
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Physiol. , 25 October 2018
Sec. Cardiac Electrophysiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.01474
This article is part of the Research Topic The Role of Calcium Handling in Heart Failure and Heart Failure Associated Arrhythmias View all 14 articles
 Marina Scardigli1,2
Marina Scardigli1,2 Cecilia Ferrantini2,3
Cecilia Ferrantini2,3 Claudia Crocini4*
Claudia Crocini4* Francesco S. Pavone1,2,5
Francesco S. Pavone1,2,5 Leonardo Sacconi1,2*
Leonardo Sacconi1,2*Asynchronous Ca2+ release promotes non-homogeneous myofilament activation, leading to mechanical dysfunction, as well as initiation of propagated calcium waves and arrhythmias. Recent advances in microscopy techniques have allowed for optical recordings of local Ca2+ fluxes and action potentials from multiple sub-cellular domains within cardiac cells with unprecedented spatial and temporal resolution. Since then, sub-cellular local information of the spatio-temporal relationship between Ca2+ release and action potential propagation have been unlocked, providing novel mechanistic insights in cardiac excitation-contraction coupling (ECC). Here, we review the promising perspectives arouse from repeatedly probing Ca2+ release at the same sub-cellular location while simultaneously probing multiple locations at the same time within a single cardiac cell. We also compare the results obtained in three different rodent models of cardiac diseases, highlighting disease-specific mechanisms. Slower local Ca2+ release has been observed in regions with defective action potential conduction in diseased cardiac cells. Moreover, significant increment of Ca2+ variability (both in time and in space) has been found in diseased cardiac cells but does not directly correlate with local electrical defects nor with the degree of structural aberrations of the cellular membrane system, suggesting a role for other players of the ECC machinery. We finally explore exciting opportunities provided by the technology for studying different cardiomyocyte populations, as well as for dissecting the mechanisms responsible for subcellular spatio-temporal variability of Ca2+ release.
In cardiac cell, Ca2+ release mediates the transduction of an action potential (AP) generated at the cellular membrane (sarcolemma) into contraction of the sarcomeres, in a process called excitation-contraction coupling (ECC) (Bers, 2002). During the AP, Dihydropyridine receptors (DHPR) located on the sarcolemma open, generating an inward Ca2+ current (IcaL), which in turns activate a massive release of Ca2+ from the sarcoplasmic reticulum (SR) through ryanodine receptor (RyR) 2 (Kushnir and Marks, 2010). This mechanism is called calcium induced calcium release (CICR) and occurs in the dyads, where RyRs are clustered in release units on the SR juxtaposing DHPRs on the sarcolemma (Scriven et al., 2013). When a DHPR opens, local [Ca2+]i rises up to 10–20 μM in less than 1 ms in the junctional cleft and triggers approximately 6–20 RyRs to release at each couplon (Bers, 2001). This increases [Ca2+]i in the cleft to 200–400 μM. Ca2+ diffuses in the cell as the spatial and temporal summation of each local Ca2+ release unit (CRU) (Wier and Balke, 1999) and activates myofilaments that are the end effector of ECC (Bers, 2002). During systole, the strength of twitch depends on SR Ca2+ release, which in turn depends on SR Ca2+ content and the extent of the Ca2+ trigger (Bassani et al., 1995; Hobai and O’Rourke, 2001). After each contraction, [Ca2+]i must decrease, so that cardiac cycle is enabled again. There are two main mechanisms to reduce [Ca2+]i: SR Ca2+-ATPase (SERCA) and sarcolemmal Na+/ Ca2+ exchanger (NCX). The first pumps Ca2+ back into the SR consuming ATP, the latter extrudes the cation in the extracellular space, exploiting the electrochemical gradient of Na+, with a stoichiometry of 3Na:1Ca (Eisner et al., 1998). This balance between trans-sarcolemmal and SR Ca2+ influx and efflux is very highly maintained and it is essential for homogeneous myofilaments contraction and relaxation. The two main features of cardiac diseases, such as contractile dysfunction and arrhythmias, are correlated with ECC abnormalities (Ter Keurs and Boyden, 2007). To ensure a homogenous contraction of the entire cardiomyocyte, the AP is rapidly propagated toward the cell core thanks to a complex network of sarcolemmal invaginations named transverse-axial tubular system (TATS) or t-tubules. In control ventricular myocytes containing t-tubules, SR Ca2+ release is spatially and temporally synchronized (Cheng et al., 1994; Haddock et al., 1999; Brette et al., 2002, 2004, 2005; Yang et al., 2002).
Ca2+ transient desynchronization can be experimentally reproduced in myocytes acutely detubulated through osmotic shock (Brette et al., 2002; Yang et al., 2002) or in cultured myocytes (Louch et al., 2004), mimicking pathological conditions in which t-tubules are disrupted (Kostin et al., 1998; He et al., 2001; Louch et al., 2006; Song et al., 2006; Heinzel et al., 2008; Dibb et al., 2009; Sacconi et al., 2012; Crocini et al., 2016c). The loss of TATS determines a Ca2+release initially triggered at sites where t-tubules are present, followed by propagation into regions without TATS, where RyR2 are orphan of their corresponding Ca2+ channels (Heinzel et al., 2002).
A common trait of ECC abnormalities consists in the loss of the perfect synchrony between membrane voltage and Ca2+ fluxes (Gomez et al., 1997). The interaction between RyR2 and Ca2+ channels is finely regulated (Ibrahim et al., 2011) as any spatio-temporal alteration may generate “waves” or “foci” leading to abrupt arrhythmias or, if persisting, to ineffective ECC and severe failure. However, electrical properties of TATS are often studied using indirect or macroscopic observations rather than on direct measurements.
In the last few years, the combination of advanced optical microscopy with novel functional reporters opened the possibility to better understand the role of TATS in both healthy and disease, by unveiling functional features with an unprecedented spatio-temporal resolution. In this regard, our group has recently developed an ultrafast random access two-photon microscope (Sacconi et al., 2012). Combining this technology with novel fluorinated voltage sensitive dyes with improved photostability (Yan et al., 2012) and calcium probes, we have simultaneously measured action potentials (APs) and intracellular calcium release at multiple sites within the TATS with sub-millisecond temporal and sub-micrometer spatial resolution. In the present work, we will review the capability of this method in providing important insights on sub-cellular alterations of calcium release and in dissecting their relationship with TATS morpho-functional defects found in different cardiac diseases.
In the last few decades, several techniques have been developed in order to get functional information of the TATS in isolated cardiomyocytes. In particular, optical methodologies have met the need to reduce invasiveness of traditional methods and have undergone a wide evolution. The major limiting factor to optically record fast physiological events, like AP and Ca2+ release in multiple site of the same cardiomyocyte, has been the scanning time that restricts recordings to only a single position of the cell at the time, i.e., line scanning approach. A strategy to overcome this limitation could consist in spending more time just to collect as many photons as possible from selected positions. We have recently implemented a new technique based on Random Access Multiphoton (RAMP) (Iyer et al., 2006) microscopy to record AP propagation and Ca2+ release at different domains of the same cardiomyocyte in a simultaneous fashion (Sacconi et al., 2012; Crocini et al., 2014a,b, 2016c). In detail, the RAMP microscope is a two-photon microscope provided with an ultrafast scanning head made of two orthogonal acousto-optic deflectors (AODs) that diffract a laser beam at a precise angle, which can be changed within a few microseconds. Based on the velocity of sound propagation in the crystal present in the AODs and the dimension of the laser beam, a commutation time of about 4 μs can be estimated between a scanning line and the next in multiplexed modality. In order to get information about the two main actors of ECC, isolated cardiomyocytes from rodents were stained with FluoForte GFP-certified, a fluorescent Ca2+ indicator, and di-4-AN(F)EPPTEA, a fluorinated voltage-sensitive dye (VSD) (Yan et al., 2012): the RAMP microscope was used to simultaneously excite both dyes and, thanks to the large Stokes shift of the fluorinated VSD, the two fluorescence signals were easily distinguished using appropriate optical tools. Figures 1A,B illustrates the real-time simultaneous optical recording of AP and Ca2+ transients in isolated cardiomyocytes at different non-contiguous domains. Isolated cardiac cells were field stimulated at room temperature (Sacconi et al., 2012; Crocini et al., 2014b) to reach steady state. Then, we performed real-time simultaneous optical recordings consisting of 10 consecutive recordings, where we probed 5–10 different tubular membrane sites per recording (TTi, i = 1 to 10) (Figure 1C). The length of scanning lines for each membrane site ranged from 2 to 10 μm with an integration time of 200 μs per line, leading to a temporal resolution of 0.4–2 ms. The signal-to-noise ratio (S/N) was sufficient to detect the presence of an AP occurring at sarcolemma and to assess the temporal features of the Ca2+ transient in the surrounding cytoplasm. Local calcium transients at each TTi were analyzed in terms of amplitude and kinetics and we calculated a coefficient of variability (CV) as the ratio between the standard deviation and the mean of a specific parameter, namely the time to peak of Ca2+ release.
