 Chunbo Yang
Chunbo Yang Sophia Kelaini
Sophia Kelaini Rachel Caines
Rachel Caines Andriana Margariti
Andriana Margariti- Centre for Experimental Medicine, Queens University Belfast, Belfast, United Kingdom
Diabetes is one of the major health care problems worldwide leading to huge suffering and burden to patients and society. Diabetes is also considered as a cardiovascular disorder because of the correlation between diabetes and an increased incidence of cardiovascular disease. Vascular endothelial cell dysfunction is a major mediator of diabetic vascular complications. It has been established that diabetes contributes to significant alteration of the gene expression profile of vascular endothelial cells. Post-transcriptional regulation by RNA binding proteins (RBPs) plays an important role in the alteration of gene expression profile under diabetic conditions. The review focuses on the roles and mechanisms of critical RBPs toward diabetic vascular endothelial dysfunction. Deeper understanding of the post- transcriptional regulation by RBPs could lead to new therapeutic strategies against diabetic manifestation in the future.
Introduction
Diabetes mellitus (DM) is a chronic, metabolic disorder characterized by hyperglycaemia due to impaired glucose homeostasis, reduced insulin activity and insulin resistance (Rother, 2007). Because of the multisystem manifestations, DM is one of the primary health care problems affecting 435 million people in 2015 around the world (Ingelfinger and Jarcho, 2017). Diabetes related complications are the 8th leading cause of death worldwide. Half of people who die from diabetic complications are under the age of 60, while the rate of incidence is equal in both sexes (Shi and Hu, 2014). There are two types of diabetes, namely, type 1 and 2. Type 1 diabetes (T1D) is caused by an autoimmune attack on the β-cells of the pancreas, which lead to pancreatic islet inflammation (insulitis), β-cell apoptosis and subsequent hyperglycemia due to low insulin production. T1D patients need daily administration of insulin and are likely to suffer ketoacidosis, coma and death (Størling and Pociot, 2017). T1D accounts for 10–15% of diabetes incidences, while Type 2 diabetes (T2D) contributes to a majority of 85–90%. T2D is characterized by insulin resistance, defects in insulin secretion, β-cell apoptosis and islet amyloid deposits. The glucose cannot be utilized by target tissues such as liver and skeletal muscle, hence the blood glucose level increased (Rawshani et al., 2017). The etiology of T2D is unknown, however, physiological, genetic, and environmental factors such as obesity, family history and pollution are known to be risk factors (Stumvoll et al., 2005). Diabetes triggers multisystemic complications such as cardiovascular diseases, neuropathy, nephropathy etc. and the patients have higher predisposition of infection, cancer, and Alzheimer's disease (Harcourt et al., 2013; Schneider et al., 2016). Nowadays people see diabetes not only as a metabolic disorder but also a cardiovascular disease because of the parallel occurrence of cardiovascular complications along with diabetes. The intimate correlation between diabetes and the predisposition of cardiovascular disease has been well-reported (Leon and Maddox, 2015). Diabetes is characterized by a two- to four- fold increased risk of cardiovascular disease while endothelial cell dysfunction is the initiating and perpetuating factor in the development of vascular complications (Brownlee, 2001; Shi and Vanhoutte, 2017).
The endothelium is the monolayer of endothelial cells (ECs) covering the lumen of blood vessels. In addition to providing a physical barrier between tissues and the circulating blood, vascular ECs play important roles in the maintenance of vascular homeostasis under physiological conditions. The endothelium is critical for the regulation of vasodilation, prevention of platelet adhesion, aggregation and thrombogenesis, as well as behavior of the underlying smooth muscle cells. The endothelium secretes various factors, regulating vessel integrity, blood vessel development, metabolism, inflammation, cell adhesion, angiogenesis, haemostasis, and vascular permeability (Sena et al., 2013).
In diabetes, endothelial functions are compromised, including increased permeability, disturbed vascular tone, aberrant angiogenesis, enhanced adhesion, and deposition of monocytes and platelets, leading to thrombogenesis. The most prevailing mechanism of endothelial dysfunction is an increase in oxidative stress and reactive oxygen species (ROS), which inactivates nitric oxide (NO) and ablates its role in regulating vascular tone as well as prevention of adhesion and aggregation of leukocytes and platelets, smooth muscle cell proliferation, inflammation and apoptosis (Vallance and Chan, 2001; Giacco and Brownlee, 2010). In addition, NO bioavailability is reduced due to down-regulation of nitric oxide synthase (eNOS), the critical enzyme in catalyzing the generation of NO from L-arginine (Sena et al., 2008).
It has been established that hyperglycaemia contributes to a significant alteration of gene expression profile in vascular ECs. High throughput assays of the transcriptome have revealed a plethora of candidate genes involved in extracellular matrix (ECM) reorganization, angiogenesis, vascular tone, inflammatory response, apoptosis, cell cycle, cell adhesion, coagulation, platelet activation etc. (Table 1) (Stenina, 2005; Ambra et al., 2014; Moradipoor et al., 2016).
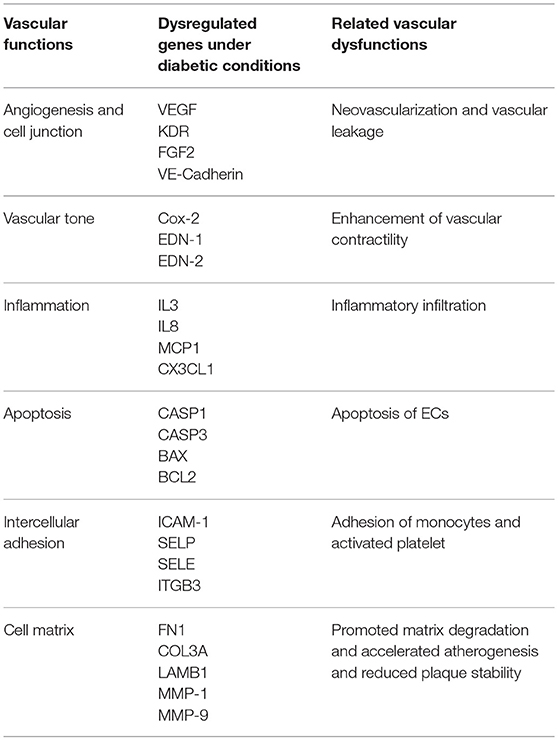
Table 1. Dysregulated genes and vascular dysfunctions under diabetic conditions.
The gene expression profile in vascular ECs is finely regulated by both transcriptional and post- transcriptional regulation systems. Post-transcriptional regulation includes processing of the pre-mRNAs toward mature mRNAs as well as mRNA transportation, quality control, mRNA decay and translational regulation etc. (Whelan et al., 2012).
There are around 424 RNA-binding proteins (RBPs) encoded by the human genome. Many of the RBPs are reportedly dysregulated in diabetes (Keene, 2007; Vanderweyde et al., 2013). For instance, HuR, hnRNP K, hnRNP F, IGF2BP2, and LIN28 are dysregulated in diabetic nephropathy, QKI, TTP and hnRNP C are related to atherosclerosis, CUGBP1, RBFOX1 and eIF4E are associated with diabetic skeletal muscle myopathy and LIN28, HuR and QKI are linked to diabetic cardiomyopathy as well (Nutter and Kuyumcu-Martinez, 2018) RBPs bind to a specific RNA sequence and/or RNA structure to form ribonucleoprotein (RNP) complexes dynamically (Gerstberger et al., 2014; Heinrich et al., 2017). RNA- binding domains (RBDs) encompassed in RBPs act as key modules for RNA recognition (Lunde et al., 2007). The most common RBDs exist in RBP of varying functions, including the hnRNPK homology (KH) domain, RNA recognition motif (RRM), double-stranded RNA-binding domain (dsRBD), zinc finger (ZnF) motif, cold shock domain (CSD) etc. (Gerstberger et al., 2014).
RBPs interact with target (pre)mRNAs at the 5′- and 3′- untranslated regions (UTRs), as well as at non-coding (intronic) and coding (exonic) regions and function in every aspect of RNA processing to produce mature mRNA and regulate mRNA localization, stability and translation (Gerstberger et al., 2014). 5′cap and 3′ poly(A) tail structures can be removed by decapping enzymes and deadenylases to cause mRNA degradation (Roy and Jacobson, 2013). Binding to cis-acting elements in the mRNA, RBPs interact with decapping or deadenylation enzymes to affect mRNA stability (Feigerlová and Battaglia-Hsu, 2017). For instance, RBPs HuR and TTP bind to the AU-rich elements (AREs) in the 3′UTRs of mRNA to promote stability or trigger decay of mRNA (Brennan and Steitz, 2001) (Figure 1). Proper function of these intricate post-transcriptional manipulations of the RNA network is essential for the vascular endothelial system. Many RBPs and RBP-regulated RNA network disruptions have been implicated in the development of diabetic dysfunction of the vascular endothelium (Scott et al., 2007). The current review focuses on five critical RNA binding proteins HuR, TTP, SRSF1, SRSF6, and QKI (Table 2).
