 Qiang Zhang1,2,3,4
Qiang Zhang1,2,3,4 Wei Dou1,2,3,4
Wei Dou1,2,3,4 Deng Pan1,2,3,4
Deng Pan1,2,3,4 Er-Hu Chen1,2,3,4
Er-Hu Chen1,2,3,4 Jin-Zhi Niu1,2,3,4
Jin-Zhi Niu1,2,3,4 Guy Smagghe1,2,3,4*
Guy Smagghe1,2,3,4* Jin-Jun Wang1,2,3,4*
Jin-Jun Wang1,2,3,4*- 1Key Laboratory of Entomology and Pest Control Engineering, College of Plant Protection, Southwest University, Chongqing, China
- 2Academy of Agricultural Sciences, Southwest University, Chongqing, China
- 3International China-Belgium Joint Laboratory on Sustainable Crop Pest Control Between Southwest University in China and Ghent University in Belgium, Chongqing, China
- 4Department of Plants and Crops, Ghent University, Ghent, Belgium
Insect metamorphosis is a complex process involving drastic morphological and physiological changes. microRNAs (miRNAs) are a class of endogenous small non-coding RNAs that play key roles in regulating various biological processes, including metamorphosis, by post-transcriptional repression of mRNAs. The oriental fruit fly, Bactrocera dorsalis, is one of the most destructive insect pests in many Asian countries and the Pacific Islands. The regulatory role of miRNAs in B. dorsalis metamorphosis is unclear. To better understand the molecular regulatory mechanisms of miRNAs in pupariation, Illumina sequencing of the wandering stage (WS), the late WS and the white puparium stage of B. dorsalis were performed. Two hundred forty-nine miRNAs, including 184 known miRNAs and 65 novel miRNAs, were obtained. Among these miRNAs, 19 miRNAs were differentially expressed in pupariation, and eight miRNAs showed relative high expression levels (>50 TPM), of which five differentially expressed miRNAs (DEMs) had target differentially expressed genes (DEGs) predicted by the expected miRNA–mRNA negative regulation pattern using the Illumina HiSeq data. Four sets of DEMs and their predicted target DEGs were confirmed by qPCR. Of the four miRNAs, two miRNAs were down-regulated: miR-981, which may target pdpc, and Bdo-novel-mir-55, which potentially regulates spsX1, psB/C, and chit3. The other two miRNAs were up-regulated: let-7a-3p, which possibly controls lap, and Bdo-novel-mir-24, which may regulate ipc and sp1/2. This study provides a useful resource to elucidate the regulatory role of miRNAs and understand the molecular mechanisms of metamorphosis.
Introduction
Metamorphosis is an important process, because pupation failure means death, and also a complex process involving extensive morphological, biochemical, and cellular changes. During larval–pupal metamorphosis, the soft cuticle of a wandering larva is transformed into a hard puparium, which involves immobilization, cuticular shrinkage, anterior retraction, longitudinal body contraction, and tanning of the cuticle (Jan and Fraenkel, 2010). In holometabolous insects, a short pulse of ecdysone peak occurs in larval–pupal metamorphosis (Dubrovsky, 2005), and the obsolete larval tissues or organs (including the integument, midgut, and fat body) undergo a remodeling process during larval–pupal metamorphosis, involving programmed cell death and histolysis of the tissues or organs to recycle intracellular components for formation of the pupal-adult organs (Yang et al., 2017; Guo S. Y. et al., 2018). The regulatory role of hormones is relatively clear during insect metamorphosis. For example, juvenile hormone (JH) and 20-hydroxyecdysone (20E) jointly regulate larval–pupal development. While both hormones are present at the larval stages, they have opposing roles in development. The role of JH is to maintain insects at the larval stage, whereas 20E induces metamorphosis. At the end of the last larval stage, the 20E titer increases while the JH titer decreases, and then the metamorphosis occurs (Liu S. et al., 2017; Zhang T. et al., 2018). Additionally, as chitin is a crucial component of the cuticle, insect metamorphosis is largely dependent on chitin degradation and synthesis, which are strictly coordinate and occur almost simultaneously during larval–pupal development (Yao et al., 2010). Thus, a series of chitin biosynthesis enzymes (such as chitin synthase) and catabolic pathway enzymes (such as chitinase) control the process of metamorphosis by regulating chitin.
However, many details of the regulatory networks involved in pupariation remain undefined. For example, it is not known how regulatory non-coding RNAs (such as long non-coding RNAs, small interfering RNAs, piwi-interacting RNAs, circular RNAs, or miRNAs) regulate pupariation. In these regulatory non-coding RNAs, miRNAs are our focus in the present study. miRNAs are 18–25 nucleotide (nt) RNAs that post-transcriptionally regulate gene expression through interactions with their target transcripts (Reynolds et al., 2017). Changes in miRNA profiles have been observed during immune responses (Garbuzov and Tatar, 2014; Yin et al., 2018), reproduction (Tariq et al., 2016; Zhang et al., 2016) and insecticide resistance (Etebari et al., 2018; Zhang Y. et al., 2018). In addition, they have been implicated as regulators of metamorphosis, involving in the JH, 20E and chitin pathways. miR-278 and let-7 regulated the early JH-response gene, Kr-h1, in Locusta migratoria (Song et al., 2018). miR-14 controlled 20E signaling gene, E75, and the ecdysone receptor, EcR-B, in Bombyx mori (Liu Z. et al., 2017). miR-173 directly targeted 20E signaling gene, Ftz-F1, and miR-2703 regulated chitin synthasegene A in Nilaparvata lugens (Li T. et al., 2017; Chen J. et al., 2018). miR-281-3p regulated EcR of Plutella xylostella (Wang et al., 2018).
The oriental fruit fly, Bactrocera dorsalis (Hendel) (Diptera: Tephritidae), is one of the most destructive pests in Southeast Asia and several Pacific Islands. They cause damage to over 250 different fruits and vegetables, including citrus, guavas, carambolas, mangos, peaches, and chili peppers (Jin et al., 2011; Wang et al., 2014). B. dorsalis is highly polyphagous and invasive, and so many countries have imposed strict quarantine restrictions (Shen et al., 2011; Khan and Akram, 2018).
As a dipteran insect, B. dorsalis is a typical holometabolous insect that undergoes larval–pupal metamorphosis. Additionally, several studies have shown that miRNAs are involved in B. dorsalis development and reproduction (Huang et al., 2014; Peng et al., 2016, 2017). Considering the diverse roles of miRNAs, some miRNAs in the regulation of gene expression during metamorphosis probably exist in B. dorsalis. Therefore, we constructed and sequenced small RNA libraries from B. dorsalis at three developmental stages, including the wandering stage (WS), the late wandering stage (LWS, the body contraction just before pupariation), and the white puparium stage (WPS). We identified known and novel miRNAs, based on the result of Illumina HiSeq, and analyzed miRNAs expression profile during the different developmental stages. Additionally, to confirm the five differentially expressed miRNAs (DEMs) and differentially expressed genes (DEGs) identified by bioinformatics analysis, the qPCR was performed. The current study provides a useful resource to elucidate the regulatory role of miRNAs and understand the molecular mechanisms of metamorphosis.
Materials and Methods
Sample Preparation and RNA Extraction
The oriental fruit fly was reared as described previously (Huang et al., 2014). Briefly, B. dorsalis were reared at 27 ± 1°C and 70 ± 5% relative humidity with a photoperiod regime of 14:10 h light:darkness. The larvae were reared on an artificial diet containing corn, yeast powder, wheat flour, and sucrose. Three replicates were performed for each of the three developmental stages (WS, LWS, and WPS), and each replicate contained eight flies. Total RNA was extracted using the RNeasy plus Micro Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. In brief, the absorbance at 260 nm was used to quantify the RNA using a NanoVue UV-Vis spectrophotometer (GE Healthcare Bio-Science, Uppsala, Sweden). The absorbance ratios of OD260/280 and OD260/230 were used to test the purity of all RNA samples. The integrity of the RNA was assessed by 1% agarose gel electrophoresis.
cDNA Library Construction and HiSeq Sequencing
A total amount of 1.5 μg RNA per sample was used as input material and made up to a volume of 6 μL with nuclease-free water for the RNA sample preparations. Sequencing libraries were generated using NEB Next Ultra small RNA Sample Library Prep Kit for Illumina (San Diego, CA, United States) following manufacturer’s recommendations, and barcodes were added to the adaptor sequences for each sample. Briefly, because of the phosphate group at the 5′ end of the small RNA and the hydroxyl group at the 3′ end, T4 RNA Ligase 1 and T4 RNA Ligase 2 (truncated), respectively, were used to ligate the adaptors at the ends of the small RNA fragments. cDNA was synthesized by reverse transcription and then amplified by PCR, and the target fragments were screened using glue separation technology. The recovered fragments were cut in the same way as the small RNA library (sRNA). After completion of library construction, the concentration of the library was determined using a Qubit 2, and then the library was diluted to 1 ng/μL. Subsequently, to guarantee the quality of the library, a 2100 Bioanalyzer (Agilent, Santa Clara, CA, United States) was used to detect the insert size, and qPCR was used to accurately quantify the effective concentration of the library.
