 Paul M. Coen
Paul M. Coen Robert V. Musci
Robert V. Musci J. Matthew Hinkley
J. Matthew Hinkley Benjamin F. Miller
Benjamin F. Miller- 1Translational Research Institute for Metabolism and Diabetes, Florida Hospital, Orlando, FL, United States
- 2Department of Health and Exercise Science, Colorado State University, Fort Collins, CO, United States
- 3Aging and Metabolism Research Program, Oklahoma Medical Research Foundation, Oklahoma City, OK, United States
Sarcopenia is the loss of muscle mass, strength, and physical function that is characteristic of aging. The progression of sarcopenia is gradual but may be accelerated by periods of muscle loss during physical inactivity secondary to illness or injury. The loss of mobility and independence and increased comorbidities associated with sarcopenia represent a major healthcare challenge for older adults. Mitochondrial dysfunction and impaired proteostatic mechanisms are important contributors to the complex etiology of sarcopenia. As such, interventions that target improving mitochondrial function and proteostatic maintenance could mitigate or treat sarcopenia. Exercise is currently the only effective option to treat sarcopenia and does so, in part, by improving mitochondrial energetics and protein turnover. Exercise interventions also serve as a discovery tool to identify molecular targets for development of alternative therapies to treat sarcopenia. In summary, we review the evidence linking mitochondria and proteostatic maintenance to sarcopenia and discuss the therapeutic potential of interventions addressing these two factors to mitigate sarcopenia.
Introduction
The Centers for Disease Control (CDC) projects that the proportion of adults older than 65 years of age in the US population will grow from 12.4 to 19.6% reflecting the aging baby boom generation and declining birth rates in the United States. Furthermore, the population of persons older than 80 years is expected to more than double from 9.3 million in 2000 to 19.5 million in 2030 (Centers for Disease Control and Prevention (CDC), 2003). This aging phenomenon is not unique to the United States since the number of individuals aged 65 years and older is expected to nearly double from 6.9% of the world population in 2000 to 12.0% by 2030 (Beard et al., 2016). Revisions to such predictions even suggest that by 2050, the population of those over the age 65 in the United States will be up to 108 million, 25.8% of the predicted population (Olshansky et al., 2009).
Since the aged population is increasing globally, the prevalence of sarcopenia, the age-related loss of skeletal muscle mass and function, is likely to increase as well. Global prevalence of sarcopenia is difficult to measure in part due to changing consensus of what constitutes the diagnosis of sarcopenia. In 1998, Baumgartner et al. defined sarcopenia as the age-associated loss of skeletal muscle mass two standard deviations below a healthy population (Baumgartner et al., 1998). Based strictly on skeletal muscle mass loss, Baumgartner and colleagues estimated that 24% of individuals less than 70 years of age have sarcopenia while 50% of those over 80 years of age had sarcopenia (Baumgartner et al., 1998). More recent analyses find widely discrepant disease incidence with NHANES data collected between 1999 and 2004 reporting that 27.8 and 19.3% of men and women at least 60 years of age were sarcopenic (Janssen et al., 2002). Other estimates are as high as 35.4 and 52.5% for women over 60 and 80 years of age, respectively, and 75.5 and 88.1% for men over 60 and 80 years of age (Batsis et al., 2014). Surprisingly, there is a paucity of data that detail the economic burden of sarcopenia, although an analysis over a decade old found that the healthcare cost of sarcopenia in the United States was an estimated $18.5 billion per year (Janssen et al., 2004).
Skeletal muscle is the largest organ in the human body and plays a key role in posture and capacity for locomotion, as well as serving as a bona fide endocrine organ (Pedersen and Febbraio, 2012). As such, skeletal muscle dysfunction has detrimental effects on many aspects of human health for older adults. Epidemiological studies have found that sarcopenia increases the overall risk for mortality (Landi et al., 2013; Batsis et al., 2014). In part, this is because sarcopenia increases the risk of developing mobility disabilities, leading to impairment in activities of daily living by twofold (Janssen et al., 2002), and risk of falls by three times (Landi et al., 2013). The loss of muscle mass and function is not exclusive to postural/locomotor muscle groups, as myopathy of key inspiratory muscles also occurs with aging, resulting in respiratory failure (Kelley and Ferreira, 2017). The increased risk of disability from respiratory failure has led to greater hospitalization of older adults (Verissimo et al., 2015; Kelley and Ferreira, 2017). The combined effect of sarcopenia and hospitalization further exacerbates muscle dysfunction, as older adults do not adequately recover from bed rest (Suetta et al., 2009, 2013; Hvid et al., 2010), which contributes to reduced functionality and ambulation upon discharge, and leads to loss of independence, nursing home placement, and increased risks of falls (Fortinsky et al., 1999; Mithal et al., 2013). In addition to physical disability, sarcopenia contributes to the development of cardiovascular and metabolic diseases because of its involvement in substrate metabolism (Batsis et al., 2015) and as an endocrine organ (Pedersen and Febbraio, 2012).
The current consensus is that to diagnose sarcopenia, one should assess walking speed or grip strength and then examine appendicular lean mass if either is below a certain cutoff value (Cruz-Jentoft et al., 2010; Fielding et al., 2011; Morley et al., 2014). If muscle mass is lower than a healthy population cutoff value, sarcopenia is diagnosed. As such, low muscle mass continues to be an important component in the definition of sarcopenia. In 2016, the World Health Organization established an ICD-10 code for sarcopenia, which will spur the development of effective therapeutic strategies and increase the recognition of the importance of maintaining muscle mass and function with age for overall human health (Anker et al., 2016).
Resistance exercise continues to be the most effective intervention against sarcopenia (Landi et al., 2014). In addition, maintenance of physical activity can delay the progression of sarcopenia (Power et al., 2016a; Lazarus and Harridge, 2017). Despite the strong support for maintaining an active lifestyle, adherence to physical activity guidelines remains low. The traditional therapeutic focus of sarcopenia treatment is to target growth-related pathways to increase muscle mass. Here, we discuss the positives of these strategies, but also build a case for targeting mitochondrial bioenergetics as a way to maintain muscle mass and function with age as summarized in Figure 1. This review will cover the etiology of muscle loss, three basic characteristics of aging that may contribute to sarcopenia, current treatments targeting mass, and how targeting mitochondria rather than mass could mitigate basic mechanisms of aging to slow sarcopenia.
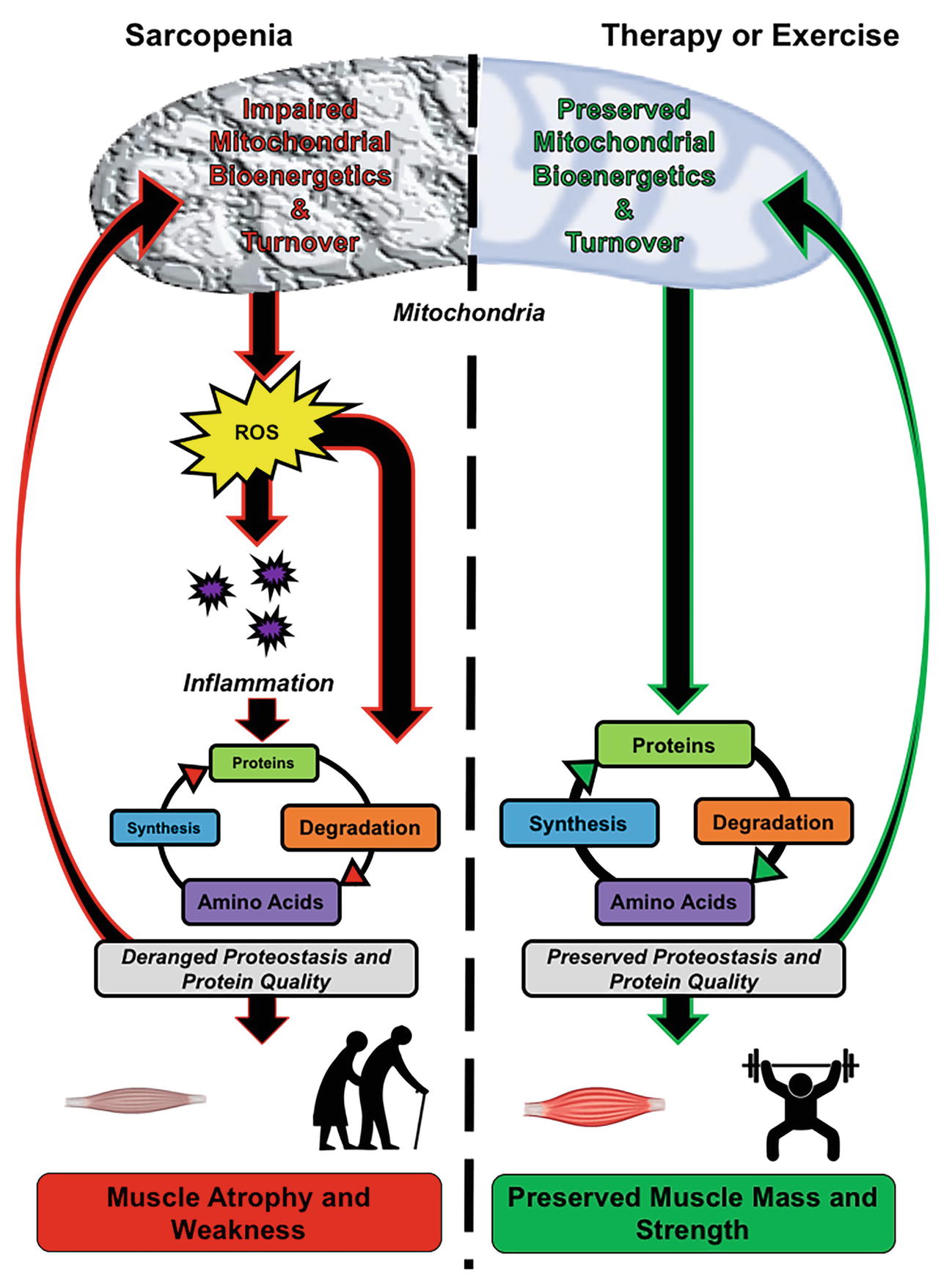
Figure 1. Role of mitochondrial bioenergetics and proteostasis in mediating skeletal muscle quality in older adults. Left panel, Sarcopenia is associated with mitochondrial dysfunction, which encompasses impaired bioenergetics and turnover. The impairment results in increased reactive oxygen species (ROS) generation and chronic low-grade inflammation, leading to impaired muscle proteostasis. The derangement in proteostasis impedes mitochondrial turnover, resulting in an accumulation of dysfunctional mitochondria and further exacerbation of organelle and tissue dysfunction. Right panel, Targeting mitochondrial bioenergetics and turnover by therapeutics and exercise impedes the age-associated rise in ROS and systemic inflammation, which results in the maintenance of muscle proteostasis. The maintained protein turnover allows for the removal of damaged proteins, such as dysfunctional mitochondria and damaged contractile proteins, while also synthesizing new functional proteins. Collectively, this leads to preservation of mitochondrial quality, muscle mass, and strength.
Mechanisms Leading to Loss of Strength and Function
Etiology of Muscle Loss
The etiology of sarcopenia is characterized by both slow, gradual loss of muscle mass over time that is propagated by acute periods of accelerated loss and poor nutrition (English and Paddon-Jones, 2010). Acute periods of muscle loss in older individuals is often met with an incomplete regain of muscle mass and strength, thus accelerating gradual sarcopenic progression (Suetta et al., 2009; White et al., 2015; Baehr et al., 2016). The inability to completely regain muscle mass and strength is common in both aging humans (Suetta et al., 2009) and animals (White et al., 2015; Baehr et al., 2016). This public health problem is particularly troublesome for older adults who comprise the majority of hospital patients in the United States (DeFrances et al., 2008; Fisher et al., 2010) and who may lose more muscle mass during bed rest (Paddon-Jones et al., 2004; Kortebein et al., 2007). Older adults do not adequately recover following bed rest without adequate rehabilitation (Suetta et al., 2009, 2013; Hvid et al., 2010), which likely contributes to their reduced functional status and ambulation upon discharge (Covinsky et al., 2003; Coker et al., 2015). It is important to target both the gradual and accelerated periods of muscle loss to mitigate the progression of sarcopenia.