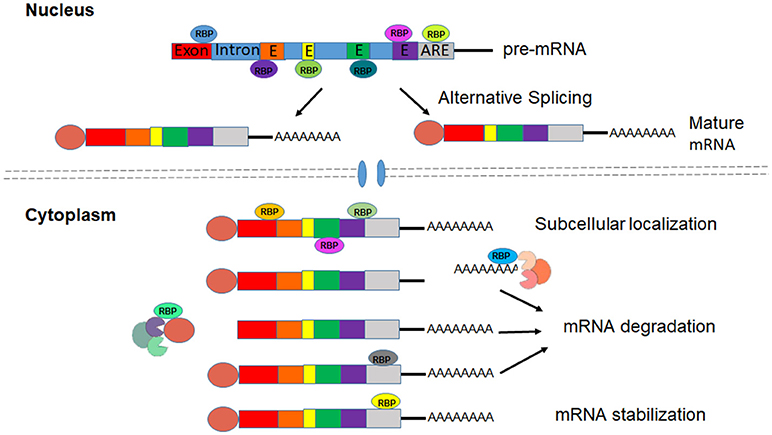
Figure 1. RBPs regulate processing, Localization and stabilization of mRNAs.
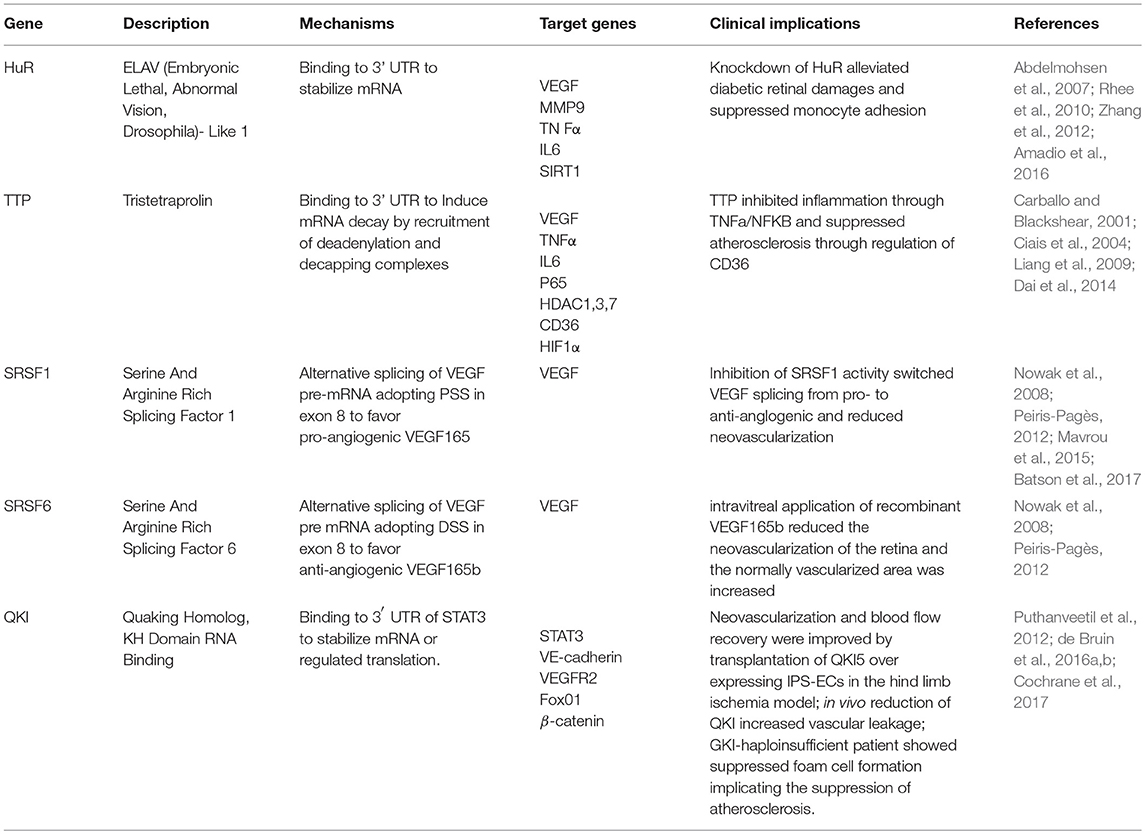
Table 2. RNA binding proteins associated with endothelial dysfunctions under diabetic conditions.
HuR
HuR is a member of the Drosophila embryonic lethal abnormal visual (ELAV) protein family that binds to mRNA PolyU- and AU- rich elements and prevents RNase mediated degradation (Chang and Hla, 2014; Pullmann and Rabb, 2014). HuR is up-regulated and activated in diabetes by various mechanisms including MiRNA regulators (MiR23 and MiR9) and protein kinase C (PKC)-mediated phosphorylation (Amadio et al., 2010; Jeyabal et al., 2016). Upon activation, HuR translocates from nucleus to the cytoplasm to bind and affect the stability and translation of target mRNAs (Govindaraju and Lee, 2013).
Vascular endothelial growth factor (VEGF) acts as a key regulator in the process of neovascularization and angiogenesis. In ECs from patients with diabetic retinopathy, HuR binds and stabilizes VEGF which triggers endothelial proliferation, migration and tube formation leading to pathological angiogenesis (Amadio et al., 2010). When streptozotocin (STZ)-induced diabetic rats were treated with intravitreal injection of lipoplexes, a Nanosystem loaded with siRNA silencing HuR expression, retinal HuR and VEGF are significantly decreased and diabetic retinal damage is alleviated (Amadio et al., 2016). Zhang et al. reported that when murine macrophages adhered to the β2 integrin ligand intercellular adhesion molecule-1 (ICAM-1), VEGF and matrix metalloproteinase-9 (MMP9) mRNAs were stabilized. Whereas, in tissue-specific HuR knockout mice, this mRNA stabilization effect was lost in bone marrow-derived macrophages. Further functional study verified the impaired recovery of blood flow and muscle neovascularization post femoral artery ligation (Zhang et al., 2012). Therefore, in contrary to its pathological angiogenic effects in diabetic retinopathy, HuR contributes to promoting the repair of damaged vascular endothelium via stabilization of VEGF.
Activation of vascular endothelial cells results in vascular diseases such as sepsis and atherosclerosis. In human pulmonary microvascular endothelial cells, HuR stabilized, and up-regulated mRNA levels of tumor necrosis factor (TNF)-induced interleukin-6 (IL-6) (Shi et al., 2012). Tiedje et al. reported that HuR binding to ARE was mandatory to stabilize and initiate translation of TNF at the endoplasmic reticulum (ER) (Tiedje et al., 2012). Cheng et al. demonstrated that HuR promoted endothelial activation by suppressing eNOS expression (Cheng et al., 2013). HuR knockdown by MiR-146a in vascular ECs negatively regulated inflammation via suppression of pro-inflammatory NF-κB, MAPK signaling pathways and downstream EGR transcription factors as well as decreases in ICAM-1, VCAM-1, and adhesion of monocytes (Rhee et al., 2010).
Sirtuin 1 (SIRT1) is the leading deacetylase in the SIRT family that serves as a protector against environmental stresses to promote cell survival (Chen et al., 2012). Vascular ECs under hyperglycaemia showed decreased SIRT1 expression. Abdelmohsen et al. reported that HuR bound to the 3′UTR of SIRT1 mRNA, stabilizing and increasing SIRT1 expression levels. Under oxidative stress, HuR was phosphorylated by ChK2 at residue Ser-100 leading to segregation of the HuR-SIRT1 mRNA complex. The degradation of separated SIRT1 mRNA coincided with the decreased SIRT1 abundance and compromised cell viability (Abdelmohsen et al., 2007).
TTP
Tristetraprolin (TTP), also known as zinc finger protein 36 homolog (ZFP36), belongs to the TIS11 family commonly containing tandem CCCH zinc fingers. TTP binds to 3′ UTR ARE region to induce destabilization and decay of mRNA by recruiting deadenylation and decapping complexes (Lai et al., 2000; Lykke-Andersen and Wagner, 2005). mRNA- decapping enzymes DCP1A and DCP2, CCR4-NOT deadenylase, the 5′-3′ exoribonuclease 1 (XRN1), exosome complex endonuclease PM-SCl75 and argonaute 2 (AGO2) are important components of the mRNA decay machinery that directly binds to TTP (Fabian et al., 2013).
Both TTP and HuR bind to ARE elements, whereas studies have shown that ARE-containing mRNAs are stabilized by HuR but destabilized by TTP. There are three members in the TTP family, TTP, TTPL1 and TTPL2 who share the activity to destabilize mRNA. TTPL1 interacts with the two ARE elements in the 3′ UTR of VEGF mRNA to trigger degradation (Ciais et al., 2004), which was opposite to the effect of HuR. In another case, HuR stabilized and up-regulated IL-6 mRNA in human pulmonary vascular ECs, whereas TTP promoted IL- 6 mRNA degradation (Sauer et al., 2006).
Apart from IL-6, a plethora of mRNAs of inflammatory modulators have been reported to be destabilized by TTP, such as interleukin, interferon and chemokine ligand family members (Xin et al., 2014). TTP was discovered to be readily stimulated by insulin (Lai et al., 1990) and was well-characterized in immune functions (Sanduja et al., 2011). TTP-deficient mice showed severe, complex inflammatory phenotype such as cachexia, arthritis and autoimmunity. Such phenotypes were ameliorated by treatment with anti-TNFα antibody or backcrossing with TNFR1 knockout mice, suggesting that TTP suppressed inflammation through inhibition of TNFα production (Taylor et al., 1996; Carballo and Blackshear, 2001).