The clustering of the barcoded samples was performed on a cBot Cluster Generation System (Illumina) using the TruSeq PE Cluster Kit v4-cBot-HS (Illumina) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina HiSeq 2500 platform and paired-end reads were generated.
Analysis of HiSeq Data
Raw sequence reads (raw data) were acquired by the Illumina HiSeq X-ten platform. Clean reads (clean data) were obtained by removing reads containing ‘N’ (an unrecognized base) at 10% or higher, reads with no adaptor sequence at the 3′ end, reads with a sequence shorter than 18 nt or more than 30 nt, low quality reads, and reads with truncated adaptor sequences at the 3′ end. The Q20, Q30, GC content and sequence duplication level of the clean data were calculated. Subsequently, using Bowtie software, the clean reads were compared with the Silva, GtRNAdb, Rfam and Repbase databases, filtering out ribosomal RNA (rRNA), small cytoplasmic RNA (scRNA), transfer RNA (tRNA), small nuclear RNA (snRNA), small nucleolar RNA (snoRNA) and repeats, to give unannotated reads. The unannotated reads were prepared for further analysis by mapping them with the B. dorsalis genome (NCBI Assembly: ASM78921v2). Based on the biological characteristics of miRNA and the mapping to the reference genome sequence, miRDeep2 (Friedlander et al., 2012) software was used to make the identification of known miRNAs with the hairpin precursors in miRBase211. Meanwhile, based on possible precursor sequences obtained by mapping reads to the genomic location, the distribution of reads on the precursor sequence and the precursor structure energy information (RNAfold), a Bayesian model was used to identify novel miRNAs. The miRNA expression levels were further quantified by the TPM (Transcripts Per Kilobase of exon model per Million mapped reads) method (Fahlgren et al., 2007). In this study, differential expression analysis of two developmental stages (WS vs. LWS and LWS vs. WPS) was performed using the DESeq R package (1.10.1). DESeq provides statistical routines for determining differential expression in digital miRNA expression data using a model based on a negative binomial distribution. miRNAs which normalized expression of 0 were modified to 0.01, and the normalized data were used to calculate a log2-ratio plot. The fold-change (log2 > 1 or log2 < -1) of miRNAs was considered to be differentially expressed.
Target Gene Prediction and Annotation
Prediction of the target genes of the miRNAs was conducted using the miRanda and RNAhybrid software packages. Gene function was annotated based on the following databases: NCBI non-redundant protein sequences (Nr), Protein family (Pfam), Clusters of Orthologous Groups of proteins (COG), a manually annotated and reviewed protein sequence database (Swiss-Prot), the evolutionary genealogy of genes: Non-supervised Orthologous Groups (eggNOG), Kyoto Encyclopedia of Genes and Genomes (KEGG Ortholog database), and Gene Ontology (GO).
Gene Ontology analysis of DEGs was implemented by the GOseq R packages based Wallenius non-central hyper-geometric distribution. For the COG annotation, the DEGs between two developmental stages (WS vs. LWS and LWS vs. WPS) were translated into amino acid sequences by using the virtual ribosome package. We used the batch web CD search tool2 to assign COG groups. For the KEGG annotation, KOBAS software (Mao et al., 2005) was used to test the statistical enrichment of differential expression genes in KEGG pathways.
Quantitative Real-Time PCR (qPCR)
In this study, qPCR was carried out to confirm the miRNAs and their predicted target genes from the Illumina sequencing results. Total RNA was extracted from the three developmental stages of WS, LWS, and WPS using TRIzol reagent.
For mRNA qPCR, reverse transcription was carried out using PrimeScript RT reagent Kit (Takara, Dalian, China) according to the manufacturer’s protocols. All the primers (Supplementary Table S1) were designed by NCBI Primer Blast3. A standard curve for each primer pair was carried out with serial dilutions of cDNA (1, 1/4, 1/16, 1/64, 1/256) to determine the amplification efficiency and cycle threshold (Ct) value. The qPCR was performed on a BIO-RAD Real-Time PCR System (Bio-Rad, Hercules, CA, United States) using the Novostar-SYBR Supermix (Novoprotein, Shanghai, China) reagent, with an initial denaturation at 95°C for 2 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. At the end of the procedure, a melting curve analysis was recorded from 60 to 95°C to ensure the specificity of each pair of primers. The amplification was conducted in a 20 μL reaction volume consisting of 10 μL of SYBR Supermix, 1 μL each of forward and reverse primers (10 μM), 7 μL of nuclease-free water, and 1 μL of cDNA. The relative expression levels were normalized using α-tubulin (GenBank Accession No. GU269902) and rps3 (XM_011212815) as internal references, as described previously (Shen et al., 2010; Wei et al., 2015).
For miRNA qPCR, the reverse transcription reagent used was the miRNA cDNA Synthesis Kit (CWBIO, Beijing, China) according to the manufacturer’s protocols. All the primers (Supplementary Table S1) were designed using the miRNA qPCR Assay Kit (CWBIO) protocol. A standard curve for each primer pair (only the forward primer was designed) was carried out with serial dilutions of cDNA (1/1, 1/2, 1/4, 1/8, 1/16) to determine the amplification efficiency and cycle threshold (Ct) value. The cycling conditions were an initial denaturation at 95°C for 10 min, followed by 45 cycles of 95°C for 15 s and 60°C for 1 min. Melting curve analysis was performed in the same way as above. The amplification was conducted in a 20 μL reaction volume consisting of 10 μL of miRNA qPCR mixture, 0.4 μL of forward and reverse primers (10 μM), 8.2 μL of nuclease-free water, and 1 μL of cDNA. The expression of U6 was used to normalize the expression of miRNA.
Statistical Analysis
qPCR experiments were performed in four biological replicates, and qBase software (Hellemans et al., 2007) was used to determine the changes in miRNA and target genes during larval–pupal metamorphosis. The qPCR and the corresponding Illumina sequencing results were analyzed with one-way analysis of variance (ANOVA), and the means were separated with Tukey’s HSD post hoc test (P < 0.05) using SPSS 23.0 software (SPSS, Chicago, IL, United States).
Results
Profile of sRNA Libraries
Three sRNA libraries were constructed from the whole body at three developmental stages (WS, LWS, and WPS) to understand the miRNA regulation involved in larval metamorphosis. The samples from each stage were sequenced in triplicate, and there was more than 11.87 M clean data for each sample after strict quality control (Supplementary Table S2). Approximately 174 million clean reads were found, including 63,799,603, 60,700,881, and 49,530,993 reads in the WS, LWS, and WPS stages, respectively. Cleaning up the Rfam reads (rRNA, scRNA, snRNA, snoRNA, tRNA) and removing repeats, the unannotated reads (41,982,118, 33,973,104, and 26,569,248 reads in the WS, LWS, and WPS stages, respectively) were obtained (Table 1) and mapped onto the B. dorsalis genome for further analysis (Supplementary Table S3). The length distribution of the clean data for all three developmental stages mostly showed one main peak at 22 nt, which is the typical length of mature miRNAs, and another smaller peak at 26–27 nt, corresponding to piRNA-like sequences (the role of these putative piRNA sequences as regulators of pupariation was excluded from further analysis).
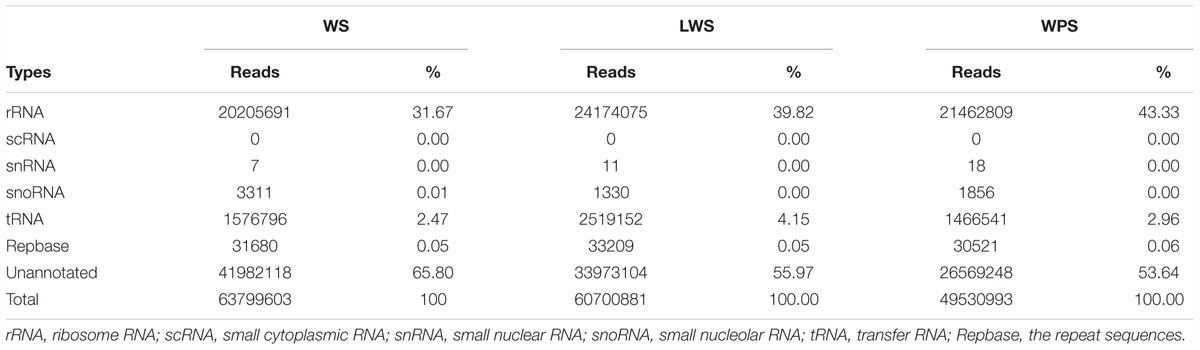
Table 1. Distribution of clean reads for different RNA categories in the libraries.