The vast majority of adults fail to meet physical activity guidelines. While 60% of adults, both European and American, self-report that they meet guidelines, objectively measured physical activity reveals that fewer than 10% of adults in the United States meet physical activity guidelines (Tucker et al., 2011; Marques et al., 2015). Moreover, sedentary behavior alone increases the risk for sarcopenia. While there are few trials in humans on the effects of lifelong sedentary behavior, studies in mice reveal lifelong sedentary behavior impairs mitochondrial function (Figueiredo et al., 2009). Moreover, a cross-sectional study in men revealed that sedentary behavior increased inflammation independent of physical activity (Parsons et al., 2017). Other lifestyle behaviors such as diet, combined with a sedentary lifestyle, also predispose individuals to increased risk for sarcopenia. An analysis of four prospective studies revealed that obesity increased the risk of developing sarcopenia by 20–162% (Stenholm et al., 2008). Indeed, sarcopenia and obesity often occur together, and evidence suggests that risk of metabolic disease and mortality increase when both are present (Wannamethee and Atkins, 2015). Obesity is characterized by inflammation and insulin resistance, both of which contribute to vascular dysfunction, as indicated by reduced endothelium-mediated vasodilation, impaired skeletal muscle perfusion, and decreased myofiber capillary density. Greater adipose infiltration into skeletal muscle is associated with the loss of skeletal muscle strength and torque (Goodpaster et al., 2001a). Lifestyle factors contribute to changes in the basic processes that lead to muscle loss.
Muscle mass is controlled by a dynamic balance of protein synthesis and degradation. A loss of muscle mass occurs when protein synthesis and degradation tip toward net degradation. Strategies to maintain muscle mass have focused on increasing protein synthesis since this is thought to be the more dynamic regulator of mass. Signaling through the mechanistic target of rapamycin (mTOR) pathway is the major regulator of protein synthesis in skeletal muscle (Bodine et al., 2001; Glass, 2005). The activation of mTOR results in activation of p70 S6K causing an increase in protein translation, and the inhibition of 4e-binding protein (4e-BP1), which is a negative regulator of the eukaryotic translation initiation factor-4e (EIF-4e). The activation of these pathways stimulates growth processes, primarily of the myofibrillar proteins. However, a focus purely on growth may not maintain protein quality as discussed later in the review.
Mechanisms of Muscle Loss—Basic Processes
Loss of Proteostasis
Proteostasis is the maintenance of protein homeostasis that refers to the location, concentration, conformation, and turnover of individual proteins (Balch et al., 2008). Proteostasis is essential for whole body and tissue function. Loss of proteostasis leads to the accumulation of damaged proteins (Levine and Stadtman, 2001; Ayyadevara et al., 2016). In skeletal muscle, impaired proteostasis could lead to the decline in quantity and quality of contractile proteins because of accumulation of damage and non-enzymatic modifications of these proteins. One such non-enzymatic modification of proteins, advanced glycation end-products (AGEs) (Semba et al., 2010) increases in concentration with age including in skeletal muscle (Haus et al., 2007). Because of cross-bridge formations, modified proteins are resistant to breakdown and accumulate and may contribute to tissue dysfunction (Barreiro and Hussain, 2010; Drenth et al., 2016) and mobility disability (Sun et al., 2012).
Since the enzymatic capacity to repair protein damage is low, protein turnover is essential to maintain the skeletal muscle proteome. Increased protein turnover should improve proteostasis by degrading damaged proteins and resynthesizing new, functional proteins. Therefore, even in the absence of muscle growth, protein turnover is a beneficial adaptation for tissue health. Protein turnover is an energetically costly process with a large proportion of this demand coming from the step of translation. Protein synthesis requires 12–72 ATP molecules per amino acid synthesized, an additional four phosphates (either ATP or GTP) per bond, and an additional 1–2 ATPs per fold (Lynch and Marinov, 2015). The energetic cost of protein breakdown remains only partially understood. Protein degradation occurs primarily through protein ubiquitination and proteasome degradation, or via the autophagy-lysosomal system. Ubiquitination requires 2 ATPs per ubiquitin tag and subsequent proteasome-mediated degradation of them requires between 100 and 200 ATPs per protein (Lynch and Marinov, 2015). Lysosomal degradation requires four ATPs for every amino acid. Altogether, protein synthesis accounts for 20% of basal metabolism and protein breakdown comprises another 5–15% of basal metabolism (Rolfe and Brown, 1997). Given the energetic demands of protein turnover, a reliable source of energetic production is essential to maintain proteostasis.
Reactive Oxygen Species
The balance between oxidant production and scavenging is essential in maintaining redox homeostasis. With age, redox homeostasis declines and leads to the progressive oxidation of cellular components, including contractile proteins, which leads to skeletal muscle dysfunction (Lourenço dos Santos et al., 2015). Evidence from studies of human skeletal muscle indicates a positive correlation between age and markers of oxidative damage including lipid peroxidation, protein carbonyl content, and 8-oxo-deoxyguanosine (8-oxo-dG), a measure of DNA oxidation (Capel et al., 2005).
Age-related impairments in mitochondrial function result in increased production of reactive oxygen species (ROS), which increases oxidative stress and contributes to the loss of proteostasis. Mitochondrial dysfunction contributes to an imbalance of reducing equivalents such as NADH, which increases ROS formation and leads to the oxidation of cellular components. Loss of structural integrity of mitochondrial supercomplexes and membranes also leads to increased ROS production and associates with impaired skeletal muscle function (Genova and Lenaz, 2014). An increasingly oxidized environment not only increases oxidative damage to cellular components, it also increases the formation of AGEs. In models such as the whole body SOD1 KO mouse (Jang et al., 2010) and the muscle-specific SOD1 KO mouse (Sakellariou et al., 2018), there is an accelerated muscle aging phenotype that resembles muscle aging in humans. Conversely, targeting oxidative stress has also shown efficacy at preventing the sarcopenic phenotype in aged mice (Lee et al., 2010; Vays et al., 2014). Therefore, these models provide support for an indirect and direct role of oxidative stress in skeletal muscle aging.
Inflammation
Inflammation is a response to a cellular disruption (infection, damage, detection of “non-self”) in an effort to facilitate a return to homeostasis. Chronic, low-grade inflammation occurs with aging due to immunosenescence and has been coined “inflammaging.” Inflammaging contributes to chronic diseases including cardiovascular disease and sarcopenia (Ferrucci and Fabbri, 2018). Pro-inflammatory cytokines including interleukin-1 (IL-1), interleukin-6 (IL-6), c-reactive protein (CRP), and tumor necrosis factor (TNF) are characteristic of inflammaging and therefore have been proposed as markers of age and disease risk (Justice et al., 2018).
Several studies have associated circulating inflammatory cytokines with decreased skeletal muscle strength. The InCHIANTI study, a prospective study of mobility in older adults, demonstrated that elevated inflammatory markers predicted handgrip and leg strength (Schrager et al., 2007). A Danish cross-sectional study indicated that inflammatory marker TNF was inversely related with lean mass (Pedersen et al., 2003). Others have found that high IL-6 (>5pg/ml) and CRP (>6.1pg/ml) levels increased the risk of losing more than 40% of muscle strength by twofold to threefold (Schaap et al., 2006). Given the evidence from corroborating studies of aging, it is likely that inflammation contributes to the etiology of sarcopenia (Chung et al., 2009).
The mechanisms by which inflammation contributes to sarcopenia are not completely understood. In a study of older male subjects, increased levels of pro-inflammatory ceramides impaired anabolic signaling following an acute bout of resistance exercise (Rivas et al., 2012) meaning that inflammation may impair normal anabolic responses. In addition, high-fat diet (HFD)-fed rodents and muscle cell culture models consistently link ceramide with exacerbated muscle atrophy or impaired anabolic signaling (Hyde et al., 2005; Roseno et al., 2015). There is also speculation that inflammatory factors such as prostaglandins impair skeletal muscle function by stimulating the generation of reactive oxygen species (ROS) (Chung et al., 2009). As an example, sphingomyelinase, an enzyme involved in the inflammatory response, stimulates the release of ROS that results in decreased contractile function as a consequence of chronic inflammation (Ferreira et al., 2010). More studies are needed to determine the mechanisms by which inflammation contributes to sarcopenia.
Interface of Mitochondria and Basic Aging Processes
The decline in skeletal muscle mitochondrial capacity with aging has been extensively studied as a contributor to slower walking speed (Coen et al., 2013; Santanasto et al., 2016; Gonzalez-Freire et al., 2018), fatigability (Santanasto et al., 2015), and sarcopenia (Joseph et al., 2012; Gouspillou et al., 2014). Evidence from the Baltimore Longitudinal Study of Aging indicates that skeletal muscle ex vivo mitochondrial respiration parallels decline in vivo oxidative capacity, cardiorespiratory fitness, and muscle strength (Gonzalez-Freire et al., 2018). While a number of cross-sectional human studies have demonstrated lower mitochondrial function with chronological age (Trounce et al., 1989; Boffoli et al., 1994; Tonkonogi et al., 2003; Short et al., 2005; Lanza et al., 2008; Porter et al., 2015a), several others have failed to observe these changes (Rasmussen et al., 2003; Hutter et al., 2007; Larsen et al., 2012b; Gouspillou et al., 2014; Gram et al., 2014). These inconsistent results may be partially due to variation in the approaches used to assess mitochondrial function. In addition, many studies of mitochondrial function in aging have not controlled for important covariates including participant physical activity levels (Boffoli et al., 1994) and adiposity (Short et al., 2005), which likely confound the relationship between mitochondrial capacity and age (Proctor et al., 1995; Hutter et al., 2007; Lanza et al., 2008; Safdar et al., 2010). For example, Distefano and colleagues demonstrated a strong, inverse correlation between age and mitochondrial function (Distefano et al., 2016). However, when controlling for fitness and adiposity, age only accounted for 1–6% of the variation observed in maximal ATP production. Regardless, there is some factor associated with the aging process that contributes to mitochondrial decline. The National Institute on Aging (NIA) recently funded the study of muscle, mobility, and aging (SOMMA), the goal of which is to determine the combination of muscle properties (energetics, autophagy, denervation, and oxidative stress) that most strongly predicts major mobility disability, declines in fitness, 400-m walking speed, and muscle mass.
Evidence from preclinical models indicates a close link between mitochondrial energetics and control of muscle mass. Release of pro-apoptotic factors (Max, 1972; Adhihetty et al., 2007), morphological alterations (fission, swelling), energy stress via reduced ATP (Romanello et al., 2010), and increased mitochondrial reactive oxygen species (ROS) emission (Adhihetty et al., 2007; Müller et al., 2007; Kavazis et al., 2009; Min et al., 2011) have all been reported during muscle atrophy in preclinical studies. In addition, for a given concentration of ADP, mitochondria from aged muscle generate more ROS than young counterparts (Holloway et al., 2018). Combined with the age-related decline in endogenous antioxidant activity, the increase in ROS emission leads to an increase in concentration of unscavenged ROS (Dai et al., 2014).
Mitochondrial ROS can depress protein synthesis by decreasing phosphorylation of 4e-BP1 and impairing mTOR assembly (Pham et al., 2000; Shenton et al., 2006; Zhang et al., 2009). Mitochondria-targeted antioxidant treatment in rodents supports a crucial role for mitochondrial ROS in mediating muscle atrophy (Min et al., 2011; Powers et al., 2011). Increased ROS production can also exacerbate muscle atrophy (Jang et al., 2010). Mitochondrial ROS stimulate proteolytic degradation pathways (autophagy and proteasome system) (Li et al., 2003; Aucello et al., 2009; McClung et al., 2009; Hussain et al., 2010) and energetic stress (reduced ATP production), which can activate the AMP kinase (AMPK)-FoxO3 pathways leading to increased expression of the ubiquitin-proteasome system and lysosome-autophagy system (Greer et al., 2007; Romanello et al., 2010). Taken together, multiple lines of compelling preclinical evidence implicate a central role for mitochondrial energetics in muscle atrophy. In addition, ROS react with and damage cellular components, including contractile proteins, which decreases proteome integrity and increases the demand for somatic maintenance.
In regard to inflammation, dysfunctional mitochondria exacerbate the detrimental effects of pro-inflammatory cytokines. Mitochondria release damage-associated molecular patterns (mito-DAMPs) that stimulate inflammatory pathways (Tezze et al., 2017). The mito-DAMPs further act on proteins such as NOD-like receptor protein 3 (NLRP3), which is involved in heart disease and aging (Salminen et al., 2012). Along with increased ROS release and pro-inflammatory signaling, dysfunctional mitochondria exacerbate inflammatory signaling that impair cellular integrity and proteostasis, which consequently leads to myocyte death (Chung et al., 2009).
Mitochondria are central to maintaining the energetic resources to preserve proteostasis. When there is a mismatch between the rate of ATP production and the demand for ATP, expendable cellular processes are sacrificed (Hou, 2013). There are three broad categories of cellular processes: metabolism, growth, and somatic maintenance (Hou et al., 2008). Processes that fall under metabolism are those that sustain life including energy production. Growth involves cellular expansion (Gregory, 2001), while somatic maintenance involves maintaining the quality of the soma (Shanley and Kirkwood, 2000). If the ability to provide ATP on demand is constrained, the cell makes trade-offs between growth and somatic maintenance since metabolism is maintained (Hou, 2013). Thus, impairment of mitochondrial function can in turn compromise proteostasis. In addition, it is conceivable, although never tested, that a focus on growth processes could compromise cellular maintenance if not matched by increased energy-producing capabilities.