NF-κB mediates the major inflammatory signal pathways and aberrant NF-κB activation is related to tissue damage and inflammatory disorders such as atherosclerosis and arthritis (Pfitzner et al., 2004; O'Neill, 2006). TTP physically interacts with the p65 subunit of NF-κB and functions as a corepressor of p65/NF-κB. Overexpression of TTP inhibited NF-κB- dependent transcription. TTP is also associated with histone deacetylases HDAC1, −3, and −7 in vivo. HDAC1 or HDAC3 knockdown by histone deacetylase inhibitors or small interfering RNA completely or partly ablated the repression of TTP on NF-κB reporter activation (Liang et al., 2009). TTP repressed the expression of inflammatory cytokines in target cells via inhibition of NF-κB transcriptional activation and destabilization of the bound cytokine mRNAs (Figure 1).
Vascular endothelial dysfunction was also observed in TTP-deficient mice (Bollmann et al., 2014). TTP−/− mice showed stabilized and up-regulated NADPH oxidase 2 mRNA, which was associated with enhanced levels of ROS and nitrogen species (RNS). The alteration of ROS and RNS level was highly related to the disruption of acetylcholine-induced NO-mediated vasorelaxation.
Zhang et al. investigated the regulation of TTP on inflammation in vascular ECs and its direct binding to target cytokine mRNAs (Zhang et al., 2013). Healthy aorta showed minimal expression of TTP which was significantly increased in ECs overlying atherosclerotic lesions. TTP upregulation was also observed in macrophage foam cells of atherosclerosis. After migration into the subendothelial arterial space, the monocytes readily differentiate into macrophages and take in modified low-density lipoprotein (LDL) to form foam cells which are essential for atherosclerosis. The uptake of oxidized low-density lipoprotein (oxLDL) by macrophages is mediated by the scavenger receptor CD36. Dai et al (Dai et al., 2014) revealed that TTP, which bound to ARE in the 3′ UTR, promoted CD36 mRNA degradation. Therefore, TTP may act as an important inhibitor of macrophage foam-cell formation to deter atherosclerosis.
Hypoxia-inducible factor 1 (HIF-1) is an important regulator of vascular ECs to direct their response to changes of environmental oxygenation. A variety of genes related to glucose metabolism and angiogenesis are regulated by HIF-1(Dewhirst et al., 2008). Chamboredon et al. analyzed the regulation of HIF-1α mRNA expression in ECs under hypoxic conditions and revealed that hypoxia-induced down-regulation of HIF-1α mRNA in ECs was mediated by TTP, which bound specifically to HIF-1α 3′ UTR. The decrease in the half-life of luciferase HIF-1α-3'UTR reporter transcript with prolonged hypoxia was mediated by TTP. While knockdown of TTP in ECs reversed the decrease of HIF-1α mRNA induced by hypoxia (Chamboredon et al., 2011).
SRSF1/6
Splicing is one of the key steps of RNA processing toward mature mRNA including intron removal from the pre-mRNA and subsequent exon ligation. By alternative splicing, selection of various subsets of exons results in different isoforms of transcripts generated from the same gene. Alternative splicing is a major origination of proteins with different functions, but in some cases abnormal RNA splicing can lead to disorders (Feero et al., 2010).
The VEGF gene plays a big part in the processes of vascularization and angiogenesis. The VEGF mRNAs are derived from eight exons to encode at least six protein isoforms. These products are termed VEGF121, VEGF165, VEGF189, etc. based on the number of amino acids, among which VEGF165 is the main isoform. When the distal splice site (DSS) in exon 8 is selected, the last exon turns to exon 8b resulting in a novel family of isoforms of VEGF, termed VEGF121b, VEGF165b, VEGF189b, etc. The first identified member of this family was VEGF165b, which is the only one whose effect on EC functions has been investigated (Bates et al., 2002). The produced proteins of the two families have different C-terminal amino acid sequences. During alternative splicing, when the proximal splice site (PSS) is selected, the C-terminal codes for CDKPRR to form VEGF165, while the selection of distal splice site (DSS) results in the C-terminal coding of SLTRKD to form VEGF165b isoform. In spite of the encompassment of receptor-binding domains, the VEGF165b isoform is not able to activate VEGF-R2. What's more, it acts as a competitor to inhibit the normal functions of VEGF165 on the regulation of EC proliferation, migration and vasodilation (Ladomery et al., 2007). Therefore, the alternative splicing products of VEGF165 and VEGF165b function as pro- or anti-angiogenic regulators separately (Peiris-Pagès, 2012). In the normal vitreous VEGF165b isoforms account for nearly two thirds of the total VEGF. In the diabetic vitreous, however, VEGF165 is significantly increased and becomes the dominant form Perrin et al. (2005). In the circumstance of diabetic retinopathy, the splicing of VEGF switches to the pro-angiogenic isoform (VEGF165) to favor vascularization in the retina.
The serine/arginine-rich (SR) proteins play critical roles in the alternative splicing of the VEGF RNA transcript. Computational sequence analysis of the VEGF gene revealed a predicted binding site for Serine/Arginine Rich Splicing Factor 1 (SRSF1) before the DSS, and a predicted SRSF6 binding site behind the DSS (Peiris-Pagès, 2012). When SRSF1 binds to the pre-mRNA of VEGF, the PSS in exon 8 is preferred to generate VEGF165 isoform. When SRSF6 binds to the pre-mRNA, the DSS dominates to form VEGF165b (Nowak et al., 2008). SRSF1 is modulated by upstream regulators SR protein kinases 1 and 2 (SRPK1/2). SPRK1 can be inhibited by small molecule inhibitors or down-regulated by RNA interference (RNAi) to block activation and nuclear transportation of SRSF1. SRSF1 inhibition consequently switches the selection of PSS to DSS and benefits the generation of anti- angiogenic VEGF165b isoform (Amin et al., 2011; Mavrou et al., 2015).
Through PKC-induced activation of SRPK1, IGF1 and TNFα support the selection of PSS in exon 8, whilst TGFβ activates p38 mitogen-activated kinases (p38 MAPK) through Clk1 and phosphorylates SRSF6, which favor the usage of DSS element (Harper and Bates, 2008; Nowak et al., 2008). Small molecule inhibitors of SRPK1, such as SRPIN340, MVRL09, and SPHINX31 are potent regulators to inhibit the binding of PSS in exon 8 by SRSF1 to elevate the level of VEGF165b through alternative splicing. In an animal model of retinal angiogenesis, application of SRPK1 inhibitor greatly inhibited activation of SRSF1, switching VEGF splicing from the pro- to anti-angiogenic form to block neovascularization (Gammons et al., 2013a,b; Batson et al., 2017). Injection of SRPK1/2 inhibitor SRPIN340 into the retina of an oxygen-induced retinopathy mouse model significantly reduced the neovascular area of the retina and the normally vascularized area was increased, which was equivalent to the effect of intravitreal application of recombinant VEGF165b. Based on these findings, modulation of VEGF165/VEGF165b balance by regulation of alternative splicing machinery may be a novel therapeutic strategy for diabetic retinopathy and other vascular disorders (Nowak et al., 2010).
QKI
Quaking (QKI) is an RNA binding protein belonging to the Signal Transduction and Activation of RNA (STAR) protein family which contain SH2 and SH3 domains, an RNA-binding motif (e.g. a KH domain) and phosphorylation sites. This implies they may play a role in the splicing of pre-mRNAs (Vernet and Artzt, 1997), mRNA nuclear exportation, mRNA stability and translation into the subsequent protein. They may also be involved in some signal transduction pathways (Justice and Hirschi, 2010). There are several QKI isoforms, three of which (QKI5, QKI6 and QKI7) have been linked to vascular development. Each of these isoforms holds different carboxy-terminal ends but matching RNA binding domains (Chénard and Richard, 2008). QKI is generally required in endothelial barrier function maintenance as it increases VE-cadherin and β-catenin expression in epithelial intercellular junctions. These three QKI isoforms are present in ECs, with QKI5 being the most abundant (de Bruin et al., 2016b). QKI gene has been discovered in functional studies to be critical in the formation and remodeling of embryonic blood vessels (Noveroske et al., 2002). The expression of QKI was observed in the yolk sac endoderm and homozygous QKk2 allele was found to be lethal to the embryos due to disrupted vasculature development.
Forkhead box O1 (FoxO1) belongs to the forkhead family of transcription factors that participates in a variety of important biological events including cell proliferation, cell death, immunologic reaction, regulation of metabolism in response to oxidative stress etc. (Puthanveetil et al., 2013). Studies have revealed the involvement of FoxO1 in the development of cardiovascular diseases and diabetes (Puthanveetil et al., 2012). In diabetes, the activated FoxO1 was associated with the dysregulation of metabolic homeostasis and activation of cell death signaling. Studies have revealed the decreased expression of QKI5 in the myocardium of ob/ob diabetic mice. When QKI5 expression was up-regulated, FoxO1 expression was repressed and the NS and ER stresses as well as ischemia/reperfusion injury alleviated. By RNA co-immunoprecipitation the interaction between QKI5 and FoxO1 mRNA was verified. When QKI5 was overexpressed the half-life of FoxO1 mRNA was shortened, suggesting the negative effect of QKI5 on FoxO1 mRNA stability.
