 Patrizia Morciano1†‡
Patrizia Morciano1†‡ Maria Laura Di Giorgio1‡
Maria Laura Di Giorgio1‡ Antonella Porrazzo1,2
Antonella Porrazzo1,2 Valerio Licursi3
Valerio Licursi3 Rodolfo Negri1,4
Rodolfo Negri1,4 Yikang Rong5
Yikang Rong5 Giovanni Cenci1,2*
Giovanni Cenci1,2*- 1Dipartimento di Biologia e Biotecnologie “Charles Darwin”, Sapienza – Università di Roma, Rome, Italy
- 2Istituto Pasteur Italia - Fondazione Cenci Bolognetti, Rome, Italy
- 3Istituto di Analisi dei Sistemi ed Informatica “Antonio Ruberti”, Consiglio Nazionale delle Ricerche, Rome, Italy
- 4Istituto di Biologia e Patologia Molecolari (IBPM) del CNR, Rome, Italy
- 5School of Life Sciences, State Key Laboratory of Biocontrol, Sun Yat-sen University, Guangzhou, China
The Citrate Lyase (ACL) is the main cytosolic enzyme that converts the citrate exported from mitochondria by the SLC25A1 carrier in Acetyl Coenzyme A (acetyl-CoA) and oxaloacetate. Acetyl-CoA is a high-energy intermediate common to a large number of metabolic processes including protein acetylation reactions. This renders ACL a key regulator of histone acetylation levels and gene expression in diverse organisms including humans. We have found that depletion of ATPCL, the Drosophila ortholog of human ACL, reduced levels of Acetyl CoA but, unlike its human counterpart, does not affect global histone acetylation and gene expression. Nevertheless, reduced ATPCL levels caused evident, although moderate, mitotic chromosome breakage suggesting that this enzyme plays a partial role in chromosome stability. These defects did not increase upon X-ray irradiation, indicating that they are not dependent on an impairment of DNA repair. Interestingly, depletion of ATPCL drastically increased the frequency of chromosome breaks (CBs) associated to mutations in scheggia, which encodes the ortholog of the mitochondrial citrate carrier SLC25A1 that is also required for chromosome integrity and histone acetylation. Our results indicate that ATPCL has a dispensable role in histone acetylation and prevents massive chromosome fragmentation when citrate efflux is altered.
Introduction
Acetyl coenzyme A (acetyl-CoA) is a high-energy intermediate common to a large number of metabolic processes, including lipogenesis and cholesterogenesis that take place in different intracellular compartments. Acetyl-CoA is also required for protein acetylation reactions, which are important in post-translation modifications of proteins such as histones.
Acetyl coenzyme A may be synthesized in mitochondria and exported to the cytosol. This transport is strictly dependent on the citrate-malate-pyruvate shuttle. For this process, mitochondrial acetyl-CoA is first condensed with oxaloacetate by citrate synthase thus producing citrate and free CoA. Citrate is then exported to the cytosol through the mitochondrial citrate carrier SLC25A1. Here, acetyl-CoA generation is mediated by the activity of the ATP-cytrate lyase (ACL), which catalyzes the ATP-dependent cleavage of mitochondrial-derived citrate into oxaloacetate and acetyl-CoA (Chypre et al., 2012). Finally, the malate dehydrogenase 1, NAD (soluble; MDH1) converts cytosolic oxaloacetate by catalyzing the NADH-dependent synthesis of malate, which is transported back to the mitochondria through SLC25A10, an inorganic phosphate/dicarboxylate antiporter. Given its key role in the production of cytosolic acetyl-CoA, it is not surprising that ACL serves several and crucial functions in mammal metabolism (Wellen et al., 2009; Lee et al., 2014, 2015; Das et al., 2015; Covarrubias et al., 2016; Deb et al., 2017; Chen et al., 2018; White et al., 2018).
Although ACL resides mainly in the endoplasmic reticulum in mammalian cells (Chypre et al., 2012), it can be found also in the nucleus (Wellen et al., 2009). Here, as citrate can diffuse through the nuclear membrane, it produces acetyl-CoA, which is directly required to promote histone acetylation and regulate gene expression in response to growth factor stimulation and during differentiation (Wellen et al., 2009). ACL has been recently shown to be phosphorylated at S455 downstream of ataxia telangiectasia mutated (ATM) and AKT following DNA damage (Sivanand et al., 2017). This phosphorylation is necessary for BRCA1 recruitment and DNA repair by homologous recombination and identifies ACL as a molecular player in the DNA damage response.