To illustrate the importance of maintaining mitochondria to mitigate the detrimental declines of basic cellular processes in sarcopenia, the neuromuscular junction (NMJ) provides an excellent example. Degradation of the NMJ leads to the denervation of muscle fibers (Jang and Van Remmen, 2011), which appears to contribute to decreased muscle size, strength, and endurance (Sundberg et al., 2018). With age, type II myofibers are more prone to denervation and are reinnervated with type I motor units (Kelly et al., 2017). Reinnervation of type I motor units leads to previously denervated type II myofiber to adopt a type I myofiber phenotype (Kelly et al., 2017). The change in innervation could contribute to the decline in skeletal muscle strength with age (Nilwik et al., 2013), as evident by the observation that older individuals have a greater proportion of type I myofibers along with lower strength (Frontera et al., 2000). Some evidence suggests that declines in NMJ quality precede the impairment of skeletal muscle function (Spendiff et al., 2016); however, more research is still needed in this area.
Oxidative damage of the NMJ promotes the loss of skeletal muscle proteostasis (Vasilaki et al., 2017). Recent data suggest that impaired redox signaling, rather than oxidative damage per se, drives denervation (McDonagh et al., 2016). Mitochondria in neurons of aged organisms appear to emit more ROS which can disrupt redox signaling by desensitizing redox sensors that are responsible for adaptation (McDonagh et al., 2016). With aging, there is decrease in the ability to activate redox-related signaling pathways, and this failure causes further oxidative damage in the neuron which promotes NMJ degradation (Vasilaki et al., 2006). Therefore, targeting mitochondrial function in the NMJ or areas surrounding the NMJ may be a mechanism to protect against age-related muscle loss.
Non-Exercise Treatments of Sarcopenia
Treating sarcopenia has revolved around therapeutic interventions to mitigate the loss of muscle mass. These therapies, which include targeting members of the transforming growth factor β (TGF-β) superfamily, testosterone, selective androgen receptor modulators (SARMS), and growth hormone (GH), among others, are currently or have been tested at various phase clinical trials (Garber, 2016; Morley, 2016). In the following sections, we will discuss the mechanism in which these therapies target muscle loss, along with their efficacy to treat sarcopenia.
Growth and differentiation factor 8 (GDF8 or Myostatin), a member of the TGF-β superfamily, and activin A are powerful negative regulators of skeletal muscle growth (Lee, 2004). Myostatin and activin A signal through the activin type II receptor (ActRIIB), which leads to activation of Smad 2/3 transcription factors, translocation to the nucleus, and activation of target genes. Myostatin negatively regulates Akt signaling preventing protein synthesis and also interferes with myoblast differentiation into myotubes and may also impair muscle growth. A number of anti-myostatin antibodies have been developed and tested in humans, all of which increase lean mass (Becker et al., 2015; Woodhouse et al., 2016; Rooks et al., 2017) and some also showed improved physical function and strength (Becker et al., 2015; Rooks et al., 2017). It was recently demonstrated that inhibition of activin A in primates enhanced muscle growth (Latres et al., 2017). However, concerns remain around the functional benefits from inhibiting signaling through ActRIIB as in myostatin knockout mice, there is a loss of specific force (Amthor et al., 2007). Inhibiting myostatin has also been linked with reduced mitochondrial capacity of skeletal muscle, poor muscle endurance, and fatigability (Mouisel et al., 2014).
Low levels of circulating testosterone in older men (otherwise known as hypogonadism) are associated with reduced lean body mass, bone mineral density, and increased fat mass (Saad et al., 2017). Mechanisms of action for testosterone include increasing protein synthesis (Wolfe et al., 2000; Ferrando et al., 2003) via Akt/mTOR activation (White et al., 2013) and reduction in adipose stem cells and activation of satellite cell recruitment (Kovacheva et al., 2010). There is strong evidence from intervention studies that treatment with testosterone is effective in increasing lean mass and reducing fat mass (Kenny et al., 2010; Srinivas-Shankar et al., 2010; Kvorning et al., 2013). However, the efficacy of testosterone to improve muscle-specific strength and physical function is less clear (Snyder et al., 1999; Saad et al., 2017). The Testosterone Trial in Older Men showed that a year of testosterone treatment in 790 older men had no benefit with respect to fatigue or walking distance (Snyder et al., 2016). Conversely, others have shown that treatment with testosterone improved muscle function (leg extension, triceps extension, biceps curl) (Ferrando et al., 2002) and grip strength (Morley et al., 1993; Sih et al., 1997) in older men with hypogonadism. Whether testosterone influences skeletal muscle mitochondrial energetics is also not clear. Testosterone deficiency is associated with reduced myocellular metabolism and mitochondrial energetics (Traish et al., 2011) and preclinical work indicates that testosterone can induce mitochondrial biogenic signaling (Usui et al., 2014). However, testosterone treatment did not alter the activity of enzymes known to regulate mitochondrial biogenesis or markers of oxidative phosphorylation and lipid metabolism in the skeletal muscle of aging men with low testosterone (Petersson et al., 2014).
Growth hormone (GH) was also considered a therapeutic for sarcopenia. There is well-described decline in activity of the GH/insulin-like growth factor-1 (IGF-1) axis in older adults (Zadik et al., 1985). In 1990, it was shown that administration of human GH to older healthy individuals could increase lean mass and it was thought at the time that GH might be an effective antiaging therapy (Rudman et al., 1990). However, subsequent studies indicated that GH increased muscle mass but not strength (Kim and Morley, 2005) and unfavorable side effects were also reported, including gynecomastia and carpal tunnel syndrome, and treated patients were more likely to experience impaired fasting glucose (Liu et al., 2007). In a cross-sectional study of healthy adults, GH/IGF-1 level corresponded to muscle mitochondrial function (by 31P MRS) (Makimura et al., 2011). However, 2 weeks of GH treatment led to a reduction in mitochondrial genes involved in β-oxidation and oxidative phosphorylation (Sjögren et al., 2007). Many of the pharmacological targets explored to date are generally efficacious in improving muscle mass. However, and perhaps germane to the lack of therapeutic effectiveness, it is less clear whether mitochondrial energetics are improved with these options.
Manipulation of diet is another approach for the treatment of sarcopenia that has received a lot of attention. Post-feeding hyperaminoacidemia is a potent stimulator of skeletal muscle protein synthesis in young adults and is blunted in older adults following ingestion of amino acid mixture, suggesting skeletal muscle of older individuals is anabolic resistant (Volpi et al., 2000). It has been proposed that a greater dietary intake of essential amino acids could help older adults maintain skeletal muscle mass (Volpi et al., 2013). However, recent studies examining increased protein intake in older individuals do not show increased lean body mass (LBM) accretion (Bhasin et al., 2018), and that the amount and quality of protein intake are not associated with muscle mass or strength in older adults (Gingrich et al., 2017). Therefore, there remain questions over whether anabolic responses from protein and amino acid intake that are classically determined over the short-term translate to long-term preservation or improvements in muscle mass.
Increasing vitamin D levels, which is commonly reduced in older adults, through dietary supplementation results in improved muscle mass and chair-stand test time, a surrogate of muscle power in older adults (Bauer et al., 2015). Interestingly, improving the vitamin D status of adults that are deficient resulted in improved mitochondrial oxidative function in skeletal muscle (Sinha et al., 2013; Rana et al., 2014). This finding suggests that improvements in muscle health by vitamin D supplementation could at least be partly due to improvements in mitochondrial oxidative phosphorylation in older adults. A greater understanding of the independent roles of these dietary components on the regulation of muscle mass and function is needed along with a focus on how they also impact mitochondrial energetics.
As illustrated, despite substantial evidence linking mitochondrial energetics to the etiology of sarcopenia, pharmacological therapies to date have not focused on mitochondrial targets but rather pathways that mediate increases in muscle mass. There are some exceptions. For example, there is currently an ongoing clinical trial testing whether metformin, an AMPK activator, enhances the response to resistance exercise (Long et al., 2017). This proposed treatment has the potential to improve cellular metabolism and mitochondrial biogenesis to improve anabolic responsiveness. Other studies have examined the effects of exogenous antioxidants, such as vitamin C, on muscle quality and oxidative stress. Vitamin C supplementation improves muscle function in at least one model of aged muscle (Ryan et al., 2010), but vitamin C is known to inhibit the redox signaling that leads to positive mitochondrial responses (Gomez-Cabrera et al., 2008; Paulsen et al., 2014; Bruns et al., 2018). Meta-analyses investigating the efficacy of long-term vitamin C supplementation conclude that supplementation actually increases the risk for disease and mortality (Bjelakovic et al., 2012). Other strategies targeting antioxidant mechanisms are promising though. For example, studies using a purported Nrf2 activator that increases endogenous antioxidants produces lifespan in male mice (Strong et al., 2016) and enhances protein synthesis during aerobic exercise training (Bruns et al., 2018).
In sum, clinical therapies that use pharmaceutical and nutritional intervention that focus solely on muscle mass regulation have been met with limited success in various phase clinical trials. Additionally, these therapies appear to have little or no influence on mitochondrial energetics. Therefore, additional interventions are needed that help preserve mitochondrial function and muscle proteostasis in older adults. On the other hand, exercise has been shown to be an effective countermeasure to age-related muscle loss. Below, we focus on how exercise training affects mitochondrial function to support the notion that targeting mitochondrial function is essential in developing effective treatments to mitigate sarcopenia.
Exercise for the Treatment of Sarcopenia
Physical Activity With Aging
Physical activity tends to decline with aging and results in difficulties in daily life activities and normal functioning (Westerterp, 2000). Data from the Health, Aging, and Body Composition (HABC) study showed that older adults who maintain higher levels of physical activity levels are protected against functional or mobility limitations in comparison to sedentary adults (Brach et al., 2004). There are several mechanisms in which maintaining physical activity throughout the lifespan protects against the onset of sarcopenia. Physical activity is a determinant of mitochondrial function, which likely is one mechanism that maintains skeletal muscle function (Larsen et al., 2012a; Tevald et al., 2014). Compared to sedentary counterparts, lifelong and masters athletes maintain skeletal muscle function concomitantly with mitochondrial function (Zampieri et al., 2014; Power et al., 2016b). Physical activity also attenuates the age-related increase in inflammation and oxidative stress, which, as highlighted, contribute to the loss of skeletal muscle mass and function (Mikkelsen et al., 2013).
Physical activity in mid-life appears to have a protective effect on physical function, as Patel et al. showed that older adults who engaged in higher levels of physical activity in mid-life performed better on the Short Physical Performance Battery (SPPB) and have reduced incidences of mobility disability than those less active in mid-life (Patel et al., 2006). Collectively, these results reveal a potential effect of exercise pre-habilitation to prevent the deleterious effects of aging and sedentary lifestyle on muscle function in older adults. The pre-habilitative effects of physical activity have led to clinical investigation into whether interventions later in life can prevent muscle dysfunction with aging. The Lifestyle Interventions and Independence for Elders (LIFE) study was designed to determine whether a physical activity intervention might confer significant physical benefits in older adults in comparison to those in a health education program. The intervention included ~150 min/wk of moderate-intensity walking, along with strength, flexibility, and balance training (Fielding et al., 2011; Pahor et al., 2014). The 6-month intervention reduced major mobility disability in older adults that participated in the physical activity protocol, as well as a reduced risk of persistent mobility disability in comparison to those in a health education program (Pahor et al., 2014). Collectively, the results of the LIFE study concluded that moderate increases in physical activity levels, which likely improve cellular bioenergetics, blunt the detrimental effects of primary aging on physical function in older adults. In the following sections, we will describe how structured exercise interventions of different modalities (i.e., strength and aerobic exercise training) lead to improvements in skeletal muscle mitochondrial health in older adults.
Resistance Exercise
Resistance exercise remains a widely prescribed means to prevent, mitigate, and even reverse sarcopenia. The efficacy of resistance exercise to stimulate lean mass accrual and increases in strength is well documented (Koopman and van Loon, 2009). It was thought that resistance exercise training had little or no effect on mitochondrial biogenesis or function. However, recent studies have shown that resistance exercise training increases mitochondrial protein fractional synthesis rates (FSRs) (Wilkinson et al., 2008; Robinson et al., 2017) and improves mitochondrial function (Porter et al., 2015b; Robinson et al., 2017). Young adults engaged in a resistance exercise program showed increases in mitochondrial enzyme activity and respiration (Porter et al., 2015b). While the changes in mitochondrial respiration are modest in comparison to endurance exercise, improvements in in vivo PCr recovery rates and oxidative capacity appear comparable in older adults engaged in either exercise intervention (Jubrias et al., 2001). Similarly, using permeabilized myofibers, resistance exercise training increases state III and maximal oxidative phosphorylation capacity in older adults, which is accompanied by improvements in ADP sensitivity (Holloway et al., 2018). Therefore, while resistance exercise directly stimulates myofibrillar protein accrual to promote strength, it may also have positive effects on mitochondrial function and proteostasis.