Previous work from our lab demonstrated that QKI5 was suppressed in heart vessels isolated from diabetic mice compared to healthy controls, suggesting a role of QKI5 in vascular dysfunction under diabetic conditions. Using the model of induced pluripotent stem (iPS) cell differentiation toward ECs, our recent paper (Cochrane et al., 2017) reported a critical role of QKI5 in the generation of ECs from iPS cells via stabilization of CD144 and activation of VEGFR2 through STAT3 signaling. RNA immunoprecipitation confirmed the direct binding of QKI5 to the 3' UTR of STAT3 to promote mRNA stability. Remarkably, angiogenesis, neovascularization, and blood flow recovery were significantly improved by transplantation of QKI5 overexpressing iPS-ECs in the animal model of experimental hind limb ischemia. These findings suggest that QKI5 down-regulation in diabetes may contribute to vascular complications resulting from EC dysfunction and probably become a novel target for therapeutic treatment of diabetes. Meanwhile, QKI5 induced the splicing factor SF3B1 during EC differentiation in a time dependent manner, implying that QKI-5 contributed to EC induction from iPSCs as an important splicing regulator.
QKI was also reported to participate in the regulation of macrophage differentiation from monocytes (de Bruin et al., 2016a). QKI showed low levels of expression in monocytes and early human atherosclerotic lesions. When naïve human monocytes were converted to macrophages with GM-CSF or M-CSF, the expression of QKI protein isoforms was significantly increased. Consistently, the examination of CD68+ macrophages in advanced atherosclerotic lesions revealed high abundance of QKI. In the QKI-haploinsufficient patient, foam cell formation was suppressed due to limited differentiation of macrophages from monocytes and subsequent reduced uptake of oxLDL along with decreased QKI expression. These studies illustrated the key role for QKI as a post-transcriptional regulator in the determination of macrophage fate and development of atherosclerosis (de Bruin et al., 2016b).
Given the observation that QKI isoforms were highly expressed in the healthy vascular endothelium in comparison with smooth muscle (van der Veer et al., 2013), de Bruin et al. studied the relevance for QKI and endothelial functions and underlying mechanisms. Both VE-cadherin and β-catenin were predicted to harbor high-affinity Quaking Response Element (QRE) (NACUAAY-N1–20-UAAY) in the 3′ UTRs of their mRNAs (Galarneau and Richard, 2005), suggesting their possible target identity of QKI which was confirmed by RNA immunoprecipitation of both mRNAs by QKI antibody. With QKI overexpression, luciferase- reporter assay with the 3′ UTRs of both genes achieved significant enhancement of luciferase activity, which was diminished when QKI was repressed. When QKI was knockdown by shRNA in the ECs, neither VE-cadherin nor β-catenin mRNA was significantly reduced, but their protein levels were decreased. For functional study, ECs with QKI knockdown failed to form a proper monolayer of high resistance, although their adherence and spread capacities were not affected. In vivo reduction of QKI resulted in a significant 40% increase of vascular leakage. Therefore, the translation of VE-cadherin and β-catenin was regulated by QKI through direct binding to 3′ UTRs. Reduced QKI expression resulted in the decrease of VE-cadherin and β-catenin proteins and subsequent impaired endothelial barrier function leading to vascular leakage.
LncRNAs and RBPs in Endothelial Cells Under Diabetic Conditions
Most of current research is centered on protein-coding RNAs, however, noncoding RNAs (ncRNAs) including long noncoding RNAs (LncRNAs) account for the majority of genome transcripts. Cooperating with RNA-binding proteins, LncRNAs participate in all aspects of biological processes and their importance is widely recognized. LncRNAs are >200 nucleotide long RNA transcripts which lack protein-coding potential (Yang et al., 2014). LncRNAs regulate gene expression at all levels through modulation of epigenetic machineries, recruitment of RNA-binding proteins, function as decoys and interact with miRNAs etc. Growing evidence has revealed the correlation of disrupted lncRNA levels and a variety of human diseases, including diabetes. A number of LncRNAs have been reported to be dysregulated under diabetic conditions, such as MALAT1, MEG3, MIAT, RNCR3, and ANRIL.
Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) was significantly increased in retinal endothelial cells of STZ-induced diabetic rats and db/db mice or high glucose treated human umbilical vein endothelial cells (HUVEC) (Liu et al., 2014; Yan et al., 2014; Puthanveetil et al., 2015) indicating its involvement in the development of diabetic retinopathy and endothelial dysfunction. Knockdown of MALAT1 by intraocular injection of shRNA ameliorated retinal vascular dysfunctions of cell barrier defects, pericyte loss, capillary degeneration, and retinal inflammation. In vitro assays revealed the decreased retinal endothelial cell proliferation, migration and tube formation with MALAT1 downregulation. It is also reported that MALAT1 knockdown decreased the level of ROS in endothelial cells under hyperglycaemic conditions (Puthanveetil et al., 2015).
MALAT1 binds to the splicing factor SRSF1 directly and influences its phosphorylation, distribution in nuclear speckle domains and alternative splicing function (Tripathi et al., 2010). Depletion of MALAT1 or SRSF1 led to the increase of anti-angiogenic VEGF isoform VEGF165b. Tube formation assay of endothelial cells in the presence of conditioned medium from modified SKBR3 cells showed that in SRSF1 and MALAT1 interfered scenarios the angiogenic capacity of ECs was significantly decreased (Pruszko et al., 2017). Liu et al. reported that MALAT1 regulated the function of endothelial cells through the p38/MAPK signaling pathway (Liu et al., 2014). The cell proliferation induced by MALAT1 was blocked by p38/MAPK pathway inhibitor SB203580 or p38 siRNA and p38 stimulated phosphorylation was blunted by MALAT1 knockdown. The crosstalk between MALAT1 and p38/MAPK pathway may become a novel strategy for the therapy of diabetes-related microvascular complications. With MALAT1 silenced, the S-phage endothelial cyclins CCNA2, CCNB1, and CCNB2 were significantly downregulated, while cell cycle inhibitory genes p21 and p27Kip1 increased (Michalik et al., 2014). More study is still required to elucidate the mechanisms of MALAT1 effects on cell signaling and cell cycle regulation.
Myocardial infarction associated transcript (MIAT) is another LncRNA identified to be upregulated under diabetic conditions in retinal endothelial cells and fibrovascular membrane of diabetic patients (Strawbridge et al., 2011; Yan et al., 2015). In vitro, MIAT knockdown inhibited EC proliferation under hyperglycaemic conditiona and in vivo downregulation of MIAT in STZ diabetic rats alleviated the retinal vascular dysfunctions of pericyte loss, vascular degeneration and inflammation. MIAT possesses a tandem UACUAAC repeat motif and binds directly to the splicing factors SRSF1, QKI, Cef3 to form nuclear bodies (Tsuiji et al., 2011; Barry et al., 2014). The UACUAAC repeat motif binds to SRSF1 with a higher affinity than the divergent branch point sequence in mammals and therefore may modulate the alternative splicing of VEGF in favor of angiogenesis. MIAT also competes with miR-150 and miR-29b in retinal endothelial cells to regulate VEGFA level and apoptosis (Yan et al., 2015; Zhang J. et al., 2017).
Antisense Noncoding RNA in the INK4 Locus (ANRIL) is a 3.8 kb antisense RNA to INK4 locus. ANRIL has been found to be associated with vascular dysfunction in diabetes (Congrains et al., 2013). ANRIL level elevation was observed in human retinal endothelial cells exposed to high glucose and also the retina of diabetic animals, while overexpression of ANRIL upregulated VEGF expression (Thomas et al., 2017; Zhang B. et al., 2017). The increased retinal microvascular permeability in diabetic mice was alleviated by ANRIL knockout, which was in consistence with the dynamics of VEGF level. In human retinal endothelial cells exposed to high glucose, the key components of PRC complex EZH2 and p300 were significantly increased, while with ANRIL knockdown the dysregulation was corrected. VEGF was found to be regulated by miR200b through PRC complex and p300 and RNA-IP assay verified the direct binding of ANRIL to EZH2 and p300, indicating that upregulated ANRIL induced VEGF generation in high glucose treated ECs through interaction with PRC complex and p300. Moreover, by recruitment of PRC complexes, ANRIL epigenetically suppressed the expression of cell cycle regulators p15 and p16 which contain an overlapping sequence with ANRIL in the promoter region (Yap et al., 2010; Kotake et al., 2011).
It was reported that ectopic expression of ANRIL promoted angiogenesis by stimulation of NFκB signaling (Zhang B. et al., 2017). Moreover, in vascular ECs, TNFα induced ANRIL expression through NFκB and ANRIL interacted with PRC-associated transcriptional factor YY1 to regulate gene expression of IL6 and IL8 (Holdt et al., 2013; Zhou et al., 2016). Thus, the ANRIL- NFκB feedback loop may serve as a target to protect endothelial cells against dysfunction and atherosclerosis.
Retinal non-coding RNA3 (RNCR3) was first identified during mouse retinal development with dynamic expression (Blackshaw et al., 2004). RNCR3 was significantly up- regulated in retinas of diabetic animals and endothelial cells upon high glucose exposure. In vivo knockdown of RNCR3 ameliorated the diabetes-induced vascular dysfunctions of acellular capillaries, vascular leakage and inflammation. In vitro knockdown of RNCR3 suppressed EC proliferation, viability, migration and tube formation (Shan et al., 2016, 2017). It was revealed that RNCR3 functioned as a competing endogenous RNA (ceRNA) to regulate KLF2 levels by sponging miR-185-5p in endothelial cells.
