GFP-fused NaV1.5 that conserves the biophysical properties of NaV1.5 (Clatot et al., 2012; Reinhard et al., 2013). Peak INa recorded in cells transfected with GFP-NaV1.5WT, with or without NaVβ1, was 1–6 nA. No INa was recorded in cells expressing GFP-NaV1.5K1493del or the non-tagged NaV1.5K1493del. Addition of a bicistronic vector expressing NaVβ1 from two species (rat or human) together with RFP as an expression reporter did not recover INa (Figure 2A), even when the amount of DNA used for transfection of GFP-NaV1.5K1493del +NaVβ1 was three-fold higher than GFP-NaV1.5WT. These results are at odds with a previous report (Zumhagen et al., 2013).
Compared with NaV1.5WT, the paternal variation NaV1.5A1924T^* did not affect INa density. In order to test whether NaV1.5K1493del has a dominant-negative effect, GFP-NaV1.5K1493del was added on top of NaV1.5WT or NaV1.5A1924T^*, in 1:1 DNA ratio. Co-expression of GFP-NaV1.5K1493del did not reduce INa density, in HEK cells (Figure 2B and Table 1).

Table 1. Current densities (pA/pF) of the normalized values presented in Figure 2B.
We used HL-1 cells, a mouse atrial cardiomyocyte tumor cell-line (Claycomb et al., 1998), to test the effect of NaV1.5K1493del on endogenous cardiac-cell INa. HL-1 cells express sodium channel subunits, predominantly the NaV1.5 α-subunit (Cerrone et al., 2014) and NaVβ1 subunit (Figure 2Ca), that recapitulate the physiological NaV1.5 stoichiometry in a cardiac cell milieu. We transfected HL-1 cells with GFP-NaV1.5WT or GFP-NaV1.5K1493del. Naïve, non-transfected, HL-1 cells showed INa of -144 ± 29 pA/pF. HL-1 cells transfected with GFP-NaV1.5WT had INa of −594 ± 84 pA/pF, while cells transfected with GFP-NaV1.5 K1493del had a basal INa of −140 ± 32 pA/pF, similar to non-transfected cells.
These findings demonstrate, in HEK and HL-1 cells, that NaV1.5K1493del is a loss-of-function mutant that does not induce a functional dominant-negative effect on INa density.
In order to understand if the basis for the loss-of-function in NaV1.5K1493del was an impairment in the protein expression or trafficking to the plasma membrane, we performed a biotinylation assay. NaV1.5WT and NaV1.5K1493del were transfected in HEK cells. Total and plasma-membrane biotin-bound NaV1.5 protein levels were quantified following Western-blot. We found a reduction in total cellular expression of NaV1.5K1493del (40 ± 7% of NaV1.5WT). The % trafficking was determined by dividing the biotinylated fraction by total fraction, normalized to % trafficking of NaV1.5WT, in the same experiment. The % trafficking of NaV1.5K1493del was not significantly different from % trafficking of NaV1.5WT (93 ± 16%) so that a similar fraction from NaV1.5 total protein was present at the plasma membrane (Figures 3A,C), suggesting that trafficking of NaV1.5K1493del to the plasma membrane was not impaired.
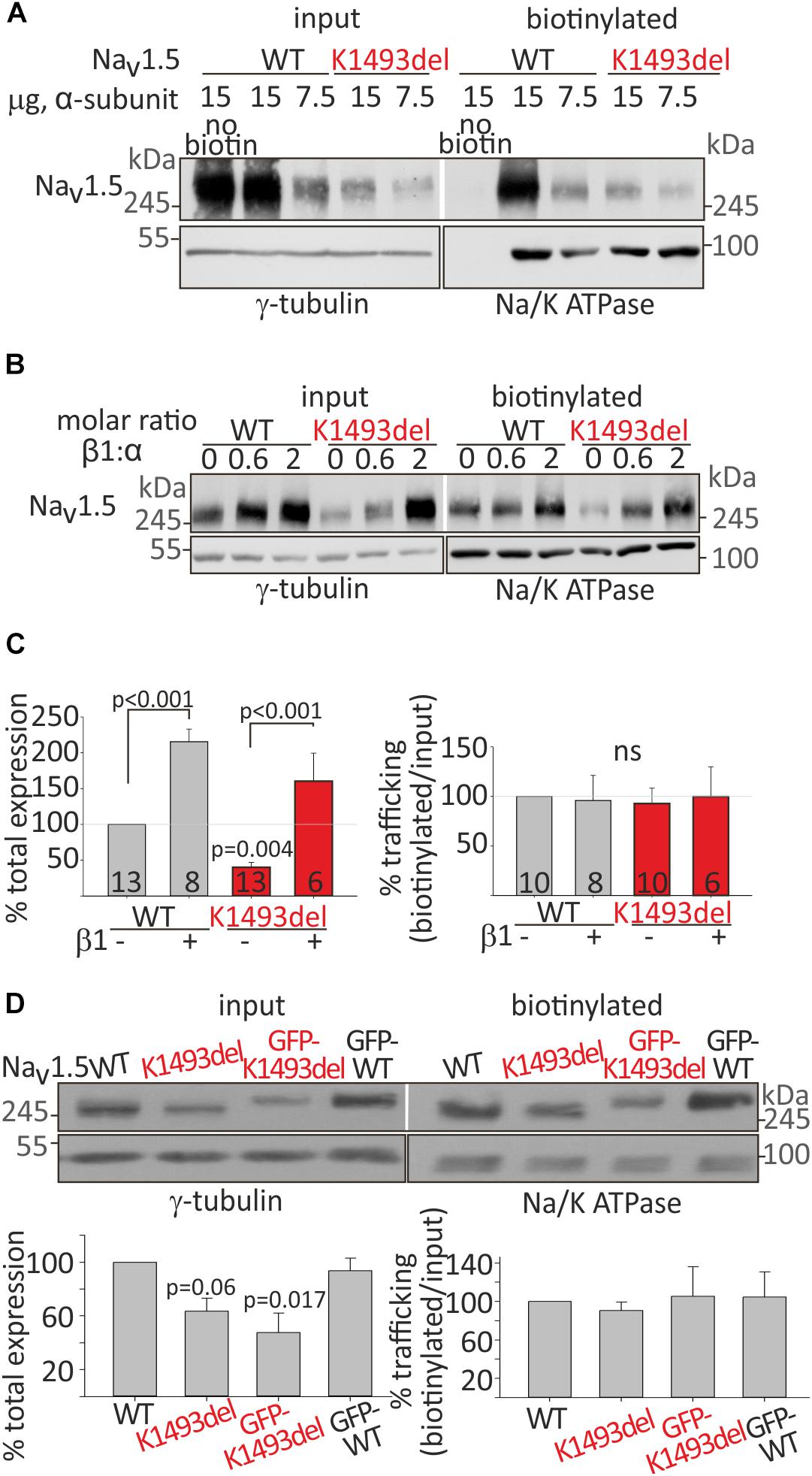
Figure 3. Cell surface and total expression of NaV1.5WT and NaV1.5K1493del. (A,B) Representative biotinylation assays. (A) HEK cells were transfected with NaV1.5WT and NaV1.5K1493del. The control “no-biotin” group shows no signal from non-biotinylated channels. (B) HEK cells transfected with NaV1.5WT and NaV1.5K1493del (7.5 μg) +/– rNaVβ1 (2.5 and 7.5 μg). (C) Summary of % total protein (left) and % trafficking (right), normalized to NaV1.5WT without NaVβ1. N = 9 experiments without NaVβ. N = 4 experiments with rNaVβ1 expressed. Groups expressing rNaVβ1 in molar ratios β:α 0.6-2 were pulled together. (D) GFP-fused and non-tagged NaV1.5WT and NaV1.5K1493del were expressed (without NaVβ subunit). This is a summary of three experiments. Statistical significance was determined by one-way ANOVA, followed by Holm–Sidak test.
NaVβ1 has been reported to improve the expression and trafficking of loss-of-function mutants of the sodium channel (Bechi et al., 2015). We examined the effect of NaVβ1 co-expression on total and cell-surface expression of NaV1.5WT and NaV1.5K1493del. Addition of NaVβ1 enhanced total expression of both NaV1.5WT and NaV1.5K1493del, with a concomitant increase in channel expression at the cell surface (Figure 3B).
A summary of total and biotinylated NaV1.5 levels, normalized to the NaV1.5WT expressed without NaVβ1 in the same experiment, is presented in Figure 3C. Co-expression of NaVβ1 increased total expression of NaV1.5WT by 215 ± 18% and NaV1.5K1493del by ∼400%, increasing the latter from 40 ± 7% to 160 ± 39% (compared to NaV1.5WT without NaVβ1). NaV1.5K1493del trafficking to the plasma membrane was similar to NaV1.5WT when NaVβ1 was co-expressed. These results support the conclusion that K1493del does not affect forward-trafficking or NaVβ1-regulated trafficking and expression (Figure 3A).
To test whether the addition of a GFP-tag to NaV1.5 N-terminus affected the expression or trafficking of NaV1.5, we performed a biotinylation experiment on GFP-tagged and non-tagged channels. GFP-fusion did not change the biogenesis properties: total expression of GFP-NaV1.5K1493del was partially reduced compared to GFP-NaV1.5WT, and GFP-tagged NaV1.5 channels were exported to the plasma membrane (Figure 3D).
In summary, K1493del partially reduced total protein expression but did not affect the plasma-membrane trafficking of NaV1.5. Co-expression of NaVβ1 restored the reduced NaV1.5K1493del cellular levels but did not restore INa (Figure 2A), thus the loss-of-function by K1493del mutation is only marginally due to a biogenesis defect.