A large amount of evidence indicate that ACL is upregulated or activated in several types of cancer (Zaidi et al., 2012b). Cancer cells rely on glucose as major carbon source for de novo fatty acid synthesis. Glycolysis generates citrate by tricarboxylic acid (TCA) cycle, the citrate is preferentially exported from mitochondria to cytosol and then cleaved by ACL to produce cytosolic acetyl-CoA, the building block for de novo lipid synthesis. Thus, by coupling energy metabolism with fatty acids synthesis, ACL plays a critical role in supporting cancer cell growth. It is not therefore unexpected that inhibition of ACL dramatically suppresses tumor cell proliferation (Zaidi et al., 2012a; Migita et al., 2013, 2014; Gao et al., 2014; Lee et al., 2015; Wang et al., 2017; Granchi, 2018).
Here we show that depletion of ATPCL, the Drosophila ortholog of human ACL, leads to a moderate frequency of chromosome breaks (CBs) indicating that this enzyme is partially required for chromosome stability in mitotic cells. Interestingly, the number of CBs is substantially enhanced when the export of citrate from mitochondria is inhibited suggesting that the ATPCL role on chromosome integrity is essential when cytosolic citrate is limited. We also show that ATPCL mutants exhibit decreased levels of Acetyl CoA but this reduction does not affect global histone acetylation and gene expression. This indicates that, unlike its human counterpart, the role of ATPCL in Drosophila histone acetylation could be redundant.
Results
The Drosophila ATP Cytrate Lyase
We previously demonstrated that perturbation in the citrate metabolism leads to genome instability in both Drosophila and human cells as consequence of reduced Acetyl-CoA production (Morciano et al., 2009). To further understand the contribution of the acetyl-CoA metabolism to the maintenance of genome stability, we focused our characterization on the ATP Cytrate Lyase. This enzyme cleaves TCA-derived citrate in oxaloacetate OAA (that in turn is converted into malate in order to re-entry in the mitochondria) and catalyzes the formation of acetyl-CoA in the presence of ATP. In Drosophila, the citrate lyase-encoding gene (CG8322, FBgn0020236) maps on 52D9-11 chromosome 2 region and consists of 10 exons (including the 5′ and 3′ UTR sequences) that, according to the FlyBase annotation, specifies four potential transcripts (namely ATPCL-RD, -RE, -RG, and -RF) that retain all exons albeit with few differences (Supplementary Text). Our RT-PCR and sequencing analysis confirmed the presence of these different transcripts (Supplementary Text). Moreover, qPCR revealed that ATPCL-RD and RF are poorly expressed with respect to ATPCL-RD/RE suggesting that ATPCL-PD/PE is the most representative ATPCL gene product (Figure 1B). Despite the differences in the exons 6, all transcripts encode almost identical ATPCL proteins that overall shares 70% of identity with the human counterpart, hACL. It is therefore conceivable that ATPCL is also functionally analogous to hACL (see also below).