Opposite to the above LncRNAs, maternally expressed gene 3 (MEG3) was reported to be decreased in retinal ECs of STZ diabetic mice (Qiu et al., 2016). In vivo knockdown of MEG3 led to retinal vascular dysfunctions of capillary degeneration, microvascular leakage and inflammation. In vitro knockdown of MEG3 in retinal vascular endothelial cells compromised EC angiogenic potential which was mediated by activation of PI3k/Akt signaling pathway.
Zhou et al. found that MEG3 interacted with p53 and overexpression of MEG3 enhanced p53 level and stimulated p53-dependent transcription implicating the involvement of p53 in the functioning of MEG3 (Zhou et al., 2007). As it is known that p53 binds to the VEGFA promoter and negatively regulates its transcription (Qin et al., 2006). Therefore, downregulation of MEG3 may contribute to the neovascularization and leakage of diabetic retinopathy through p53 suppression and subsequent induction of VEGF.
RBP-Based Therapies and Future Perspective
As RBP-regulated RNA networks play a critical role in the development of diabetic vascular manifestations, targeting the candidate RBPs or RBP-RNA interactions could be a promising therapeutic strategy against diabetic vascular endothelial dysfunction.
Some RBPs per se are promising treatments for diabetic disorders. For instance, tristetraprolin (TTP) acts against diabetic inflammation and atherosclerosis through degradation of pro-inflammatory cytokines. TTP knockout mice showed overexpression of the potent pro-inflammatory cytokine TNFα and severe inflammatory phenotypes (Carballo et al., 1998). Inversely, TTP overexpression exerted profound suppression effect on inflammatory disease models (Patial et al., 2016). Kirkwood et al. reported the study on a rat model of periodontitis induced by intra-oral injection of LPS. When TTP overexpression was achieved by local application of an adenovirus expression vector, the complications of bone loss and inflammatory infiltration were significantly alleviated and the level of local cytokines was markedly reduced (Patil et al., 2008). Diabetic vascular complications feature inflammatory events and TNFα is implicated as an important mediator cytokine of inflammation in diabetes. Therefore, the discoveries from animal studies implies the possible beneficial effects of TTP application on diabetic inflammation conditions.
Under various pathologic conditions such as age-related macular degeneration (AMD) and diabetic retinopathy (DR), VEGF serves as a key mitogen to stimulate angiogenesis. Anti-VEGF antibodies have emerged as clinical treatment against AMD and DR. However, intravitreal injection of VEGF antibodies may harbor the risks of various complications, such as infection, inflammation and vitreous hemorrhage (Ventrice et al., 2013). Therefore, interest has grown to invent more effective drugs (e.g., small molecules) and safer methods (e.g., eye drop, ointment) of drug delivery to deal with neovascularization complications. Inhibition of serine-arginine protein kinase 1 (SRPK1) promotes the switch from the pro-angiogenic isoform VEGF165 to the anti-angiogenic isoform VEGF165b and suppresses pathologic angiogenesis (Dong et al., 2013). Using a computational protocol combined with a pharmacophore-based database search, Morooka et al. identified a new small molecule, SRPIN803, that inhibits both casein kinase 2 (CK2) and SRPK1 to suppress VEGF generation synergistically. In a laser-induced choroidal neovascularization mouse model, topical administration of SRPIN803 substantially suppressed intraocular neovascularization, suggesting SRPIN803 as a promising therapeutic drug for pathologic angiogenesis. (Morooka et al., 2015).
In diabetic conditions, highly expressed HuR translocates from nucleus to cytoplasm to bind and stabilize VEGF which triggers pathological angiogenesis. Amadio et al. carried out intravitreal injection of nanosystems loaded with siRNA against HuR (lipoplexes) to treat streptozotocin (STZ)-induced diabetic retinopathy in rats. The results showed that retinal HuR and VEGF were significantly silenced by HuR siRNA treatment and diabetic retinal damage was rescued (Amadio et al., 2016).
Anti-sense oligos (ASOs) are short single-stranded deoxyribonucleotides complementary to sense strand nucleic acid sequences. ASOs bind to target RNA sites and regulate the expression of genes by several mechanisms, including modulation of RNA stability, modification of RBP binding to RNA, regulation of RNA splicing, and mRNA translation (Lundin et al., 2015; Bishop, 2017). Some ASOs have progressed to human clinical trials for disease treatment, including cancer, diabetes, neurodegenerative disorders, and muscular dystrophy. Alternative polyadenylation of the KDR gene gives rise to two protein products with different functions, membrane-bound KDR (mbKDR) and soluble KDR (sKDR). sKDR functions as antagonist of lymphangiogenesis due to the lack of a tyrosine kinase domain. Accordingly, an antisense morpholino oligomer was designed complementary to the exon 13-intron 13 junction sequence to increase sKDR at the expense of mbKDR, thereby suppressing both haemangiogenesis and lymphangiogenesis. Uehara et al. demonstrated the suppression of laser choroidal neovascularization by intravitreal morpholino injection. Furthermore, subconjunctival application of the morpholino significantly inhibited corneal angiogenesis, lymphangiogenesis as well as graft rejection after transplantation in the mouse cornea. (Uehara et al., 2013). In conclusion, the post-transcriptional dysregulation of gene expression by RBPs plays important roles in the development and progression of diabetic endothelial dysfunction. The emergence and advancement of high throughput technologies enables the identification of RNA targets of RBPs, which offers novel insight into the mechanisms of diabetic disorders. Our deepened understanding of the mechanisms of RNA transcripts, as well as the critical regulatory roles played by RBPs enables the potential to provide novel therapeutic options for diabetic patients. In terms of the designation and application of RBP-RNA based therapeutic strategies, caution is necessary to ensure the requirement of safety and accuracy is fulfilled. The advances on specificity and efficacy of RBP-RNA treatments are currently being investigated. The future will see the possible inclusion of these smart therapeutic modalities in the therapeutic field to combat diabetic complications.
Author Contributions
CY conception and design, manuscript writing. SK and RC revision and approval of manuscript. AM conception and design, financial support, final approval of manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Grants from BBSRC and the British Heart Foundation.
References
Abdelmohsen, K., Pullmann, R. Jr., Lal, A., Kim, H. H., Galban, S., Yang, X., et al. (2007). Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol. Cell 25, 543–557. doi: 10.1016/j.molcel.2007.01.011
Amadio, M., Bucolo, C., Leggio, G. M., Drago, F., Govoni, S., and Pascale, A. (2010). The PKCbeta/HuR/VEGF pathway in diabetic retinopathy. Biochem. Pharmacol. 80, 1230–1237. doi: 10.1016/j.bcp.2010.06.033
Amadio, M., Pascale, A., Cupri, S., Pignatello, R., Osera, C. D., Agata, V., et al. (2016). Nanosystems based on siRNA silencing HuR expression counteract diabetic retinopathy in rat. Pharmacol. Res. 111, 713–720. doi: 10.1016/j.phrs.2016.07.042
Ambra, R., Manca, S., Palumbo, M. C., Leoni, G., Natarelli, L., De Marco, A., et al. (2014). Transcriptome analysis of human primary endothelial cells (HUVEC) from umbilical cords of gestational diabetic mothers reveals candidate sites for an epigenetic modulation of specific gene expression. Genomics 103, 337–348. doi: 10.1016/j.ygeno.2014.03.003
Amin, E. M., Oltean, S., Hua, J., Gammons, M. V., Hamdollah-Zadeh, M., Welsh, G. I., et al. (2011). WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 20, 768–780. doi: 10.1016/j.ccr.2011.10.016
Barry, G., Briggs, J. A., Vanichkina, D. P., Poth, E. M., Beveridge, N. J., Ratnu, V. S., et al. (2014). The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol. Psychiatry 19, 486–494. doi: 10.1038/mp.2013.45
Bates, D. O., Cui, T. G., Doughty, J. M., Winkler, M., Sugiono, M., Shields, J. D., et al. (2002). VEGF165b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 62, 4123–4131.