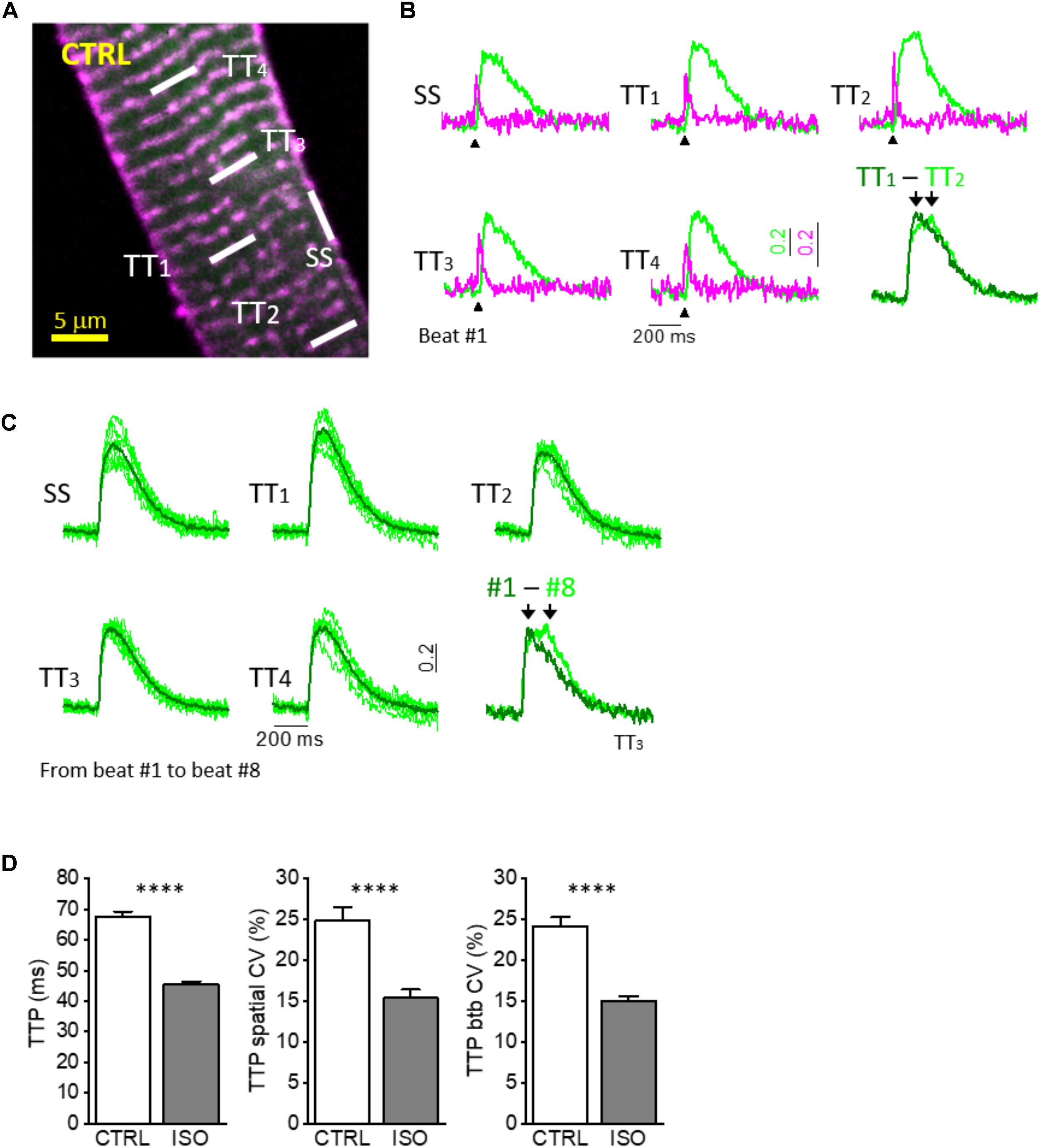
FIGURE 1. Simultaneous multisite voltage and Ca2+ recordings. (A) Two-photon fluorescence (TPF) image of a stained rat ventricular myocyte: sarcolemma in magenta (di-4-ANE(F)PTEA) and [Ca2+]i in green (GFP-certified Fluoforte). Modified from Crocini et al. (2014b). (B) Normalized fluorescence traces (ΔF/F0) simultaneously recorded from the scanned sites indicated in white in A: surface sarcolemma (SS) and four T-tubules (TTi). AP is elicited at 200 ms (black arrowheads). Membrane voltage (magenta) and [Ca2+]i (green). The last is a superimposition of two simultaneous Ca2+ transients. Modified from Crocini et al. (2014b). (C) Superimposition of ten subsequent Ca2+ transients recorded from five scanned sites in a CTRL cardiomyocyte. The last is a superimposition of one scanned t-tubule obtained from different subsequent beating (recording #1 and recording #8). (D) Graphs showing mean values for Ca2+ transient time-to-peak (TTP), time-to-peak spatial variability coefficient (TTP spatial CV), and time-to-peak beat-to-beat variability coefficient (TTP btb CV) of Ca2+ release. Data reported as mean ± SEM from 122 TTs (27 CTRL cells from 10 rats) and 48 TTs (11 ISO cells from 3 rats). Student’s t-test applied, ∗∗∗∗P < 0.0001.
Of note, by averaging the 10 consecutive RAMP measurements performed at each site, we have demonstrated the tight electrical coupling between surface sarcolemma (SS) and t-tubules (TTs) (Sacconi et al., 2012), as well as the perfect overlap of Ca2+ transients originating close to SS and TTs during steady-state stimulations in control ventricular cardiomyocytes (Crocini et al., 2014b). These results have demonstrated the uniformity of Ca2+ release across the entire cardiomyocyte and highlighted the crucial role of the TATS in propagating AP into the whole cell ensuring a synchronous Ca2+ release. Exploiting the possibility to simultaneously and consecutively perform several measurements of selected regions, we obtained information regarding Ca2+ release variability in both space and time. These two parameters appeared to be of peculiar interest:
1. The spatial CV of time to peak (spatial CV of TTP), that is the ratio of the standard deviation to the mean between TTi = 1 to 10 recorded during a single activation (e.g., trial 1 or trial 2 or trial 3, etc.).
2. The beat-to-beat CV of time to peak (btb CV of TTP), that is the ratio of the standard deviation to the mean between consecutive activations (trial 1, trial 2, trial 3 etc.) calculated at a specific membrane site (e.g., TT1 or TT2 or TT3, etc.).
Regarding the mechanisms underlying the observed spatio-temporal variability, we first hypothesized that the presence of a non-negligible spatial CV (approximately 25%; Figures 1C,D) between far apart TTi in control rat ventricular cardiomyocytes could be related to heterogeneous functional or structural characteristics at different calcium release sites. Non-homogeneous phosphorylation levels of RYR2 or DHPRs in space, for instance, or variability in the dyadic cleft dimensions/structure could lead to a large spatial CV even in control cells. This hypothesis was denied when we observed a quantitatively similar variability of Ca2+ release in time, i.e., when we compared consecutive activations at the same single t-tubule site. In control rat ventricular cardiomyocytes btb CV of TTP was also in the order of 25% (Figures 1C,D) On a beat-to-beat basis, no functional or structural changes at the calcium release sites may occur that could account for a relatively large (25%) CV and, in our view, the sole possible mechanism underlying the phenomenon is the stochastic nature of Ca2+ release itself, that our RAMP technique appears able to detect. Indeed, only a few Ca2+ release units (CRUs) reside within the volume probed by each scanned line (5–10 CRUs in ∼10 μm3) (Franzini-Armstrong et al., 1999; Soeller et al., 2007). A typical CRU hosts about 50–200 RyRs and 10–25 Ca2+ release channels (Franzini-Armstrong et al., 1999). During normal twitch activation (at low inotropic levels) only a small fraction of RyR2 is recruited (e.g., of the total ∼3 million of RyR2 in the cell, less than 2–5% would open during a regular twitch) (Wier et al., 1994; Bers, 2001). This low open probability (Po) of the RyR2 explains the high variability of Ca2+ release that we observed. We expected that by increasing RyR2 Po the spatial and btb CV of TTP would decrease. In order to enhance RyR2 Po, we performed measurements in the presence of a β-adrenergic agonist, isoproterenol (ISO, 10-7 M), that acutely promotes RyR2 hyperphosphorylation at Protein Kinase A (PKA) sites, thus prolonging the duration of RyR2 opening (RyR2 ton) and enhancing RyR2 cytosolic and luminal Ca2+ sensitivity (Eisner et al., 2004; Ferrantini et al., 2016). As expected, we found that both spatial and btb CV of TTP were markedly reduced under ISO: the extent of the reduction (Figure 1D) was similar for the two parameters. This result suggests that the local variability of TTP (in space and time) is tightly related to the functional state of RyR2s and reflects the stochastic nature of their openings. Of note, to some extent, the synchronization of local Ca2+ release under ISO could be attributed to a more intense trigger (i.e., slower ICaL inactivation), and further experiments with a direct RyR2 agonist would be helpful to define the reciprocal role of the two mechanisms (Ca2+ channels vs. RyR2 PKA-mediated phosphorylation).
