 Xiaoming Lian
Xiaoming Lian Sandra Beer-Hammer2
Sandra Beer-Hammer2 Evi Kostenis
Evi Kostenis Bernd Nürnberg
Bernd Nürnberg Maik Gollasch
Maik Gollasch
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 04 September 2018
Sec. Integrative Physiology
Volume 9 - 2018 | https://doi.org/10.3389/fphys.2018.01234
This article is part of the Research TopicAdipose Tissue and the Cardiovascular System: Biology and Clinical ImplicationsView all 4 articles
Background: Relaxins are small peptide hormones, which are novel candidate molecules that play important roles in cardiometablic syndrome. Relaxins are structurally related to the insulin hormone superfamily, which provide vasodilatory effects by activation of G-protein-coupled relaxin receptors (RXFPs) and stimulation of endogenous nitric oxide (NO) generation. Recently, relaxin could be demonstrated to activate Gi proteins and phosphoinositide 3-kinase (PI3K) pathways in cultured endothelial cells in vitro. However, the contribution of the Gi-PI3K pathway and their individual components in relaxin-dependent relaxation of intact arteries remains elusive.
Methods: We used Gαi2- (Gnai2-/-) and Gαi3-deficient (Gnai3-/-) mice, pharmacological tools and wire myography to study G-protein-coupled signaling pathways involved in relaxation of mouse isolated mesenteric arteries by relaxins. Human relaxin-1, relaxin-2, and relaxin-3 were tested.
Results: Relaxin-2 (∼50% relaxation at 10-11 M) was the most potent vasodilatory relaxin in mouse mesenteric arteries, compared to relaxin-1 and relaxin-3. The vasodilatory effects of relaxin-2 were inhibited by removal of the endothelium or treatment of the vessels with N (G)-nitro-L-arginine methyl ester (L-NAME, endothelial nitric oxide synthase (eNOS) inhibitor) or simazine (RXFP1 inhibitor). The vasodilatory effects of relaxin-2 were absent in arteries of mice treated with pertussis toxin (PTX). They were also absent in arteries isolated from Gnai2-/- mice, but not from Gnai3-/- mice. The effects were not affected by FR900359 (Gαq protein inhibitor) or PI-103 (PI3Kα inhibitor), but inhibited by TGX-221 (PI3Kβ inhibitor) or AS-252424 (PI3Kγ inhibitor). Simazine did not influence the anti-contractile effect of perivascular adipose tissue.
Conclusion: Our data indicate that relaxin-2 produces endothelium- and NO-dependent relaxation of mouse mesenteric arteries by activation of RXFP1 coupled to Gi2-PI3K-eNOS pathway. Targeting vasodilatory Gi-protein-coupled RXFP1 pathways may provide promising opportunities for drug discovery in endothelial dysfunction and cardiometabolic disease.
Cardiometabolic syndrome is a combination of metabolic dysfunctions mainly characterized by insulin resistance, impaired glucose tolerance, dyslipidemia, hypertension, and central adiposity. Relaxins were initially viewed as pregnancy hormones because originally identified to be produced by the ovary corpus luteum in pregnant women (Bell et al., 1987). Later relaxins were also found to be produced in the heart, in the mammary gland, in the endometrium (Kakouris et al., 1992; Du et al., 2010), in the placental trophoblast cells and in the prostate (Hansell et al., 1991). Relaxins are structurally related to the insulin hormone superfamily, consisting of two peptide chains derived from a common processor that are linked by two disulfide bridges. They act through different relaxin family peptide receptors (RXFPs) and signaling pathways (Dessauer and Nguyen, 2005). Humans express three forms of relaxins encoded by three separate genes, that is, relaxin-1 (RLX1), relaxin-2 (RLX2), and relaxin-3 (RLX3) genes. Relaxin-1 and relaxin-2 are the major circulating relaxin isoforms in humans and other mammals (Grossman and Frishman, 2010), while relaxin-3 is expressed at high levels in the nucleus incertus in human and rodent brains, where it has been postulated to act locally as a neuropeptide (Goto et al., 2001).
In recent years, there has been increasing interest in the possible role of relaxins in cardiometabolic syndrome. Relaxin treatment reduces food intake in rats (McGowan et al., 2010), reverses insulin resistance and restored endothelial-dependent vasodilatation in high-fat-diet mice (Bonner et al., 2013). Moreover, RXFPs have been suggested to represent potential targets for anti-anxiety and anti-obesity drugs (Halls et al., 2007). Meanwhile, serelaxin (RLX030), the drug represents the recombinant form of human relaxin-2, also shows promising effects in the therapeutic process in patients with hypertension (Papadopoulos et al., 2013), acute heart failure (AHF) (Teerlink et al., 2013), and ischemic heart disease (Parikh et al., 2013), most likely through its vasodilatory, antifibrotic, and antigenic properties (Conrad, 2010).