Batson, J., Toop, H. D., Redondo, C., Babaei-Jadidi, R., Chaikuad, A., Wearmouth, S. F., et al. (2017). Development of potent, selective SRPK1 inhibitors as potential topical therapeutics for neovascular eye disease. ACS Chem. Biol. 12, 825–832. doi: 10.1021/acschembio.6b01048
Bishop, K. M. (2017). Progress and promise of antisense oligonucleotide therapeutics for central nervous system diseases. Neuropharmacology 120, 56–62. doi: 10.1016/j.neuropharm.2016.12.015
Blackshaw, S., Harpavat, S., Trimarchi, J., Cai, L., Huang, H., Kuo, W. P., et al. (2004). Genomic analysis of mouse retinal development. PLoS Biol. 2:E247. doi: 10.1371/journal.pbio.0020247
Bollmann, F., Wu, Z., Oelze, M., Siuda, D., Xia, N., Henke, J., et al. (2014). Endothelial dysfunction in tristetraprolin- deficient mice is not caused by enhanced tumor necrosis factor-alpha expression. J. Biol. Chem. 289, 15653–15665. doi: 10.1074/jbc.M114.566984
Brennan, C. M., and Steitz, J. A. (2001). HuR and mRNA stability. Cell. Mol. Life Sci. 58, 266–277. doi: 10.1007/PL00000854
Brownlee, M. (2001). Biochemistry and molecular cell biology of diabetic complications. Nature 414, 813–820. doi: 10.1038/414813a
Carballo, E., and Blackshear, P. J. (2001). Roles of tumor necrosis factor-alpha receptor subtypes in the pathogenesis of the tristetraprolin-deficiency syndrome. Blood 98, 2389–2395. doi: 10.1182/blood.V98.8.2389
Carballo, E., Lai, W. S., and Blackshear, P. J. (1998). Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science 281, 1001–1005. doi: 10.1126/science.281.5379.1001
Chamboredon, S., Ciais, D., Desroches-Castan, A., Savi, P., Bono, F., Feige, J. J., et al. (2011). Hypoxia-inducible factor-1alpha mRNA: a new target for destabilization by tristetraprolin in endothelial cells. Mol. Biol. Cell 22, 3366–3378. doi: 10.1091/mbc.e10-07-0617
Chang, S. H., and Hla, T. (2014). Post-transcriptional gene regulation by HuR and microRNAs in angiogenesis. Curr. Opin. Hematol. 21, 235–240. doi: 10.1097/MOH.0000000000000040
Chen, X., Yang, H. H., Huangfu, Y. C., Wang, W. K., Liu, Y., Ni, Y. X., et al. (2012). Molecular epidemiologic analysis of Staphylococcus aureus isolated from four burn centers. Burns 38, 738–742. doi: 10.1016/j.burns.2011.12.023
Chénard, C. A., and Richard, S. (2008). New implications for the QUAKING RNA binding protein in human disease. J. Neurosci. Res. 86, 233–242. doi: 10.1002/jnr.21485
Cheng, H. S., Sivachandran, N., Lau, A., Boudreau, E., Zhao, J. L., Baltimore, D., et al. (2013). MicroRNA-146 represses endothelial activation by inhibiting pro-inflammatory pathways. EMBO Mol. Med. 5, 1017–1034. doi: 10.1002/emmm.201202318
Ciais, D., Cherradi, N., Bailly, S., Grenier, E., Berra, E., Pouyssegur, J., et al. (2004). Destabilization of vascular endothelial growth factor mRNA by the zinc-finger protein TIS11b. Oncogene 23, 8673–8680. doi: 10.1038/sj.onc.1207939
Cochrane, A., Kelaini, S., Tsifaki, M., Bojdo, J., Vila-Gonzalez, M., Drehmer, D., et al. (2017). Quaking is a key regulator of endothelial cell differentiation, neovascularization, and angiogenesis. Stem Cells 35, 952–966. doi: 10.1002/stem.2594
Congrains, A., Kamide, K., Ohishi, M., and Rakugi, H. (2013). ANRIL: molecular mechanisms and implications in human health. Int. J. Mol. Sci. 14, 1278–1292. doi: 10.3390/ijms14011278
Dai, X. Y., Cai, Y., Sun, W., Ding, Y., Wang, W., Kong, W., et al. (2014). Intermedin inhibits macrophage foam-cell formation via tristetraprolin- mediated decay of CD36 mRNA. Cardiovasc. Res. 101, 297–305. doi: 10.1093/cvr/cvt254
de Bruin, R. G., Shiue, L., Prins, J., de Boer, H. C., Singh, A., Fagg, W. S., et al. (2016a). Quaking promotes monocyte differentiation into pro-atherogenic macrophages by controlling pre-mRNA splicing and gene expression. Nat. Commun. 7:10846. doi: 10.1038/ncomms10846
de Bruin, R. G., van der Veer, E. P., Prins, J., Lee, D. H., Dane, M. J., Zhang, H., et al. (2016b). The RNA-binding protein quaking maintains endothelial barrier function and affects VE-cadherin and beta-catenin protein expression. Sci. Rep. 6:21643. doi: 10.1038/srep21643
Dewhirst, M. W., Cao, Y., and Moeller, B. (2008). Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer 8, 425–437. doi: 10.1038/nrc2397
Dong, Z., Noda, K., Kanda, A., Fukuhara, J., Ando, R., Murata, M. Ishida, et al. (2013). Specific inhibition of serine/arginine-rich protein kinase attenuates choroidal neovascularization. Mol. Vis. 19, 536–543.
Fabian, M. R., Frank, F., Rouya, C., Siddiqui, N., Lai, W. S., Karetnikov, A., et al. (2013). Structural basis for the recruitment of the human CCR4- NOT deadenylase complex by tristetraprolin. Nat. Struct. Mol. Biol. 20, 735–739. doi: 10.1038/nsmb.2572
Feero, W. G., Guttmacher, A. E., and Collins, F. S. (2010). Genomic medicine–an updated primer. N. Engl. J. Med. 362, 2001–2011. doi: 10.1056/NEJMra0907175
Feigerlová, E., and Battaglia-Hsu, S. F. (2017). Role of post-transcriptional regulation of mRNA stability in renal pathophysiology: focus on chronic kidney disease. FASEB J. 31, 457–468. doi: 10.1096/fj.201601087RR
Galarneau, A., and Richard, S. (2005). Target RNA motif and target mRNAs of the Quaking STAR protein. Nat. Struct. Mol. Biol. 12, 691–698. doi: 10.1038/nsmb963
Gammons, M. V., Dick, A. D., Harper, S. J., and Bates, D. O. (2013a). SRPK1 inhibition modulates VEGF splicing to reduce pathological neovascularization in a rat model of retinopathy of prematurity. Invest. Ophthalmol. Vis. Sci. 54, 5797–5806. doi: 10.1167/iovs.13-11634
Gammons, M. V., Fedorov, O., Ivison, D., Du, C., Clark, T., Hopkins, C., et al. (2013b). Topical antiangiogenic SRPK1 inhibitors reduce choroidal neovascularization in rodent models of exudative AMD. Invest. Ophthalmol. Vis. Sci. 54, 6052–6062. doi: 10.1167/iovs.13-12422
Gerstberger, S., Hafner, M., and Tuschl, T. (2014). A census of human RNA-binding proteins. Nat. Rev. Genet. 15, 829–845. doi: 10.1038/nrg3813
Giacco, F., and Brownlee, M. (2010). Oxidative stress and diabetic complications. Circ. Res. 107, 1058–1070. doi: 10.1161/CIRCRESAHA.110.223545
Govindaraju, S., and Lee, B. S. (2013). Adaptive and maladaptive expression of the mRNA regulatory protein HuR. World J. Biol. Chem. 4, 111–118. doi: 10.4331/wjbc.v4.i4.111
Harcourt, B. E., Penfold, S. A., and Forbes, J. M. (2013). Coming full circle in diabetes mellitus: from complications to initiation. Nat. Rev. Endocrinol. 9, 113–123. doi: 10.1038/nrendo.2012.236
Harper, S. J., and Bates, D. O. (2008). VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat. Rev. Cancer 8, 880–887. doi: 10.1038/nrc2505
Heinrich, S., Derrer, C. P., Lari, A., Weis, K., and Montpetit, B. (2017). Temporal and spatial regulation of mRNA export: single particle RNA-imaging provides new tools and insights. Bioessays 39:1600124. doi: 10.1002/bies.201600124
Holdt, L. M., Hoffmann, S., Sass, K., Langenberger, D., Scholz, M., Krohn, K., et al. (2013). Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLoS Genet. 9:e1003588. doi: 10.1371/journal.pgen.1003588
Ingelfinger, J. R., and Jarcho, J. A. (2017). Increase in the incidence of diabetes and its implications. N. Engl. J. Med. 376, 1473–1474. doi: 10.1056/NEJMe1616575
Jeyabal, P., Thandavarayan, R. A., Joladarashi, D., Suresh Babu, S., Krishnamurthy, S., Bhimaraj, A., et al. (2016). MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochem. Biophys. Res. Commun. 471, 423–429. doi: 10.1016/j.bbrc.2016.02.065
Justice, M. J., and Hirschi, K. K. (2010). The role of quaking in mammalian embryonic development. Adv. Exp. Med. Biol. 693, 82–92. doi: 10.1007/978-1-4419-7005-3_6
Keene, J. D. (2007). RNA regulons: coordination of post-transcriptional events. Nat. Rev. Genet. 8, 533–543. doi: 10.1038/nrg2111
Kotake, Y., Nakagawa, T., Kitagawa, K., Suzuki, S., Liu, N., Kitagawa, M., et al. (2011). Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 30, 1956–1962. doi: 10.1038/onc.2010.568
Ladomery, M. R., Harper, S. J., and Bates, D. O. (2007). Alternative splicing in angiogenesis: the vascular endothelial growth factor paradigm. Cancer Lett. 249, 133–142. doi: 10.1016/j.canlet.2006.08.015
Lai, W. S., Carballo, E., Thorn, J. M., Kennington, E. A., and Blackshear, P. J. (2000). Interactions of CCCH zinc finger proteins with mRNA. Binding of tristetraprolin-related zinc finger proteins to Au-rich elements and destabilization of mRNA. J. Biol. Chem. 275, 17827–17837. doi: 10.1074/jbc.M001696200
Lai, W. S., Stumpo, D. J., and Blackshear, P. J. (1990). Rapid insulin-stimulated accumulation of an mRNA encoding a proline-rich protein. J. Biol. Chem. 265, 16556–16563.