We wanted to examine whether the non-conducting channel NaV1.5K1493del affects the current of the co-expressed NaV1.5A1924T^* variant, consistent with the compound heterozygosity observed in the patient. We tested Ca2+-dependent gating properties of INa in view of previous studies that included K1493 and A1924 residues in structural elements that bind the Ca2+-sensor CaM: DIII-IV linker and the CT, respectively. When the INa conducting variants NaV1.5A1924T^* or NaV1.5WT were co-expressed with the non-conducting variant NaV1.5K1493del, the latter was expressed as a GFP-fused channel to attest its co-expression in cells that displayed INa. Co-expression of GFP-NaV1.5K1493del resulted in a 3 mV depolarizing shift of the activation curve of NaV1.5A1924T^* in the presence of [10 μM Ca2+]in but not in the absence of [Ca2+]in. Co-expression of GFP-NaV1.5K1493del with NaV1.5WT resulted in a similar 3.5 mV depolarizing shift of the activation curve, only in [10 μM Ca2+]in (Figure 4A and Table 2). Although modest, the depolarizing shift in the activation curve in the presence of Ca2+ was significant. The similar current amplitudes (Table 2) and the similar effect in the two unrelated groups argue against the possibility of a recording artifact being the reason for the observed shift.
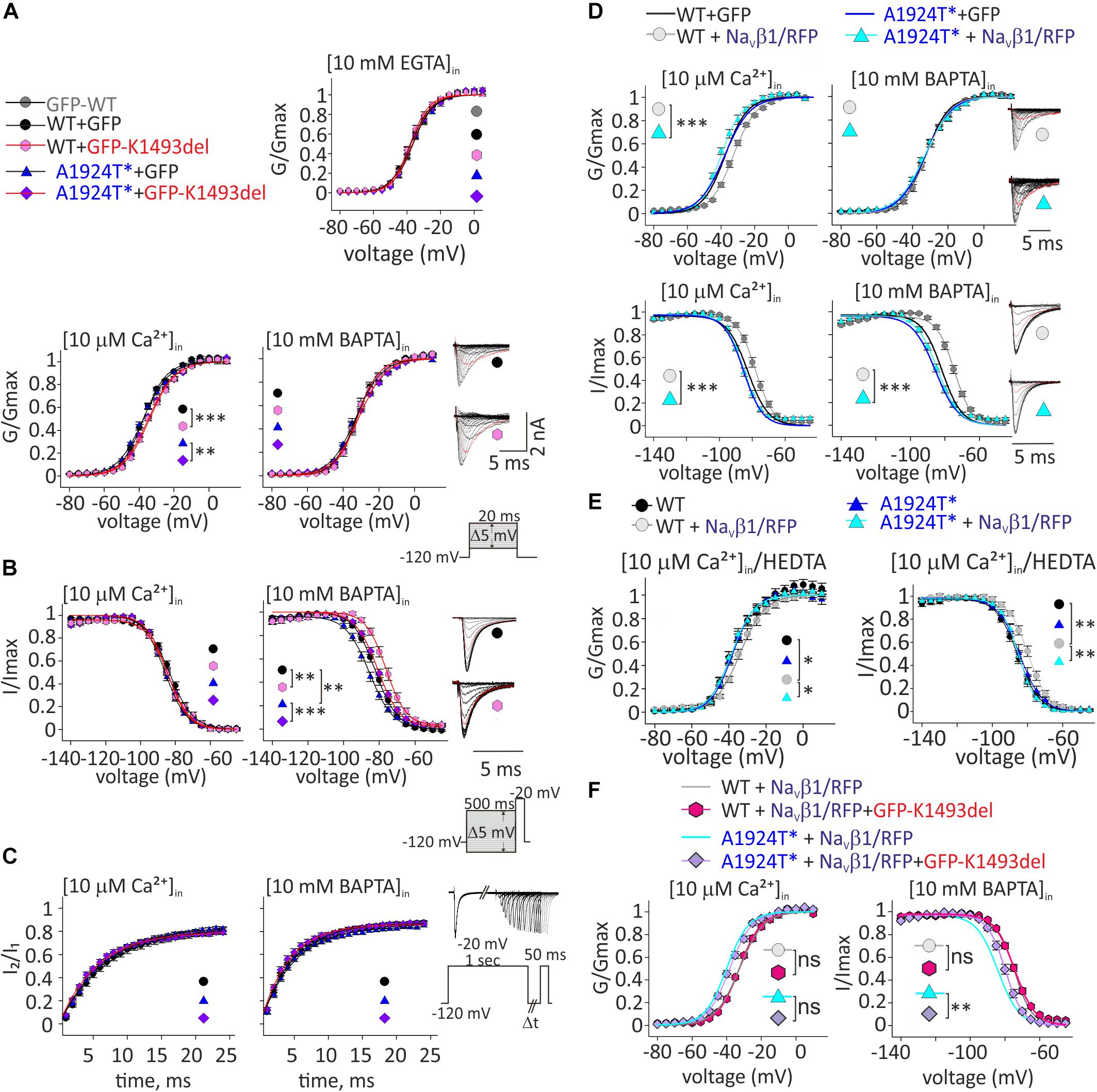
Figure 4. Ca2+-dependent gating properties of NaV1.5 variants. (A) Activation curves of conductance-voltage relationship. Current traces recorded in [10 μM Ca2+]in, and –40 mV trace is red. (B) SSI curves of voltage-dependent channel availability. Current traces during the test pulse at –20 mV are with (10 mM BAPTA)in. The test-pulse trace that follows –80 mV pre-pulse is red. (C) Recovery from inactivation. (D) Activation (top) and SSI (bottom) curves of NaV1.5WT +GFP and NaV1.5A1924T^*+GFP co-expressing rNaVβ1/RFP were plotted with the fitted curves of these groups without rNaVβ1/RFP (presented in A, B). Current traces recorded in [10 μM Ca2+]in. The –40 mV trace during activation and the test-pulse that follows the –80 mV pre-pulse during SSI are red. (E) Activation (left) and SSI (right) curves with 10 μM Ca2+ chelated with HEDTA in the internal solution. (F) Activation (left) and SSI (right) curves of NaV1.5 variants co-expressing rNaVβ1/RFP and GFP-NaV1.5K1493del were plotted with the fitted curves of these groups without GFP-NaV1.5K1493del (presented in D). Statistical significance of Va was determined by t-test: ns, not significant;*p < 0.05; ∗∗p < 0.01; ∗∗∗, p < 0.001 (see Table 2).
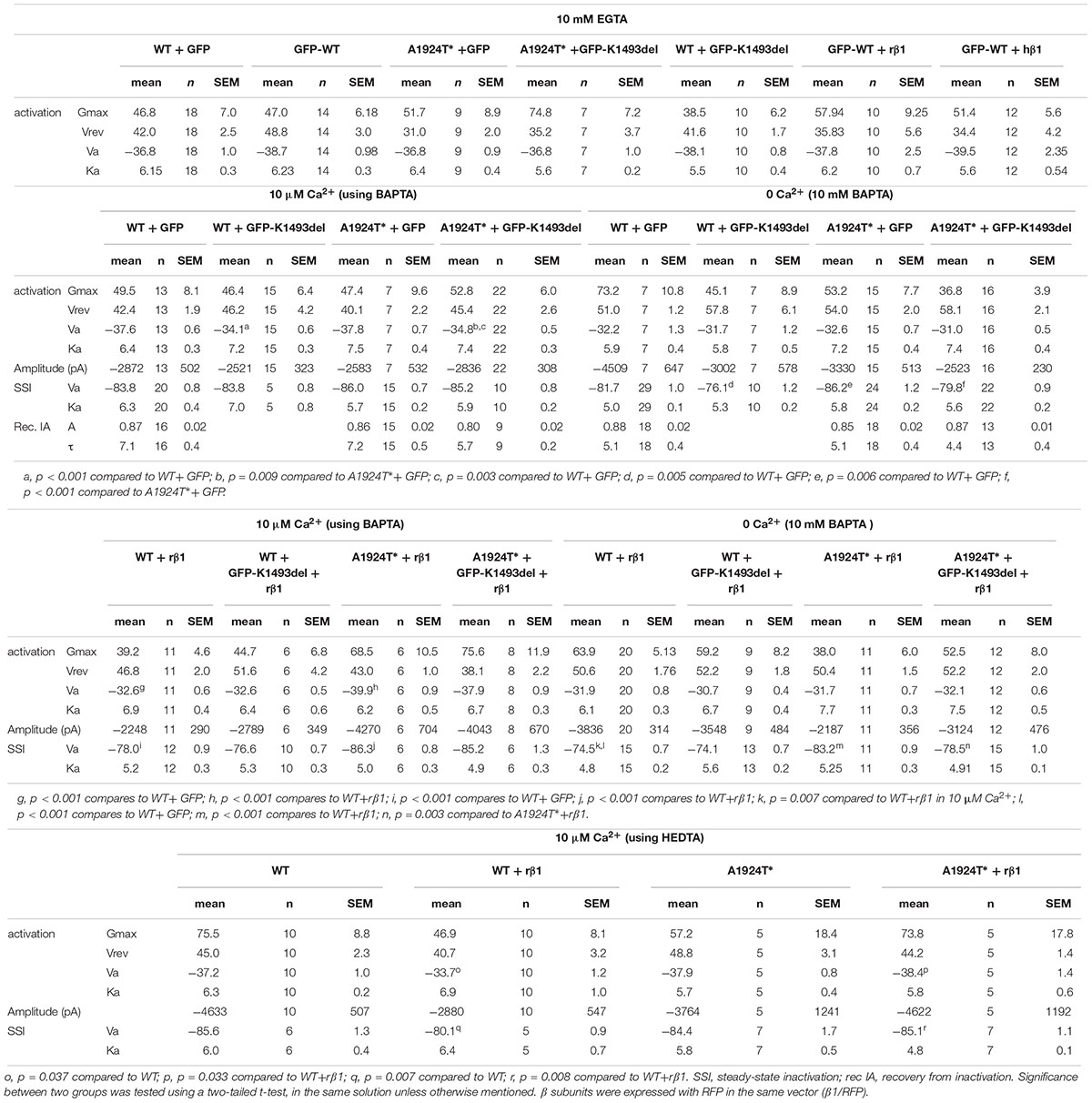
Table 2. Gating properties of NaV1.5 in different Ca2+ chelation.