The ability of our method to probe the spatiotemporal relationship between Ca2+ and electrical activity was then explored in three models of cardiac disease characterized by TATS morpho-functional remodeling.
All animal handling and procedures were performed in accordance with the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes and conformed to the principles and regulations as described in the editorial by Grundy (2015). The experimental protocol was approved by the Italian Ministry of Health (protocol number 647/2015-PR).
We compared the following pathological settings:
• a rat model of ischemic heart failure (HF-ischemic), in which myocardial infarction was induced by ligation of the left anterior coronary artery as previously described (Lyon et al., 2009); the animals were studied 8–10 weeks after the ischemic damage, when echocardiographic signs of heart failure were (such as left ventricle (LV) dilation and reduced LV contractility) easily revealed.
• a rat model of left ventricular hypertrophy induced by spontaneous hypertension (i.e., spontaneously hypertensive rats, SHR). Notably we analyzed the late-stage of the disease, when LV dilation and failure occurred. Thus, this model represents a second example of overt heart failure (HF-SHR) (Okamoto and Aoki, 1963).
• a mouse model of hypertrophic cardiomyopathy (HCM), expressing a human mutated cardiac troponin T (deletion of a codon at position 160 of the protein, cTnT Δ160E), associated with high risk of sudden cardiac death and severe diastolic dysfunction in patients (Pasquale et al., 2012; Coppini et al., 2014). Mice employed were 6–8 months old, reflecting early stages of the disease, when LV function is compensated (Moore et al., 2013).
In order to quantify the loss of TATS transverse components (TT) from confocal images of isolated cardiomyocytes, we used AutoTT, a software developed specifically for quantitative analysis of the TATS architecture (Guo and Song, 2014). Cardiomyocytes isolated from the three pathological models introduced above showed different levels of t-tubular remodeling (Figure 2). In particular, we found a variable reduction of TT density, more accentuated in the HF-ischemic model (HF-ischemic > HF-SHR > HCM). Disruption of TT in failing cardiomyocyte was previously reported in a number of animal models and in human myocytes (He et al., 2001; Song et al., 2006). In a rat model of left ventricular hypertrophy, the loss of t-tubules was described as a marker of the transition from compensated hypertrophy to HF (Wei et al., 2010). Our genetic-based model of LV hypertrophy (HCM) demonstrates that t-tubule disruption can occur also during the compensated stages of LV hypertrophy (Crocini et al., 2016c), highlighting the “detubulation” process as an early event of cardiomyocyte remodeling.
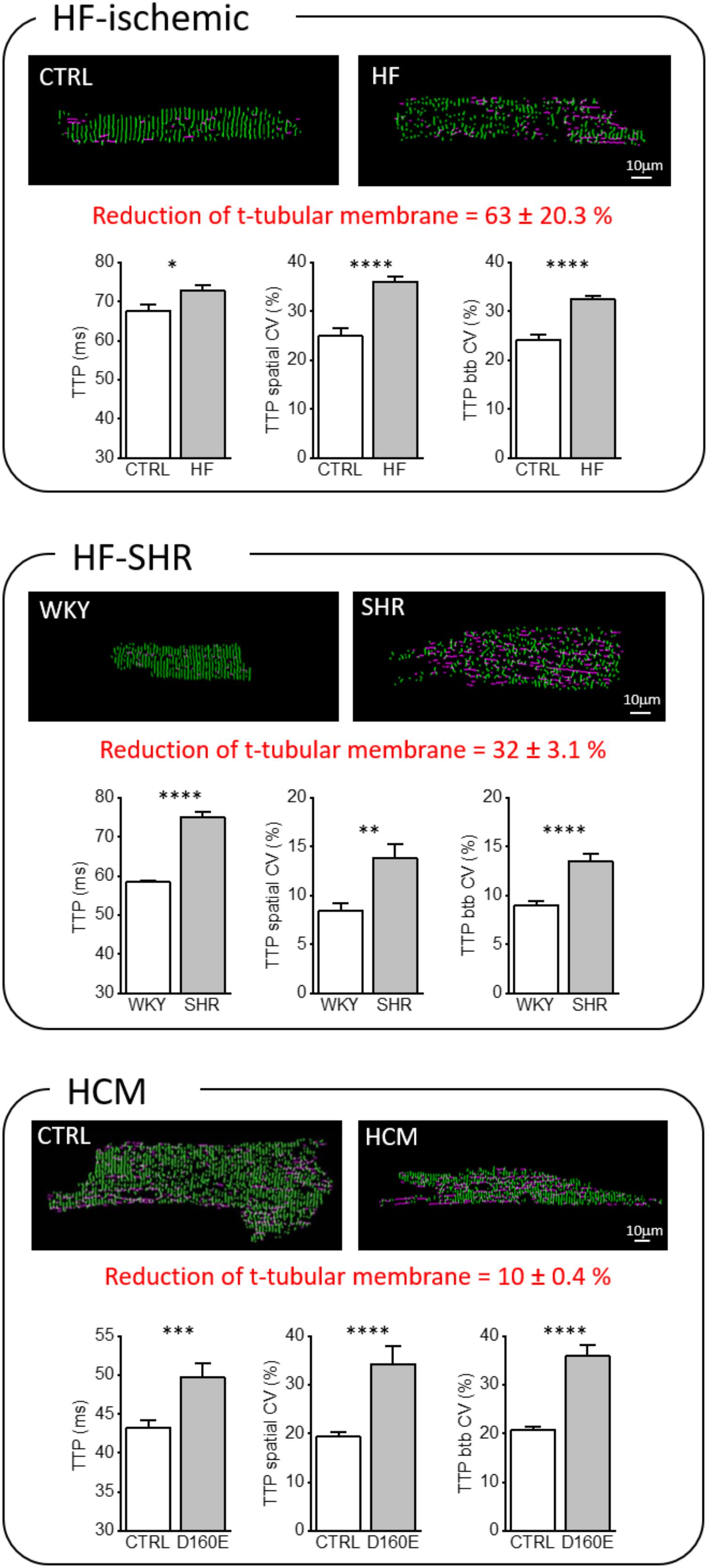
FIGURE 2. Transverse-axial tubular system architecture and Ca2+ kinetics in ischemic heart failure (HF-ischemic), spontaneously hypertensive rats in overt heart failure (HF-SHR), and hypertrophic cardiomyopathy (HCM). On the top, representative images of cellsshowing skeletonized T-tubular system, obtained with AUTO-TT. Transverse elements are shown in green and axial ones in magenta. On the bottom, Graphs showing mean values for Ca2+ transient time-to-peak (TTP), time-to-peak spatial variability coefficient (TTP spatial CV), and time-to-peak beat-to-beat variability coefficient (TTP btb CV) of Ca2+ release. Data reported as mean ± SEM from 122 TTs (27 CTRL cells from 10 rats), 364 TTs (59 HF cells from 12 rats), 87 TTs (25 WKY cells from 8 rats), 87 TTs (23 SHR cells from 8 rats), 101 TTs (28 CTRL cells from 10 mice), and 66 TTs (17 Δ160E cells from 7 mice). Auto TT images of: HF-SHR from Scardigli et al. (2017); of CTRL and HCM from Crocini et al. (2016c). Student’s t-test applied, ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001, ∗∗∗∗P < 0.0001.