Leon, B. M., and Maddox, T. M. (2015). Diabetes and cardiovascular disease: epidemiology, biological mechanisms, treatment recommendations and future research. World J. Diabetes 6, 1246–1258. doi: 10.4239/wjd.v6.i13.1246
Liang, J., Lei, T., Song, Y., Yanes, N., Qi, Y., and Fu, M. (2009). RNA-destabilizing factor tristetraprolin negatively regulates NF-kappaB signaling. J. Biol. Chem. 284, 29383–29390. doi: 10.1074/jbc.M109.024745
Liu, J. Y., Yao, J., Li, X. M., Song, Y. C., Wang, X. Q., Li, Y. J., et al. (2014). Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis. 5:e1506. doi: 10.1038/cddis.2014.466
Lunde, B. M., Moore, C., and Varani, G. (2007). RNA-binding proteins: modular design for efficient function. Nat. Rev. Mol. Cell Biol. 8, 479–490. doi: 10.1038/nrm2178
Lundin, K. E., Gissberg, O., and Smith, C. I. (2015). Oligonucleotide therapies: the past and the present. Hum. Gene Ther. 26, 475–485. doi: 10.1089/hum.2015.070
Lykke-Andersen, J., and Wagner, E. (2005). Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev. 19, 351–361. doi: 10.1101/gad.1282305
Mavrou, A., Brakspear, K., Hamdollah-Zadeh, M., Damodaran, G., Babaei-Jadidi, R., Oxley, J., et al. (2015). Serine-arginine protein kinase 1 (SRPK1) inhibition as a potential novel targeted therapeutic strategy in prostate cancer. Oncogene 34, 4311–4319. doi: 10.1038/onc.2014.360
Michalik, K. M., You, X., Manavski, Y., Doddaballapur, A., Zornig, M., Braun, T., et al. (2014). Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ. Res. 114, 1389–1397. doi: 10.1161/CIRCRESAHA.114.303265
Moradipoor, S., Ismail, P., Etemad, A., Wan Sulaiman, W. A., and Ahmadloo, S. (2016). Expression profiling of genes related to endothelial cells biology in patients with type 2 diabetes and patients with prediabetes. Biomed Res. Int. 2016:1845638. doi: 10.1155/2016/1845638
Morooka, S., Hoshina, M., Kii, I., Okabe, T., Kojima, H., Inoue, N., et al. (2015). Identification of a dual inhibitor of SRPK1 and CK2 that attenuates pathological angiogenesis of macular degeneration in mice. Mol. Pharmacol. 88, 316–325. doi: 10.1124/mol.114.097345
Noveroske, J. K., Lai, L., Gaussin, V., Northrop, J. L., Nakamura, H., Hirschi, K. K., et al. (2002). Quaking is essential for blood vessel development. Genesis 32, 218–230. doi: 10.1002/gene.10060
Nowak, D. G., Amin, E. M., Rennel, E. S., Hoareau-Aveilla, C., Gammons, M., Damodoran, G., et al. (2010). Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: a novel therapeutic strategy for angiogenesis. J. Biol. Chem. 285, 5532–5540. doi: 10.1074/jbc.M109.074930
Nowak, D. G., Woolard, J., Amin, E. M., Konopatskaya, O., Saleem, M. A., Churchill, A. J., et al. (2008). Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J. Cell Sci. 121(Pt 20), 3487–3495. doi: 10.1242/jcs.016410
Nutter, C. A., and Kuyumcu-Martinez, M. N. (2018). Emerging roles of RNA-binding proteins in diabetes and their therapeutic potential in diabetic complications. Wiley Interdiscip. Rev. RNA 9:e1459. doi: 10.1002/wrna.1459
O'Neill, L. A. (2006). Targeting signal transduction as a strategy to treat inflammatory diseases. Nat. Rev. Drug Discov. 5, 549–563. doi: 10.1038/nrd2070
Patial, S., Curtis, A. D. II., Lai, W. S., Stumpo, D. J., Hill, G. D., Flake, G. P., et al. (2016). Enhanced stability of tristetraprolin mRNA protects mice against immune-mediated inflammatory pathologies. Proc. Natl. Acad. Sci. U.S.A. 113, 1865–1870. doi: 10.1073/pnas.1519906113
Patil, C. S., Liu, M., Zhao, W., Coatney, D. D., Li, F., VanTubergen, E. A., et al. (2008). Targeting mRNA stability arrests inflammatory bone loss. Mol. Ther. 16, 1657–1664. doi: 10.1038/mt.2008.163
Peiris-Pagès, M. (2012). The role of VEGF 165b in pathophysiology. Cell Adh. Migr. 6, 561–568. doi: 10.4161/cam.22439
Perrin, R. M., Konopatskaya, O., Qiu, Y., Harper, S., Bates, D. O., and Churchill, A. J. (2005). Diabetic retinopathy is associated with a switch in splicing from anti- to pro-angiogenic isoforms of vascular endothelial growth factor. Diabetologia 48, 2422–2427. doi: 10.1007/s00125-005-1951-8
Pfitzner, E., Kliem, S., Baus, D., and Litterst, C. M. (2004). The role of STATs in inflammation and inflammatory diseases. Curr. Pharm. Des. 10, 2839–2850. doi: 10.2174/1381612043383638
Pruszko, M., Milano, E., Forcato, M., Donzelli, S., Ganci, F., Di Agostino, S., et al. (2017). The mutant p53-ID4 complex controls VEGFA isoforms by recruiting lncRNA MALAT1. EMBO Rep. 18, 1331–1351. doi: 10.15252/embr.201643370
Pullmann, R. Jr., and Rabb, H. (2014). HuR and other turnover- and translation-regulatory RNA-binding proteins: implications for the kidney. Am. J. Physiol. Renal Physiol. 306, F569–F576. doi: 10.1152/ajprenal.00270.2013
Puthanveetil, P., Chen, S., Feng, B., Gautam, A., and Chakrabarti, S. (2015). Long non-coding RNA MALAT1 regulates hyperglycaemia induced inflammatory process in the endothelial cells. J. Cell. Mol. Med. 19, 1418–1425. doi: 10.1111/jcmm.12576
Puthanveetil, P., Wan, A., and Rodrigues, B. (2013). FoxO1 is crucial for sustaining cardiomyocyte metabolism and cell survival. Cardiovasc. Res. 97, 393–403. doi: 10.1093/cvr/cvs426
Puthanveetil, P., Zhang, D., Wang, Y., Wang, F., Wan, A., Abrahani, A., et al. (2012). Diabetes triggers a PARP1 mediated death pathway in the heart through participation of FoxO1. J. Mol. Cell. Cardiol. 53, 677–686. doi: 10.1016/j.yjmcc.2012.08.013
Qin, G., Kishore, R., Dolan, C. M., Silver, M., Wecker, A., Luedemann, C. N., et al. (2006). Cell cycle regulator E2F1 modulates angiogenesis via p53-dependent transcriptional control of VEGF. Proc. Natl. Acad. Sci. U.S.A. 103, 11015–11020. doi: 10.1073/pnas.0509533103
Qiu, G. Z., Tian, W., Fu, H. T., Li, C. P., and Liu, B. (2016). Long noncoding RNA-MEG3 is involved in diabetes mellitus-related microvascular dysfunction. Biochem. Biophys. Res. Commun. 471, 135–141. doi: 10.1016/j.bbrc.2016.01.164
Rawshani, A., Rawshani, A., and Gudbjornsdottir, S. (2017). Mortality and cardiovascular disease in Type 1 and Type 2 diabetes. N. Engl. J. Med. 377, 300–301. doi: 10.1056/NEJMc1706292
Rhee, W. J., Ni, C. W., Zheng, Z., Chang, K., Jo, H., and Bao, G. (2010). HuR regulates the expression of stress-sensitive genes and mediates inflammatory response in human umbilical vein endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 107, 6858–6863. doi: 10.1073/pnas.1000444107
Rother, K. I. (2007). Diabetes treatment–bridging the divide. N. Engl. J. Med. 356, 1499–1501. doi: 10.1056/NEJMp078030
Roy, B., and Jacobson, A. (2013). The intimate relationships of mRNA decay and translation. Trends Genet. 29, 691–699. doi: 10.1016/j.tig.2013.09.002
Sanduja, S., Blanco, F. F., and Dixon, D. A. (2011). The roles of TTP and BRF proteins in regulated mRNA decay. Wiley Interdiscip. Rev. RNA 2, 42–57. doi: 10.1002/wrna.28
Sauer, I., Schaljo, B., Vogl, C., Gattermeier, I., Kolbe, T., Muller, M., et al. (2006). Interferons limit inflammatory responses by induction of tristetraprolin. Blood 107, 4790–4797. doi: 10.1182/blood-2005-07-3058
Schneider, A. L., Kalyani, R. R., Golden, S., Stearns, S. C., Wruck, L., Yeh, H. C., et al. (2016). Diabetes and prediabetes and risk of hospitalization: the Atherosclerosis Risk in Communities (ARIC) Study. Diabetes Care 39, 772–779. doi: 10.2337/dc15-1335
Scott, L. J., Mohlke, K. L., Bonnycastle, L. L., Willer, C. J., Li, Y., Duren, W. L., et al. (2007). A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316, 1341–1345. doi: 10.1126/science.1142382
Sena, C. M., Nunes, E., Louro, T., Proenca, T., Fernandes, R., Boarder, M. R., et al. (2008). Effects of alpha-lipoic acid on endothelial function in aged diabetic and high-fat fed rats. Br. J. Pharmacol. 153, 894–906. doi: 10.1038/sj.bjp.0707474