Novel training regimens, such as power training, have been proposed to improve muscle function in older adults. Power training has received significant interest to prevent the sarcopenic phenotype, due to its targeting of type II myofibers, which are more prone to atrophy in older adults (Purves-Smith et al., 2014; St-Jean Pelletier et al., 2017). Indeed, clinical studies have shown that power training improves muscle function in older adults that is comparable or slightly greater than traditional resistance exercise training (Reid et al., 2008; Tschopp et al., 2011). Though preservation of mitochondrial function through physical activity is an important factor in the prevention of sarcopenia, the effects of power training on organelle bioenergetics is unclear.
While resistance exercise remains an effective intervention to combat sarcopenia, there are several considerations when prescribing resistance exercise training to older individuals. These concerns have led to alternative resistance training programs that focus on low load. It appears that low-load, high-repetition exercise is equally effective as high-load exercise in stimulating muscle hypertrophy in healthy individuals (Schoenfeld et al., 2017). Blood flow restricted resistance exercise is a second modification that may reduce the load necessary for positive adaptations. The premise is that a metabolic stress (blood flow restriction) may stimulate both mitochondrial and hypertrophy responses. Even with low load and number of repetitions, blood flow restricted exercise can positively influence motor unit activation and hypertrophy (Farup et al., 2015). More studies on blood flow restriction are necessary to determine if there are additional mitochondrial adaptations beyond traditional resistance exercise.
Aerobic Exercise
Aerobic exercise is generally not appreciated as a stimulator of hypertrophy; however, there is evidence that it can lead to muscle hypertrophy. In older adults, aerobic exercise training improves myofiber size and strength, as well as whole muscle size and strength (Harber et al., 2012). Therefore, aerobic exercise stimulates muscle growth either directly or indirectly.
Nearly half a century ago, Holloszy first documented that aerobic exercise increases mitochondrial content (Holloszy, 1967). Since then, research has consistently documented that aerobic exercise improves both mitochondrial content and function (Menshikova et al., 2006; Jacobs and Lundby, 2013; Zampieri et al., 2014). Aerobic exercise increases mitochondrial turnover since it increases both mitochondrial biogenesis (protein synthesis) (Wilkinson et al., 2008; Scalzo et al., 2014) and mitophagy (mitochondrial-specific autophagy) (Drake et al., 2015). The improvement in the rate of ATP production from aerobic exercise training suggests that more energy is available to maintain proteostasis. Additionally, improvement in mitochondrial efficiency (reduction in ROS generated per oxygen consumed or ATP generated) suggests that there is less oxidative stress and damage, which would in turn improve the quality of the proteome. In all, aerobic exercise mediated improvements in mitochondrial function likely protects against sarcopenia (Musci et al., 2017).
Enhanced mitochondrial content and function can sustain greater energetic flux and oxidation of substrates. Increased energetic flux can decrease the accumulation of lipotoxic intermediates that promote inflammation and oxidative stress (Goodpaster et al., 2001b; Coen and Goodpaster, 2012). Exercise-induced improvements in substrate flux thus decrease inflammation and oxidatively modified proteins (Corcoran et al., 2007; Fabbri et al., 2016). It has been shown that long-term aerobic exercise training in diabetic individuals can return mitochondrial function to that of a lean individual (Konopka et al., 2015). Importantly, aerobic exercise improves the ability of the muscle to provide energy on demand. Having energy on demand reduces compromises between growth and somatic maintenance that comes during periods of energy shortage. Thus, aerobic exercise improves the ability to maintain cellular integrity and adaptation to stress (Ristow and Schmeisser, 2014).
Exercise as a Discovery Tool (MOTRPAC)
Exercise and physical activity enhance various aspects of human health and prevent metabolic and neurological diseases as well as cancer. In 2016, the National Institutes of Health (NIH) common fund dedicated $170 million to the molecular transducers of physical activity consortium (MoTrPAC). The goal is to identify the molecular networks in key tissues that are changed in response to acute and chronic exercise (Neufer et al., 2015). Elucidating how acute molecular responses to exercise integrate over time and improve health outcomes will unveil novel mechanisms contributing to health and disease processes and identify potential new therapeutic targets to aid in the prevention and treatment of disease. Older adults will be included in the study cohort and so the molecular networks that are altered in muscle with aging will be captured, in addition to the effects of exercise on mitochondrial and proteostasis molecular networks. From this study, there is enormous potential for us to further understand how endurance and resistance exercise improve mitochondrial function and muscle health in older adults.
Future Directions
Additional studies are needed to directly answer the question of whether treatments that target mitochondrial adaptations, such as aerobic exercise, are sufficient to maintain muscle mass with age. From these studies, the formulation of practical guidelines that refine recommendations of frequency, intensity, and duration of such activities should be derived. After establishing the appropriate evidence, it will be important to integrate important modulating factors such as medications (Robinson et al., 2009) and dietary factors (Smith et al., 2015) for long-term outcomes. Although there is strong preclinical and clinical support for targeting mitochondria with exercise, more studies are needed to directly support its efficacy to mitigate sarcopenia.
Summary
If one considers a few of the primary underlying contributors to the loss of muscle mass with age, that is, loss of proteostasis, inflammation, and oxidative stress, it is apparent that mitochondria should be a target to mitigate sarcopenia (Figure 1). Unfortunately, to date, the field has primarily focused on directly stimulating growth processes rather than basic processes that create an environment that is favorable for growth processes. In other tissue types such as cardiac, hepatic, and nervous, it is well recognized that maintaining cellular energetics is of primary importance for maintaining tissue quality with age. Although skeletal muscle mitochondria are viewed as important for metabolism, the role of mitochondria in maintaining muscle mass is less appreciated. In our opinion, targeting mitochondria with exercise or other treatments is an effective way to treat sarcopenia and one that is still underexplored.
Author Contributions
BM conceived of the review. PC, RM, MH, and BM wrote the review. PC and MH created the figure. PC, RM, MH, and BM proofread the review.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adhihetty, P. J., O’Leary, M. F. N., Chabi, B., Wicks, K. L., and Hood, D. A. (2007). Effect of denervation on mitochondrially mediated apoptosis in skeletal muscle. J. Appl. Physiol. 102, 1143–1151. doi: 10.1152/japplphysiol.00768.2006
Amthor, H., Macharia, R., Navarrete, R., Schuelke, M., Brown, S. C., Otto, A., et al. (2007). Lack of myostatin results in excessive muscle growth but impaired force generation. Proc. Natl. Acad. Sci. USA. 104, 1835–1840. doi: 10.1073/pnas.0604893104
Anker, S. D., Morley, J. E., and Haehling von, S. (2016). Welcome to the ICD-10 code for sarcopenia. J. Cachexia Sarcopenia Muscle 7, 512–514. doi: 10.1002/jcsm.12147
Aucello, M., Dobrowolny, G., and Musarò, A. (2009). Localized accumulation of oxidative stress causes muscle atrophy through activation of an autophagic pathway. Autophagy 5, 527–529. doi: 10.4161/auto.5.4.7962
Ayyadevara, S., Balasubramaniam, M., Suri, P., Mackintosh, S. G., Tackett, A. J., Sullivan, D. H., et al. (2016). Proteins that accumulate with age in human skeletal-muscle aggregates contribute to declines in muscle mass and function in Caenorhabditis elegans. Aging 8, 3486–3497. doi: 10.18632/aging.101141
Baehr, L. M., West, D. W. D., Marcotte, G., Marshall, A. G., De Sousa, L. G., Baar, K., et al. (2016). Age-related deficits in skeletal muscle recovery following disuse are associated with neuromuscular junction instability and ER stress, not impaired protein synthesis. Aging 8, 127–146. doi: 10.18632/aging.100879
Balch, W. E., Morimoto, R. I., Dillin, A., and Kelly, J. W. (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. doi: 10.1126/science.1141448
Barreiro, E., and Hussain, S. N. A. (2010). Protein carbonylation in skeletal muscles: impact on function. Antioxid. Redox Signal. 12, 417–429. doi: 10.1089/ars.2009.2808
Batsis, J. A., Mackenzie, T. A., Barre, L. K., Lopez-Jimenez, F., and Bartels, S. J. (2014). Sarcopenia, sarcopenic obesity and mortality in older adults: results from the National Health and Nutrition Examination Survey III. Eur. J. Clin. Nutr. 68, 1001–1007. doi: 10.1038/ejcn.2014.117
Batsis, J. A., Mackenzie, T. A., Lopez-Jimenez, F., and Bartels, S. J. (2015). Sarcopenia, sarcopenic obesity, and functional impairments in older adults: national health and nutrition examination surveys 1999-2004. Nutr. Res. 35, 1031–1039. doi: 10.1016/j.nutres.2015.09.003
Bauer, J. M., Verlaan, S., Bautmans, I., Brandt, K., Donini, L. M., Maggio, M., et al. (2015). Effects of a vitamin D and leucine-enriched whey protein nutritional supplement on measures of sarcopenia in older adults, the PROVIDE study: a randomized, double-blind, placebo-controlled trial. J. Am. Med. Dir. Assoc. 16, 740–747. doi: 10.1016/j.jamda.2015.05.021
Baumgartner, R. N., Koehler, K. M., Gallagher, D., Romero, L., Heymsfield, S. B., Ross, R. R., et al. (1998). Epidemiology of sarcopenia among the elderly in New Mexico. Am. J. Epidemiol. 147, 755–763. doi: 10.1093/oxfordjournals.aje.a009520
Beard, J. R., Officer, A. M., and Cassels, A. K. (2016). The World Report on Ageing and Health. Gerontologist 56, S163–S166. doi: 10.1093/geront/gnw037
Becker, C., Lord, S. R., Studenski, S. A., Warden, S. J., Fielding, R. A., Recknor, C. P., et al. (2015). Myostatin antibody (LY2495655) in older weak fallers: a proof-of-concept, randomised, phase 2 trial. Lancet Diabetes Endocrinol. 3, 948–957. doi: 10.1016/S2213-8587(15)00298-3
Bhasin, S., Apovian, C. M., Travison, T. G., Pencina, K., Moore, L. L., Huang, G., et al. (2018). Effect of protein intake on lean body mass in functionally limited older men: a randomized clinical trial. JAMA Intern. Med. 178:530. doi: 10.1001/jamainternmed.2018.0008
Bjelakovic, G., Nikolova, D., Gluud, L. L., Simonetti, R. G., and Gluud, C. (2012). Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst. Rev. 336:CD007176. doi: 10.1002/14651858.CD007176.pub2
Bodine, S. C., Stitt, T. N., Gonzalez, M., Kline, W. O., Stover, G. L., Bauerlein, R., et al. (2001). Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 3, 1014–1019. doi: 10.1038/ncb1101-1014
Boffoli, D., Scacco, S. C., Vergari, R., Solarino, G., Santacroce, G., and Papa, S. (1994). Decline with age of the respiratory chain activity in human skeletal muscle. Biochim. Biophys. Acta 1226, 73–82.