Identification of Known and Novel miRNAs
By filtering the data set, the unannotated reads aligned to the B. dorsalis reference genome were analyzed. The name of the miRNA with the most reads was used to represent that miRNA and other variants (Peng et al., 2016). Two hundred forty-nine mature miRNAs were obtained, including 184 known miRNAs and 65 novel miRNAs (Supplementary Table S4). Specifically, the samples from WS (244 miNRAs), LWS (235 miRNAs) and WPS (221 miRNAs) yielded 179, 174, and 160 known miRNAs and 65, 61, and 61 novel miRNAs, respectively (Figure 1). The known and novel miRNAs among the three libraries were compared to search for any overlaps, and 152 known and 59 novel miRNAs were observed in all three libraries (Supplementary Table S4).
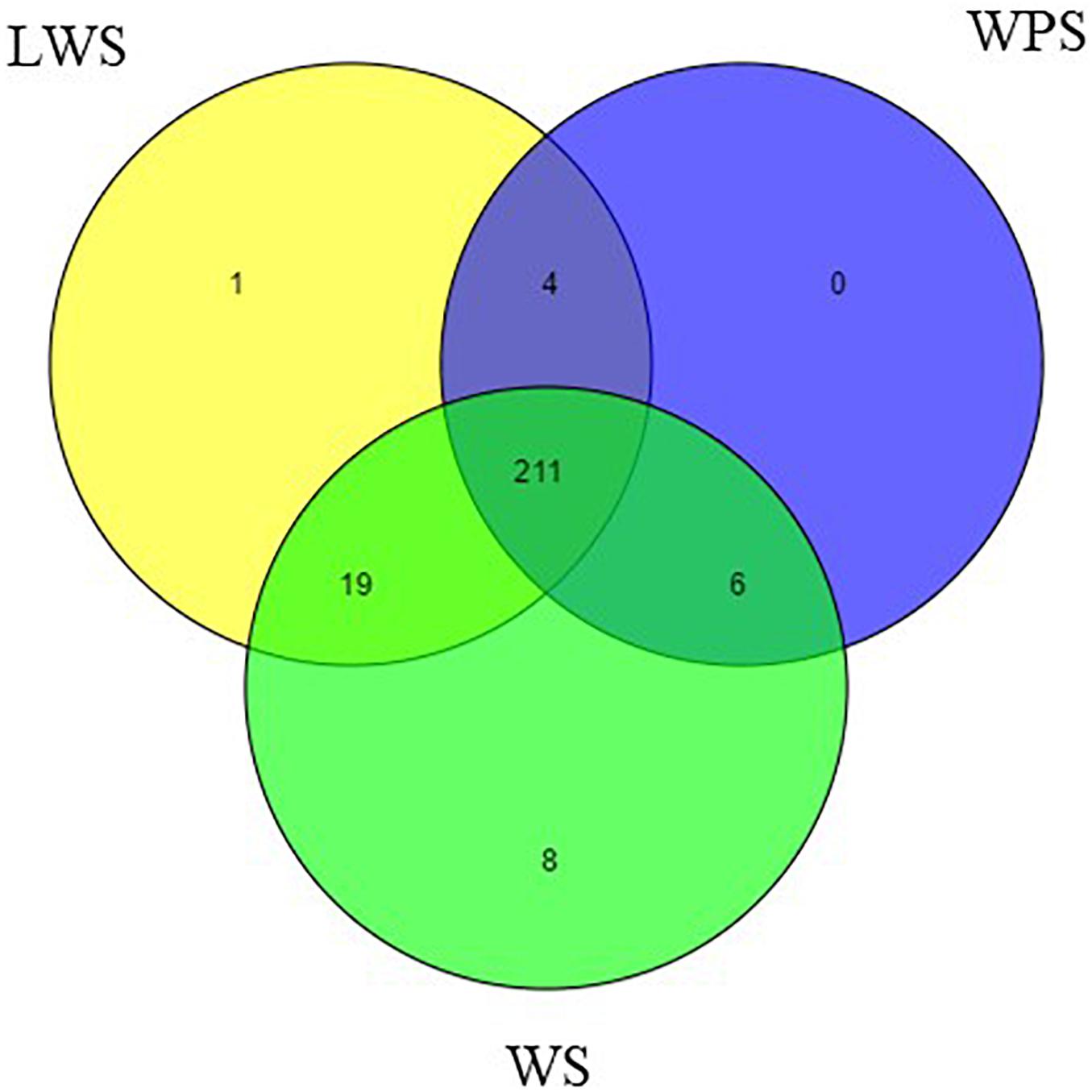
Figure 1. Venn diagram representing the distribution of miRNA in the WS, LWS, and WPS stages of Bactrocera dorsalis.
Differential Expression of miRNA
To study the differential expression of miRNAs in the three developmental stages (WS, LWS, and WPS) of B. dorsalis, pairwise comparison of the DEMs was conducted, and 89 miRNAs (42 up-regulated and 47 down-regulated, WS vs. LWS) and 81 miRNAs (39 up-regulated and 42 down-regulated, LWS vs. WPS) were differentially expressed (Supplementary Table S5). By filtering the low expression miRNAs (the expression level of any one stage is less than five TPM in the three stages), a total of 19 DEMs was obtained in the current study. Three DEMs (let-7a-3p, miR-2-3p, and Bdo-novel-mir-55) shared between both two comparisons and 16 DEMs were specific (14 and 2 DEMs in WS vs. LWS and LWS vs. WPS, respectively) (Supplementary Table S6). A cluster heat map was subsequently adopted to show the differential expression of B. dorsalis miRNAs over the three stages (Figure 2). Eight DEMs showed relative high expression levels (>50 TPM in any comparison stage), where three DEMs (let-7a-3p, miR-2-3p, and Bdo-novel-mir-55) were shared between both two comparisons, miR-282-5p, miR-981, Bdo-novel-mir-24 and Bdo-novel-mir-44 were specific to WS vs. LWS, and miR-100 was unique to LWS vs. WPS.
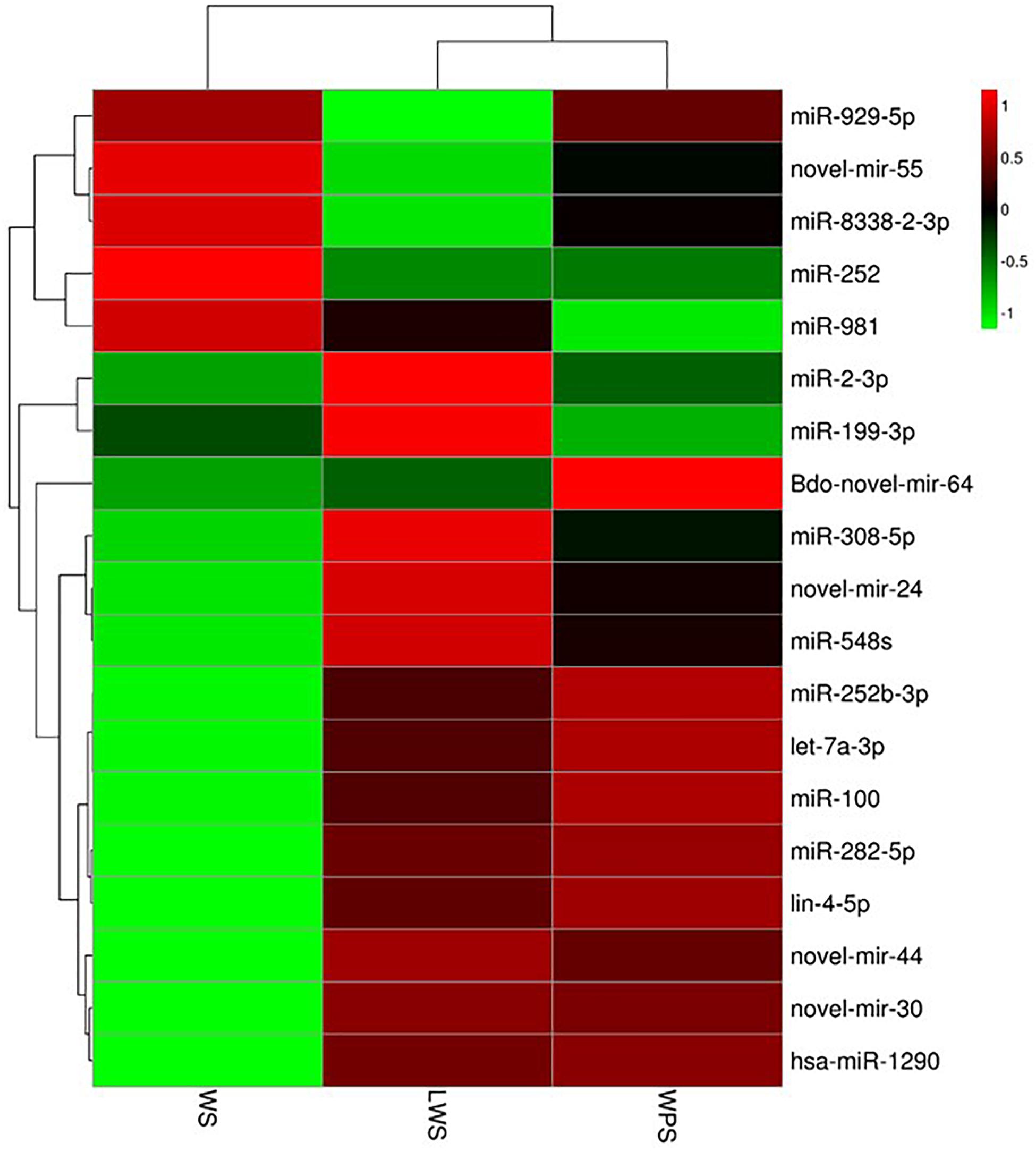
Figure 2. Cluster heat map showing the differential expression of B. dorsalis miRNAs over the three stages. (Parameter setting: data adopt the logarithm: log10 and normalization; clustering by row and column).