As previously reported with NaV1.5A1924T (Potet et al., 2009), the SSI curve of NaV1.5A1924T^* shifts to hyperpolarized voltages compared to NaV1.5WT, in the absence, but not in the presence, of [Ca2+]in. Co-expression of GFP-NaV1.5K1493del caused a depolarizing shift of SSI of NaV1.5WT and NaV1.5A1924T^* by 5.6 and 6.4 mV, respectively, in the absence of [Ca2+]in, but did not affect SSI properties in the presence of [10 μM Ca2+]in (Figure 4B and Table 2). Recovery from inactivation induced by a 1 s depolarizing pulse was not affected by GFP-NaV1.5K1493del co-expression (Figure 4C and Table 2).
To conclude, expression of non-conductive GFP-NaV1.5K1493del altered the gating properties of co-expressed conducting channels, in a Ca2+-dependent manner: a slight decrease in the voltage dependency of activation curve in [10 μM Ca2+]in, and an increase in NaV1.5 availability following inactivation, in the absence of [Ca2+]in.
Since NaV1.5K1493del plasma membrane expression was ∼40% of NaV1.5WT, and NaVβ1 enhanced NaV1.5K1493del membrane expression (Figure 3C), we decided to examine the Ca2+-dependent gating effects of functional channels (NaV1.5WT or NaV1.5A1924T^*) co-expressed with NaVβ1/RFP and GFP-NaV1.5K1493del.
NaVβ1 expression induced a Ca2+-independent depolarizing shift in NaV1.5WT SSI (Wingo et al., 2004), nevertheless, we observed a Ca2+-dependent regulation of the activation. NaVβ1- expression induced a depolarizing shift in NaV1.5WT activation curve, in the presence of [10 μM Ca2+]in but not in the absence of [Ca2+]in (Table 2 and Figure 4D). A1924T* mutation eliminated NaVβ1-dependent effects on gating. Co-expression of NaVβ1 did not shift the activation or SSI curves of NaV1.5A1924T^*, in either the absence or presence of [Ca2+]in (Figures 4D,E and Table 2). The same effect was observed when 10 μM Ca2+ were chelated with HEDTA instead of BAPTA (Figure 4E).
Co-expression of GFP-NaV1.5K1493del had no significant effect on activation or SSI curves of NaV1.5WT +NaVβ1, in either the absence or presence of [Ca2+]in. Nevertheless, GFP-NaV1.5K1493del co-expression right-shifted the SSI curve of NaV1.5A1924T^* + NaVβ1, only in the absence of [Ca2+]in (Figure 4F and Table 2), similar to the effect recorded without NaVβ1 (Figure 4B). GFP-NaV1.5K1493del co-expression did not significantly change the activation properties of NaV1.5A1924T^* + NaVβ1 (Table 2). Thus, the mutation A1924T* blunted NaVβ1-induced gating regulation and partially restored the effects that were induced by GFP-NaV1.5K1493del co-expression.
We suggest that NaVβ1 directly affects NaV1.5 Ca2+-dependent gating in addition to its role in expression regulation. Our results demonstrate, for the first time, that NaVβ1-regulates NaV1.5 gating in a Ca2+-dependent manner, and that NaVβ1-induced regulation mechanism includes the A1924 residue. In summary, NaV1.5 Ca2+-sensitivity involves multiple elements, including both CaM-interacting elements: CT and DIII-IV, and possibly a protein complex that includes more than one NaV1.5 channel.
We wanted to examine CaM-interaction with NaV1.5 variants, since CaM directly interacts with NaV1.5 at the two domains where variants were found in the proband: A1924T located in the conserved IQ-domain in the cytoplasmic CT (Gabelli et al., 2014; Wang et al., 2014), and K1493del in the proximal segment of the cytoplasmic DIII-IV linker (Potet et al., 2009; Sarhan et al., 2012; Johnson et al., 2018; Figure 1C). We explored the interactions of CaM with NaV1.5 expressed in HEK cells by a pull-down essay using CaM-coated agarose beads.
CaM interaction in the presence of 2 mM Ca2+ (Ca2+/CaM interaction) and in the absence of Ca2+ (apo-CaM interaction in 10 mM EGTA) with NaV1.5WT and mutants in the same experiment, were normalized to Ca2+/CaM-NaV1.5WT interaction. Co-expression of NaVβ1 enabled comparable protein expression levels of NaV1.5K1493del and NaV1.5WT. A three-fold reduction in CaM-NaV1.5WT interaction in the absence of Ca2+ (from 100% with Ca2+ to 35 ± 13% without Ca2+), and a two-fold reduction in CaM-NaV1.5A1924T^* interaction in the absence of Ca2+ (from 108 ± 8% with Ca2+ to 51 ± 6% without Ca2+) were observed (Figure 5A). Ca2+/CaM interaction with NaV1.5K1493del were strongly reduced (20 ± 5% of NaV1.5WT). The Ca2+/CaM and apo-CaM interaction with NaV1.5K1493del were not significantly different, indicating an impaired Ca2+-dependent interaction (Figure 5A).
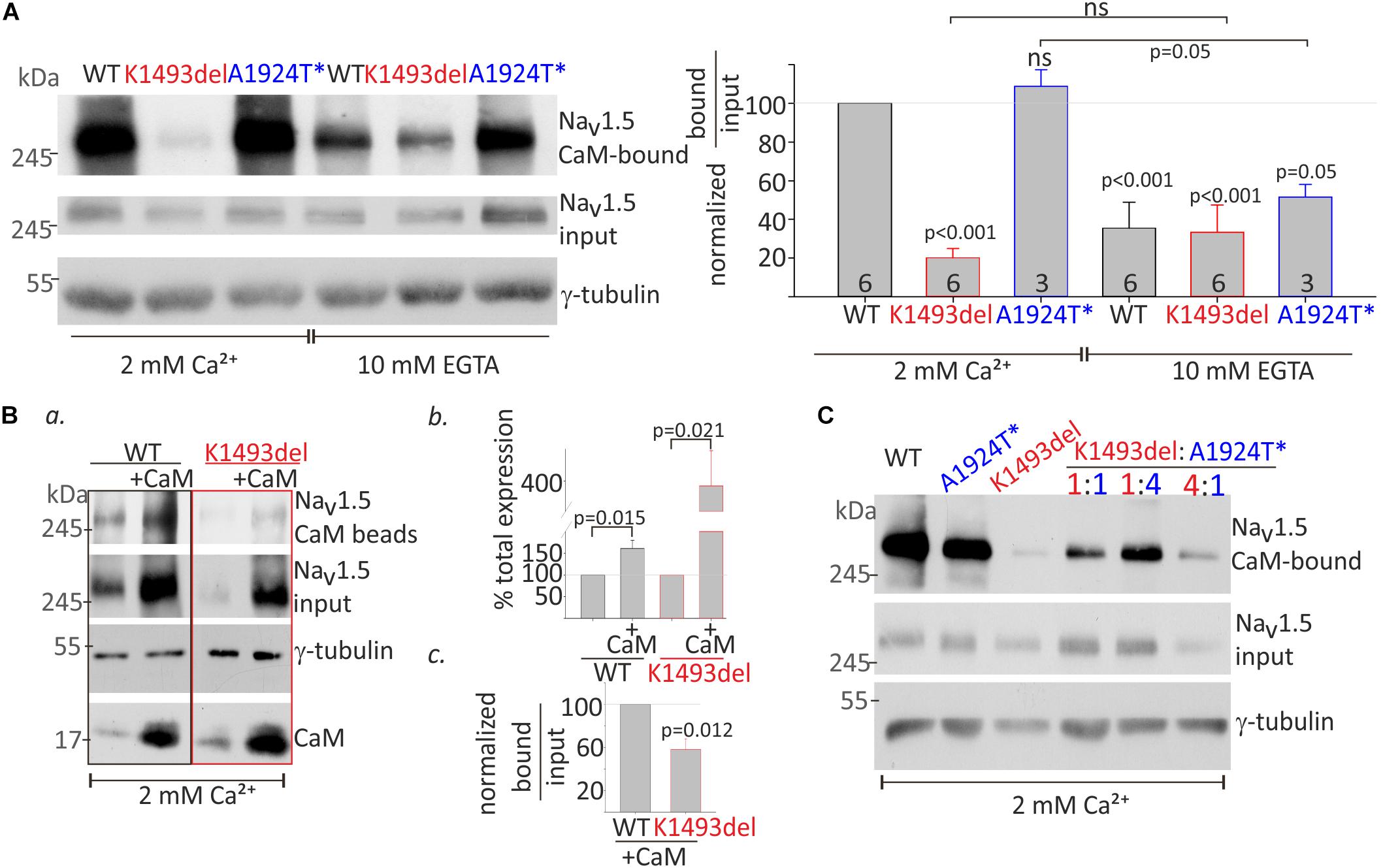
Figure 5. CaM-interaction with NaV1.5 variants (A). HEK cells were transfected with NaV1.5WT, NaV1.5K1493del, and NaV1.5A1924T^* together with rNaVβ1. Lysates from transfected groups were pulled-down with CaM-agarose beads in Ca2+ and EGTA. Left, a representative Western blot from a pull-down assay. Right, the CaM-bound fraction was normalized to input level, and presented as % of NaV1.5WT in 2 mM Ca2+, in the same experiment. Statistical significance was determined by one-way ANOVA followed by Holm–Sidak test. P-value compared to NaV1.5WT in 2 mM Ca2+ is indicated above each bar. (B) NaV1.5WT and NaV1.5K1493del with or without co-expressed CaM were pulled-down with CaM-agarose beads in 2 mM Ca2+. (a) CaM- bound NaV1.5, and total input levels of NaV1.5, CaM and γ-tubulin were detected in Western-blot. (b) Input NaV1.5+CaM levels were normalized to NaV1.5 without CaM, from the same experiment, N = 4. (c) CaM-bound fractions were normalized to input levels, and are presented as % of NaV1.5WT+CaM in the same experiment, N = 3. Statistical significance was estimated by t-test. (C) HEK cells transfected with equal total NaV1.5 α-subunit (7.5 μg; ratio 1:1, 3.75 μg of each construct; ratio 1:4, 1.5, and 6 μg of the constructs indicated) together with rNaVβ1. Lysates from transfected groups were pulled-down with CaM-agarose beads in 2 mM Ca2+. This is a representative experiment out of two.