Relaxins are capable of inducing vasodilation in human (Fisher et al., 2002) and rodent microvessels (Conrad, 2010). The effects are mediated via specific G-protein-coupled receptors (GPCRs), the relaxin family peptide receptor (RXFP) 1–4 (Bathgate et al., 2006). RXFP1 is widely expressed in heart, kidney, lung, liver, blood vessels, and various areas of the brain. This GPCR is considered to be the fundamental RXFP receptor to mediate relaxin effects in the cardiovascular system by complex mechanisms and intracellular signaling pathways (Bani-Sacchi et al., 1995; Nistri and Bani, 2003). Cell culture experiments indicate that relaxin could activate endothelial nitric oxide synthase (eNOS) via a pertussis toxin (PTX)-sensitive Gi-PI3K-dependent pathway (Halls et al., 2006; Novak et al., 2006; van der Westhuizen et al., 2008). However, it is unknown whether this pathway is relevant to vasodilation of intact vessels. It is also unknown which G proteins are involved in this pathway. RXFP2 activates adenylate cyclase in recombinant systems, but physiological responses are sensitive to pertussis toxin. RXFP3 and RXFP4 resemble more conventional peptide ligand receptors and both inhibit adenylate cyclase, and in addition RXFP3 activates Erk1/2 signaling in vitro (Bathgate et al., 2006). Although Gi-proteins have been suggested to play important roles in cardiovascular disease, in particular in ischemia reperfusion injury (Eisen et al., 2004), the involvements of specific Gi isoform(s) [Gαi1, Gαi2, and/or Gαi3] and vasodilatory GPCRs are unknown. This is particularly important since there are only few reports that GPCRs are capable to utilize Gi-coupled signaling pathways to cause vasodilation (i.e., for bradykinin, beta2 adrenergic agonists, thrombin) (Liao and Homcy, 1993; Ciccarelli et al., 2007; Vanhoutte et al., 2017). It is therefore not surprising that current research is focused on the identification of novel compounds and GPCRs which can utilize Gi-signaling pathways to produce potent relaxations. Since relaxins are endogenous hormones, which could exhibit vascular effects via Gi protein-coupled pathways (Halls et al., 2006; Novak et al., 2006; van der Westhuizen et al., 2008), our study was aimed to examine the putative vasodilatory effect of relaxins and the involved G- and PI3K-dependent signaling pathways. In this study, we compared the sensitivity of the three human relaxins 1–3 in eliciting relaxation of mouse mesenteric arteries and tested the hypothesis that the NO-dependent vasodilatory effect of relaxins is mediated by the activation of endothelial RXFP1 receptors, which are coupled to vasodilatory Gi2-PI3K-eNOS signaling pathways. Finally, we also tested whether the RXFP1 pathway is involved in the periadventitial control of arterial tone by perivascular adipose tissue (PVAT).
Experiments were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and the protocols were previously approved by the local Animal Care and Use Committee from Berlin LAGeSo (G0132/14). Animal experiments used 10- to 14-week-old mice of either sex and were housed in groups of four to six animals in cages with nesting material, mouse lodges, and open access to water and feed, at 23°C with a 12 h/12 h circadian cycle. Most experiments were performed using male wild-type (WT, C57BL/6N) mice. To define Gαi isoforms involved in relaxin effects, we used female Gαi2-deficient (Gnai2-/-) and Gαi3-deficient (Gnai3-/-) mice and respective littermate (+/+) controls. The generation and basal phenotypic characterization of Gαi2-deficient and Gαi3-deficient mice are described elsewhere (Rudolph et al., 1995; Gohla et al., 2007; Ezan et al., 2013; Wiege et al., 2013).
Male wild-type C57BL/6 mice (20–25 g, 8–12 weeks) were maintained according to national guidelines for animal care at the animal facility. Mice were injected intraperitoneally with 150 μg/kg body weight pertussis toxin (PTX) or NaCl solution (0.9 %) as vehicle control 48 h before use (Köhler et al., 2014).
