 Shanna Hamilton
Shanna Hamilton Dmitry Terentyev
Dmitry Terentyev- 1Department of Medicine, The Warren Alpert Medical School of Brown University, Providence, RI, United States
- 2Cardiovascular Research Center, Rhode Island Hospital, Providence, RI, United States
A rapid growth in the incidence of diabetes and obesity has transpired to a major heath issue and economic burden in the postindustrial world, with more than 29 million patients affected in the United States alone. Cardiovascular defects have been established as the leading cause of mortality and morbidity of diabetic patients. Over the last decade, significant progress has been made in delineating mechanisms responsible for the diminished cardiac contractile function and enhanced propensity for malignant cardiac arrhythmias characteristic of diabetic disease. Rhythmic cardiac contractility relies upon the precise interplay between several cellular Ca2+ transport protein complexes including plasmalemmal L-type Ca2+ channels (LTCC), Na+-Ca2+ exchanger (NCX1), Sarco/endoplasmic Reticulum (SR) Ca2+-ATPase (SERCa2a) and ryanodine receptors (RyR2s), the SR Ca2+ release channels. Here we provide an overview of changes in Ca2+ homeostasis in diabetic ventricular myocytes and discuss the therapeutic potential of targeting Ca2+ handling proteins in the prevention of diabetes-associated cardiomyopathy and arrhythmogenesis.
Introduction
Heart failure (HF) and sudden cardiac death (SCD) due to malignant ventricular arrhythmias remain a major cause of mortality and morbidity in the developed world, in part due to alarming growth in the rates of obesity and diabetes (Benjamin et al., 2018). Diabetic patients have a two-fold increased risk for SCD and approximately 70 % suffer cardiovascular complications (Chugh et al., 2008; Laakso, 2008; Spooner, 2008; Bergner and Goldberger, 2010; Siscovick et al., 2010; Vasiliadis et al., 2014). Defective intracellular Ca2+ homeostasis has been established as a key contributor to diabetes-related cardiac dysfunction and enhanced arrhythmogenesis independent of coronary heart disease or hypertension (Belke and Dillmann, 2004; Aneja et al., 2008; Junttila et al., 2010; Pappone and Santinelli, 2010; Axelsen et al., 2015; Singh et al., 2018).
Diabetes is a chronic metabolic disorder characterized by hyperglycemia and reduced glucose utilization due to defective insulin secretion or action (American Diabetes Association, 2009). Type 1 diabetes is caused by the autoimmune destruction of pancreatic β-cells and a deficiency in insulin production. Linked with increasing rates of obesity, the more prevalent Type 2 diabetes is caused by cellular resistance to insulin and the failure of β-cells to compensate. In the diabetic heart, mitochondrial energetics are altered, with a switch of metabolic substrate from glucose to fatty acids (Boudina and Abel, 2010; Bertero and Maack, 2018). An imbalance between energy production and substrate utilization results in increased myocardial oxygen consumption and lipotoxicity, leading to mitochondrial dysfunction and reduced cardiac efficiency (Bhatti et al., 2017; Bertero and Maack, 2018). Altered mitochondrial energetics also drive changes in morphology and enhance the emission of reactive oxygen species (ROS), resulting in increased oxidative stress of the myocardium (Shen et al., 2004; Dabkowski et al., 2009; Joubert et al., 2018). It is well established that cardiovascular complications are common in both types of diabetes (Laakso, 2008; Bergner and Goldberger, 2010).
In the heart the process of excitation-contraction (EC) coupling drives cyclic changes in intracellular Ca2+ concentration [Ca2+], leading to rhythmic contraction and relaxation of cardiomyocytes in response to variable metabolic demand (Bers, 2002). Cardiac contractility is precisely regulated by ion channels and exchangers that maintain beat-to-beat Ca2+ concentrations in the steady state, whereby Ca2+ influx must equal Ca2+ efflux (Eisner D. et al., 2013). Depolarization of the sarcolemma and activation of voltage-dependent L-type Ca2+ channels (LTCCs) leads to Ca2+ influx into the cytosol. This small influx subsequently triggers a much larger Ca2+-induced Ca2+ release (CICR; Fabiato, 1985) from the sarcoplasmic reticulum (SR) Ca2+ stores through ryanodine receptors (RyR2s). The significant global increase in cytosolic [Ca2+], known as the Ca2+ transient, activates contractile machinery and leads to muscle contraction. For relaxation to then occur, Ca2+ must be sequestered and the intracellular [Ca2+] decreased. This is primarily by extrusion from the cytosol via the Na+/Ca2+ exchanger (NCX1), or resequestration into the SR via the sarco/endoplasmic reticulum-ATPase (SERCa2a).
Abnormal function of components of Ca2+ cycling machinery has been implicated in reduced contractility and proarrhythmic electrical instabilities in a variety of inherited and acquired cardiac diseases including HF and diabetic cardiomyopathy (Lagadic-Gossmann et al., 1996; Aneja et al., 2008; Jia et al., 2018). Reduction of SR Ca2+ release during systole contributes to diminished contraction, while enhancement of spontaneous Ca2+ release promotes early and delayed after-depolarizations (i.e., EADs and DADs respectively) of sarcolemma implicated in initiation of triggered activity in the heart (Landstrom et al., 2017). In addition to trigger for arrhythmia initiation, abnormal Ca2+ cycling contributes to arrhythmia substrate to maintain and perpetuate it via beat-to-beat alternations of electrical activity of the heart, i.e., alternans (Edwards and Blatter, 2014).
Despite many similarities in diabetes-related remodeling of Ca2+ homeostasis in comparison to that in HF, there are also some differences. This may also be complicated by the various animal models and species used to study the condition, highlighted in the review of King (2012). Models of Type 1 diabetes include chemically induced hyperglycemia by injection of streptozotocin (STZ) or alloxan (Szkudelski, 2001), as well as animals with genetically induced β-cell destruction (Mathews et al., 2002). Type 2 diabetes is modeled in both obese and non-obese animals. Genetically obese models include ob/ob, db/db and Zucker diabetic fatty (ZDF) hyperglycemic rodents, while obesity can also be induced by high fat diet (HFD) or high-sucrose diet (King and Bowe, 2016). Although larger animal models have been studied more recently (Xie et al., 2013; Zhang et al., 2017; Liang et al., 2018; Yang et al., 2018), most research investigating diabetes-related ventricular arrhythmias to date has been performed on rodents and remains limited. Conversely, functional alterations of Ca2+ handling proteins and EC coupling in HF have been extensively researched over several decades, in both small and large animal models as well as failing human cardiomyocytes (Hasenfuss et al., 1994; Studer et al., 1994; Schmidt et al., 1999; Louch et al., 2004; Sossalla et al., 2010; Crossman et al., 2011; Ottolia et al., 2013; Zima et al., 2014; Gorski et al., 2015; Høydal et al., 2018).
To place defective Ca2+ homeostasis in the context of our current understanding of EC coupling in cardiac disease, this review summarizes the changes and contribution of major cardiac Ca2+ handling proteins LTCC, RyR2, SERCa2a, and NCX1 to the reduced cardiac contractility observed in both HF and diabetes. We discuss the role of perturbed EC coupling in arrhythmogenesis in diabetes and the potential of targeting Ca2+ handling proteins as an anti-arrhythmic strategy.
L-Type Ca2+ Channel
Ca2+ influx though voltage-dependent L-type Ca2+ channels (LTCC) during action potential initiates Ca2+ release from the sarcoplasmic reticulum (SR). The LTCC consists of the pore forming subunit α1c, and regulatory subunits α2/δ and β2 (Muralidharan et al., 2017). C-terminus associated calmodulin (CaM) confers Ca2+-dependent inactivation of the channel (Peterson et al., 1999; Zühlke et al., 1999). Activity of LTCC can be increased by PKA phosphorylation (Leach et al., 1996; Bünemann et al., 1999). Ca2+-dependent inactivation of LTCC can be lessened by CaMKII-phosphorylation, a process activated under oxidizing conditions (Xie et al., 2009). In addition, evidence suggests that the Ca2+ channel can be directly activated during oxidative stress, and Cysteine 543 of α1c subunit confers redox sensitivity (Muralidharan et al., 2017; Wilson et al., 2018). Clusters of ≤10 channels are primarily localized in T-tubules in the sites of contact with junctional SR, i.e., dyads, opposing clusters of RyR2 Ca2+ release channels (Inoue and Bridge, 2003). Such distribution ensures efficiency of Ca2+ release initiation during EC coupling.
L-Type Ca2+ Channel and Cardiac Arrhythmia
Abnormal LTCC function has been implicated in arrhythmogenesis. Gain of function mutations of Cav1.2α1c, as well as loss of function mutation of CaM (reduced Ca2+ sensitivity) were linked to hereditary Long QT syndrome type 8 and 14 (Venetucci et al., 2012; Crotti et al., 2013; Marsman et al., 2014). Changes in activation and inactivation parameters leading to widening of so called “window” current were linked to enhanced propensity of reactivation during late phases of AP and thereby generation of early after depolarizations (EADs) (Weiss et al., 2010). Reduction in LTCC expression levels is thought to promote arrhythmogenic Ca2+ alternans via reduced fidelity of channel coupling with RyR2s (Harvey and Hell, 2013). Interestingly, reduced LTCC expression levels in disease states are not always reflected by reduced current. For example, in ventricular cardiomyocytes from human failing hearts ICa was similar to controls, despite of a significant decrease in α1c expression levels, likely due to enhanced phosphorylation by PKA (Chen et al., 2002). Also, fidelity of LTCC-RyR2 coupling can be reduced due to structural remodeling and loss of T-tubules as in hypertrophy, myocardial infarct and HF (Wei et al., 2010).
L-Type Ca2+ Channel in Diabetes
The majority of studies using various models of diabetes did not find statistically significant changes in ICa with a few exceptions (Pereira et al., 2006; Lu et al., 2007). Pereira et al. (2006) showed that in db/db mice (Type 2), the decrease in ICa originates from a reduced number of channels in the sarcolemma. Similar results were obtained in the Akita mouse model (Type 1, Lu et al., 2007). In both models, steady state activation of ICa was shifted to more positive voltages which is expected to reduce ‘window current.’ However, in the latter model steady state inactivation was found shifted even further to the right, resulting in larger “window current.” The information as to whether LTCCs in diabetes undergo posttranslational modifications, or if their distribution with regard to RyR2s is altered, is scarce. While Shao et al. (2012) saw no T-tubular remodeling in STZ-diabetic rats, diminished T-tubular density was observed in db/db mice (Stølen et al., 2009). These findings, along with changes in LTCC in HF, are summarized in Table 1.
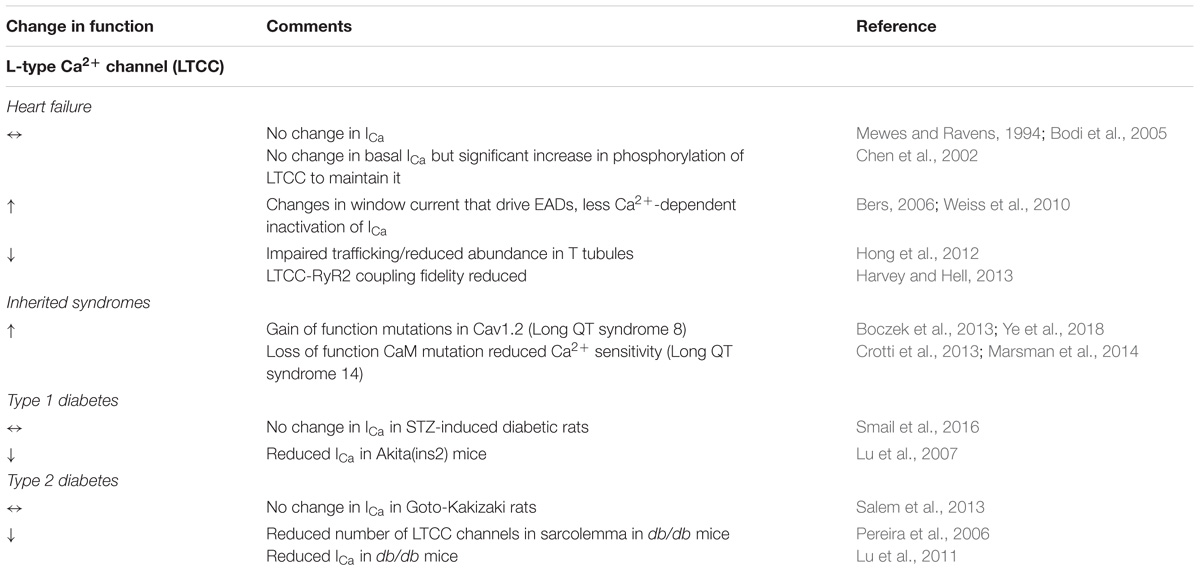
TABLE 1. Changes in LTCC in HF, inherited syndromes and diabetes.
The Ryanodine Receptor
The cardiac SR Ca2+ release channel, RyR2, is a large 2.2 MDa homotetramer consisting of four 565 kDa subunits (Tunwell et al., 1996). While a variety of physiological ligands can modulate RyR2 channel activity including Mg2+ and ATP, Ca2+ is the primary effector of function (Bers, 2002; Fill and Copello, 2002). During EC coupling, the LTCC-mediated influx of cytosolic Ca2+ and increase in [Ca2+] drives activation of other RyR2 channels within the cardiomyocyte via CICR (Fabiato, 1985). Activation of single RyR2 clusters consisting of 8–100 channels (Baddeley et al., 2009) generates a local increase in the concentration of cytosolic Ca2+, known as a Ca2+ spark (Cheng et al., 1996). The summation of Ca2+ sparks produced by activated RyR2 clusters throughout the cardiomyocyte leads to a global Ca2+ transient that initiates muscle contraction (Cheng et al., 1996).
The positive feedback nature of CICR means it is a self-regenerating process that would be inherently unstable without some mechanism for termination of RyR2-mediated Ca2+ release (Fill and Copello, 2002; Kunitomo and Terentyev, 2011). Several candidate mechanisms have been proposed. While it was originally thought that the binding of Ca2+ to cytosolic low affinity sites on the channel during Ca2+ release inactivated RyR2 channels (Fabiato, 1985), it is now apparent that cytosolic Ca2+ plays a limited role in the termination of CICR. There is an accumulation of evidence that demonstrates RyR2 responds to luminal Ca2+ concentrations (Sitsapesan and Williams, 1994; Lukyanenko et al., 1996; Györke and Györke, 1998; Terentyev et al., 2002), with Ca2+ spark termination occurring when SR [Ca2+] falls to a certain level (Brochet et al., 2005; Terentyev et al., 2008; Zima et al., 2008). Depletion may cause unbinding of Ca2+ from luminal activation sites and drive closing of the channel [i.e., deactivation (Jiang et al., 2007; Terentyev et al., 2008)]. More recently, in a hypothesis termed ‘induction decay’ or ‘pernicious attrition’ (Gillespie and Fill, 2013; Laver et al., 2013), it was hypothesized that by decreasing local concentrations of cytosolic Ca2+, a reduction in unitary current via RyR2 breaks the positive-feedback loop of CICR within a cluster. Another phenomenon related to termination is refractoriness of SR Ca2+ release, a period after RyR2 activation and deactivation during which another Ca2+ release event cannot occur (Sham et al., 1998; Szentesi et al., 2004; Györke and Terentyev, 2008). While it was thought that the refractory state persists until SR [Ca2+] is recovered to a critical level, SR Ca2+ load was shown to recover to pre-release levels long before spontaneous Ca2+ wave initiation (Belevych et al., 2012). Mechanisms that determine the refractoriness of CICR remain incompletely understood.
While RyR2 channels open and release Ca2+ from the SR in response to LTCC-mediated Ca2+ influx, channels are not completely closed and have a finite open probability, leading to substantial Ca2+ leak during diastole (Bers, 2014; Eisner et al., 2017). This Ca2+ leak, measurable as Ca2+ sparks, plays an important physiological role in determining the appropriate SR Ca2+ load of cardiomyocytes and threshold for SR Ca2+ release (Cheng et al., 1996; Eisner et al., 2017).
Multiple accessory proteins have been shown to coimmunoprecipitate with RyR2, indicative that the channels exist as large macromolecular complexes (Bers, 2004; Meissner, 2017). The Ca2+ binding protein calmodulin (CaM) directly associates with and regulates RyR2 channels (Samsó and Wagenknecht, 2002; Bers, 2004), while auxilliary proteins CSQ, triadin (TRDN) and junctin (JUN) form the luminal Ca2+ sensor of RyR2 within the SR (Györke et al., 2004; Györke and Terentyev, 2008). FK506 binding protein 12.6 (FKBP12.6) is also an accessory protein of RyR2, and the interaction was proposed to stabilize the channel, preventing spontaneous Ca2+ release and SR Ca2+ leak, although this phenomenon was not observed by many others (Marx et al., 2000; Prestle et al., 2001; George et al., 2003; Goonasekera et al., 2005; Wehrens et al., 2005).
During β-adrenergic stimulation as part of the ‘fight or flight’ response, several EC coupling proteins are targets for posttranslational modification, with phosphorylation well established as a regulatory mechanism modulates ion channel activity with positive chronotropic and inotropic effects to enhance cardiac function (Bers, 2002). The RyR2 channel complex includes a network of associated kinases (protein kinase A (PKA) and Ca2+-calmodulin dependent protein kinase II (CaMKII)) and phosphatases (PP1, PP2A, and PP2B) that dynamically and reversibly modulate its phosphorylation state (Belevych et al., 2011a; Niggli et al., 2013; Terentyev and Hamilton, 2016). Three major phosphorylation sites have been identified – PKA-specific Serine 2808 (S2808) and Serine 2031 (S2031), and CaMKII-specific Serine 2814 (S2814) (Witcher et al., 1991; Wehrens et al., 2004b; Xiao et al., 2005).
Another major posttranslational modification of RyR2 function is oxidation (Zima and Mazurek, 2016). To maintain a fine-tuned balance of reduced and oxidized proteins within the cardiomyocytes, there are multiple sources of ROS as well as antioxidant defense components (Xu et al., 1998). There are approximately 21 cysteine residues within RyR2 that are reduced under physiological conditions, and the channel is known to be susceptible to reversible redox modification (Xu et al., 1998; Dulhunty et al., 2000). At the single channel level, oxidation of RyR2 increases channel open probability and increases the sensitivity to activating Ca2+, while reducing agents have opposite effects (Boraso and Williams, 1994; Xu et al., 1998; Salama et al., 2000).