Analysis of Differentially Expressed miRNAs and Potential Target mRNAs
According to the sequence information of the known and newly predicted miRNAs, miRNA target gene prediction was conducted. There were 12 (9 up-regulated and 3 down-regulated) and 4 up-regulated DEMs giving 132 and 25 predicted target genes in WS vs. LWS and LWS vs. WPS, respectively (Supplementary Table S7). These were analyzed in combination with the expression data of the differentially expressed target genes; the expression data is from Chen E. H. et al. (2018). There were 10 (7 up-regulated and 3 down-regulated) DEMs associated with 29 predicted DEGs in WS vs. LWS but no one in LWS vs. WPS (Supplementary Table S8). Taking into account the expected pattern of negative regulation between miRNA and mRNA, this resulted in 8 (6 up-regulated and 2 down-regulated) miRNAs associated with 18 predicted DEGs in WS vs. LWS (Supplementary Table S9). Among these DEMs with the predicted DEGs, there were five miRNAs with high expression levels, including three up-regulated miRNAs, let-7a-3p, Bdo-novel-mir-24 and Bdo-novel-mir-44, and two down-regulated miRNA, miR-981 and Bdo-novel-mir-55 that were in WS vs. LWS. Meanwhile, the five miRNAs were significantly different in their expression with P-value < 0.05.
GO, COG, and KEGG Analysis of the Target Genes of the Differentially Expressed miRNAs
To explore the potential function of the DEMs during pupariation, seven databases were used to identify functional modules of the predicted target genes. These databases were GO, KEGG, Nr, COG, Pfam, Swiss-Prot, and eggNOG. There were 132 and 25 predicted target genes in WS vs. LWS and LWS vs. WPS, respectively, that were successfully annotated (Supplementary Table S7). GO, COG, and KEGG analysis was then performed on the predicted DEGs. Because there was no predicted DEG in LWS vs. WPS, the above analysis was only conducted in WS vs. LWS. For the GO classification, the predicted DEGs of the DEMs were divided into three ontologies: biological process, cellular component, and molecular function, including 44 annotations for each comparison. The most enriched functions included ‘Single-organism process,’ ‘Cellular process,’ ‘Metabolic process,’ ‘Binding,’ and ‘Cell’ (Supplementary Figure S1). Furthermore, COG functional classification was performed to compare the enriched functional categories of the predicted DEGs among the three stages using the rpstBlastn program. When mapping the predicted DEGs into the COG database, 11 genes were annotated (Supplementary Table S7). The annotations of the predicted DEGs were mainly focused on ‘general function prediction only,’ followed by ‘Carbohydrate transport and metabolism,’ ‘Amino acid transport and metabolism,’ ‘Replication, recombination and repair,’ and ‘Signal transduction mechanisms’ (Supplementary Figure S2). Additionally, KEGG analysis of the predicted DEGs was used to increase our understanding of the biological functions of the DEMs in metabolism and development of during B. dorsalis pupariation. The predicted DEGs were mainly enriched in ‘ECM-receptor interaction,’ followed by ‘Amino sugar and nucleotide sugar metabolism,’ ‘Drug metabolism – cytochrome P450,’ and ‘Glycine, serine, and threonine metabolism’ (Supplementary Figure S3). Furthermore, the Nr annotation of the DEGs of the five highly expressed DEMs included PP2C-like domain-containing protein CG9801 (pdpc), corresponding to miR-981; serine proteinase stubble isoform X1 (spsX1), protein skeletor, isoforms B/C (psB/C), and probable chitinase 3 (chit3), all corresponding to Bdo-novel-mir-55; lysosomal aspartic protease-like (lap), corresponding to let-7a-3p; collagen alpha-1(IV) chain (cac), corresponding to Bdo-novel-mir-44; and putative inorganic phosphate cotransporter (ipc) and serine proteases 1/2-like (sp1/2), both corresponding to Bdo-novel-mir-24 (Table 2).
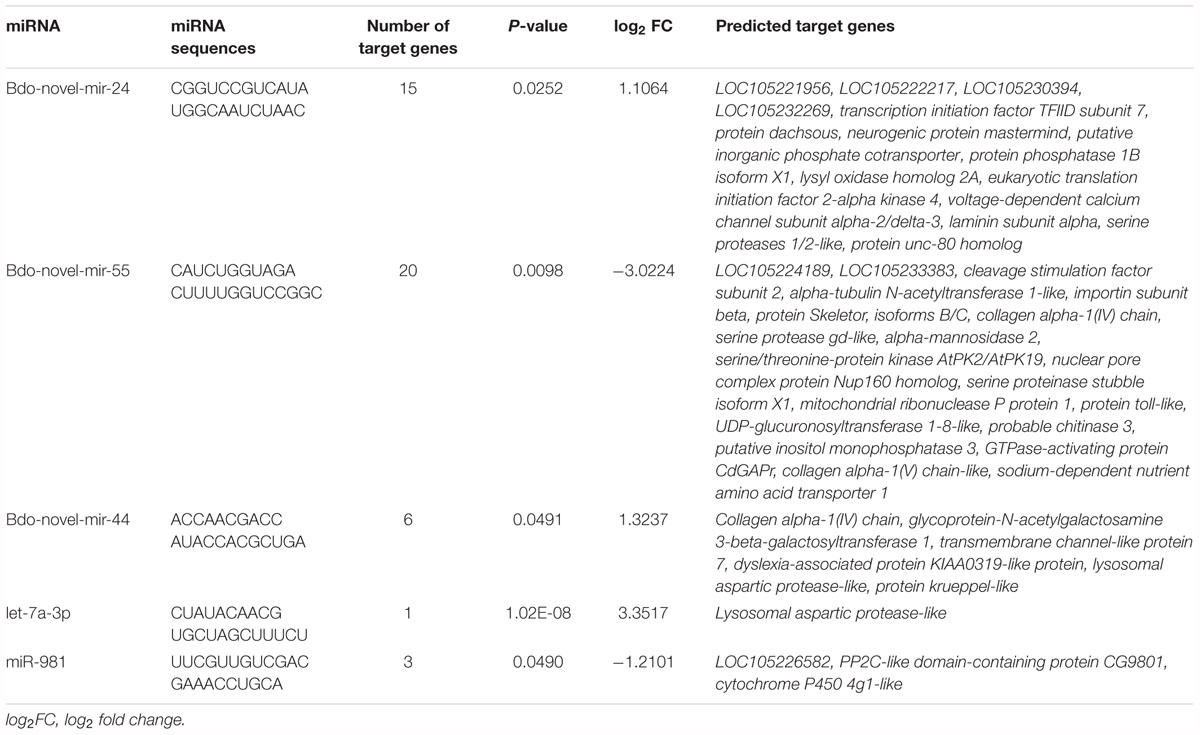
Table 2. Predicted targets of five high differential expression miRNAs in pupariation of Bactrocera dorsalis.
Analysis of Selected miRNA–mRNA Expression Patterns
qPCR was used to analyze the miRNA–mRNA expression patterns and validate the Illumina sequencing results. Five DEMs showed relative high expression levels (>50 TPM) with their eight predicted DEGs, in accordance with the expected miRNA–mRNA negative regulation pattern. The results showed that miR-981 and Bdo-novel-miR-55 were significantly down-regulated upon pupariation and that their predicted target genes pdpc, spsX1, psB/C, and chit3 were significantly up-regulated during pupariation. A significant increase in the expression of let-7a-3p and Bdo-novel-mir-24 was observed upon pupariation, and the predicted target genes of those miRNAs, lap, ipc, and sp1/2, were significantly down-regulated during pupariation. It was observed that Bdo-novel-mir-44 was not significantly changed upon pupariation while its predicted target gene cac was significantly down-regulated during this progress (Figure 3). The qPCR results were consistent with the Illumina sequencing results. Furthermore, the miRNA and mRNA expression levels were almost unanimous in the LWS and WPS stages between the qPCR and Illumina sequencing results.