CaM co-expression enhanced total-expression of NaV1.5WT by 161 ± 18% and NaV1.5K1493del by 387 ± 93% (Figures 5Ba,b), suggesting that despite a reduction in Ca2+/CaM interaction, CaM regulates the expression levels of NaV1.5K1493del. Upregulation of NaV1.5K1493del expression in the presence of over-expressed CaM enabled quantification of CaM-NaV1.5K1493del interaction. The ratio of NaV1.5 bound-to-input levels in 2 mM Ca2+ was lower in NaV1.5K1493del compared to NaV1.5WT (58 ± 10%, Figures 5Ba,c). Thus, CaM over-expression was not able to fully restore the reduction in Ca2+/CaM-NaV1.5K1493del interaction.
We wanted to determine whether the combination of the two mutated channels, as presented in the patient, affects the overall binding of the expressed channels to CaM. We hypothesized that a cross-talk between NaV1.5 proteins would result in cooperative CaM-interaction and that reduced Ca2+/CaM-interaction in NaV1.5K1493del would interfere with overall Ca2+/CaM-interaction. Our results did not support this hypothesis. The extent of overall channel binding to CaM beads in 2 mM Ca2+ varied proportionally to the ratio of expressed NaV1.5K1493del and NaV1.5A1924T^*: the more NaV1.5K1493del – the less overall CaM binding (Figure 5C).
The lysine doublet in position 1492-3 is positively charged and is located in a region rich with polar and charged residues that could potentially contribute to protein-protein interactions (Figure 6A). These residues are located in a conserved helical structure, downstream of the IFM motif that confers fast inactivation (West et al., 1992). We hypothesized that a salt-bridge could affect channel function and CaM interaction. To test this, we created three NaV1.5 mutants, where the lysine in position 1493 was mutated to the neutral residue alanine, NaV1.5K1493A, the negative residue glutamate, NaV1.5K1493E, or another positive residue, arginine, NaV1.5K1493R.
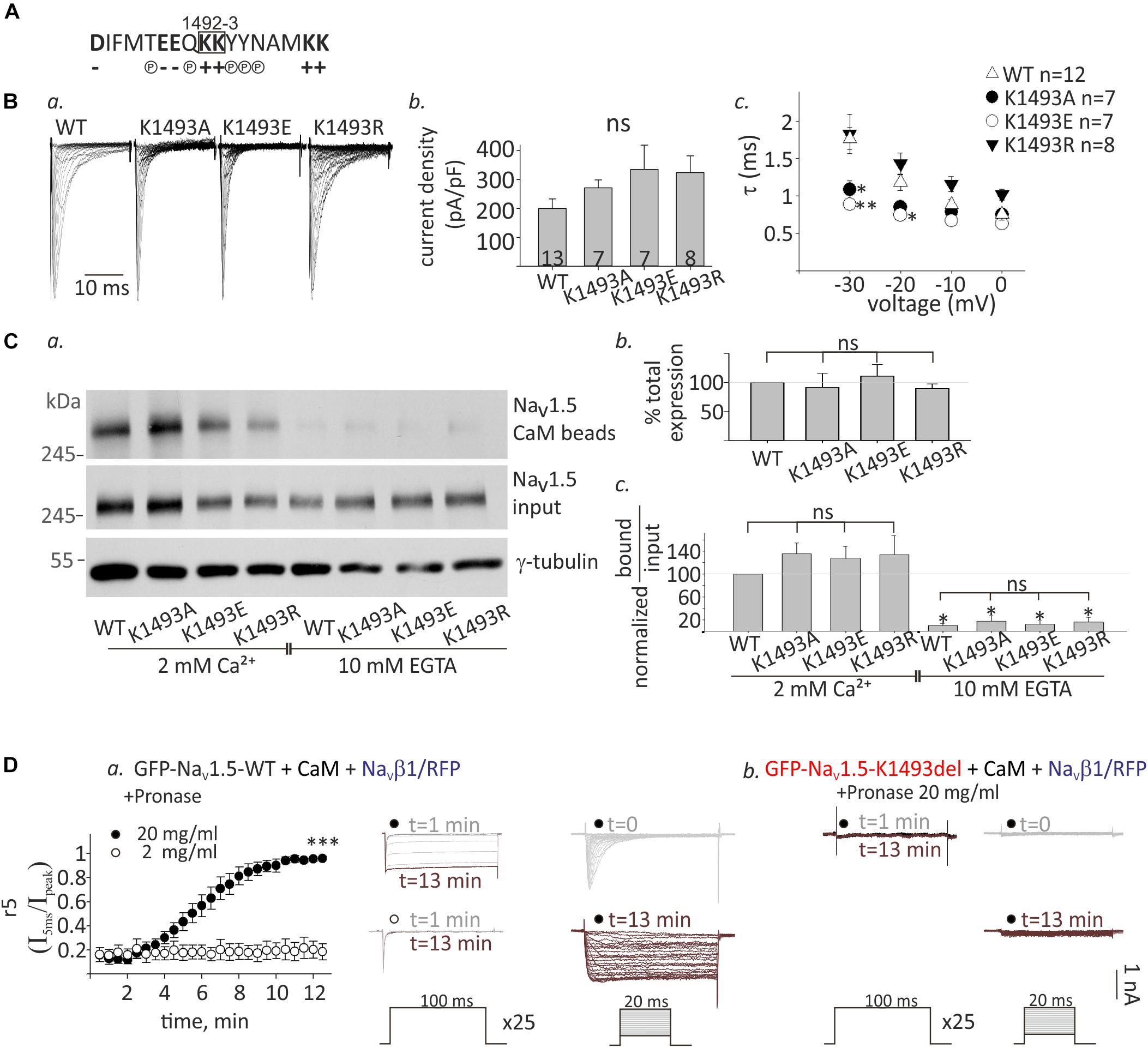
Figure 6. Functional and biochemical role of K1493 residue. (A) The amino acid sequence of residues 1484–1500 in NaV1.5. The double lysines 1492-3 are marked in a square. Amino acid’s charge (+ or–) or polarity (P) are indicated. (B) NaV1.5 variants co-expressed with rNaVβ1. (a) Representative INa traces recorded in (10 mM EGTA)in. (b) Maximal current densities. (c) Time constant (τ) obtained by fitting a single exponential to the decay phase of the currents in (10 mM EGTA)in at four voltages. Statistical significance was determined by one-way ANOVA followed by Holm–Sidak test (compared to NaV1.5WT). (C) Cells were transfected with NaV1.5K1493A/E/R together with rNaVβ1. (a) Example of a pull-down experiment with CaM-agarose beads, in Ca2+ and EGTA. (b) The total expression as quantified in EGTA, and normalized to NaV1.5WT (N = 3). (c) Pull-down experiments with CaM-agarose beads. The CaM-bound fraction was normalized to input level and quantified as % of NaV1.5WT in 2 mM Ca2+ (N = 3). Statistical significance was determined by one-way ANOVA, followed by Holm–Sidak test, compared to WT in 2 mM Ca2+. ∗∗p < 0.01; *p < 0.05. (D) GFP-NaV1.5WT (a) or GFP-NaV1.5K1493del (b) coexpressed with rNaVβ1/RFP and CaM in HEK cells. 2 or 20 mg/ml pronase were added to (10 mM EGTA)in pipette solution. t indicates the time from the beginning of the recording. (a) Currents elicited from –120 mV to –20 mV for 100 ms, every 30 s. Between pulses, the cells were held at –120 mV. Inactivation was evaluated by r5, which is the fraction of current that remained after 5 ms at –20 mV (I5ms/Ipeak), n = 5 in each group (left). Representative currents during repetitive steps to –20 mV (middle), and activation protocols in the same cell, at different time points (right). (b) Recording from cells expressing GFP-NaV1.5K1493del with 20 mg/ml pronase in the pipette solution. Currents during repetitive depolarization (left) and activation protocols (right) at different time points are presented. This is a representative cell out of 12 cells.
Analysis of NaV1.5 currents revealed that all three mutated channels were functional (Figures 6Ba,b). However, inactivation properties were altered. Using a single-exponential fit to estimate the inactivation time constant, τ, we found that when the positive charge in residue 1493 was conserved (K1493R), inactivation rate was similar to WT, as previously reported (Li et al., 2009). However, the inactivation rate was increased when K1493 was changed to an uncharged or a negatively charged residue (K1493A/E, respectively, Figure 6Bc). These results indicate that the electric charge in position 1493 is important for fast-inactivation kinetic properties, but not for the total function.