The Ryanodine Receptor and Cardiac Arrhythmia
Under pathophysiological conditions, diastolic Ca2+ leak from the SR is increased, thus exceeding the critical threshold level and increasing RyR2 channel activation, which in turn activates other channels and results in proarrhythmic diastolic Ca2+ waves (Cheng et al., 1996). This subsequently activates NCX1, driving a net inward current that gives rise to delayed after depolarizations (DADs) and arrhythmias (Ferrier et al., 1973; Mechmann and Pott, 1986; Landstrom et al., 2017). Evidence suggests that increased refractory period shortening, thus increased RyR2-mediated SR Ca2+ leak, promotes Ca2+-dependent arrhythmias in failing hearts (Belevych et al., 2012; Brunello et al., 2013; Cooper et al., 2013).
Abnormal Ca2+ release is the driving force for arrhythmia observed in patients with catecholaminergic polymorphic ventricular tachycardia (CPVT), a condition characterized by pathogenic mutations in RyR2 (Priori et al., 2002; Priori and Chen, 2011), as well as in associated accessory proteins including CSQ (Terentyev et al., 2006; Nyegaard et al., 2012; Roux-Buisson et al., 2012). The condition presents under conditions of enhanced catecholaminergic drive, in the absence of any structural defects in the heart (Priori et al., 2002; Tester et al., 2005). Premature ventricular contractions (PVCs) that often manifest during exercise or periods of stress can degenerate into polymorphic or bidirectional ventricular tachycardia (VT) or fibrillation (VF) and subsequently lead to SCD. Mutations in RyR2 associated with CPVT usually cause increased SR Ca2+ leak that is exacerbated by β-adrenergic stimulation, but both gain- and loss-of-function mutations have been reported (Wehrens et al., 2003; Loaiza et al., 2013; Zhao et al., 2015; Landstrom et al., 2017; Uehara et al., 2017). Mutations in CSQ cause reduction or complete loss of protein expression, and without CSQ-mediated Ca2+ buffering within the SR, RyR2 channels are prone to spontaneous activation, offering a trigger for arrhythmia (Yano and Zarain-Herzberg, 1994; Lahat et al., 2001; Knollmann et al., 2006; Faggioni et al., 2012).
The significance of altered PKA-mediated phosphorylation of RyR2 function in the pathogenesis of cardiac arrhythmia remains controversial. Hyperphosphorylation of RyR2 at S2808 was originally hypothesized to cause dissociation of FKBP12.6, destabilizing the channel and thus increasing open channel probability (Marx et al., 2000). However, the significance of both phosphorylation at this site and the role of PKA-mediated phosphorylation on the function of RyR2 remains a disputed subject (Jiang et al., 2002; Benkusky et al., 2007; Curran et al., 2007; Belevych et al., 2011b; Bovo et al., 2017). The role of CaMKII-mediated Ry2 phosphorylation in modulating channel function is more strongly supported, with consensus that activation of CaMKII as opposed to PKA increases SR Ca2+ leak (Ai et al., 2005; Curran et al., 2007; Niggli et al., 2013), although this is not universal (Wehrens et al., 2006; Yang et al., 2007). It is also well established that chronic CaMKII activity in cardiac disease is a major regulator of RyR2 function with arrhythmogenic consequences (Ai et al., 2005; Zhang et al., 2005; Terentyev et al., 2009; Belevych et al., 2012; Respress et al., 2012; Uchinoumi et al., 2016). Additionally, oxidation as a posttranslational modification may alter RyR2 function in cardiac disease. In the healthy heart, oxidation may serve to transiently enhance Ca2+ release, increasing cardiac output (Niggli et al., 2013). However, in conditions of severe oxidative stress such as HF, increased RyR2 oxidation by ROS can lead to RyR2 activation and increased proarrhythmic SR Ca2+ leak (Mochizuki et al., 2007; Belevych et al., 2011a,b). Modulation of RyR2 activity by ROS can also be indirect, via the oxidation of CaMKII and subsequent increased CaMKII-mediated phosphorylation of the channel (Chelu et al., 2009; Anderson, 2015).
The SR Ca2+ load is thought to be a critical factor of cardiac alternans, a repetitive beat-to-beat fluctuation in cellular repolarization at a constant heart rate that closely linked to development of ventricular arrhythmias (Edwards and Blatter, 2014). Any impairment in the refractoriness of RyR2-mediated Ca2+ release or the recovery after channel inactivation may facilitate the onset of alternans, due to a reduction in the subsequent Ca2+ transient amplitude (Edwards and Blatter, 2014). These properties of RyR2 have been shown to be a major component underlying generation of alternans in both computational and experimental studies (Sobie et al., 2006; Nivala and Qu, 2012; Shkryl et al., 2012; Alvarez-Lacalle et al., 2013; Sun et al., 2018).
The Ryanodine Receptor in Diabetes
It is well established that increased SR Ca2+ leak due to enhanced RyR2 channel activity significantly contributes to arrhythmogenic potential in diabetic cardiomyopathy.
Injection of STZ destroys insulin-producing β cells and has long been used for the generation of Type 1 diabetes phenotypes. Early studies of RyR2-mediated Ca2+ release in diabetes utilized STZ-diabetic rat models. Work of Yu et al. (1994) showed that cardiomyocytes from STZ-diabetic rats had reduced maximum rates of shortening and relengthening, as well as depressed SR Ca2+ content. Using [3H] ryanodine binding assay as an indication of RyR2 channel functionality, isolated SR membranes from diabetic rats showed reduced high-affinity binding sites. Bidasee et al. (2001) also reported decreased [3H] ryanodine binding in 6-week post STZ injection, but posited this was due to dysfunctional RyR2 rather than a decrease in protein expression or mRNA level as observed in Teshima et al. (2000) and Choi et al. (2002). Interestingly, Zhao et al. (2014) found that Ca2+ spark frequency showed a gradual decline in correlation with progression of STZ-diabetes, with significant differences between 4-week and 12-week post-injection groups.
Yaras et al. (2005) importantly showed increased Ca2+ spark frequency in cardiomyocytes from STZ-diabetic rat, with a reduced Ca2+ transient amplitude and depressed SR Ca2+ loading. This was accompanied by significantly increased phosphorylation of RyR2 at S2808 and a ∼40% decrease in FKBP12.6 association. Similar findings were reported by other groups, where these phenomena were suggested to underscore depressed SR Ca2+ release in STZ-diabetic cardiomyocytes (Shao et al., 2007, 2009; Tuncay et al., 2014), indicative that Ca2+ leak via hyperactive RyR2 contributes to the disease phenotype. Later work showed that exercise training for 4 weeks could attenuate this, reducing S2808 phosphorylation and increase levels of FKBP12.6 expression (Shao et al., 2009). However, the functional role of PKA-mediated RyR2 phosphorylation remains controversial and many studies have shown phosphorylation at the S2808 site does not modulate channel activity in other cardiac disease states (Xiao et al., 2004; Guo et al., 2010). An increase in endogenous CaMKII-mediated phosphorylation of RyR2 has also been implicated in the aberrant Ca2+ handling observed in STZ-diabetic rats (Netticadan et al., 2001) and db/db mice (Stølen et al., 2009). Conversely, Tian et al. (2011) posited that gain-of-function changes in RyR2 function observed in single channel recordings were independent of phosphorylation at either S2808 or S2814 sites. Instead, the increase in open channel probability and 20% reduction in conductance was attributed to increased responsiveness to cytoplasmic activators including Ca2+, alterations in the threshold for activation by luminal Ca2+, and a blunted response to physiological inhibitors.
Other posttranslational modifications of RyR2 in Type 1 diabetes have also been suggested to underscore channel dysfunction. Bidasee et al. (2003a) suggested RyR2 dysfunction, evidenced by decreased [3H] ryanodine binding in STZ-diabetic rats, was in part due to formation of disulfide bonds between adjacent sulfhydryl groups. The same group also showed that non-cross-linking advanced glycation end products (AGEs) on RyR2 are significantly increased in diabetic heart tissue, and this increase could be partially attenuated with insulin treatment (Bidasee et al., 2003b). Extensive carbonylation of RyR2 by increased reactive carbonyl species (RCS) in STZ- diabetic rats was suggested to reduce the responsiveness of RyR2 to cytoplasmic Ca2+. While expression levels of RyR2 remained unchanged, there was an increase in non-functional RyR2 channels, but also an increase in the activity of others. The increased heterogeneity of RyR2 channels was posited to increase spontaneous and dyssynchronous SR Ca2+ release in isolated cardiomyocytes, thus providing a trigger for arrhythmia. Treatment of diabetic rats with RCS scavengers attenuated spontaneous SR Ca2+ release, reduced RyR2 carbonylation and normalized channel functionality.
Fewer studies have investigated changes in RyR2-mediated Ca2+ handling in models of Type 2 diabetes. In the non-failing myocardium of type 2 diabetic patients RyR2 protein expression was decreased, while mRNA levels were decreased in the Goto-Kakizaki model (Reuter et al., 2008; Gaber et al., 2014). In a prediabetic model of metabolic syndrome, whereby dogs were chronically fed a high fat diet (HFD), phosphorylation of RyR2 at S2808 was significantly elevated and the channel’s ability to bind [3H] ryanodine significantly depressed in the ventricles compared to healthy controls, while no changes in RyR2 mRNA or protein expression were observed (Dinçer et al., 2006). Okatan et al. (2016) also observed increased RyR2 phosphorylation at S2808 in rats with high sucrose diet-induced metabolic syndrome, accompanied by reduced FKBP12.6 expression. Through studies of electric-field stimulated intracellular Ca2+ handling, cardiomyocytes isolated from rats with metabolic syndrome showed significantly increased SR Ca2+ leak, depressed SR Ca2+ loading and reduced Ca2+ transient amplitude vs. controls. This data is suggestive that alterations in RyR2 function and SR Ca2+ release may be an important mechanism of early cardiac dysfunction in insulin resistance and diabetes development.
Abnormal intracellular lipid concentration is a hallmark of both obesity and diabetes, and Joseph et al. (2016) recently studied Ca2+ handling in cardiomyocytes from a transgenic model of cardiac lipid overload, with peroxisome proliferator-activated receptor-γ (PPARg) overexpression. This revealed increased Ca2+ spark activity compared to controls that could be reduced by application of antioxidant mitoTEMPO. A significant increase in mitochondrial oxidative stress was suggested to increase RyR2 oxidation and subsequent SR Ca2+ release. In 8-week mice with HFD-induced obesity, Sánchez et al. (2018) reported a shift in the distribution of single RyR2 channel responsiveness to activating cytosolic [Ca2+], whereby channels were much more active to those isolated from control mice. No changes were observed in RyR2 expression levels or phosphorylation status at S2808 or S2814 sites. Instead, this phenomenon was attributed to significantly increased RyR2 oxidation in HFD-mice, implicating the diabetes-related increase in oxidative stress in abnormal Ca2+ handling. Changes in RyR2 in HF and diabetes are summarized in Table 2.
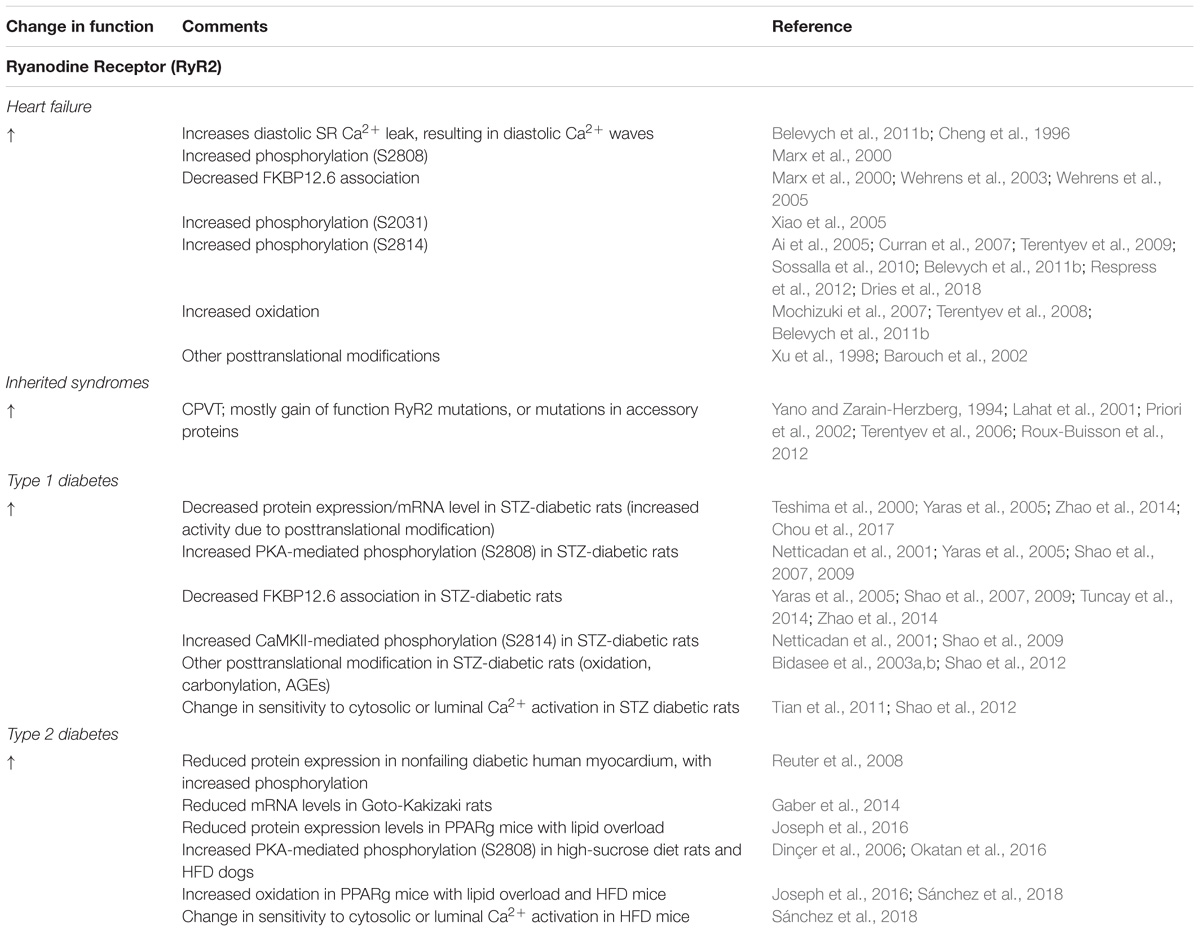
TABLE 2. Changes in RyR2 in HF, inherited syndromes and diabetes.
Sarco/Endoplasmic Reticulum Ca2+-Atpase
For relaxation to occur, intracellular [Ca2+] is decreased primarily via sequestration into the SR by SERCa2a, the primary cardiac SERCa isoform (Bers, 2002). While SERCa2a interacts with multiple proteins (including calreticulin, HRC, PP1, S100A, sarcolipin, SUMO), the most important regulator of function is phospholamban (PLB) (Kranias and Hajjar, 2012). Unphosphorylated PLB has an inhibitory effect on SERCa2a activity, lowering affinity of the pump for Ca2+. Conversely phosphorylation of PLB, either by PKA at Serine 16 (S16) or CaMKII at Threonine 17 (T17), relieves SERCa2a inhibition and increases activity (Simmerman et al., 1986). Oxidative thiol modification of SERCa2a at Cysteine 674 also enhances function (Lancel et al., 2009). Upregulated SERCa2a function is the primary mechanism for positive lusitropic (accelerated relaxation) and inotropic (increased contraction) responses during β-adrenergic stimulation (Fearnley et al., 2011; Vervliet et al., 2018).
Sarco/Endoplasmic Reticulum Ca2+-ATPase and Cardiac Arrhythmia
In HF, impaired SERCa2a expression and activity blunts Ca2+ transient amplitude and rate of decay (Winslow et al., 1999). Reduced sequestration of Ca2+ into the SR drives Ca2+ extrusion from the cardiomyocyte via NCX1. This generates a net inward depolarizing current that can prolong action potential and thus facilitate triggered activity (O’Rourke et al., 1999; Bers et al., 2002). Diminished SR Ca2+ uptake by SERCa2a is also associated with the initiation of cardiac alternans (Merchant and Armoundas, 2012; Nivala and Qu, 2012).
While it may seem counterintuitive to increase SR Ca2+ uptake as a therapeutic strategy, given that SR Ca2+ overload may exacerbate SR Ca2+ leak through RyR2, accumulated evidence suggests the contrary. Upregulation of SERCa2a in small and large animal studies has been shown to protect against development of arrhythmias, improve contractile function and normalize intracellular Ca2+ handling (Meyer and Dillmann, 1998; del Monte et al., 2001, 2004; Suarez et al., 2004; Prunier et al., 2008; Lyon et al., 2011; Fernandez-Tenorio and Niggli, 2018). Multicenter SERCa2a gene therapy trials in humans with HF have been completed, but with limited success (Jaski et al., 2009; Jessup et al., 2011; Zsebo et al., 2014; Greenberg et al., 2016). Increased SERCa2a activity also suppressed cardiac alternans in both computational models and experimental studies (Cutler et al., 2009; Stary et al., 2016).
Sarco/Endoplasmic Reticulum Ca2+-ATPase in Diabetes
Diminished SR Ca2+ uptake has been identified as a primary mechanism for decreased cardiac contractility observed in diabetic cardiomyopathy (Ganguly et al., 1983). Dysfunction of SERCa2a has been established at early stages of type 1 diabetes development and is primarily ascribed to decreased mRNA levels or expression of the protein, resulting in reduced Ca2+ transient amplitudes and a slower rate of transient decay (Ganguly et al., 1983; Trost et al., 2002; Bidasee et al., 2004; Hu et al., 2005; Lacombe et al., 2007). Increased oxidative stress and intracellular ROS concentrations in diabetic hearts can reduce SERCa2a activity by oxidizing Cysteine 674, as well as interfering with the ATP binding site (Xu et al., 1997; Ying et al., 2008). Other molecular mechanisms suggested to drive SERCa2a downregulation include increased carbonylation, glycation and O-GlcNAcylation (Bidasee et al., 2004; Hu et al., 2005; Shao et al., 2011). Changes in PLB expression and phosphorylation have also been reported in STZ-diabetic rats, but this finding is not universal (Wold et al., 2005). Gene therapy with recombinant PLB antibody, which could mimic PLB phosphorylation thus activate SERCa2a, was also shown to increase the rate of whole heart relaxation, contraction and pressure development in a diabetic mouse model and cardiomyopathic hamsters (Meyer et al., 2004; Dieterle et al., 2005).
