 Ronald Wilders
Ronald Wilders- Department of Medical Biology, Amsterdam University Medical Centers, Amsterdam, Netherlands
Background: Carriers of the E161K mutation in the SCN5A gene, encoding the NaV1.5 pore-forming α-subunit of the ion channel carrying the fast sodium current (INa), show sinus bradycardia and occasional exit block. Voltage clamp experiments in mammalian expression systems revealed a mutation-induced 2.5- to 4-fold reduction in INa peak current density as well as a +19 mV shift and reduced steepness of the steady-state activation curve. The highly common H558R polymorphism in NaV1.5 limits this shift to +13 mV, but also introduces a -10 mV shift in steady-state inactivation.
Aim: We assessed the cellular mechanism by which the E161K mutation causes sinus node dysfunction in heterozygous mutation carriers as well as the potential role of the H558R polymorphism.
Methods: We incorporated the mutation-induced changes in INa into the Fabbri-Severi model of a single human sinoatrial node cell and the Maleckar et al. human atrial cell model, and carried out simulations under control conditions and over a wide range of acetylcholine levels.
Results: In absence of the H558R polymorphism, the E161K mutation increased the basic cycle length of the sinoatrial node cell from 813 to 866 ms. In the simulated presence of 10 and 25 nM acetylcholine, basic cycle length increased from 1027 to 1131 and from 1448 to 1795 ms, respectively. The increase in cycle length was the result of a significant slowing of diastolic depolarization. The mutation-induced reduction in INa window current had reduced the contribution of the mutant component of INa to the net membrane current during diastolic depolarization to effectively zero. Highly similar results were obtained in presence of the H558R polymorphism. Atrial excitability was reduced, both in absence and presence of the H558R polymorphism, as reflected by an increase in threshold stimulus current and a concomitant decrease in capacitive current of the atrial cell.
Conclusion: We conclude that the experimentally identified mutation-induced changes in INa can explain the clinically observed sinus bradycardia and potentially the occasional exit block. Furthermore, we conclude that the common H558R polymorphism does not significantly alter the effects of the E161K mutation and can thus not explain the reduced penetrance of the E161K mutation.
Introduction
The “fast sodium current“ (INa), which flows through NaV1.5 sodium channels, is a key player in the electrical activity of the human heart (Zimmer et al., 2014), where it is responsible for the fast >150 V/s upstroke of individual atrial and ventricular cardio-myocytes (Workman et al., 2001; O’Hara et al., 2011). The cardiac-specific NaV1.5 protein, encoded by the SCN5A gene, is the pore-forming α-subunit of the channel (Remme, 2013). INa has also been observed in patch-clamp recordings from isolated human sinoatrial (SA) node cells (Verkerk et al., 2009), in line with the observation by Chandler et al. (2009) that NaV1.5 is expressed in human SA nodal tissue. Accordingly, INa was included in the comprehensive computational model of a single human SA nodal cell that was recently developed by Fabbri et al. (2017) and is known as the Fabbri-Severi model.
Mutations in genes encoding ion channel-related proteins may result in inherited arrhythmia disorders, in particular the long QT syndrome (LQTS) and the Brugada syndrome (BrS). Gain-of-function and loss-of-function mutations in SCN5A underlie LQTS type 3 (LQT3) and BrS type 1 (BrS1), respectively (Brugada et al., 2018; Giudicessi et al., 2018; Wilde and Amin, 2018). Mutations in SCN5A may also lead to overlapping phenotypes (SCN5A overlap syndromes), where clinical characteristics of LQTS, BrS, and other arrhythmia syndromes, e.g., conduction disease and sinus node dysfunction, coexist in a single family or even in a single patient (Remme et al., 2008; Giudicessi et al., 2018). Sinus node dysfunction is a relatively common observation in both LQT3 and BrS1 patients (Hayashi et al., 2018; Wilders and Verkerk, 2018). It should be noted that the “gain of function” in case of LQT3 refers to the persistent INa that prolongs the ventricular action potential and thus causes QT interval prolongation. If an LQT3 mutation is associated with sinus bradycardia, this mutation shows a concomitant “loss of function” in the voltage range of diastolic depolarization (Wilders and Verkerk, 2018).
In 2005, Smits et al. reported the clinical and biophysical features of a novel sodium channel mutation, E161K, i.e., the replacement of the highly conserved acidic residue glutamic acid (E) with the basic residue lysine (K) at position 161 of the NaV1.5 protein (p.Glu161Lys; c.481G > A) (Smits et al., 2005). This loss-of-function mutation was identified in individuals of two non-related families. Affected patients had a complex clinical phenotype with symptoms of bradycardia, sinus node dysfunction, generalized conduction disease or BrS, or various combinations thereof. Sinus node dysfunction was observed in 8 out of 14 mutation carriers. Twenty-four-hour Holter recordings from 10 mutation carriers revealed that their absolute minimum heart rate, but not their maximum heart rate, was significantly lower compared to controls (39 ± 1 vs. 51 ± 0.6 beats/min, mean ± SEM). Furthermore, the incidence of signs of sinus node dysfunction in E161K mutation carriers was particularly high at night, when vagal tone is dominant.
Smits et al. (2005) assessed the biophysical effects of the E161K mutation by transfecting E161K or wild-type sodium channel α-subunit into tsA201 cells, together with wild-type β1-subunit. Voltage clamp experiments on the transfected cells revealed a 2.5-fold reduction in peak INa at -20 mV for E161K sodium channels compared to wild-type channels. Furthermore, the steady-state activation curve of the mutant channels showed a +11.9 mV shift of its half-maximal activation potential (V1/2) compared to wild-type (-30.7 ± 0.8 vs. -42.6 ± 1.4 mV, respectively). Also, the steepness of this curve was slightly reduced; its slope factor (k) amounted to 7.9 ± 0.3 vs. 6.7 ± 0.4 mV, respectively. Voltage dependence of steady-state inactivation, recovery from inactivation, and development of slow inactivation were not affected by the E161K mutation. Of note, these data were all obtained in H558 background, i.e., with histidine (H) at position 558 of the NaV1.5 protein.
Iwasa et al. (2000) were the first to report on the c.1673A > G single nucleotide polymorphism (“SNP”) in the SCN5A gene, which is responsible for the replacement of histidine (H) with arginine (R) at position 558 of the NaV1.5 protein (p.His558Arg or H558R), in relation to familial LQTS. Ackerman et al. (2004) showed that H558R is the most common polymorphism in SCN5A and that this variant is present in all four ethnic groups, albeit at a significantly lower prevalence in Asians. In blacks, whites, and Hispanics, the prevalence of R558 instead of H558 amounts to 20–30%, whereas this prevalence is near 10% among Asians (Ackerman et al., 2004). Over the years, it has become clear that the H558R polymorphism can either mitigate or aggravate the effects of specific mutations in SCN5A. For example, this polymorphism has mitigating effects on the mutations M1766L (Ye et al., 2003) and P2006A (Shinlapawittayatorn et al., 2011), but aggravating effects on the mutations G400A (Hu et al., 2007) and A572D (Tester et al., 2010).
In 2010, Gui et al. (2010a,b) demonstrated that the H558R polymorphism also affects the E161K mutation. They carried out voltage clamp experiments on HEK-293 cells transfected with E161K mutant or wild-type sodium channel α-subunit. Like Smits et al. (2005), they observed a reduction in peak INa for E161K sodium channels compared to wild-type channels, both in H558 and R558 background. In either case, this reduction was approximately 4-fold, which is more pronounced than the 2.5-fold reduction in the study by Smits et al. (2005). With a value of ≈19 mV, the positive shift in V1/2 of the steady-state activation curve in H558 background was also more pronounced. The steepness of this curve showed a reduction similar to that observed by Smits et al. (2005). In R558 background, the reduction in peak INa and steepness of the steady-state activation curve were both similar to those in H558 background (Gui et al., 2010b). However, the positive shift in V1/2 of the steady-state activation curve was less pronounced, with a value of ≈13 mV rather than ≈19 mV. However, the potentially mitigating effects of this less pronounced shift in R558 background was counteracted by a -10 mV shift in V1/2 of the steady-state inactivation curve, which was absent in H558 background.
To assess the mechanism by which the E161K mutation causes sinus node dysfunction as well as the potential role of the H558R polymorphism, we incorporated the mutation-induced changes in INa into the Fabbri-Severi model of a single human SA node cell (Fabbri et al., 2017). Furthermore, we incorporated these changes into the Maleckar et al. human atrial cell model (Maleckar et al., 2008, 2009) to assess the effects of the E161K mutation on atrial excitability, which may also play a role in sinus node dysfunction.
Materials and Methods
Implementation of the E161K Mutation and H558R Polymorphism
Effects of the heterozygous E161K mutation in SCN5A were implemented in the CellML code (Cuellar et al., 2003) of the Fabbri-Severi human SA nodal cell model (Fabbri et al., 2017) by scaling the fully-activated conductance of INa (gNa) and shifting the steady-state INa activation and/or inactivation curves, as detailed below, based on the experimental data from literature described in the Introduction and summarized in Table 1. The slight change in steepness of the steady-state activation curve was also included. These modifications were applied to half of the intrinsic INa channels, thus representing the heterozygous nature of the mutation, taking into account that a functional INa channel is built with a single NaV1.5 protein.