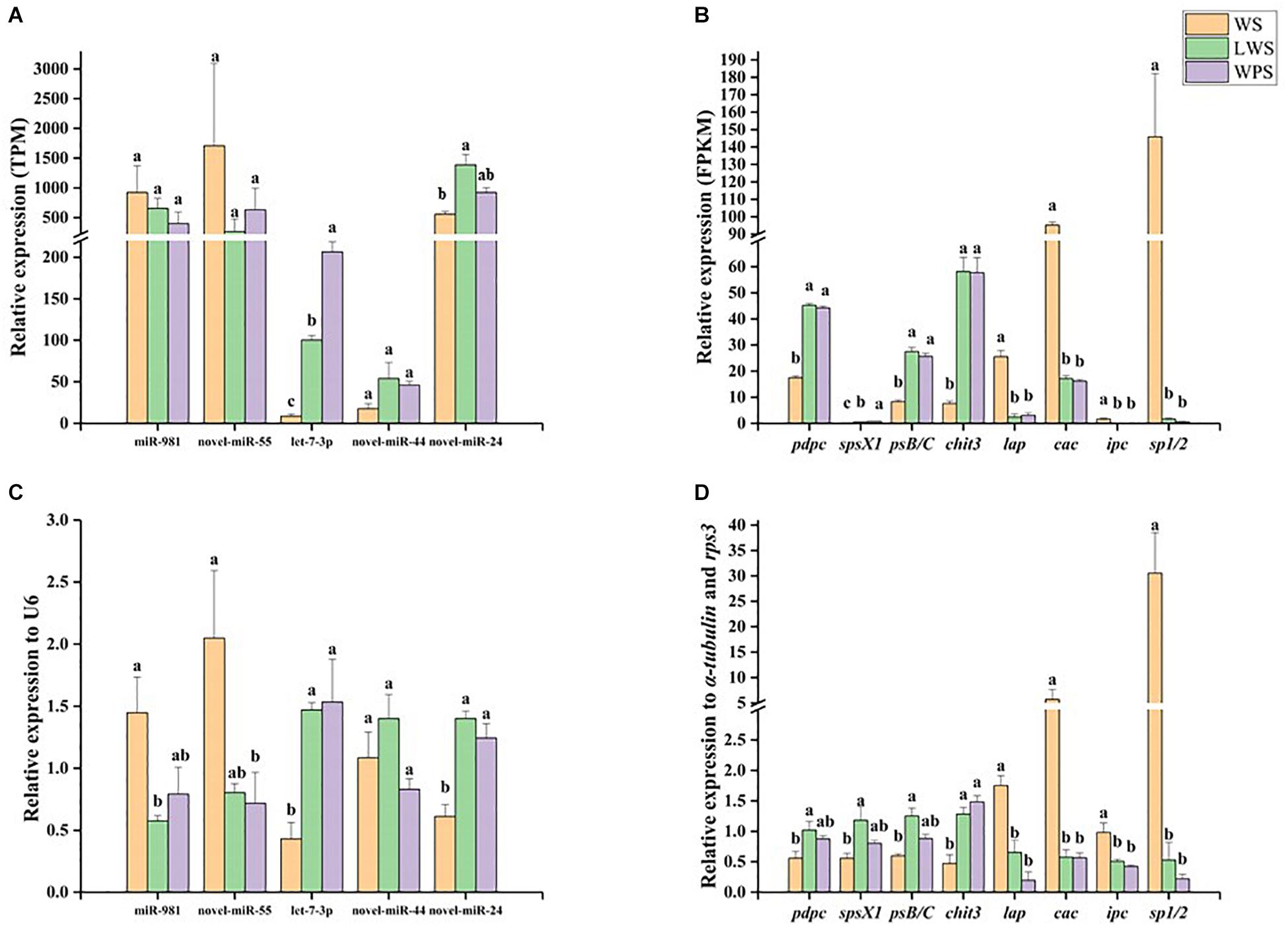
Figure 3. The expression patterns of differentially expressed miRNAs and their predicted differentially expressed mRNA targets were analyzed by quantitative real-time PCR (qPCR) in wandering stage (WS), late wandering stage (LWS, the body contraction just before pupariation) and white puparium stage (WPS) of Bactrocera dorsalis. The results of Illumina sequencing also showed here. They included (A) relative expression of five miRNAs in Illumina sequencing, (B) relative expression of eight targets in Illumina sequencing, (C) relative expression of five miRNAs in qPCR, (D) relative expression of eight targets in qPCR. The qPCR and Illumina sequencing results were analyzed with one-way analysis of variance (ANOVA), and the means were separated with Tukey’s HSD post hoc test (P < 0.05). Error bars show the SE. Different letters indicate statistically significant difference among the three stages (P < 0.05).
Discussion
The characteristic role of miRNAs is the post-transcriptional regulation of gene expression that occurs following duplex formation between a transcript and a complementary miRNA, causing subsequent degradation of target mRNAs or inhibition of translational machinery (Bartel, 2009; Belles, 2017). Previous studies have reported insect miRNAs participating in metamorphosis by regulating genes such as the gene encoding the JH-regulated serine protease JHA15 (Lucas et al., 2015), the ecdysone signaling pathway E75 gene (Peng et al., 2017), and chitin synthase (Li T. et al., 2017). In the present study, we obtained miRNAs at the three stages of metamorphosis development and detected five highly expressed miRNAs and their eight potential target genes, which may participate in the regulation of metamorphosis development in B. dorsalis.
In this study, we identified B. dorsalis miRNAs at three stages of metamorphosis development, WS, LWS and WPS, using deep sequencing. We detected 249 miRNAs, including 184 known miRNAs and 65 novel miRNAs. This result is comparable to the findings of Tariq et al. (2016), who reported 172 known and 78 novel miRNAs from the testes of fully mature, immature, and middle-aged adult flies of B. dorsalis. Our data shows a small difference in novel miRNAs when compared with the report by Peng et al. (2016), in which data analysis revealed 183 known and 120 novel miRNAs from the whole body of females and males, the ovaries and the testes of B. dorsalis. Our data has detected significantly more miRNAs than the early report by Huang et al. (2014), in which only 123 known and 60 novel miRNAs were identified among different developmental stages (eggs, larvae, pupae, and adults) in B. dorsalis. We were able to detect increased numbers of known and novel miRNAs in our study compared to Huang et al. (2014) because of the availability of the whole genome sequence (WGS) of B. dorsalis, and compared with Peng et al. (2016) because of the upgraded miRBASE database.
When pairwise comparison of the DEMsbetween the three developmental stages (WS vs. LWS and LWS vs. WPS) was carried out, 19 miRNAs were found which eight miRNAs showed relative high expression levels. Highly expressed DEMs are of interest, for example, Reynolds et al. (2017) selected high abundance DEMs from the transcriptome data and other conditional miRNAs to study further. In our study, eight highly expressed levels DEMs, miR-2-3p, miR-282-5p, let-7a-3p, miR-981, miR-100, Bdo-novel-mir-24, Bdo-novel-mir-44 and Bdo-novel-mir-55, were detected during pupariation of B. dorsalis, which indicates that they may have regulatory functions. Previous studies have discussed these known DEMs in other insects. The miR-2 family targets awd and fng to regulate wing morphogenesis in the holometabolous species, B. mori (Ling et al., 2015) and regulates insect metamorphosis by controlling the JH signaling pathway gene Kr-h1 in the hemimetabolous species, Blattella germanica (Lozano et al., 2015). miR-282 regulates the nervous system-specific adenylate cyclase (rutabaga) to control the viability, longevity, and egg production of Drosophila melanogaster (Vilmos et al., 2013), and has been shown by Solexa sequencing to be differentially expressed between foraging and dancing honey bees, Apis mellifera, and to target the genes associated with kinases, neural function, synaptotagmin, and energy (Li et al., 2012). miR-958 inhibits Toll signaling and Drosomycin expression by direct targeting of Toll and Dif in D. melanogaster (Li S. et al., 2017). miR-100 regulates wing morphogenesis and may be related to the Broad complex gene, an early 20E response gene in the ecdysteroid cascade, in B. germanica (Rubio and Belles, 2013). Therefore, the eight DEMs, combined with their predicted target mRNAs, may be associated with the regulation of metamorphosis.
Using the miRanda and RNAhybrid software packages, the target genes of the DEMs detected during metamorphosis development were predicted. There were 12 and 4 DEMs with predicted target genes in WS vs. LWS and LWS vs. WPS, respectively. Although some DEMs were not predicted to have mRNA target genes, this does not mean that they have no regulatory role, and they may work through other regulatory factors (such as long non-coding RNAs and circular RNAs) to influence pupariation. Braconi et al. (2011) showed that miRNA-29 can regulate expression of the long non-coding RNA gene MEG3, and Ebbesen et al. (2016) observed that circular RNAs can function as miRNA sponges acting as protein decoys, or transcriptional regulators of mRNA. When the miRNAs were differentially expressed during metamorphosis development, their predicted mRNA target genes were also differentially expressed, indicating that the miRNAs may participate in this progress, especially when the DEMs and their predicted target genes were observed in a miRNA–mRNA negative regulation pattern, as this is the typical pattern seen between miRNAs and their target genes. For instance, miR-71 and miR-263 negatively regulate their target genes chitin synthase 1 and chitinase 10, respectively, to control locust molting in L. migratoria (Yang et al., 2016); miRNA-252-5p negatively controls the target gene Abelson interacting protein to decrease the protein levels of cyclins A and B, controlling the cell cycle in Drosophila (Lim et al., 2018). Similarly, miR-9a directly negatively regulates the target gene adenylyl cyclase 2 in L. migratoria (Guo X. et al., 2018). There were no DEMs and corresponding DEGs in the comparison of LWS vs. WPS once this negative regulation pattern was considered. This is probably because the two stages are so close that there are few differences between LWS and WPS, and there were few differentially expressed genes in the RNA-seq analysis for the comparison of the two stages (Chen E. H. et al., 2018). The subsequent qPCR results also showed that there were no significant differences between the verified miRNAs and mRNAs at the two stages.