The pathophysiological role of SERCa2a in cardiomyopathy observed in type 2 diabetes remains to be fully elucidated, although activity is mostly reduced in various models (Zarain-Herzberg et al., 2014). Decreased SERCa2a mRNA levels were observed in the obese fa/fa rats while levels of protein, but not mRNA, were reduced in Otsuka Long-Evans Tokushima Fatty (OLETF) rats. No changes in protein expression were observed in the db/db mouse, but rather an increased PLB expression was posited to account for diminished SR Ca2+ uptake in this model. Stølen et al. (2009) reported reduced SERCa2a protein expression, increased PLB phosphorylation and overall reduced SERCa2a Ca2+ uptake in db/db mice. Conversely, increased expression of SERCa2a and reduced PLB mRNA was observed in ZDF rats, which could be further increased with insulin treatment in a concentration-dependent manner (Fredersdorf et al., 2012). Upregulation of SR Ca2+ uptake via SERCa2a may offer protection in early phases of disease development, countering volume overload and impaired relaxation (Fredersdorf et al., 2012; Zarain-Herzberg et al., 2014). Changes in SERCa2a expression and function in both HF and diabetes are summarized in Table 3.

TABLE 3. Changes in SERCa2a in HF, inherited syndromes and diabetes.
Na+/Ca2+ Exchanger
To maintain the cardiac contraction cycle, equal amount of Ca2+ that enters the cell though LTCCs must be removed to the extracellular milieu. The Na+/Ca2+ exchanger is the main route for Ca2+ extrusion from myocytes. Cardiac NCX (NCX1, 110 kDa) is activated by intracellular Ca2+ in submicromolar range transporting one Ca2+ ion in exchange to thee Na2+ ions (Despa and Bers, 2013). In the early phases of action potential at voltages more positive than reversal potential of NCX1, a small amount of Ca2+ enters the cell. This “primes” RyR2 clusters for activation during subsequent openings of LTCCs, enhancing efficiency of CICR (Neco et al., 2010). At later stages of action potential during the Ca2+ transient, NCX1 works in a forward mode extruding Ca2+, and, since it is electrogenic, contributes to depolarization. Increase in intracellular [Na2+] can significantly increase NCX1-mediated Ca2+ influx and reduce removal. Although NCX1 activity was demonstrated being increased in the presence of oxidants (Kuster et al., 2010), evidence also exists that enhanced production of ROS leads to NCX1 inhibition (Liu and O’Rourke, 2013).
Na+/Ca2+ Exchanger and Cardiac Arrhythmia
Enhanced expression and activity of NCX1 is thought to be one of the major causes of increased arrhythmogenesis in HF (Pogwizd et al., 1999; Bers et al., 2002). NCX1 translates intracellular [Ca2+] during spontaneous Ca2+ waves into depolarizations, i.e., DADs, which can lead to activation of Na2+ channels and extrasystolic action potentials. During systole enhanced NCX1 prolongs APD allowing LTCCs to reactivate thereby contributing to generation of EADs. Increased NCX1-mediated Ca2+ influx during reverse mode due to increased intracellular [Na2+] characteristic of HF or ischemia may increase cytosolic Ca2+ and thereby activity of RyR2s (Satoh et al., 2000; Szepesi et al., 2015). Pharmacological inhibition of NCX1 substantially reduced triggered activity in various models of ventricular arrhythmia with perturbed Ca2+ homeostasis (Bourgonje et al., 2013; Jost et al., 2013; Nagy et al., 2014; Zhong et al., 2018). Experiments using mouse ventricular myocytes with reduced NCX1 demonstrated that genetic inhibition of NCX1 suppressed arrhythmogenic after depolarizations (Bögeholz et al., 2015), while transgenic overexpression promoted generation of EADs due to prolonged repolarization and spontaneous action potentials (Pott et al., 2012).
Na+/Ca2+ Exchanger in Diabetes
The levels and activity of NCX1 vary in different diabetes models. Alloxan or STZ- injected mice with diabetes type 1 showed reduction in NCX1 activity, reduced expression and mRNA levels (Makino et al., 1987; Pierce et al., 1990; Golfman et al., 1998; Hattori et al., 2000). A number of studies shows increased intracellular [Na+] in myocytes from diabetic hearts which enhances NCX1-mediated Ca2+ influx and reduces extrusion (Doliba et al., 2000; Wickley et al., 2007), but reduced NCX1 expression levels have also been reported (Bilginoglu et al., 2013). Belke et al. (2004) using the diabetes type 2 db/db mouse model showed no difference in NCX1 activity. In db/db mice, Stølen et al. (2009) reported increased NCX1 function. Insulin-resistant sucrose-fed rats with diastolic dysfunction showed normal expression and function of NCX1 (Wold et al., 2005). No changes in INCX were reported in high fat diet mouse model (Ricci et al., 2006). An increase in mRNA levels of NCX1 was shown in human patients with diabetes type 2 (Ashrafi et al., 2017). A similar increase was demonstrated in the mouse model with lipid overload (Joseph et al., 2016) and diabetes type 1 Akita(ins2) mouse model of cardiomyopathy (LaRocca et al., 2012). Alterations in NCX1 expression and function in HF and diabetes are summarized in Table 4.

TABLE 4. Changes in NCX1 in HF, inherited syndromes and diabetes.
Mechanisms of Ca2+ Dependent Arrhythmia in Diabetes
The enhanced propensity to ventricular tachyarrhythmias in diabetic and obese patients is well established (Tse et al., 2016). The arrhythmic potential increases relatively early in the course of development of diabetic cardiomyopathy before the onset of systolic dysfunction. Lacombe et al. (2007) showed dramatic increase in EADs and DADs in ventricular myocytes from diabetic rats with diastolic dysfunction 8 weeks after STZ injection. Sommese et al. (2016) demonstrated enhanced arrhythmogenesis in vivo in fructose rich diet (FRD) prediabetic mice. The authors showed increased activity of RyR2 manifested in enhanced frequency of spontaneous Ca2+ waves in field-stimulated myocytes. Mice expressing SR-targeted CaMKII inhibitor peptide AIP showed less ectopic activity that WT FRD mice, and in rats FRD-mediated increase in pro-arrhythmic spontaneous Ca2+ release was eliminated by incubation of myocytes with pharmacological inhibitor of CaMKII KN93 or antioxidant Tempol. The authors conclude that RyR2 phosphorylation by ROS-activated CaMKII at CaMKII site S2814 plays a major role in diabetes-related arrhythmogenesis. Interestingly, the authors did not find changes in RyR2 phosphorylation at PKA site S2808. Earlier work by Erickson et al. (2013) linked diabetic hyperglycemia-mediated activation of CaMKII with Ca2+-dependent triggered activity in rats. The authors proposed that acute hyperglycemia causes covalent modification of CaMKII by O-linked N-acetylglucosamine (O-GlcNAc), resulting in prolonged activation of CaMKII and subsequent increase in RyR2 CaMKII phosphorylation and thereby its activity.
Diabetes is associated with oxidative stress and mitochondrial dysfunction plays significant role in enhanced production of ROS (Akar, 2013; Xie et al., 2013; Teshima et al., 2014). It is postulated that a micro-domain between the SR and mitochondria allows for control by Ca2+ of mitochondrial function and SR Ca2+ handling machinery by mitochondrial ROS (Ruiz-Meana et al., 2010; Eisner V. et al., 2013; Lopez-Cristosto et al., 2017). The close proximity of mitochondria to SR Ca2+ release sites (∼37–270 nm; Sharma et al., 2000) facilitates not only mitochondrial Ca2+ uptake and subsequent alterations in mitochondrial function (Csordás et al., 2001; Dorn and Maack, 2013), but also ROS-mediated modification of RyR2 and SERCa2a, both of which are redox sensitive (Zima and Blatter, 2006). In the PPARg overexpression mouse, it was demonstrated that oxidative stress due to mitochondrial dysfunction causes increased SR Ca2+ leak by oxidizing RyR2 channels (Joseph et al., 2016). This promoted ventricular ectopy, which was significantly reduced in vivo by a mitochondrial-targeted antioxidant mitoTEMPO. Sánchez et al. (2018) reported increased incidences of PVCs and non-sustained VT in HFD-mice vs. controls and this phenomenon was attributed to significantly increased RyR2 oxidation. When antioxidant apocynin was provided in the drinking water, appearance of ventricular arrhythmias in this model was completely abolished. Interestingly, results from Fauconnier et al. (2007) and Llano-Diez et al. (2016) suggest that in ob/ob and HFD mice at certain stages of disease development, mitochondrial ROS emission is reduced. Fauconnier et al. (2007) proposed that during prolonged exposure to increased fatty acid levels due to the switch in mitochondrial substrate utilization, cardiomyocytes from the ob/ob mouse adapt to preferential use of fatty acids for metabolism, so further exposure to fatty acids improved intracellular Ca2+ homeostasis in this model.
Increased NCX1 activity has also been identified as a mechanism promoting arrhythmia via enhancement of depolarization during spontaneous SR Ca2+ release events (Pott et al., 2011). Joseph et al. (2016) showed that in the PPARg overexpression model, NCX1 was upregulated. Importantly, in left ventricular tissue samples from type 2 diabetes human patients NCX1 expression was found significantly higher than in control patients with hypertrophy (Ashrafi et al., 2017).
Direct evidence that diabetes-related aberrant Ca2+ homeostasis can contribute to reentrant mechanisms of arrhythmia was obtained by Chou et al. (2017) who showed that optically mapped hearts from obese db/db mice exhibit concordant Ca2+/Vm alternans at lower stimulation frequencies than controls. The authors attributed it to lower expression levels of RyR2 and enhanced activity of CaMKII. Interestingly, previous studies using this model showed that the expression levels of LTCCs are substantially reduced (Pereira et al., 2006), while RyR2- mediated leak enhanced; and SERCA2a function severely depressed because of enhanced expression of PLB (Belke et al., 2004). Indeed, reduced fidelity of LTCC/RyR2 coupling and impaired ability to re-sequester Ca2+ into the SR are thought to be major underlying causes for beat-to-beat alternations of intracellular Ca2+ transient amplitude (Edwards and Blatter, 2014).
Therapeutic Strategies to Improve Ca2+ Homeostasis in Diabetes
Profound changes in cardiac Ca2+ homeostasis were reported in multiple animal models of diabetes at various states of disease progression, yet mechanisms underlying Ca2+ mishandling remain to be fully understood. While many experimental studies have sought to elucidate mechanisms underlying the enhanced propensity for arrhythmia in insulin-dependent models of type 1 diabetes, those driving arrhythmogenesis in type 2 diabetes are far less investigated. Given the prevalence of acquired diabetes is drastically increasing due to higher rates of obesity in the developed world, future studies are necessary to understand this complex phenotype.
Insulin Replacement
Strategies to manage diabetes include insulin replacement and aerobic exercise regimes (Winnick et al., 2008). Insulin elicits positive inotropic effects on the myocardium, inducing SERCa2a activity (Pierce et al., 1985), while endurance exercise has been shown to improve intracellular Ca2+ cycling and protect against oxidative stress in diabetic animal models (Kim et al., 1996; Shao et al., 2009; Stølen et al., 2009). Combined therapy was shown to increase expression of Ca2+ handling proteins, restore Ca2+ handling and improve basal cardiac function in type 1 diabetic rats (Le Douairon Lahaye et al., 2012; da Silva et al., 2015). However, due to the multifaceted nature of the disease, diabetes remains a prime risk factor for cardiovascular disease despite beneficial effects of these frontline treatments (Leon and Maddox, 2015).
Inhibition of the Renin-Angiotensin System
During the progression of diabetic cardiomyopathy, hyperglycemia is known to increase activity of the renin-angiotensin system (RAS) (Sechi et al., 1994; Ohishi, 2018). Angiotensin II has been shown to have direct effects on cardiomyocytes, with activation of its receptor (AT1) increasing generation of oxidants and increasing intracellular [Ca2+], as well as activating protein kinase C (PKC) and PKA (Malhotra et al., 1997; de Lannoy et al., 1998; Dostal, 2000; Raimondi et al., 2004). Angiotensin-converting enzyme (ACE) inhibitors and AT1 blockers are used to prevent hypertension and cardiovascular disease in diabetic patients (Mancia et al., 2013; Singh et al., 2018). Antagonism of angiotensin II was demonstrated to alleviate diminished SERCa2a activity in HF models (Okuda et al., 2004; Gassanov et al., 2006), while administration of AT1 blockers to STZ-diabetic rat cardiomyocytes reduced cellular oxidative stress as well as phosphorylation levels of RyR2, improving Ca2+ homeostasis (Privratsky et al., 2003; Yaras et al., 2007; Ozdemir et al., 2009).
β-Blockade
Current and conventional treatments of diabetes are limited in terms of preventing ventricular arrhythmias and SCD. Blockade of the β-adrenergic stimulation cascade remains a primary treatment of HF in a bid to reduce arrhythmogenic responses, including those of Ca2+ handling proteins (Klapholz, 2009; Rehsia and Dhalla, 2010). However, use of antagonists in diabetic patients remains controversial, given β-blockers have been associated with increased risk for cardiovascular events in diabetic patients and can have hypoglycemic side effects (Casiglia and Tikhonoff, 2017; Tsujimoto et al., 2017).
Targeting LTCC or NCX1
As it stands, inhibition of LTCC or NCX1 does not appear to be an appropriate strategy in ameliorating Ca2+ mishandling in diabetic hearts. Current density of LTCC is reported as unaltered or reduced in diabetic models (Pereira et al., 2006; Lu et al., 2007). A reduction of ICa in this setting would diminish the trigger for RyR2-mediated Ca2+ release and thereby may exacerbate systolic dysfunction and increase propensity to Ca2+ alternans. Pharmacological inhibition of NCX1 is generally viewed as beneficial since it reduces Ca2+ influx during reverse mode limiting Ca2+ overload and attenuates depolarization during forward mode reducing triggered activity (Antoons et al., 2012). However, if NCX1 is already diminished as shown in many models of diabetes (Makino et al., 1987; Pierce et al., 1990; Golfman et al., 1998; Hattori et al., 2000) additional inhibition was proven to be detrimental leading to adverse accumulation of Ca2+ in cytosol and cell death (Bögeholz et al., 2017).
Targeting SERCa2a
SERCa2a overexpression and/or enhancement may be a more suitable approach to ameliorate reduced contractility in diabetic cardiomyopathy, given the majority of studies report diminished pump activity. Insulin treatment has been shown to restore SERCa2a expression levels and improve Ca2+ homeostasis in obese type 2 diabetic rats (Fredersdorf et al., 2012). Transgenic overexpression of SERCa2a (or SERCa1a) in diabetic models increased SR Ca2+ uptake and attenuated diminished contractile function (Trost et al., 2002; Vetter et al., 2002; Waller et al., 2015). As previously discussed, studies using adenoviral mediated SERCa2a gene transfer in both small and large animal models of HF demonstrated similar results (del Monte et al., 2001, 2004; Lyon et al., 2011). While gene transfer via adeno-associated virus showed promise in the first human trial, it has shown limited success in others. Improvements and advances in gene therapy technology are likely to facilitate efficient and effective strategies to treat cardiac disease in the future (Ishikawa and Hajjar, 2017).
To enhance SERCa2a activity one could also modulate the inhibitory action of the accessory protein PLB. Ablation or knockdown of PLB has been demonstrated to suppress pro-arrhythmic Ca2+ waves generation in a model of CPVT (Bai et al., 2013), improve mortality rates in CSQ-transgenic mice [severe HF model, (Kaneko et al., 2016)] and importantly, improve contractile function in failing human cardiomyocytes (del Monte et al., 2002). However, ablation has not alleviated HF development in all models and a mutant form of PLB unable to inhibit SERCa has been linked to lethal dilated cardiomyopathy in humans (Haghighi et al., 2003; Song et al., 2003; Sipido and Vangheluwe, 2010; Zhang et al., 2010).
Targeting RyR2
Abnormally high activity of RyR2 is the most universal finding demonstrated across several models of diabetes. Compounds thought to modulate RyR2 including JTV-519 (K201), carvedilol, dantrolene and tetracaine analogs have previously been tested for therapeutic potential (Kaneko et al., 1997; Wehrens et al., 2004a; Kobayashi et al., 2010; Zhou et al., 2011; Zhang et al., 2015). However, there remains a need for drugs without off-target effects and significant effort is currently being made to identify small novel modulators of the channel that will prevent arrhythmogenic Ca2+ leak (Li et al., 2017; Rebbeck et al., 2017).
Stabilization of RyR2-mediated Ca release can be achieved indirectly by targeting CaMKII, given chronic activity in cardiac disease has been linked to RyR2 channel dysfunction (Ai et al., 2005; Uchinoumi et al., 2016; Zhang, 2017). It has been demonstrated that blockade of CaMKII and inhibition of RyR2 phosphorylation in cardiac disease improves intracellular Ca2+ homeostasis and attenuates arrhythmogenesis (Ather et al., 2013; Tzimas et al., 2015; Uchinoumi et al., 2016), including in a rat model of type 2 diabetes (Sommese et al., 2016), but this is also not a universal finding (Chakraborty et al., 2014). Alternatively, stabilization could be achieved by a reduction of RyR2 oxidation. In mouse and rat models of type 2 diabetes, it was demonstrated that treatment with ROS scavengers protects against spontaneous Ca2+ release events, blunting diastolic dysfunction and arrhythmogenesis in vivo (Shao et al., 2011; Joseph et al., 2016; Sommese et al., 2016; Sánchez et al., 2018). Antioxidants may also reduce ROS-dependent CaMKII activation, hence reduce RyR2 phosphorylation and elevated SR Ca2+ leak (Luczak and Anderson, 2014; Sommese et al., 2016; Uchinoumi et al., 2016). While increased oxidative stress and ROS concentrations are a hallmark of multiple cardiac disease states including diabetes, usage of ROS scavengers as a therapeutic strategy is not straightforward because certain levels of intracellular ROS are essential for many physiological processes and the ability to target antioxidants to specific subcellular compartments remains limited (Zima et al., 2014; Dietl and Maack, 2017).
