
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 12 October 2018
Sec. Vascular Physiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.01236
This article is part of the Research TopicAxonal Guidance Molecules in Mature Vascular BiologyView all 6 articles
 Shuhong Hu1,2*
Shuhong Hu1,2* Li Zhu1,2*
Li Zhu1,2*Semaphorins are a large family of secreted, transmembrane, or GPI-anchored proteins initially identified as axon guidance cues signaling through their receptors, neuropilins, and plexins. Emerging evidence suggests that beyond the guidance, they also function in a broad spectrum of pathophysiological conditions, including atherosclerosis, a vascular inflammatory disease. Particular semaphorin members have been demonstrated to participate in atherosclerosis via eliciting endothelial dysfunction, leukocyte infiltration, monocyte-macrophage retention, platelet hyperreactivity, and neovascularization. In this review, we focus on the role of those semaphorin family members in the development of atherosclerosis and highlight the mechanistic relevance of semaphorins to atherogenesis.
Atherosclerosis is a chronic inflammatory process initiated by endothelial dysfunction and the secretion of cytokines and chemokines, and it allows the subendothelial accumulation of low-density lipoprotein (LDL) (Gibson et al., 2018). The deposited LDL is then oxidized and becomes oxidized LDL (ox-LDL) that potently stimulates cytokine release from intimal endothelial cells and facilitates leukocyte adhesion and transmigration (Singh et al., 2002; Paoletti et al., 2004). The infiltrated monocytes differentiate into macrophages that uptake oxLDL and other forms of modified LDL (Rader and Pure, 2005), subsequently forming foam cells that are the constitutes of fatty streaks (Siegel-Axel et al., 2008). Accumulating immune cells including macrophages and T cells fuel immune response and inflammation, promoting the transition from a fatty streak to a complex atherosclerotic plaque (Jonasson et al., 1986; Samson et al., 2012). The ultimate stage of atherosclerotic disease is hallmarked by plaque rupture and thrombus formation that may lead to acute fatal coronary syndrome (ACS), myocardial infarction (MI), and stroke. To date, a vast number of proteins have been shown to participate in the onset and progression of atherosclerosis, including those from plasma, blood cells, and the vascular wall, that fall into the classical class of molecules that have established current understanding of atherogenesis (Geovanini and Libby, 2018). However, additional molecules are being identified, shedding new light on the modulation and control of atherosclerosis.
Recently, emerging evidence suggests the semaphorin family in atherosclerosis plays a role. Semaphorins are a large family of transmembrane (including GPI-anchored) or secreted proteins that were originally identified as indispensable regulators of neuron-axonal guidance (Kolodkin et al., 1993). Their molecular structure consists of a highly conserved sema domain, a plexin–semaphorin–integrin (PSI) domain, and distinct protein domains that further define semaphorins, including immunoglobulin-like (Ig), thrombospondin, and basic C-terminal domains (Zhou et al., 2008). More than 20 semaphorin family members have been identified that fall into eight classes (Semaphorin Nomenclature Committee, 1999; Wannemacher et al., 2011). Classes 1 and 2 belong to invertebrates, and classes 3 through 7 belong to vertebrates while class 8 is viral-encoded. In addition, classes 1 and 4–7 are membrane-associated, whereas those in classes 2, 3, and 8 are in a secreted form (Capparuccia and Tamagnone, 2009; Figure 1). Two groups of proteins, including plexins and neuropilins (Npns or Nrps), have been identified as the primary semaphorin receptors (Tamagnone et al., 1999). It is worthy to note that nine plexins that also contain the extracellular sema domains were found in vertebrates and fall into classes A–D (Wannemacher et al., 2011). Several recent studies on semaphorin structure confirmed that each sema domain of a semaphorin homodimer binds to a plexin-sema domain to promote plexin dimerization for signal transduction (Janssen et al., 2010; Nogi et al., 2010; Wannemacher et al., 2011; Figure 1). Plexins are associated with different co-receptors in distinct tissues to allow semaphorins to exert pleiotropic functions (Alto and Terman, 2017). Although semaphorins were originally identified as axon guidance molecules required for axon pointing to proper targets (Kolodkin et al., 1993), cumulative findings indicate that they are indispensable in diverse physiological processes (Zhou et al., 2008), including cardiomyogenesis (Toyofuku and Kikutani, 2007; Toyofuku et al., 2008), tumor neovascularization, metastasis (Giordano et al., 2002; Neufeld and Kessler, 2008; Capparuccia and Tamagnone, 2009), osteoclastogenesis (Takegahara et al., 2006), angiogenesis (Serini et al., 2003; Gu et al., 2005; Toyofuku et al., 2007), and immunomodulation (Kikutani and Kumanogoh, 2003; Suzuki et al., 2008; Nishide and Kumanogoh, 2018). In this review, we focus on the role of semaphorins and their receptors in the setting of atherosclerosis development. Attention will be placed on several semaphorin family members for their role in semaphorin-receptor signaling during atherogenesis.
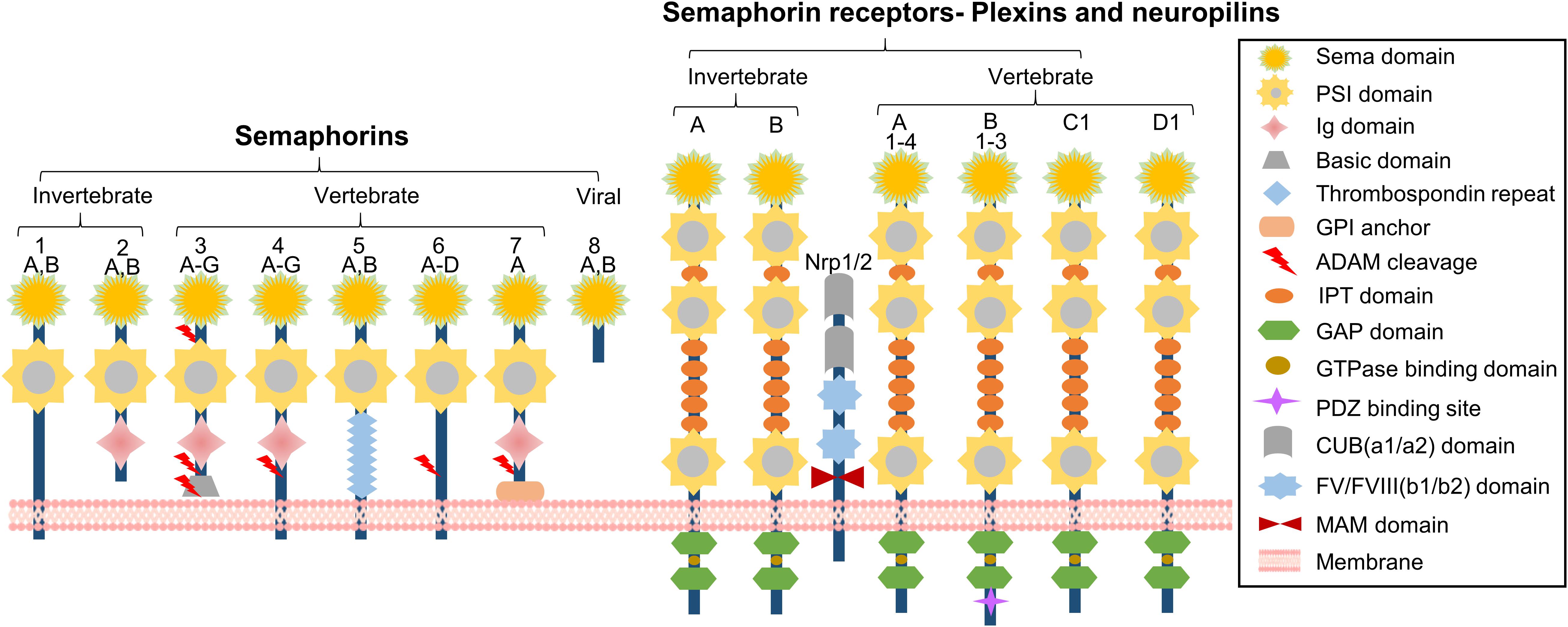
FIGURE 1. Schematic representation of semaphorins and semaphorin receptors. The semaphorin family can be divided into eight classes. Classes 1 and 2 are found in invertebrates and classes 3–7 belong to vertebrates. Classes 2 and 3, and the viral semaphorin 8 are secreted whereas classes 4–6 are transmembrane proteins. Class 7 is the only GPI-anchored protein. Each semaphorin consists of a large sema domain, a plexin-sema-integrin domain (PSI), immunoglobulin (Ig)-like domains, and thrombospondin repeats. Semaphorins signal through their receptors, plexins. Plexins A and B are found in invertebrates. Vertebrates have plexins A1–4, plexins B1–3, PlexinC1, and PlexinD1. Plexins contain a sema domain, a PSI domain, and an Ig-like, plexins, transcription factors (IPT) domain. Structurally, the cytoplasmic domain of the plexin contains two GTPase activating protein (GAP) domains, including one GTPase-binding domain and one PDZ domain (postsynaptic protein PSD-95/SAP90, the Drosophila septate junction protein Discs-large, and the tight junction protein ZO-1 domain) (B-type plexins only). In invertebrates, semaphorins 1 and 2 signal through Plexin A and Plexin B. In vertebrates, semaphorins 3, 5, and 6 signal via Plexin A, while semaphorin 3 requires a combination of neuropilins (Nrp1 or Nrp2) for signal transduction. Neuropilin is a transmembrane receptor composed of two complement-like (CUB) domains, two FV/FVIII clotting factor-like domains, one meprin-like MAM domain, and a short cytoplasmic tail.