To further understand the potential function of the DEMs during pupariation, their predicted target genes were annotated. A GO classification map was generated by gene enrichment analysis of the DEMs and their predicted target DEGs. This classification map could be useful for further study into how the DEMs are related to metamorphosis development. Considering of previously reported genes related to insect pupariation, the DEGs were expected to be mapped to the genes involved in 20E, JH, chitin pathways, cuticular proteins and etc. The RNAi-mediated knockdown of ecdysone synthesis pathway genes (CYP306A1 and CYP314A1) caused lethality and slowed down ecdysis during Leptinotarsa decemlineata nymphal stages (Kong et al., 2014; Wan et al., 2014). Silencing of both E74 isoforms (E74A and E74B) caused failure of ecdysis, and most beetles after LdE74 RNAi remained as prepupae while a few larvae finally became deformed pupae, with shortened antennae and legs in L. decemlineata (Xu et al., 2018). RNA interference-mediated knockdown of juvenile hormone acid O-methyltransferase (JHAMT) (Minakuchi et al., 2008) or Kr-h1 (Minakuchi et al., 2009) in the Tribolium castaneum larval stage both resulted in precocious larval–pupal metamorphosis. In Spodoptera exigua, injection of chitinase dsRNA at the larval–pupal stage caused lethal phenotype with visible pupal cuticle under the old larval cuticle, indicating that chitinase participated in the degradation of old cuticle during insect pupariation (Zhang et al., 2012). When silencing chitin synthase 1 in third-instar larvae of B. dorsalis, the larvae were trapped their old cuticle and died without pupation (Yang et al., 2013). In a recently published paper about B. dorsalis, some key DEGs were annotated in RNA-seq analysis during pupariation, for example, 20E biosynthesis (CYP314a1) and signaling pathway genes (E74), JH synthesis (farnesoic acid O-methyl transferase), chitinases (chitinase 3 and 5), and serine proteases (Chen E. H. et al., 2018). Unexpectedly, the predicted target DEGs in the current study were largely unsuccessfully mapped to those genes above. One of the reasons might be that some genes related to insect pupariation are not regulated by miRNAs. A previous study pointed out that only about 30% of human genes are regulated by microRNAs (Lewis et al., 2005). Namely, the majority of organism genes are not regulated by microRNAs. In addition, other crucial genes were not significant different (|log2Ratio| < 1) in expression during pupariation, for example, 20E biosynthesis (CYP315a1), JH synthesis (3-Hydroxy-3-methylglutaryl-CoA reductase) and signaling pathway genes (Met and Kr-h1), chitin synthases (glucose-6-phosphate isomerase) (Chen E. H. et al., 2018). Furthermore, the miRNAs regulation is temporal during growth and development stages (Belles, 2017). Nevertheless, serine proteases and chitinase reported in this process were annotated in predicted target genes of DEMs. Among the Nr annotation of the predicted DEGs of the five highly expressed DEMs, two of the genes, spsX1 (Paskewitz et al., 1999) and sp1/2, were related to serine proteases, which have been suggested to be involved in integument remodeling during metamorphosis (Gu et al., 2013; Liu H. W. et al., 2017). However, these two genes showed contrasting changes in expression levels (spsX1 was up-regulated and sp1/2 was down-regulated), indicating that the serine proteases may be regulated by different miRNAs. During metamorphosis, high expression levels of serine proteases in the epidermis may be required to break down the cuticle proteins and release the chitin so that it can be degraded (Gu et al., 2013). Chitinase 3, a member of the chitinase family, is responsible for chitin degradation. During pupariation, the chitin needs to be degraded and replaced by newly synthesized chitin. Here, we found that Bdo-novel-mir-55 probably targets chitinase 3 to control B. dorsalis metamorphosis. Chitinase can be regulated by miRNAs and the regulation has been confirmed in previous studies. For example, miR-263 mediates the gene encoding chitinase 10 in L. migratoria (Yang et al., 2016), and miR-24 targets chitinase to regulate the molting process of Helicoverpa armigera (Agrawal et al., 2013). Furthermore, the genes protein skeletor (Sengupta et al., 2016) and lap (Rabossi et al., 2000) have been reported to be involved in metamorphosis. As the majority of these predicted target genes are related to the process of metamorphosis development, in combination with the qPCR results, we hypothesize that miR-981 may target pdpc, Bdo-novel-mir-55 potentially regulates spsX1, psB/C and chit3, let-7a-3p possibly targets lap, and Bdo-novel-mir-24 may regulate ipc and sp1/2 to govern the metamorphosis of B. dorsalis. Undoubtedly, the relationship of regulation between DEMs and predicted target genes deserves validating further. The miRNAs-mediated mechanism underlying pupariation is complex and the systematic interpretation deserves further work (i.e., definition of crucial development node of pupariation).
Conclusion
In this study, HiSeq combined with RNA-seq analysis was performed to elucidate the molecular regulatory mechanisms of small RNAs in B. dorsalis during pupariation. One hundred twenty-eight miRNAs were differentially expressed, which indicates that these DEMs may play important regulatory roles in the process of pupariation. The numbers of up-regulated and down-regulated DEMs were approximately even. There were eight miRNAs showing relative high expression levels (>50 TPM), of which five DEMs had predicted DEGs according to the expected miRNA–mRNA negative regulation pattern using the DESeq R algorithm, and four pairs of DEMs and their predicted DEGs were confirmed by qPCR. Of these four miRNAs, two miRNAs were down-regulated, with miR-981 potentially targeting pdpc and Bdo-novel-mir-55 potentially regulating spsX1, psB/C and chit3, and the other two miRNAs were up-regulated, with let-7a-3p possibly controlling lap and Bdo-novel-mir-24 perhaps managing ipc and sp1/2. Overall, our study provides basic miRNA expression data that will aid the exploration of the regulatory mechanisms of metamorphosis by miRNAs in B. dorsalis. Verification of the regulatory relationships between these miRNAs and their target genes is needed to further uncover the possible roles of these miRNAs in B. dorsalis pupariation.
Author Contributions
QZ, WD, E-HC, J-ZN, and GS contributed conception and design of the study. QZ and DP organized the database. QZ and J-JW performed the statistical analysis. QZ wrote the first draft of the manuscript. QZ, WD, J-ZN, J-JW, and GS wrote sections of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 31872031 and 31672030), the Chongqing Research Program of Basic Research and Frontier Technology (Grant No. cstc2018jcyjAX0285), the Foundation Project of Southwest University (Grant No. SWU114049), and the earmarked fund for the Modern Agroindustry (Citrus) Technology Research System of China (Grant No. CARS-26).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We sincerely thank Yoonseong Park (Department of Entomology, Kansas State University, Manhattan, KS, United States) for useful comments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2019.00301/full#supplementary-material
FIGURE S1 | GO classification analysis for predicted differentially expressed genes (DEGs) of differentially expressed miRNAs (DEMs) in WS vs. LWS. (All secondary level was under the backgrounds of the predicted DEGs and the predicted target genes of DEMs).
FIGURE S2 | COG classification analysis for predicted differentially expressed genes (DEGs) of differentially expressed miRNAs (DEMs) in WS vs. LWS.
FIGURE S3 | KEGG classification analysis for predicted differentially expressed genes (DEGs) of differentially expressed miRNAs (DEMs) in WS vs. LWS.
TABLE S1 | Primers of miRNA and mRNA used in our study.
TABLE S2 | Overview of sRNA sequencing data.
TABLE S3 | Mapping information of unannotated reads with reference genome.
TABLE S4 | List of total identification, respective and overlapped known and novel miRNAs in three developmental stages of Bactrocera dorsalis.
TABLE S5 | List of differentially expressed miRNAs in WS vs. LWS and LWS vs. WPS of Bactrocera dorsalis.
TABLE S6 | List of 19 differentially expressed miRNAs in the pupariation of Bactrocera dorsalis.
TABLE S7 | List of annotation of differentially expressed miRNAs in WS vs. LWS and LWS vs. WPS of Bactrocera dorsalis.
TABLE S8 | List of differentially expressed miRNAs with changed predicted target mRNAs in WS vs. LWS of Bactrocera dorsalis.
TABLE S9 | List of miRNAs and predicted target mRNAs coinciding with the miRNA–mRNA negative regulation pattern in WS vs. LWS of Bactrocera dorsalis.