Most of the studies regarding membrane architecture in pathological conditions have been conducted with lipophilic membrane-selective fluorescent dyes and reported a reduction of t-tubule staining. Two distinct possibilities underlie this observation. First, t-tubules may seal off from the surface membrane and remain as (permanent or transitory) sealed-off intracellular membrane structures. Alternatively, t-tubules could be truly depleted from the “diseased” cells with a real loss of membrane domains and associated proteins. Discrimination between these possibilities would be crucial to acquire new insights about pathological t-tubular turnover, but rather difficult to explore experimentally. We instead focused our attention on the function of the “residual” remodeled TATS elements. Besides a reduction of transverse components, remodeled TATS show: (1) a reduction in the number of t-tubular openings on the surface (mouths) (Wagner et al., 2012), (2) a greater proportion of tubules running in the longitudinal and oblique directions (Louch et al., 2006; Swift et al., 2008), (3) increased mean t-tubular diameter and length (Maron et al., 1975; Kaprielian et al., 2000), and (4) tubular proliferation, with enhanced t-tubular tortuosity, increased number of constrictions and branches (Kostin et al., 1998). These geometrical changes, characterized with nanometric resolution by STED microscopy (Wagner et al., 2012; Kohl et al., 2013), go hand in hand with structural alterations of the dyadic cleft, as well as changes of membrane channels density and function. In failing myocytes, an expansion of the dyadic cleft occurs, resulting in a greater distance between Ca2+ channels and RyR2 (Gomez et al., 1997; Wei et al., 2010; van Oort et al., 2011). Recently, an increased variability in dyadic cleft distance has been also described (Wei et al., 2010). There is a general consensus that ICaL density is unchanged in HF and HCM cardiomyocytes when measured during voltage-clamp steps (Bers, 2006; Coppini et al., 2013), but the number of Ca2+channels is reportedly reduced (He et al., 2001). Increased single Ca2+channel activity appears to maintain normal Ca2+ current density in HF (Schroder et al., 1998), and this may result from increased phosphorylation of the Ca2+channel by PKA and/or Ca2+/calmodulin kinase II (CaMKII) (Chen et al., 2002). Similarly, RyR2 density in HF and HCM cardiomyocytes appear to be unchanged (Gomez et al., 2001; Benitah et al., 2002; Coppini et al., 2013) but channel function is enhanced by post-translational modifications. For instance, chronic “hyper-phosphorylation” of RyR2 by PKA (Marx et al., 2000) and CaMKII (Ai et al., 2005; Curran et al., 2010; Grimm et al., 2015; Pereira et al., 2017) that occurs in HF causes dissociation of FKBP12.6 from RyR2, destabilizes the RyR2 complex and causes functional uncoupling of neighboring RyRs.
In addition to phosphorylation (Marx et al., 2000; Braz et al., 2004; Wehrens et al., 2004), RYR2 is regulated by other post-translational modifications such as oxidation and S-nitrosylation (Xu et al., 1998; Eu et al., 2000; Barouch et al., 2002; Santos et al., 2011) as well as accessory and structural proteins (e.g., triadin, junctin, and junctophilin-2) (van Oort et al., 2011). Every level of RyR2 regulation can be impaired in HF (Yano et al., 2005), with the functional effect of destabilizing RYR2 closed states.
Thus, while arrays of RyR2s normally tend to open and close together, RyR2 functional uncoupling in failing myocytes is proposed to result in a greater open probability during diastole and desynchronised channels opening during systole. Increased dyadic cleft distance associated with impaired Ca2+ channels and, more importantly, impaired RyR2 function in the CRUs may lead to a general disruption of local Ca2+ release synchronicity in remodeled TATS.
Using the RAMP microscope, we have directly addressed this issue and observed that a number of functional abnormalities are common in “residual” TATS of HF-ischemic, HF-SHR, and HCM cardiomyocytes: Ca2+ transient TTPs are prolonged and both the spatial and btb CV (of TTPs) are increased (Figure 2). We believe that the structural TATS and dyadic changes combined with the impaired RyR2 regulation described above play a major role in the increased local variability of TTP observed in the three diseases. However, exploiting the possibility of RAMP technique to simultaneously map local TATS electrical activity and measure corresponding Ca2+ fluxes, we found that the increased TTPs could be also correlated to electrical abnormalities occurring at the single t-tubular level (Sacconi et al., 2012; Crocini et al., 2014b; 2016c; Scardigli et al., 2017).
Our laboratory has demonstrated that loss of the TATS leads to alteration of AP propagation. A number of t-tubules fail to propagate AP from the surface sarcolemma into the cell core (failing T-tubule; AP-) (Figure 3) in diseased cardiomyocytes. In two different rat models of heart failure, HF-ischemic and HF-SHR, (6.3 ± 1.3)% and (8.5 ± 2.9)% of t-tubules, respectively, were not able to conduct the AP, while a regular stimulated AP occured on the surface sarcolemma and neighbor t-tubules. Interestingly, in cTnT Δ160E ventricular cardiomyocytes, we observed (23 ± 5)% of AP-failing t-tubules, while TATS morphological alterations were minimal (Crocini et al., 2016c). This result suggests that the number of failing t-tubules is not correlated with the degree of lost t-tubular elements, but is due to local ultrastructural alterations (Crocini et al., 2016b; Scardigli et al., 2017). Further support comes from our findings regarding acutely detubulated cardiomyocytes, in which a dramatic detachment of t-tubules was associated with only (12 ± 4)% of failing t-tubules among the remaining connected elements (Sacconi et al., 2012). Based on the pathological substrate, electrical defects may profoundly impair local Ca2+ release determining significant differences of Ca2+ release TTP between coupled (AP+) and uncoupled (AP-) t-tubules. Ca2+ transient TTPs were significantly prolonged in AP- as compared to corresponding AP+ in HF-ischemic and HCM myocytes. These results strongly suggest that the Ca2+ transient detected at AP- tubules originates from the Ca2+ signal propagating from neighboring electrically coupled sites (AP+). This phenomenon was confirmed by a follow-up work, in which we employed a β-adrenergic agonist in HF-ischemic myocytes (Crocini et al., 2016a). At variance with AP+ tubules, Ca2+ transient TTPs at AP- sites were not affected by β-adrenergic stimulation, validating the propagative Ca2+-cascade. In fact, β-adrenergic signaling does not regulate the velocity of Ca2+ propagation in the cell.