There are seven members of secreted class 3 semaphorins, named Sema3A through Sema3G, which were found to express in the nervous system (Hashimoto et al., 2004; Hou et al., 2008), immune system (Ji et al., 2009), lung (Martin-Satue and Blanco, 1999), osteogenesis (Ryynanen et al., 2017), and cancer cells (Herman and Meadows, 2007; Karayan-Tapon et al., 2008). They function through their receptors, the neuropilins and plexins, forming complexes in which neuropilins serve as the ligand-binding moiety and plexins act as the signal transduction component (Sharma et al., 2012b). The extensive expression pattern of class 3 semaphorins suggests their other functions beyond the nervous system. Sema3A, first designated as collapsin-1, was found to regulate stimulation-induced growth cone slump (Luo et al., 1993) and repulsion of neuron axon growth in the nervous system (Kolodkin et al., 1997; Tamagnone and Comoglio, 2000; Dent et al., 2004). Nevertheless, emerging evidence indicates that Sema3A branches out beyond the nervous system, especially in the regulation of the immune system. Sema3A suppresses B- and T-cell proliferation and activation by inhibiting actin cytoskeleton reorganization and alleviating generation of proinflammatory cytokines (Lepelletier et al., 2006; Vadasz and Toubi, 2014). In addition, it induces apoptosis of endothelial cells (Guttmann-Raviv et al., 2007) and monocyte-derived macrophages through its receptors containing neuropilin and PlexinA (Moretti et al., 2008; Vadasz et al., 2013). Furthermore, Sema3A functions as an inhibitor of integrin function (Serini et al., 2003) and tube formation in endothelial cells (Guttmann-Raviv et al., 2007).
Recently, van Gils et al. (2013) revealed that endothelial Sema3A modulates leukocyte rolling, adhesion, and transmigration into the subendothelial wall during atherogenesis. They examined Sema3A expression in the inner and outer curvatures of the aortic arch of low-density-lipoprotein receptor-/- (LDLR-/-) mice following a western diet for 2 weeks and showed that Sema3A is abundantly expressed by endothelial cells in the antiatherogenic greater curvature, but little was detected in the proatherogenic lesser curvature. In in vitro analysis, Sema3A expression was obviously downregulated in human coronary artery endothelial cells (HCAECs) exposed to oscillatory (atheroprone) flow, proatherogenic factors, and proinflammatory cytokines. Sema3A acts as a barrier to prevent monocyte migration into the arterial intima as leukocyte rolling and adhesion to the endothelium was increased when Sema3A was inhibited using blocking peptides or blocking its receptor neuropilin-1. These data imply that Sema3A functions as a negative regulator of monocyte invasion, a priming step in the process of atherogenesis, alleviating inflammation progression and atherosclerosis development (Figure 2). In addition, endothelial Sema3A was reported to bind platelets and suppress platelet activation by inhibiting αIIbβ3 integrin-dependent spreading and granule releasing (Kashiwagi et al., 2005). Whether its antiplatelet effect contributes to atherosclerosis development is not clear.
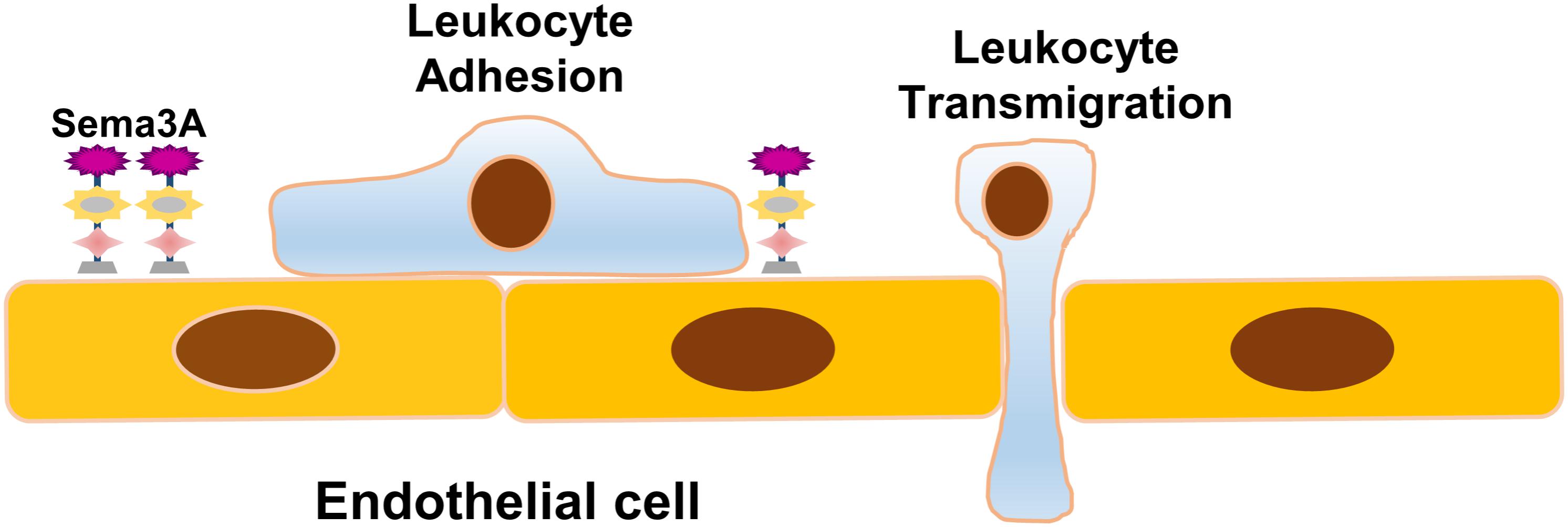
FIGURE 2. Schematic representation of the role of Sema3A in atherosclerosis. Sema3A functions as a natural barrier to prevent monocyte migration into the arterial intima under basal conditions. Sema3A is abundantly expressed by endothelial cells in the atheroresistent greater curvature while little is expressed in the lesser aortic curvature. The downregulation of Sema3A in the d-flow-injured endothelial cells leads to inflammatory leukocyte trafficking into the intima, an initial essential step in the process of atherogenesis, and accelerates inflammation and atherosclerosis development.
Semaphorin 3E (Sema3E), originally defined as M-SemaH, was identified in tumor cells and involved in embryonic development (Christensen et al., 1998). In the nervous system, Sema3E functions as a negative regulator of retinal ganglion cell axon growth and induces the collapse of retinal ganglion cell (RGC) axons via cGMP signaling (Steinbach et al., 2002). Further studies showed that the functions of Sema3E are associated with axon regeneration after injury (Sharma et al., 2012a; Bribian et al., 2014) and vascular development and remodeling (Gu et al., 2005; Kim et al., 2011; Oh and Gu, 2013). Sema3E suppresses endothelial cell motility and tube formation through the inhibition of VEGF-mediated Akt phosphorylation via PlexinD1 (Moriya et al., 2010; Aghajanian et al., 2014). Sema3E-PlexinD1 signaling in endothelial cells relies on the GTPase-activating protein (GAP) activity to modulate cytoskeleton reorganization and cellular migration and adhesion (Sakurai et al., 2010).
Accumulating evidence suggests that the migration and proliferation of vascular smooth muscle cells (VSMCs) are crucial processes in neointimal formation, while neointimal hyperplasia is a pivotal pathophysiological process that contributes to atherosclerosis (Dzau et al., 2002; Owens et al., 2004; Gomez and Owens, 2012). To examine the role of Sema3E in VSMC migration and proliferation during neointimal formation, Wu et al. (2017) found that abundant Sema3E was expressed in alpha-smooth muscle actin (α-SMA)-positive VSMCs of mouse and human arteries under normal condition, while its expression in VSMCs of atherosclerotic plaques was markedly deceased as compared with the normal aortic arteries. Moreover, they found that Sema3E inhibited VSMC migration and neointimal formation in a dose-dependent manner in vitro. Mechanistically, Sema3E inactivated Rap1-Akt signaling pathways to suppress VSMC migration and proliferation via the PlexinD1 receptor. Therefore, Sema3E plays a negative regulatory role in the process of neointimal hyperplasia (Figure 3A).
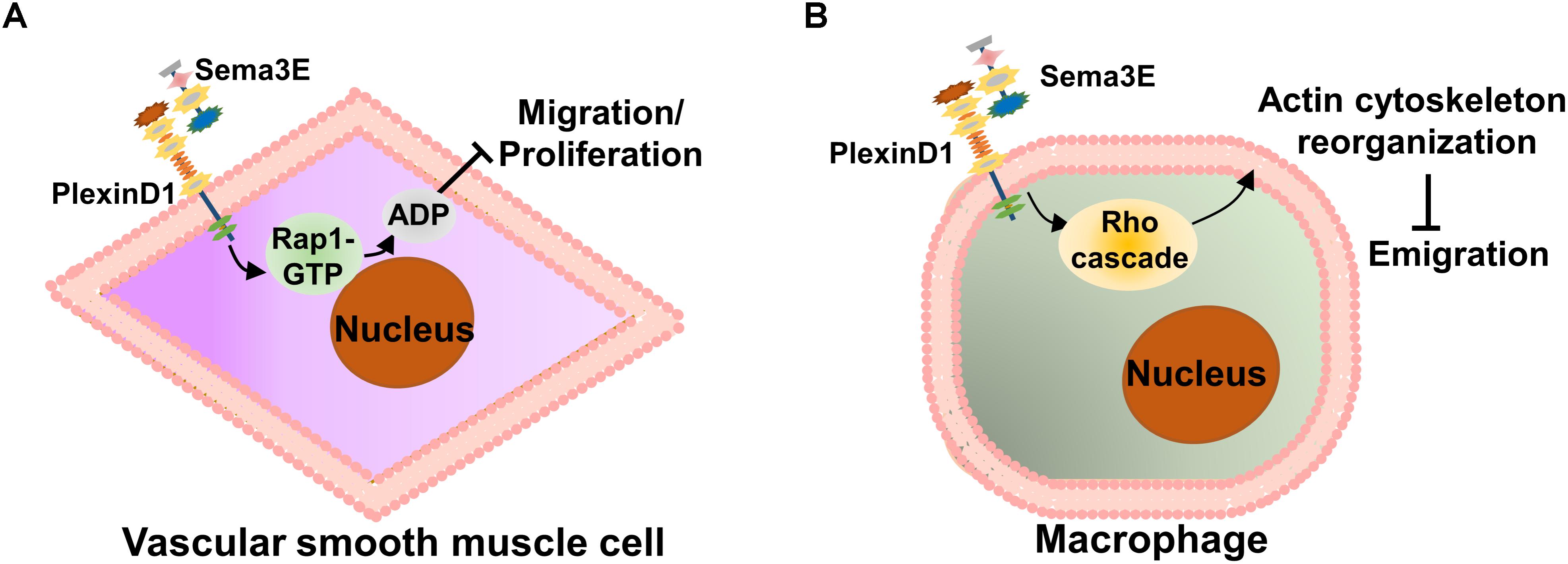
FIGURE 3. Schematic representation of the role of Sema3E in atherosclerosis. (A) In the early stage of atherosclerosis, Sema3E and its receptor PlexinD1 are remarkably decreased in atherosclerotic plaques. Sema3E inhibits VSMC migration and proliferation by inactivating Rap1-AKT signaling pathways via binding to PlexinD1 in the process of neointimal formation. (B) Sema3E and its receptor PlexinD1 are remarkably increased in monocyte-macrophages of advanced atherosclerotic plaques. Sema3E inhibits the directional emigration of macrophages by disrupting the Rho GTPase signaling cascade and actin cytoskeleton reorganization via binding to PlexinD1, which promotes monocyte-macrophage retention, exacerbating atherosclerosis.