Footnotes
- ^ http://www.mirbase.org/
- ^ http://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi
- ^ http://www.ncbi.nlm.nih.gov/tools/primer-blast/
References
Agrawal, N., Sachdev, B., Rodrigues, J., Sree, K. S., and Bhatnagar, R. K. (2013). Development associated profiling of chitinase and microRNA of Helicoverpa armigera identified chitinase repressive microRNA. Sci. Rep. 3:2292. doi: 10.1038/srep02292
Bartel, D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. doi: 10.1016/j.cell.2009.01.002
Belles, X. (2017). MicroRNAs and the evolution of insect metamorphosis. Annu. Rev. Entomol. 62, 111–125. doi: 10.1146/annurev-ento-031616-034925
Braconi, C., Kogure, T., Valeri, N., Huang, N., Nuovo, G., Costinean, S., et al. (2011). microRNA-29 can regulate expression of the long non-coding RNA gene MEG3 in hepatocellular cancer. Oncogene 30, 4750–4756. doi: 10.1038/onc.2011.193
Chen, E. H., Hou, Q. L., Dou, W., Wei, D. D., Yue, Y., Yang, R. L., et al. (2018). RNA-seq analysis of gene expression changes during pupariation in Bactrocera dorsalis (Hendel) (Diptera: Tephritidae). BMC Genomics 19:693. doi: 10.1186/s12864-018-5077-z
Chen, J., Li, T. C., Pang, R., Yue, X. Z., Hu, J., and Zhang, W. Q. (2018). Genome-wide screening and functional analysis reveal that the specific microRNA nlu-miR-173 regulates molting by targeting Ftz-F1 in Nilaparvata lugens. Front. Physiol. 9:1854. doi: 10.3389/fphys.2018.01854
Dubrovsky, E. B. (2005). Hormonal cross talk in insect development. Trends Endocri. Met. 16, 6–11. doi: 10.1016/j.tem.2004.11.003
Ebbesen, K. K., Kjems, J., and Hansen, T. B. (2016). Circular RNAs: Identification, biogenesis and function. Biochim. Biophys. Acta 1859, 163–168. doi: 10.1016/j.bbagrm.2015.07.007
Etebari, K., Afrad, M. H., Tang, B., Silva, R., Furlong, M. J., and Asgari, S. (2018). Involvement of microRNA miR-2b-3p in regulation of metabolic resistance to insecticides in Plutella xylostella. Insect Mol. Biol. 27, 478–491. doi: 10.1111/imb.12387
Fahlgren, N., Howell, M. D., Kasschau, K. D., Chapman, E. J., Sullivan, C. M., Cumbie, J. S., et al. (2007). High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS One 2:e219. doi: 10.1371/journal.pone.0000219
Friedlander, M. R., Mackowiak, S. D., Li, N., Chen, W., and Rajewsky, N. (2012). miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 40, 37–52. doi: 10.1093/nar/gkr688
Garbuzov, A., and Tatar, M. (2014). Hormonal regulation of Drosophila microRNA let-7 and miR-125 that target innate immunity. Fly 4, 306–311. doi: 10.4161/fly.4.4.13008
Gu, J., Huang, L. X., Gong, Y. J., Zheng, S. C., Liu, L., Huang, L. H., et al. (2013). De novo characterization of transcriptome and gene expression dynamics in epidermis during the larval-pupal metamorphosis of common cutworm. Insect Biochem. Mol. Biol. 43, 794–808. doi: 10.1016/j.ibmb.2013.06.001
Guo, S. Y., Wu, W. M., Li, S. Y., Liu, Y., Ruan, Z. F., Ye, M. Q., et al. (2018). 20-Hydroxyecdysone-upregulated proteases involved in Bombyx larval fat body destruction. Insect Mol. Biol. 27, 724–738. doi: 10.1111/imb.12511
Guo, X., Ma, Z., Du, B., Li, T., Li, W., Xu, L., et al. (2018). Dop1 enhances conspecific olfactory attraction by inhibiting miR-9a maturation in locusts. Nat. Commun. 9:1193. doi: 10.1038/s41467-018-03437-z
Hellemans, J., Mortier, G., De Paepe, A., Speleman, F., and Vandesompele, J. (2007). qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 8:R19. doi: 10.1186/gb-2007-8-2-r19
Huang, Y., Dou, W., Liu, B., Wei, D., Liao, C. Y., Smagghe, G., et al. (2014). Deep sequencing of small RNA libraries reveals dynamic expression patterns of microRNAs in multiple developmental stages of Bactrocera dorsalis. Insect Mol. Biol. 23, 656–667. doi: 10.1111/imb.12111
Jan, Z., and Fraenkel, G. (2010). The mechanism of puparium formation in flies. J. Exp. Zool. Part B 179, 315–323. doi: 10.1002/jez.1401790304
Jin, T., Zeng, L., Lin, Y., Lu, Y., and Liang, G. (2011). Insecticide resistance of the oriental fruit fly, Bactrocera dorsalis (Hendel) (Diptera: Tephritidae), in mainland China. Pest Manag. Sci. 67, 370–376. doi: 10.1002/ps.2076
Khan, H. A. A., and Akram, W. (2018). Trichlorfon and spinosad resistance survey and preliminary determination of the resistance mechanism in Pakistani field strains of Bactrocera dorsalis. Sci. Rep. 8:11223. doi: 10.1038/s41598-018-29622-0
Kong, Y., Liu, X. P., Wan, P. J., Shi, X. Q., Guo, W. C., and Li, G. Q. (2014). The P450 enzyme Shade mediates the hydroxylation of ecdysone to 20-hydroxyecdysone in the colorado potato beetle, Leptinotarsa decemlineata. Insect Mol. Biol. 23, 632–643. doi: 10.1111/imb.12115
Lewis, B. P., Burge, C. B., and Bartel, D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20. doi: 10.1016/j.cell.2004.12.035
Li, L., Liu, F., Li, W., Li, Z., Pan, J., Yan, L., et al. (2012). Differences in microRNAs and their expressions between foraging and dancing honey bees, Apis mellifera L. J. Insect Physiol. 58, 1438–1443. doi: 10.1016/j.jinsphys.2012.08.008
Li, S., Li, Y., Shen, L., Jin, P., Chen, L., and Ma, F. (2017). miR-958 inhibits toll signaling and drosomycin expression via direct targeting of toll and dif in Drosophila melanogaster. Am. J. Physiol. Cell Physiol. 312, C103–C110. doi: 10.1152/ajpcell.00251.2016
Li, T., Chen, J., Fan, X., Chen, W., and Zhang, W. (2017). MicroRNA and dsRNA targeting chitin synthase A reveal a great potential for pest management of the hemipteran insect Nilaparvata lugens. Pest Manag. Sci. 73, 1529–1537. doi: 10.1002/ps.4492
Lim, D. H., Lee, S., Han, J. Y., Choi, M. S., Hong, J. S., Seong, Y., et al. (2018). Ecdysone-responsive microRNA-252-5p controls the cell cycle by targeting abi in Drosophila. FASEB J. 32, 4519–4533. doi: 10.1096/fj.201701185RR
Ling, L., Ge, X., Li, Z., Zeng, B., Xu, J., Chen, X., et al. (2015). MiR-2 family targets awd and fng to regulate wing morphogenesis in Bombyx mori. RNA Biol. 12, 742–748. doi: 10.1080/15476286.2015.1048957
Liu, H. W., Wang, L. L., Meng, Z., Tang, X., Li, Y. S., Xia, Q. Y., et al. (2017). A clip domain serine protease involved in moulting in the silkworm, Bombyx mori: cloning, characterization, expression patterns and functional analysis. Insect Mol. Biol. 26, 507–521. doi: 10.1111/imb.12312
Liu, S., Li, K., Gao, Y., Liu, X., Chen, W., Ge, W., et al. (2017). Antagonistic actions of juvenile hormone and 20-hydroxyecdysone within the ring gland determine developmental transitions in Drosophila. Proc. Natl. Acad. Sci. U.S.A. 115, 139–144. doi: 10.1073/pnas.1716897115
Liu, Z., Ling, L., Xu, J., Zeng, B., Huang, Y., Shang, P., et al. (2017). MicroRNA-14 regulates larval development time in Bombyx mori. Insect Biochem. Mol. Biol. 93, 57–65. doi: 10.1016/j.ibmb.2017.12.009
Lozano, J., Montanez, R., and Belles, X. (2015). MiR-2 family regulates insect metamorphosis by controlling the juvenile hormone signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 112, 3740–3745. doi: 10.1073/pnas.1418522112
Lucas, K. J., Zhao, B., Roy, S., Gervaise, A. L., and Raikhel, A. S. (2015). Mosquito-specific microRNA-1890 targets the juvenile hormone-regulated serine protease JHA15 in the female mosquito gut. RNA Biol. 12, 1383–1390. doi: 10.1080/15476286.2015.1101525
Mao, X. Z., Cai, T., Olyarchuk, J. G., and Wei, L. P. (2005). Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793. doi: 10.1093/bioinformatics/bti430