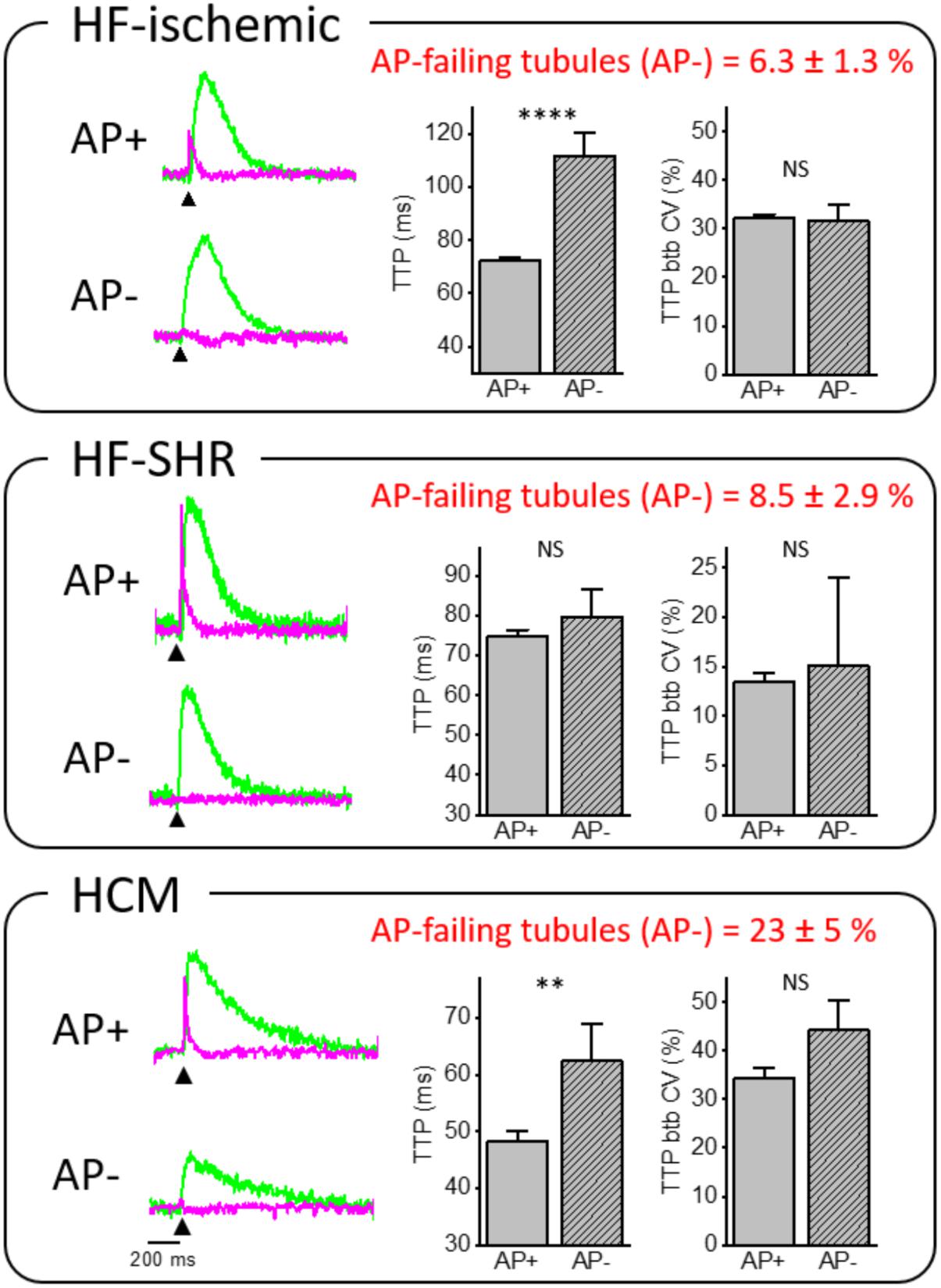
FIGURE 3. Relationship between electrical defects and Ca2+ kinetics in ischemic heart failure (HF-ischemic), spontaneously hypertensive rats in overt heart failure (HF-SHR), and hypertrophic cardiomyopathy (HCM). On the left, normalized fluorescence traces (ΔF/F0) of voltage (magenta) and [Ca2+]i (green) recorded from the two scanned sites. An action potential (elicited at 200 ms, black arrowheads) is clearly visible in AP+ but absent in AP– within the same cell. On the right, graphs showing mean values for Ca2+ transient time-to-peak (TTP) and time-to-peak beat-to-beat variability coefficient (TTP btb CV) of Ca2+ release in AP+ and AP–. Data reported as mean ± SEM from 341 AP+ and 23 AP– (59 HF cells from 12 rats), 80 AP+ and 7 AP– (23 SHR cells from 8 rats), and 51 AP+ and 15 AP– (17 Δ160E cells from 7 mice). Traces of HCM from Crocini et al. (2016c) Student’s t-test applied, NS P > 0.05, ∗∗P < 0.01, ∗∗∗∗P < 0.0001.
Interestingly, btb CV of TTP was not affected by the electrical coupling of the corresponding t-tubule in all of the disease settings we have investigated. Importantly, this result indicates that local Ca2+ release variability measured with our technique is determined by RyR2 functional state rather than propagative Ca2+-cascade from functioning AP+ sites to AP-. Again, this result has been confirmed by acute treatment of HF-ischemic myocytes with β-adrenergic agonist (Crocini et al., 2016a), in which btb CV of TTP was reduced upon β-adrenergic stimulation. We can conclude that β-adrenergic signaling can regulate RyR2 functional states, resulting in a reduced CV in any location of the cell. Differently from HF-ischemic and HCM, Ca2+ kinetics did not show a statistical difference between AP+ and AP- in SHR/HF cardiomyocytes. This result remains of difficult interpretation and may be related to the pronounced delay of the overall Ca2+ transient that involves the whole cell in SHR (Bing et al., 1991).
We employed the RAMP microscope to simultaneously and consecutively measure local TATS activity and obtain information regarding Ca2+ release kinetics and variability of Ca2+ transient time to peak both in space (spatial CV) and in time (btb CV). We found that, in control cardiomyocytes, space and btb CV of TTP are similar one other (approximately ∼25%) and are both reduced by half upon isoproterenol treatment. Starting from the consideration that a relative small number of RyR2 are located in the scanned regions, our results in control cardiomyocytes suggest that the local variability of TTP may be tightly related to the stochastic opening of RyR2s and is influenced by the channel functional state. The beat-to-beat variability at single membrane domains strongly exclude the presence of systematic structural or functional factors underlying the variability in space. The stochastic nature of Ca2+ release is further confirmed by the fact that mean local Ca2+ transients, resulting from averaging 10 consecutively measurements, perfectly overlap in terms of amplitude and kinetics between SS and TT. In our view, the fact that an average “in time” abolishes the spatial variability is a further demonstration that the mechanisms underlying this variability is the stochasticity of Ca2+ release.
We next studied the electrical function and associated Ca2+ fluxes of “residual” structurally remodeled TATS in three pathological conditions (HF-ischemic, HF-SHR, HCM) and found a number of common changes as well as disease-specific alterations: prolonged local Ca2+ transients TTPs and markedly increased spatial and temporal variability of TTPs. Despite different numbers, we found a population of “residual” t-tubules failing to propagating AP in all three pathological settings. We found two main results. The first (expected) was an increased spatial and btb CV of TTPs in disease remodeled TATS. The second (unexpected) was that the electrical coupling of t-tubules (AP+ and AP-) does not influence the variability of Ca2+ release.
At glance, the spatial CV appears to be the more relevant parameter in terms of functional implications for non-homogenous sarcomere activation and the generation of pro-arrhythmogenic triggers through mechano-electric feedback. In diseased myocardium, where t-tubules are structurally and functionally remodeled and the spatial variability of Ca2+ release is increased, the coexistence of “in parallel” myofibril layers with different timing of initial activation would result in reduced force development and overall slower kinetics. However, we can speculate that the increased temporal variability of Ca2+ release may generate also the coexistence of “in series” myofibrils with different timing of activation, eventually promoting arrhythmias by favoring AP alternans. These new pro-arrhythmogenic mechanisms that occur in remodeled t-tubules may be added to those previously described (Orchard et al., 2013; Crocini et al., 2014a).
A number of mechanisms may contribute to the increased spatial and btb CV of TTPs of remodeled TATS, including dyadic structural changes and alterations of Ca2+ channels density and regulation. However, the absence of difference in btb CV of TTPs between AP+ and AP-, conditions where local ICaL triggers are present (AP+) and absent (AP-), respectively, exclude a major role of Ca2+ channels function and indicates that the main player in the local variability of Ca2+ release is the RyR2 functional state. While RyR2 function resulting from targeted single post-translational modification (e.g., the effects of single phosphorylation at specific sites) has been largely studied in channels re-expressed in bilayers, the complex RyR2 alterations that occur in cardiac diseases cannot be reproduced in vitro. Preserving the manifold context of pathological settings is therefore essential to accurately investigate disease-related channel alternations. Thanks to our RAMP approach, a novel set of information may become accessible, allowing for unprecedented exploration of RyR2 function in living cardiomyocytes. Distinct modulation of different subpopulations of RyR2 clusters, those coupled to the t-tubules (dyadic RyR2) and those non-coupled (corbular RyR2), has been recently highlighted in a number of studies (Dries et al., 2013, 2016). CaMKII-dependent potentiation of RyR2 activity, for instance, is restricted to coupled RyRs in the dyadic cleft (Dries et al., 2013), while PKA phosphorylation occur at all RyRs concurrently, including those “non-coupled” to the sarcolemmal membrane (Dries et al., 2013, 2016). In addition to the subcellular microdomain compartmentalization, the phosphorylations by CaMKII and PKA differently affect RyR2 gating as well as its modulation by a number of factors (e.g., cytosolic and luminal calcium, regulatory proteins) (Bers, 2001). As an example, the mean open probability (Po) is increased by both kinases, but the effects on channel open times (To) and channel closed times (Tc) are specific and may directly affect the stochasticity of Ca2+ release and its subcellular spatio-temporal variability. The scenario of RyR2 subpopulations at different microdomains is even more complex in cells that exhibit a t-tubular network poor of transverse elements but enriched of other components (e.g., axial tubules, AT).
In the absence of abundant transverse membrane invaginations, as in atrial cells or in disease-remodeled ventricular cardiomyocytes, we can configure the presence of at list other three populations of RyR2 in addition to those mentioned above:
- RyR2s that are coupled to axial tubules, well represented in atrial cells.
- RyR2s that are “orphaned” of their t-tubules because of t-tubule disruption in disease-associated structural remodeling.