In the later stage of atherosclerosis, as part of the resolution phase of acute inflammation, normally activated macrophages in the lesion emigrate from the site of local inflammation to the draining lymphatic vessels (Bellingan et al., 1996). However, unlike other inflammatory conditions, atherosclerotic cholesterol-laden macrophages (foam cells) persist in the arterial wall and the ability of emigration appears to be impaired (Llodrsa et al., 2004; Trogan et al., 2006). To date, the mechanisms by which these cells are retained in plaques remain poorly understood. Wanschel et al. (2013) investigated the dynamics of Sema3E expression in atherosclerosis using a model of atherosclerosis regression and identified Sema3E in atherosclerotic lesions, which are co-localized with infiltrated macrophages. Functionally, they also showed that Sema3E contributes to monocyte-macrophage invasion and accumulation in plaques. They further demonstrated that the decreased Sema3E mRNA level was accompanied by a downregulation of plaque macrophage accumulation and an increase in the proportion of M2 subtype macrophages. Besides, they showed that Sema3E regulates monocyte-macrophage retention and that its expression reduces along with plaque regression (Wanschel et al., 2013). Using bone marrow-derived macrophages (BMDM) in vitro, Sema3E expression was further investigated in macrophage polarization, which showed that Sema3E was highly upregulated in proinflammatory M1 subtype macrophages and that Sema3E inhibits the directional emigration of macrophages by disrupting the Rho GTPase signaling cascade, actin cytoskeleton reorganization, and polarization via binding to PlexinD1 (Wanschel et al., 2013). Thus, Sema3E appears to function as an inducer of monocyte-macrophage infiltration and an inhibitor of macrophage emigration and thus may accelerate atherosclerosis by promoting monocyte-macrophage retention and long-term inflammation (Figure 3B).
Semaphorin 4D (Sema4D/CD100) is a 150-kD transmembrane protein that belongs to the semaphorin class 4. It was first perceived as an “immune system semaphorin” (Wang et al., 2001) and indispensably takes part in the immune responses (Kumanogoh and Kikutani, 2003). Two types of receptors with distinct binding affinities have been identified for Sema4D. PlexinB1 is a receptor that has been shown to have a relatively high affinity for Sema4D (Maestrini et al., 1996; Tamagnone et al., 1999), while CD72, expressed at prominent levels in lymphocytes, is a low-affinity receptor for Sema4D. The interaction between Sema4D and its receptor CD72 is critical for immune regulation (Chapoval et al., 2017), including lymphocyte activation, production of antibody, and autoimmune diseases (Wang et al., 2001).
Sema4D is the first semaphorin family member that was shown to participate in the development of atherosclerosis (Zhu et al., 2009). This finding was largely based on the previous observations that Sema4D promotes platelet activation in vitro and accelerates thrombus formation and growth (Zhu et al., 2007; Wannemacher et al., 2010). Using Sema4D-deficient LDLR-/- mice, Zhu et al. (2007, 2009) investigated whether eliminating Sema4D-dependent events could provide a means of platelet hyperactivity reduction in the setting of dyslipidemia. They found that the loss of Sema4D expression ameliorates the effects of dyslipidemia on the platelet function in vivo and ex vivo, and reduces atherosclerotic lesions in hyperlipidemic mice on a high-fat, high-cholesterol diet for 3 or 6 months (Zhu et al., 2009) (Figure 4A). Platelet is not the unique source of Sema4D expression as T cells, B cells, and monocytes/microphages have been reported to express Sema4D (Bougeret et al., 1992; Hall et al., 1996). Luque et al. (2013, 2015) showed that Sema4D is expressed in plaque macrophages and foam cells and mediates monocyte-endothelial cell adhesion by interacting with its receptors, PlexinB2 and PlexinB1, implying that non-platelet Sema4D may participate in atherogenesis as well. However, Zhu et al. (2007, 2009) did not observe the reduction in the infiltration of Sema4D-expressing cells, including T cells, B cells, and monocytes/macrophages in atherosclerotic lesions when investigating whether the reduction of hyperlipidemia is by the global absence of Sema4D. Whether the role of Sema4D in the platelet function is directly related to its ability to enhance atherogenesis needs to be examined by a megakaryocyte-selective deletion of Sema4D.
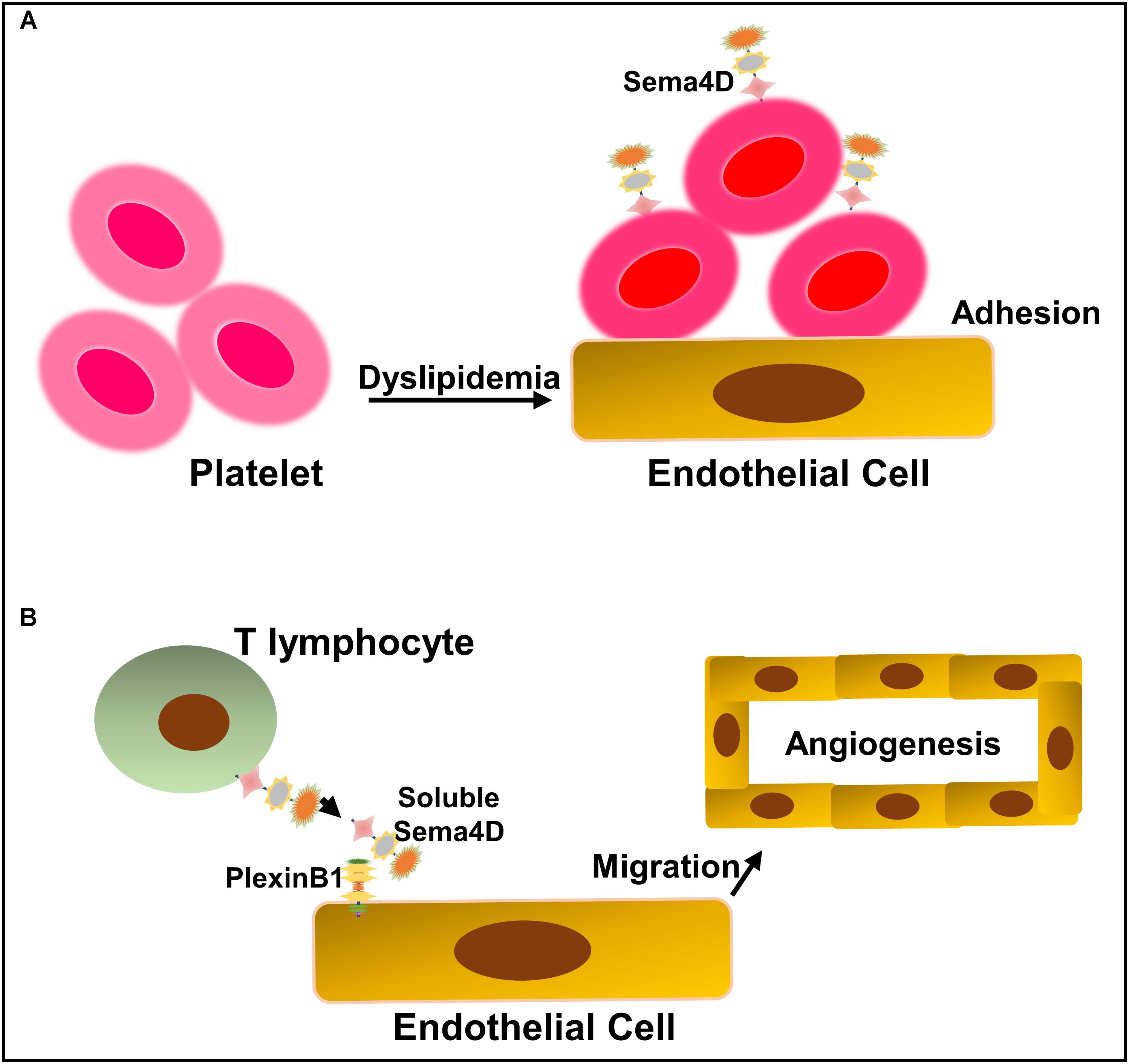
FIGURE 4. Schematic representation of Sema4D and its receptor PlexinB1 in the development of atherogenesis. (A) Dyslipidemia upregulates platelet sensitivity, making them more likely to become activated in response to vascular injury. Sema4D enhances platelet hyperactivity, which may lead to platelet adhesion and transmigration into the intima, thus accelerating the development of atherosclerosis. (B) Sema4D expresses on the surface of invasive lymphocytes in atherosclerotic lesions and interacts with PlexinB1 on the endothelial cells, resulting in endothelial cell activation, macrophage infiltration, and intraplaque angiogenesis.
The neovasculature originates from the adventitia to nourish blood vessels and the thickened atherosclerotic intima (Kumamoto et al., 1995; Moreno et al., 2006). Neovascularization in atherosclerotic plaques is thought to exacerbate atherosclerosis by fueling plaques with metalloproteases from blood, resulting in plaque rupture and thrombosis (Jonsson-Rylander et al., 2005). Therefore, neovascularization is considered to be one of the major causes of atherosclerotic plaque growth and destabilization (Barger et al., 1984). Subsequent to the role of Sema4D in enhancing platelet hyperreactivity in atherosclerosis, Yukawa et al. (2010) reported that it accelerates atherosclerosis by facilitating intimal neovascularization and monocyte-macrophage infiltration in apolipoprotein E (ApoE)-deficient mice. Furthermore, they found that Sema4D was expressed in the invasive lymphocytes of atherosclerotic lesions and that the degree of plaque neovascularization in Sema4D-/- ApoE-/- mice was significantly reduced. They concluded that Sema4D plays an integral role in the development of atherosclerosis by promoting neovascularization of the intima, facilitating macrophage infiltration in atherosclerotic plaques.