Brach, J. S., Simonsick, E. M., Kritchevsky, S., Yaffe, K., and Newman, A. B. Health, Aging and Body Composition Study Research Group. (2004). The association between physical function and lifestyle activity and exercise in the health, aging and body composition study. J. Am. Geriatr. Soc. 52, 502–509. doi: 10.1111/j.1532-5415.2004.52154.x
Bruns, D. R., Ehrlicher, S. E., Khademi, S., Biela, L. M., Peelor, F. F., Miller, B. F., et al. (2018). Differential effects of vitamin C or protandim on skeletal muscle adaptation to exercise. J. Appl. Physiol. 509, 661–671. doi: 10.1152/japplphysiol.00277.2018
Capel, F., Rimbert, V., Lioger, D., Diot, A., Rousset, P., Mirand, P. P., et al. (2005). Due to reverse electron transfer, mitochondrial H2O2 release increases with age in human vastus lateralis muscle although oxidative capacity is preserved. Mech. Ageing Dev. 126, 505–511. doi: 10.1016/j.mad.2004.11.001
Centers for Disease Control and Prevention (CDC). (2003). Trends in aging—United States and worldwide. MMWR Morb. Mortal. Wkly Rep. 52, 101–104. Available at: https://www.cdc.gov/mmwr/preview/mmwrhtml/mm5206a2.htm
Chung, H. Y., Cesari, M., Anton, S., Marzetti, E., Giovannini, S., Seo, A. Y., et al. (2009). Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res. Rev. 8, 18–30. doi: 10.1016/j.arr.2008.07.002
Coen, P. M., and Goodpaster, B. H. (2012). Role of intramyocelluar lipids in human health. Trends Endocrinol. Metab. 23, 391–398. doi: 10.1016/j.tem.2012.05.009
Coen, P. M., Jubrias, S. A., Distefano, G., Amati, F., Mackey, D. C., Glynn, N. W., et al. (2013). Skeletal muscle mitochondrial energetics are associated with maximal aerobic capacity and walking speed in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 68, 447–455. doi: 10.1093/gerona/gls196
Coker, R. H., Hays, N. P., Williams, R. H., Wolfe, R. R., and Evans, W. J. (2015). Bed rest promotes reductions in walking speed, functional parameters, and aerobic fitness in older, healthy adults. J. Gerontol. A Biol. Sci. Med. Sci. 70, 91–96. doi: 10.1093/gerona/glu123
Corcoran, M. P., Lamon-Fava, S., and Fielding, R. A. (2007). Skeletal muscle lipid deposition and insulin resistance: effect of dietary fatty acids and exercise. Am. J. Clin. Nutr. 85, 662–677. doi: 10.1093/ajcn/85.3.662
Covinsky, K. E., Palmer, R. M., Fortinsky, R. H., Counsell, S. R., Stewart, A. L., Kresevic, D., et al. (2003). Loss of independence in activities of daily living in older adults hospitalized with medical illnesses: increased vulnerability with age. J. Am. Geriatr. Soc. 51, 451–458. doi: 10.1046/j.1532-5415.2003.51152.x
Cruz-Jentoft, A. J., Baeyens, J. P., Bauer, J. M., Boirie, Y., Cederholm, T., Landi, F., et al. (2010). Sarcopenia: European consensus on definition and diagnosis: report of the European Working Group on Sarcopenia in Older People (Oxford University Press). Age and Ageing 39, 412–423. doi: 10.1093/ageing/afq034
Dai, D.-F., Chiao, Y. A., Marcinek, D. J., Szeto, H. H., and Rabinovitch, P. S. (2014). Mitochondrial oxidative stress in aging and healthspan. Longev Healthspan. 3:6. doi: 10.1186/2046-2395-3-6
DeFrances, C. J., Lucas, C. A., Buie, V. C., and Golosinskiy, A. (2008). 2006 National Hospital Discharge Survey. Natl. Health Stat. Rep. 39, 1–20. Available at: https://www.cdc.gov/nchs/data/nhsr/nhsr005.pdf
Distefano, G., Standley, R. A., Dubé, J. J., Carnero, E. A., Ritov, V. B., Stefanovic-Racic, M., et al. (2016). Chronological age does not influence ex-vivo mitochondrial respiration and quality control in skeletal muscle. J. Gerontol. A Biol. Sci. Med. Sci. 72, 535–542. doi: 10.1093/gerona/glw102
Drake, J. C., Wilson, R. J., and Yan, Z. (2015). Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. FASEB J. 30, 13–22. doi: 10.1096/fj.15-276337
Drenth, H., Zuidema, S., Bunt, S., Bautmans, I., van der Schans, C., and Hobbelen, H. (2016). The contribution of advanced glycation end product (AGE) accumulation to the decline in motor function. Eur. Rev. Aging Phys. Act. 13:3. doi: 10.1186/s11556-016-0163-1
English, K. L., and Paddon-Jones, D. (2010). Protecting muscle mass and function in older adults during bed rest. Curr. Opin. Clin. Nutr. Metab. Care 13, 34–39. doi: 10.1097/MCO.0b013e328333aa66
Fabbri, E., Yang, A., Simonsick, E. M., Chia, C. W., Zoli, M., Haughey, N. J., et al. (2016). Circulating ceramides are inversely associated with cardiorespiratory fitness in participants aged 54-96 years from the Baltimore Longitudinal Study of Aging. Aging Cell 15, 825–831. doi: 10.1111/acel.12491
Farup, J., de Paoli, F., Bjerg, K., Riis, S., Ringgard, S., and Vissing, K. (2015). Blood flow restricted and traditional resistance training performed to fatigue produce equal muscle hypertrophy. Scand. J. Med. Sci. Sports 25, 754–763. doi: 10.1111/sms.12396
Ferrando, A. A., Sheffield-Moore, M., Paddon-Jones, D., Wolfe, R. R., and Urban, R. J. (2003). Differential anabolic effects of testosterone and amino acid feeding in older men. J. Clin. Endocrinol. Metab. 88, 358–362. doi: 10.1210/jc.2002-021041
Ferrando, A. A., Sheffield-Moore, M., Yeckel, C. W., Gilkison, C., Jiang, J., Achacosa, A., et al. (2002). Testosterone administration to older men improves muscle function: molecular and physiological mechanisms. Am. J. Physiol. Endocrinol. Metab. 282, E601–E607. doi: 10.1152/ajpendo.00362.2001
Ferreira, L. F., Moylan, J. S., Gilliam, L. A. A., Smith, J. D., Nikolova-Karakashian, M., and Reid, M. B. (2010). Sphingomyelinase stimulates oxidant signaling to weaken skeletal muscle and promote fatigue. AJP: Cell Physiology 299, C552–C560. doi: 10.1152/ajpcell.00065.2010
Ferrucci, L., and Fabbri, E. (2018). Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 15, 505–522. doi: 10.1038/s41569-018-0064-2
Fielding, R. A., Vellas, B., Evans, W. J., Bhasin, S., Morley, J. E., Newman, A. B., et al. (2011). Sarcopenia: an undiagnosed condition in older adults. Current consensus definition: prevalence, etiology, and consequences. International working group on sarcopenia. J. Am. Med. Dir. Assoc. 12, 249–256. doi: 10.1016/j.jamda.2011.01.003
Figueiredo, P. A., Powers, S. K., Ferreira, R. M., Amado, F., Appell, H. J., and Duarte, J. A. (2009). Impact of lifelong sedentary behavior on mitochondrial function of mice skeletal muscle. J. Gerontol. A Biol. Sci. Med. Sci. 64, 927–939. doi: 10.1093/gerona/glp066
Fisher, S. R., Kuo, Y.-F., Graham, J. E., Ottenbacher, K. J., and Ostir, G. V. (2010). Early ambulation and length of stay in older adults hospitalized for acute illness. Arch. Intern. Med. 170, 1942–1943. doi: 10.1001/archinternmed.2010.422
Fortinsky, R. H., Covinsky, K. E., Palmer, R. M., and Landefeld, C. S. (1999). Effects of functional status changes before and during hospitalization on nursing home admission of older adults. J. Gerontol. A Biol. Sci. Med. Sci. 54, M521–M526.
Frontera, W. R., Hughes, V. A., Fielding, R. A., Fiatarone, M. A., Evans, W. J., and Roubenoff, R. (2000). Aging of skeletal muscle: a 12-yr longitudinal study. J. Appl. Physiol. 88, 1321–1326. doi: 10.1152/jappl.2000.88.4.1321
Genova, M. L., and Lenaz, G. (2014). Functional role of mitochondrial respiratory supercomplexes. Biochim. Biophys. Acta 1837, 427–443. doi: 10.1016/j.bbabio.2013.11.002
Gingrich, A., Spiegel, A., Kob, R., Schoene, D., Skurk, T., Hauner, H., et al. (2017). Amount, distribution, and quality of protein intake are not associated with muscle mass, strength, and power in healthy older adults without functional limitations—an enable study. Nutrients 9:1358. doi: 10.3390/nu9121358
Glass, D. J. (2005). Skeletal muscle hypertrophy and atrophy signaling pathways. Int. J. Biochem. Cell Biol. 37, 1974–1984. doi: 10.1016/j.biocel.2005.04.018
Gomez-Cabrera, M.-C., Domenech, E., Romagnoli, M., Arduini, A., Borras, C., Pallardo, F. V., et al. (2008). Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am. J. Clin. Nutr. 87, 142–149. doi: 10.1093/ajcn/87.1.142
Gonzalez-Freire, M., Scalzo, P., D’Agostino, J., Moore, Z. A., Diaz Ruiz, A., Fabbri, E., et al. (2018). Skeletal muscle ex vivo mitochondrial respiration parallels decline in vivo oxidative capacity, cardiorespiratory fitness, and muscle strength: the Baltimore Longitudinal Study of Aging. Aging Cell 6:e12725. doi: 10.1111/acel.12725
Goodpaster, B. H., Carlson, C. L., Visser, M., Kelley, D. E., Scherzinger, A., Harris, T. B., et al. (2001a). Attenuation of skeletal muscle and strength in the elderly: the Health ABC Study. J. Appl. Physiol. 90, 2157–2165. doi: 10.1152/jappl.2001.90.6.2157
Goodpaster, B. H., He, J., Watkins, S., and Kelley, D. E. (2001b). Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J. Clin. Endocrinol. Metab. 86, 5755–5761. doi: 10.1210/jcem.86.12.8075
Gouspillou, G., Sgarioto, N., Kapchinsky, S., Purves-Smith, F., Norris, B., Pion, C. H., et al. (2014). Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J. 28, 1621–1633. doi: 10.1096/fj.13-242750
Gram, M., Vigelsø, A., Yokota, T., Hansen, C. N., Helge, J. W., Hey-Mogensen, M., et al. (2014). Two weeks of one-leg immobilization decreases skeletal muscle respiratory capacity equally in young and elderly men. Exp. Gerontol. 58, 269–278. doi: 10.1016/j.exger.2014.08.013
Greer, E. L., Oskoui, P. R., Banko, M. R., Maniar, J. M., Gygi, M. P., Gygi, S. P., et al. (2007). The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 282, 30107–30119. doi: 10.1074/jbc.M705325200
Gregory, T. R. (2001). Coincidence, coevolution, or causation? DNA content, cell size, and the C-value enigma. Biol. Rev. Camb. Philos. Soc. 76, 65–101. doi: 10.1017/S1464793100005595
Harber, M. P., Konopka, A. R., Undem, M. K., Hinkley, J. M., Minchev, K., Kaminsky, L. A., et al. (2012). Aerobic exercise training induces skeletal muscle hypertrophy and age-dependent adaptations in myofiber function in young and older men. J. Appl. Physiol. 1495–1504. doi: 10.1152/japplphysiol.00786.2012
Haus, J. M., Carrithers, J. A., Trappe, S. W., and Trappe, T. A. (2007). Collagen, cross-linking, and advanced glycation end products in aging human skeletal muscle. J. Appl. Physiol. 103, 2068–2076. doi: 10.1152/japplphysiol.00670.2007
Holloszy, J. O. (1967). Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J. Biol. Chem. 242, 2278–2282.
Holloway, G. P., Holwerda, A. M., Miotto, P. M., Dirks, M. L., Verdijk, L. B., and van Loon, L. J. C. (2018). Age-associated impairments in mitochondrial ADP sensitivity contribute to redox stress in senescent human skeletal muscle. Cell Rep. 22, 2837–2848. doi: 10.1016/j.celrep.2018.02.069
Hou, C. (2013). The energy trade-off between growth and longevity. Mech. Ageing Dev. 134, 373–380. doi: 10.1016/j.mad.2013.07.001
Hou, C., Zuo, W., Moses, M. E., Woodruff, W. H., Brown, J. H., and West, G. B. (2008). Energy uptake and allocation during ontogeny. Science 322, 736–739. doi: 10.1126/science.1162302
Hussain, S. N. A., Mofarrahi, M., Sigala, I., Kim, H. C., Vassilakopoulos, T., Maltais, F., et al. (2010). Mechanical ventilation-induced diaphragm disuse in humans triggers autophagy. Am. J. Respir. Crit. Care Med. 182, 1377–1386. doi: 10.1164/rccm.201002-0234OC
Hutter, E., Skovbro, M., Lener, B., Prats, C., Rabøl, R., Dela, F., et al. (2007). Oxidative stress and mitochondrial impairment can be separated from lipofuscin accumulation in aged human skeletal muscle. Aging Cell 6, 245–256. doi: 10.1111/j.1474-9726.2007.00282.x
Hvid, L., Aagaard, P., Justesen, L., Bayer, M. L., Andersen, J. L., Ørtenblad, N., et al. (2010). Effects of aging on muscle mechanical function and muscle fiber morphology during short-term immobilization and subsequent retraining. J. Appl. Physiol. 109, 1628–1634. doi: 10.1152/japplphysiol.00637.2010
Hyde, R., Hajduch, E., Powell, D. J., Taylor, P. M., and Hundal, H. S. (2005). Ceramide down-regulates system A amino acid transport and protein synthesis in rat skeletal muscle cells. FASEB J. 19, 461–463. doi: 10.1096/fj.04-2284fje
Jacobs, R. A., and Lundby, C. (2013). Mitochondria express enhanced quality as well as quantity in association with aerobic fitness across recreationally active individuals up to elite athletes. J. Appl. Physiol. 114, 344–350. doi: 10.1152/japplphysiol.01081.2012
Jang, Y. C., and Van Remmen, H. (2011). Age-associated alterations of the neuromuscular junction. Exp. Gerontol. 46, 193–198. doi: 10.1016/j.exger.2010.08.029
Jang, Y. C., Lustgarten, M. S., Liu, Y., Müller, F. L., Bhattacharya, A., Liang, H., et al. (2010). Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. 24, 1376–1390. doi: 10.1096/fj.09-146308
Janssen, I., Heymsfield, S. B., and Ross, R. (2002). Low relative skeletal muscle mass (sarcopenia) in older persons is associated with functional impairment and physical disability. J. Am. Geriatr. Soc. 50, 889–896.