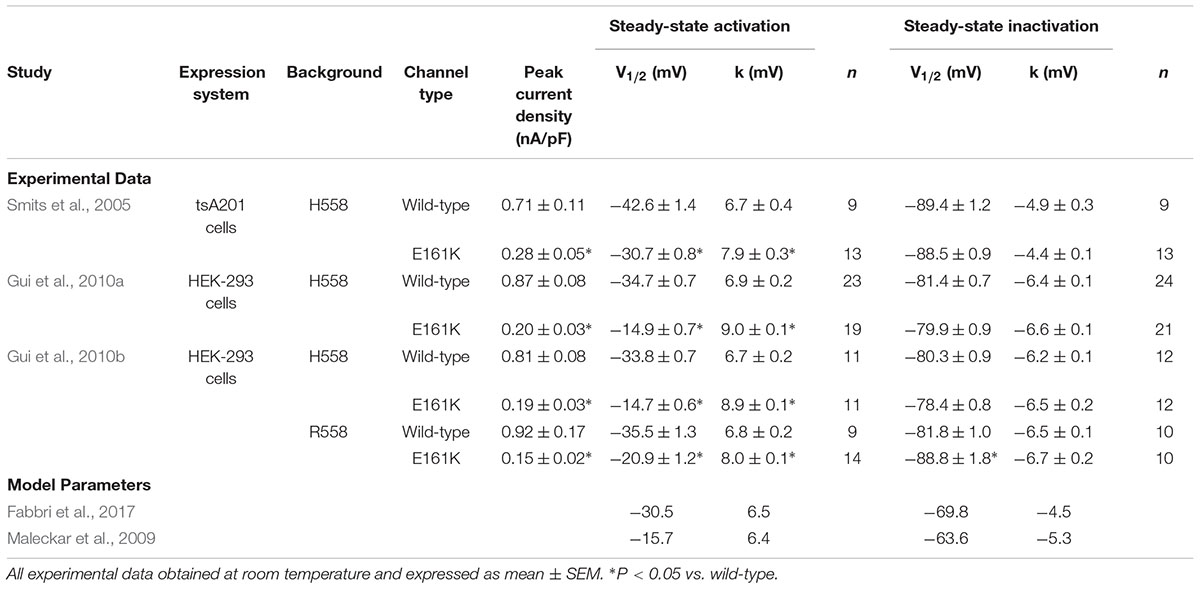
Table 1. Experimental data on biophysical effects of the E161K mutation in SCN5A.
Identical changes were applied to the Maleckar et al. (2009) human atrial cell model. The latter model, which is also known as the “human atrial myocyte with new repolarization” (hAMr) model, was selected because it includes well-validated equations for the acetylcholine-activated potassium current IK,ACh (Maleckar et al., 2008), thus allowing simulations over a wide range of acetylcholine levels. Action potentials were elicited with a 1 ms, ≈50% suprathreshold stimulus current at a frequency of 1 Hz. The threshold stimulus current amplitude was determined by increasing the stimulus current amplitude in 0.1 pA/pF steps until a train of 200 action potentials could successfully be elicited. To prevent slow drifts in ion concentrations, the intracellular Na+ and K+ concentrations were fixed, as were the cleft ion concentrations.
In H558 background, the E161K mutation was implemented by shifting the steady-state activation curve of the model INa by +19 mV. In R558 background, this curve was shifted by +13 mV, together with a -10 mV shift in the steady-state inactivation curve. In either background, a 50% decrease in gNa was applied to arrive at an almost 4-fold decrease in peak current density of mutant INa during voltage clamp simulations. Furthermore, the slope factor of the steady-state activation curve of the model INa was increased by 20% to account for mutation-induced decrease in steepness of this curve.
Computer Simulations
The CellML code of both models, as available from the CellML Model Repository (Lloyd et al., 2008), was edited and run in version 0.9.31.1409 of the Windows based Cellular Open Resource (COR) environment (Garny et al., 2003b). All simulations were run for a sufficiently long time, i.e., for the duration of a train of 200 action potentials, to reach steady-state behavior. Data from the final ten action potentials were used for analysis.
Results
Characteristics of the SA Nodal Fast Sodium Current
First, we characterized INa of the Fabbri-Severi model of a single human SA nodal pacemaker cell (Fabbri et al., 2017) and its effect on the spontaneous activity of this cell. Figure 1A shows the steady-state activation and inactivation curves of INa (left) and the associated “window” (area of overlap; right). The window is in the voltage range of diastolic depolarization, between -60 and -40 mV. Because of the relatively slow changes in membrane potential of the SA nodal cell and the relatively fast kinetics of INa, this window is the major determinant of the course of INa during the action potential, of course in combination with the fully-activated conductance of INa (gNa), which amounts to 22.3 nS in the Fabbri-Severi model. The associated steady-state current-voltage relationship of INa is shown in Figure 1B.
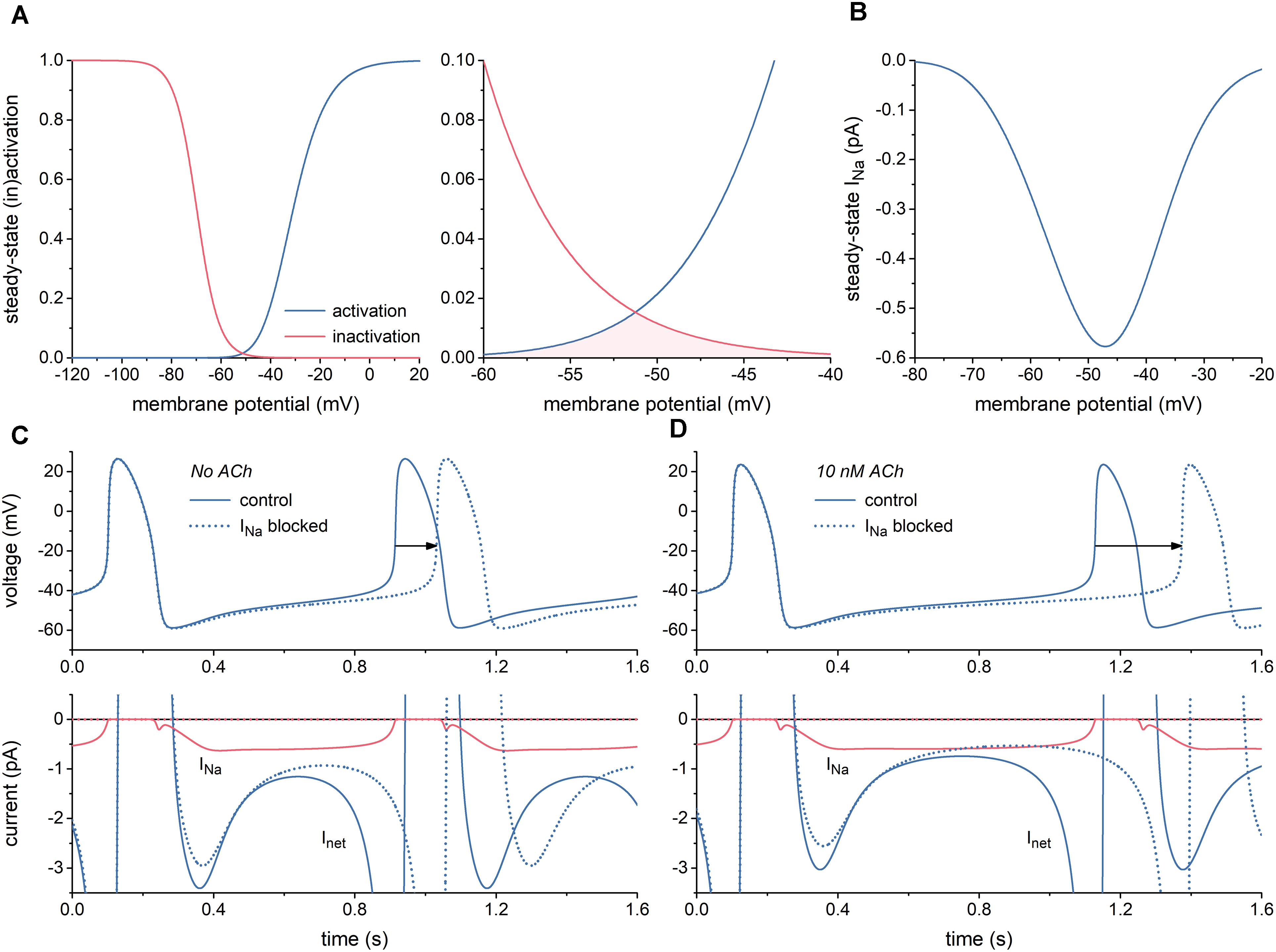
Figure 1. Characteristics of the fast sodium current (INa) of the Fabbri-Severi model of a human SA nodal pacemaker cell. (A) Steady-state activation and inactivation curves (blue and red curves, respectively) of INa (left) and associated “window” of overlap (shaded area) in the voltage range between -60 and -40 mV (right). (B) Current–voltage relationship of steady-state INa. (C) Effect of full block of INa in the absence of acetylcholine (ACh). Spontaneous action potentials (top) and associated net membrane current (Inet) and INa (bottom). Horizontal arrow indicates increase in cycle length. (D) Effect of full block of INa during simulated administration of 10 nM ACh. Spontaneous action potentials (top) and associated Inet and INa (bottom). Horizontal arrow indicates increase in cycle length.
With a value near 0.6 pA, the amplitude of INa during diastolic depolarization is small. However, this amplitude is relatively large in comparison to that of the net membrane current (Inet) (Figure 1C, solid lines). It is therefore not surprising that full block of INa results in a 117-ms increase in cycle length from 813 to 930 ms (Figure 1C, dotted lines), corresponding with a 13% decrease in beating rate from 74 to 64 beats/min. In the simulated presence of 10 nM acetylcholine (ACh), the amplitude of Inet during diastolic depolarization becomes considerably smaller, whereas that of INa does barely change. Accordingly, full block of INa now results in a more prominent increase in cycle length, by 245 ms from 1027 to 1272 ms (Figure 1D), corresponding with a 19% decrease in beating rate from 58 to 47 beats/min. Of note, INa hardly affects the cycle length through a change in action potential duration.
Now that the simulations of Figure 1 had shown that changes in INa may modify the cycle length of the Fabbri-Severi model cell to a considerable extent, we first assessed the changes in peak and window INa caused by the E161K mutation in SCN5A, either in H558 or R558 background. Next, we tested the effects of these changes on the spontaneous pacemaker activity of the SA nodal model cell.
Effects of the E161K Mutation in SCN5A on Biophysical Properties of INa
The left panel of Figure 2A illustrates the changes in the steady-state INa activation and inactivation curves due to the E161K mutation in H558 background. These changes are limited to a +19 mV shift in the steady-state INa activation curve and a slight decrease in steepness of this curve, which is poorly discernible in Figure 2A. As a result, the window of overlap is significantly reduced and shifted to less negative values of membrane potential, now roughly ranging from -50 to -30 mV (Figure 2A, right) and thus limiting the contribution of INa to diastolic depolarization. This is reflected in the current–voltage relationship of steady-state INa (Figure 2B, solid line), which is strongly reduced in comparison with wild-type INa.