Targeting accessory proteins of RyR2 also offers therapeutic potential. Adenoviral overexpression of sorcin in the hearts of diabetic mice improved contractile function and increased the Ca2+ transient amplitude of isolated rat cardiomyocytes (Suarez et al., 2004). In recent work of Liu et al. (2018) it was demonstrated that gene transfer of modified CaM prolonged refractoriness of RyR2-mediated SR Ca2+ release, abolishing ventricular arrhythmias observed in a mouse model of CPVT.
Conclusion
In conclusion, changes in cardiac Ca2+ homeostasis vary across different models of diabetes and obesity. Diabetes is a progressive disease and therefore results from different laboratories using the same model can differ. Longitudinal studies are warranted to resolve the ongoing discrepancies. Although there are many similarities with HF, there are substantial differences in Ca2+ handling in diabetic cardiomyopathy so the treatment strategies could be different.
Abnormalities in Ca2+ cycling sufficient to increase arrhythmic potential appear very early during disease progression of diabetes. The most consistent finding even at very early stages of disease is enhanced RyR2 activity, making it an attractive therapeutic target. Future studies are needed to identify the most suitable approaches for RyR2 stabilization in diabetes. SERCa2a is also attractive target, especially at the later stages of disease progression and cardiomyopathy development. However, it remains unclear whether SERCa2a function is depressed at the early stages when arrhythmic risk is already high. The emerging concept of a mitochondria-SR microdomain and its potential as a therapeutic target may warrant investigation, given mitochondrial dysfunction is a well-established in diabetes. However, levels of mitochondrial ROS emission should be confirmed at different stages of disease, given work of some laboratories suggesting there may be an adaptive improvement in metabolism of fatty acids and thus a reduction in ROS emission. Furthermore, there remains a need for studies using larger animal models of type 2 diabetes with physiology more analogous to that of humans.
Author Contributions
SH and DT obtained funding, conceived of, and wrote the manuscript.
Funding
This work was supported by American Heart Association Grant #18POST33960456 to SH and NIH Grant HL128507 to DT.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ai, X., Curran, J. W., Shannon, T. R., Bers, D. M., and Pogwizd, S. M. (2005). Ca2 + /calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2 + leak in heart failure. Circ. Res. 97, 1314–1322. doi: 10.1161/01.RES.0000194329.41863.89
Akar, F. G. (2013). Mitochondrial targets for arrhythmia suppression: is there a role for pharmacological intervention? J. Interv. Card Electrophysiol. 37, 249–258. doi: 10.1007/s10840-013-9809-3
Allo, S. N., Lincoln, T. M., Wilson, G. L., Green, F. J., Watanabe, A. M., and Schaffer, S. W. (1991). Non-insulin-dependent diabetes-induced defects in cardiac cellular calcium regulation. Am. J. Physiol. 260, C1165–C1171. doi: 10.1152/ajpcell.1991.260.6.C1165
Alvarez-Lacalle, E., Cantalapiedra, I. R., Peñaranda, A., Cinca, J., Hove-Madsen, L., and Echebarria, B. (2013). Dependency of calcium alternans on ryanodine receptor refractoriness. PLoS One 8:e55042. doi: 10.1371/journal.pone.0055042
American Diabetes Association. (2009). Diagnosis, and classification of diabetes mellitus. Diabetes Care 32(Suppl. 1), S62–S67. doi: 10.2337/dc09-S062
Anderson, M. E. (2015). Oxidant stress promotes disease by activating CaMKII. J. Mol. Cell. Cardiol. 89, 160–167. doi: 10.1016/j.yjmcc.2015.10.014
Aneja, A., Tang, W. H., Bansilal, S., Garcia, M. J., and Farkouh, M. E. (2008). Diabetic cardiomyopathy: insights into pathogenesis, diagnostic challenges, and therapeutic options. Am. J. Med. 121, 748–757. doi: 10.1016/j.amjmed.2008.03.046
Antoons, G., Willems, R., and Sipido, K. R. (2012). Alternative strategies in arrhythmia therapy: evaluation of Na/Ca exchange as an anti-arrhythmic target. Pharmacol. Ther. 134, 26–42. doi: 10.1016/j.pharmthera.2011.12.001
Armoundas, A. A., Rose, J., Aggarwal, R., Stuyvers, B. D., O’Rourke, B., Kass, D. A., et al. (2007). Cellular and molecular determinants of altered Ca2 + handling in the failing rabbit heart: primary defects in SR Ca2 + uptake and release mechanisms. Am. J. Physiol. Heart Circ. Physiol. 292, H1607–H1618. doi: 10.1152/ajpheart.00525.2006
Ashrafi, R., Modi, P., Oo, A. Y., Pullan, D. M., Jian, K., Zhang, H., et al. (2017). Arrhythmogenic gene remodelling in elderly patients with type 2 diabetes with aortic stenosis and normal left ventricular ejection fraction. Exp. Physiol. 102, 1424–1434. doi: 10.1113/EP086412
Ather, S., Wang, W., Wang, Q., Li, N., Anderson, M. E., and Wehrens, X. H. (2013). Inhibition of CaMKII phosphorylation of RyR2 prevents inducible ventricular arrhythmias in mice with Duchenne muscular dystrophy. Heart Rhythm. 10, 592–599. doi: 10.1016/j.hrthm.2012.12.016
Axelsen, L. N., Calloe, K., Braunstein, T. H., Riemann, M., Hofgaard, J. P., Liang, B., et al. (2015). Diet-induced pre-diabetes slows cardiac conductance and promotes arrhythmogenesis. Cardiovasc. Diabetol. 14:87. doi: 10.1186/s12933-015-0246-8
Baddeley, D., Jayasinghe, I. D., Lam, L., Rossberger, S., Cannell, M. B., and Soeller, C. (2009). Optical single-channel resolution imaging of the ryanodine receptor distribution in rat cardiac myocytes. Proc Natl Acad Sci U.S.A. 106, 22275–22280. doi: 10.1073/pnas.0908971106
Bai, Y., Jones, P. P., Guo, J., Zhong, X., Clark, R. B., Zhou, Q., et al. (2013). Phospholamban knockout breaks arrhythmogenic Ca2+ waves and suppresses catecholaminergic polymorphic ventricular tachycardia in mice. Circ. Res. 113, 517–526. doi: 10.1161/CIRCRESAHA.113.301678
Barouch, L. A., Harrison, R. W., Skaf, M. W., Rosas, G. O., Cappola, T. P., Kobeissi, Z. A., et al. (2002). Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature 416, 337–339. doi: 10.1038/416337a
Belevych, A. E., Sansom, S. E., Terentyeva, R., Ho, H. T., Nishijima, Y., Martin, M. M., et al. (2011a). MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS One 6:e28324. doi: 10.1371/journal.pone.0028324
Belevych, A. E., Terentyev, D., Terentyeva, R., Ho, H. T., Györke, I., Bonilla, I. M., et al. (2012). Shortened Ca2 + signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ. Res. 110, 569–577. doi: 10.1161/CIRCRESAHA.111.260455
Belevych, A. E., Terentyev, D., Terentyeva, R., Nishijima, Y., Sridhar, A., Hamlin, R. L., et al. (2011b). The relationship between arrhythmogenesis and impaired contractility in heart failure: role of altered ryanodine receptor function. Cardiovasc. Res. 90, 493–502. doi: 10.1093/cvr/cvr025
Belke, D. D., and Dillmann, W. H. (2004). Altered cardiac calcium handling in diabetes. Curr. Hypertens. Rep. 6, 424–429. doi: 10.1007/s11906-004-0035-3
Belke, D. D., Swanson, E. A., and Dillmann, W. H. (2004). Decreased sarcoplasmic reticulum activity and contractility in diabetic db/db mouse heart. Diabetes 53, 3201–3208. doi: 10.2337/diabetes.53.12.3201
Benjamin, E. J., Virani, S. S., Callaway, C. W., Chamberlain, A. M., Chang, A. R., Cheng, S., et al. (2018). Heart disease and stroke statistics-2018 update: a report from the American heart association. Circulation 137, e67–e492. doi: 10.1161/CIR.0000000000000558
Benkusky, N. A., Weber, C. S., Scherman, J. A., Farrell, E. F., Hacker, T. A., John, M. C., et al. (2007). Intact beta-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circ. Res. 101, 819–829. doi: 10.1161/CIRCRESAHA.107.153007
Bergner, D. W., and Goldberger, J. J. (2010). Diabetes mellitus and sudden cardiac death: what are the data? Cardiol. J. 17, 117–129.
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. doi: 10.1038/415198a
Bers, D. M. (2004). Macromolecular complexes regulating cardiac ryanodine receptor function. J. Mol. Cell. Cardiol. 37, 417–429. doi: 10.1016/j.yjmcc.2004.05.026
Bers, D. M. (2006). Altered cardiac myocyte Ca regulation in heart failure. Physiology 21, 380–387. doi: 10.1152/physiol.00019.2006
Bers, D. M. (2014). Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu. Rev. Physiol. 76, 107–127. doi: 10.1146/annurev-physiol-020911-153308
Bers, D. M., Pogwizd, S. M., and Schlotthauer, K. (2002). Upregulated Na/Ca exchange is involved in both contractile dysfunction and arrhythmogenesis in heart failure. Basic Res. Cardiol. 97(Suppl. 1), I36–I42. doi: 10.1007/s003950200027
Bertero, E., and Maack, C. (2018). Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 15, 457–470. doi: 10.1038/s41569-018-0044-6
Bhatti, J. S., Bhatti, G. K., and Reddy, P. H. (2017). Mitochondrial dysfunction and oxidative stress in metabolic disorders - A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta 1863, 1066–1077. doi: 10.1016/j.bbadis.2016.11.010
Bidasee, K. R., Dinçer, U. D., and Besch, H. R. Jr. (2001). Ryanodine receptor dysfunction in hearts of streptozotocin-induced diabetic rats. Mol. Pharmacol. 60, 1356–1364. doi: 10.1124/mol.60.6.1356
Bidasee, K. R., Nallani, K., Besch, H. R. Jr., and Dinçer, U. D. (2003a). Streptozotocin-induced diabetes increases disulfide bond formation on cardiac ryanodine receptor (RyR2). J. Pharmacol. Exp. Ther. 305, 989–998. doi: 10.1124/jpet.102.046201
Bidasee, K. R., Nallani, K., Yu, Y., Cocklin, R. R., Zhang, Y., Wang, M., et al. (2003b). Chronic diabetes increases advanced glycation end products on cardiac ryanodine receptors/calcium-release channels. Diabetes 52,1825–1836.
Bidasee, K. R., Zhang, Y., Shao, C. H., Wang, M., Patel, K. P., Dinçer, U. D., et al. (2004). Diabetes increases formation of advanced glycation end products on Sarco(endo)plasmic reticulum Ca2+ -ATPase. Diabetes 53, 463–473. doi: 10.2337/diabetes.53.2.463
Bilginoglu, A., Kandilci, H. B., and Turan, B. (2013). Intracellular levels of Na(+) and TTX-sensitive Na(+) channel current in diabetic rat ventricular cardiomyocytes. Cardiovasc. Toxicol. 13, 138–147. doi: 10.1007/s12012-012-9192-9
Boczek, N. J., Best, J. M., Tester, D. J., Giudicessi, J. R., Middha, S., Evans, J. M., et al. (2013). Exome sequencing and systems biology converge to identify novel mutations in the L-type calcium channel, CACNA1C, linked to autosomal dominant long QT syndrome. Circ. Cardiovasc. Genet. 6, 279–289. doi: 10.1161/CIRCGENETICS.113.000138
Bodi, I., Mikala, G., Koch, S. E., Akhter, S. A., and Schwartz, A. (2005). The L-type calcium channel in the heart: the beat goes on. J. Clin. Invest. 115, 3306–3317. doi: 10.1172/JCI27167
Bögeholz, N., Pauls, P., Bauer, B. K., Schulte, J. S., Dechering, D. G., Frommeyer, G., et al. (2015). Suppression of early and late afterdepolarizations by heterozygous knockout of the Na + /Ca2 + exchanger in a murine model. Circ. Arrhythm. Electrophysiol. 8, 1210–1218. doi: 10.1161/CIRCEP.115.002927
Bögeholz, N., Schulte, J. S., Kaese, S., Bauer, B. K., Pauls, P., Dechering, D. G., et al. (2017). The effects of SEA0400 on Ca2 + transient amplitude and proarrhythmia depend on the Na + /Ca2 + exchanger expression level in murine models. Front. Pharmacol. 21:649. doi: 10.3389/fphar.2017.00649
Boraso, A., and Williams, A. J. (1994). Modification of the gating of the cardiac sarcoplasmic reticulum Ca(2 + )-release channel by H2O2 and dithiothreitol. Am. J. Physiol. 267(3 Pt 2), H1010–H1016. doi: 10.1152/ajpheart.1994.267.3.H1010
Boudina, S., and Abel, E. D. (2010). Diabetic cardiomyopathy, causes and effects. Rev. Endocr. Metab. Disord. 11, 31–39. doi: 10.1007/s11154-010-9131-7
Bourgonje, V. J., Vos, M. A., Ozdemir, S., Doisne, N., Acsai, K., Varro, A., et al. (2013). Combined Na( + )/Ca(2 + ) exchanger and L-type calcium channel block as a potential strategy to suppress arrhythmias and maintain ventricular function. Circ. Arrhythm. Electrophysiol. 6, 371–379. doi: 10.1161/CIRCEP.113.000322
Bovo, E., Huke, S., Blatter, L. A., and Zima, A. V. (2017). The effect of PKA-mediated phosphorylation of ryanodine receptor on SR Ca2 + leak in ventricular myocytes. J. Mol. Cell. Cardiol. 104, 9–16. doi: 10.1016/j.yjmcc.2017.01.015
Brochet, D. X., Yang, D., Di Maio, A., Lederer, W. J., Franzini-Armstrong, C., and Cheng, H. (2005). Ca2 + blinks: rapid nanoscopic store calcium signaling. Proc. Natl. Acad. Sci. U.S.A. 102, 3099–3104. doi: 10.1073/pnas.0500059102
Brunello, L., Slabaugh, J. L., Radwanski, P. B., Ho, H. T., Belevych, A. E., Lou, Q., et al. (2013). Decreased RyR2 refractoriness determines myocardial synchronization of aberrant Ca2 + release in a genetic model of arrhythmia. Proc. Natl. Acad. Sci. U.S.A. 110, 10312–10317. doi: 10.1073/pnas.1300052110
Bünemann, M., Gerhardstein, B. L., Gao, T., and Hosey, M. M. (1999). Functional regulation of L-type calcium channels via protein kinase A-mediated phosphorylation of the beta(2) subunit. J. Biol. Chem. 26, 33851–33854. doi: 10.1074/jbc.274.48.33851
Casiglia, E., and Tikhonoff, V. (2017). Long-standing problem of β-blocker-elicited hypoglycemia in diabetes mellitus. Hypertension 70, 42–43. doi: 10.1161/HYPERTENSIONAHA.117.09378
Chakraborty, A., Pasek, D. A., Huang, T. Q., Gomez, A. C., Yamaguchi, N., Anderson, M. E., et al. (2014). Inhibition of CaMKII does not attenuate cardiac hypertrophy in mice with dysfunctional ryanodine receptor. PLoS One 9:e104338. doi: 10.1371/journal.pone.0104338
Chelu, M. G., Sarma, S., Sood, S., Wang, S., van Oort, R. J., Skapura, D. G., et al. (2009). Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2 + leak promotes atrial fibrillation in mice. J. Clin. Invest. 119, 1940–1951. doi: 10.1172/JCI37059
Chen, X., Piacentino, V. III, Furukawa, S., Goldman, B., Margulies, K. B., and Houser, S. R. (2002). L-type Ca2 + channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ. Res. 20, 517–524. doi: 10.1161/01.RES.0000033988.13062.7C
Cheng, H., Lederer, M. R., Lederer, W. J., and Cannell, M. B. (1996). Calcium sparks and [Ca2 + ]i waves in cardiac myocytes. Am. J. Physiol. 270(1 Pt 1), C148–C159. doi: 10.1152/ajpcell.1996.270.1.C148
Choi, K. M., Zhong, Y., Hoit, B. D., Grupp, I. L., Hahn, H., Dilly, K. W., et al. (2002). Defective intracellular Ca(2 + ) signaling contributes to cardiomyopathy in Type 1 diabetic rats. Am. J. Physiol. Heart Circ. Physiol. 283, H1398–H1408. doi: 10.1152/ajpheart.00313.2002
Chou, C. C., Ho, C. T., Lee, H. L., Chu, Y., Yen, T. H., Wen, M. S., et al. (2017). Roles of impaired intracellular calcium cycling in arrhythmogenicity of diabetic mouse model. Pacing Clin. Electrophysiol. 40, 1087–1095. doi: 10.1111/pace.13166
Chugh, S. S., Reinier, K., Teodorescu, C., Evanado, A., Kehr, E., Al Samara, M., et al. (2008). Epidemiology of sudden cardiac death: clinical and research implications. Prog. Cardiovasc. Dis. 51, 213–228. doi: 10.1016/j.pcad.2008.06.003
Cooper, L. L., Li, W., Lu, Y., Centracchio, J., Terentyeva, R., Koren, G., et al. (2013). Redox modification of ryanodine receptors by mitochondria-derived reactive oxygen species contributes to aberrant Ca2 + handling in ageing rabbit hearts. J. Physiol. 591, 5895–5911. doi: 10.1113/jphysiol.2013.260521
Crossman, D. J., Ruygrok, P. N., Soeller, C., and Cannell, M. B. (2011). Changes in the organization of excitation-contraction coupling structures in failing human heart. PLoS One 6:e17901. doi: 10.1371/journal.pone.0017901
Crotti, L., Johnson, C. N., Graf, E., De Ferrari, G. M., Cuneo, B. F., Ovadia, M., et al. (2013). Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 127, 1009–1017. doi: 10.1161/CIRCULATIONAHA.112.001216
Csordás, G., Thomas, A. P., and Hajnóczky, G. (2001). Calcium signal transmission between ryanodine receptors and mitochondria in cardiac muscle. Trends Cardiovasc. Med. 11, 269–275. doi: 10.1016/S1050-1738(01)00123-2
Curran, J., Hinton, M. J., Ríos, E., Bers, D. M., and Shannon, T. R. (2007). Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ. Res. 100, 391–398. doi: 10.1161/01.RES.0000258172.74570.e6
Currie, S., and Smith, G. L. (1999). Enhanced phosphorylation of phospholamban and downregulation of sarco/endoplasmic reticulum Ca2 + ATPase type 2 (SERCA 2) in cardiac sarcoplasmic reticulum from rabbits with heart failure. Cardiovasc. Res. 41, 135–146. doi: 10.1016/S0008-6363(98)00241-7
Cutler, M. J., Wan, X., Laurita, K. R., Hajjar, R. J., and Rosenbaum, D. S. (2009). Targeted SERCA2a gene expression identifies molecular mechanism and therapeutic target for arrhythmogenic cardiac alternans. Circ. Arrhythm. Electrophysiol. 2, 686–694. doi: 10.1161/CIRCEP.109.863118
da Silva, M. F., Natali, A. J., da Silva, E., Gomes, G. J., Teodoro, B. G., Cunha, D. N., et al. (2015). Attenuation of Ca2 + homeostasis, oxidative stress, and mitochondrial dysfunctions in diabetic rat heart: insulin therapy or aerobic exercise? J. Appl. Physiol. 119, 148–156. doi: 10.1152/japplphysiol.00915.2014
Dabkowski, E. R., Williamson, C. L., Bukowski, V. C., Chapman, R. S., Leonard, S. S., Peer, C. J., et al. (2009). Diabetic cardiomyopathy-associated dysfunction in spatially distinct mitochondrial subpopulations. Am. J. Physiol. Heart Circ. Physiol. 296, H359–H369. doi: 10.1152/ajpheart.00467.2008
de Lannoy, L. M., Danser, A. H., Bouhuizen, A. M., Saxena, P. R., and Schalekamp, M. A. (1998). Localization and production of angiotensin II in the isolated perfused rat heart. Hypertension 31, 1111–1117. doi: 10.1161/01.HYP.31.5.1111
del Monte, F., Hajjar, R. J., and Harding, S. E. (2001). Overwhelming evidence of the beneficial effects of SERCA gene transfer in heart failure. Circ. Res. 88, E66–E67. doi: 10.1161/hh1101.092004
del Monte, F., Harding, S. E., Dec, G. W., Gwathmey, J. K., and Hajjar, R. J. (2002). Targeting phospholamban by gene transfer in human heart failure. Circulation 105, 904–907. doi: 10.1161/hc0802.105564
del Monte, F., Lebeche, D., Guerrero, J. L., Tsuji, T., Doye, A. A., Gwathmey, J. K., et al. (2004). Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc. Natl. Acad. Sci. U.S.A. 101, 5622–5627. doi: 10.1073/pnas.0305778101
Despa, S., and Bers, D. M. (2013). Na+ transport in the normal and failing heart - remember the balance. J. Mol. Cell. Cardiol. 61, 2–10. doi: 10.1016/j.yjmcc.2013.04.011
Despa, S., Islam, M. A., Weber, C. R., Pogwizd, S. M., and Bers, D. M. (2002). Intracellular Na( + ) concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 105, 2543–2548. doi: 10.1161/01.CIR.0000016701.85760.97
Dieterle, T., Meyer, M., Gu, Y., Belke, D. D., Swanson, E., Iwatate, M., et al. (2005). Gene transfer of a phospholamban-targeted antibody improves calcium handling and cardiac function in heart failure. Cardiovasc. Res. 67, 678–688. doi: 10.1016/j.cardiores.2005.04.029
Dietl, A., and Maack, C. (2017). Targeting mitochondrial calcium handling and reactive oxygen species in heart failure. Curr. Heart Fail. Rep. 14, 338–349. doi: 10.1007/s11897-017-0347-7
Dinçer, U. D., Araiza, A., Knudson, J. D., Shao, C. H., Bidasee, K. R., and Tune, J. D. (2006). Dysfunction of cardiac ryanodine receptors in the metabolic syndrome. J. Mol. Cell. Cardiol. 41, 108–114. doi: 10.1016/j.yjmcc.2006.04.018
Doliba, N. M., Babsky, A. M., Wehrli, S. L., Ivanics, T. M., Friedman, M. F., and Osbakken, M. D. (2000). Metabolic control of sodium transport in streptozotocin-induced diabetic rat hearts. Biochemistry 65, 502–508.