- RyR2s that are coupled to non-propagating (AP-) t-tubules, as observed in disease-remodeled myocytes and described in detail above.
The study of subcellular spatio-temporal variability of Ca2+ release with the RAMP technique may provide significant insights to clarify the functional characteristics of these RyR2 populations. For instance, in mouse atrial myocytes, Ca2+ release from the SR in axial tubules is approximately two times faster at the center of the cell as compared to Ca2+ release at the surface. Rapid Ca2+ release correlated with colocalization of highly phosphorylated (highphos) RyR2 clusters at junctions between axial tubules and SR junctions (Brandenburg et al., 2016). Novel evidence of atrial “super-hub” Ca2+ signaling has been recently reported across different species (Brandenburg et al., 2018), possibly establishing a new paradigm for atrial Ca2+ release. We can speculate that the spatio-temporal variability of Ca2+ release could be reduced at the highphos RyR2 clusters and our approach may help establishing the role of axial tubules, highphos RyR2 clusters, and non-junctional Ca2+ release. RAMP measurements can be extended to the intact tissue in order to study subcellular Ca2+ release spatio-temporal variability in various cells at the same time (Sacconi et al., 2012; Ferrantini et al., 2014). These studies could include human myocardium, whose characteristics are only partly reproduced by murine models, thus providing an even further interesting perspective to dissect RyR2 function in a physiological environment. In fact, recent studies in failing human hearts agree on the reduction of t-tubule density, which was two to three times lower in failing ventricular myocytes from HF patients with different aetiologies than in healthy donors (Cannell et al., 2006; Lyon et al., 2009). Loss of t-tubules have been also described in patients with genetic-based cardiomyopathies (Maron et al., 1975; Kaprielian et al., 2000; Ferrantini et al., 2018). We believe that our studies on rodent models of HF and congenital cardiac diseases could (qualitatively) reflect the spatio-temporal variability of Ca2+ release that may occur in human cardiomyocytes. Though, direct RAMP measurements of Ca2+ release in human t-tubules are needed, and may represent the most exciting perspective for this technology.
MS analyzed the data. MS, CC, CF, FP, and LS wrote the paper.
This work was supported by the European Union Horizon 2020 research and innovation program under grant agreement no. 654148 Laserlab-Europe, by the Italian Ministry for Education, University and Research in the framework of the Flagship Project NanoMAX, by the Italian Ministry of Health (WFR GR-2011-02350583), by Telethon–Italy (GGP13162), by Ente Cassa di Risparmio di Firenze (private foundation), and by FAS-Salute ToRSADE project. CC holds a long-term fellowship from the Human Frontiers Science Program Organization (LT001449/2017-L).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling Editor declared a shared affiliation, though no other collaboration, with several of the authors CF, FP, and LS at the time of review.
Ai, X., Curran, J. W., Shannon, T. R., Bers, D. M., and Pogwizd, S. M. (2005). Ca2 + /calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2 + leak in heart failure. Circ. Res. 97, 1314–1322. doi: 10.1161/01.RES.0000194329.41863.89
Barouch, L. A., Harrison, R. W., Skaf, M. W., Rosas, G. O., Cappola, T. P., Kobeissi, Z. A., et al. (2002). Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature 416, 337–339.
Bassani, J. W., Yuan, W., and Bers, D. M. (1995). Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am. J. Physiol. 268(5 Pt 1), C1313–C1319. doi: 10.1152/ajpcell.1995.268.5.C1313
Benitah, J. P., Kerfant, B. G., Vassort, G., Richard, S., and Gomez, A. M. (2002). Altered communication between L-type calcium channels and ryanodine receptors in heart failure. Front. Biosci. 7:e21. doi: 10.2741/benitah
Bers, D. M. (2001). Excitation-Contraction Coupling and Cardiac Contractile Force. Norwell, MA: Kluwer Academic Publishers. doi: 10.1007/978-94-010-0658-3
Bers, D. M. (2002). Cardiac excitation-contraction coupling. Nature 415, 198–205. doi: 10.1038/415198a
Bers, D. M. (2006). Altered cardiac myocyte Ca regulation in heart failure. Physiology 21, 380–387. doi: 10.1152/physiol.00019.2006
Bing, O. H., Brooks, W. W., Conrad, C. H., Sen, S., Perreault, C. L., and Morgan, J. P. (1991). Intracellular calcium transients in myocardium from spontaneously hypertensive rats during the transition to heart failure. Circ. Res. 68, 1390–1400. doi: 10.1161/01.RES.68.5.1390
Brandenburg, S., Kohl, T., Williams, G. S., Gusev, K., Wagner, E., Rog-Zielinska, E. A., et al. (2016). Axial tubule junctions control rapid calcium signaling in atria. J. Clin. Invest. 126, 3999–4015. doi: 10.1172/JCI88241
Brandenburg, S., Pawlowitz, J., Fakuade, F. E., Kownatzki-Danger, D., Kohl, T., Mitronova, G., et al. (2018). Axial tubule junctions activate atrial Ca2+ release across specie. Front. Physiol. 9:1227. doi: 10.3389/fphys.2018.01227
Braz, J. C., Gregory, K., Pathak, A., Zhao, W., Sahin, B., Klevitsky, R., et al. (2004). PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat. Med. 10, 248–254. doi: 10.1038/nm1000
Brette, F., Despa, S., Bers, D. M., and Orchard, C. H. (2005). Spatiotemporal characteristics of SR Ca(2 + ) uptake and release in detubulated rat ventricular myocytes. J. Mol. Cell Cardiol. 39, 804–812. doi: 10.1016/j.yjmcc.2005.08.005
Brette, F., Komukai, K., and Orchard, C. H. (2002). Validation of formamide as a detubulation agent in isolated rat cardiac cells. Am. J. Physiol. Heart Circ. Physiol. 283, H1720–H1728. doi: 10.1152/ajpheart.00347.2002
Brette, F., Rodriguez, P., Komukai, K., Colyer, J., and Orchard, C. H. (2004). beta-adrenergic stimulation restores the Ca transient of ventricular myocytes lacking t-tubules. J. Mol. Cell Cardiol. 36, 265–275. doi: 10.1016/j.yjmcc.2003.11.002
Cannell, M. B., Crossman, D. J., and Soeller, C. (2006). Effect of changes in action potential spike configuration, junctional sarcoplasmic reticulum micro-architecture and altered t-tubule structure in human heart failure. J. Muscle Res. Cell Motil. 27, 297–306. doi: 10.1007/s10974-006-9089-y
Chen, X., Piacentino, V. III, Furukawa, S., Goldman, B., Margulies, K. B., and Houser, S. R. (2002). L-type Ca2 + channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ. Res. 91, 517–524. doi: 10.1161/01.RES.0000033988.13062.7C
Cheng, H., Cannell, M. B., and Lederer, W. J. (1994). Propagation of excitation-contraction coupling into ventricular myocytes. Pflugers Arch. 428, 415–417. doi: 10.1007/BF00724526
Coppini, R., Ferrantini, C., Aiazzi, A., Mazzoni, L., Sartiani, L., Mugelli, A., et al. (2014). Isolation and functional characterization of human ventricular cardiomyocytes from fresh surgical samples. J. Vis. Exp. 21:86. doi: 10.3791/51116
Coppini, R., Ferrantini, C., Yao, L., Fan, P., Del Lungo, M., Stillitano, F., et al. (2013). Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation 127, 575–584. doi: 10.1161/CIRCULATIONAHA.112.134932
Crocini, C., Coppini, R., Ferrantini, C., Pavone, F. S., and Sacconi, L. (2014a). Functional cardiac imaging by random access microscopy. Front. Physiol. 5:403. doi: 10.3389/fphys.2014.00403
Crocini, C., Coppini, R., Ferrantini, C., Yan, P., Loew, L. M., Tesi, C., et al. (2014b). Defects in T-tubular electrical activity underlie local alterations of calcium release in heart failure. Proc. Natl. Acad. Sci. U.S.A. 111, 15196–15201. doi: 10.1073/pnas.1411557111
Crocini, C., Coppini, R., Ferrantini, C., Yan, P., Loew, L. M., Poggesi, C., et al. (2016a). T-tubular electrical defects contribute to blunted beta-adrenergic response in heart failure. Int. J. Mol. Sci. 17, 1410–1471. doi: 10.3390/ijms17091471