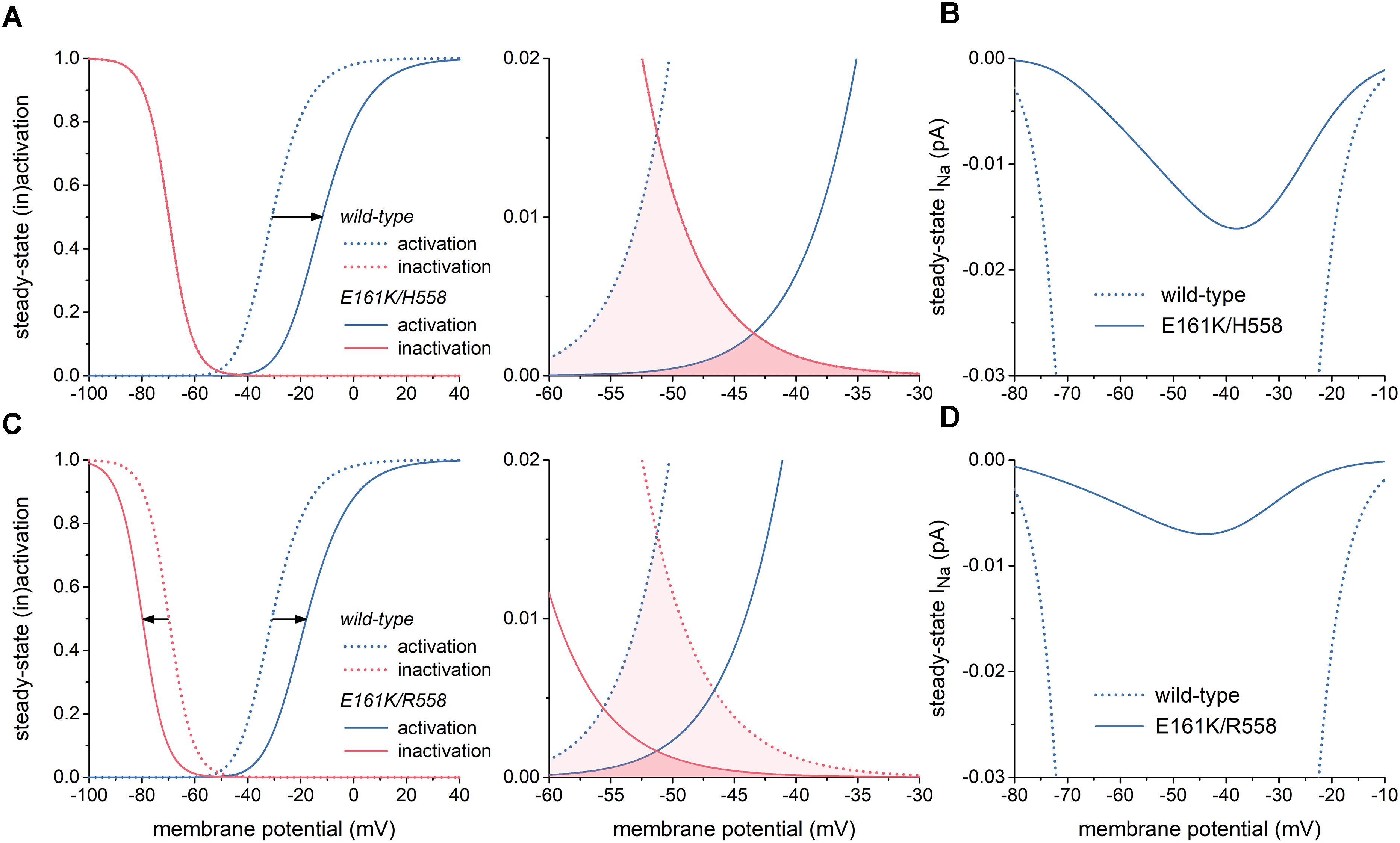
Figure 2. Effects of the E161K mutation in SCN5A, either in H558 or R558 background, on biophysical properties of the fast sodium current (INa) of the Fabbri-Severi model of a human SA nodal pacemaker cell. (A) Shift and change in steepness of the steady-state activation curve (left) and effect on “window” (right) in H558 background. Horizontal arrow indicates the +19 mV shift. (B) Resulting effect on current–voltage relationship of steady-state INa in H558 background. (C) Shift and change in steepness of the steady-state activation curve and shift of steady-state inactivation curve (left) and effect on “window” (right) in R558 background. Horizontal arrows indicate the +13 and -10 mV shifts. (D) Resulting effect on current–voltage relationship of steady-state INa in R558 background.
In R558 background, the shift in the steady-state INa activation curve is less pronounced (+13 vs. +19 mV), but accompanied by a -10 mV shift in the steady-state inactivation curve (Figure 2C, left). As a result, the window of overlap is smaller than in H558 background (Figure 2C, right). However, it better fits with the voltage range of diastolic depolarization. Yet, the steady-state INa in this voltage range is even more reduced in comparison with wild-type INa than in H558 background (Figure 2D, solid line).
Effects of the E161K Mutation in SCN5A on INa in Voltage Clamp Experiments
As set out in the Introduction, voltage clamp experiments on SCN5A channels expressed in tsA201 and HEK-293 cells (Smits et al., 2005; Gui et al., 2010a,b) had revealed that the E161K mutation induces a 2.5- to 4-fold decrease in INa peak current density, both in H558 and R558 background. To assess the extent to which this reduction is due to the changes in the INa steady-state activation and inactivation curves per se, we reconstructed current traces in response to voltage clamp steps from a holding potential of -120 mV to test potentials ranging from -100 to +50 mV. Figure 3A shows examples of wild-type and mutant current traces at test potentials of -40, -20, and 0 mV. These already demonstrate that the peak current density of the wild-type channels is higher than that of mutant channels, despite the identical value of gNa used in the voltage clamp simulations. Figure 3B summarizes the simulation data. The changes in the steady-state activation and inactivation curves per se reduce the peak current density by ≈35%. To arrive at an almost 4-fold decrease in INa peak current density, in line with the experimental observations, we reduced the mutant gNa by a factor of 2 in our further simulations. Thus we split the original gNa of 22.3 nS of the Fabbri-Severi model cell into 11.15 nS for the wild-type INa channels and 5.575 nS for the E161K mutant INa channels, in either background.
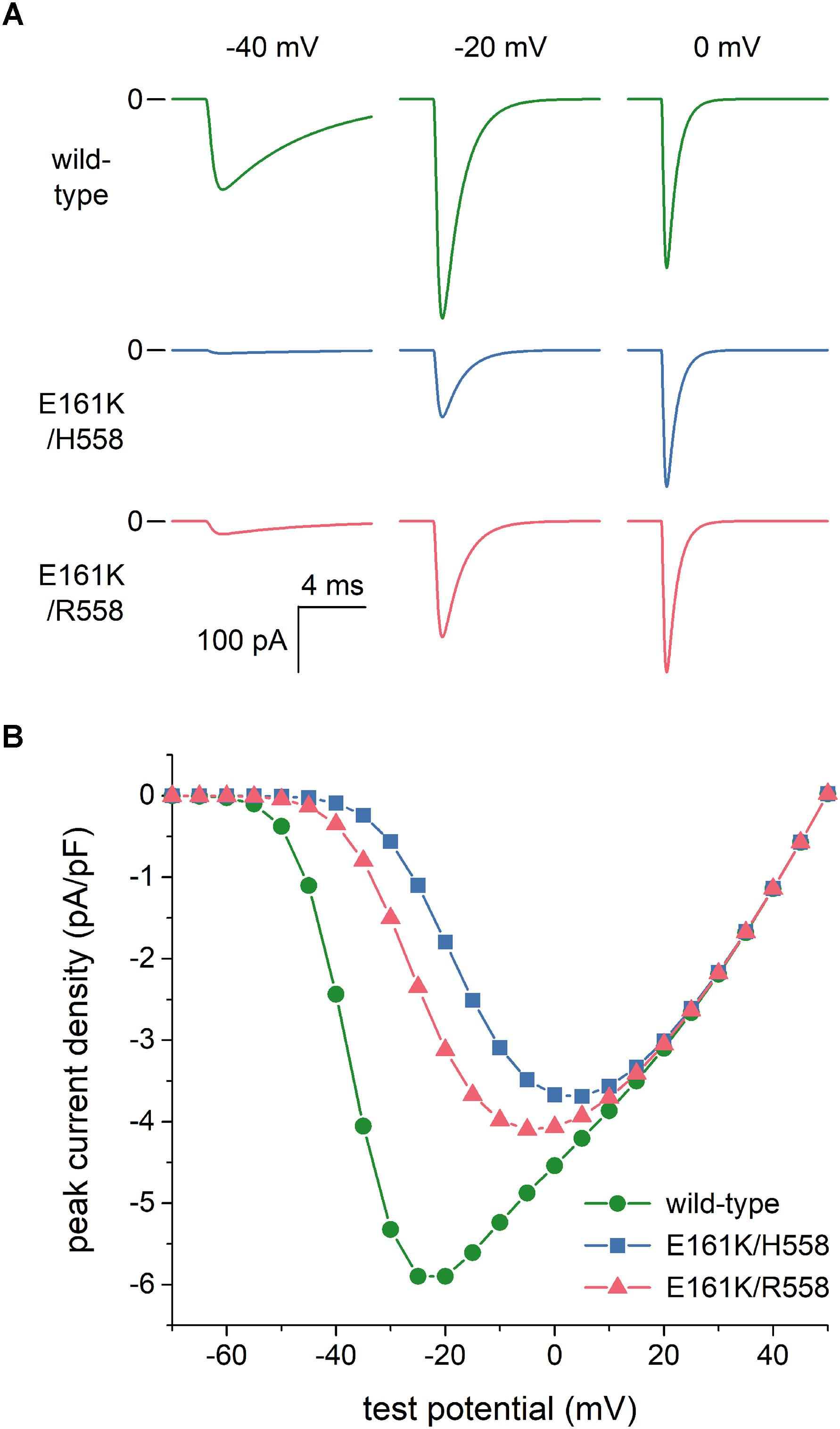
Figure 3. Effects of the E161K mutation in SCN5A, either in H558 or R558 background, on the fast sodium current (INa) of the Fabbri-Severi model of a human SA nodal pacemaker cell during simulated voltage clamp experiments. (A) Control (“wild-type”) and mutant INa during voltage clamp steps from a holding potential of -120 mV to test potentials of -40, -20, and 0 mV. (B) Current–voltage relationship of peak INa, as derived from these simulated voltage clamp experiments.