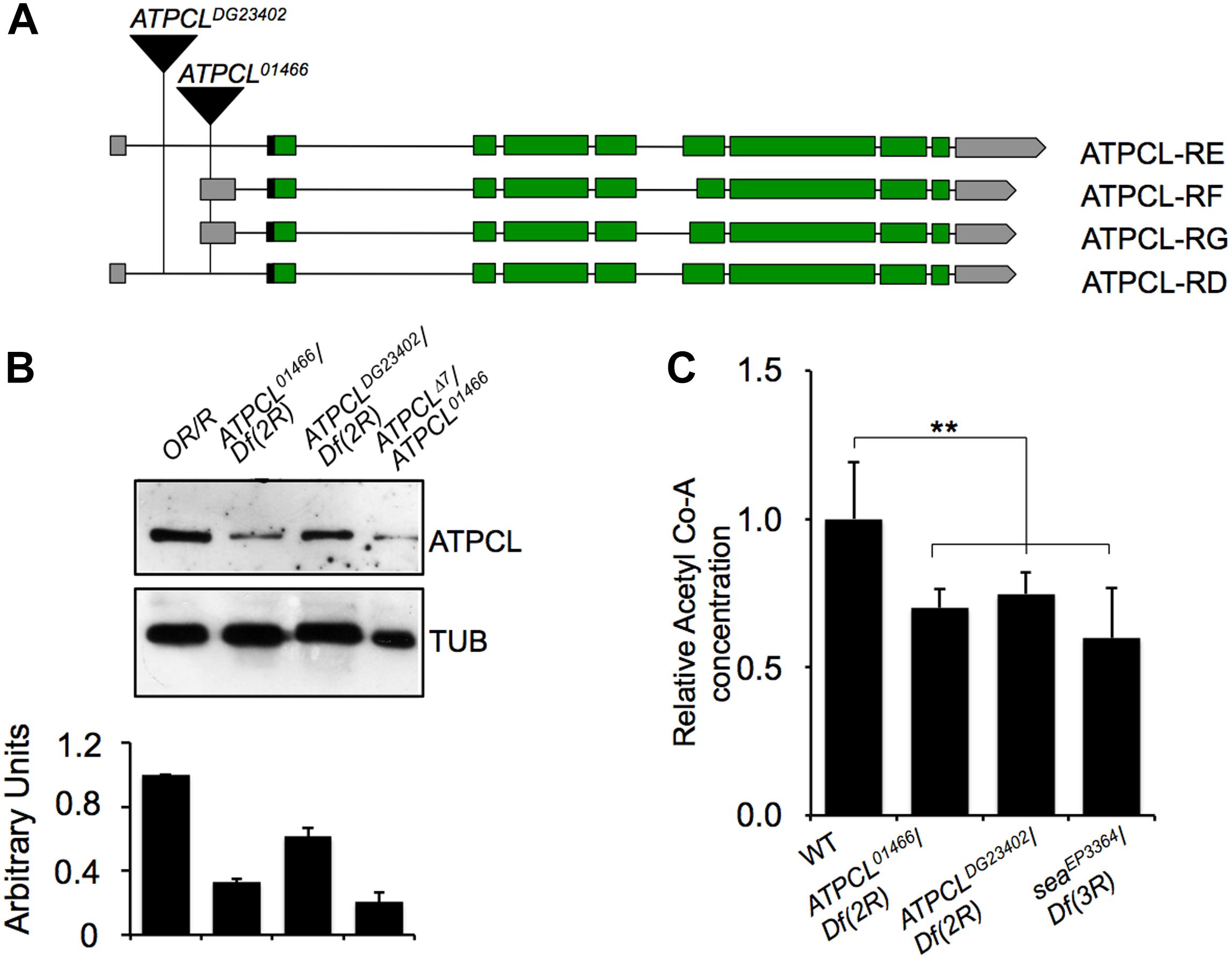
Figure 1. Molecular analysis of ATPCL mutations. (A) An analysis of the published DNA sequence reveals that ATPCL-encoding gene CG8322 specifies four potential transcripts RD, RE, RF and RG. The black triangles indicate the position of the P{PZ}ATPCL01466 and P{wHy}ATPCLDG23402 insertions in the ATPCL locus referred as ATPCL01466 and ATPCLDG23402, respectively. Green boxes: encoding exons; gray boxes: UTR-containing exons; horizontal black lines: introns. (B) Western blot of third instar larvae brain extracts from control (OR/R) and ATPCL mutants. The anti-ATPCL antibody recognizes a ∼130 KDa band which is significantly reduced in all ATPCL mutant combinations. Df (2R) is the Df(2R)Exel7138 deficiency that removes ATPCL. Tubulin is used as a loading control. The bottom figure illustrates the quantification analysis of ATPCL levels, which has been based on band intensities of three independent WBs (biological replicates). Bars indicate standard deviation. (C) Reduced levels of Acetyl CoA in ATPCL mutants. Columns indicate the percentage of Acetyl-CoA quantification (pmol/ml) in both ATPCLDG23402 and ATPCLDG23402 hemizygotes with respect to OR/R (control; ∗p < 0.01, Student t-test). Bars show SD.