Sena, C. M., Pereira, A. M., and Seica, R. (2013). Endothelial dysfunction - a major mediator of diabetic vascular disease. Biochim. Biophys. Acta 1832, 2216–2231. doi: 10.1016/j.bbadis.2013.08.006
Shan, K., Jiang, Q., Wang, X. Q., Wang, Y. N., Yang, H., Yao, M. D., et al. (2016). Role of long non-coding RNA-RNCR3 in atherosclerosis-related vascular dysfunction. Cell Death Dis. 7, e2248. doi: 10.1038/cddis.2016.145
Shan, K., Li, C. P., Liu, C., Liu, X., and Yan, B. (2017). RNCR3: A regulator of diabetes mellitus- related retinal microvascular dysfunction. Biochem. Biophys. Res. Commun. 482, 777–783. doi: 10.1016/j.bbrc.2016.11.110
Shi, J. X., Su, X., Xu, J., Zhang, W. Y., and Shi, Y. (2012). HuR post-transcriptionally regulates TNF-alpha-induced IL-6 expression in human pulmonary microvascular endothelial cells mainly via tristetraprolin. Respir. Physiol. Neurobiol. 181, 154–161. doi: 10.1016/j.resp.2012.02.011
Shi, Y., and Hu, F. B. (2014). The global implications of diabetes and cancer. Lancet 383, 1947–1948. doi: 10.1016/S0140-6736(14)60886-2
Shi, Y., and Vanhoutte, P. M. (2017). Macro- and microvascular endothelial dysfunction in diabetes. J. Diabetes 9, 434–449. doi: 10.1111/1753-0407.12521
Stenina, O. I. (2005). Regulation of vascular genes by glucose. Curr. Pharm. Des. 11, 2367–2381. doi: 10.2174/1381612054367283
Størling, J., and Pociot, F. (2017). Type 1 diabetes candidate genes linked to pancreatic islet cell inflammation and beta-cell apoptosis. Genes 8:72. doi: 10.3390/genes8020072
Strawbridge, R. J., Dupuis, J., Prokopenko, I., Barker, A., Ahlqvist, E., Rybin, D., et al. (2011). Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes 60, 2624–2634. doi: 10.2337/db11-0415
Stumvoll, M., Goldstein, B. J., and van Haeften, T. W. (2005). Type 2 diabetes: principles of pathogenesis and therapy. Lancet 365, 1333–1346. doi: 10.1016/S0140-6736(05)61032-X
Taylor, G. A., Carballo, E., Lee, D. M., Lai, W. S., Thompson, M. J., Patel, D. D., et al. (1996). A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 4, 445–454. doi: 10.1016/S1074-7613(00)80411-2
Thomas, A. A., Feng, B., and Chakrabarti, S. (2017). ANRIL: A Regulator of VEGF in Diabetic Retinopathy. Invest. Ophthalmol. Vis. Sci. 58, 470–480. doi: 10.1167/iovs.16-20569
Tiedje, C., Ronkina, N., Tehrani, M., Dhamija, S., Laass, K., Holtmann, H., et al. (2012). The p38/MK2-driven exchange between tristetraprolin and HuR regulates AU-rich element-dependent translation. PLoS Genet. 8:e1002977. doi: 10.1371/journal.pgen.1002977
Tripathi, V., Ellis, J. D., Shen, Z., Song, D. Y., Pan, Q., Watt, A. T., et al. (2010). The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 39, 925–938. doi: 10.1016/j.molcel.2010.08.011
Tsuiji, H., Yoshimoto, R., Hasegawa, Y., Furuno, M., Yoshida, M., and Nakagawa, S. (2011). Competition between a noncoding exon and introns: Gomafu contains tandem UACUAAC repeats and associates with splicing factor-1. Genes Cells 16, 479–490. doi: 10.1111/j.1365-2443.2011.01502.x
Uehara, H., Cho, Y., Simonis, J., Cahoon, J., Archer, B., Luo, L., et al. (2013). Dual suppression of hemangiogenesis and lymphangiogenesis by splice-shifting morpholinos targeting vascular endothelial growth factor receptor 2 (KDR). FASEB J. 27, 76–85. doi: 10.1096/fj.12-213835
Vallance, P., and Chan, N. (2001). Endothelial function and nitric oxide: clinical relevance. Heart 85, 342–350. doi: 10.1136/heart.85.3.342
van der Veer, E. P., de Bruin, R. G., Kraaijeveld, A. O., de Vries, M. R., Bot, I., Pera, T., et al. (2013). Quaking, an RNA-binding protein, is a critical regulator of vascular smooth muscle cell phenotype. Circ. Res. 113, 1065–1075. doi: 10.1161/CIRCRESAHA.113.301302
Vanderweyde, T., Youmans, K., Liu-Yesucevitz, L., and Wolozin, B. (2013). Role of stress granules and RNA-binding proteins in neurodegeneration: a mini-review. Gerontology 59, 524–533. doi: 10.1159/000354170
Ventrice, P., Leporini, C., Aloe, J. F., Greco, E., Leuzzi, G., Marrazzo, G., et al. (2013). Anti-vascular endothelial growth factor drugs safety and efficacy in ophthalmic diseases. J. Pharmacol. Pharmacother. 4(Suppl. 1), S38–S42. doi: 10.4103/0976-500X.120947
Vernet, C., and Artzt, K. (1997). STAR, a gene family involved in signal transduction and activation of RNA. Trends Genet. 13, 479–484. doi: 10.1016/S0168-9525(97)01269-9
Whelan, J. T., Hollis, S. E., Cha, D. S., Asch, A. S., and Lee, M. H. (2012). Post-transcriptional regulation of the Ras-ERK/MAPK signaling pathway. J. Cell. Physiol. 227, 1235–1241. doi: 10.1002/jcp.22899
Xin, H., Deng, K., and Fu, M. (2014). Post-transcriptional gene regulation by RNA-binding proteins in vascular endothelial dysfunction. Sci. China Life Sci. 57, 836–844. doi: 10.1007/s11427-014-4703-5
Yan, B., Tao, Z. F., Li, X. M., Zhang, H., Yao, J., and Jiang, Q. (2014). Aberrant expression of long noncoding RNAs in early diabetic retinopathy. Invest. Ophthalmol. Vis. Sci. 55, 941–951. doi: 10.1167/iovs.13-13221
Yan, B., Yao, J., Liu, J. Y., Li, X. M., Wang, X. Q., Li, Y. J., et al. (2015). lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circ. Res. 116, 1143–1156. doi: 10.1161/CIRCRESAHA.116.305510
Yang, L., Froberg, J. E., and Lee, J. T. (2014). Long noncoding RNAs: fresh perspectives into the RNA world. Trends Biochem. Sci. 39, 35–43. doi: 10.1016/j.tibs.2013.10.002
Yap, K. L., Li, S., Munoz-Cabello, A. M., Raguz, S., Zeng, L., Mujtaba, S., et al. (2010). Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 38, 662–674. doi: 10.1016/j.molcel.2010.03.021
Zhang, B., Wang, D., Ji, T. F., Shi, L., and Yu, J. L. (2017). Overexpression of lncRNA ANRIL up-regulates VEGF expression and promotes angiogenesis of diabetes mellitus combined with cerebral infarction by activating NF-kappaB signaling pathway in a rat model. Oncotarget 8, 17347–17359. doi: 10.18632/oncotarget.14468
Zhang, H., Taylor, W. R., Joseph, G., Caracciolo, V., Gonzales, D. M., Sidell, N., et al. (2013). mRNA-binding protein ZFP36 is expressed in atherosclerotic lesions and reduces inflammation in aortic endothelial cells. Arterioscler. Thromb. Vasc. Biol. 33, 1212–1220. doi: 10.1161/ATVBAHA.113.301496
Zhang, J., Chen, M., Chen, J., Lin, S., Cai, D., Chen, C., et al. (2017). Long non-coding RNA MIAT acts as a biomarker in diabetic retinopathy by absorbing miR-29b and regulating cell apoptosis. Biosci. Rep. 37:BSR20170036. doi: 10.1042/BSR20170036
Zhang, J., Modi, Y., Yarovinsky, T., Yu, J., Collinge, M., Kyriakides, T., et al. (2012). Macrophage beta2 integrin-mediated, HuR-dependent stabilization of angiogenic factor-encoding mRNAs in inflammatory angiogenesis. Am. J. Pathol. 180, 1751–1760. doi: 10.1016/j.ajpath.2011.12.025
Zhou, X., Han, X., Wittfeldt, A., Sun, J., Liu, C., Wang, X., et al. (2016). Long non-coding RNA ANRIL regulates inflammatory responses as a novel component of NF-kappaB pathway. RNA Biol. 13, 98–108. doi: 10.1080/15476286.2015.1122164
Keywords: RNA binding protein, diabetes, vascular endothelia, dysfunction, therapy
Citation: Yang C, Kelaini S, Caines R and Margariti A (2018) RBPs Play Important Roles in Vascular Endothelial Dysfunction Under Diabetic Conditions. Front. Physiol. 9:1310. doi: 10.3389/fphys.2018.01310
Received: 14 July 2018; Accepted: 30 August 2018;
Published: 20 September 2018.
Edited by:
Lars Maegdefessel, Karolinska Institutet (KI), SwedenReviewed by:
Suowen Xu, University of Rochester, United StatesMichael Autieri, Temple University, United States
Copyright © 2018 Yang, Kelaini, Caines and Margariti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andriana Margariti, a.margariti@qub.ac.uk