Effects of the E161K Mutation on SA Nodal Pacemaker Activity
We applied the mutation-induced changes in INa, i.e., the shifts in steady-state activation and inactivation curves, the reduction in steepness of the steady-state activation curve, and the reduction in fully-activated conductance, to half of the INa channels in the Fabbri-Severi model to assess the effects of the heterozygous E161K mutation on the spontaneous pacemaker activity of a human SA nodal cell. Figures 4A–G, shows the effects on the action potential (Figure 4A), intracellular calcium concentration (Figure 4B) and underlying membrane currents (Figures 4C–G) under control conditions (no ACh), whereas the corresponding simulation data in the presence of 10 nM ACh are shown in Figures 4H–N. The simulated administration of ACh to the Fabbri-Severi model cell does not only activate the acetylcholine-activated potassium current IK,ACh, which is zero under control conditions, but also reduces the hyperpolarization-activated “funny current” If (through a -4.95 mV shift in its voltage dependence of activation), the L-type calcium current ICaL (through a 3.1% decrease in its maximal conductance), and the rate of Ca2+ uptake into the sarcoplasmic reticulum (SR) by the SERCA pump (through a 7.0% decrease in its maximum activity). Of note, the changes in ICaL and Ca2+ uptake rate have only marginal effects at this level of ACh (Fabbri et al., 2017).
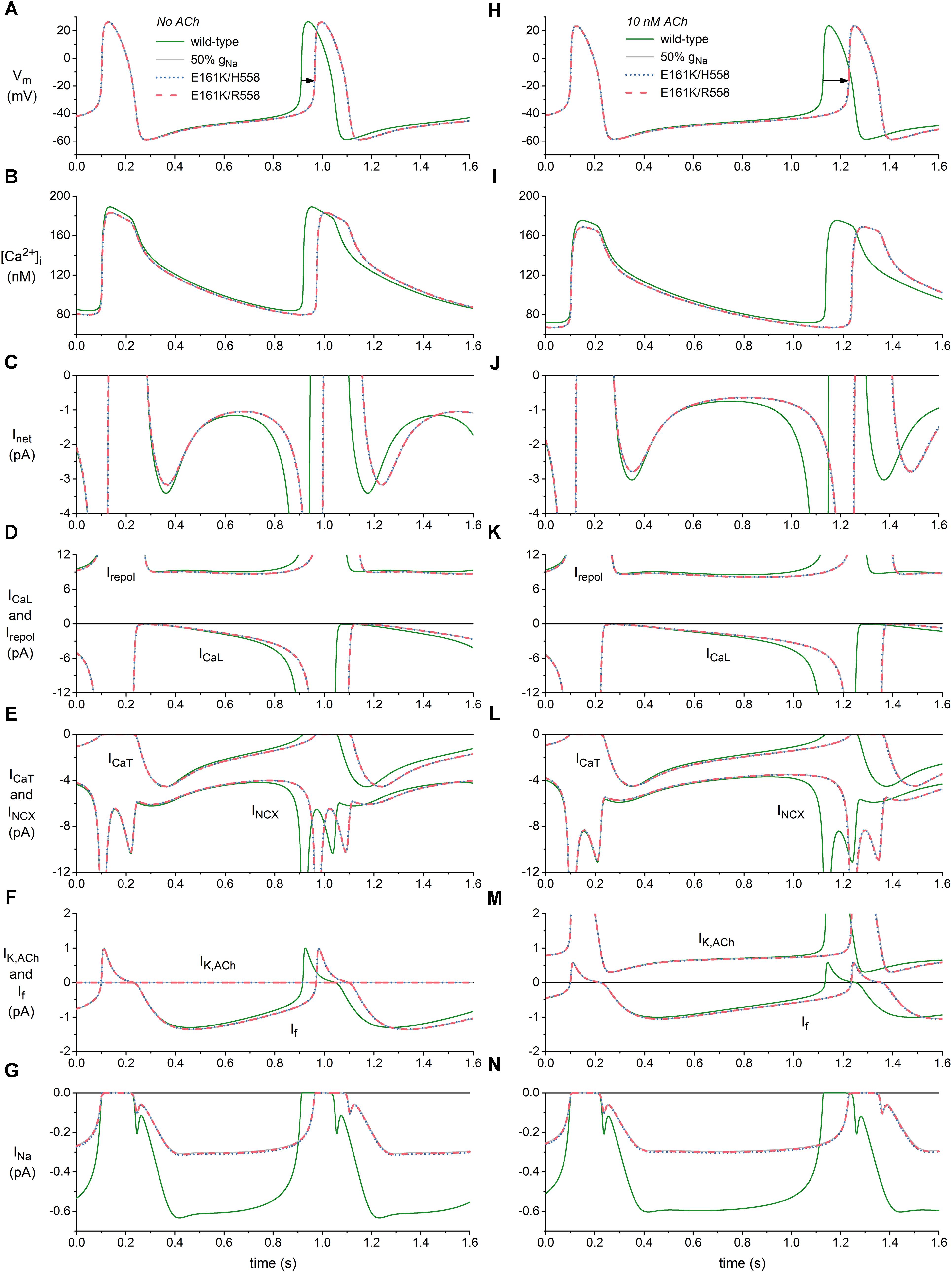
Figure 4. Effects of the E161K mutation in SCN5A, either in H558 or R558 background, on the spontaneous activity of the Fabbri-Severi model of a human SA nodal pacemaker cell. (A) Membrane potential (Vm), and associated (B) intracellular calcium concentration ([Ca2+]i), (C) net membrane current (Inet), (D) L-type calcium current (ICaL) and total repolarizing current (Irepol), (E) T-type calcium current (ICaT) and sodium-calcium exchange current (INCX), (F) ACh-activated potassium current (IK,ACh) and hyperpolarization-activated “funny current” (If), and (G) fast sodium current (INa) in the absence of ACh. (H) Vm, (I) [Ca2+]i, (J) Inet, (K) ICaL and Irepol, (L) ICaT and INCX, (M) IK,ACh, and If, and (N) INa during simulated administration of 10 nM ACh. Solid gray lines show the effect of a 50% reduction in fully-activated conductance of INa (gNa). Horizontal arrows indicate increase in cycle length. Irepol consists of IK,ACh, the rapid delayed rectifier potassium current IKr, the slow delayed rectifier potassium current IKs, the ultrarapid delayed rectifier potassium current IKur, the transient outward potassium current Ito, and the sodium-potassium pump current INaK.
The main effect of the application of ACh is a prolongation of the basic cycle length from 813 to 1027 ms (Figures 4A,H, wild-type traces), which is associated with a decrease in the intracellular calcium concentration [Ca2+]i (Figures 4B,I, wild-type traces), and sodium-calcium exchange current INCX during diastolic depolarization (Figures 4E,L, wild-type traces). If is also reduced (Figures 4F,M), as a result of the direct effect of ACh on its voltage dependence of activation. The remaining inward currents, i.e., the L-type calcium current ICaL (Figures 4D,K), T-type calcium current ICaT (Figures 4E,L) and INa (Figures 4G,N), are not largely affected. The total repolarizing current Irepol is also hardly affected, with an almost constant value of ≈9 pA during diastolic depolarization (Figures 4D,K). This is because it includes the ACh-activated IK,ACh, which is also shown separately (Figures 4F,M), in addition to the rapid delayed rectifier potassium current IKr, the slow delayed rectifier potassium current IKs, the ultrarapid delayed rectifier potassium current IKur, the transient outward potassium current Ito, and the sodium-potassium pump current INaK, which are not shown separately in Figure 4. The reduction in the “pacemaker currents” If and INCX (Lakatta and DiFrancesco, 2009), in combination with the minor changes in other inward currents as well as Irepol, result in the reduction in the net membrane current Inet (Figures 4C,J) that underlies the observed increase in cycle length.
In absence of ACh, the mutation-induced reduction in INa (Figure 4G) causes a reduction in Inet, which becomes most prominent during the second half of diastolic depolarization (Figure 4C) and in turn results in a prolongation of the cycle length by 53 and 54 ms in H558 and R558 background, respectively (Figure 4A, horizontal arrow), from the basic cycle length of 813 ms, corresponding with a 6% decrease in beating rate from 74 to 69 beats/min in either background. Simulations with the wild-type model in which gNa is reduced by 50% (labeled “50% gNa”), thus simply blocking 50% of the channels, result in a cycle length prolongation of 55 ms and traces that are barely distinguishable from the mutant ones, thus demonstrating that the contribution of the mutant component of INa to total INa is almost zero.
In the simulated presence of 10 nM ACh, the effects of the mutation are more pronounced, as reflected by the prolongation of the cycle length by 104 ms in H558 background and 105 ms in R558 background (Figure 4H, horizontal arrow), from the basic cycle length of 1027 ms, corresponding with a 9% decrease in beating rate from 58 to 53 beats/min in either background. Again, the contribution of the mutant component of INa to total INa is almost zero, as demonstrated by the highly similar prolongation of cycle length, by 107 ms, in case of 50% gNa. The more pronounced effect of the highly similar reduction in INa in the simulated presence of 10 nM ACh (Figures 4G,N) can be explained by its occurrence in the setting of a smaller Inet during diastolic depolarization (Figures 4C,J).
As illustrated in Figure 5A, the mutation-induced increase in cycle length increases with increasing levels of ACh. At 25 nM, this increase amounts to 347 and 355 ms in H558 and R558 background, respectively, from a basic cycle length of 1448 ms, slightly less than the increase of 361 ms in case of 50% gNa. In terms of beating rate (Figure 5B), an ACh level of 25 nM results in a rate of 41 beats/min in case of wild-type sodium channels and 33 beats/min in case of the heterozygous E161K mutation, in either background. The percent decrease in beating rate relative to wild type is shown in Figure 5C. In absence of ACh (Figure 5C, leftmost bars), the E161K mutation results in a 6.1% decrease in beating rate in H558 background and 6.2% decrease in R558 background. This percentage increases with increasing levels of ACh. In the simulated presence of 10 nM ACh, the decrease in beating rate amounts to 9.2 and 9.3%, respectively. At an ACh level of 25 nM (Figure 5C, rightmost bars), the mutation-induced decrease in beating rate is almost 20%, with values of 19.3% in H558 background and 19.7% in R558 background.