The mechanism for the role of Sema4D in angiogenesis has been well studied. It is not likely to be mediated by upregulating VEGF or angiogenin (Conrotto et al., 2005). A study on tumor-induced angiogenesis showed that Sema4D may function as a direct inducer of endothelial cell migration to promote neovascularization instead of promoting endothelial cell growth like VEGF. It has been found that angiogenesis has a spatial preference for atherosclerosis (Moulton, 2001; Moulton et al., 2003). Therefore, unlike VEGF, Sema4D may regulate neovascularization in atherosclerotic plaques by modulating local endothelial cell migration. In-depth studies showed that Sema4D exerts proangiogenic efficacy by binding to PlexinB1 that is expressed on the surface of endothelial cells. Instead of activating the Rho kinase pathway, the binding of Sema4D to PlexinBl activates the Met receptor for signal transduction (Basile et al., 2004; Conrotto et al., 2005). Interestingly, R-Ras, whose activity is downregulated during the signal transduction of Sema4D to PlexinBl, also functions to inhibit intimal growth and tumor angiogenesis (Komatsu and Ruoslahti, 2005). Similarly, studies conducted by Yukawa et al. (2010) indicated that cleaved Sema4D from infiltrated T lymphocytes may promote atherosclerosis by inducing the migration of regional endothelial cells to form new blood vessels, causing macrophage infiltration and atherosclerosis development (Figure 4B).
Semaphorin 7A (Sema7A) is the only membrane-associated glycosylphosphatidylinositol (GPI)-anchored semaphorin (Xu et al., 1998) with an N-terminal seven-bladed β-propeller sema domain, a plexin-semaphorin-integrin domain, an immunoglobulin-like domain, and a C-terminal GPI-anchoring domain (Lange et al., 1998; Liu et al., 2010). The sema domain contains an RGD motif commonly found in integrin-binding proteins (Pasterkamp et al., 2003). The expression of Sema7A and its known membrane receptors, integrin β1 and PlexinC1, were found in neurons (Pasterkamp et al., 2007), endothelial cells (Morote-Garcia et al., 2012; Hu et al., 2018), platelets (Jaimes et al., 2012), monocytes (Holmes et al., 2002), T cells (Czopik et al., 2006; Suzuki et al., 2007), dendritic cells (DCs) (Scott et al., 2008), lung fibroblasts (Esnault et al., 2017), and cancer cells (Garcia-Areas et al., 2013, 2014, 2017). The expression and binding properties of Sema7A suggest that it could play an important role in both the adult nervous system and immune function modulation (Xu et al., 1998). It has been reported that Sema7A complexes with the semaphorin-binding module of PlexinC1. Two PlexinC1 molecules are symmetrically bridged by Sema7A dimers, in which the Sema7A and PlexinC1 β propellers interact (Liu et al., 2010).
The first evidence for Sema7A in the nervous system was provided by the work on axon outgrowth in the olfactory system. Sema7A promotes the outgrowth of olfactory bulb axons and is required for the development of the lateral olfactory tract that carries olfactory bulb axons (Pasterkamp et al., 2003). Increasing evidence suggests that Sema7A plays important roles beyond the nervous system. Sema7A binding to PlexinC1 mediates tumor growth (Scott et al., 2009), while Sema7A binding to integrin β1 initiates T-lymphocyte-mediated inflammatory-immune response (Suzuki et al., 2007). More recently, Sema7A is implied in endothelial homeostasis. When exposed to hypoxia, Sema7A expression is significantly upregulated in vascular endothelial cells and promotes neutrophil adhesion and transmigration to the subendothelial layer (Morote-Garcia et al., 2012). Besides, Sema7A was reported to stimulate monocyte chemotaxis and cytokine production. Suzuki et al. (2007) showed that Sema7A, which is highly upregulated on activated T lymphocytes, irritates monocytes and macrophages to produce inflammatory cytokines via integrin α1β1.
Given the wide expression of Sema7A and its receptors in blood cells (Garcia-Areas et al., 2013) and its well-known function in immunity and inflammation, Hu et al. (2018) recently hypothesized that Sema7A may be an inflammatory responsive protein in the endothelial cells and examined the role of Sema7A in atherogenesis using ApoE-/- mice. They showed that Sema7A deficiency attenuates plaque formation in ApoE-/- mice on a high-fat diet, with reduced accumulation of macrophages in the plaques (Hu et al., 2018). Interestingly, the reduction in the plaque size was primarily in the aortic arch exposed to d-flow. Further studies showed that Sema7A is upregulated in the lesser curvature of mouse aortic arch endothelium, carotid artery exposed to d-flow in mice subjected to partial carotid artery ligation (PCL), and human umbilical venous endothelial cells (HUVECs) under oscillatory shear stress, potentially associated with the inhibition of cAMP response element-binding protein (CREB) signaling. Functionally, Sema7A deficiency remarkably reduced leukocyte rolling and adhesion on TNF-α-primed endothelium and leukocyte–endothelium interaction. Mechanistically, Sema7A overexpression enhances intercellular cell adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) expression and THP-1 adhesion to HUVECs in a β1 integrin-dependent manner that involves its downstream FAK/MAPK/NF-κB signaling pathway. They concluded that Sema7A upregulation by d-flow mediates endothelial dysfunction and atherosclerosis in a β1 integrin-dependent manner (Figure 5).
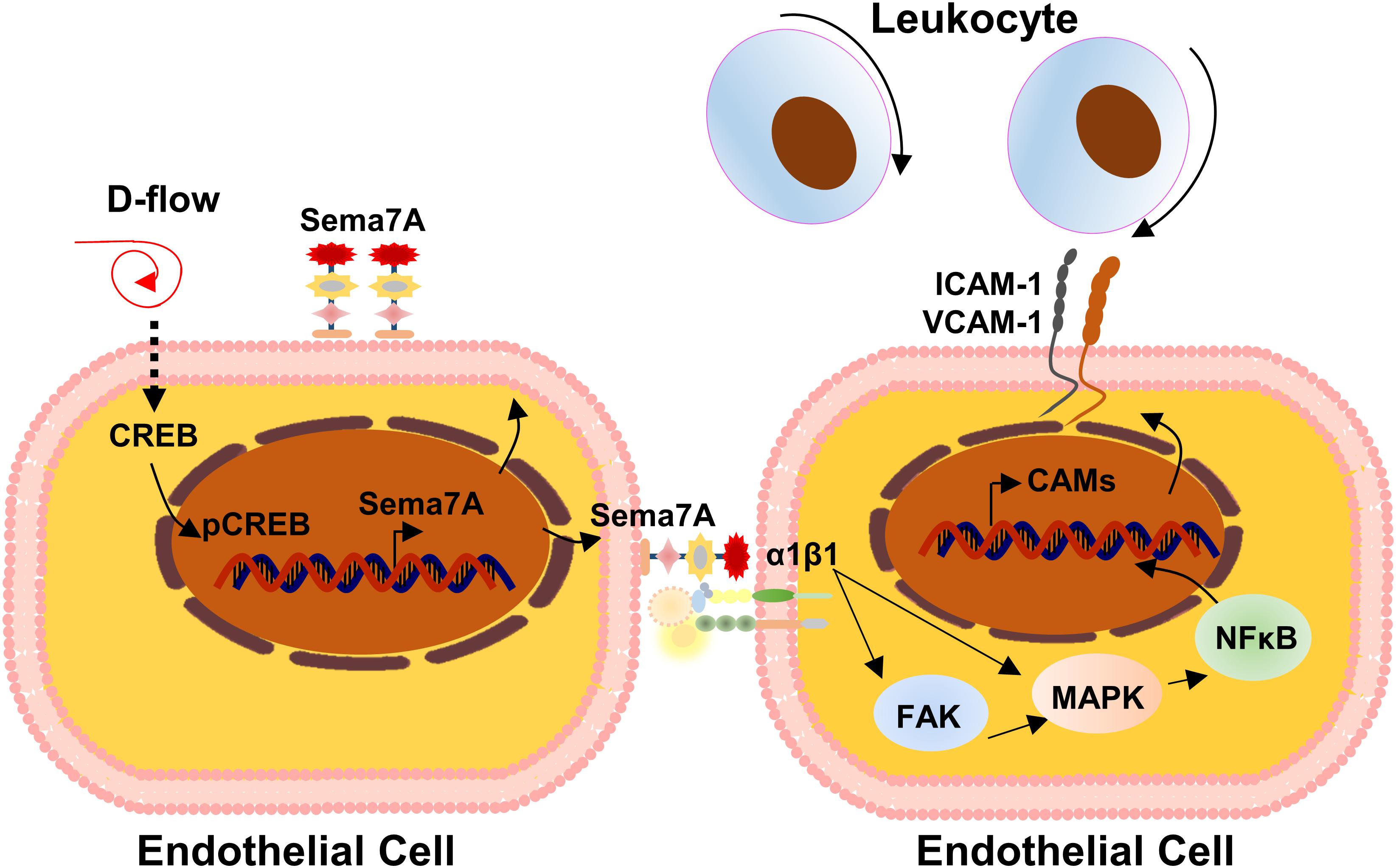
FIGURE 5. Mechanistic diagram of Sema7A in atherosclerosis. Endothelial Sema7A is upregulated in response to d-flow in the lesser curvature of mouse aortic arch, which is potentially regulated by the inhibition of cAMP response element-binding protein (CREB) signaling. Sema7A promotes the expression of adhesion molecule ICAM-1/VCAM-1 via NF-κB activation and leukocyte recruitment through integrin β1, accelerating atherosclerosis.
In addition to Sema3A, Sema3E, Sema4D, and Sema7A that have been extensively described for their role in the development of atherosclerosis, other members of the semaphorin family are potentially involved in atherogenesis due to their role in endothelial activation and dysfunction, neovascularization, and immune response. Semaphorin 6A (Sema6A), a transmembrane semaphorin protein expressed in endothelial cells, sustains the homeostasis of endothelial cells by regulating the vascular endothelial growth factor receptor 2 (VEGFR2) expression and signaling (Segarra et al., 2012). Segarra et al. (2012) identified Sema6A as a crucial regulator of vascular development and potential therapeutic target for ocular pathologies by contributing to the formation and remodeling of hyaloid vessels, indicating that Sema6A may be involved in angiogenesis and vessel remodeling that play indispensable roles in advanced plaque development.