We obtained from the Bloomington stock center two putative P-element induced ATPCL mutant alleles, ATPCL01466 and ATPCLDG23042. Our PCR analysis confirmed that ATPCL01466 and ATPCLDG23042 are located within the 5′ UTR of the gene at position -340 and -1167 upstream of the translation initiation site, respectively (Figure 1A). ATPCL01466 homozygotes and ATPCL01466/ Df(2R)Exel7138 hemizygotes (Df(2R)Exel7138 is a deficiency in the 52D1-52D12 polytene region that removes ATPCL) were late lethal while either ATPCLDG23042 homozygotes and hemizygotes displayed semi-lethality. By mobilizing the ATPCLDG23042 P-element, we generated an additional lethal ATPCL mutant allele (Δ7), which potentially bears partial deletions of the gene and severely affected ATPCL expression (see below). This suggests that loss of function of ATPCL might prevent development as it occurs also for ACL knock-out mice (Beigneux et al., 2004). Finally, the expression of a wild type UAS ATPCL transgene under the control of a TubGal4 promoter in ATPCLDG23042 or ATPCL01466 mutant background rescued the late lethality and CBs (see section “Materials and Methods”) confirming that both phenotypes are due to lesions in the ATPCL gene.
We have generated a guinea pig anti ATPCL polyclonal antibody that in Western blotting (WB) analysis on larval brain extracts detected a band of expected size (∼130 KDa), which decreased in ATPCL mutants (Figure 1B). ATPCL01466/ATPCLΔ7 trans heterozygotes yielded to the strongest reduction (∼80%) of expression of the ATPCL protein indicating that this mutant allele combination represents the most severe condition (Figure 1B). Immunofluorescence (IF) on larval neuroblasts with the same anti-ATPCL antibody revealed that ATPCL exhibits a punctuate localization pattern both in the cytoplasm and nucleus, which is absent from mutant cells (Supplementary Figure 2). However, as cells enter mitosis, ATPCL is excluded from the portion of nucleus containing chromatin and is retained predominantly in the cytoplasm. This pattern is clearly visible in either metaphase or ana-telophase in which ATPCL is enriched in the entire cell except the portion surrounding the dividing chromosomes (Supplementary Figure 2). Thus, like its human counterpart (Wellen et al., 2009), ATPCL localizes to the cytoplasm and the nucleus in interphase. Moreover, the nuclear localization changes during mitosis.
Finally, our fluorometric analysis revealed that Acetyl-CoA levels are ∼40% reduced in ATPCL mutants with respect to wild-type (1.30 pm/μl) indicating that, similarly to its mammalian counterpart, ATPCL is required for Acetyl-CoA synthesis (Figure 1C).
The ATPCL Role in Mitosis
In the citrate-dependent Acetyl CoA biosynthesis, ACL works downstream the citrate mitochondria carrier SLC25A1 (see section “Introduction”). As SLC25A1 and Scheggia (Sea, the SLC25A1 fly ortholog) are required for chromosome integrity in mammalian and Drosophila cells, respectively (Morciano et al., 2009), we asked if also ATPCL was involved in the same process. The analysis of DAPI-stained colchicine-treated mitotic neuroblasts from the different ATPCL mutant larval brains revealed that ATPCL mutants exhibited a moderate, although statistically significant, number of CBs (2.5–4.5%; Total Cells = 250; Figure 2). Moreover 2 Gy X-ray exposure did not enhance the number of CBs of ATPCL mutants indicating that they do not result from defective DNA repair (Figure 2B). However, WB and IF analyses using anti-acetylated H4 and H3 histones indicated that the overall histone acetylation in all ATPCL mutants was not significantly affected (Supplementary Figure 3). Although it cannot be ruled out the possibility that reduction of histone acetylation occurs at damaged sites, our observations suggest that chromosome defects are not dependent on a general reduction of histone acetylation. We then verified whether ATPCL and scheggia (sea) could genetically interact. The cytological analysis of mitotic metaphases from late lethal ATPCL sea double mutants revealed that CBs frequency is synergistically increased (∼46%; Total Cells = 180) with respect to the sum of break frequencies of both ATPCL (4.5%) and sea ∼25%; (Total Cells = 200; Figure 2B). This indicates that ATPCL prevents genome instability when certain intracellular metabolites (i.e., citrate) are limited.