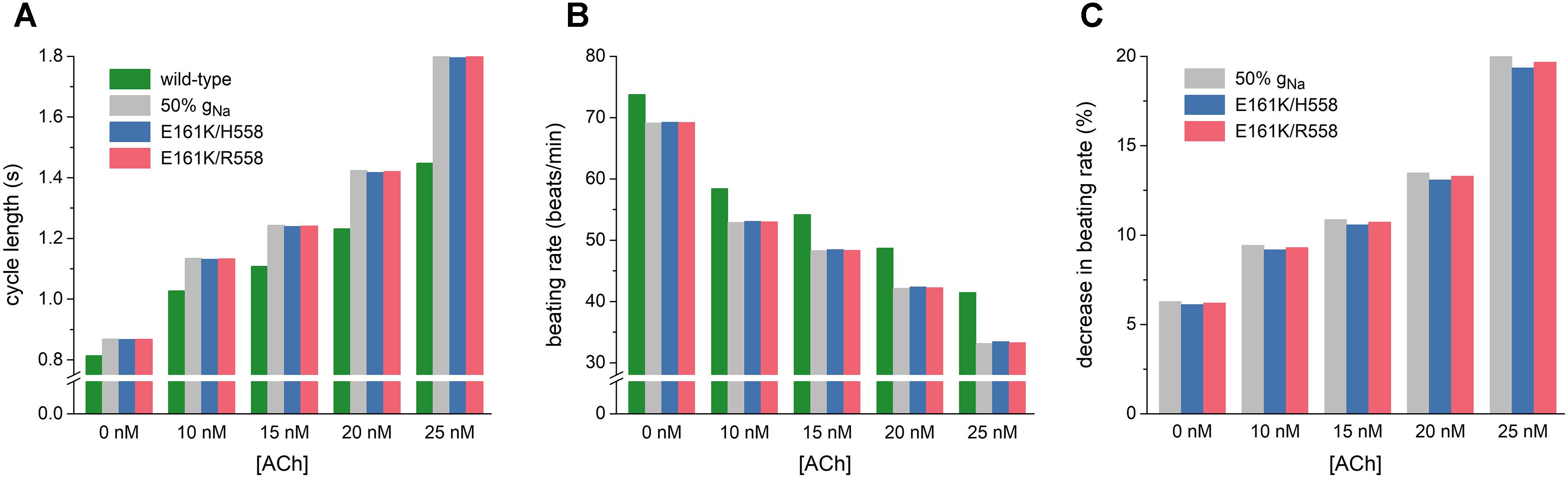
Figure 5. Bradycardic effects of the E161K mutation in SCN5A, either in H558 or R558 background, on the Fabbri-Severi model of a human SA nodal pacemaker cell at different levels of ACh (0–25 nM). (A) Cycle length. (B) Associated beating rate. (C) Percent decrease in beating rate relative to wild-type. Solid gray bars show the effect of a 50% reduction in fully-activated conductance of INa (gNa). Note the breaks in the vertical axes of (A,B).
Effects of the E161K Mutation on Atrial Excitability
Sinus node dysfunction may also be related to changes in atrial excitability, potentially leading to sinus node exit block or atrial conduction block. Therefore, we assessed the effects of the E161K mutation on atrial excitability using the Maleckar et al. human atrial cell model (Maleckar et al., 2009), which we used in combination with its well-validated equations for IK,ACh (Maleckar et al., 2008). Changes in INa, simulating a heterozygous mutation in SCN5A, were implemented as in the Fabbri-Severi model. Of note, the effect of ACh on the Maleckar et al. model cell is limited to the activation of IK,ACh.
Figure 6A shows atrial action potentials elicited at 1 Hz with a stimulus current of 1 ms duration under control conditions (no ACh). Both variants of the E161K mutation result in a reduction in the action potential overshoot and a less rapid activation, indicative of a decrease in capacitive current of the atrial cell (Figure 6A, inset). The associated INa, which is shown in Figure 6B, is approximately halved or – because the slower activation allows a larger fraction of the channels to inactivate during the activation process – even more than halved. In the simulated presence of 10 nM ACh, the resting membrane potential becomes more negative with a value of -79 mV vs. -74 mV (Figure 6C), which results in a larger INa because less sodium channels are inactivated at this more negative resting potential (Figure 6D). This does, however, not imply that action potential generation is facilitated in presence of ACh since the distance to action potential threshold is increased and an additional outward current, i.e., IK,ACh, is activated. Again, activation is slowed and the underlying INa is approximately halved (Figures 6C,D, insets).
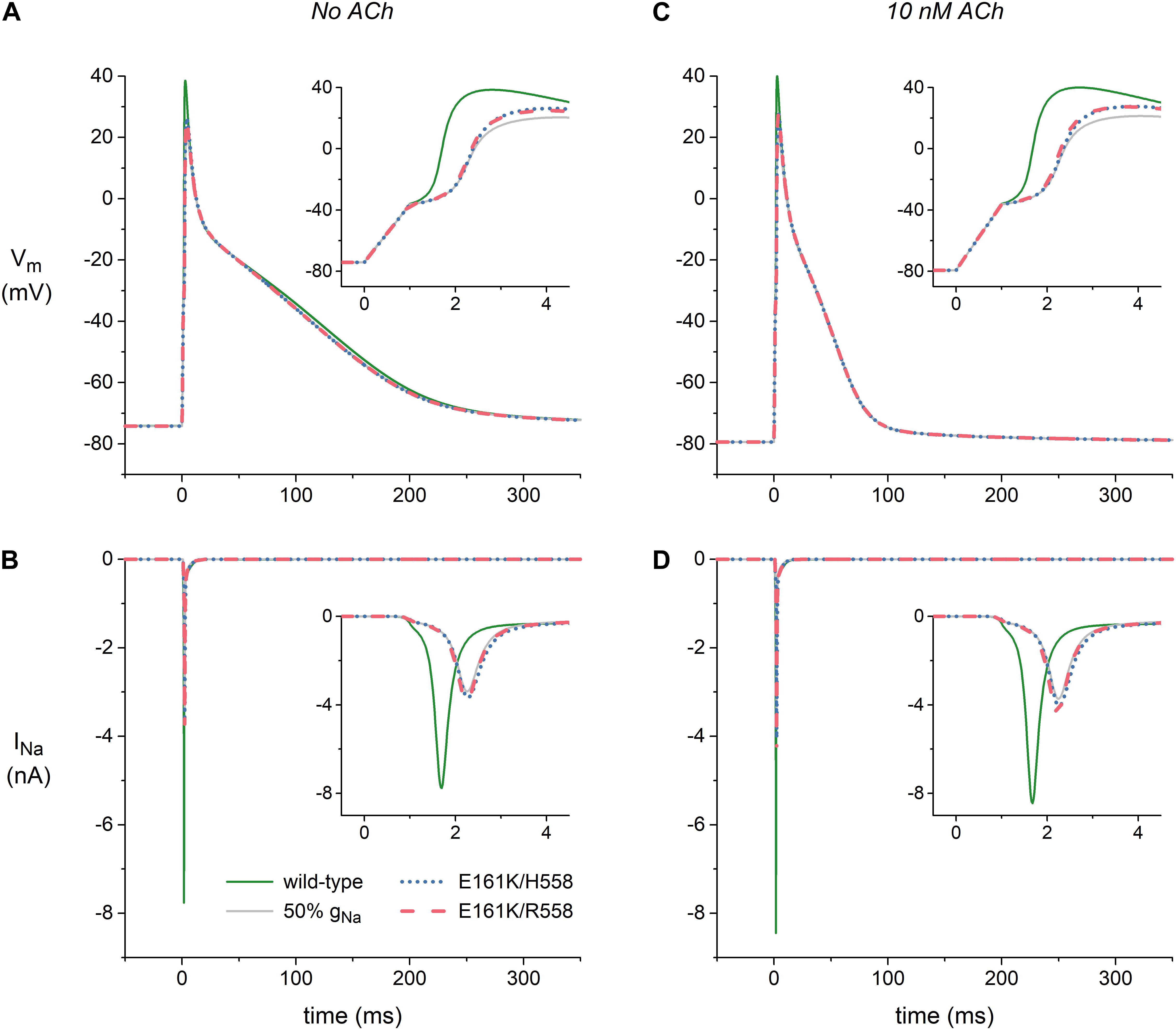
Figure 6. Effects of the E161K mutation in SCN5A, either in H558 or R558 background, on the action potential and fast sodium current (INa) of the Maleckar et al. model of a human atrial myocyte. (A) Action potentials elicited at 1 Hz and (B) associated fast sodium current (INa) in the absence of ACh. (C) Action potentials elicited at 1 Hz and (D) associated INa during simulated administration of 10 nM ACh. Insets show membrane potential (Vm) and INa on an expanded time scale. Solid gray lines show the effect of a 50% reduction in fully-activated conductance of INa (gNa).
Atrial excitability was characterized, over a wide range of concentrations of ACh, by determining the threshold stimulus current of the atrial cell as well as its maximum upstroke velocity, as a direct measure of the capacitive current. Threshold stimulus current was determined with 1 ms stimuli of increasing amplitude that were applied at a frequency of 1 Hz. Results are shown in Figure 7A. The threshold increases with increasing levels of ACh and is 5–8% higher in case of the E161K mutation. Although barely visible in Figure 7A, the threshold in R558 background, with the smaller shift of the steady-state activation curve, is smaller than in H558 background.