Atherosclerosis is the consequence of a chronic inflammatory response in the arterial wall and the activation of the innate immune system has been recognized to be fundamentally involved in atherogenesis (Chavez-Sanchez et al., 2014). More recently, evidence has been provided that adaptive immunity is also involved in the initiation and progression of atherosclerosis (Luchtefeld et al., 2010). In particular, Sema4A has been investigated extensively. Expressed on dendritic cells and B-lymphocytes, Sema4A increases the activity of T lymphocytes (Suzuki and Kumanogoh, 2006), including cell activation and differentiation, and regulates the generation of T lymphocytes both in vitro and in vivo (Kumanogoh et al., 2002). However, there is no report on whether Sema4A participates in atherosclerosis. Like Sema4A, Sema4D, Sema3A, and Sema7A, reported to participate in atherosclerosis, are also widely investigated in immune cells. Sema4D has been shown to be critical for B-lymphocyte (Kuklina et al., 2017) and dendritic cell activation (Wang et al., 2001; Suzuki et al., 2003). Sema3A is highly expressed on many immune cells and responsible for T-cell and dendritic cell activation, inhibiting T-cell proliferation and proinflammatory cytokines production (Lepelletier et al., 2006; Vadasz and Toubi, 2014). Suzuki et al. (2007) reported that Sema7A, abundantly expressed on activated T lymphocytes, may potently stimulate monocyte-macrophages at the immunological synapse where Sema7A protein accumulates at the contact site between T lymphocytes and macrophages. Therefore, semaphorins in the immune system may provide a link between immune response and atherosclerosis. How such mechanisms of semaphorins would be started up in response to inflammatory injury in the context of atherosclerosis remains to be investigated.
Semaphorins, netrins, slits, and ephrins make up the four families of neuronal guidance cues that regulate neuronal growth and migration via multiple signaling pathways. Growing evidence suggests that members from other families participate in the regulation of atherosclerosis. Similar to Sema3A, netrin-1 was found to be involved in the initiation of atherosclerosis. Netrin-1 was detected by immunostaining in endothelial cells of the greater curvature but was significantly downregulated in endothelial cells at the lesser curvature of the aortic arch. In vitro, netrin-1 suppresses leukocyte recruitment by inhibiting the production of proatherogenic chemokines monocyte chemotactic protein-1 (MCP-1) and fractalkine. A consistent result was observed in the adhesion of monocytes to endothelial cells, suggesting that netrin-1 functions as a negative regulator of atherosclerosis (van Gils et al., 2013). Besides, recent studies showed that Slit2 inhibits monocyte adhesion on activated human endothelial cells and chemotaxis of monocytes to chemokines stromal cell-derived factor-1 (SDF-1) and MCP-1. In addition, the inhibition of monocyte recruitment by supplementing Slit2 was found to delay atheroprogression in mice (Mukovozov et al., 2015). On the other hand, ephrinB2 was demonstrated to be upregulated in endothelial cells by proatherogenic stimuli and remarkably promote monocyte recruitment (Poitz et al., 2015).
Growing evidence indicates that semaphorins have distinct biological activities, which are not only limited to the nervous system but also branching into other pathophysiological processes, including inflammation, immune response, angiogenesis, and especially atherogenesis, of which several mechanisms work in concert to modulate the initiation, progression, and regression of atherosclerosis. When exposed to disturbed blood flow or injured by inflammatory factors, endothelial cells upregulate Sema7A and downregulate Sema3A while VSMCs reduce Sema3E expression. The upregulation of Sema7A and downregulation of Sema3A and Sema3E function as proatherogenic factors to induce cell adhesion and transmigration via several mechanotransduction pathways. Meanwhile, inflammatory stimulation may induce platelet Sema4D activation, enhancing leukocyte adhesion and transmigration into the subendothelium to accelerate the development of atherosclerosis. In the plaques, Sema4D may shed from activated T lymphocytes and interact with PlexinB1 on endothelial cells, causing endothelial cell activation, leukocyte migration, and intraplaque angiogenesis. Subsequently, Sema3E interacts with PlexinD1 on macrophages to impair their emigration from advanced plaques, thereby promoting macrophage retention and chronic inflammation. Upon plaque rupture, platelet Sema4D binds to leukocytes and injured endothelial cells to exacerbate thrombosis and inflammation (Figure 6). Future investigation using the state-of-the-art technologies on the role of additional semaphorin family members in atherogenesis, their potential network regulation in atherosclerosis and their association with other proatherosclerotic or antiatherosclerotic factors will not only shed light on the full picture of the functional role of semaphorins in atherogenesis but also identify potential therapeutic targets and pharmacological interventions for atherosclerosis.
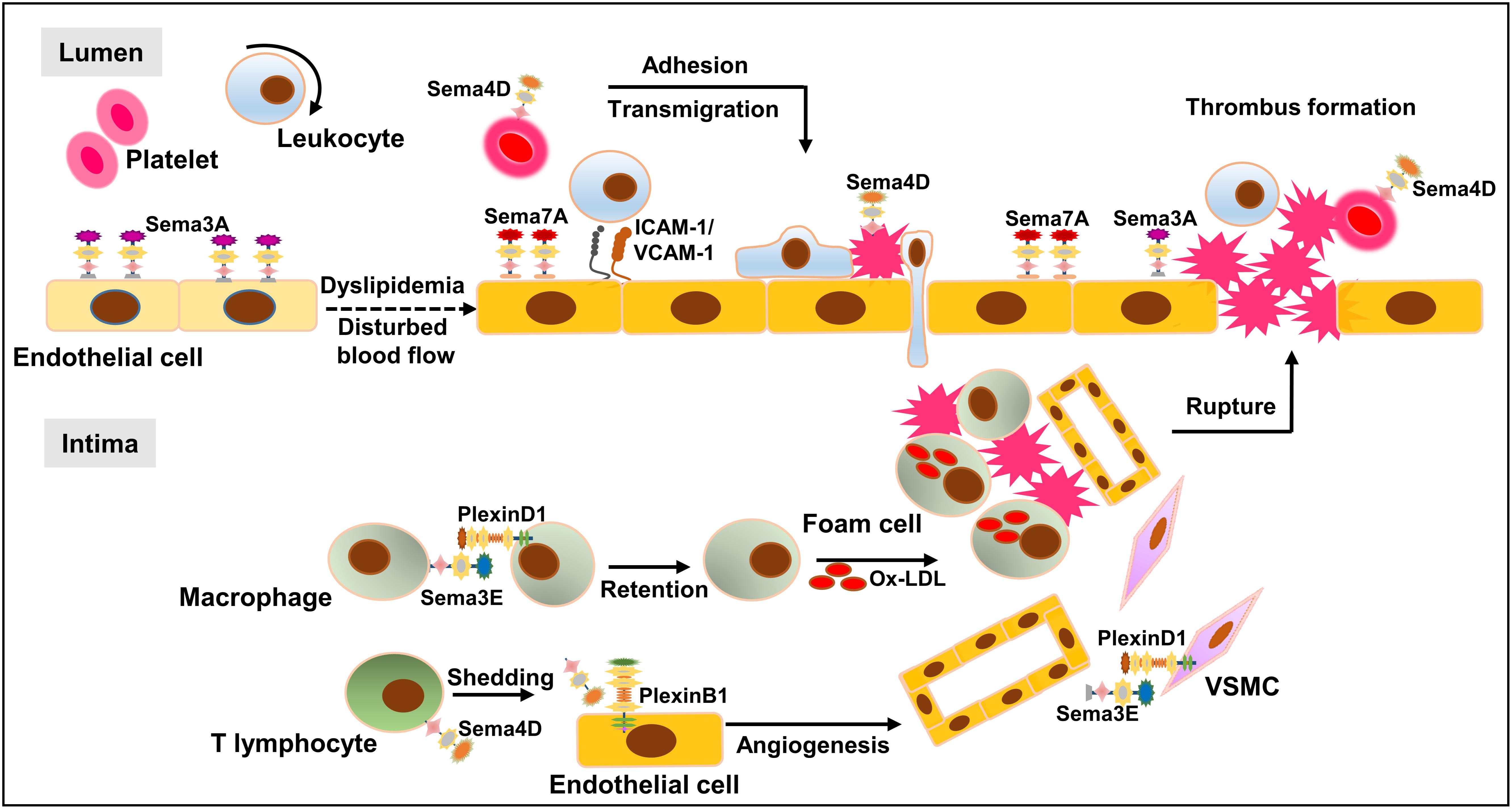
FIGURE 6. Network regulation of semaphorins in atherosclerosis. When exposed to oxLDL or injured by disturbed blood flow, endothelial cells undergo major phenotypic changes, e.g., upregulation of inflammatory cytokines, leading to leukocyte adhesion and infiltration into the intima wall. Accumulating immune cells including macrophages and T cells fuel the immune response, leading to the transition of a fatty streak to a complex atherosclerotic plaque. Plaque rupture and thrombus formation may lead to fatal cardio-cerebrovascular events. D-flow upregulates endothelial Sema7A and downregulates endothelial Sema3A and VSMC-Sema3E, leading to the infiltration of inflammatory leukocytes and VSMCs. Following cell infiltration and plaque formation, intraplaque Sema4D, shedding from activated T lymphocytes, interacts with PlexinB1 on endothelial cells, resulting in endothelial cell migration and intraplaque angiogenesis. In the advanced plaque, Sema3E interacts with PlexinD1 on macrophages to regulate macrophage emigration. Upon plaque rupture, platelet Sema4D binds leukocytes as well as injured endothelial cells to support thrombus formation, exacerbating the pathological process.
SH selected topics and wrote the review. LZ reviewed the manuscript and modified the content. All authors critically read and commented on the manuscript.