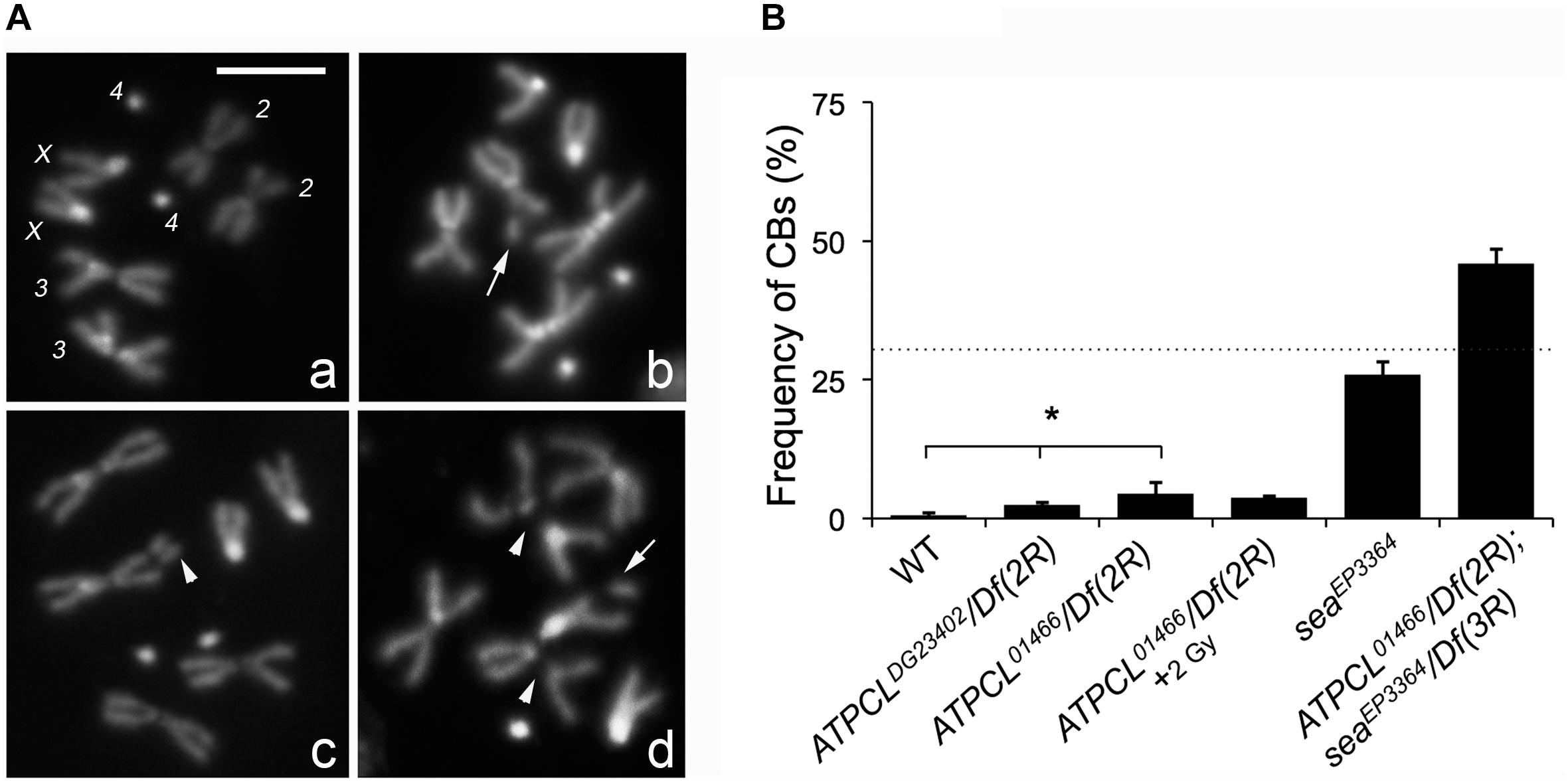
Figure 2. Depletion of ATPCL yields to chromosome breaks (CBs). (A) DAPI-stained female metaphases from larval neuroblasts of control (Oregon R, a), ATPCL mutants (b,c) and ATPCL, sea double mutants (d). Numbers in (a) indicate chromosomes X, 2, 3, and 4 of a normal female caryotype. White arrows and arrowheads indicated chromatid and CBs, respectively. Bar: 5 μm. (B) Frequency of CBs in ATPCL mutants. All ATPCL mutants exhibit a moderated (although statistically significant) frequency of CBs, which are almost absent in control cells (∗p < 0.05, Student t-test). CBs frequency is not influenced by a 2 Gy X-ray irradiation. Note that the frequency of CBs in ATPCL; sea double mutants is higher than the sum of CB frequencies observed in either ATPCL or sea single mutants (indicated by the dotted line) suggesting a synergistic effect. ∼200 cells (4 slides) for mutant combinations and ∼500 (5 slides) for Oregon R (control) were analyzed. See text for further details. Bars refer to SD.