Expression analysis showed that the total protein levels of the three mutants NaV1.5K1493A/E/R were not significantly different from NaV1.5WT (Figures 6Ca,b). CaM-interaction with NaV1.5K1493A/E/R was Ca2+-dependent, similar to NaV1.5WT (Figures 6Ca,c). This set of experiments demonstrates that, unlike the robust effect of the K1493 deletion mutation, the biochemical and functional characteristics of NaV1.5 are not highly dependent on the electric charge in K1493 residue.
We further explored the mechanism of NaV1.5K1493del loss-of-function in view of the mutation location, in NaV1.5 inactivation gate. We hypothesized that NaV1.5K1493del is constitutively inactivated resulting in a channel-pore block. To address this hypothesis, we used the protease pronase that selectively destroys the inactivation of sodium channels while leaving activation intact (Armstrong et al., 1973). HEK cells were transfected with GFP-NaV1.5WT or GFP-NaV1.5K1493del together with both CaM and NaVβ1/RFP to allow maximal expression of NaV1.5 variants. Dialysis of 20 mg/ml pronase into the cell through the patch pipette gradually relieved the inactivation of NaV1.5WT INa until its complete elimination after 12 min. The effect on INa inactivation was dose-dependent and 2 mg/ml pronase did not eliminate the inactivation in 12 min (Figure 6Da). Cells expressing GFP-NaV1.5K1493del and NaVβ1/RFP in addition to CaM had no INa. Dialysis of 20 mg/ml pronase, in the same protocol used for NaV1.5WT, did not restore NaV1.5K1493del current. We conclude that the reasons for NaV1.5K1493del loss-of-function are beyond a change in the inactivation properties, and probably involve structural and additional functional perturbations.
Sinus-bradycardia and cardiac conduction-disease in the proband were associated with novel heterozygous SCN5A variants composition, K1493del in DIII-IV linker and A1924T* in the CT of NaV1.5. Deletion of K1493 caused a complete loss of NaV1.5 function. Surprisingly, the expression of the non-conducting NaV1.5K1493del affected Ca2+-dependent gating properties of co-expressed conducting channels. Moreover, Ca2+-dependent CaM-NaV1.5 interaction was impaired in NaV1.5K1493del. A Ca2+-dependent NaVβ1 modulation that was characterized in NaV1.5WT currents, was impaired in the NaV1.5A1924T^* variant. These results highlight the significance of NaV1.5 DIII-IV linker in channel function and CaM-interaction and suggest that the Ca2+-sensing machinery of NaV1.5 involves NaVβ1 and more than one monomeric NaV1.5 channel.
Sodium channelopathies usually occur due to disruption of two mechanisms: gating and/or biogenesis, a process that includes synthesis, folding, assembly of the macromolecular complex, trafficking to the plasma membrane and localization in cell surface compartments (Chen-Izu et al., 2015). Co-expression of the auxiliary proteins NaVβ1 and CaM increased NaV1.5WT and NaV1.5K1493del total expression (Figures 3B, 5Bb), possibly by serving as chaperons, suggesting a proper biogenesis regulation of NaV1.5K1493del by these proteins. The NaVβ1 and the ubiquitous CaM are expressed throughout the heart and the cardiac conduction system (O’malley and Isom, 2015). Thus, we assume that expression levels of NaV1.5K1493del in the human heart are close to the physiological levels of NaV1.5WT.
The non-conducting channel NaV1.5K1493del did not exert a dominant-negative effect on the macroscopic current density of NaV1.5WT or NaV1.5A1924T^* in HEK cells, or on endogenous INa of HL-1 atrial cells. Hence, NaV1.5K1493del does not impair biogenesis of other sodium channels, a mechanism reported in other NaV1.5 mutants (Keller et al., 2005; Clatot et al., 2012; Hoshi et al., 2014).
Deletion of K1493 resulted in a loss of INa. Changing K1493 electrostatic properties did not reduce INa but only modified inactivation kinetics (Figure 6B). Based on the location of the mutation in the inactivation gate, a constitutive inactivation state could account for NaV1.5K1493del loss-of-function. However, pronase, a protease that relieves the inactivation of sodium channels following the digestion of DIII-IV linker (Stühmer et al., 1989), did not restore NaV1.5K1493del currents (Figure 6D). Possible explanations are, first, that K1493del mutation altered NaV1.5 DIII-IV linker conformation to hinder the access of pronase to its target sequence. In an inactivated state, the IFM motif acts as a latch of a hinged-lid that docks within the pore. If K1493del does not allow a release of the latch in resting potentials, then the channel remains locked in an inactivated state, and the substrate of pronase might not be exposed to the cytoplasm. A second possible explanation is that K1493 deletion not only destabilized the inactivation gate but also lead to a major deformation that obstructed the pore’s cytosolic mouth. In this case, a twisted DIII-IV linker conformation results in a global interference in the protein structure and distal interaction with other NaV1.5 cytosolic elements. Both options imply that the lysines in position 1492-3 are pivotal residues in NaV1.5 gate and that removal of one lysine alters fundamental functional and structural elements in the channel gate.
Unlike a previous report (Zumhagen et al., 2013), we were unable to record INa in HEK cells expressing NaV1.5K1493del. In the previous and the current reports, NaV1.5K1493del was expressed in HEK cells, and the patch-clamp solutions and experimental conditions were essentially similar. We tested several mechanisms that may have contributed to the discrepancy: (1) Impaired cellular expression: total and surface expression of NaV1.5K1493del was confirmed by Western blot and biotinylation assay (Figure 3). (2) Low transfection of NaV1.5 α-subunit: currents were measured in cells expressing GFP-labeled α-subunit (NaV1.5WT and NaV1.5K1493del), and the amount of DNA used for transfection of NaV1.5K1493del was three-fold higher. (3) Functional variability between NaVβ1 species: NaVβ1 subunit from two species were used (rat and human; the human NaVβ1 was previously used). (4) Low expression of NaVβ1 subunit: NaVβ1 was co-expressed with a fluorescent marker in a bicistronic vector. (5) Finally, the coding sequences of the constructs were fully sequenced, and two separately constructed α-subunit mutants (NaV1.5K1493del and GFP-NaV1.5K1493del) were tested. In summary, our results show that NaV1.5K1493del is a loss-of-function mutation due to a gating rather than a biogenesis defect, and the reason for the discrepancy with the previous report could not be determined.
The Ca2+-sensing machinery of NaV1.5 may include the DIII-IV linker, CT and the Ca2+ sensor CaM. Interaction of CaM with DIII-IV linker was studied mainly using small peptides (Kim et al., 2004; Potet et al., 2009; Sarhan et al., 2012; Yan H. et al., 2017; Johnson et al., 2018), but the relevance of DIII-IV linker to the overall CaM-binding complex in the full NaV1.5 protein is unclear. The binding affinities of NaV1.5-CT peptide to CaM were not Ca2+-sensitive (Wang et al., 2014), while Ca2+-dependent enhancement in CaM binding was measured with DIII-IV linker segments (Sarhan et al., 2009; Sarhan et al., 2012; Johnson et al., 2018). We detected a novel property of CaM interaction with full NaV1.5 expressed in cells: Ca2+/CaM was stronger than apo-CaM interaction, in NaV1.5WT and the variants NaV1.5A1924T^* (Figure 5A) and NaV1.5K1493A/E/R (Figure 6C) when using pull-down assay. K1493del blunted the Ca2+-dependent CaM-interaction: Ca2+/CaM-NaV1.5K1493del interaction was similar to apo-CaM-interaction with NaV1.5WT or NaV1.5K1493del. These results highlight the role of DIII-IV linker in CaM binding complex, suggesting that the Ca2+-dependent CaM-interaction with NaV1.5 DIII-IV linker contributes, directly or indirectly, to the overall enhancement in Ca2+/CaM-NaV1.5 interaction.
A1924T mutation reduced Ca2+/CaM-interaction in a CT peptide (Wang et al., 2014). However, the length of NaV1.5 CaM-interacting segments was shown to be crucial for determining binding affinities to CaM [(Wang et al., 2014; Johnson et al., 2018) vs. (Sarhan et al., 2012)]. When we pulled-down the full-length expressed NaV1.5, the interaction between Ca2+/CaM and NaV1.5A1924T^* was not weakened compared to NaV1.5WT, unlike the reports in CT segments. However, in the absence of Ca2+, NaV1.5A1924T^*-CaM interaction was slightly higher compared to NaV1.5WT, so the “net” Ca2+-dependent change in CaM interaction was diminished. Our results point that the binding of CaM to a native NaV1.5 channel is probably determined by multiple segments, e.g., both the CT and the DIII-IV linker. CaM affinities to separate segments may not reflect the full dynamic interaction. Understanding the mode of integration of all NaV1.5-CaM binding elements within the complete channel protein is indispensable for resolving the mechanism of CaM-regulation in a physiological context. Pioneering cryogenic electron microscopy (cryo-EM) studies were able to capture the cytosolic complex of sodium channels (Shen et al., 2017; Yan Z. et al., 2017), and together with functional and molecular information the understanding of the complex and dynamic Ca2+-dependent regulation will be refined.
Several studies demonstrated that NaV1.5 is regulated by Ca2+ (Deschenes et al., 2002; Tan et al., 2002; Kim et al., 2004; Wingo et al., 2004; Biswas et al., 2009; Glynn et al., 2015; Gabelli et al., 2016; Abdelsayed et al., 2017), but the molecular details and the physiological relevance remains highly controversial [e.g. (Ben-Johny et al., 2014)]. We showed that NaV1.5K1493del is located in the plasma membrane as a non-conducting channel. If individual NaV1.5 monomers gate independently of others, the “silent” mutant NaV1.5K1493del would not have contributed to overall macroscopic current properties when expressed with other channels in the same cell. Strikingly, expression of the non-conducting mutant channel affected the Ca2+-dependent gating of macroscopic current arising from co-expressed conducting channels. Co-expression of NaV1.5K1493del similarly altered the Ca2+-dependent gating properties of both NaV1.5WT and NaV1.5A1924T^*: a depolarization shift in the voltage-dependent activation with [Ca2+]in and SSI-curve in the absence of [Ca2+]in.