This work was supported by grants from the Natural Science Foundation of China (81620108001, 81870325, and 91739302), and the Priority Academic Program Development of Jiangsu Higher Education Institutions of China to LZ.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aghajanian, H., Choi, C., Ho, V. C., Gupta, M., Singh, M. K., and Epstein, J. A. (2014). Semaphorin 3d and semaphorin 3e direct endothelial motility through distinct molecular signaling pathways. J. Biol. Chem. 289, 17971–17979. doi: 10.1074/jbc.M113.544833
Alto, L. T., and Terman, J. R. (2017). Semaphorins and their signaling mechanisms. Methods Mol. Biol. 1493, 1–25. doi: 10.1007/978-1-4939-6448-2_1
Barger, A. C., Beeuwkes, R. III, Lainey, L. L., and Silverman, K. J. (1984). Hypothesis: vasa vasorum and neovascularization of human coronary arteries, a possible role in the pathophysiology of atherosclerosis. N. Engl. J. Med. 310, 175–177. doi: 10.1056/NEJM198401193100307
Basile, J. R., Barac, A., Zhu, T., Guan, K. L., and Gutkind, J. S. (2004). Class IV semaphorins promote angiogenesis by stimulating Rho-initiated pathways through plexin-B. Cancer Res. 64, 5212–5224. doi: 10.1158/0008-5472.CAN-04-0126
Bellingan, G. J., Caldwell, H., Howie, S. E., Dransfield, I., and Haslett, C. (1996). In vivo fate of the inflammatory macrophage during the resolution of inflammation: inflammatory macrophages do not die locally, but emigrate to the draining lymph nodes. J. Immunol. 157, 2577–2585.
Bougeret, C., Mansur, I. G., Dastot, H., Schmid, M., Mahouy, G., Bensussan, A., et al. (1992). Increased surface expression of a newly identified 150-kDa dimer early after human T lymphocyte activation. J. Immunol. 148, 318–323.
Bribian, A., Nocentini, S., Llorens, F., Gil, V., Mire, E., Reginensi, D., et al. (2014). Sema3E/PlexinD1 regulates the migration of hem-derived Cajal-Retzius cells in developing cerebral cortex. Nat. Commun. 5:4265. doi: 10.1038/ncomms5265
Capparuccia, L., and Tamagnone, L. (2009). Semaphorin signaling in cancer cells and in cells of the tumor microenvironment–two sides of a coin. J. Cell Sci. 122(Pt 11), 1723–1736. doi: 10.1242/jcs.030197
Chapoval, S. P., Vadasz, Z., Chapoval, A. I., and Toubi, E. (2017). Semaphorins 4A and 4D in chronic inflammatory diseases. Inflamm. Res. 66, 111–117. doi: 10.1007/s00011-016-0983-5
Chavez-Sanchez, L., Espinosa-Luna, J. E., Chavez-Rueda, K., Legorreta-Haquet, M. V., Montoya-Diaz, E., and Blanco-Favela, F. (2014). Innate immune system cells in atherosclerosis. Arch. Med. Res. 45, 1–14. doi: 10.1016/j.arcmed.2013.11.007
Christensen, C. R., Klingelhofer, J., Tarabykina, S., Hulgaard, E. F., Kramerov, D., and Lukanidin, E. (1998). Transcription of a novel mouse semaphorin gene, M-semaH, correlates with the metastatic ability of mouse tumor cell lines. Cancer Res. 58, 1238–1244.
Conrotto, P., Valdembri, D., Corso, S., Serini, G., Tamagnone, L., Comoglio, P. M., et al. (2005). Sema4D induces angiogenesis through met recruitment by Plexin B1. Blood 105, 4321–4329. doi: 10.1182/blood-2004-07-2885
Czopik, A. K., Bynoe, M. S., Palm, N., Raine, C. S., and Medzhitov, R. (2006). Semaphorin 7A is a negative regulator of T cell responses. Immunity 24, 591–600. doi: 10.1016/j.immuni.2006.03.013
Dent, E. W., Barnes, A. M., Tang, F., and Kalil, K. (2004). Netrin-1 and semaphorin 3A promote or inhibit cortical axon branching, respectively, by reorganization of the cytoskeleton. J. Neurosci. 24, 3002–3012. doi: 10.1523/JNEUROSCI.4963-03.2004
Dzau, V. J., Braun-Dullaeus, R. C., and Sedding, D. G. (2002). Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat. Med. 8, 1249–1256. doi: 10.1038/nm1102-1249
Esnault, S., Torr, E. E., Bernau, K., Johansson, M. W., Kelly, E. A., Sandbo, N., et al. (2017). Endogenous semaphorin-7A impedes human lung fibroblast differentiation. PLoS One 12:e0170207. doi: 10.1371/journal.pone.0170207
Garcia-Areas, R., Libreros, S., Amat, S., Keating, P., Carrio, R., Robinson, P., et al. (2014). Semaphorin7A promotes tumor growth and exerts a pro-angiogenic effect in macrophages of mammary tumor-bearing mice. Front. Physiol. 5:17. doi: 10.3389/fphys.2014.00017
Garcia-Areas, R., Libreros, S., and Iragavarapu-Charyulu, V. (2013). Semaphorin 7A: branching beyond axonal guidance and into immunity. Immunol. Res. 57, 81–85. doi: 10.1007/s12026-013-8460-5
Garcia-Areas, R., Libreros, S., Simoes, M., Castro-Silva, C., Gazaniga, N., Amat, S., et al. (2017). Suppression of tumor-derived semaphorin 7A and genetic ablation of host-derived semaphorin 7A impairs tumor progression in a murine model of advanced breast carcinoma. Int. J. Oncol. 51, 1395–1404. doi: 10.3892/ijo.2017.4144
Geovanini, G. R., and Libby, P. (2018). Atherosclerosis and inflammation: overview and updates. Clin. Sci. (Lond.) 132, 1243–1252. doi: 10.1042/CS20180306
Gibson, M. S., Domingues, N., and Vieira, O. V. (2018). Lipid and non-lipid factors affecting macrophage dysfunction and inflammation in atherosclerosis. Front. Physiol. 9:654. doi: 10.3389/fphys.2018.00654
Giordano, S., Corso, S., Conrotto, P., Artigiani, S., Gilestro, G., Barberis, D., et al. (2002). The semaphorin 4D receptor controls invasive growth by coupling with Met. Nat. Cell Biol. 4, 720–724. doi: 10.1038/ncb843
Gomez, D., and Owens, G. K. (2012). Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 95, 156–164. doi: 10.1093/cvr/cvs115
Gu, C., Yoshida, Y., Livet, J., Reimert, D. V., Mann, F., Merte, J., et al. (2005). Semaphorin 3E and plexin-D1 control vascular pattern independently of neuropilins. Science 307, 265–268. doi: 10.1126/science.1105416
Guttmann-Raviv, N., Shraga-Heled, N., Varshavsky, A., Guimaraes-Sternberg, C., Kessler, O., and Neufeld, G. (2007). Semaphorin-3A and semaphorin-3F work together to repel endothelial cells and to inhibit their survival by induction of apoptosis. J. Biol. Chem. 282, 26294–26305. doi: 10.1074/jbc.M609711200
Hall, K. T., Boumsell, L., Schultze, J. L., Boussiotis, V. A., Dorfman, D. M., Cardoso, A. A., et al. (1996). Human CD100, a novel leukocyte semaphorin that promotes B-cell aggregation and differentiation. Proc. Natl. Acad. Sci. U.S.A. 93, 11780–11785. doi: 10.1073/pnas.93.21.11780
Hashimoto, M., Ino, H., Koda, M., Murakami, M., Yoshinaga, K., Yamazaki, M., et al. (2004). Regulation of semaphorin 3A expression in neurons of the rat spinal cord and cerebral cortex after transection injury. Acta Neuropathol. 107, 250–256. doi: 10.1007/s00401-003-0805-z
Herman, J. G., and Meadows, G. G. (2007). Increased class 3 semaphorin expression modulates the invasive and adhesive properties of prostate cancer cells. Int. J. Oncol. 30, 1231–1238. doi: 10.3892/ijo.30.5.1231
Holmes, S., Downs, A. M., Fosberry, A., Hayes, P. D., Michalovich, D., Murdoch, P., et al. (2002). Sema7A is a potent monocyte stimulator. Scand. J. Immunol. 56, 270–275. doi: 10.1046/j.1365-3083.2002.01129.x
Hou, S. T., Keklikian, A., Slinn, J., O’Hare, M., Jiang, S. X., and Aylsworth, A. (2008). Sustained up-regulation of semaphorin 3A, Neuropilin1, and doublecortin expression in ischemic mouse brain during long-term recovery. Biochem. Biophys. Res. Commun. 367, 109–115. doi: 10.1016/j.bbrc.2007.12.103
Hu, S., Liu, Y., You, T., Heath, J., Xu, L., Zheng, X., et al. (2018). Vascular semaphorin 7A upregulation by disturbed flow promotes atherosclerosis through endothelial beta1 integrin. Arterioscler. Thromb. Vasc. Biol. 38, 335–343. doi: 10.1161/ATVBAHA.117.310491
Jaimes, Y., Gras, C., Goudeva, L., Buchholz, S., Eiz-Vesper, B., Seltsam, A., et al. (2012). Semaphorin 7A inhibits platelet production from CD34 + progenitor cells. J. Thromb. Haemost. 10, 1100–1108. doi: 10.1111/j.1538-7836.2012.04708.x
Janssen, B. J., Robinson, R. A., Perez-Branguli, F., Bell, C. H., Mitchell, K. J., Siebold, C., et al. (2010). Structural basis of semaphorin-plexin signalling. Nature 467, 1118–1122. doi: 10.1038/nature09468
Ji, J. D., Park-Min, K. H., and Ivashkiv, L. B. (2009). Expression and function of semaphorin 3A and its receptors in human monocyte-derived macrophages. Hum. Immunol. 70, 211–217. doi: 10.1016/j.humimm.2009.01.026
Jonasson, L., Holm, J., Skalli, O., Bondjers, G., and Hansson, G. K. (1986). Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis 6, 131–138. doi: 10.1161/01.ATV.6.2.131
Jonsson-Rylander, A. C., Nilsson, T., Fritsche-Danielson, R., Hammarstrom, A., Behrendt, M., Andersson, J. O., et al. (2005). Role of ADAMTS-1 in atherosclerosis: remodeling of carotid artery, immunohistochemistry, and proteolysis of versican. Arterioscler. Thromb. Vasc. Biol. 25, 180–185. doi: 10.1161/01.ATV.0000150045.27127.37
Karayan-Tapon, L., Wager, M., Guilhot, J., Levillain, P., Marquant, C., Clarhaut, J., et al. (2008). Semaphorin, neuropilin and VEGF expression in glial tumours: SEMA3G, a prognostic marker? Br. J. Cancer 99, 1153–1160. doi: 10.1038/sj.bjc.6604641
Kashiwagi, H., Shiraga, M., Kato, H., Kamae, T., Yamamoto, N., Tadokoro, S., et al. (2005). Negative regulation of platelet function by a secreted cell repulsive protein, semaphorin 3A. Blood 106, 913–921. doi: 10.1182/blood-2004-10-4092
Kikutani, H., and Kumanogoh, A. (2003). Semaphorins in interactions between T cells and antigen-presenting cells. Nat. Rev. Immunol. 3, 159–167. doi: 10.1038/nri1003
Kim, J., Oh, W. J., Gaiano, N., Yoshida, Y., and Gu, C. (2011). Semaphorin 3E-Plexin-D1 signaling regulates VEGF function in developmental angiogenesis via a feedback mechanism. Genes Dev. 25, 1399–1411. doi: 10.1101/gad.2042011
Kolodkin, A. L., Levengood, D. V., Rowe, E. G., Tai, Y. T., Giger, R. J., and Ginty, D. D. (1997). Neuropilin is a semaphorin III receptor. Cell 90, 753–762. doi: 10.1016/S0092-8674(00)80535-8