ATPCL Is Not Required for Global Gene Transcription
We next asked whether loss of ATPCL could at least influence the expression of potential ATPCL-responsive mitotic genes. Thus we extracted RNA from either wild-type or ATPCL mutant brains, labeled, and hybridized to CDMC 14K Drosophila arrays (Canadian Drosophila Microarray Center, Toronto, Canada). Surprisingly, only ∼5% (772) of the genes significantly changed their transcriptional profile indicating that loss of ATPCL only marginally affected global gene transcription. Of these, approximately 23% (182) were > 2-fold either down- or up-regulated (Supplementary Table S1). Gene ontology (GO) revealed a clear functional bias toward brain development and behavior gene categories indicating a potential requirement of ATPCL in the regulation of nervous system genes (Supplementary Figure 4). However, based on these results, we cannot discriminate whether the change in gene expression is a direct consequence of the loss of histone acetylation rather than that of secondary responses to metabolic alterations induced by loss of acetyl-CoA. Yet, as microarrays were probed with larval brain RNA we cannot exclude that the observed trends on brain development might reflect a tissue-specific function of ATPCL in gene regulation.
Discussion
Here we have molecularly characterized for the first time ATPCL that encodes the Drosophila ortholog of human ATP citrate lyase. We have shown that, consistently with previous findings in other organisms including mammals, mutations in the ATPCL reduce cytosolic acetyl-CoA confirming that ACL has an evolutionarily conserved role in Acetyl-CoA biosynthesis. However, despite Drosophila ATPCL mutants exhibit an evident, although moderate, mitotic chromosome breakage phenotype, depletion of ATPCL, unlike human cells, has not a general impact on histone acetylation in flies suggesting that it role in histone acetylation is either partially redundant in Drosophila or compensated by alternative pathways (i.e., from acetate by acyl-CoA synthetases). However, it cannot be ruled out the possibility that ATPCL could be required for the acetylation of either nuclear or cytoplasmic factors, instead of histones, that in turn are required for chromosome integrity. Alternatively, as ATPCL plays a direct role in the homeostasis of lipids, whose modifications (i.e., peroxidation) are also required to modulate DNA repair (Tudek et al., 2017), it is possible that ATPCL mutants defects on chromosome integrity could arise as a consequence of protein acetylation-independent activity. Proteomic studies will be fundamental to verify these hypotheses.
We have previously demonstrated that mutations in the mitochondrial citrate carrier Sea/SLC25A1, led to frequent CBs in Drosophila mitotic cells (Morciano et al., 2009). The finding that perturbation of ATPCL, a key component of the pathway that from citrate generates Acetyl-CoA, affected mitosis only marginally was therefore unexpected. Moreover, our WB analysis revealed that, unlike Sea/SLC25A1, ATPCL is not required for the acetylation of histones, which could be required for chromosome integrity. However, this does not exclude that a decreased acetyl-CoA concentration could affect the local kinetics of HAT-dependent histone acetylation at selected genomic loci and/or damaged sites thus leading to specific changes in histone acetylation. Finally, our transcriptomic analysis that indicates that depletion of ATPCL has a modest modulatory effect on transcription, limited to few classes of genes (although with a bias toward brain development functions), further sustains the view that loss of ATPCL has a minimal effect on bulk chromatin histone acetylation.