Previous studies provided evidence that NaV1.5 proteins are in physical proximity when expressed in cells (Clatot et al., 2012; Mercier et al., 2012; Clatot et al., 2018). This interaction is indirect, via 14-3-3 protein (Clatot et al., 2017). Functional cooperation of the gate has been demonstrated when loss-of-function NaV1.5 mutant impaired NaV1.5WT gating by a dominant-negative mechanism (Clatot et al., 2018) while several other mutations showed a dominant-negative effect via defective biogenesis that suggests co-trafficking of several channels [reviewed in (Sottas and Abriel, 2016)]. Here, the expression of NaV1.5K1493del did not cause a dominant-negative loss-of-function effect (Figures 2B,C). We propose that the loss of Ca2+/CaM-interaction due to K1493del mutation prompted changes in the Ca2+-dependent gating of co-expressed conducting channels. Thus, our results support a cooperative gating regulation of multimeric-NaV1.5 complex and suggest that this mechanism involves a Ca2+/CaM-regulated component. To note, CaM plays a critical role in the functional coupling of the structurally homologous calcium channel, CaV1.2 (Dixon et al., 2015). In summary, the mutation K1493del underlines the role of DIII-IV linker in Ca2+-dependent regulation of coupled NaV1.5 channels. The involvement of CaM and the details of Ca2+-dependent regulation of NaV1.5’s cooperative gating mechanism are yet to be determined.
The interaction between monomeric NaV1.5 during channel biogenesis is mediated by NaVβ1 (Mercier et al., 2012), but the involvement of NaVβ1 in the gating of coupled channels or in Ca2+-dependent mechanisms have not been elucidated. The reported effects of NaVβ1 on NaV1.5 gating parameters in mammalian expression systems are conflicting, generally suggesting that the cellular environment is critical for channel function (Calhoun and Isom, 2014). Similar to previous reports (An et al., 1998; Wingo et al., 2004; Zhu et al., 2017) we showed that NaVβ1 co-expression right-shifted NaV1.5WT SSI curve (Figure 4D, bottom). Further, we showed that NaVβ1-induced a right-shift in NaV1.5WT activation curve in the presence, but not in the absence, of Ca2+ (Figure 4D, top). This provides an indication for the role of Ca2+ in NaVβ1 regulation.
GFP-NaV1.5K1493del altered NaV1.5WT gating properties only when NaVβ1 was not expressed, whereas in the presence of NaVβ1, GFP-NaV1.5K1493del expression did not affect gating. NaVβ1 was shown to modulate the voltage-sensor of NaV1.5 domain IV (DIV, Figure 1C), and is believed to localize in close proximity to DIII-IV linker (Yan Z. et al., 2017; Zhu et al., 2017). Thus, we propose that NaVβ1 is intricately involved in the Ca2+-dependent gating regulation, and can modify the contribution of DIII-IV linker to NaV1.5 channels’ cooperative-gating modulation.
Both effects of NaVβ1 on NaV1.5WT were eliminated in NaV1.5A1924T^*: the gating modulations and the loss of NaV1.5K1493del effect, indicating that A1924 residue may mediate NaVβ1 regulation. Interestingly, structural dimers were found in the CT of NaV1.5. In the dimer, position 1924 is located in the interface of NaV1.5 CT dimer, and A1924T mutation was shown to attenuate these interactions (Gabelli et al., 2014). In view of the structural and functional data, we suggest that cytosolic interaction between NaVβ1 and the IQ domain(s) in NaV1.5 cytosolic CT, including A1924 residue, are involved in the regulation of NaV1.5-gate. Since both DIII-IV linker and A1924T are thought to be included in the CaM-NaV1.5 interaction complex, we postulate that NaVβ1 is part of a cytosolic CaM-interaction complex and a dynamic modulator of a complexed Ca2+-regulated gate that comprises DIII-IV linker and the CT of multiple NaV1.5 channels.
Compound heterozygosity in SCN5A was associated with increased arrhythmic expression compared to heterozygotes in the same family. The clinical phenotype included severe bradycardia and conduction disease, which represents a reduction in electric activity of both the sinus-atria and the conduction system. Among the two mutations that were detected in the proband, the most striking biophysical feature is the loss-of-function due to K1493del mutation, which would lead to a 50% reduction in INa and haploinsufficiency. The changes in gating that arise from the second NaV1.5 variant, A1924T*, are expected to have a relatively mild impact on cardiac rhythm but might have added to the pathological expression on the background of NaV1.5K1493del.
The physiological implication of Ca2+ modulation of NaV1.5 is heart-rate dependent. During low-to-normal heart rate, INa transient of the cardiac action-potential precedes the Ca2+ transient and senses low Ca2+ levels. Repetitive and fast Ca2+ transients, during tachycardia, are expected to reveal Ca2+-dependent conformational changes in protein complex formation that are limited by the association/dissociation rate, like NaV1.5-CaM direct interaction, and other distal Ca2+-induced pathways.
NaV1.5 channels are physiologically modulated by NaVβ1 in cardiac cells (Edokobi and Isom, 2018). Interestingly, A1924T* mutant blunted NaVβ1- induced gating modulation. Close to resting potential and in the absence of Ca2+, the availability of NaV1.5A1924T^* + NaVβ1 channels to open is two times lower than that of NaV1.5WT + NaVβ1, which is expected to reduce the action-potential upstroke during low-to-normal heart rate (Figure 4D), and may explain the BrS phenotype reported in A1924T carriers (Rook et al., 1999). The addition of NaV1.5K1493del on top of NaV1.5A1924T^* + NaVβ1 increased the number of NaV1.5 channels available to open at rest (Figure 4F, right). This restoration of SSI properties may have moderated the development of BrS ECG pattern in the compound-heterozygote in our study, although a low penetration of A1924T phenotype cannot be ruled out.
In high Ca2+, the window current of NaV1.5A1924T^* + NaVβ1 is shifted to hyperpolarized voltages comparing to NaV1.5WT, resulting in increased NaV1.5 excitability. We speculate that this property can facilitate conduction and increase the propensity for development of exercise-induced atrial flutter with rapid ventricular response, on the background of bradycardia and conduction disease, that was demonstrated in the compound heterozygote in this study (Figure 1A, right).
It is noteworthy that certain loss-of-function mutations are expressed in BrS phenotype, e.g., A1924T, while others are associated with slow cardiac conduction and bradycardia, without BrS ECG pattern, e.g., K1493del [see also (Hu et al., 2010; Park et al., 2015)]. Possibly, the impaired mechanism that results in a loss-of-function plays a critical role in the downstream expression of the syndrome.
Extrapolation of the biophysical properties of mutated channels to disease expression, in the presented proband, is limited since we cannot determine the relative expression of each allele in the compound heterozygote cardiac cells, and thus we cannot determine the relative functional contribution of each mutated channel. In addition, the role of the polymorphism V1251M was not studied. The use of induced pluripotent stem-cells derived cardiomyocytes (iPSC-CMs), originated from the proband’s cells, would be an attractive approach to investigate how the composite of mutations with distinct biophysical properties results in the cardiac phenotype, in a complex molecular environment, and in dynamic (Ca2+) cycles.
We show that K1493del mutation induces a complete loss-of-function of NaV1.5 due to gating, rather than biogenesis, defect. The effect of K1493 deletion is independent of the electric charge in this position. K1493del is associated with impaired CaM-interaction and Ca2+-dependent modulation of sodium currents. NaVβ1 gating regulation is Ca2+-dependent and involves the NaV1.5 CT. These results highlight the role of NaV1.5 DIII-IV linker in NaV1.5-CaM-interaction and support a coupling between NaV1.5 channels that is implicated in a Ca2+-dependent gating mechanism.
The proband and his parents gave written informed consents for both the clinical and genetic studies, which were approved by the Institutional Ethics-Committee of the Sheba Medical Center, Tel-Hashomer (approval 2853/03).
SO and EN designed the research. SO planned the experiments, performed patch-clamp experiments, interpreted the data, and wrote the manuscript. EN, RB, DL, and MG clinically evaluated the patient. MG acquired the financial support for the project. LV performed the biochemical experiments. EM prepared the DNA constructs, interpreted the data, and edited the manuscript. CA, JC, and EB performed and analyzed the genetic screen and edited the manuscript.
This work was supported by the Rothschild Caesarea Foundation and Dizengoff Trading Company (1952).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Dr. Ronit S. Cherki for performing preliminary experiments. They also thank Ms. Elaine Finkelstein, Dr. Moshe Giladi, Dr. Moran Rubinstein, Dr. Sharon Weiss, Dr. Dahlia Palevski, and especially Prof. Nathan Dascal and Dr. Samuel Frere for reading the manuscript and for their helpful comments. The authors are grateful to Prof. T. Zimmer (Jena, Germany) for providing GFP-NaV1.5 construct, Prof. L. Isom (Ann Arbor, MI, United States) for providing hβ1 construct, Prof. W.A. Catterall (Seattle, United States) for providing rβ1 construct and Prof. J. Adelman (Portland, United States) for providing CaM construct.