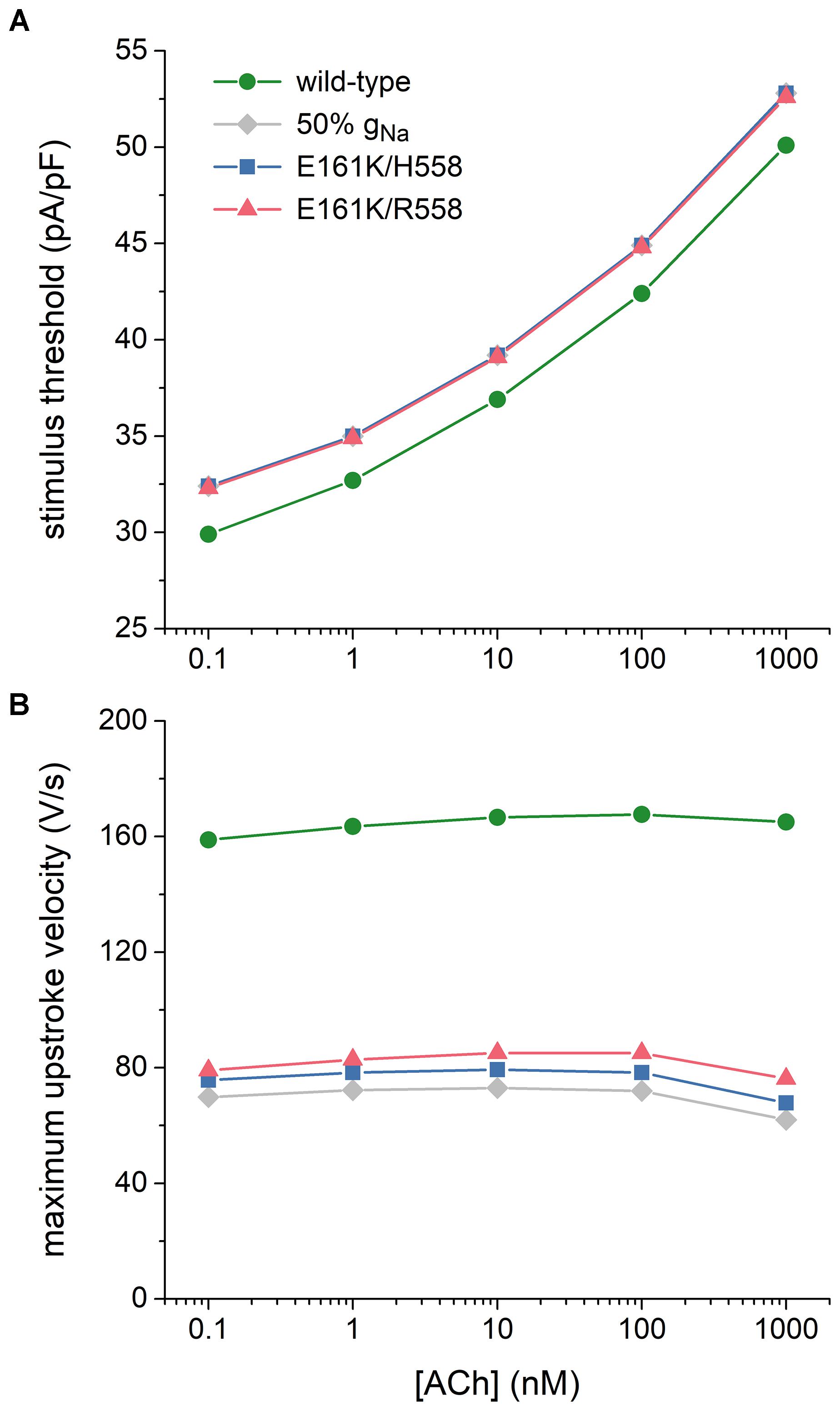
Figure 7. Effects of the E161K mutation in SCN5A, either in H558 or R558 background, on the excitability of the Maleckar et al. model of a human atrial myocyte. (A) Threshold stimulus current amplitude for a stimulus of 1 ms duration and (B) maximum upstroke velocity at ACh concentrations ranging from 0.1 nM to 1 μM. Gray diamonds show the effect of a 50% reduction in fully-activated conductance of INa (gNa). Note the logarithmic abscissa scale.
Maximum upstroke velocity was determined by eliciting action potentials with a 1 ms, ≈50% suprathreshold stimulus current at a frequency of 1 Hz. As shown in Figure 7B, it is not strongly dependent on the level of ACh. Upstroke velocity is approximately halved, or even more than halved, in case of the E161K mutation. As for the threshold stimulus current, the mutation in R558 background is associated with (slightly) less strong effects than in H558 background. In contrast with the SA nodal cell, the mutant component of INa is not effectively zero, as revealed by the larger INa (Figures 6B,D, insets) and larger maximum upstroke velocity (Figure 7B) in comparison with the 50% gNa simulations.
Discussion
General Discussion
Our simulations demonstrate that the E161K mutation in SCN5A hampers both impulse generation and impulse propagation through its effects on the electrophysiological properties of human SA nodal and atrial cells. Generation of the SA nodal action potential is hampered by the strong decrease in INa during diastolic depolarization and the associated decrease in Inet, in particular in presence of ACh (Figures 4, 5). Impulse propagation, as comprehensively reviewed by Kléber and Rudy (2004), is hampered by the current-to-load mismatch that results from, on the one hand, the increase in threshold stimulus current and, on the other hand, the decrease in capacitive current of the atrial cell (Figures 6, 7). The highly common H558R polymorphism in SCN5A does not have a major effect on the outcome of the simulations, despite its effects on the biophysical properties of the E161K mutant channels (Figures 2, 3).
The E161K mutation has been subject of simulations since its discovery in 2005 (Smits et al., 2005). Smits et al. (2005) showed that the E161K mutation impairs conduction in linear strands of atrial and ventricular cells, using the human atrial and ventricular cell models by Courtemanche et al. (1998) and Priebe and Beuckelmann (1998), respectively. Simulations were based on the observations by Smits et al. (2005) on E161K channels expressed in tsA201 cells. Mutation effects were relatively mild in comparison to later observations by Gui et al. (2010a,b) and obtained in H558 background. Smits et al. (2005) also carried out simulations on SA nodal cells, using the model of a peripheral rabbit SA nodal cell by Zhang et al. (2000), including equations for IK,ACh (Zhang et al., 2002). Despite the largely different time course of the human SA nodal action potential, with a much smaller INa, a much longer diastolic phase, and a significantly less negative maximum diastolic potential, the main findings of the present study are similar to those of Smits et al. (2005) regarding the SA nodal action potential: the E161K mutation causes a decrease in diastolic depolarization rate that results in an increase in cycle length, which is much more pronounced in presence of ACh.
Butters et al. (2010) studied the effects of several mutations in SCN5A, including E161K, on cardiac pacemaking in a two-dimensional model of sino-atrial tissue based on a reconstruction of a single slice of the rabbit right atrium, using modified versions of the single cell models of rabbit SA nodal and atrial cells by Zhang et al. (2000, 2002) and Lindblad et al. (1996), respectively. Their simulations of the E161K mutation were again based on the observations by Smits et al. (2005). The mutation slowed down pacemaking and this effect was more pronounced in the presence of ACh. Furthermore, a conduction block in the direction toward the atrial septum occurred in the absence of ACh, while conduction in the direction toward the crista terminalis was sustained. With 15 nM ACh, exit block occurred in both directions and the SA node was unable to drive the surrounding atrium.
In the present study, we carried out simulations with the use of comprehensive well-validated human single cell models and implemented effects of the E161K mutation based on the experimental data by Gui et al. (2010a,b). Furthermore, we assessed the role of the highly common H558R polymorphism, also based on the experimental data by Gui et al. (2010a,b). We focused on cellular mechanisms and refrained from using two- or even three-dimensional models of sino-atrial interaction, with their coupling interactions between heterogeneous cells, because results of such simulations are critically dependent on the exact geometry and cell distribution (Garny et al., 2003a), which have not been fully elucidated.
Our simulations show that the effects of the E161K mutation are so large, both in H558 and R558 background, that the contribution of mutant INa to total INa of the SA nodal cell is effectively zero. Accordingly, it is not surprising that E161K mutation carriers show sinus node dysfunction as do carriers of several mutations in SCN5A that are associated with a complete or almost complete loss of function of the mutant channels, as reviewed by Lei et al. (2007, 2008) and Remme et al. (2008). In many cases, the sinus node dysfunction shows a reduced penetrance, as for the E161K mutation, where 8 out of 14 mutation carriers show the disease. Reduced penetrance is a common finding in primary electrical diseases, including those associated with mutations in SCN5A (Remme, 2013; Robyns et al., 2014). Several factors, including co-inherited genetic variants and alternative splicing sites, may play a role. However, our simulations suggest that it is highly unlikely that the common H558R polymorphism can explain the reduced penetrance of the E161K mutation.
Limitations
Unfortunately, experimental data on the effects of the E161K mutation are limited to voltage clamp data from mutant SCN5A channels in mammalian expression systems, viz. tsA201 and HEK-293 cells, obtained at room temperature and in the presence (Smits et al., 2005) or absence (Gui et al., 2010a,b) of a wild-type β-subunit. There are some quantitative differences in the experimental data, potentially due to differences in cell type or presence of β-subunit, with a more severe pattern in the data of Gui et al. (2010a,b), with a +19 mV shift of the steady-state activation curve in H558 background vs. the +11.9 mV shift reported by Smits et al. (2005) and a 4-fold vs. 2.5-fold reduction in peak current density. However, the latter difference may partly be explained by the larger shift in the steady-state activation curve. In our simulations, we have assumed that the experimentally observed effects of the mutation in mammalian expression systems at room temperature also hold for cardiac cells at the physiological temperature of 37°C. We cannot rule out that recordings at a more close-to-physiological temperature would have revealed additional effects of the E161K mutation, as has been the case for several other BrS-related mutations in SCN5A expressed in mammalian cell lines, like T1620M and Y1795H (Dumaine et al., 1999; Rivolta et al., 2001). Furthermore, we have to keep in mind that the cardiac sodium channel is part of a macromolecular complex that does not only comprise the α- and β-subunits, but also several other proteins that may regulate channel activity, such as ankyrin, caveolin and syntrophin, and may act differently in case of mutant channels (Ruan et al., 2009). Thus, it is uncertain to which extent the experimental data on the effects of the E161K mutation acquired in mammalian expression systems represent the mutant-induced changes that occur in human cardiac cells.