Crocini, C., Ferrantini, C., Coppini, R., and Sacconi, L. (2016b). Electrical defects of the transverse-axial tubular system in cardiac diseases. J. Physiol. 595, 3815–3822. doi: 10.1113/JP273042
Crocini, C., Ferrantini, C., Scardigli, M., Coppini, R., Mazzoni, L., Lazzeri, E., et al. (2016c). Novel insights on the relationship between T-tubular defects and contractile dysfunction in a mouse model of hypertrophic cardiomyopathy. J. Mol. Cell Cardiol. 91, 42–51. doi: 10.1016/j.yjmcc.2015.12.013
Curran, J., Brown, K. H., Santiago, D. J., Pogwizd, S., Bers, D. M., and Shannon, T. R. (2010). Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca(2 + )-calmodulin-dependent protein kinase II. J. Mol. Cell Cardiol. 49, 25–32. doi: 10.1016/j.yjmcc.2010.03.013
Dibb, K. M., Clarke, J. D., Horn, M. A., Richards, M. A., Graham, H. K., Eisner, D. A., et al. (2009). Characterization of an extensive transverse tubular network in sheep atrial myocytes and its depletion in heart failure. Circ. Heart Fail. 2, 482–489. doi: 10.1161/CIRCHEARTFAILURE.109.852228
Dries, E., Bito, V., Lenaerts, I., Antoons, G., Sipido, K. R., and Macquaide, N. (2013). Selective modulation of coupled ryanodine receptors during microdomain activation of calcium/calmodulin-dependent kinase II in the dyadic cleft. Circ. Res. 113, 1242–1252. doi: 10.1161/CIRCRESAHA.113.301896
Dries, E., Santiago, D. J., Johnson, D. M., Gilbert, G., Holemans, P., Korte, S. M., et al. (2016). Calcium/calmodulin-dependent kinase II and nitric oxide synthase 1-dependent modulation of ryanodine receptors during beta-adrenergic stimulation is restricted to the dyadic cleft. J. Physiol. 594, 5923–5939. doi: 10.1113/JP271965
Eisner, D. A., Diaz, M. E., O’Neill, S. C., and Trafford, A. W. (2004). Physiological and pathological modulation of ryanodine receptor function in cardiac muscle. Cell Calcium 35, 583–589. doi: 10.1016/j.ceca.2004.01.012
Eisner, D. A., Trafford, A. W., Diaz, M. E., Overend, C. L., and O’Neill, S. C. (1998). The control of Ca release from the cardiac sarcoplasmic reticulum: regulation versus autoregulation. Cardiovasc. Res. 38, 589–604. doi: 10.1016/S0008-6363(98)00062-5
Eu, J. P., Sun, J., Xu, L., Stamler, J. S., and Meissner, G. (2000). The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell 102, 499–509. doi: 10.1016/S0092-8674(00)00054-4
Ferrantini, C., Coppini, R., Sacconi, L., Tosi, B., Zhang, M. L., Wang, G. L., et al. (2014). Impact of detubulation on force and kinetics of cardiac muscle contraction. J. Gen. Physiol. 143, 783–797. doi: 10.1085/jgp.201311125
Ferrantini, C., Coppini, R., Scellini, B., Ferrara, C., Pioner, J. M., Mazzoni, L., et al. (2016). R4496C RyR2 mutation impairs atrial and ventricular contractility. J. Gen. Physiol. 147, 39–52. doi: 10.1085/jgp.201511450
Ferrantini, C., Pioner, J. M., Mazzoni, L., Gentile, F., Tosi, B., Rossi, A., et al. (2018). Late sodium current inhibitors to treat exercise-induced obstruction in hypertrophic cardiomyopathy: an in vitro study in human myocardium. Br. J. Pharmacol. 175, 2635–2652. doi: 10.1111/bph.14223
Franzini-Armstrong, C., Protasi, F., and Ramesh, V. (1999). Shape, size, and distribution of Ca(2 + ) release units and couplons in skeletal and cardiac muscles. Biophys. J. 77, 1528–1539. doi: 10.1016/S0006-3495(99)77000-1
Gomez, A. M., Guatimosim, S., Dilly, K. W., Vassort, G., and Lederer, W. J. (2001). Heart failure after myocardial infarction: altered excitation-contraction coupling. Circulation 104, 688–693. doi: 10.1161/hc3201.092285
Gomez, A. M., Valdivia, H. H., Cheng, H., Lederer, M. R., Santana, L. F., Cannell, M. B., et al. (1997). Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science 276, 800–806. doi: 10.1126/science.276.5313.800
Grimm, M., Ling, H., Willeford, A., Pereira, L., Gray, C. B., Erickson, J. R., et al. (2015). CaMKIIdelta mediates beta-adrenergic effects on RyR2 phosphorylation and SR Ca(2 + ) leak and the pathophysiological response to chronic beta-adrenergic stimulation. J. Mol. Cell Cardiol. 85, 282–291. doi: 10.1016/j.yjmcc.2015.06.007
Grundy, D. (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J. Physiol. 593, 2547–2549. doi: 10.1113/JP270818
Guo, A., and Song, L. S. (2014). AutoTT: automated detection and analysis of T-tubule architecture in cardiomyocytes. Biophys. J. 106, 2729–2736. doi: 10.1016/j.bpj.2014.05.013
Haddock, P. S., Coetzee, W. A., Cho, E., Porter, L., Katoh, H., Bers, D. M., et al. (1999). Subcellular [Ca2 + ]i gradients during excitation-contraction coupling in newborn rabbit ventricular myocytes. Circ. Res. 85, 415–427. doi: 10.1161/01.RES.85.5.415
He, J., Conklin, M. W., Foell, J. D., Wolff, M. R., Haworth, R. A., Coronado, R., et al. (2001). Reduction in density of transverse tubules and L-type Ca(2 + ) channels in canine tachycardia-induced heart failure. Cardiovasc. Res. 49, 298–307. doi: 10.1016/S0008-6363(00)00256-X
Heinzel, F. R., Bito, V., Biesmans, L., Wu, M., Detre, E., von Wegner, F., et al. (2008). Remodeling of T-tubules and reduced synchrony of Ca2 + release in myocytes from chronically ischemic myocardium. Circ. Res. 102, 338–346. doi: 10.1161/CIRCRESAHA.107.160085
Heinzel, F. R., Bito, V., Volders, P. G., Antoons, G., Mubagwa, K., and Sipido, K. R. (2002). Spatial and temporal inhomogeneities during Ca2 + release from the sarcoplasmic reticulum in pig ventricular myocytes. Circ. Res. 91, 1023–1030. doi: 10.1161/01.RES.0000045940.67060.DD
Hobai, I. A., and O’Rourke, B. (2001). Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation 103, 1577–1584. doi: 10.1161/01.CIR.103.11.1577
Ibrahim, M., Gorelik, J., Yacoub, M. H., and Terracciano, C. M. (2011). The structure and function of cardiac t-tubules in health and disease. Proc. Biol. Sci. 278, 2714–2723. doi: 10.1098/rspb.2011.0624
Iyer, V., Hoogland, T. M., and Saggau, P. (2006). Fast functional imaging of single neurons using random-access multiphoton (RAMP) microscopy. J. Neurophysiol. 95, 535–545. doi: 10.1152/jn.00865.2005
Kaprielian, R. R., Stevenson, S., Rothery, S. M., Cullen, M. J., and Severs, N. J. (2000). Distinct patterns of dystrophin organization in myocyte sarcolemma and transverse tubules of normal and diseased human myocardium. Circulation 101, 2586–2594. doi: 10.1161/01.CIR.101.22.2586
Kohl, T., Westphal, V., Hell, S. W., and Lehnart, S. E. (2013). Superresolution microscopy in heart - cardiac nanoscopy. J. Mol. Cell Cardiol. 58, 13–21. doi: 10.1016/j.yjmcc.2012.11.016
Kostin, S., Scholz, D., Shimada, T., Maeno, Y., Mollnau, H., Hein, S., et al. (1998). The internal and external protein scaffold of the T-tubular system in cardiomyocytes. Cell Tissue Res. 294, 449–460. doi: 10.1007/s004410051196
Kushnir, A., and Marks, A. R. (2010). The ryanodine receptor in cardiac physiology and disease. Adv. Pharmacol. 59, 1–30. doi: 10.1016/S1054-3589(10)59001-X
Louch, W. E., Bito, V., Heinzel, F. R., Macianskiene, R., Vanhaecke, J., Flameng, W., et al. (2004). Reduced synchrony of Ca2 + release with loss of T-tubules-a comparison to Ca2 + release in human failing cardiomyocytes. Cardiovasc. Res. 62, 63–73. doi: 10.1016/j.cardiores.2003.12.031
Louch, W. E., Mork, H. K., Sexton, J., Stromme, T. A., Laake, P., Sjaastad, I., et al. (2006). T-tubule disorganization and reduced synchrony of Ca2 + release in murine cardiomyocytes following myocardial infarction. J. Physiol. 574(Pt 2), 519–533. doi: 10.1113/jphysiol.2006.107227
Lyon, A. R., MacLeod, K. T., Zhang, Y., Garcia, E., Kanda, G. K., Lab, M. J., et al. (2009). Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc. Natl. Acad. Sci. U.S.A. 106, 6854–6859. doi: 10.1073/pnas.0809777106
Maron, B. J., Ferrans, V. J., and Roberts, W. C. (1975). Ultrastructural features of degenerated cardiac muscle cells in patients with cardiac hypertrophy. Am. J. Pathol. 79, 387–434.