The different effect of loss of Sea and ATPCL on Drosophila chromosome integrity is intriguing. This is not due to the molecular nature of mutant alleles of either sea or ATPCL, which are almost genetically null and exhibited a strong reduction of corresponding transcripts. It is conceivable that citrate deprivation in sea mutants causes much greater effects than reduction of Acetyl-CoA in ATPCL mutants. Indeed, citrate may play different tissue-specific metabolic roles and function as a general chelator of physiologically important cations. Thus its deprivation in the cytosol may compromise several intracellular pathways. Citrate reduction as consequence of inhibition of Sea/SLC25A1 transport activity might also inhibit ATPCL activity and/or expression thus exacerbating the ATPCL mutant phenotype. It has been recently demonstrated that in Drosophila citrate may indeed regulate a feedback mechanism that coordinates intracellular metabolism with glycolysis and TCA cycle (Li et al., 2018). It could be important to understand whether this metabolic feedback loop may also involve ATPCL. Our findings that ATPCL depletion in sea mutants yields to a synergic CBs phenotype suggest that ATPCL prevents chromosome fragmentation when citrate metabolism is impaired.
The mild phenotype associated with loss of ATPCL in Drosophila does not preclude the possibility that also the overexpression of ATPCL may impact Drosophila somatic cell proliferation. Overexpression of the human ortholog ACL supports cancer growth by fueling the glucose-dependent de novo lipogenesis. Whether a similar histone-acetylation independent effect takes place also in Drosophila will be an interesting issue to address in the future.
Author Contributions
PM, MDG, and AP performed gene characterization and mutants analysis. VL and RN carried out the microarray analysis. YR supervised gene characterization and discussed the data. GC supervised the experiments and wrote the manuscript.
Funding
This work has been supported by a grant from Italian Association for Cancer Research (AIRC, IG8589), Fondazione Cenci Bolognetti-Istituto Pasteur Roma and Pasteur Institute of Paris (PTR N. 24-17) to GC.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Sara Saraniero for her help in the qRNA analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2019.00383/full#supplementary-material
References
Beigneux, A. P., Kosinski, C., Gavino, B., Horton, J. D., Skarnes, W. C., and Young, S. G. (2004). ATP-citrate lyase deficiency in the mouse. J. Biol. Chem. 279, 9557–9564. doi: 10.1074/jbc.M310512200
Chen, L. Y., Lotz, M., Terkeltaub, R., and Liu-Bryan, R. (2018). Modulation of matrix metabolism by ATP-citrate lyase in articular chondrocytes. J. Biol. Chem. 293, 12259–12270. doi: 10.1074/jbc.RA118.002261
Chypre, M., Zaidi, N., and Smans, K. (2012). ATP-citrate lyase: a mini-review. Biochem. Biophys. Res. Commun. 422, 1–4. doi: 10.1016/j.bbrc.2012.04.144
Covarrubias, A. J., Aksoylar, H. I., Yu, J., Snyder, N. W., Worth, A. J., Iyer, S. S., et al. (2016). Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. eLife 5:e11612. doi: 10.7554/eLife.11612
Das, S., Morvan, F., Jourde, B., Meier, V., Kahle, P., Brebbia, P., et al. (2015). ATP citrate lyase improves mitochondrial function in skeletal muscle. Cell Metab. 21, 868–876. doi: 10.1016/j.cmet.2015.05.006
Deb, D. K., Chen, Y., Sun, J., Wang, Y., and Li, Y. C. (2017). ATP-citrate lyase is essential for high glucose-induced histone hyperacetylation and fibrogenic gene upregulation in mesangial cells. Am. J. Physiol. Renal. Physiol. 313, F423–F429. doi: 10.1152/ajprenal.00029.2017
Gao, Y., Islam, M. S., Tian, J., Lui, V. W., and Xiao, D. (2014). Inactivation of ATP citrate lyase by Cucurbitacin B: a bioactive compound from cucumber, inhibits prostate cancer growth. Cancer Lett. 349, 15–25. doi: 10.1016/j.canlet.2014.03.015
Granchi, C. (2018). ATP citrate lyase (ACLY) inhibitors: an anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur. J. Med. Chem. 157, 1276–1291. doi: 10.1016/j.ejmech.2018.09.001