Janssen, I., Shepard, D. S., Katzmarzyk, P. T., and Roubenoff, R. (2004). The healthcare costs of sarcopenia in the United States. J. Am. Geriatr. Soc. 52, 80–85.
Joseph, A.-M., Adhihetty, P. J., Buford, T. W., Wohlgemuth, S. E., Lees, H. A., Nguyen, L. M. D., et al. (2012). The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high- and low-functioning elderly individuals. Aging Cell 11, 801–809. doi: 10.1111/j.1474-9726.2012.00844.x
Jubrias, S. A., Esselman, P. C., Price, L. B., Cress, M. E., and Conley, K. E. (2001). Large energetic adaptations of elderly muscle to resistance and endurance training. J. Appl. Physiol. 90, 1663–1670. doi: 10.1152/jappl.2001.90.5.1663
Justice, J. N., Ferrucci, L., Newman, A. B., Aroda, V. R., Bahnson, J. L., Divers, J., et al. (2018). A framework for selection of blood-based biomarkers for geroscience-guided clinical trials: report from the TAME Biomarkers Workgroup. Geroscience 29, 419–436. doi: 10.1007/s11357-018-0042-y
Kavazis, A. N., Talbert, E. E., Smuder, A. J., Hudson, M. B., Nelson, W. B., and Powers, S. K. (2009). Mechanical ventilation induces diaphragmatic mitochondrial dysfunction and increased oxidant production. Free Radic. Biol. Med. 46, 842–850. doi: 10.1016/j.freeradbiomed.2009.01.002
Kelley, R. C., and Ferreira, L. F. (2017). Diaphragm abnormalities in heart failure and aging: mechanisms and integration of cardiovascular and respiratory pathophysiology. Heart Fail. Rev. 22, 191–207. doi: 10.1007/s10741-016-9549-4
Kelly, N. A., Hammond, K. G., Stec, M. J., Bickel, C. S., Windham, S. T., Tuggle, S. C., et al. (2017). Quantification and characterization of grouped type I myofibers in human aging. Muscle Nerve. 57, E52–E59. doi: 10.1002/mus.25711
Kenny, A. M., Kleppinger, A., Annis, K., Rathier, M., Browner, B., Judge, J. O., et al. (2010). Effects of transdermal testosterone on bone and muscle in older men with low bioavailable testosterone levels, low bone mass, and physical frailty. J. Am. Geriatr. Soc. 58, 1134–1143. doi: 10.1111/j.1532-5415.2010.02865.x
Kim, M. J., and Morley, J. E. (2005). The hormonal fountains of youth: myth or reality? J. Endocrinol. Invest. 28(Suppl. 11), 5–14.
Konopka, A. R., Asante, A., Lanza, I. R., Robinson, M. M., Johnson, M. L., Dalla Man, C., et al. (2015). Defects in mitochondrial efficiency and H2O2 emissions in obese women are restored to a lean phenotype with aerobic exercise training. Diabetes 64, 2104–2115. doi: 10.2337/db14-1701
Koopman, R., and van Loon, L. J. C. (2009). Aging, exercise, and muscle protein metabolism. J. Appl. Physiol. 106, 2040–2048. doi: 10.1152/japplphysiol.91551.2008
Kortebein, P., Ferrando, A., Lombeida, J., Wolfe, R., and Evans, W. J. (2007). Effect of 10 days of bed rest on skeletal muscle in healthy older adults. JAMA 297, 1772–1774. doi: 10.1001/jama.297.16.1772-b
Kovacheva, E. L., Hikim, A. P. S., Shen, R., Sinha, I., and Sinha-Hikim, I. (2010). Testosterone supplementation reverses sarcopenia in aging through regulation of myostatin, c-Jun NH2-terminal kinase, Notch, and Akt signaling pathways. Endocrinology 151, 628–638. doi: 10.1210/en.2009-1177
Kvorning, T., Christensen, L. L., Madsen, K., Nielsen, J. L., Gejl, K. D., Brixen, K., et al. (2013). Mechanical muscle function and lean body mass during supervised strength training and testosterone therapy in aging men with low-normal testosterone levels. J. Am. Geriatr. Soc. 61, 957–962. doi: 10.1111/jgs.12279
Landi, F., Cruz-Jentoft, A. J., Liperoti, R., Russo, A., Giovannini, S., Tosato, M., et al. (2013). Sarcopenia and mortality risk in frail older persons aged 80 years and older: results from ilSIRENTE study. Age Ageing 42, 203–209. doi: 10.1093/ageing/afs194
Landi, F., Marzetti, E., Martone, A. M., Bernabei, R., and Onder, G. (2014). Exercise as a remedy for sarcopenia. Curr. Opin. Clin. Nutr. Metab. Care 17, 25–31. doi: 10.1097/MCO.0000000000000018
Lanza, I. R., Short, D. K., Short, K. R., Raghavakaimal, S., Basu, R., Joyner, M. J., et al. (2008). Endurance exercise as a countermeasure for aging. Diabetes 57, 2933–2942. doi: 10.2337/db08-0349
Larsen, R. G., Callahan, D. M., Foulis, S. A., and Kent-Braun, J. A. (2012a). Age-related changes in oxidative capacity differ between locomotory muscles and are associated with physical activity behavior. Appl. Physiol. Nutr. Metab. 37, 88–99. doi: 10.1139/h11-135
Larsen, S., Hey Mogensen, M., Rabøl, R., Stride, N., Helge, J. W., and Dela, F. (2012b). The influence of age and aerobic fitness: effects on mitochondrial respiration in skeletal muscle. Acta Physiol. 205, 423–432. doi: 10.1111/j.1748-1716.2012.02408.x
Latres, E., Mastaitis, J., Fury, W., Miloscio, L., Trejos, J., Pangilinan, J., et al. (2017). Activin A more prominently regulates muscle mass in primates than does GDF8. Nat. Commun. 8:15153. doi: 10.1038/ncomms15153
Lazarus, N. R., and Harridge, S. D. R. (2017). Declining performance of master athletes: silhouettes of the trajectory of healthy human ageing? J. Physiol. 595, 2941–2948. doi: 10.1113/JP272443
Lee, H.-Y., Choi, C. S., Birkenfeld, A. L., Alves, T. C., Jornayvaz, F. R., Jurczak, M. J., et al. (2010). Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab. 12, 668–674. doi: 10.1016/j.cmet.2010.11.004
Lee, S.-J. (2004). Regulation of muscle mass by myostatin. Annu. Rev. Cell Dev. Biol. 20, 61–86. doi: 10.1146/annurev.cellbio.20.012103.135836
Levine, R. L., and Stadtman, E. R. (2001). Oxidative modification of proteins during aging. Exp. Gerontol. 36, 1495–1502. doi: 10.1016/S0531-5565(01)00135-8
Li, Y.-P., Chen, Y., Li, A. S., and Reid, M. B. (2003). Hydrogen peroxide stimulates ubiquitin-conjugating activity and expression of genes for specific E2 and E3 proteins in skeletal muscle myotubes. Am. J. Physiol. Cell Physiol. 285, C806–C812. doi: 10.1152/ajpcell.00129.2003
Liu, H., Bravata, D. M., Olkin, I., Nayak, S., Roberts, B., Garber, A. M., et al. (2007). Systematic review: the safety and efficacy of growth hormone in the healthy elderly. Ann. Intern. Med. 146, 104–115.
Long, D. E., Peck, B. D., Martz, J. L., Tuggle, S. C., Bush, H. M., McGwin, G., et al. (2017). Metformin to augment strength training effective response in seniors (MASTERS): study protocol for a randomized controlled trial. Trials 18:192. doi: 10.1186/s13063-017-1932-5
Lourenço dos Santos, S., Baraibar, M. A., Lundberg, S., Eeg-Olofsson, O., Larsson, L., and Friguet, B. (2015). Oxidative proteome alterations during skeletal muscle ageing. Redox Biol. 5, 267–274. doi: 10.1016/j.redox.2015.05.006
Lynch, M., and Marinov, G. K. (2015). The bioenergetic costs of a gene. Proc. Natl. Acad. Sci. 112, 15690–15695. doi: 10.1073/pnas.1514974112
Makimura, H., Stanley, T. L., Sun, N., Hrovat, M. I., Systrom, D. M., and Grinspoon, S. K. (2011). The association of growth hormone parameters with skeletal muscle phosphocreatine recovery in adult men. J. Clin. Endocrinol. Metab. 96, 817–823. doi: 10.1210/jc.2010-2264
Marques, A., Sarmento, H., Martins, J., and Saboga Nunes, L. (2015). Prevalence of physical activity in European adults—Compliance with the World Health Organization’s physical activity guidelines. Prev. Med. 81, 333–338. doi: 10.1016/j.ypmed.2015.09.018
Max, S. R. (1972). Disuse atrophy of skeletal muscle: loss of functional activity of mitochondria. Biochem. Biophys. Res. Commun. 46, 1394–1398. doi: 10.1016/S0006-291X(72)80130-X
McClung, J. M., Judge, A. R., Talbert, E. E., and Powers, S. K. (2009). Calpain-1 is required for hydrogen peroxide-induced myotube atrophy. AJP: Cell Physiology 296, C363–C371. doi: 10.1152/ajpcell.00497.2008
McDonagh, B., Scullion, S. M., Vasilaki, A., Pollock, N., McArdle, A., and Jackson, M. J. (2016). Ageing-induced changes in the redox status of peripheral motor nerves imply an effect on redox signalling rather than oxidative damage. Free Radic. Biol. Med. 94, 27–35. doi: 10.1016/j.freeradbiomed.2016.02.008
Menshikova, E. V., Ritov, V. B., Fairfull, L., Ferrell, R. E., Kelley, D. E., and Goodpaster, B. H. (2006). Effects of exercise on mitochondrial content and function in aging human skeletal muscle. J. Gerontol. A Biol. Sci. Med. Sci. 61, 534–540. doi: 10.1093/gerona/61.6.534
Mikkelsen, U. R., Couppé, C., Karlsen, A., Grosset, J. F., Schjerling, P., Mackey, A. L., et al. (2013). Life-long endurance exercise in humans: circulating levels of inflammatory markers and leg muscle size. Mech. Ageing Dev. 134, 531–540. doi: 10.1016/j.mad.2013.11.004
Min, K., Smuder, A. J., Kwon, O.-S., Kavazis, A. N., Szeto, H. H., and Powers, S. K. (2011). Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J. Appl. Physiol. 111, 1459–1466. doi: 10.1152/japplphysiol.00591.2011
Mithal, A., Bonjour, J. P., Boonen, S., Burckhardt, P., Degens, H., Fuleihan, H., et al. (2013). Impact of nutrition on muscle mass, strength, and performance in older adults. Osteoporos. Int. 24, 1555–1566. doi: 10.1007/s00198-012-2236-y
Morley, J. E. (2016). Pharmacologic options for the treatment of sarcopenia. Calcif. Tissue Int. 98, 319–333. doi: 10.1007/s00223-015-0022-5
Morley, J. E., Anker, S. D., and Haehling von, S. (2014). Prevalence, incidence, and clinical impact of sarcopenia: facts, numbers, and epidemiology—update 2014. J. Cachexia Sarcopenia Muscle 5, 253–259. doi: 10.1007/s13539-014-0161-y
Morley, J. E., Kaiser, F. E., Kraenzle, D., Jensen, J., Houston, K., Mattammal, M., et al. (1993). Effects of testosterone replacement therapy in old hypogonadal males: a preliminary study. J. Am. Geriatr. Soc. 41, 149–152.