Marx, S. O., Reiken, S., Hisamatsu, Y., Jayaraman, T., Burkhoff, D., Rosemblit, N., et al. (2000). PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 101, 365–376. doi: 10.1016/S0092-8674(00)80847-8
Moore, R. K., Grinspan, L. T., Jimenez, J., Guinto, P. J., Ertz-Berger, B., and Tardiff, J. C. (2013). HCM-linked 160E cardiac troponin T mutation causes unique progressive structural and molecular ventricular remodeling in transgenic mice. J. Mol. Cell Cardiol. 58, 188–198. doi: 10.1016/j.yjmcc.2013.02.004
Okamoto, K., and Aoki, K. (1963). Development of a strain of spontaneously hypertensive rats. Jpn. Circ. J. 27, 282–293. doi: 10.1253/jcj.27.282
Orchard, C. H., Bryant, S. M., and James, A. F. (2013). Do t-tubules play a role in arrhythmogenesis in cardiac ventricular myocytes? J. Physiol. 591, 4141–4147. doi: 10.1113/jphysiol.2013.254540
Pasquale, F., Syrris, P., Kaski, J. P., Mogensen, J., McKenna, W. J., and Elliott, P. (2012). Long-term outcomes in hypertrophic cardiomyopathy caused by mutations in the cardiac troponin T gene. Circ. Cardiovasc. Genet. 5, 10–17. doi: 10.1161/CIRCGENETICS.111.959973
Pereira, L., Bare, D. J., Galice, S., Shannon, T. R., and Bers, D. M. (2017). beta-Adrenergic induced SR Ca(2 + ) leak is mediated by an Epac-NOS pathway. J. Mol. Cell Cardiol. 108, 8–16. doi: 10.1016/j.yjmcc.2017.04.005
Sacconi, L., Ferrantini, C., Lotti, J., Coppini, R., Yan, P., Loew, L. M., et al. (2012). Action potential propagation in transverse-axial tubular system is impaired in heart failure. Proc. Natl. Acad. Sci. U.S.A. 109, 5815–5819. doi: 10.1073/pnas.1120188109
Santos, C. X., Anilkumar, N., Zhang, M., Brewer, A. C., and Shah, A. M. (2011). Redox signaling in cardiac myocytes. Free Radic. Biol. Med. 50, 777–793. doi: 10.1016/j.freeradbiomed.2011.01.003
Scardigli, M., Crocini, C., Ferrantini, C., Gabbrielli, T., Silvestri, L., Coppini, R., et al. (2017). Quantitative assessment of passive electrical properties of the cardiac T-tubular system by FRAP microscopy. Proc. Natl. Acad. Sci. U.S.A. 114, 5737–5742. doi: 10.1073/pnas.1702188114
Schroder, F., Handrock, R., Beuckelmann, D. J., Hirt, S., Hullin, R., Priebe, L., et al. (1998). Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation 98, 969–976. doi: 10.1161/01.CIR.98.10.969
Scriven, D. R., Asghari, P., and Moore, E. D. (2013). Microarchitecture of the dyad. Cardiovasc. Res. 98, 169–176. doi: 10.1093/cvr/cvt025
Soeller, C., Crossman, D., Gilbert, R., and Cannell, M. B. (2007). Analysis of ryanodine receptor clusters in rat and human cardiac myocytes. Proc. Natl. Acad. Sci. U.S.A. 104, 14958–14963. doi: 10.1073/pnas.0703016104
Song, L. S., Sobie, E. A., McCulle, S., Lederer, W. J., Balke, C. W., and Cheng, H. (2006). Orphaned ryanodine receptors in the failing heart. Proc. Natl. Acad. Sci. U.S.A. 103, 4305–4310. doi: 10.1073/pnas.0509324103
Swift, F., Birkeland, J. A., Tovsrud, N., Enger, U. H., Aronsen, J. M., Louch, W. E., et al. (2008). Altered Na + /Ca2 + -exchanger activity due to downregulation of Na + /K + -ATPase alpha2-isoform in heart failure. Cardiovasc. Res. 78, 71–78. doi: 10.1093/cvr/cvn013
Ter Keurs, H. E., and Boyden, P. A. (2007). Calcium and arrhythmogenesis. Physiol. Rev. 87, 457–506. doi: 10.1152/physrev.00011.2006
van Oort, R. J., Garbino, A., Wang, W., Dixit, S. S., Landstrom, A. P., Gaur, N., et al. (2011). Disrupted junctional membrane complexes and hyperactive ryanodine receptors after acute junctophilin knockdown in mice. Circulation 123, 979–988. doi: 10.1161/CIRCULATIONAHA.110.006437
Wagner, E., Lauterbach, M. A., Kohl, T., Westphal, V., Williams, G. S., Steinbrecher, J. H., et al. (2012). Stimulated emission depletion live-cell super-resolution imaging shows proliferative remodeling of T-tubule membrane structures after myocardial infarction. Circ. Res. 111, 402–414. doi: 10.1161/CIRCRESAHA.112.274530
Wehrens, X. H., Lehnart, S. E., Reiken, S. R., and Marks, A. R. (2004). Ca2 + /calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circ. Res. 94, e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2
Wei, S., Guo, A., Chen, B., Kutschke, W., Xie, Y. P., Zimmerman, K., et al. (2010). T-tubule remodeling during transition from hypertrophy to heart failure. Circ. Res. 107, 520–531. doi: 10.1161/CIRCRESAHA.109.212324
Wier, W. G., and Balke, C. W. (1999). Ca(2 + ) release mechanisms, Ca(2 + ) sparks, and local control of excitation-contraction coupling in normal heart muscle. Circ. Res. 85, 770–776. doi: 10.1161/01.RES.85.9.770
Wier, W. G., Egan, T. M., Lopez-Lopez, J. R., and Balke, C. W. (1994). Local control of excitation-contraction coupling in rat heart cells. J. Physiol. 474, 463–471. doi: 10.1113/jphysiol.1994.sp020037
Xu, L., Eu, J. P., Meissner, G., and Stamler, J. S. (1998). Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279, 234–237. doi: 10.1126/science.279.5348.234
Yan, P., Acker, C. D., Zhou, W. L., Lee, P., Bollensdorff, C., Negrean, A., et al. (2012). Palette of fluorinated voltage-sensitive hemicyanine dyes. Proc. Natl. Acad. Sci. U.S.A. 109, 20443–20448. doi: 10.1073/pnas.1214850109
Yang, Z., Pascarel, C., Steele, D. S., Komukai, K., Brette, F., and Orchard, C. H. (2002). Na + -Ca2 + exchange activity is localized in the T-tubules of rat ventricular myocytes. Circ. Res. 91, 315–322. doi: 10.1161/01.RES.0000030180.06028.23
Keywords: t-tubule, excitation-contraction coupling, calcium imaging, voltage imaging, microscopy
Citation: Scardigli M, Ferrantini C, Crocini C, Pavone FS and Sacconi L (2018) Interplay Between Sub-Cellular Alterations of Calcium Release and T-Tubular Defects in Cardiac Diseases. Front. Physiol. 9:1474. doi: 10.3389/fphys.2018.01474
Received: 10 July 2018; Accepted: 28 September 2018;
Published: 25 October 2018.
Edited by:
Alessandro Mugelli, Università degli Studi di Firenze, ItalyReviewed by:
Peter Kohl, Imperial College London, United KingdomCopyright © 2018 Scardigli, Ferrantini, Crocini, Pavone and Sacconi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudia Crocini, Y2xhdWRpYS5jcm9jaW5pQGNvbG9yYWRvLmVkdQ== Leonardo Sacconi, c2FjY29uaUBsZW5zLnVuaWZpLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.