Abdelsayed, M., Baruteau, A. E., Gibbs, K., Sanatani, S., Krahn, A. D., Probst, V., et al. (2017). Differential calcium sensitivity in NaV 1.5 mixed syndrome mutants. J. Physiol. 595, 6165–6186. doi: 10.1113/JP274536
Abriel, H. (2010). Cardiac sodium channel Na(v)1.5 and interacting proteins: physiology and pathophysiology. J. Mol. Cell. Cardiol. 48, 2–11. doi: 10.1016/j.yjmcc.2009.08.025
An, R. H., Wang, X. L., Kerem, B., Benhorin, J., Medina, A., Goldmit, M., et al. (1998). Novel LQT-3 mutation affects Na+ channel activity through interactions between alpha- and beta1-subunits. Circ. Res. 83, 141–146. doi: 10.1161/01.res.83.2.141
Armstrong, C. M., Bezanilla, F., and Rojas, E. (1973). Destruction of sodium conductance inactivation in squid axons perfused with pronase. J. Gen. Physiol. 62, 375–391. doi: 10.1085/jgp.62.4.375
Bechi, G., Rusconi, R., Cestele, S., Striano, P., Franceschetti, S., and Mantegazza, M. (2015). Rescuable folding defective NaV1.1 (SCN1A) mutants in epilepsy: properties, occurrence, and novel rescuing strategy with peptides targeted to the endoplasmic reticulum. Neurobiol. Dis. 75, 100–114. doi: 10.1016/j.nbd.2014.12.028
Ben-Johny, M., Yang, P. S., Niu, J., Yang, W., Joshi-Mukherjee, R., and Yue, D. T. (2014). Conservation of Ca2+/calmodulin regulation across Na and Ca2+ channels. Cell 157, 1657–1670. doi: 10.1016/j.cell.2014.04.035
Benson, D. W., Wang, D. W., Dyment, M., Knilans, T. K., Fish, F. A., Strieper, M. J., et al. (2003). Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J. Clin. Invest. 112, 1019–1028. doi: 10.1172/jci18062
Biswas, S., Disilvestre, D., Tian, Y., Halperin, V. L., and Tomaselli, G. F. (2009). Calcium-mediated dual-mode regulation of cardiac sodium channel gating. Circ. Res. 104, 870–878. doi: 10.1161/CIRCRESAHA.108.193565
Calhoun, J. D., and Isom, L. L. (2014). The role of non-pore-forming beta subunits in physiology and pathophysiology of voltage-gated sodium channels. Handb. Exp. Pharmacol. 221, 51–89. doi: 10.1007/978-3-642-41588-3_4
Cerrone, M., Lin, X., Zhang, M., Agullo-Pascual, E., Pfenniger, A., Chkourko Gusky, H., et al. (2014). Missense mutations in plakophilin-2 cause sodium current deficit and associate with a brugada syndrome phenotype. Circulation 129, 1092–1103. doi: 10.1161/CIRCULATIONAHA.113.003077
Chen-Izu, Y., Shaw, R. M., Pitt, G. S., Yarov-Yarovoy, V., Sack, J. T., Abriel, H., et al. (2015). Na+ channel function, regulation, structure, trafficking and sequestration. J. Physiol. 593, 1347–1360. doi: 10.1113/jphysiol.2014.281428
Chiang, D. Y., Kim, J. J., Valdes, S. O., De La Uz, C., Fan, Y., Orcutt, J., et al. (2015). Loss-of-function SCN5A mutations associated with sinus node dysfunction, atrial arrhythmias, and poor pacemaker capture. Circ. Arrhythm. Electrophysiol. 8, 1105–1112. doi: 10.1161/CIRCEP.115.003098
Clatot, J., Hoshi, M., Wan, X., Liu, H., Jain, A., Shinlapawittayatorn, K., et al. (2017). Voltage-gated sodium channels assemble and gate as dimers. Nat. Commun. 8:2077. doi: 10.1038/s41467-017-02262-0
Clatot, J., Zheng, Y., Girardeau, A., Liu, H., Laurita, K. R., Marionneau, C., et al. (2018). Mutant voltage-gated sodium channels can exert a dominant-negative effect through coupled gating. Am. J. Physiol. Heart Circ. Physiol. 315, H1250–H1257.
Clatot, J., Ziyadeh-Isleem, A., Maugenre, S., Denjoy, I., Liu, H., Dilanian, G., et al. (2012). Dominant-negative effect of SCN5A N-terminal mutations through the interaction of Na(v)1.5 alpha-subunits. Cardiovasc. Res. 96, 53–63. doi: 10.1093/cvr/cvs211
Claycomb, W. C., Lanson, N. A. Jr., Stallworth, B. S., Egeland, D. B., Delcarpio, J. B., Bahinski, A., et al. (1998). HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. U.S.A. 95, 2979–2984. doi: 10.1073/pnas.95.6.2979
Deschenes, I., Neyroud, N., Disilvestre, D., Marban, E., Yue, D. T., and Tomaselli, G. F. (2002). Isoform-specific modulation of voltage-gated Na(+) channels by calmodulin. Circ. Res. 90, E49–E57.
Dixon, R. E., Moreno, C. M., Yuan, C., Opitz-Araya, X., Binder, M. D., Navedo, M. F., et al. (2015). Graded Ca(2)(+)/calmodulin-dependent coupling of voltage-gated CaV1.2 channels. eLife 4:e05608. doi: 10.7554/eLife.05608
Edokobi, N., and Isom, L. L. (2018). Voltage-gated sodium channel beta1/beta1b subunits regulate cardiac physiology and pathophysiology. Front. Physiol. 9:351. doi: 10.3389/fphys.2018.00351
Gabelli, S. B., Boto, A., Kuhns, V. H., Bianchet, M. A., Farinelli, F., Aripirala, S., et al. (2014). Regulation of the NaV1.5 cytoplasmic domain by calmodulin. Nat. Commun. 5:5126. doi: 10.1038/ncomms6126
Gabelli, S. B., Yoder, J. B., Tomaselli, G. F., and Amzel, L. M. (2016). Calmodulin and Ca(2+) control of voltage gated Na(+) channels. Channels 10, 45–54. doi: 10.1080/19336950.2015.1075677
Glynn, P., Musa, H., Wu, X., Unudurthi, S. D., Little, S., Qian, L., et al. (2015). Voltage-gated sodium channel phosphorylation at Ser571 regulates late current, arrhythmia, and cardiac function in vivo. Circulation 132, 567–577. doi: 10.1161/CIRCULATIONAHA.114.015218
Gui, J., Wang, T., Jones, R. P., Trump, D., Zimmer, T., and Lei, M. (2010). Multiple loss-of-function mechanisms contribute to SCN5A-related familial sick sinus syndrome. PLoS One 5:e10985. doi: 10.1371/journal.pone.0010985
Holst, A. G., Liang, B., Jespersen, T., Bundgaard, H., Haunso, S., Svendsen, J. H., et al. (2010). Sick sinus syndrome, progressive cardiac conduction disease, atrial flutter and ventricular tachycardia caused by a novel SCN5A mutation. Cardiology 115, 311–316. doi: 10.1159/000312747
Hoshi, M., Du, X. X., Shinlapawittayatorn, K., Liu, H., Chai, S., Wan, X., et al. (2014). Brugada syndrome disease phenotype explained in apparently benign sodium channel mutations. Circ. Cardiovasc. Genet. 7, 123–131. doi: 10.1161/CIRCGENETICS.113.000292
Hu, D., Barajas-Martinez, H., Nesterenko, V. V., Pfeiffer, R., Guerchicoff, A., Cordeiro, J. M., et al. (2010). Dual variation in SCN5A and CACNB2b underlies the development of cardiac conduction disease without brugada syndrome. Pacing Clin. Electrophysiol. 33, 274–285. doi: 10.1111/j.1540-8159.2009.02642.x
Johnson, C. N. (2019). Calcium modulation of cardiac sodium channels. J. Physiol. [Epub ahead of print]Google Scholar
Johnson, C. N., Potet, F., Thompson, M. K., Kroncke, B. M., Glazer, A. M., Voehler, M. W., et al. (2018). A mechanism of calmodulin modulation of the human cardiac sodium channel. Structure 26:e683. doi: 10.1016/j.str.2018.03.005
Kapplinger, J. D., Giudicessi, J. R., Ye, D., Tester, D. J., Callis, T. E., Valdivia, C. R., et al. (2015). Enhanced classification of brugada syndrome-associated and long-QT syndrome-associated genetic variants in the SCN5A-encoded Na(v)1.5 cardiac sodium channel. Circ. Cardiovasc. Genet. 8, 582–595. doi: 10.1161/CIRCGENETICS.114.000831
Keller, D. I., Rougier, J. S., Kucera, J. P., Benammar, N., Fressart, V., Guicheney, P., et al. (2005). Brugada syndrome and fever: genetic and molecular characterization of patients carrying SCN5A mutations. Cardiovasc. Res. 67, 510–519. doi: 10.1016/j.cardiores.2005.03.024
Kim, J., Ghosh, S., Liu, H., Tateyama, M., Kass, R. S., and Pitt, G. S. (2004). Calmodulin mediates Ca2+ sensitivity of sodium channels. J. Biol. Chem. 279, 45004–45012. doi: 10.1074/jbc.m407286200
Kodama, I., Nikmaram, M. R., Boyett, M. R., Suzuki, R., Honjo, H., and Owen, J. M. (1997). Regional differences in the role of the Ca2+ and Na+ currents in pacemaker activity in the sinoatrial node. Am. J. Physiol. 272, H2793–H2806.