Kolodkin, A. L., Matthes, D. J., and Goodman, C. S. (1993). The semaphorin genes encode a family of transmembrane and secreted growth cone guidance molecules. Cell 75, 1389–1399. doi: 10.1016/0092-8674(93)90625-Z
Komatsu, M., and Ruoslahti, E. (2005). R-Ras is a global regulator of vascular regeneration that suppresses intimal hyperplasia and tumor angiogenesis. Nat. Med. 11, 1346–1350. doi: 10.1038/nm1324
Kuklina, E. M., Nekrasova, I. V., and Valieva, Y. V. (2017). Involvement of semaphorin (Sema4D) in T-dependent activation of B cells. Bull. Exp. Biol. Med. 163, 447–450. doi: 10.1007/s10517-017-3825-8
Kumamoto, M., Nakashima, Y., and Sueishi, K. (1995). Intimal neovascularization in human coronary atherosclerosis: its origin and pathophysiological significance. Hum. Pathol. 26, 450–456. doi: 10.1016/0046-8177(95)90148-5
Kumanogoh, A., and Kikutani, H. (2003). Immune semaphorins: a new area of semaphorin research. J. Cell Sci. 116(Pt 17), 3463–3470. doi: 10.1242/jcs.00674
Kumanogoh, A., Marukawa, S., Suzuki, K., Takegahara, N., Watanabe, C., Ch’ng, E., et al. (2002). Class IV semaphorin Sema4A enhances T-cell activation and interacts with Tim-2. Nature 419, 629–633. doi: 10.1038/nature01037
Lange, C., Liehr, T., Goen, M., Gebhart, E., Fleckenstein, B., and Ensser, A. (1998). New eukaryotic semaphorins with close homology to semaphorins of DNA viruses. Genomics 51, 340–350. doi: 10.1006/geno.1998.5256
Lepelletier, Y., Moura, I. C., Hadj-Slimane, R., Renand, A., Fiorentino, S., Baude, C., et al. (2006). Immunosuppressive role of semaphorin-3A on T cell proliferation is mediated by inhibition of actin cytoskeleton reorganization. Eur. J. Immunol. 36, 1782–1793. doi: 10.1002/eji.200535601
Liu, H., Juo, Z. S., Shim, A. H., Focia, P. J., Chen, X., Garcia, K. C., et al. (2010). Structural basis of semaphorin-plexin recognition and viral mimicry from Sema7A and A39R complexes with PlexinC1. Cell 142, 749–761. doi: 10.1016/j.cell.2010.07.040
Llodra, J., Angeli, V., Liu, J., Trogan, E., Fisher, E. A., and Randolph, G. J. (2004). Emigration of monocyte-derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc. Natl. Acad. Sci. U.S.A. 101, 11779–11784. doi: 10.1073/pnas.0403259101
Luchtefeld, M., Grothusen, C., Gagalick, A., Jagavelu, K., Schuett, H., Tietge, U. J., et al. (2010). Chemokine receptor 7 knockout attenuates atherosclerotic plaque development. Circulation 122, 1621–1628. doi: 10.1161/CIRCULATIONAHA.110.956730
Luo, Y., Raible, D., and Raper, J. A. (1993). Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell 75, 217–227. doi: 10.1016/0092-8674(93)80064-L
Luque, M. C., Gutierrez, P. S., Debbas, V., Kalil, J., and Stolf, B. S. (2015). CD100 and plexins B2 and B1 mediate monocyte-endothelial cell adhesion and might take part in atherogenesis. Mol. Immunol. 67(2 Pt B), 559–567. doi: 10.1016/j.molimm.2015.07.028
Luque, M. C., Gutierrez, P. S., Debbas, V., Martins, W. K., Puech-Leao, P., Porto, G., et al. (2013). Phage display identification of CD100 in human atherosclerotic plaque macrophages and foam cells. PLoS One 8:e75772. doi: 10.1371/journal.pone.0075772
Maestrini, E., Tamagnone, L., Longati, P., Cremona, O., Gulisano, M., Bione, S., et al. (1996). A family of transmembrane proteins with homology to the MET-hepatocyte growth factor receptor. Proc. Natl. Acad. Sci. U.S.A. 93, 674–678. doi: 10.1073/pnas.93.2.674
Martin-Satue, M., and Blanco, J. (1999). Identification of semaphorin E gene expression in metastatic human lung adenocarcinoma cells by mRNA differential display. J. Surg. Oncol. 72, 18–23. doi: 10.1002/(SICI)1096-9098(199909)72:1<18::AID-JSO5>3.0.CO;2-P
Moreno, P. R., Purushothaman, K. R., Sirol, M., Levy, A. P., and Fuster, V. (2006). Neovascularization in human atherosclerosis. Circulation 113, 2245–2252. doi: 10.1161/CIRCULATIONAHA.105.578955
Moretti, S., Procopio, A., Lazzarini, R., Rippo, M. R., Testa, R., Marra, M., et al. (2008). Semaphorin3A signaling controls Fas (CD95)-mediated apoptosis by promoting Fas translocation into lipid rafts. Blood 111, 2290–2299. doi: 10.1182/blood-2007-06-096529
Moriya, J., Minamino, T., Tateno, K., Okada, S., Uemura, A., Shimizu, I., et al. (2010). Inhibition of semaphorin as a novel strategy for therapeutic angiogenesis. Circ. Res. 106, 391–398. doi: 10.1161/CIRCRESAHA.109.210815
Morote-Garcia, J. C., Napiwotzky, D., Kohler, D., and Rosenberger, P. (2012). Endothelial semaphorin 7A promotes neutrophil migration during hypoxia. Proc. Natl. Acad. Sci. U.S.A. 109, 14146–14151. doi: 10.1073/pnas.1202165109
Moulton, K. S. (2001). Plaque angiogenesis and atherosclerosis. Curr. Atheroscler. Rep. 3, 225–233. doi: 10.1007/s11883-001-0065-0
Moulton, K. S., Vakili, K., Zurakowski, D., Soliman, M., Butterfield, C., Sylvin, E., et al. (2003). Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc. Natl. Acad. Sci. U.S.A. 100, 4736–4741. doi: 10.1073/pnas.0730843100
Mukovozov, I., Huang, Y. W., Zhang, Q., Liu, G. Y., Siu, A., Sokolskyy, Y., et al. (2015). The neurorepellent Slit2 inhibits postadhesion stabilization of monocytes tethered to vascular endothelial cells. J. Immunol. 195, 3334–3344. doi: 10.4049/jimmunol.1500640
Neufeld, G., and Kessler, O. (2008). The semaphorins: versatile regulators of tumour progression and tumour angiogenesis. Nat. Rev. Cancer 8, 632–645. doi: 10.1038/nrc2404
Nishide, M., and Kumanogoh, A. (2018). The role of semaphorins in immune responses and autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 14, 19–31. doi: 10.1038/nrrheum.2017.201
Nogi, T., Yasui, N., Mihara, E., Matsunaga, Y., Noda, M., Yamashita, N., et al. (2010). Structural basis for semaphorin signalling through the plexin receptor. Nature 467, 1123–1127. doi: 10.1038/nature09473
Oh, W. J., and Gu, C. (2013). The role and mechanism-of-action of Sema3E and Plexin-D1 in vascular and neural development. Semin. Cell Dev. Biol. 24, 156–162. doi: 10.1016/j.semcdb.2012.12.001
Owens, G. K., Kumar, M. S., and Wamhoff, B. R. (2004). Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84, 767–801. doi: 10.1152/physrev.00041.2003
Paoletti, R., Gotto, A. M. Jr., and Hajjar, D. P. (2004). Inflammation in atherosclerosis and implications for therapy. Circulation 109(23 Suppl. 1), III20–III26. doi: 10.1161/01.CIR.0000131514.71167.2e
Pasterkamp, R. J., Kolk, S. M., Hellemons, A. J., and Kolodkin, A. L. (2007). Expression patterns of semaphorin7A and plexinC1 during rat neural development suggest roles in axon guidance and neuronal migration. BMC Dev. Biol. 7:98. doi: 10.1186/1471-213X-7-98
Pasterkamp, R. J., Peschon, J. J., Spriggs, M. K., and Kolodkin, A. L. (2003). Semaphorin 7A promotes axon outgrowth through integrins and MAPKs. Nature 424, 398–405. doi: 10.1038/nature01790
Poitz, D. M., Ende, G., Stutz, B., Augstein, A., Friedrichs, J., Brunssen, C., et al. (2015). EphrinB2/EphA4-mediated activation of endothelial cells increases monocyte adhesion. Mol. Immunol. 68(2 Pt C), 648–656. doi: 10.1016/j.molimm.2015.10.009
Rader, D. J., and Pure, E. (2005). Lipoproteins, macrophage function, and atherosclerosis: beyond the foam cell? Cell Metab. 1, 223–230. doi: 10.1016/j.cmet.2005.03.005
Ryynanen, J., Kriebitzsch, C., Meyer, M. B., Janssens, I., Pike, J. W., Verlinden, L., et al. (2017). Class 3 semaphorins are transcriptionally regulated by 1,25(OH)2D3 in osteoblasts. J. Steroid Biochem. Mol. Biol. 173, 185–193. doi: 10.1016/j.jsbmb.2017.02.005
Sakurai, A., Gavard, J., Annas-Linhares, Y., Basile, J. R., Amornphimoltham, P., Palmby, T. R., et al. (2010). Semaphorin 3E initiates antiangiogenic signaling through plexin D1 by regulating Arf6 and R-Ras. Mol. Cell. Biol. 30, 3086–3098. doi: 10.1128/MCB.01652-09
Samson, S., Mundkur, L., and Kakkar, V. V. (2012). Immune response to lipoproteins in atherosclerosis. Cholesterol 2012:571846. doi: 10.1155/2012/571846
Scott, G. A., McClelland, L. A., and Fricke, A. F. (2008). Semaphorin 7a promotes spreading and dendricity in human melanocytes through beta1-integrins. J. Invest. Dermatol. 128, 151–161. doi: 10.1038/sj.jid.5700974
Scott, G. A., McClelland, L. A., Fricke, A. F., and Fender, A. (2009). Plexin C1, a receptor for semaphorin 7a, inactivates cofilin and is a potential tumor suppressor for melanoma progression. J. Invest. Dermatol. 129, 954–963. doi: 10.1038/jid.2008.329
Segarra, M., Ohnuki, H., Maric, D., Salvucci, O., Hou, X., Kumar, A., et al. (2012). Semaphorin 6A regulates angiogenesis by modulating VEGF signaling. Blood 120, 4104–4115. doi: 10.1182/blood-2012-02-410076
Semaphorin Nomenclature Committee (1999). Unified nomenclature for the semaphorins/collapsins. Cell 97, 551–552. doi: 10.1016/S0092-8674(00)80766-7
Serini, G., Valdembri, D., Zanivan, S., Morterra, G., Burkhardt, C., Caccavari, F., et al. (2003). Class 3 semaphorins control vascular morphogenesis by inhibiting integrin function. Nature 424, 391–397. doi: 10.1038/nature01784
Sharma, A., Pollett, M. A., Plant, G. W., and Harvey, A. R. (2012a). Changes in mRNA expression of class 3 semaphorins and their receptors in the adult rat retino-collicular system after unilateral optic nerve injury. Invest. Ophthalmol. Vis. Sci. 53, 8367–8377. doi: 10.1167/iovs.12-10799
Sharma, A., Verhaagen, J., and Harvey, A. R. (2012b). Receptor complexes for each of the class 3 semaphorins. Front. Cell Neurosci. 6:28. doi: 10.3389/fncel.2012.00028
Siegel-Axel, D., Daub, K., Seizer, P., Lindemann, S., and Gawaz, M. (2008). Platelet lipoprotein interplay: trigger of foam cell formation and driver of atherosclerosis. Cardiovasc. Res. 78, 8–17. doi: 10.1093/cvr/cvn015
Singh, R. B., Mengi, S. A., Xu, Y. J., Arneja, A. S., and Dhalla, N. S. (2002). Pathogenesis of atherosclerosis: a multifactorial process. Exp. Clin. Cardiol. 7, 40–53.