In our simulations, it is assumed that the Fabbri-Severi model of a human SA nodal cell (Fabbri et al., 2017) and the Maleckar et al. model of a human atrial cell (Maleckar et al., 2009) are fully representative of the biophysical properties of the corresponding real cells. However, experimental data on the biophysical properties of cardiac cells, and human cells in particular, are often incomplete or obtained under non-physiological conditions. Functional data on INa in human SA nodal cells, for example, are limited to accidental observations by Verkerk et al. (2009) when performing voltage clamp experiments on single human SA nodal pacemaker cells to record If. Accordingly, INa was included in the Fabbri-Severi model, but simply adopted from its parent model, i.e., the model of a rabbit SA nodal cell by Severi et al. (2012), who, in turn, used the equations of the SA nodal cell model by Noble et al. (1989). The latter equations were adopted from the model of SA nodal cell electrical activity by Noble and Noble (1984), who based their equations on the model of cardiac electrical activity by DiFrancesco and Noble (1985). The description of INa in the latter model is essentially that of Hodgkin and Huxley (1952), but with modifications based on data from experiments on rabbit cardiac Purkinje fibers at 10–26°C (Colatsky, 1980) and from isolated rat ventricular myocytes at 20–22°C (Brown et al., 1981). Activation and inactivation curves were shifted along the voltage axis to account for their observed temperature dependence, whereas rate constants were scaled to arrive at time constant values near the experimentally observed ones. The INa equations in the Maleckar et al. (2009) model are adopted from the model of an adult human atrial cell by Nygren et al. (1998), who used the voltage clamp data of Sakakibara et al. (1992) on INa in isolated human atrial myocytes at 17 ± 1°C to construct their model equations. Activation and inactivation curves were shifted in the positive direction along the voltage axis to reach a realistic activation threshold and stable resting potential. The time constants of activation and inactivation are very similar to those of the rabbit atrial cell model of Lindblad et al. (1996), who based their mathematical description of INa on the voltage clamp data of Wendt et al. (1992), acquired from cultured rabbit atrial myocytes at 17°C, and INa of the DiFrancesco-Noble model (DiFrancesco and Noble, 1985). Activation kinetics of INa were modified from those of the DiFrancesco-Noble model with the use of a Q10 of 1.7.
In our simulations, it is also assumed that mutation effects are limited to half of the intrinsic INa channels, thus representing the heterozygous nature of the mutation, taking into account that a functional INa channel is built with a single NaV1.5 protein. This assumption is challenged by recent experimental findings of Clatot et al. (2018), who demonstrated that wild-type and mutant NaV1.5 proteins can form dimers, enabling coupled gating of wild-type and mutant NaV1.5 sodium channels that can be responsible for a dominant negative effect of the mutation. Thus, the cellular effects of the E161K mutation could be more severe than anticipated.
The action of ACh in the Fabbri-Severi model is, apart from the activation of IK,ACh, limited to suppression of If, ICaL, and Ca2+ uptake into the SR, thus simplifying the intracellular SR-based calcium clock signaling cascade through adenylyl cyclase, cyclic AMP and protein kinase A (Maltsev et al., 2014; Behar et al., 2016). This cascade is important for a detailed understanding of the action of ACh. Yet, in the present study, where the focus is on the effects of the E161K mutation in SCN5A and possible effects of the H558R polymorphism, it is sufficient that the suppression by ACh of If, ICaL, and Ca2+ uptake into the SR, and that of INCX through the reduced intracellular calcium level, are included, in addition to the activation of IK,ACh.
One should be careful in the interpretation of experimental data, not only because of the above considerations, but also because changes in biophysical parameters are not necessarily independent. This is nicely demonstrated by the decrease in peak current density shown in Figure 3. Such decrease is often interpreted as a reduction in the number of channels or a reduction in their single conductance. In this case, however, the E161K mutant-induced changes in steady-state activation and inactivation curves per se reduce the peak current density by ≈35%. This effect may, at least partly, explain the apparent discrepancy between the ≈30% decrease in cell surface expression of E161K/H558 channels in HEK-293 cells and the > 70% decrease in peak current density reported by Gui et al. (2010a). Similarly, an apparently slower inactivation of E161K/H558 channels at membrane potentials ranging from -40 to 0 mV can, at least partly, be explained by the +19 mV shift in their steady-state activation curve (Gui et al., 2010a). Because experimentally observed changes in time constants of (fast) inactivation or recovery from inactivation, if any, are already small, no attempts were made to include these changes in the simulations of the present study.
Conclusion
We conclude that the experimentally identified mutation-induced changes in INa can explain the clinically observed sinus bradycardia and potentially the occasional exit block. Furthermore, we conclude that the common H558R polymorphism does not significantly alter the effects of the E161K mutation and can thus not explain the reduced penetrance of the E161K mutation.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the author, without undue reservation, to any qualified researcher.
Author Contributions
RW designed the experiments, acquired, analyzed and interpreted the data, and drafted, edited, and approved the manuscript.
Funding
This study was supported by the Amsterdam University Medical Centers, location Academic Medical Center.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank the staff of the Departments of Medical Biology and Experimental Cardiology for their valuable contributions.
References
Ackerman, M. J., Splawski, I., Makielski, J. C., Tester, D. J., Will, M. L., Timothy, K. W., et al. (2004). Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm 1, 600–607. doi: 10.1016/j.hrthm.2004.07.013
Behar, J., Ganesan, A., Zhang, J., and Yaniv, Y. (2016). The autonomic nervous system regulates the heart rate through cAMP-PKA dependent and independent coupled-clock pacemaker cell mechanisms. Front. Physiol. 7:419. doi: 10.3389/fphys.2016.00419
Brown, A. M., Lee, K. S., and Powell, T. (1981). Sodium current in single rat heart muscle cells. J. Physiol. 318, 479–500. doi: 10.1113/jphysiol.1981.sp013879
Brugada, J., Campuzano, O., Arbelo, E., Sarquella-Brugada, G., and Brugada, R. (2018). Present status of Brugada syndrome: JACC State-of-the-art review. J. Am. Coll. Cardiol. 72, 1046–1059. doi: 10.1016/j.jacc.2018.06.037
Butters, T. D., Aslanidi, O. V., Inada, S., Boyett, M. R., Hancox, J. C., Lei, M., et al. (2010). Mechanistic links between Na+ channel (SCN5A) mutations and impaired cardiac pacemaking in sick sinus syndrome. Circ. Res. 107, 126–137. doi: 10.1161/CIRCRESAHA.110.219949
Chandler, N. J., Greener, I. D., Tellez, J. O., Inada, S., Musa, H., Molenaar, P., et al. (2009). Molecular architecture of the human sinus node: insights into the function of the cardiac pacemaker. Circulation 119, 1562–1575. doi: 10.1161/CIRCULATIONAHA.108.804369
Clatot, J., Zheng, Y., Girardeau, A., Liu, H., Laurita, K. R., Marionneau, C., et al. (2018). Mutant voltage-gated Na+ channels can exert a dominant negative effect through coupled gating. Am. J. Physiol. Heart Circ. Physiol. 315, H1250–H1257. doi: 10.1152/ajpheart.00721.2017
Colatsky, T. J. (1980). Voltage clamp measurements of sodium channel properties in rabbit cardiac Purkinje fibres. J. Physiol. 305, 215–234. doi: 10.1113/jphysiol.1980.sp013359
Courtemanche, M., Ramirez, R. J., and Nattel, S. (1998). Ionic mechanisms underlying human atrial action potential properties: insights from a mathematical model. Am. J. Physiol. 275, H301–H321. doi: 10.1152/ajpheart.1998.275.1.H301
Cuellar, A. A., Lloyd, C. M., Nielsen, P. F., Bullivant, D. P., Nickerson, D. P., and Hunter, P. J. (2003). An overview of CellML 1.1, a biological model description language. Simulation 79, 740–747. doi: 10.1177/0037549703040939
DiFrancesco, D., and Noble, D. (1985). A model of cardiac electrical activity incorporating ionic pumps and concentration changes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 307, 353–398. doi: 10.1098/rstb.1985.0001
Dumaine, R., Towbin, J. A., Brugada, P., Vatta, M., Nesterenko, D. V., Nesterenko, V. V., et al. (1999). Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ. Res. 85, 803–809. doi: 10.1161/01.RES.85.9.803
Fabbri, A., Fantini, M., Wilders, R., and Severi, S. (2017). Computational analysis of the human sinus node action potential: model development and effects of mutations. J. Physiol. 595, 2365–2396. doi: 10.1113/JP273259
Garny, A., Hunter, P. J., Noble, D., Boyett, M. R., and Kohl, P. (2003a). “Rabbit sino-atrial node modeling: from single cell to tissue structure,” in Proceedings of the 25th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Piscataway, NJ: IEEE, 28–31. doi: 10.1109/IEMBS.2003.1279489