Lee, J. H., Jang, H., Lee, S. M., Lee, J. E., Choi, J., Kim, T. W., et al. (2015). ATP-citrate lyase regulates cellular senescence via an AMPK- and p53-dependent pathway. FEBS J. 282, 361–371. doi: 10.1111/febs.13139
Lee, J. V., Carrer, A., Shah, S., Snyder, N. W., Wei, S., Venneti, S., et al. (2014). Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 20, 306–319. doi: 10.1016/j.cmet.2014.06.004
Li, H., Hurlburt, A. J., and Tennessen, J. M. (2018). A Drosophila model of combined D-2- and L-2-hydroxyglutaric aciduria reveals a mechanism linking mitochondrial citrate export with oncometabolite accumulation. Dis. Model. Mech. 11:dmm035337. doi: 10.1242/dmm.035337
Migita, T., Okabe, S., Ikeda, K., Igarashi, S., Sugawara, S., Tomida, A., et al. (2013). Inhibition of ATP citrate lyase induces an anticancer effect via reactive oxygen species: AMPK as a predictive biomarker for therapeutic impact. Am. J. Pathol. 182, 1800–1810. doi: 10.1016/j.ajpath.2013.01.048
Migita, T., Okabe, S., Ikeda, K., Igarashi, S., Sugawara, S., Tomida, A., et al. (2014). Inhibition of ATP citrate lyase induces triglyceride accumulation with altered fatty acid composition in cancer cells. Int. J. Cancer 135, 37–47. doi: 10.1002/ijc.28652
Morciano, P., Carrisi, C., Capobianco, L., Mannini, L., Burgio, G., Cestra, G., et al. (2009). A conserved role for the mitochondrial citrate transporter Sea/SLC25A1 in the maintenance of chromosome integrity. Hum. Mol. Genet. 18, 4180–4188. doi: 10.1093/hmg/ddp370
Sivanand, S., Rhoades, S., Jiang, Q., Lee, J. V., Benci, J., Zhang, J., et al. (2017). Nuclear Acetyl-CoA production by ACLY promotes homologous recombination. Mol. Cell 67, 252–265.e6. doi: 10.1016/j.molcel.2017.06.008
Tudek, B., Zdżalik-Bielecka, D., Tudek, A., Kosicki, K., Fabisiewicz, A., and Speina, E. (2017). Lipid peroxidation in face of DNA damage, DNA repair and other cellular processes. Free Radic. Biol. Med. 107, 77–89. doi: 10.1016/j.freeradbiomed.2016.11.043
Wang, D., Yin, L., Wei, J., Yang, Z., and Jiang, G. (2017). ATP citrate lyase is increased in human breast cancer, depletion of which promotes apoptosis. Tumour Biol. 39:1010428317698338. doi: 10.1177/1010428317698338
Wellen, K. E., Hatzivassiliou, G., Sachdeva, U. M., Bui, T. V., Cross, J. R., and Thompson, C. B. (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. doi: 10.1126/science.1164097
White, P. J., Mcgarrah, R. W., Grimsrud, P. A., Tso, S. C., Yang, W. H., Haldeman, J. M., et al. (2018). The BCKDH kinase and phosphatase integrate BCAA and lipid metabolism via regulation of ATP-citrate lyase. Cell Metab. 27, 1281–1293.e7. doi: 10.1016/j.cmet.2018.04.015
Zaidi, N., Royaux, I., Swinnen, J. V., and Smans, K. (2012a). ATP citrate lyase knockdown induces growth arrest and apoptosis through different cell- and environment-dependent mechanisms. Mol. Cancer Ther. 11, 1925–1935. doi: 10.1158/1535-7163.MCT-12-0095
Keywords: citrate lyase, Drosophila chromosomes, histone acetylation, acetyl-CoA, Drosophila
Citation: Morciano P, Di Giorgio ML, Porrazzo A, Licursi V, Negri R, Rong Y and Cenci G (2019) Depletion of ATP-Citrate Lyase (ATPCL) Affects Chromosome Integrity Without Altering Histone Acetylation in Drosophila Mitotic Cells. Front. Physiol. 10:383. doi: 10.3389/fphys.2019.00383
Received: 29 December 2018; Accepted: 21 March 2019;
Published: 04 April 2019.
Edited by:
Paola Cusumano, University of Padua, ItalyReviewed by:
Aram Megighian, University of Padua, ItalyAngelique Christine Paulk, Massachusetts General Hospital and Harvard Medical School, United States
Copyright © 2019 Morciano, Di Giorgio, Porrazzo, Licursi, Negri, Rong and Cenci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanni Cenci, Z2lvdmFubmkuY2VuY2lAdW5pcm9tYTEuaXQ=
†Present address: Patrizia Morciano, INFN – Laboratori Nazionali del Gran Sasso, L’Aquila,Italy
‡These authors have contributed equally to this work