Steinbach, K., Volkmer, H., and Schlosshauer, B. (2002). Semaphorin 3E/collapsin-5 inhibits growing retinal axons. Exp. Cell Res. 279, 52–61. doi: 10.1006/excr.2002.5595
Suzuki, K., and Kumanogoh, A. (2006). [Semaphorins in immune responses: involvement of Sema4A in T-cell priming and Th1/Th2 regulation]. Seikagaku 78, 141–145.
Suzuki, K., Kumanogoh, A., and Kikutani, H. (2003). CD100/Sema4D, a lymphocyte semaphorin involved in the regulation of humoral and cellular immune responses. Cytokine Growth Fact. Rev. 14, 17–24. doi: 10.1016/S1359-6101(02)00073-4
Suzuki, K., Kumanogoh, A., and Kikutani, H. (2008). Semaphorins and their receptors in immune cell interactions. Nat. Immunol. 9, 17–23. doi: 10.1038/ni1553
Suzuki, K., Okuno, T., Yamamoto, M., Pasterkamp, R. J., Takegahara, N., Takamatsu, H., et al. (2007). Semaphorin 7A initiates T-cell-mediated inflammatory responses through alpha1beta1 integrin. Nature 446, 680–684. doi: 10.1038/nature05652
Takegahara, N., Takamatsu, H., Toyofuku, T., Tsujimura, T., Okuno, T., Yukawa, K., et al. (2006). Plexin-A1 and its interaction with DAP12 in immune responses and bone homeostasis. Nat. Cell Biol. 8, 615–622. doi: 10.1038/ncb1416
Tamagnone, L., Artigiani, S., Chen, H., He, Z., Ming, G. I., Song, H., et al. (1999). Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell 99, 71–80. doi: 10.1016/S0092-8674(00)80063-X
Tamagnone, L., and Comoglio, P. M. (2000). Signalling by semaphorin receptors: cell guidance and beyond. Trends Cell Biol. 10, 377–383. doi: 10.1016/S0962-8924(00)01816-X
Toyofuku, T., and Kikutani, H. (2007). Semaphorin signaling during cardiac development. Adv. Exp. Med. Biol. 600, 109–117. doi: 10.1007/978-0-387-70956-7_9
Toyofuku, T., Yabuki, M., Kamei, J., Kamei, M., Makino, N., Kumanogoh, A., et al. (2007). Semaphorin-4A, an activator for T-cell-mediated immunity, suppresses angiogenesis via Plexin-D1. EMBO J. 26, 1373–1384. doi: 10.1038/sj.emboj.7601589
Toyofuku, T., Yoshida, J., Sugimoto, T., Yamamoto, M., Makino, N., Takamatsu, H., et al. (2008). Repulsive and attractive semaphorins cooperate to direct the navigation of cardiac neural crest cells. Dev. Biol. 321, 251–262. doi: 10.1016/j.ydbio.2008.06.028
Trogan, E., Feig, J. E., Dogan, S., Rothblat, G. H., Angeli, V., Tacke, F., et al. (2006). Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 103, 3781–3786. doi: 10.1073/pnas.0511043103
Vadasz, Z., Haj, T., Kessel, A., and Toubi, E. (2013). B-regulatory cells in autoimmunity and immune mediated inflammation. FEBS Lett. 587, 2074–2078. doi: 10.1016/j.febslet.2013.05.023
Vadasz, Z., and Toubi, E. (2014). Semaphorins: their dual role in regulating immune-mediated diseases. Clin. Rev. Allergy Immunol. 47, 17–25. doi: 10.1007/s12016-013-8360-4
van Gils, J. M., Ramkhelawon, B., Fernandes, L., Stewart, M. C., Guo, L., Seibert, T., et al. (2013). Endothelial expression of guidance cues in vessel wall homeostasis dysregulation under proatherosclerotic conditions. Arterioscler. Thromb. Vasc. Biol. 33, 911–919. doi: 10.1161/ATVBAHA.112.301155
Wang, X., Kumanogoh, A., Watanabe, C., Shi, W., Yoshida, K., and Kikutani, H. (2001). Functional soluble CD100/Sema4D released from activated lymphocytes: possible role in normal and pathologic immune responses. Blood 97, 3498–3504. doi: 10.1182/blood.V97.11.3498
Wannemacher, K. M., Wang, L., Zhu, L., and Brass, L. F. (2011). The role of semaphorins and their receptors in platelets: lessons learned from neuronal and immune synapses. Platelets 22, 461–465. doi: 10.3109/09537104.2011.561891
Wannemacher, K. M., Zhu, L., Jiang, H., Fong, K. P., Stalker, T. J., Lee, D., et al. (2010). Diminished contact-dependent reinforcement of Syk activation underlies impaired thrombus growth in mice lacking Semaphorin 4D. Blood 116, 5707–5715. doi: 10.1182/blood-2010-04-279943
Wanschel, A., Seibert, T., Hewing, B., Ramkhelawon, B., Ray, T. D., van Gils, J. M., et al. (2013). Neuroimmune guidance cue emaphorin 3E is expressed in atherosclerotic plaques and regulates macrophage retention. Arterioscler. Thromb. Vasc. Biol. 33, 886–893. doi: 10.1161/ATVBAHA.112.300941
Wu, J. H., Li, Y., Zhou, Y. F., Haslam, J., Elvis, O. N., Mao, L., et al. (2017). Semaphorin-3E attenuates neointimal formation via suppressing VSMCs migration and proliferation. Cardiovasc. Res. 113, 1763–1775. doi: 10.1093/cvr/cvx190
Xu, X., Ng, S., Wu, Z. L., Nguyen, D., Homburger, S., Seidel-Dugan, C., et al. (1998). Human semaphorin K1 is glycosylphosphatidylinositol-linked and defines a new subfamily of viral-related semaphorins. J. Biol. Chem. 273, 22428–22434. doi: 10.1074/jbc.273.35.22428
Yukawa, K., Tanaka, T., Kishino, M., Yoshida, K., Takeuchi, N., Ito, T., et al. (2010). Deletion of Sema4D gene reduces intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Int. J. Mol. Med. 26, 39–44. doi: 10.3892/ijmm_00000432
Zhou, Y., Gunput, R. A., and Pasterkamp, R. J. (2008). Semaphorin signaling: progress made and promises ahead. Trends Biochem. Sci. 33, 161–170. doi: 10.1016/j.tibs.2008.01.006
Zhu, L., Bergmeier, W., Wu, J., Jiang, H., Stalker, T. J., Cieslak, M., et al. (2007). Regulated surface expression and shedding support a dual role for semaphorin 4D in platelet responses to vascular injury. Proc. Natl. Acad. Sci. U.S.A. 104, 1621–1626. doi: 10.1073/pnas.0606344104
Zhu, L., Stalker, T. J., Fong, K. P., Jiang, H., Tran, A., Crichton, I., et al. (2009). Disruption of SEMA4D ameliorates platelet hypersensitivity in dyslipidemia and confers protection against the development of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 29, 1039–1045. doi: 10.1161/ATVBAHA.109.185405
Keywords: Semaphorins, atherosclerosis, inflammation, cell infiltration, angiogenesis
Citation: Hu S and Zhu L (2018) Semaphorins and Their Receptors: From Axonal Guidance to Atherosclerosis. Front. Physiol. 9:1236. doi: 10.3389/fphys.2018.01236
Received: 27 May 2018; Accepted: 15 August 2018;
Published: 12 October 2018.
Edited by:
Vincenzo Lionetti, Scuola Sant’Anna di Studi Avanzati, ItalyReviewed by:
Suowen Xu, University of Rochester, United StatesCopyright © 2018 Hu and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuhong Hu, aHVzaHVob25nMTk5MDEwMTBAMTI2LmNvbQ== Li Zhu, emh1bEBzdWRhLmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.