Mouisel, E., Relizani, K., Mille-Hamard, L., Denis, R., Hourdé, C., Agbulut, O., et al. (2014). Myostatin is a key mediator between energy metabolism and endurance capacity of skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 307, R444–R454. doi: 10.1152/ajpregu.00377.2013
Musci, R. V., Hamilton, K. L., and Miller, B. F. (2017). Targeting mitochondrial function and proteostasis to mitigate dynapenia. Eur. J. Appl. Physiol. 104, 1–9. doi: 10.1007/s00421-017-3730-x
Müller, F. L., Song, W., Jang, Y. C., Liu, Y., Sabia, M., Richardson, A., et al. (2007). Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R1159–R1168. doi: 10.1152/ajpregu.00767.2006
Neufer, P. D., Bamman, M. M., Muoio, D. M., Bouchard, C., Cooper, D. M., Goodpaster, B. H., et al. (2015). Understanding the cellular and molecular mechanisms of physical activity-induced health benefits. Cell Metab. 22, 4–11. doi: 10.1016/j.cmet.2015.05.011
Nilwik, R., Snijders, T., Leenders, M., Groen, B. B. L., van Kranenburg, J., Verdijk, L. B., et al. (2013). The decline in skeletal muscle mass with aging is mainly attributed to a reduction in type II muscle fiber size. Exp. Gerontol. 48, 492–498. doi: 10.1016/j.exger.2013.02.012
Olshansky, S. J., Goldman, D. P., Zheng, Y., and Rowe, J. W. (2009). Aging in America in the twenty-first century: demographic forecasts from the MacArthur Foundation Research Network on an Aging Society. Milbank Q. 87, 842–862. doi: 10.1111/j.1468-0009.2009.00581.x
Paddon-Jones, D., Sheffield-Moore, M., Urban, R. J., Sanford, A. P., Aarsland, A., Wolfe, R. R., et al. (2004). Essential amino acid and carbohydrate supplementation ameliorates muscle protein loss in humans during 28 days bedrest. J. Clin. Endocrinol. Metab. 89, 4351–4358. doi: 10.1210/jc.2003-032159
Pahor, M., Guralnik, J. M., Ambrosius, W. T., Blair, S., Bonds, D. E., Church, T. S., et al. (2014). Effect of structured physical activity on prevention of major mobility disability in older adults: the LIFE study randomized clinical trial. JAMA 311, 2387–2396. doi: 10.1001/jama.2014.5616
Parsons, T. J., Sartini, C., Welsh, P., Sattar, N., Ash, S., Lennon, L. T., et al. (2017). Physical activity, sedentary behavior, and inflammatory and hemostatic markers in men. Med. Sci. Sports Exerc. 49, 459–465. doi: 10.1249/MSS.0000000000001113
Patel, K. V., Coppin, A. K., Manini, T. M., Lauretani, F., Bandinelli, S., Ferrucci, L., et al. (2006). Midlife physical activity and mobility in older age: the InCHIANTI study. Am. J. Prev. Med. 31, 217–224. doi: 10.1016/j.amepre.2006.05.005
Paulsen, G., Cumming, K. T., Holden, G., Hallén, J., Rønnestad, B. R., Sveen, O., et al. (2014). Vitamin C and E supplementation hampers cellular adaptation to endurance training in humans: a double-blind randomized controlled trial. J. Physiol. 592, 5391–5408. doi: 10.1113/jphysiol.2013.267419
Pedersen, B. K., and Febbraio, M. A. (2012). Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 8, 457–465. doi: 10.1038/nrendo.2012.49
Pedersen, M., Bruunsgaard, H., Weis, N., Hendel, H. W., Andreassen, B. U., Eldrup, E., et al. (2003). Circulating levels of TNF-alpha and IL-6-relation to truncal fat mass and muscle mass in healthy elderly individuals and in patients with type-2 diabetes. Mech. Ageing Dev. 124, 495–502. doi: 10.1016/S0047-6374(03)00027-7
Petersson, S. J., Christensen, L. L., Kristensen, J. M., Kruse, R., Andersen, M., and Højlund, K. (2014). Effect of testosterone on markers of mitochondrial oxidative phosphorylation and lipid metabolism in muscle of aging men with subnormal bioavailable testosterone. Eur. J. Endocrinol. 171, 77–88. doi: 10.1530/EJE-14-0006
Pham, F. H., Sugden, P. H., and Clerk, A. (2000). Regulation of protein kinase B and 4E-BP1 by oxidative stress in cardiac myocytes. Circ. Res. 86, 1252–1258. doi: 10.1161/01.RES.86.12.1252
Porter, C., Hurren, N. M., Cotter, M. V., Bhattarai, N., Reidy, P. T., Dillon, E. L., et al. (2015a). Mitochondrial respiratory capacity and coupling control decline with age in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 309, E224–E232. doi: 10.1152/ajpendo.00125.2015
Porter, C., Reidy, P. T., Bhattarai, N., Sidossis, L. S., and Rasmussen, B. B. (2015b). Resistance exercise training alters mitochondrial function in human skeletal muscle. Med. Sci. Sports Exerc. 47, 1922–1931. doi: 10.1249/MSS.0000000000000605
Power, G. A., Allen, M. D., Gilmore, K. J., Stashuk, D. W., Doherty, T. J., Hepple, R. T., et al. (2016a). Motor unit number and transmission stability in octogenarian world class athletes: can age-related deficits be outrun? J. Appl. Physiol. 121, 1013–1020. doi: 10.1152/japplphysiol.00149.2016
Power, G. A., Minozzo, F. C., Spendiff, S., Filion, M. E., Konokhova, Y., Purves-Smith, M. F., et al. (2016b). Reduction in single muscle fiber rate of force development with aging is not attenuated in world class older masters athletes. AJP: Cell Physiology 310, C318–C327. doi: 10.1152/ajpcell.00289.2015
Powers, S. K., Hudson, M. B., Nelson, W. B., Talbert, E. E., Min, K., Szeto, H. H., et al. (2011). Mitochondria-targeted antioxidants protect against mechanical ventilation-induced diaphragm weakness. Crit. Care Med. 39, 1749–1759. doi: 10.1097/CCM.0b013e3182190b62
Proctor, D. N., Sinning, W. E., Walro, J. M., Sieck, G. C., and Lemon, P. W. (1995). Oxidative capacity of human muscle fiber types: effects of age and training status. J. Appl. Physiol. 78, 2033–2038. doi: 10.1152/jappl.1995.78.6.2033
Purves-Smith, F. M., Sgarioto, N., and Hepple, R. T. (2014). Fiber typing in aging muscle. Exerc. Sport Sci. Rev. 42, 45–52. doi: 10.1249/JES.0000000000000012
Rana, P., Marwaha, R. K., Kumar, P., Narang, A., Devi, M. M., Tripathi, R. P., et al. (2014). Effect of vitamin D supplementation on muscle energy phospho-metabolites: a 31P magnetic resonance spectroscopy-based pilot study. Endocr. Res. 39, 152–156. doi: 10.3109/07435800.2013.865210
Rasmussen, U. F., Krustrup, P., Kjaer, M., and Rasmussen, H. N. (2003). Human skeletal muscle mitochondrial metabolism in youth and senescence: no signs of functional changes in ATP formation and mitochondrial oxidative capacity. Pflugers Arch. 446, 270–278. doi: 10.1007/s00424-003-1022-2
Reid, K. F., Callahan, D. M., Carabello, R. J., Phillips, E. M., Frontera, W. R., and Fielding, R. A. (2008). Lower extremity power training in elderly subjects with mobility limitations: a randomized controlled trial. Aging Clin. Exp. Res. 20, 337–343. doi: 10.1007/BF03324865
Ristow, M., and Schmeisser, K. (2014). Mitohormesis: promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose-Response 12, 288–341. doi: 10.2203/dose-response.13-035.Ristow
Rivas, D. A., Morris, E. P., Haran, P. H., Pasha, E. P., Morais, M. D. S., Dolnikowski, G. G., et al. (2012). Increased ceramide content and NFκB signaling may contribute to the attenuation of anabolic signaling after resistance exercise in aged males. J. Appl. Physiol. 113, 1727–1736. doi: 10.1152/japplphysiol.00412.2012
Robinson, M. M., Dasari, S., Konopka, A. R., Johnson, M. L., Manjunatha, S., Esponda, R. R., et al. (2017). Enhanced protein translation underlies improved metabolic and physical adaptations to different exercise training modes in young and old humans. Cell Metab. 25, 581–592. doi: 10.1016/j.cmet.2017.02.009
Robinson, M. M., Hamilton, K. L., and Miller, B. F. (2009). The interactions of some commonly consumed drugs with mitochondrial adaptations to exercise. J. Appl. Physiol. 107, 8–16. doi: 10.1152/japplphysiol.00343.2009
Rolfe, D. F., and Brown, G. C. (1997). Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 77, 731–758. doi: 10.1152/physrev.1997.77.3.731
Romanello, V., Guadagnin, E., Gomes, L., Roder, I., Sandri, C., Petersen, Y., et al. (2010). Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 29, 1774–1785. doi: 10.1038/emboj.2010.60
Rooks, D., Praestgaard, J., Hariry, S., Laurent, D., Petricoul, O., Perry, R. G., et al. (2017). Treatment of sarcopenia with Bimagrumab: results from a phase II, randomized, controlled, proof-of-concept study. J. Am. Geriatr. Soc. 65, 1988–1995. doi: 10.1111/jgs.14927
Roseno, S. L., Davis, P. R., Bollinger, L. M., Powell, J. J. S., Witczak, C. A., and Brault, J. J. (2015). Short-term, high-fat diet accelerates disuse atrophy and protein degradation in a muscle-specific manner in mice. Nutr. Metab. 12:39. doi: 10.1186/s12986-015-0037-y
Rudman, D., Feller, A. G., Nagraj, H. S., Gergans, G. A., Lalitha, P. Y., Goldberg, A. F., et al. (1990). Effects of human growth hormone in men over 60 years old. N. Engl. J. Med. 323, 1–6. doi: 10.1056/NEJM199007053230101
Ryan, M. J., Dudash, H. J., Docherty, M., Geronilla, K. B., Baker, B. A., Haff, G. G., et al. (2010). Vitamin E and C supplementation reduces oxidative stress, improves antioxidant enzymes and positive muscle work in chronically loaded muscles of aged rats. Exp. Gerontol. 45, 882–895. doi: 10.1016/j.exger.2010.08.002
Saad, F., Röhrig, G., Haehling von, S., and Traish, A. (2017). Testosterone deficiency and testosterone treatment in older men. Gerontology 63, 144–156. doi: 10.1159/000452499
Safdar, A., Hamadeh, M. J., Kaczor, J. J., Raha, S., deBeer, J., and Tarnopolsky, M. A. (2010). Aberrant mitochondrial homeostasis in the skeletal muscle of sedentary older adults. PLoS One 5:e10778. doi: 10.1371/journal.pone.0010778
Sakellariou, G. K., McDonagh, B., Porter, H., Giakoumaki, I. I., Earl, K. E., Nye, G. A., et al. (2018). Comparison of whole body SOD1 knockout with muscle-specific SOD1 knockout mice reveals a role for nerve redox signaling in regulation of degenerative pathways in skeletal muscle. Antioxid. Redox Signal. 28, 275–295. doi: 10.1089/ars.2017.7249
Salminen, A., Ojala, J., Kaarniranta, K., and Kauppinen, A. (2012). Mitochondrial dysfunction and oxidative stress activate inflammasomes: impact on the aging process and age-related diseases. Cell. Mol. Life Sci. 69, 2999–3013. doi: 10.1007/s00018-012-0962-0
Santanasto, A. J., Coen, P. M., Glynn, N. W., Conley, K. E., Jubrias, S. A., Amati, F., et al. (2016). The relationship between mitochondrial function and walking performance in older adults with a wide range of physical function. Exp. Gerontol. 81, 1–7. doi: 10.1016/j.exger.2016.04.002
Santanasto, A. J., Glynn, N. W., Jubrias, S. A., Conley, K. E., Boudreau, R. M., Amati, F., et al. (2015). Skeletal muscle mitochondrial function and fatigability in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 70, 1379–1385. doi: 10.1093/gerona/glu134
Scalzo, R. L., Peltonen, G. L., Binns, S. E., Shankaran, M., Giordano, G. R., Hartley, D. A., et al. (2014). Greater muscle protein synthesis and mitochondrial biogenesis in males compared with females during sprint interval training. FASEB J. 28, 2705–2714. doi: 10.1096/fj.13-246595
Schaap, L. A., Schaap, L. A., Pluijm, S. M. F., Pluijm, S. M., Deeg, D. J., Deeg, D. J. H., et al. (2006). Inflammatory markers and loss of muscle mass (sarcopenia) and strength. Am. J. Med. 119, 526.e9–526.e17. doi: 10.1016/j.amjmed.2005.10.049
Schoenfeld, B. J., Grgic, J., Ogborn, D., and Krieger, J. W. (2017). Strength and hypertrophy adaptations between low- vs. high-load resistance training: a systematic review and meta-analysis. J. Strength Cond. Res. 31, 3508–3523. doi: 10.1519/JSC.0000000000002200