Dorn, G. W., and Maack, C. (2013). SR and mitochondria: calcium cross-talk between kissing cousins. J. Mol. Cell. Cardiol. 55, 42–49. doi: 10.1016/j.yjmcc.2012.07.015
Dostal, D. E. (2000). The cardiac renin-angiotensin system: novel signaling mechanisms related to cardiac growth and function. Regul. Pept. 28, 1–11. doi: 10.1016/S0167-0115(99)00123-8
Dries, E., Santiago, D. J., Gilbert, G., Lenaerts, I., Vandenberk, B., Nagaraju, C. K., et al. (2018). Hyperactive ryanodine receptors in human heart failure and ischaemic cardiomyopathy reside outside of couplons. Cardiovasc. Res. 114, 1512–1524. doi: 10.1093/cvr/cvy088
Dulhunty, A., Haarmann, C., Green, D., and Hart, J. (2000). How many cysteine residues regulate ryanodine receptor channel activity? Antioxid. Redox Signal. 2, 27–34. doi: 10.1089/ars.2000.2.1-27
Edwards, J. N., and Blatter, L. A. (2014). Cardiac alternans and intracellular calcium cycling. Clin. Exp. Pharmacol. Physiol. 41, 524–532. doi: 10.1111/1440-1681.12231
Eisner, D., Bode, E., Venetucci, L., and Trafford, A. (2013). Calcium flux balance in the heart. J. Mol. Cell. Cardiol. 58, 110–117. doi: 10.1016/j.yjmcc.2012.11.017
Eisner, V., Csordás, G., and Hajnóczky, G. (2013). Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle - pivotal roles in Ca2+ and reactive oxygen species signaling. J. Cell. Sci. 126(Pt 14), 2965–2978. doi: 10.1242/jcs.093609
Eisner, D. A., Caldwell, J. L., Kistamás, K., and Trafford, A. W. (2017). Calcium and excitation-contraction coupling in the heart. Circ. Res. 121, 181–195. doi: 10.1161/CIRCRESAHA.117.310230
Erickson, J. R., Pereira, L., Wang, L., Han, G., Ferguson, A., Dao, K., et al. (2013). Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502, 372–376. doi: 10.1038/nature12537
Fabiato, A. (1985). Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J. Gen. Physiol. 85, 247–289. doi: 10.1085/jgp.85.2.247
Faggioni, M., Kryshtal, D. O., and Knollmann, B. C. (2012). Calsequestrin mutations and catecholaminergic polymorphic ventricular tachycardia. Pediatr. Cardiol. 33, 959–967. doi: 10.1007/s00246-012-0256-1
Fauconnier, J., Andersson, D. C., Zhang, S. J., Lanner, J. T., Wibom, R., and Katz, A. (2007). Effects of palmitate on Ca(2 + ) handling in adult control and ob/ob cardiomyocytes: impact of mitochondrial reactive oxygen species. Diabetes 56, 1136–1142. doi: 10.2337/db06-0739
Fearnley, C. J., Roderick, H. L., and Bootman, M. D. (2011). Calcium signaling in cardiac myocytes. Cold Spring Harb Perspect. Biol. 3:a004242. doi: 10.1101/cshperspect.a004242
Fernandez-Tenorio, M., and Niggli, E. (2018). Stabilization of Ca2 + signaling in cardiac muscle by stimulation of SERCA. J. Mol. Cell. Cardiol. 119, 87–95. doi: 10.1016/j.yjmcc.2018.04.015
Ferrier, G. R., Saunders, J. H., and Mendez, C. (1973). A cellular mechanism for the generation of ventricular arrhythmias by acetylstrophanthidin. Circ. Res. 32, 600–609. doi: 10.1161/01.RES.32.5.600
Fill, M., and Copello, J. A. (2002). Ryanodine receptor calcium release channels. Physiol. Rev. 82, 893–922. doi: 10.1152/physrev.00013.2002
Fredersdorf, S., Thumann, C., Zimmermann, W. H., Vetter, R., Graf, T., Luchner, A., et al. (2012). Increased myocardial SERCA expression in early type 2 diabetes mellitus is insulin dependent: In vivo and in vitro data. Cardiovasc. Diabetol. 11:57. doi: 10.1186/1475-2840-11-57
Gaber, E. M., Jayaprakash, P., Qureshi, M. A., Parekh, K., Oz, M., Adrian, T. E., et al. (2014). Effects of a sucrose-enriched diet on the pattern of gene expression, contraction and Ca(2 + ) transport in Goto-Kakizaki type 2 diabetic rat heart. Exp. Physiol. 99, 881–893. doi: 10.1113/expphysiol.2013.077594
Ganguly, P. K., Pierce, G. N., Dhalla, K. S., and Dhalla, N. S. (1983). Defective sarcoplasmic reticular calcium transport in diabetic cardiomyopathy. Am. J. Physiol. 244, E528–E535. doi: 10.1152/ajpendo.1983.244.6.E528
Gassanov, N., Brandt, M. C., Michels, G., Lindner, M., Er, F., and Hoppe, U. C. (2006). Angiotensin II-induced changes of calcium sparks and ionic currents in human atrial myocytes: potential role for early remodeling in atrial fibrillation. Cell Calcium 39, 175–186. doi: 10.1016/j.ceca.2005.10.008
George, C. H., Sorathia, R., Bertrand, B. M., and Lai, F. A. (2003). In situ modulation of the human cardiac ryanodine receptor (hRyR2) by FKBP12.6. Biochem. J. 370(Pt 2), 579–589. doi: 10.1042/BJ20021433
Gillespie, D., and Fill, M. (2013). Pernicious attrition and inter-RyR2 CICR current control in cardiac muscle. J. Mol. Cell. Cardiol. 58, 53–58. doi: 10.1016/j.yjmcc.2013.01.011
Golfman, L., Dixon, I. M., Takeda, N., Lukas, A., Dakshinamurti, K., and Dhalla, N. S. (1998). Cardiac sarcolemmal Na( + )-Ca2 + exchange and Na( + )-K + ATPase activities and gene expression in alloxan-induced diabetes in rats. Mol. Cell. Biochem. 188, 91–101. doi: 10.1023/A:1006824623496
Goonasekera, S. A., Chen, S. R., and Dirksen, R. T. (2005). Reconstitution of local Ca2 + signaling between cardiac L-type Ca2 + channels and ryanodine receptors: insights into regulation by FKBP12.6. Am. J. Physiol. Cell Physiol. 289, C1476–C1484. doi: 10.1152/ajpcell.00250.2005
Gorski, P. A., Ceholski, D. K., and Hajjar, R. J. (2015). Altered myocardial calcium cycling and energetics in heart failure–a rational approach for disease treatment. Cell Metab. 21, 183–194. doi: 10.1016/j.cmet.2015.01.005
Greenberg, B., Butler, J., Felker, G. M., Ponikowski, P., Voors, A. A., Desai, A. S., et al. (2016). Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 387, 1178–1186. doi: 10.1016/S0140-6736(16)00082-9
Guo, T., Cornea, R. L., Huke, S., Camors, E., Yang, Y., Picht, E., et al. (2010). Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on Ca sparks. Circ. Res. 106, 1743–1752. doi: 10.1161/CIRCRESAHA.110.219816
Györke, I., and Györke, S. (1998). Regulation of the cardiac ryanodine receptor channel by luminal Ca2 + involves luminal Ca2 + sensing sites. Biophys. J. 75, 2801–2810. doi: 10.1016/S0006-3495(98)77723-9
Györke, I., Hester, N., Jones, L. R., and Györke, S. (2004). The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys. J. 86, 2121–2128. doi: 10.1016/S0006-3495(04)74271-X
Györke, S., and Terentyev, D. (2008). Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc. Res. 77, 245–255. doi: 10.1093/cvr/cvm038
Haghighi, K., Kolokathis, F., Gramolini, A. O., Waggoner, J. R., Pater, L., Lynch, R. A., et al. (2006). A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc. Natl. Acad. Sci. U.S.A. 103, 1388–1393. doi: 10.1073/pnas.0510519103
Haghighi, K., Kolokathis, F., Pater, L., Lynch, R. A., Asahi, M., Gramolini, A. O., et al. (2003). Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J. Clin. Invest. 111, 869–876. doi: 10.1172/JCI17892
Harvey, R. D., and Hell, J. W. (2013). CaV1.2 signaling complexes in the heart. J. Mol. Cell. Cardiol. 58, 143–152. doi: 10.1016/j.yjmcc.2012.12.006
Hasenfuss, G., Reinecke, H., Studer, R., Meyer, M., Pieske, B., Holtz, J., et al. (1994). Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2 + )-ATPase in failing and nonfailing human myocardium. Circ. Res. 75, 434–442. doi: 10.1161/01.RES.75.3.434
Hattori, Y., Matsuda, N., Kimura, J., Ishitani, T., Tamada, A., Gando, S., et al. (2000). Diminished function and expression of the cardiac Na + -Ca2 + exchanger in diabetic rats: implication in Ca2 + overload. J. Physiol. 15(527 Pt 1), 85–94. doi: 10.1111/j.1469-7793.2000.00085.x
Hong, T. T., Smyth, J. W., Chu, K. Y., Vogan, J. M., Fong, T. S., Jensen, B. C., et al. (2012). BIN1 is reduced and Cav1.2 trafficking is impaired in human failing cardiomyocytes. Heart Rhythm. 9, 812–820. doi: 10.1016/j.hrthm.2011.11.055
Høydal, M. A., Kirkeby-Garstad, I., Karevold, A., Wiseth, R., Haaverstad, R., Wahba, A., et al. (2018). Human cardiomyocyte calcium handling and transverse tubules in mid-stage of post-myocardial-infarction heart failure. ESC Heart Fail. 5, 332–342. doi: 10.1002/ehf2.12271
Hu, Y., Belke, D., Suarez, J., Swanson, E., Clark, R., Hoshijima, M., et al. (2005). Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circ. Res. 96, 1006–1013. doi: 10.1161/01.RES.0000165478.06813.58
Inoue, M., and Bridge, J. H. (2003). Ca2 + sparks in rabbit ventricular myocytes evoked by action potentials: involvement of clusters of L-type Ca2 + channels. Circ. Res. 92, 532–538. doi: 10.1161/01.RES.0000064175.70693.EC
Ishikawa, K., and Hajjar, R. J. (2017). Current methods in cardiac gene therapy: overview. Methods Mol. Biol. 1521, 3–14. doi: 10.1007/978-1-4939-6588-5_1
Jaski, B. E., Jessup, M. L., Mancini, D. M., Cappola, T. P., Pauly, D. F., Greenberg, B., et al. (2009). Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J. Card Fail. 15, 171–181. doi: 10.1016/j.cardfail.2009.01.013
Jessup, M., Greenberg, B., Mancini, D., Cappola, T., Pauly, D. F., Jaski, B., et al. (2011). Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2 + -ATPase in patients with advanced heart failure. Circulation 124, 304–313. doi: 10.1161/CIRCULATIONAHA.111.022889
Jia, G., Hill, M. A., and Sowers, J. R. (2018). Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ. Res. 122, 624–638. doi: 10.1161/CIRCRESAHA.117.311586
Jiang, D., Chen, W., Wang, R., Zhang, L., and Chen, S. R. (2007). Loss of luminal Ca2 + activation in the cardiac ryanodine receptor is associated with ventricular fibrillation and sudden death. Proc. Natl. Acad. Sci. U.S.A. 104, 18309–18314. doi: 10.1073/pnas.0706573104
Jiang, M. T., Lokuta, A. J., Farrell, E. F., Wolff, M. R., Haworth, R. A., and Valdivia, H. H. (2002). Abnormal Ca2 + release, but normal ryanodine receptors, in canine and human heart failure. Circ. Res. 29, 1015–1022. doi: 10.1161/01.RES.0000043663.08689.05
Joseph, L. C., Subramanyam, P., Radlicz, C., Trent, C. M., Iyer, V., Colecraft, H. M., et al. (2016). Mitochondrial oxidative stress during cardiac lipid overload causes intracellular calcium leak and arrhythmia. Heart Rhythm. 13, 1699–1706. doi: 10.1016/j.hrthm.2016.05.002
Jost, N., Nagy, N., Corici, C., Kohajda, Z., Horváth, A., Acsai, K., et al. (2013). ORM-10103, a novel specific inhibitor of the Na + /Ca2 + exchanger, decreases early and delayed afterdepolarizations in the canine heart. Br J Pharmacol. 170, 768–778. doi: 10.1111/bph.12228
Joubert, M., Manrique, A., Cariou, B., and Prieur, X. (2018). Diabetes-related cardiomyopathy: The sweet story of glucose overload from epidemiology to cellular pathways. Diabetes Metab. doi: 10.1016/j.diabet.2018.07.003 [Epub ahead of print].