Lei, M., Huang, C. L., and Zhang, Y. (2008). Genetic Na+ channelopathies and sinus node dysfunction. Prog. Biophys. Mol. Biol. 98, 171–178. doi: 10.1016/j.pbiomolbio.2008.10.003
Li, Q., Huang, H., Liu, G., Lam, K., Rutberg, J., Green, M. S., et al. (2009). Gain-of-function mutation of Nav1.5 in atrial fibrillation enhances cellular excitability and lowers the threshold for action potential firing. Biochem. Biophys. Res. Commun. 380, 132–137. doi: 10.1016/j.bbrc.2009.01.052
Liu, M., Yang, K. C., and Dudley, S. C. Jr. (2016). Cardiac sodium channel mutations: why so many phenotypes? Curr. Top. Membr. 78, 513–559. doi: 10.1016/bs.ctm.2015.12.004
Lopez, K. N., Decker, J. A., Friedman, R. A., and Kim, J. J. (2011). Homozygous mutation in SCN5A associated with atrial quiescence, recalcitrant arrhythmias, and poor capture thresholds. Heart Rhythm. 8, 471–473. doi: 10.1016/j.hrthm.2010.10.014
Mercier, A., Clement, R., Harnois, T., Bourmeyster, N., Faivre, J. F., Findlay, I., et al. (2012). The beta1-subunit of Na(v)1.5 cardiac sodium channel is required for a dominant negative effect through alpha-alpha interaction. PLoS One 7:e48690. doi: 10.1371/journal.pone.0048690
Milanesi, R., Bucchi, A., and Baruscotti, M. (2015). The genetic basis for inherited forms of sinoatrial dysfunction and atrioventricular node dysfunction. J. Interv. Card Electrophysiol. 43, 121–134. doi: 10.1007/s10840-015-9998-z
O’malley, H. A., and Isom, L. L. (2015). Sodium channel beta subunits: emerging targets in channelopathies. Annu. Rev. Physiol. 77, 481–504. doi: 10.1146/annurev-physiol-021014-071846
Park, D. S., Cerrone, M., Morley, G., Vasquez, C., Fowler, S., Liu, N., et al. (2015). Genetically engineered SCN5A mutant pig hearts exhibit conduction defects and arrhythmias. J. Clin. Invest. 125, 403–412. doi: 10.1172/JCI76919
Pitt, G. S., and Lee, S. Y. (2016). Current view on regulation of voltage-gated sodium channels by calcium and auxiliary proteins. Protein Sci. 25, 1573–1584. doi: 10.1002/pro.2960
Poelzing, S., Forleo, C., Samodell, M., Dudash, L., Sorrentino, S., Anaclerio, M., et al. (2006). SCN5A polymorphism restores trafficking of a brugada syndrome mutation on a separate gene. Circulation 114, 368–376. doi: 10.1161/circulationaha.105.601294
Potet, F., Chagot, B., Anghelescu, M., Viswanathan, P. C., Stepanovic, S. Z., Kupershmidt, S., et al. (2009). Functional interactions between distinct sodium channel cytoplasmic domains through the action of calmodulin. J. Biol. Chem. 284, 8846–8854. doi: 10.1074/jbc.M806871200
Reinhard, K., Rougier, J. S., Ogrodnik, J., and Abriel, H. (2013). Electrophysiological properties of mouse and epitope-tagged human cardiac sodium channel Na v1.5 expressed in HEK293 cells. F1000 Res. 2:48. doi: 10.12688/f1000research.2-48.v2
Rook, M. B., Bezzina Alshinawi, C., Groenewegen, W. A., Van Gelder, I. C., Van Ginneken, A. C., Jongsma, H. J., et al. (1999). Human SCN5A gene mutations alter cardiac sodium channel kinetics and are associated with the brugada syndrome. Cardiovasc. Res. 44, 507–517. doi: 10.1016/s0008-6363(99)00350-8
Sarhan, M. F., Tung, C. C., Van Petegem, F., and Ahern, C. A. (2012). Crystallographic basis for calcium regulation of sodium channels. Proc. Natl. Acad. Sci. U.S.A. 109, 3558–3563. doi: 10.1073/pnas.1114748109
Sarhan, M. F., Van Petegem, F., and Ahern, C. A. (2009). A double tyrosine motif in the cardiac sodium channel domain III-IV linker couples calcium-dependent calmodulin binding to inactivation gating. J. Biol. Chem. 284, 33265–33274. doi: 10.1074/jbc.M109.052910
Schott, J. J., Alshinawi, C., Kyndt, F., Probst, V., Hoorntje, T. M., Hulsbeek, M., et al. (1999). Cardiac conduction defects associate with mutations in SCN5A. Nat. Genet. 23, 20–21. doi: 10.1038/12618
Shen, H., Zhou, Q., Pan, X., Li, Z., Wu, J., and Yan, N. (2017). Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 355:eaal4326. doi: 10.1126/science.aal4326
Sottas, V., and Abriel, H. (2016). Negative-dominance phenomenon with genetic variants of the cardiac sodium channel Nav1.5. Biochim. Biophys. Acta 1863, 1791–1798. doi: 10.1016/j.bbamcr.2016.02.013
Stühmer, W., Conti, F., Suzuki, H., Wang, X., Noda, M., Yahagi, N., et al. (1989). Structural parts involved in activation and inactivation of the sodium channel. Nature 339, 597–603. doi: 10.1038/339597a0
Tan, H. L., Bink-Boelkens, M. T., Bezzina, C. R., Viswanathan, P. C., Beaufort-Krol, G. C., Van Tintelen, P. J., et al. (2001). A sodium-channel mutation causes isolated cardiac conduction disease. Nature 409, 1043–1047. doi: 10.1038/35059090
Tan, H. L., Kupershmidt, S., Zhang, R., Stepanovic, S., Roden, D. M., Wilde, A. A., et al. (2002). A calcium sensor in the sodium channel modulates cardiac excitability. Nature 415, 442–447. doi: 10.1038/415442a
Van Petegem, F., Lobo, P. A., and Ahern, C. A. (2012). Seeing the forest through the trees: towards a unified view on physiological calcium regulation of voltage-gated sodium channels. Biophys. J. 103, 2243–2251. doi: 10.1016/j.bpj.2012.10.020
Verkerk, A. O., Amin, A. S., and Remme, C. A. (2018). Disease modifiers of inherited SCN5A channelopathy. Front. Cardiovasc. Med. 5:137. doi: 10.3389/fcvm.2018.00137
Wang, C., Chung, B. C., Yan, H., Wang, H. G., Lee, S. Y., and Pitt, G. S. (2014). Structural analyses of Ca(2)(+)/CaM interaction with NaV channel C-termini reveal mechanisms of calcium-dependent regulation. Nat. Commun. 5:4896. doi: 10.1038/ncomms5896
West, J. W., Patton, D. E., Scheuer, T., Wang, Y., Goldin, A. L., and Catterall, W. A. (1992). A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc. Natl. Acad. Sci. U.S.A. 89, 10910–10914. doi: 10.1073/pnas.89.22.10910
Wingo, T. L., Shah, V. N., Anderson, M. E., Lybrand, T. P., Chazin, W. J., and Balser, J. R. (2004). An EF-hand in the sodium channel couples intracellular calcium to cardiac excitability. Nat. Struct. Mol. Biol. 11, 219–225. doi: 10.1038/nsmb737
Yan, H., Wang, C., Marx, S. O., and Pitt, G. S. (2017). Calmodulin limits pathogenic Na+ channel persistent current. J. Gen. Physiol. 149, 277–293. doi: 10.1085/jgp.201611721
Yan, Z., Zhou, Q., Wang, L., Wu, J., Zhao, Y., Huang, G., et al. (2017). Structure of the Nav1.4-beta1 complex from electric Eel. Cell 170:e411. doi: 10.1016/j.cell.2017.06.039
Zhu, W., Voelker, T. L., Varga, Z., Schubert, A. R., Nerbonne, J. M., and Silva, J. R. (2017). Mechanisms of noncovalent beta subunit regulation of NaV channel gating. J. Gen. Physiol. [Epub ahead of print]
Zimmer, T., Biskup, C., Dugarmaa, S., Vogel, F., Steinbis, M., Bohle, T., et al. (2002). Functional expression of GFP-linked human heart sodium channel (hH1) and subcellular localization of the a subunit in HEK293 cells and dog cardiac myocytes. J. Membr. Biol. 186, 1–12. doi: 10.1007/s00232-001-0130-1
Ziyadeh-Isleem, A., Clatot, J., Duchatelet, S., Gandjbakhch, E., Denjoy, I., Hidden-Lucet, F., et al. (2014). A truncating SCN5A mutation combined with genetic variability causes sick sinus syndrome and early atrial fibrillation. Heart Rhythm 11, 1015–1023. doi: 10.1016/j.hrthm.2014.02.021
Zumhagen, S., Veldkamp, M. W., Stallmeyer, B., Baartscheer, A., Eckardt, L., Paul, M., et al. (2013). A heterozygous deletion mutation in the cardiac sodium channel gene SCN5A with loss- and gain-of-function characteristics manifests as isolated conduction disease, without signs of brugada or long QT syndrome. PLoS One 8:e67963. doi: 10.1371/journal.pone.0067963
Keywords: β1-subunit, calmodulin, cardiac arrhythmia, channelopathies, DIII-IV linker, heart, SCN5A, sodium current
Citation: Nof E, Vysochek L, Meisel E, Burashnikov E, Antzelevitch C, Clatot J, Beinart R, Luria D, Glikson M and Oz S (2019) Mutations in NaV1.5 Reveal Calcium-Calmodulin Regulation of Sodium Channel. Front. Physiol. 10:700. doi: 10.3389/fphys.2019.00700
Received: 04 February 2019; Accepted: 20 May 2019;
Published: 05 June 2019.
Edited by:
Ademuyiwa S. Aromolaran, SUNY Downstate Medical Center, United StatesReviewed by:
Brian P. Delisle, University of Kentucky, United StatesCopyright © 2019 Nof, Vysochek, Meisel, Burashnikov, Antzelevitch, Clatot, Beinart, Luria, Glikson and Oz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shimrit Oz, c2hpbXJpdC5vekBzaGViYS5oZWFsdGguZ292Lmls; c2hpbXJpdC5vei5mckBnbWFpbC5jb20=
†Present address: David Luria, Hadassah Medical Center, Jerusalem, Israel; Michael Glikson, Shaare Zedek Medical Center, Jerusalem, Israel
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.