Schrager, M. A., Metter, E. J., Simonsick, E., Ble, A., Bandinelli, S., Lauretani, F., et al. (2007). Sarcopenic obesity and inflammation in the InCHIANTI study. J. Appl. Physiol. 102, 919–925. doi: 10.1152/japplphysiol.00627.2006
Semba, R. D., Nicklett, E. J., and Ferrucci, L. (2010). Does accumulation of advanced glycation end products contribute to the aging phenotype? J. Gerontol. A Biol. Sci. Med. Sci. 65, 963–975. doi: 10.1093/gerona/glq074
Shanley, D. P., and Kirkwood, T. B. (2000). Calorie restriction and aging: a life-history analysis. Evolution 54, 740–750. doi: 10.2307/2640568
Shenton, D., Smirnova, J. B., Selley, J. N., Carroll, K., Hubbard, S. J., Pavitt, G. D., et al. (2006). Global translational responses to oxidative stress impact upon multiple levels of protein synthesis. J. Biol. Chem. 281, 29011–29021. doi: 10.1074/jbc.M601545200
Short, K. R., Bigelow, M. L., Kahl, J., Singh, R., Coenen-Schimke, J., Raghavakaimal, S., et al. (2005). Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA. 102, 5618–5623. doi: 10.1073/pnas.0501559102
Sih, R., Morley, J. E., Kaiser, F. E., Perry, H. M., Patrick, P., and Ross, C. (1997). Testosterone replacement in older hypogonadal men: a 12-month randomized controlled trial. J. Clin. Endocrinol. Metab. 82, 1661–1667. doi: 10.1210/jcem.82.6.3988
Sinha, A., Hollingsworth, K. G., Ball, S., and Cheetham, T. (2013). Improving the vitamin D status of vitamin D deficient adults is associated with improved mitochondrial oxidative function in skeletal muscle. J. Clin. Endocrinol. Metab. 98, E509–E513. doi: 10.1210/jc.2012-3592
Sjögren, K., Leung, K.-C., Kaplan, W., Gardiner-Garden, M., Gibney, J., and Ho, K. K. Y. (2007). Growth hormone regulation of metabolic gene expression in muscle: a microarray study in hypopituitary men. Am. J. Physiol. Endocrinol. Metab. 293, E364–E371. doi: 10.1152/ajpendo.00054.2007
Smith, G. I., Julliand, S., Reeds, D. N., Sinacore, D. R., Klein, S., and Mittendorfer, B. (2015). Fish oil-derived n-3 PUFA therapy increases muscle mass and function in healthy older adults. Am. J. Clin. Nutr. 102, 115–122. doi: 10.3945/ajcn.114.105833
Snyder, P. J., Bhasin, S., Cunningham, G. R., Matsumoto, A. M., Stephens-Shields, A. J., Cauley, J. A., et al. (2016). Effects of testosterone treatment in older men. N. Engl. J. Med. 374, 611–624. doi: 10.1056/NEJMoa1506119
Snyder, P. J., Peachey, H., Hannoush, P., Berlin, J. A., Loh, L., Lenrow, D. A., et al. (1999). Effect of testosterone treatment on body composition and muscle strength in men over 65 years of age. J. Clin. Endocrinol. Metab. 84, 2647–2653. doi: 10.1210/jcem.84.8.5885
Spendiff, S., Vuda, M., Gouspillou, G., Aare, S., Perez, A., Morais, J. A., et al. (2016). Denervation drives mitochondrial dysfunction in skeletal muscle of octogenarians. J. Physiol. 594, 7361–7379. doi: 10.1113/JP272487
Srinivas-Shankar, U., Roberts, S. A., Connolly, M. J., O’Connell, M. D. L., Adams, J. E., Oldham, J. A., et al. (2010). Effects of testosterone on muscle strength, physical function, body composition, and quality of life in intermediate-frail and frail elderly men: a randomized, double-blind, placebo-controlled study. J. Clin. Endocrinol. Metab. 95, 639–650. doi: 10.1210/jc.2009-1251
St-Jean Pelletier, F., Pion, C. H., Leduc-Gaudet, J.-P., Sgarioto, N., Zovilé, I., Barbat-Artigas, S., et al. (2017). The impact of ageing, physical activity, and pre-frailty on skeletal muscle phenotype, mitochondrial content, and intramyocellular lipids in men. J. Cachexia Sarcopenia Muscle 8, 213–228. doi: 10.1002/jcsm.12139
Stenholm, S., Harris, T. B., Rantanen, T., Visser, M., Kritchevsky, S. B., and Ferrucci, L. (2008). Sarcopenic obesity: definition, cause and consequences. Curr. Opin. Clin. Nutr. Metab. Care 11, 693–700. doi: 10.1097/MCO.0b013e328312c37d
Strong, R., Miller, R. A., Antebi, A., Astle, C. M., Bogue, M., Denzel, M. S., et al. (2016). Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an α-glucosidase inhibitor or a Nrf2-inducer. Aging Cell 15, 872–884. doi: 10.1111/acel.12496
Suetta, C., Frandsen, U., Mackey, A. L., Jensen, L., Hvid, L. G., Bayer, M. L., et al. (2013). Ageing is associated with diminished muscle re-growth and myogenic precursor cell expansion early after immobility-induced atrophy in human skeletal muscle. J. Physiol. 591, 3789–3804. doi: 10.1113/jphysiol.2013.257121
Suetta, C., Hvid, L. G., Justesen, L., Christensen, U., Neergaard, K., Simonsen, L., et al. (2009). Effects of aging on human skeletal muscle after immobilization and retraining. J. Appl. Physiol. 107, 1172–1180. doi: 10.1152/japplphysiol.00290.2009
Sun, K., Semba, R. D., Fried, L. P., Schaumberg, D. A., Ferrucci, L., and Varadhan, R. (2012). Elevated serum carboxymethyl-Lysine, an advanced glycation end product, predicts severe walking disability in older eomen: the Women’s Health and Aging Study I. J Aging Res. 2012:586385. doi: 10.1155/2012/586385
Sundberg, C. W., Kuplic, A., Hassanlouei, H., and Hunter, S. K. (2018). Mechanisms for the age-related increase in fatigability of the knee extensors in old and very old adults. J. Appl. Physiol. 125, 146–158. doi: 10.1152/japplphysiol.01141.2017
Tevald, M. A., Foulis, S. A., and Kent, J. A. (2014). Effect of age on in vivo oxidative capacity in two locomotory muscles of the leg. Age 36: 9713. doi: 10.1007/s11357-014-9713-5
Tezze, C., Romanello, V., Desbats, M. A., Fadini, G. P., Albiero, M., Favaro, G., et al. (2017). Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab. 25, 1374–1389.e6. doi: 10.1016/j.cmet.2017.04.021
Tonkonogi, M., Fernström, M., Walsh, B., Ji, L. L., Rooyackers, O., Hammarqvist, F., et al. (2003). Reduced oxidative power but unchanged antioxidative capacity in skeletal muscle from aged humans. Pflugers Arch. 446, 261–269. doi: 10.1007/s00424-003-1044-9
Traish, A. M., Abdallah, B., and Yu, G. (2011). Androgen deficiency and mitochondrial dysfunction: implications for fatigue, muscle dysfunction, insulin resistance, diabetes, and cardiovascular disease. Horm. Mol. Biol. Clin. Invest. 8, 431–444. doi: 10.1515/HMBCI.2011.132
Trounce, I., Byrne, E., and Marzuki, S. (1989). Decline in skeletal muscle mitochondrial respiratory chain function: possible factor in ageing. Lancet 1, 637–639. doi: 10.1016/S0140-6736(89)92143-0
Tschopp, M., Sattelmayer, M. K., and Hilfiker, R. (2011). Is power training or conventional resistance training better for function in elderly persons? A meta-analysis. Age Ageing 40, 549–556. doi: 10.1093/ageing/afr005
Tucker, J. M., Welk, G. J., and Beyler, N. K. (2011). Physical activity in U.S.: adults compliance with the Physical Activity Guidelines for Americans. Am. J. Prev. Med. 40, 454–461. doi: 10.1016/j.amepre.2010.12.016
Usui, T., Kajita, K., Kajita, T., Mori, I., Hanamoto, T., Ikeda, T., et al. (2014). Elevated mitochondrial biogenesis in skeletal muscle is associated with testosterone-induced body weight loss in male mice. FEBS Lett. 588, 1935–1941. doi: 10.1016/j.febslet.2014.03.051
Vasilaki, A., McArdle, F., Iwanejko, L. M., and McArdle, A. (2006). Adaptive responses of mouse skeletal muscle to contractile activity: the effect of age. Mech. Ageing Dev. 127, 830–839. doi: 10.1016/j.mad.2006.08.004
Vasilaki, A., Richardson, A., Van Remmen, H., Brooks, S. V., Larkin, L., McArdle, A., et al. (2017). Role of nerve-muscle interactions and reactive oxygen species in regulation of muscle proteostasis with ageing. J. Physiol. 595, 6409–6415. doi: 10.1113/JP274336
Vays, V. B., Eldarov, C. M., Vangely, I. M., Kolosova, N. G., Bakeeva, L. E., and Skulachev, V. P. (2014). Antioxidant SkQ1 delays sarcopenia-associated damage of mitochondrial ultrastructure. Aging 6, 140–148. doi: 10.18632/aging.100636
Verissimo, P., Casalaspo, T. J. A., Gonçalves, L. H. R., Yang, A. S. Y., Eid, R. C., and Timenetsky, K. T. (2015). High prevalence of respiratory muscle weakness in hospitalized acute heart failure elderly patients. PLoS One 10:e0118218. doi: 10.1371/journal.pone.0118218
Volpi, E., Campbell, W. W., Dwyer, J. T., Johnson, M. A., Jensen, G. L., Morley, J. E., et al. (2013). Is the optimal level of protein intake for older adults greater than the recommended dietary allowance? J. Gerontol. A Biol. Sci. Med. Sci. 68, 677–681. doi: 10.1093/gerona/gls229
Volpi, E., Mittendorfer, B., Rasmussen, B. B., and Wolfe, R. R. (2000). The response of muscle protein anabolism to combined hyperaminoacidemia and glucose-induced hyperinsulinemia is impaired in the elderly. J. Clin. Endocrinol. Metab. 85, 4481–4490. doi: 10.1210/jcem.85.12.7021
Wannamethee, S. G., and Atkins, J. L. (2015). Muscle loss and obesity: the health implications of sarcopenia and sarcopenic obesity. Proc. Nutr. Soc. 74, 405–412. doi: 10.1017/S002966511500169X
Westerterp, K. R. (2000). Daily physical activity and ageing. Curr. Opin. Clin. Nutr. Metab. Care 3, 485–488.
White, J. P., Gao, S., Puppa, M. J., Sato, S., Welle, S. L., and Carson, J. A. (2013). Testosterone regulation of Akt/mTORC1/FoxO3a signaling in skeletal muscle. Mol. Cell. Endocrinol. 365, 174–186. doi: 10.1016/j.mce.2012.10.019
White, J. R., Confides, A. L., Moore-Reed, S., Hoch, J. M., and Dupont-Versteegden, E. E. (2015). Regrowth after skeletal muscle atrophy is impaired in aged rats, despite similar responses in signaling pathways. Exp. Gerontol. 64, 17–32. doi: 10.1016/j.exger.2015.02.007
Wilkinson, S. B., Phillips, S. M., Atherton, P. J., Patel, R., Yarasheski, K. E., Tarnopolsky, M. A., et al. (2008). Differential effects of resistance and endurance exercise in the fed state on signalling molecule phosphorylation and protein synthesis in human muscle. J. Physiol. 586, 3701–3717. doi: 10.1113/jphysiol.2008.153916
Wolfe, R., Ferrando, A., Sheffield-Moore, M., and Urban, R. (2000). Testosterone and muscle protein metabolism. Mayo Clin. Proc. 75, discussion S59–S60.
Woodhouse, L., Gandhi, R., Warden, S. J., Poiraudeau, S., Myers, S. L., Benson, C. T., et al. (2016). A phase 2 randomized study investigating the efficacy and safety of myostatin antibody LY2495655 versus placebo in patients undergoing elective total hip arthroplasty. J. Frailty Aging 5, 62–70. doi: 10.14283/jfa.2016.81
Zadik, Z., Chalew, S. A., McCarter, R. J., Meistas, M., and Kowarski, A. A. (1985). The influence of age on the 24-hour integrated concentration of growth hormone in normal individuals. J. Clin. Endocrinol. Metab. 60, 513–516. doi: 10.1210/jcem-60-3-513
Zampieri, S., Pietrangelo, L., Loefler, S., Fruhmann, H., Vogelauer, M., Burggraf, S., et al. (2014). Lifelong physical exercise delays age-associated skeletal muscle decline. J. Gerontol. A Biol. Sci. Med. Sci. 70, 163–173. doi: 10.1093/gerona/glu006
Keywords: skeletal muscle, aging, sarcopenia, exercise, mitochondria, treatment
Citation: Coen PM, Musci RV, Hinkley JM and Miller BF (2019) Mitochondria as a Target for Mitigating Sarcopenia. Front. Physiol. 9:1883. doi: 10.3389/fphys.2018.01883
Edited by:
Martin Burtscher, University of Innsbruck, AustriaReviewed by:
Barbara Strasser, University of Regensburg, GermanyGiorgos K. Sakkas, University of St Mark & St John, United Kingdom
Copyright © 2019 Coen, Musci, Hinkley and Miller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benjamin F. Miller, QmVuamFtaW4tbWlsbGVyQG9tcmYub3Jn