Junttila, M. J., Barthel, P., Myerburg, R. J., Mäkikallio, T. H., Bauer, A., Ulm, K., et al. (2010). Sudden cardiac death after myocardial infarction in patients with type 2 diabetes. Heart Rhythm. 7, 1396–1403. doi: 10.1016/j.hrthm.2010.07.031
Kaneko, M., Hashikami, K., Yamamoto, S., Matsumoto, H., and Nishimoto, T. (2016). Phospholamban ablation using CRISPR/Cas9 system improves mortality in a murine heart failure model. PLoS One 11:e0168486. doi: 10.1371/journal.pone.0168486
Kaneko, N., Matsuda, R., Toda, M., and Shimamoto, K. (1997). Inhibition of annexin V-dependent Ca2 + movement in large unilamellar vesicles by K201, a new 1,4-benzothiazepine derivative. Biochim. Biophys. Acta 13, 1–7. doi: 10.1016/S0005-2736(97)00132-6
Kim, J. D., Yu, B. P., McCarter, R. J., Lee, S. Y., and Herlihy, J. T. (1996). Exercise and diet modulate cardiac lipid peroxidation and antioxidant defenses. Free Radic. Biol. Med. 20, 83–88. doi: 10.1016/0891-5849(95)02023-3
King, A., and Bowe, J. (2016). Animal models for diabetes: understanding the pathogenesis and finding new treatments. Biochem. Pharmacol. 99, 1–10. doi: 10.1016/j.bcp.2015.08.108
King, A. J. F. (2012). The use of animal models in diabetes research. Br. J. Pharmacol. 166, 877–894. doi: 10.1111/j.1476-5381.2012.01911.x
Kiss, E., Ball, N. A., Kranias, E. G., and Walsh, R. A. (1995). Differential changes in cardiac phospholamban and sarcoplasmic reticular Ca(2 + )-ATPase protein levels. Effects on Ca2 + transport and mechanics in compensated pressure-overload hypertrophy and congestive heart failure. Circ. Res. 77, 759–764. doi: 10.1161/01.RES.77.4.759
Klapholz, M. (2009). Beta-blocker use for the stages of heart failure. Mayo Clin. Proc. 84, 718–729. doi: 10.4065/84.8.718
Knollmann, B. C., Chopra, N., Hlaing, T., Akin, B., Yang, T., Ettensohn, K., et al. (2006). Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2 + release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Invest. 116, 2510–2520. doi: 10.1172/JCI29128
Kobayashi, S., Yano, M., Uchinoumi, H., Suetomi, T., Susa, T., Ono, M., et al. (2010). Dantrolene, a therapeutic agent for malignant hyperthermia, inhibits catecholaminergic polymorphic ventricular tachycardia in a RyR2(R2474S/ + ) knock-in mouse model. Circ. J. 74, 2579–2584. doi: 10.1253/circj.CJ-10-0680
Kranias, E. G., and Hajjar, R. J. (2012). Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circ. Res. 110, 1646–1660. doi: 10.1161/CIRCRESAHA.111.259754
Kunitomo, Y., and Terentyev, D. (2011). How to stop the fire? Control of Ca2+-induced Ca2+ release in cardiac muscle. J. Physiol. 589(Pt 24), 5899–5900. doi: 10.1113/jphysiol.2011.222554
Kuster, G. M., Lancel, S., Zhang, J., Communal, C., Trucillo, M. P., Lim, C. C., et al. (2010). Redox-mediated reciprocal regulation of SERCA and Na + -Ca2 + exchanger contributes to sarcoplasmic reticulum Ca2 + depletion in cardiac myocytes. Free Radic. Biol Med. 48, 1182–1187. doi: 10.1016/j.freeradbiomed.2010.01.038
Laakso, M. (2008). Diabetes as a ’cardiovascular disease equivalent’: implications for treatment. Nat. Clin. Pract. Cardiovasc. Med. 5, 682–683. doi: 10.1038/ncpcardio1344
Lacombe, V. A., Viatchenko-Karpinski, S., Terentyev, D., Sridhar, A., Emani, S., Bonagura, J. D., et al. (2007). Mechanisms of impaired calcium handling underlying subclinical diastolic dysfunction in diabetes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R1787–R1797. doi: 10.1152/ajpregu.00059.2007
Lagadic-Gossmann, D., Buckler, K. J., Le Prigent, K., and Feuvray, D. (1996). Altered Ca2 + handling in ventricular myocytes isolated from diabetic rats. Am. J. Physiol. 270(5 Pt 2), H1529–H1537. doi: 10.1152/ajpheart.1996.270.5.H1529
Lahat, H., Pras, E., Olender, T., Avidan, N., Ben-Asher, E., Man, O., et al. (2001). A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am. J. Hum. Genet. 69, 1378–1384. doi: 10.1086/324565
Lancel, S., Zhang, J., Evangelista, A., Trucillo, M. P., Tong, X., Siwik, D. A., et al. (2009). Nitroxyl activates SERCA in cardiac myocytes via glutathiolation of cysteine 674. Circ. Res. 104, 720–723. doi: 10.1161/CIRCRESAHA.108.188441
Landstrom, A. P., Dobrev, D., and Wehrens, X. H. T. (2017). Calcium signaling and cardiac arrhythmias. Circ. Res. 120, 1969–1993. doi: 10.1161/CIRCRESAHA.117.310083
LaRocca, T. J., Fabris, F., Chen, J., Benhayon, D., Zhang, S., McCollum, L., et al. (2012). Na + /Ca2 + exchanger-1 protects against systolic failure in the Akitains2 model of diabetic cardiomyopathy via a CXCR4/NF-κB pathway. Am. J. Physiol. Heart Circ. Physiol. 303, H353–H367. doi: 10.1152/ajpheart.01198.2011
Laver, D. R., Kong, C. H., Imtiaz, M. S., and Cannell, M. B. (2013). Termination of calcium-induced calcium release by induction decay: an emergent property of stochastic channel gating and molecular scale architecture. J. Mol. Cell. Cardiol. 54, 98–100. doi: 10.1016/j.yjmcc.2012.10.009
Le Douairon Lahaye, S., Gratas-Delamarche, A., Malardé, L., Zguira, S., Vincent, S., Lemoine Morel, S., et al. (2012). Combined insulin treatment and intense exercise training improved basal cardiac function and Ca(2 + )-cycling proteins expression in type 1 diabetic rats. Appl. Physiol. Nutr. Metab. 37, 53–62. doi: 10.1139/h11-127
Leach, R. N., Brickley, K., and Norman, R. I. (1996). Cyclic AMP-dependent protein kinase phosphorylates residues in the C-terminal domain of the cardiac L-type calcium channel alpha1 subunit. Biochim. Biophys. Acta. 11, 205–212. doi: 10.1016/0005-2736(96)00013-2
Leon, B. M., and Maddox, T. M. (2015). Diabetes and cardiovascular disease: Epidemiology, biological mechanisms, treatment recommendations and future research. World J. Diabetes 6, 1246–1258. doi: 10.4239/wjd.v6.i13.1246
Li, N., Wang, Q., Sibrian-Vazquez, M., Klipp, R. C., Reynolds, J. O., Word, T. A., et al. (2017). Treatment of catecholaminergic polymorphic ventricular tachycardia in mice using novel RyR2-modifying drugs. Int. J. Cardiol. 15, 668–673. doi: 10.1016/j.ijcard.2016.10.078
Liang, X., Zhang, Q., Wang, X., Yuan, M., Zhang, Y., Xu, Z., et al. (2018). Reactive oxygen species mediated oxidative stress links diabetes and atrial fibrillation. Mol. Med. Rep. 17, 4933–4940. doi: 10.3892/mmr.2018.8472
Liu, B1 Walton, S. D., Ho, H. T., Belevych, A. E., Tikunova, S. B., Bonilla, I., et al. (2018). Gene transfer of engineered calmodulin alleviates ventricular arrhythmias in a calsequestrin-associated mouse model of catecholaminergic polymorphic ventricular tachycardia. J. Am. Heart Assoc. 7:e008155. doi: 10.1161/JAHA.117.008155
Liu, T., and O’Rourke, B. (2013). Regulation of the Na + /Ca2 + exchanger by pyridine nucleotide redox potential in ventricular myocytes. J. Biol. Chem. 288, 31984–31992. doi: 10.1074/jbc.M113.496588
Llano-Diez, M., Sinclair, J., Yamada, T., Zong, M., Fauconnier, J., Zhang, S. J., et al. (2016). The role of reactive oxygen species in β-adrenergic signaling in cardiomyocytes from mice with the metabolic syndrome. PLoS One 11:e0167090. doi: 10.1371/journal.pone.0167090
Loaiza, R., Benkusky, N. A., Powers, P. P., Hacker, T., Noujaim, S., Ackerman, M. J., et al. (2013). Heterogeneity of ryanodine receptor dysfunction in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 112, 298–308. doi: 10.1161/CIRCRESAHA.112.274803
Lopaschuk, G. D., Katz, S., and McNeill, J. H. (1983). The effect of alloxan- and streptozotocin-induced diabetes on calcium transport in rat cardiac sarcoplasmic reticulum. The possible involvement of long chain acylcarnitines. Can. J. Physiol. Pharmacol. 61, 439–448. doi: 10.1139/y83-068
Lopez-Crisosto, C., Pennanen, C., Vasquez-Trincado, C., Morales, P. E., Bravo-Sagua, R., Quest, A. F. G., et al. (2017). Sarcoplasmic reticulum-mitochondria communication in cardiovascular pathophysiology. Nat. Rev. Cardiol. 14, 342–360. doi: 10.1038/nrcardio.2017.23
Louch, W. E., Bito, V., Heinzel, F. R., Macianskiene, R., Vanhaecke, J., Flameng, W., et al. (2004). Reduced synchrony of Ca2 + release with loss of T-tubules-a comparison to Ca2 + release in human failing cardiomyocytes. Cardiovasc. Res. 62, 63–73. doi: 10.1016/j.cardiores.2003.12.031
Lu, Z., Ballou, L. M., Jiang, Y. P., Cohen, I. S., and Lin, R. Z. (2011). Restoration of defective L-type Ca2 + current in cardiac myocytes of type 2 diabetic db/db mice by Akt and PKC-ι. J. Cardiovasc. Pharmacol. 58, 439–445. doi: 10.1097/FJC.0b013e318228e68c
Lu, Z., Jiang, Y. P., Xu, X. H., Ballou, L. M., Cohen, I. S., and Lin, R. Z. (2007). Decreased L-type Ca2 + current in cardiac myocytes of type 1 diabetic Akita mice due to reduced phosphatidylinositol 3-kinase signaling. Diabetes Metab. Res. Rev. 56, 2780–2789. doi: 10.2337/db06-1629
Luczak, E. D., and Anderson, M. E. (2014). CaMKII oxidative activation and the pathogenesis of cardiac disease. J. Mol. Cell. Cardiol. 73, 112–116. doi: 10.1016/j.yjmcc.2014.02.004
Lukyanenko, V., Györke, I., and Györke, S. (1996). Regulation of calcium release by calcium inside the sarcoplasmic reticulum in ventricular myocytes. Pflugers Arch. 432, 1047–1054. doi: 10.1007/s004240050233
Lyon, A. R., Bannister, M. L., Collins, T., Pearce, E., Sepehripour, A. H., Dubb, S. S., et al. (2011). SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ. Arrhythm. Electrophysiol. 4, 362–372. doi: 10.1161/CIRCEP.110.961615
Makino, N., Dhalla, K. S., Elimban, V., and Dhalla, N. S. (1987). Sarcolemmal Ca2 + transport in streptozotocin-induced diabetic cardiomyopathy in rats. Am. J. Physiol. 253(2 Pt 1), E202–E207. doi: 10.1152/ajpendo.1987.253.2.E202
Malhotra, A., Reich, D., Reich, D., Nakouzi, A., Sanghi, V., Geenen, D. L., et al. (1997). Experimental diabetes is associated with functional activation of protein kinase C epsilon and phosphorylation of troponin I in the heart, which are prevented by angiotensin II receptor blockade. Circ. Res. 81, 1027–1033. doi: 10.1161/01.RES.81.6.1027
Mancia, G., Fagard, R., Narkiewicz, K., Redón, J., Zanchetti, A., Böhm, M., et al. (2013). 2013 ESH/ESC Guidelines for the management of arterial hypertension: the Task Force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J. Hypertens. 31, 1281–1357. doi: 10.1097/01.hjh.0000431740.32696.cc
Marsman, R. F., Barc, J., Beekman, L., Alders, M., Dooijes, D., and van den Wijngaard, A. (2014). A mutation in CALM1 encoding calmodulin in familial idiopathic ventricular fibrillation in childhood and adolescence. J. Am. Coll. Cardiol. 63, 259–266. doi: 10.1016/j.jacc.2013.07.091
Marx, S. O., Reiken, S., Hisamatsu, Y., Jayaraman, T., Burkhoff, D., Rosemblit, N., et al. (2000). PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101, 365–376. doi: 10.1016/S0092-8674(00)80847-8
Mathews, C. E., Langley, S. H., and Leiter, E. H. (2002). New mouse model to study islet transplantation in insulin-dependent diabetes mellitus. Transplantation 73, 1333–1336. doi: 10.1097/00007890-200204270-00024
Mechmann, S., and Pott, L. (1986). Identification of Na-Ca exchange current in single cardiac myocytes. Nature 319, 597–599. doi: 10.1038/319597a0
Meissner, G. (2017). The structural basis of ryanodine receptor ion channel function. J. Gen. Physiol. 149, 1065–1089. doi: 10.1085/jgp.201711878
Merchant, F. M., and Armoundas, A. A. (2012). Role of substrate and triggers in the genesis of cardiac alternans, from the myocyte to the whole heart: implications for therapy. Circulation 125, 539–549. doi: 10.1161/CIRCULATIONAHA.111.033563
Mewes, T., and Ravens, U. (1994). L-type calcium currents of human myocytes from ventricle of non-failing and failing hearts and from atrium. J. Mol. Cell. Cardiol. 26, 1307–1320. doi: 10.1006/jmcc.1994.1149
Meyer, M., Belke, D. D., Trost, S. U., Swanson, E., Dieterle, T., Scott, B., et al. (2004). A recombinant antibody increases cardiac contractility by mimicking phospholamban phosphorylation. FASEB J. 18, 1312–1314. doi: 10.1096/fj.03-1231fje
Meyer, M., and Dillmann, W. H. (1998). Sarcoplasmic reticulum Ca(2 + )-ATPase overexpression by adenovirus mediated gene transfer and in transgenic mice. Cardiovasc. Res. 37, 360–366. doi: 10.1016/S0008-6363(97)00270-8
Mochizuki, M., Yano, M., Oda, T., Tateishi, H., Kobayashi, S., Yamamoto, T., et al. (2007). Scavenging free radicals by low-dose carvedilol prevents redox-dependent Ca2 + leak via stabilization of ryanodine receptor in heart failure. J. Am. Coll. Cardiol. 49, 1722–1732. doi: 10.1016/j.jacc.2007.01.064
Muralidharan, P., Cserne Szappanos, H., Ingley, E., and Hool, L. C. (2017). The cardiac L-type calcium channel alpha subunit is a target for direct redox modification during oxidative stress-the role of cysteine residues in the alpha interacting domain. Clin. Exp. Pharmacol. Physiol. 44(Suppl. 1), 46–54. doi: 10.1111/1440-1681.12750
Nagy, N., Kormos, A., Kohajda, Z., Szebeni,Á, Szepesi, J., Pollesello, P., et al. (2014). Selective Na( + ) /Ca(2 + ) exchanger inhibition prevents Ca(2 + ) overload-induced triggered arrhythmias. Br. J. Pharmacol. 171, 5665–5681. doi: 10.1111/bph.12867
Neco, P., Rose, B., Huynh, N., Zhang, R., Bridge, J. H., Philipson, K. D., et al. (2010). Sodium-calcium exchange is essential for effective triggering of calcium release in mouse heart. Biophys. J. 99, 755–764. doi: 10.1016/j.bpj.2010.04.071
Netticadan, T., Temsah, R. M., Kent, A., Elimban, V., and Dhalla, N. S. (2001). Depressed levels of Ca2 + -cycling proteins may underlie sarcoplasmic reticulum dysfunction in the diabetic heart. Diabetes 50, 2133–2138. doi: 10.2337/diabetes.50.9.2133
Niggli, E., Ullrich, N. D., Gutierrez, D., Kyrychenko, S., Poláková, E., and Shirokova, N. (2013). Posttranslational modifications of cardiac ryanodine receptors: Ca(2 + ) signaling and EC-coupling. Biochim. Biophys. Acta 1833, 866–875. doi: 10.1016/j.bbamcr.2012.08.016
Nivala, M., and Qu, Z. (2012). Calcium alternans in a couplon network model of ventricular myocytes: role of sarcoplasmic reticulum load. Am. J. Physiol. Heart Circ. Physiol. 303, H341–H352. doi: 10.1152/ajpheart.00302.2012
Nyegaard, M., Overgaard, M. T., Søndergaard, M. T., Vranas, M., Behr, E. R., Hildebrandt, L. L., et al. (2012). Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am. J. Hum. Genet. 91, 703–712. doi: 10.1016/j.ajhg.2012.08.015
Ohishi, M. (2018). Hypertension with diabetes mellitus: physiology and pathology. Hypertens. Res. 41, 389–393. doi: 10.1038/s41440-018-0034-4
Okatan, E. N., Durak, A. T., and Turan, B. (2016). Electrophysiological basis of metabolic-syndrome-induced cardiac dysfunction. Can. J. Physiol. Pharmacol. 94, 1064–1073. doi: 10.1139/cjpp-2015-0531
Okuda, S., Yano, M., Doi, M., Oda, T., Tokuhisa, T., Kohno, M., et al. (2004). Valsartan restores sarcoplasmic reticulum function with no appreciable effect on resting cardiac function in pacing-induced heart failure. Circulation 109, 911–919. doi: 10.1161/01.CIR.0000115526.92541.D2
O’Rourke, B., Kass, D. A., Tomaselli, G. F., Kääb, S., Tunin, R., and Marbán, E. (1999). Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure. I: experimental studies. Circ. Res. 84, 562–570. doi: 10.1161/01.RES.84.5.562
Ottolia, M., Torres, N., Bridge, J. H., Philipson, K. D., and Goldhaber, J. I. (2013). Na/Ca exchange and contraction of the heart. J. Mol. Cell. Cardiol. 61, 28–33. doi: 10.1016/j.yjmcc.2013.06.001
Ozdemir, S., Tandogan, B., Ulusu, N. N., and Turan, B. (2009). Angiotensin II receptor blockage prevents diabetes-induced oxidative damage in rat heart. Folia Biol. 55, 11–16.
Pappone, C., and Santinelli, V. (2010). Cardiac electrophysiology in diabetes. Minerva Cardioangiol. 58, 269–276.
Pereira, L., Matthes, J., Schuster, I., Valdivia, H. H., Herzig, S., Richard, S., et al. (2006). Mechanisms of [Ca2 + ]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes 55, 608–615. doi: 10.2337/diabetes.55.03.06.db05-1284
Peterson, B. Z., DeMaria, C. D., Adelman, J. P., and Yue, D. T. (1999). Calmodulin is the Ca2 + sensor for Ca2 + -dependent inactivation of L-type calcium channels. Neuron 22, 549–558. doi: 10.1016/S0896-6273(00)80709-6
Piacentino, V. III, Weber, C. R., Chen, X., Weisser-Thomas, J., Margulies, K. B., Bers, D. M., et al. (2003). Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ. Res. 92, 651–658. doi: 10.1161/01.RES.0000062469.83985.9B
Pierce, G. N., Ganguly, P. K., Dzurba, A., and Dhalla, N. S. (1985). Modification of the function of cardiac subcellular organelles by insulin. Adv. Myocardiol. 6, 113–125.