Minakuchi, C., Namiki, T., and Shinoda, T. (2009). Kruppel homolog 1, an early juvenile hormone-response gene downstream of Methoprene-tolerant, mediates its anti-metamorphic action in the red flour beetle Tribolium castaneum. Dev. Biol. 325, 341–350. doi: 10.1016/j.ydbio.2008.10.016
Minakuchi, C., Namiki, T., Yoshiyama, M., and Shinoda, T. (2008). RNAi-mediated knockdown of juvenile hormone acid O-methyltransferase gene causes precocious metamorphosis in the red flour beetle Tribolium castaneum. FEBS J. 275, 2919–2931. doi: 10.1111/j.1742-4658.2008.06428.x
Paskewitz, S. M., Reese-Stardy, S., and Gorman, M. J. (1999). An easter-like serine protease from Anopheles gambiae exhibits changes in transcript abundance following immune challenge. Insect Mol. Biol. 8, 329–337. doi: 10.1046/j.1365-2583.1999.83124.x
Peng, W., Tariq, K., Xie, J., and Zhang, H. (2016). Identification and characterization of sex-biased microRNAs in Bactrocera dorsalis (Hendel). PLoS One 11:e0159591. doi: 10.1371/journal.pone.0159591
Peng, W., Zheng, W. W., Tariq, K., Yu, S. N., and Zhang, H. Y. (2017). MicroRNA Let-7 targets the ecdysone signaling pathway E75 gene to control larval-pupal development in Bactrocera dorsalis. Insect Sci. 26, 229–239. doi: 10.1111/1744-7917.12542
Rabossi, A., Acion, L., and Quesada-Allue, L. A. (2000). Metamorphosis-associated proteolysis in Ceratitis capitata. Entomol. Exp. Appl. 94, 57–65. doi: 10.1046/j.1570-7458.2000.00604.x
Reynolds, J. A., Peyton, J. T., and Denlinger, D. L. (2017). Changes in microRNA abundance may regulate diapause in the flesh fly, Sarcophaga bullata. Insect Biochem. Mol. Biol. 84, 1–14. doi: 10.1016/j.ibmb.2017.03.002
Rubio, M., and Belles, X. (2013). Subtle roles of microRNAs let-7, miR-100 and miR-125 on wing morphogenesis in hemimetabolan metamorphosis. J. Insect Physiol. 59, 1089–1094. doi: 10.1016/j.jinsphys.2013.09.003
Sengupta, S., Rath, U., Yao, C., Zavortink, M., Wang, C., Girton, J., et al. (2016). Digitor/dASCIZ has multiple roles in Drosophila development. PLoS One 11:e0166829. doi: 10.1371/journal.pone.0166829
Shen, G. M., Dou, W., Niu, J. Z., Jiang, H. B., Yang, W. J., Jia, F. X., et al. (2011). Transcriptome analysis of the oriental fruit fly (Bactrocera dorsalis). PLoS One 6:e29127. doi: 10.1371/journal.pone.0029127
Shen, G. M., Jiang, H. B., Wang, X. N., and Wang, J. J. (2010). Evaluation of endogenous references for gene expression profiling in different tissues of the oriental fruit fly Bactrocera dorsalis (Diptera: Tephritidae). BMC Mol. Biol. 11:76. doi: 10.1186/1471-2199-11-76
Song, J. S., Li, W. W., Zhao, H. H., Gao, L. L., Fan, Y. N., and Zhou, S. T. (2018). The microRNAs let-7 and miR-278 regulate insect metamorphosis and oogenesis by targeting the juvenile hormone early-response gene Kruppel-homolog 1. Development 145:dev170670. doi: 10.1242/dev.170670
Tariq, K., Metzendorf, C., Peng, W., Sohail, S., and Zhang, H. (2016). miR-8-3p regulates mitoferrin in the testes of Bactrocera dorsalis to ensure normal spermatogenesis. Sci. Rep. 6:22565. doi: 10.1038/srep22565
Vilmos, P., Bujna, A., Szuperak, M., Havelda, Z., Varallyay, E., Szabad, J., et al. (2013). Viability, longevity, and egg production of Drosophila melanogaster are regulated by the miR-282 microRNA. Genetics 195, 469–480. doi: 10.1534/genetics.113.153585
Wan, P. J., Jia, S., Li, N., Fan, J. M., and Li, G. Q. (2014). The putative Halloween gene phantom involved in ecdysteroidogenesis in the white-backed planthopper Sogatella furcifera. Gene 548, 112–118. doi: 10.1016/j.gene.2014.07.023
Wang, H. X., Jin, L., Peng, T., Zhang, H., Chen, Q., and Hua, Y. (2014). Identification of cultivable bacteria in the intestinal tract of Bactrocera dorsalis from three different populations and determination of their attractive potential. Pest Manag. Sci. 70, 80–87. doi: 10.1002/ps.3528
Wang, Z. Z., Ye, X. Q., Shi, M., Li, F., Wang, Z. H., Zhou, Y. N., et al. (2018). Parasitic insect-derived miRNAs modulate host development. Nat. Commun. 9:2205. doi: 10.1038/s41467-018-04504-1
Wei, D., Li, H. M., Yang, W. J., Wei, D. D., Dou, W., Huang, Y., et al. (2015). Transcriptome profiling of the testis reveals genes involved in spermatogenesis and marker discovery in the oriental fruit fly, Bactrocera dorsalis. Insect Mol. Biol. 24, 41–57. doi: 10.1111/imb.12134
Xu, Q. Y., Meng, Q. W., Deng, P., Fu, K. Y., Guo, W. C., and Li, G. Q. (2018). Requirement of Leptinotarsa decemlineata gene within the 74EF puff for larval-pupal metamorphosis and appendage growth. Insect Mol. Biol. 27, 439–453. doi: 10.1111/imb.12384
Yang, C., Lin, X. W., and Xu, W. H. (2017). Cathepsin L participates in the remodeling of the midgut through dissociation of midgut cells and activation of apoptosis via caspase-1. Insect Biochem. Mol. Biol. 82, 21–30. doi: 10.1016/j.ibmb.2017.01.010
Yang, M., Wang, Y., Jiang, F., Song, T., Wang, H., Liu, Q., et al. (2016). miR-71 and miR-263 jointly regulate target genes chitin synthase and chitinase to control locust molting. PLoS Genet. 12:e1006257. doi: 10.1371/journal.pgen.1006257
Yang, W. J., Xu, K. K., Cong, L., and Wang, J. J. (2013). Identification, mRNA expression, and functional analysis of chitin synthase 1 gene and its two alternative splicing variants in oriental fruit fly, Bactrocera dorsalis. Int. J Biol. Sci. 9, 331–342. doi: 10.7150/ijbs.6022
Yao, Q., Zhang, D., Tang, B., Chen, J., Lu, L., and Zhang, W. (2010). Identification of 20-hydroxyecdysone late-response genes in the chitin biosynthesis pathway. PLoS One 5:e14058. doi: 10.1371/journal.pone.0014058
Yin, C., Li, M., Hu, J., Lang, K., Chen, Q., Liu, J., et al. (2018). The genomic features of parasitism, polyembryony and immune evasion in the endoparasitic wasp Macrocentrus cingulum. BMC Genomics 19:420. doi: 10.1186/s12864-018-4783-x
Zhang, D. W., Chen, J., Yao, Q., Pan, Z. Q., Chen, J., and Zhang, W. Q. (2012). Functional analysis of two chitinase genes during the pupation and eclosion stages of the beet armyworm Spodoptera exigua by RNA interference. Arch. Insect Biochem. Physiol. 79, 220–234. doi: 10.1002/arch.21018
Zhang, T., Song, W., Li, Z., Qian, W., Wei, L., Yang, Y., et al. (2018). Kruppel homolog 1 represses insect ecdysone biosynthesis by directly inhibiting the transcription of steroidogenic enzymes. Proc. Natl. Acad. Sci. U.S.A. 115, 3960–3965. doi: 10.1073/pnas.1800435115
Zhang, Y., Feng, K., Hu, J., Shi, L., Wei, P., Xu, Z., et al. (2018). A microRNA-1 gene, tci-miR-1-3p, is involved in cyflumetofen resistance by targeting a glutathione S-transferase gene, TCGSTM4, in Tetranychus cinnabarinus. Insect Mol. Biol. 27, 352–364. doi: 10.1111/imb.12375
Keywords: miRNAs, Bactrocera dorsalis, pupariation, metamorphosis, gene expression
Citation: Zhang Q, Dou W, Pan D, Chen E-H, Niu J-Z, Smagghe G and Wang J-J (2019) Genome-Wide Analysis of MicroRNAs in Relation to Pupariation in Oriental Fruit Fly. Front. Physiol. 10:301. doi: 10.3389/fphys.2019.00301
Received: 07 December 2018; Accepted: 07 March 2019;
Published: 22 March 2019.
Edited by:
Xanthe Vafopoulou, York University, CanadaReviewed by:
Pin-Jun Wan, China National Rice Research Institute (CAAS), ChinaYou Ming Hou, Fujian Agriculture and Forestry University, China
Copyright © 2019 Zhang, Dou, Pan, Chen, Niu, Smagghe and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guy Smagghe, Z3V5LnNtYWdnaGVAdWdlbnQuYmU= Jin-Jun Wang, d2FuZ2ppbmp1bkBzd3UuZWR1LmNu