Garny, A., Kohl, P., and Noble, D. (2003b). Cellular Open Resource (COR): a public CellML based environment for modelling biological function. Int. J. Bifurcat. Chaos 13, 3579–3590. doi: 10.1142/S021812740300882X
Giudicessi, J. R., Wilde, A. A. M., and Ackerman, M. J. (2018). The genetic architecture of long QT syndrome: a critical reappraisal. Trends Cardiovasc. Med. 28, 453–464. doi: 10.1016/j.tcm.2018.03.003
Gui, J., Wang, T., Jones, R. P. O., Trump, D., Zimmer, T., and Lei, M. (2010a). Multiple loss-of-function mechanisms contribute to SCN5A-related familial sick sinus syndrome. PLoS One 5:e10985. doi: 10.1371/journal.pone.0010985
Gui, J., Wang, T., Trump, D., Zimmer, T., and Lei, M. (2010b). Mutation-specific effects of polymorphism H558R in SCN5A-related sick sinus syndrome. J. Cardiovasc. Electrophysiol. 21, 564–573. doi: 10.1111/j.1540-8167.2010.01762.x
Hayashi, H., Sumiyoshi, M., Nakazato, Y., and Daida, H. (2018). Brugada syndrome and sinus node dysfunction. J. Arrhythm 4, 216–221. doi: 10.1002/joa3.12046
Hodgkin, A. L., and Huxley, A. F. (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117, 500–544. doi: 10.1113/jphysiol.1952.sp004764
Hu, D., Viskin, S., Oliva, A., Carrier, T., Cordeiro, J. M., Barajas-Martinez, H., et al. (2007). Novel mutation in the SCN5A gene associated with arrhythmic storm development during acute myocardial infarction. Heart Rhythm 4, 1072–1080. doi: 10.1016/j.hrthm.2007.03.040
Iwasa, H., Itoh, T., Nagai, R., Nakamura, Y., and Tanaka, T. (2000). Twenty single nucleotide polymorphisms (SNPs) and their allelic frequencies in four genes that are responsible for familial long QT syndrome in the Japanese population. J. Hum. Genet. 45, 182–183. doi: 10.1007/s100380050207
Kléber, A. G., and Rudy, Y. (2004). Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol. Rev. 84, 431–488. doi: 10.1152/physrev.00025.2003
Lakatta, E. G., and DiFrancesco, D. (2009). What keeps us ticking: a funny current, a calcium clock, or both? J. Mol. Cell. Cardiol. 47, 157–170. doi: 10.1016/j.yjmcc.2009.03.022
Lei, M., Huang, C. L.-H., and Zhang, Y. (2008). Genetic Na+ channelopathies and sinus node dysfunction. Prog. Biophys. Mol. Biol. 98, 171–178. doi: 10.1016/j.pbiomolbio.2008.10.003
Lei, M., Zhang, H., Grace, A. A., and Huang, C. L.-H. (2007). SCN5A and sinoatrial node pacemaker function. Cardiovasc. Res. 74, 356–365. doi: 10.1016/j.cardiores.2007.01.009
Lindblad, D. S., Murphey, C. R., Clark, J. W., and Giles, W. R. (1996). A model of the action potential and underlying membrane currents in a rabbit atrial cell. Am. J. Physiol. 271, H1666–H1696. doi: 10.1152/ajpheart.1996.271.4.H1666
Lloyd, C. M., Lawson, J. R., Hunter, P. J., and Nielsen, P. F. (2008). The CellML model repository. Bioinformatics 24, 2122–2123. doi: 10.1093/bioinformatics/btn390
Maleckar, M. M., Greenstein, J. L., Giles, W. R., and Trayanova, N. A. (2009). K+ current changes account for the rate dependence of the action potential in the human atrial myocyte. Am. J. Physiol. Heart Circ. Physiol. 297, H1398–H1410. doi: 10.1152/ajpheart.00411.2009
Maleckar, M. M., Greenstein, J. L., Trayanova, N. A., and Giles, W. R. (2008). Mathematical simulations of ligand-gated and cell-type specific effects on the action potential of human atrium. Prog. Biophys. Mol. Biol. 98, 161–170. doi: 10.1016/j.pbiomolbio.2009.01.010
Maltsev, V. A., Yaniv, Y., Maltsev, A. V., Stern, M. D., and Lakatta, E. G. (2014). Modern perspectives on numerical modeling of cardiac pacemaker cell. J. Pharmacol. Sci. 125, 6–38. doi: 10.1254/jphs.13R04CR
Noble, D., DiFrancesco, D., and Denyer, J. C. (1989). “Ionic mechanisms in normal and abnormal cardiac pacemaker activity,” in Neuronal and Cellular Oscillators, ed. J. W. Jacklet (New York, NY: Marcel Dekker Inc.), 59–85.
Noble, D., and Noble, S. J. (1984). A model of sino-atrial node electrical activity based on a modification of the DiFrancesco-Noble (1984) equations. Proc. R. Soc. Lond. B Biol. Sci. 222, 295–304. doi: 10.1098/rspb.1984.0065
Nygren, A., Fiset, C., Firek, L., Clark, J. W., Lindblad, D. S., Clark, R. B., et al. (1998). Mathematical model of an adult human atrial cell: the role of K+ currents in repolarization. Circ. Res. 82, 63–81. doi: 10.1161/01.RES.82.1.63
O’Hara, T., Virág, L., Varró, A., and Rudy, Y. (2011). Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput. Biol. 7:e1002061. doi: 10.1371/journal.pcbi.1002061
Priebe, L., and Beuckelmann, D. J. (1998). Simulation study of cellular electric properties in heart failure. Circ. Res. 82, 1206–1223. doi: 10.1161/01.RES.82.11.1206
Remme, C. A. (2013). Cardiac sodium channelopathy associated with SCN5A mutations: electrophysiological, molecular and genetic aspects. J. Physiol. 591, 4099–4116. doi: 10.1113/jphysiol.2013.256461
Remme, C. A., Wilde, A. A. M., and Bezzina, C. R. (2008). Cardiac sodium channel overlap syndromes: different faces of SCN5A mutations. Trends Cardiovasc. Med. 18, 78–87. doi: 10.1016/j.tcm.2008.01.002
Rivolta, I., Abriel, H., Tateyama, M., Liu, H., Memmi, M., Vardas, P., et al. (2001). Inherited Brugada and long QT-3 syndrome mutations of a single residue of the cardiac sodium channel confer distinct channel and clinical phenotypes. J. Biol. Chem. 276, 30623–30630. doi: 10.1074/jbc.M104471200
Robyns, T., Nuyens, D., Van Casteren, L., Corveleyn, A., De Ravel, T., Heidbuchel, H., et al. (2014). Reduced penetrance and variable expression of SCN5A mutations and the importance of co-inherited genetic variants: case report and review of the literature. Indian Pacing Electrophysiol. J. 14, 133–149. doi: 10.1016/S0972-6292(16)30754-9
Ruan, Y., Liu, N., and Priori, S. G. (2009). Sodium channel mutations and arrhythmias. Nat. Rev. Cardiol. 6, 337–348. doi: 10.1038/nrcardio.2009.44
Sakakibara, Y., Wasserstrom, J. A., Furukawa, T., Jia, H., Arentzen, C. E., Hartz, R. S., et al. (1992). Characterization of the sodium current in single human atrial myocytes. Circ. Res. 71, 535–546. doi: 10.1161/01.RES.71.3.535
Severi, S., Fantini, M., Charawi, L. A., and DiFrancesco, D. (2012). An updated computational model of rabbit sinoatrial action potential to investigate the mechanisms of heart rate modulation. J. Physiol. 590, 4483–4499. doi: 10.1113/jphysiol.2012.229435
Shinlapawittayatorn, K., Du, X. X., Liu, H., Ficker, E., Kaufman, E. S., and Deschênes, I. (2011). A common SCN5A polymorphism modulates the biophysical defects of SCN5A mutations. Heart Rhythm 8, 455–462. doi: 10.1016/j.hrthm.2010.11.034
Smits, J. P. P., Koopmann, T. T., Wilders, R., Veldkamp, M. W., Opthof, T., Bhuiyan, Z. A., et al. (2005). A mutation in the human cardiac sodium channel (E161K) contributes to sick sinus syndrome, conduction disease and Brugada syndrome in two families. J. Mol. Cell. Cardiol. 38, 969–981. doi: 10.1016/j.yjmcc.2005.02.024
Tester, D. J., Valdivia, C., Harris-Kerr, C., Alders, M., Salisbury, B. A., Wilde, A. A. M., et al. (2010). Epidemiologic, molecular, and functional evidence suggest A572D-SCN5A should not be considered an independent LQT3-susceptibility mutation. Heart Rhythm 7, 912–919. doi: 10.1016/j.hrthm.2010.04.014
Verkerk, A. O., Wilders, R., Van Borren, M. M. G. J., and Tan, H. L. (2009). Is sodium current present in human sinoatrial node cells? Int. J. Biol. Sci. 5, 201–204. doi: 10.7150/ijbs.5.201
Wendt, D. J., Starmer, C. F., and Grant, A. O. (1992). Na channel kinetics remain stable during perforated-patch recordings. Am. J. Physiol. 263, C1234–C1240. doi: 10.1152/ajpcell.1992.263.6.C1234
Wilde, A. A. M., and Amin, A. S. (2018). Clinical spectrum of SCN5A mutations: long QT syndrome, Brugada syndrome, and cardiomyopathy. JACC Clin. Electrophysiol. 4, 569–579. doi: 10.1016/j.jacep.2018.03.006
Wilders, R., and Verkerk, A. O. (2018). Long QT syndrome and sinus bradycardia—a mini review. Front. Cardiovasc. Med. 5:106. doi: 10.3389/fcvm.2018.00106
Workman, A. J., Kane, K. A., and Rankin, A. C. (2001). The contribution of ionic currents to changes in refractoriness of human atrial myocytes associated with chronic atrial fibrillation. Cardiovasc. Res. 52, 226–235. doi: 10.1016/S0008-6363(01)00380-7
Ye, B., Valdivia, C. R., Ackerman, M. J., and Makielski, J. C. (2003). A common human SCN5A polymorphism modifies expression of an arrhythmia causing mutation. Physiol. Genomics 12, 187–193. doi: 10.1152/physiolgenomics.00117.2002
Zhang, H., Holden, A. V., Kodama, I., Honjo, H., Lei, M., Varghese, T., et al. (2000). Mathematical models of action potentials in the periphery and center of the rabbit sinoatrial node. Am. J. Physiol. Heart Circ. Physiol. 279, H397–H421. doi: 10.1152/ajpheart.2000.279.1.H397
Zhang, H., Holden, A. V., Noble, D., and Boyett, M. R. (2002). Analysis of the chronotropic effect of acetylcholine on sinoatrial node cells. J. Cardiovasc. Electrophysiol. 13, 465–474. doi: 10.1046/j.1540-8167.2002.00465.x
Keywords: computer simulations, electrophysiology, genetics, human, ion channel, sinus bradycardia, sinus node dysfunction, sodium current
Citation: Wilders R (2018) Cellular Mechanisms of Sinus Node Dysfunction in Carriers of the SCN5A-E161K Mutation and Role of the H558R Polymorphism. Front. Physiol. 9:1795. doi: 10.3389/fphys.2018.01795
Received: 11 September 2018; Accepted: 29 November 2018;
Published: 18 December 2018.
Edited by:
Javier Saiz, Universitat Politècnica de València, SpainReviewed by:
Yael Yaniv, Technion – Israel Institute of Technology, IsraelArun V. Holden, University of Leeds, United Kingdom
Copyright © 2018 Wilders. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ronald Wilders, r.wilders@amc.uva.nl