Pierce, G. N., Ramjiawan, B., Dhalla, N. S., and Ferrari, R. (1990). Na( + )-H + exchange in cardiac sarcolemmal vesicles isolated from diabetic rats. Am. J. Physiol. 258(1 Pt 2), H255–H261. doi: 10.1152/ajpheart.1990.258.1.H255
Pieske, B., Kretschmann, B., Meyer, M., Holubarsch, C., Weirich, J., Posival, H., et al. (1995). Alterations in intracellular calcium handling associated with the inverse force-frequency relation in human dilated cardiomyopathy. Circulation 92, 1169–1178. doi: 10.1161/01.CIR.92.5.1169
Pogwizd, S. M., Qi, M., Yuan, W., Samarel, A. M., and Bers, D. M. (1999). Upregulation of Na( + )/Ca(2 + ) exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ. Res. 85, 1009–1019. doi: 10.1161/01.RES.85.11.1009
Pogwizd, S. M., Schlotthauer, K., Li, L., Yuan, W., and Bers, D. M. (2001). Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current, and residual beta-adrenergic responsiveness. Circ. Res. 88, 1159–1167. doi: 10.1161/hh1101.091193
Pott, C., Eckardt, L., and Goldhaber, J. I. (2011). Triple threat: the Na + /Ca2 + exchanger in the pathophysiology of cardiac arrhythmia, ischemia and heart failure. Curr. Drug Targets 12, 737–747. doi: 10.2174/138945011795378559
Pott, C., Muszynski, A., Ruhe, M., Bögeholz, N., Schulte, J. S., Milberg, P., et al. (2012). Proarrhythmia in a non-failing murine model of cardiac-specific Na + /Ca2 + exchanger overexpression: whole heart and cellular mechanisms. Basic Res. Cardiol. 107:247. doi: 10.1007/s00395-012-0247-7
Prestle, J., Janssen, P. M., Janssen, A. P., Zeitz, O., Lehnart, S. E., Bruce, L., et al. (2001). Overexpression of FK506-binding protein FKBP12.6 in cardiomyocytes reduces ryanodine receptor-mediated Ca(2 + ) leak from the sarcoplasmic reticulum and increases contractility. Circ. Res. 88, 188–194. doi: 10.1161/01.RES.88.2.188
Priori, S. G., and Chen, S. R. (2011). Inherited dysfunction of sarcoplasmic reticulum Ca2 + handling and arrhythmogenesis. Circ. Res. 108, 871–883. doi: 10.1161/CIRCRESAHA.110.226845
Priori, S. G., Napolitano, C., Memmi, M., Colombi, B., Drago, F., Gasparini, M., et al. (2002). Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2, 69–74. doi: 10.1161/01.CIR.0000020013.73106.D8
Privratsky, J. R., Wold, L. E., Sowers, J. R., Quinn, M. T., and Ren, J. (2003). AT1 blockade prevents glucose-induced cardiac dysfunction in ventricular myocytes: role of the AT1 receptor and NADPH oxidase. Hypertension 42, 206–212. doi: 10.1161/01.HYP.0000082814.62655.85
Prunier, F., Kawase, Y., Gianni, D., Scapin, C., Danik, S. B., Ellinor, P. T., et al. (2008). Prevention of ventricular arrhythmias with sarcoplasmic reticulum Ca2 + ATPase pump overexpression in a porcine model of ischemia reperfusion. Circulation 118, 614–624. doi: 10.1161/CIRCULATIONAHA.108.770883
Raimondi, L., De Paoli, P., Mannucci, E., Lonardo, G., Sartiani, L., Banchelli, G., et al. (2004). Restoration of cardiomyocyte functional properties by angiotensin II receptor blockade in diabetic rats. Diabetes 53, 1927–1933. doi: 10.2337/diabetes.53.7.1927
Rebbeck, R. T., Essawy, M. M., Nitu, F. R., Grant, B. D., Gillispie, G. D., Thomas, D. D., et al. (2017). High-throughput screens to discover small-molecule modulators of ryanodine receptor calcium release channels. SLAS Discov. 22, 176–186. doi: 10.1177/1087057116674312
Respress, J. L., van Oort, R. J., Li, N., Rolim, N., Dixit, S. S., deAlmeida, A., et al. (2012). Role of RyR2 phosphorylation at S2814 during heart failure progression. Circ. Res. 110, 1474–1483. doi: 10.1161/CIRCRESAHA.112.268094
Reuter, H., Grönke, S., Adam, C., Ribati, M., Brabender, J., Zobel, C., et al. (2008). Sarcoplasmic Ca2 + release is prolonged in nonfailing myocardium of diabetic patients. Mol. Cell. Biochem. 308, 141–149. doi: 10.1007/s11010-007-9622-3
Ricci, E., Smallwood, S., Chouabe, C., Mertani, H. C., Raccurt, M., Morel, G., et al. (2006). Electrophysiological characterization of left ventricular myocytes from obese Sprague-Dawley rat. Obesity 14, 778–786. doi: 10.1038/oby.2006.90
Roux-Buisson, N., Cacheux, M., Fourest-Lieuvin, A., Fauconnier, J., Brocard, J., Denjoy, I., et al. (2012). Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum. Mol. Genet. 21, 2759–2767. doi: 10.1093/hmg/dds104
Ruiz-Meana, M., Fernandez-Sanz, C., and Garcia-Dorado, D. (2010). The SR-mitochondria interaction: a new player in cardiac pathophysiology. Cardiovasc. Res. 88, 30–39. doi: 10.1093/cvr/cvq225
Salama, G., Menshikova, E. V., and Abramson, J. J. (2000). Molecular interaction between nitric oxide and ryanodine receptors of skeletal and cardiac sarcoplasmic reticulum. Antioxid. Redox. Signal. 2, 5–16. doi: 10.1089/ars.2000.2.1-5
Salem, K. A., Qureshi, M. A., Sydorenko, V., Parekh, K., Jayaprakash, P., Iqbal, T., et al. (2013). Effects of exercise training on excitation-contraction coupling and related mRNA expression in hearts of Goto-Kakizaki type 2 diabetic rats. Mol. Cell. Biochem. 380, 83–96. doi: 10.1007/s11010-013-1662-2
Samsó, M., and Wagenknecht, T. (2002). Apocalmodulin and Ca2 + -calmodulin bind to neighboring locations on the ryanodine receptor. J. Biol. Chem. 11, 1349–1353. doi: 10.1074/jbc.M109196200
Sánchez, G., Araneda, F., Peña, J. P., Finkelstein, J. P., Riquelme, J. A., Montecinos, L., et al. (2018). High-fat-diet-induced obesity produces spontaneous ventricular arrhythmias and increases the activity of ryanodine receptors in mice. Int. J. Mol. Sci. 19:E533. doi: 10.3390/ijms19020533
Satoh, H., Ginsburg, K. S., Qing, K., Terada, H., Hayashi, H., and Bers, D. M. (2000). KB-R7943 block of Ca(2 + ) influx via Na( + )/Ca(2 + ) exchange does not alter twitches or glycoside inotropy but prevents Ca(2 + ) overload in rat ventricular myocytes. Circulation 101, 1441–1446. doi: 10.1161/01.CIR.101.12.1441
Schmidt, U., Hajjar, R. J., Helm, P. A., Kim, C. S., Doye, A. A., and Gwathmey, J. K. (1998). Contribution of abnormal sarcoplasmic reticulum ATPase activity to systolic and diastolic dysfunction in human heart failure. J. Mol. Cell. Cardiol. 30, 1929–1937. doi: 10.1006/jmcc.1998.0748
Schmidt, U., Hajjar, R. J., Kim, C. S., Lebeche, D., Doye, A. A., and Gwathmey, J. K. (1999). Human heart failure: cAMP stimulation of SR Ca(2 + )-ATPase activity and phosphorylation level of phospholamban. Am. J. Physiol. 277(2 Pt 2), H474–H480. doi: 10.1152/ajpheart.1999.277.2.H474
Schmitt, J. P., Kamisago, M., Asahi, M., Li, G. H., Ahmad, F., Mende, U., et al. (2003). Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 299, 1410–1413. doi: 10.1126/science.1081578
Schwinger, R. H., Bölck, B., Münch, G., Brixius, K., Müller-Ehmsen, J., and Erdmann, E. (1998). cAMP-dependent protein kinase A-stimulated sarcoplasmic reticulum function in heart failure. Ann. N. Y. Acad. Sci. 853, 240–250. doi: 10.1111/j.1749-6632.1998.tb08272.x
Schwinger, R. H., Münch, G., Bölck, B., Karczewski, P., Krause, E. G., and Erdmann, E. (1999). Reduced Ca(2 + )-sensitivity of SERCA 2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation. J. Mol. Cell. Cardiol. 31, 479–491. doi: 10.1006/jmcc.1998.0897
Sechi, L. A., Griffin, C. A., and Schambelan, M. (1994). The cardiac renin-angiotensin system in STZ-induced diabetes. Diabetes 43, 1180–1184. doi: 10.2337/diab.43.10.1180
Sham, J. S., Song, L. S., Chen, Y., Deng, L. H., Stern, M. D., Lakatta, E. G., et al. (1998). Termination of Ca2 + release by a local inactivation of ryanodine receptors in cardiac myocytes. Proc. Natl Acad. Sci. U.S.A. 95, 15096–15101. doi: 10.1073/pnas.95.25.15096
Shao, C. H., Capek, H. L., Patel, K. P., Wang, M., Tang, K., DeSouza, C., et al. (2011). Carbonylation contributes to SERCA2a activity loss and diastolic dysfunction in a rat model of type 1 diabetes. Diabetes 60, 947–959. doi: 10.2337/db10-1145
Shao, C. H., Rozanski, G. J., Patel, K. P., and Bidasee, K. R. (2007). Dyssynchronous (non-uniform) Ca2 + release in myocytes from streptozotocin-induced diabetic rats. J. Mol. Cell. Cardiol. 42, 234–246. doi: 10.1016/j.yjmcc.2006.08.018
Shao, C. H., Tian, C., Ouyang, S., Moore, C. J., Alomar, F., Nemet, I., et al. (2012). Carbonylation induces heterogeneity in cardiac ryanodine receptor function in diabetes mellitus. Mol. Pharmacol. 82, 383–399. doi: 10.1124/mol.112.078352
Shao, C. H., Wehrens, X. H., Wyatt, T. A., Parbhu, S., Rozanski, G. J., Patel, K. P., et al. (2009). Exercise training during diabetes attenuates cardiac ryanodine receptor dysregulation. J. Appl. Physiol. 106, 1280–1292. doi: 10.1152/japplphysiol.91280.2008
Sharma, V. K., Ramesh, V., Franzini-Armstrong, C., and Sheu, S. S. (2000). Transport of Ca2 + from sarcoplasmic reticulum to mitochondria in rat ventricular myocytes. J. Bioenerg. Biomembr. 32, 97–104. doi: 10.1023/A:1005520714221
Shen, X., Zheng, S., Thongboonkerd, V., Xu, M., Pierce, W. M. Jr., Klein, J. B., et al. (2004). Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes. Am. J. Physiol. Endocrinol. Metab. 287, E896–E905. doi: 10.1152/ajpendo.00047.2004
Shkryl, V. M., Maxwell, J. T., Domeier, T. L., and Blatter, L. A. (2012). Refractoriness of sarcoplasmic reticulum Ca2 + release determines Ca2 + alternans in atrial myocytes. Am. J. Physiol. Heart Circ. Physiol. 302, H2310–H2320. doi: 10.1152/ajpheart.00079.2012
Simmerman, H. K., Collins, J. H., Theibert, J. L., Wegener, A. D., and Jones, L. R. (1986). Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. J. Biol. Chem. 261, 13333–13341.
Singh, R. M., Waqar, T., Howarth, F. C., Adeghate, E., Bidasee, K., and Singh, J. (2018). Hyperglycemia-induced cardiac contractile dysfunction in the diabetic heart. Heart Fail. Rev. 23, 37–54. doi: 10.1007/s10741-017-9663-y
Sipido, K. R., and Vangheluwe, P. (2010). Targeting sarcoplasmic reticulum Ca2 + uptake to improve heart failure: hit or miss. Circ. Res. 106, 230–233. doi: 10.1161/CIRCRESAHA.109.210740
Siscovick, D. S., Sotoodehnia, N., Rea, T. D., Raghunathan, T. E., Jouven, X., and Lemaitre, R. N. (2010). Type 2 diabetes mellitus and the risk of sudden cardiac arrest in the community. Rev. Endocr. Metab. Disord. 11, 53–59. doi: 10.1007/s11154-010-9133-5
Sitsapesan, R., and Williams, A. J. (1994). Regulation of the gating of the sheep cardiac sarcoplasmic reticulum Ca(2 + )-release channel by luminal Ca2 +. J. Membr. Biol. 137, 215–226. doi: 10.1007/BF00232590
Smail, M. M., Qureshi, M. A., Shmygol, A., Oz, M., Singh, J., Sydorenko, V., et al. (2016). Regional effects of streptozotocin-induced diabetes on shortening and calcium transport in epicardial and endocardial myocytes from rat left ventricle. Physiol. Rep. 4:e13034. doi: 10.14814/phy2.13034
Sobie, E. A., Song, L. S., and Lederer, W. J. (2006). Restitution of Ca(2 + ) release and vulnerability to arrhythmias. J. Cardiovasc. Electrophysiol. 17(Suppl. 1), S64–S70. doi: 10.1111/j.1540-8167.2006.00385.x
Sommese, L., Valverde, C. A., Blanco, P., Castro, M. C., Rueda, O. V., Kaetzel, M., et al. (2016). Ryanodine receptor phosphorylation by CaMKII promotes spontaneous Ca(2 + ) release events in a rodent model of early stage diabetes: the arrhythmogenic substrate. Int. J. Cardiol. 202, 394–406. doi: 10.1016/j.ijcard.2015.09.022
Song, Q., Schmidt, A. G., Hahn, H. S., Carr, A. N., Frank, B., Pater, L., et al. (2003). Rescue of cardiomyocyte dysfunction by phospholamban ablation does not prevent ventricular failure in genetic hypertrophy. J. Clin. Invest. 111, 859–867. doi: 10.1172/JCI16738
Sossalla, S., Fluschnik, N., Schotola, H., Ort, K. R., Neef, S., Schulte, T., et al. (2010). Inhibition of elevated Ca2 + /calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ. Res. 107, 1150–1161. doi: 10.1161/CIRCRESAHA.110.220418
Spooner, P. M. (2008). Sudden cardiac death: influence of diabetes. Diabetes Obes. Metab. 10, 523–532. doi: 10.1111/j.1463-1326.2007.00723.x
Stary, V., Puppala, D., Scherrer-Crosbie, M., Dillmann, W. H., and Armoundas, A. A. (2016). SERCA2a upregulation ameliorates cellular alternans induced by metabolic inhibition. J. Appl. Physiol. 120, 865–875. doi: 10.1152/japplphysiol.00588.2015
Stølen, T. O., Høydal, M. A., Kemi, O. J., Catalucci, D., Ceci, M., Aasum, E., et al. (2009). Interval training normalizes cardiomyocyte function, diastolic Ca2 + control, and SR Ca2 + release synchronicity in a mouse model of diabetic cardiomyopathy. Circ. Res. 105, 527–536. doi: 10.1161/CIRCRESAHA.109.199810
Studer, R., Reinecke, H., Bilger, J., Eschenhagen, T., Böhm, M., Hasenfuss, G., et al. (1994). Gene expression of the cardiac Na( + )-Ca2 + exchanger in end-stage human heart failure. Circ. Res. 75, 443–453. doi: 10.1161/01.RES.75.3.443
Suarez, J., Belke, D. D., Gloss, B., Dieterle, T., McDonough, P. M., Kim, Y. K., et al. (2004). In vivo adenoviral transfer of sorcin reverses cardiac contractile abnormalities of diabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 286, H68–H75. doi: 10.1152/ajpheart.00245.2003
Sun, B., Wei, J., Zhong, X., Guo, W., Yao, J., Wang, R., et al. (2018). The cardiac ryanodine receptor, but not sarcoplasmic reticulum Ca2 + -ATPase, is a major determinant of Ca2 + alternans in intact mouse hearts. J. Biol. Chem. 293:jbc.RA118.003760. doi: 10.1074/jbc.RA118.003760
Szentesi, P., Pignier, C., Egger, M., Kranias, E. G., and Niggli, E. (2004). Sarcoplasmic reticulum Ca2 + refilling controls recovery from Ca2 + -induced Ca2 + release refractoriness in heart muscle. Circ. Res. 95, 807–813. doi: 10.1161/01.RES.0000146029.80463.7d
Szepesi, J., Acsai, K., Sebok, Z., Prorok, J., Pollesello, P., Levijoki, J., et al. (2015). Comparison of the efficiency of Na + /Ca2 + exchanger or Na + /H + exchanger inhibition and their combination in reducing coronary reperfusion-induced arrhythmias. J Physiol. Pharmacol. 66, 215–226.
Szkudelski, T. (2001). The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol. Res. 50, 537–546.
Terentyev, D., Belevych, A. E., Terentyeva, R., Martin, M. M., Malana, G. E., Kuhn, D. E., et al. (2009). miR-1 overexpression enhances Ca(2 + ) release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56alpha and causing CaMKII-dependent hyperphosphorylation of RyR2. Circ. Res. 104, 514–521. doi: 10.1161/CIRCRESAHA.108.181651
Terentyev, D., and Hamilton, S. (2016). Regulation of sarcoplasmic reticulum Ca2 + release by serine-threonine phosphatases in the heart. J. Mol. Cell. Cardiol. 101, 156–164. doi: 10.1016/j.yjmcc.2016.08.020
Terentyev, D., Kubalova, Z., Valle, G., Nori, A., Vedamoorthyrao, S., Terentyeva, R., et al. (2008). Modulation of SR Ca release by luminal Ca and calsequestrin in cardiac myocytes: effects of CASQ2 mutations linked to sudden cardiac death. Biophys. J. 95, 2037–2048. doi: 10.1529/biophysj.107.128249
Terentyev, D., Nori, A., Santoro, M., Viatchenko-Karpinski, S., Kubalova, Z., Györke, I., et al. (2006). Abnormal interactions of calsequestrin with the ryanodine receptor calcium release channel complex linked to exercise-induced sudden cardiac death. Circ. Res. 98, 1151–1158. doi: 10.1161/01.RES.0000220647.93982.08
Terentyev, D., Viatchenko-Karpinski, S., Valdivia, H. H., Escobar, A. L., and Györke, S. (2002). Luminal Ca2+ controls termination and refractory behavior of Ca2+ -induced Ca2+ release in cardiac myocytes. Circ. Res. 6, 414–420. doi: 10.1161/01.RES.0000032490.04207.BD
Teshima, Y., Takahashi, N., Nishio, S., Saito, S., Kondo, H., Fukui, A., et al. (2014). Production of reactive oxygen species in the diabetic heart. Roles of mitochondria and NADPH oxidase. Circ. J. 78, 300–306. doi: 10.1253/circj.CJ-13-1187
Teshima, Y., Takahashi, N., Saikawa, T., Hara, M., Yasunaga, S., Hidaka, S., et al. (2000). Diminished expression of sarcoplasmic reticulum Ca(2 + )-ATPase and ryanodine sensitive Ca(2 + )Channel mRNA in streptozotocin-induced diabetic rat heart. J. Mol. Cell. Cardiol. 32, 655–664. doi: 10.1006/jmcc.2000.1107
Tester, D. J., Kopplin, L. J., Will, M. L., and Ackerman, M. J. (2005). Spectrum and prevalence of cardiac ryanodine receptor (RyR2) mutations in a cohort of unrelated patients referred explicitly for long QT syndrome genetic testing. Heart Rhythm. 2, 1099–1105. doi: 10.1016/j.hrthm.2005.07.012
Tian, C., Shao, C. H., Moore, C. J., Kutty, S., Walseth, T., DeSouza, C., et al. (2011). Gain of function of cardiac ryanodine receptor in a rat model of type 1 diabetes. Cardiovasc. Res. 91, 300–309. doi: 10.1093/cvr/cvr076
Trost, S. U., Belke, D. D., Bluhm, W. F., Meyer, M., Swanson, E., and Dillmann, W. H. (2002). Overexpression of the sarcoplasmic reticulum Ca(2+)-ATPase improves myocardial contractility in diabetic cardiomyopathy. Diabetes 51, 1166–1171. doi: 10.2337/diabetes.51.4.1166
Tse, G., Lai, E. T., Tse, V., and Yeo, J. M. (2016). Molecular and electrophysiological mechanisms underlying cardiac arrhythmogenesis in diabetes mellitus. J. Diabetes Res. 2016:2848759. doi: 10.1155/2016/2848759
Tsujimoto, T., Sugiyama, T., Shapiro, M. F., Noda, M., and Kajio, H. (2017). Risk of cardiovascular events in patients with diabetes mellitus on β-blockers. Hypertension 70, 103–110. doi: 10.1161/HYPERTENSIONAHA.117.09259
Tuncay, E., Okatan, E. N., Toy, A., and Turan, B. (2014). Enhancement of cellular antioxidant-defence preserves diastolic dysfunction via regulation of both diastolic Zn2 + and Ca2 + and prevention of RyR2-leak in hyperglycemic cardiomyocytes. Oxid. Med. Cell. Longev. 2014:290381. doi: 10.1155/2014/290381
Tunwell, R. E., Wickenden, C., Bertrand, B. M., Shevchenko, V. I., Walsh, M. B., Allen, P. D., et al. (1996). The human cardiac muscle ryanodine receptor-calcium release channel: identification, primary structure and topological analysis. Biochem. J. 318( Pt 2), 477–487. doi: 10.1042/bj3180477
Tzimas, C., Terrovitis, J., Lehnart, S. E., Kranias, E. G., and Sanoudou, D. (2015). Calcium/calmodulin-dependent protein kinase II (CaMKII) inhibition ameliorates arrhythmias elicited by junctin ablation under stress conditions. Heart Rhythm. 12, 1599–1610. doi: 10.1016/j.hrthm.2015.03.043
Uchinoumi, H., Yang, Y., Oda, T., Li, N., Alsina, K. M., Puglisi, J. L., et al. (2016). CaMKII-dependent phosphorylation of RyR2 promotes targetable pathological RyR2 conformational shift. J. Mol. Cell. Cardiol. 98, 62–72. doi: 10.1016/j.yjmcc.2016.06.007
Uehara, A., Murayama, T., Yasukochi, M., Fill, M., Horie, M., Okamoto, T., et al. (2017). Extensive Ca2 + leak through K4750Q cardiac ryanodine receptors caused by cytosolic and luminal Ca2 + hypersensitivity. J. Gen. Physiol. 149, 199–218. doi: 10.1085/jgp.201611624
Vasiliadis, I., Kolovou, G., Mavrogeni, S., Nair, D. R., and Mikhailidis, D. P. (2014). Sudden cardiac death and diabetes mellitus. J. Diabetes Complications 28, 573–579. doi: 10.1016/j.jdiacomp.2014.02.003
Venetucci, L., Denegri, M., Napolitano, C., and Priori, S. G. (2012). Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat. Rev. Cardiol. 9, 561–575. doi: 10.1038/nrcardio.2012.93
Vervliet, T., Robinson, E. L., and Roderick, H. L. (2018). Lnc’ing Ca2 +. SERCA and cardiac disease. Cell Calcium 72, 132–134. doi: 10.1016/j.ceca.2018.05.005
Vetter, R., Rehfeld, U., Reissfelder, C., Weiss, W., Wagner, K. D., Günther, J., et al. (2002). Transgenic overexpression of the sarcoplasmic reticulum Ca2 + ATPase improves reticular Ca2 + handling in normal and diabetic rat hearts. FASEB J. 16, 1657–1659. doi: 10.1096/fj.01-1019fje
Waller, A. P., Kalyanasundaram, A., Hayes, S., Periasamy, M., and Lacombe, V. A. (2015). Sarcoplasmic reticulum Ca2 + ATPase pump is a major regulator of glucose transport in the healthy and diabetic heart. Biochim. Biophys. Acta 1852, 873–881. doi: 10.1016/j.bbadis.2015.01.009
Weber, C. R., Piacentino, V. III, Houser, S. R., and Bers, D. M. (2003). Dynamic regulation of sodium/calcium exchange function in human heart failure. Circulation 108, 2224–2229. doi: 10.1161/01.CIR.0000095274.72486.94
Wehrens, X. H., Lehnart, S. E., Huang, F., Vest, J. A., Reiken, S. R., Mohler, P. J., et al. (2003). FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 27, 829–840. doi: 10.1016/S0092-8674(03)00434-3
Wehrens, X. H., Lehnart, S. E., Reiken, S., van der Nagel, R., Morales, R., Sun, J., et al. (2005). Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proc. Natl. Acad. Sci. U.S.A. 102, 9607–9612. doi: 10.1073/pnas.0500353102
Wehrens, X. H., Lehnart, S. E., Reiken, S., Vest, J. A., Wronska, A., and Marks, A. R. (2006). Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc. Natl. Acad. Sci. U.S.A. 103, 511–518. doi: 10.1073/pnas.0510113103
Wehrens, X. H., Lehnart, S. E., Reiken, S. R., Deng, S. X., Vest, J. A., Cervantes, D., et al. (2004a). Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science 304, 292–296. doi: 10.1126/science.1094301
Wehrens, X. H., Lehnart, S. E., Reiken, S. R., and Marks, A. R. (2004b). Ca2 + /calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 94, e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2
Wei, S., Guo, A., Chen, B., Kutschke, W., Xie, Y. P., Zimmerman, K., et al. (2010). T-tubule remodeling during transition from hypertrophy to heart failure. Circ. Res. 107, 520–531. doi: 10.1161/CIRCRESAHA.109.212324
Weiss, J. N., Garfinkel, A., Karagueuzian, H. S., Chen, P. S., and Qu, Z. (2010). Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. 7, 1891–1899. doi: 10.1016/j.hrthm.2010.09.017
Wickley, P. J., Shiga, T., Murray, P. A., and Damron, D. S. (2007). Propofol modulates Na + -Ca2 + exchange activity via activation of protein kinase C in diabetic cardiomyocytes. Anesthesiology 106, 302–311. doi: 10.1097/00000542-200702000-00019
Wilson, A. J., Gill, E. K., Abudalo, R. A., Edgar, K. S., Watson, C. J., and Grieve, D. J. (2018). Reactive oxygen species signalling in the diabetic heart: emerging prospect for therapeutic targeting. Heart 104, 293–299. doi: 10.1136/heartjnl-2017-311448
Winnick, J. J., Sherman, W. M., Habash, D. L., Stout, M. B., Failla, M. L., Belury, M. A., et al. (2008). Short-term aerobic exercise training in obese humans with type 2 diabetes mellitus improves whole-body insulin sensitivity through gains in peripheral, not hepatic insulin sensitivity. J. Clin. Endocrinol. Metab. 93, 771–778. doi: 10.1210/jc.2007-1524
Winslow, R. L., Rice, J., Jafri, S., Marbán, E., and O’Rourke, B. (1999). Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure. II: model studies. Circ. Res. 19, 571–586. doi: 10.1161/01.RES.84.5.571
Witcher, D. R., Kovacs, R. J., Schulman, H., Cefali, D. C., and Jones, L. R. (1991). Unique phosphorylation site on the cardiac ryanodine receptor regulates calcium channel activity. J. Biol. Chem. 15, 11144–11152.
Wold, L. E., Dutta, K., Mason, M. M., Ren, J., Cala, S. E., Schwanke, M. L., et al. (2005). Impaired SERCA function contributes to cardiomyocyte dysfunction in insulin resistant rats. J. Mol. Cell. Cardiol. 39, 297–307. doi: 10.1016/j.yjmcc.2005.03.014
Xiao, B., Jiang, M. T., Zhao, M., Yang, D., Sutherland, C., Lai, F. A., et al. (2005). Characterization of a novel PKA phosphorylation site, serine-2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circ. Res. 96, 847–855. doi: 10.1161/01.RES.0000163276.26083.e8
Xiao, B., Sutherland, C., Walsh, M. P., and Chen, S. R. (2004). Protein kinase A phosphorylation at serine-2808 of the cardiac Ca2 + -release channel (ryanodine receptor) does not dissociate 12.6-kDa FK506-binding protein (FKBP12.6). Circ. Res. 94, 487–495. doi: 10.1161/01.RES.0000115945.89741.22
Xie, C., Biary, N., Tocchetti, C. G., Aon, M. A., Paolocci, N., Kauffman, J., et al. (2013). Glutathione oxidation unmasks proarrhythmic vulnerability of chronically hyperglycemic guinea pigs. Am. J. Physiol. Heart Circ. Physiol. 304, H916–H926. doi: 10.1152/ajpheart.00026.2012
Xie, L. H., Chen, F., Karagueuzian, H. S., and Weiss, J. N. (2009). Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling. Circ. Res. 104, 79–86. doi: 10.1161/CIRCRESAHA.108.183475
Xu, K. Y., Zweier, J. L., and Becker, L. C. (1997). Hydroxyl radical inhibits sarcoplasmic reticulum Ca(2 + )-ATPase function by direct attack on the ATP binding site. Circ. Res. 80, 76–81. doi: 10.1161/01.RES.80.1.76
Xu, L., Eu, J. P., Meissner, G., and Stamler, J. S. (1998). Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 9, 234–237. doi: 10.1126/science.279.5348.234
Yang, D., Zhu, W. Z., Xiao, B., Brochet, D. X., Chen, S. R., Lakatta, E. G., et al. (2007). Ca2 + /calmodulin kinase II-dependent phosphorylation of ryanodine receptors suppresses Ca2 + sparks and Ca2 + waves in cardiac myocytes. Circ. Res. 100, 399–407. doi: 10.1161/01.RES.0000258022.13090.55
Yang, Y., Zhao, J., Qiu, J., Li, J., Liang, X., Zhang, Z., et al. (2018). Xanthine oxidase inhibitor allopurinol prevents oxidative stress-mediated atrial remodeling in alloxan-induced diabetes mellitus rabbits. J. Am. Heart Assoc. 7:e008807. doi: 10.1161/JAHA.118.008807
Yano, K., and Zarain-Herzberg, A. (1994). Sarcoplasmic reticulum calsequestrins: structural and functional properties. Mol. Cell. Biochem. 15, 61–70. doi: 10.1007/BF00925961
Yaras, N., Bilginoglu, A., Vassort, G., and Turan, B. (2007). Restoration of diabetes-induced abnormal local Ca2 + release in cardiomyocytes by angiotensin II receptor blockade. Am. J. Physiol. Heart Circ. Physiol. 292, H912–H920. doi: 10.1152/ajpheart.00824.2006
Yaras, N., Ugur, M., Ozdemir, S., Gurdal, H., Purali, N., Lacampagne, A., et al. (2005). Effects of diabetes on ryanodine receptor Ca release channel (RyR2) and Ca2 + homeostasis in rat heart. Diabetes Metab. Res. Rev. 54, 3082–3088. doi: 10.2337/diabetes.54.11.3082
Ye, D., Tester, D. J., Zhou, W., Papagiannis, J., and Ackerman, M. J. (2018). A pore-localizing CACNA1C-E1115K missense mutation, identified in a patient with idiopathic qt prolongation, bradycardia, and autism spectrum disorder, converts the l-type calcium channel into a hybrid non-selective monovalent cation channel. Heart Rhythm. doi: 10.1016/j.hrthm.2018.08.030 [Epub ahead of print].
Ying, J., Sharov, V., Xu, S., Jiang, B., Gerrity, R., Schöneich, C., et al. (2008). Cysteine-674 oxidation and degradation of sarcoplasmic reticulum Ca(2 + ) ATPase in diabetic pig aorta. Free Radic. Biol. Med. 45, 756–762. doi: 10.1016/j.freeradbiomed.2008.05.029
Yu, Z., Tibbits, G. F., and McNeill, J. H. (1994). Cellular functions of diabetic cardiomyocytes: contractility, rapid-cooling contracture, and ryanodine binding. Am. J Physiol. 266(5 Pt 2), H2082–H2089. doi: 10.1152/ajpheart.1994.266.5.H2082
Zarain-Herzberg, A., García-Rivas, G., and Estrada-Avilés, R. (2014). Regulation of SERCA pumps expression in diabetes. Cell Calcium 56, 302–310. doi: 10.1016/j.ceca.2014.09.005
Zhang, J., Zhou, Q., Smith, C. D., Chen, H., Tan, Z., Chen, B., et al. (2015). Non-β-blocking R-carvedilol enantiomer suppresses Ca2 + waves and stress-induced ventricular tachyarrhythmia without lowering heart rate or blood pressure. Biochem. J. 470, 233–242. doi: 10.1042/BJ20150548
Zhang, P. (2017). CaMKII: the molecular villain that aggravates cardiovascular disease. Exp. Ther. Med. 13, 815–820. doi: 10.3892/etm.2017.4034
Zhang, R., Khoo, M. S., Wu, Y., Yang, Y., Grueter, C. E., Ni, G., et al. (2005). Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 11, 409–417. doi: 10.1038/nm1215
Zhang, T., Guo, T., Mishra, S., Dalton, N. D., Kranias, E. G., Peterson, K. L., et al. (2010). Phospholamban ablation rescues sarcoplasmic reticulum Ca(2 + ) handling but exacerbates cardiac dysfunction in CaMKIIdelta(C) transgenic mice. Circ. Res. 106, 354–362. doi: 10.1161/CIRCRESAHA.109.207423
Zhang, X., Zhang, Z., Zhao, Y., Jiang, N., Qiu, J., Yang, Y., et al. (2017). Alogliptin, a dipeptidyl peptidase-4 inhibitor, alleviates atrial remodeling and improves mitochondrial function and biogenesis in diabetic rabbits. J. Am. Heart Assoc. 6:e005945. doi: 10.1161/JAHA.117.005945
Zhao, S. M., Wang, Y. L., Guo, C. Y., Chen, J. L., and Wu, Y. Q. (2014). Progressive decay of Ca2 + homeostasis in the development of diabetic cardiomyopathy. Cardiovasc. Diabetol. 13:75. doi: 10.1186/1475-2840-13-75
Zhao, Y. T., Valdivia, C. R., Gurrola, G. B., Powers, P. P., Willis, B. C., Moss, R. L., et al. (2015). Arrhythmogenesis in a catecholaminergic polymorphic ventricular tachycardia mutation that depresses ryanodine receptor function. Proc. Natl. Acad. Sci. U.S.A. 112, E1669–E1677. doi: 10.1073/pnas.1419795112
Zhong, M., Rees, C. M., Terentyev, D., Choi, B. R., Koren, G., and Karma, A. (2018). NCX-mediated subcellular Ca2 + dynamics underlying early afterdepolarizations in LQT2 cardiomyocytes. Biophys. J. 115, 1019–1032. doi: 10.1016/j.bpj.2018.08.004
Zhou, Q., Xiao, J., Jiang, D., Wang, R., Vembaiyan, K., Wang, A., et al. (2011). Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2 + release. Nat. Med. 17, 1003–1009. doi: 10.1038/nm.2406
Zima, A. V., and Blatter, L. A. (2006). Redox regulation of cardiac calcium channels and transporters. Cardiovasc. Res. 71, 310–321. doi: 10.1016/j.cardiores.2006.02.019
Zima, A. V., Bovo, E., Mazurek, S. R., Rochira, J. A., Li, W., and Terentyev, D. (2014). Ca handling during excitation-contraction coupling in heart failure. Pflugers Arch. 466, 1129–1137. doi: 10.1007/s00424-014-1469-3
Zima, A. V., and Mazurek, S. R. (2016). Functional impact of ryanodine receptor oxidation on intracellular calcium regulation in the heart. Rev. Physiol. Biochem. Pharmacol. 171, 39–62. doi: 10.1007/112_2016_2
Zima, A. V., Picht, E., Bers, D. M., and Blatter, L. A. (2008). Termination of cardiac Ca2 + sparks: role of intra-SR [Ca2 + ], release flux, and intra-SR Ca2 + diffusion. Circ. Res. 103, e105–e115. doi: 10.1161/CIRCRESAHA.107.183236
Zsebo, K., Yaroshinsky, A., Rudy, J. J., Wagner, K., Greenberg, B., Jessup, M., et al. (2014). Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ. Res. 114, 101–108. doi: 10.1161/CIRCRESAHA.113.302421
Keywords: Ca2+-dependent cardiac arrhythmia, diabetes, heart failure, L-type Ca2+ channels, Na+-Ca2+ exchanger type 1, ryanodine receptor type 2, sarco/endoplasmic reticulum Ca2+-ATPase type 2a
Citation: Hamilton S and Terentyev D (2018) Proarrhythmic Remodeling of Calcium Homeostasis in Cardiac Disease; Implications for Diabetes and Obesity. Front. Physiol. 9:1517. doi: 10.3389/fphys.2018.01517
Received: 16 July 2018; Accepted: 09 October 2018;
Published: 30 October 2018.
Edited by:
John Pearce Morrow, Columbia University, United StatesReviewed by:
Matthew W. Kay, The George Washington University, United StatesXun Ai Ai, Rush University Medical Center, United States
Copyright © 2018 Hamilton and Terentyev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dmitry Terentyev, ZG1pdHJ5X3RlcmVudHlldkBicm93bi5lZHU=