The second branches of mesenteric arteries were isolated from mice under inhalation anesthesia with isoflurane and killed by cervical dislocation. The vessels were then quickly transferred to cold (4°C) and oxygenated (95% O2/5% CO2) physiological salt solution (PSS) containing (in mmol/L): 119 NaCl, 4.7 KCl, 1.2 KH2PO4, 25 NaHCO3, 1.2 MgSO4, 11.1 glucose, 1.6 CaCl2, and then dissected into 2 mm rings whereby perivascular fat and connective tissue were either intact [(+) PVAT or removed (-) PVAT] without damaging the adventitia. Each ring was positioned between two stainless steel wires (diameter 0.0394 mm) in a 5-mL organ bath of a Small Vessel Myograph (DMT 610M, Danish Myo Technology, Aarhus, Denmark) (Fésüs et al., 2007). The software Chart5 (AD Instruments, Ltd., Spechbach, Germany) was used for data acquisition and display. The rings were pre-contracted and equilibrated for 30 min until a stable resting tension has been acquired. In some vessels, the endothelium was removed mechanically by a whisker or an air bubble (Fésüs et al., 2007). Endothelium integrity or functional removal was confirmed by the presence or absence, respectively, of the relaxant response to 1 μM acetylcholine (ACh) on phenylephrine (PE 1 μM) pre-contracted arteries. Following PSS wash, the pharmacological drugs were applied. After a waiting period of 30 min, PE and subsequently relaxin-1 or -2 or -3 or vehicle (PSS) was added to the bath solution. Relaxations induced by relaxins were expressed as percentage relaxations obtained with ACh (100%) or as percentage relaxations of PE contractions. Contractions induced by PE were expressed as percentage tension obtained with KCl-PSS (100%) containing (in mmol/L): 63.7 NaCl, 60 KCl, 1.2 KH2PO4, 25 NaHCO3, 1.2 Mg2SO4, 11.1 glucose, and 1.6 CaCl2. During the experiments, relaxin was applied for at least 5 min, and data were recorded to ensure that relaxins achieved their maximal effects (Willcox et al., 2013). All drugs were added to the bath solution (PSS).
FR900359 was isolated as described previously (Schrage et al., 2015). Salts and drugs have been purchased from Sigma-Aldrich (Germany) with the exception of PTX and TGX 221, which were obtained from Merck Millipore (Calbiochem) (Germany) and PI-103 from Enzo Life Sciences. The PI3K inhibitors TGX-221, AS-252424, and PI-103 were administered at submaximal concentrations to achieve biological effects (Ali et al., 2008; Tsvetkov et al., 2016a). All drugs were freshly dissolved on the day of the experiment according to the material sheet. The relaxin peptides have been purchased from Sigma-Aldrich (Germany). Relaxins were dissolved in water. The following concentrations were used: relaxin-1 at 10 pM or at 100 pM, relaxin-2 at 10 pM or at 100 pM or from 1 pM to 10 nM, relaxin-3 at 10 pM or at 100 pM.
Results are presented as mean ± SEM. Data were analyzed statistically using the GraphPad Prism 7 software (GraphPad Software, San Diego, CA, United States). Unpaired Student’s t-tests or ANOVA were used where appropriate. A value of P less than 0.05 was considered statistically significant; n represents the number of arteries tested.
We first evaluated the vasoactive properties of the three different relaxins, that is, relaxin-1, relaxin-2, and relaxin-3. Isolated mesenteric artery rings were pre-contracted by phenylephrine (PE 1 μM) and exposed to acetylcholine (ACh 1 μM) for control. After wash-out of these substances, the vessels were re-exposed to PE and subsequently incubated with relaxin-1, relaxin-2, or relaxin-3 (at 10-10 M each) in separate vessels (Willcox et al., 2013) (Figure 1). All three relaxins produced relaxations (Figure 2A). However, relaxin-2, at the same concentration tested, was more effective than relaxin-1 or relaxin-3 in producing vasodilatory effects (Figure 2A). Importantly, this increased efficiency for relaxin-2 was even more pronounced at 10-fold lower concentrations (10-11 M; Figure 2B). Compared to ACh, relaxations in response to relaxin-1, relaxin-2, and relaxin-3 were slow and delayed (Figure 1). In order to exclude the possibility that relaxations by relaxins result from a spontaneous loss of the pre-contraction level, we performed control experiments with vehicle (PSS) in arteries with and without endothelium. Our results argue against this possibility (Figure 2). Removal of the endothelium abolished both ACh and relaxin-2 relaxations (Figure 1A, right). These data indicate that relaxins are powerful peptide hormones to produce relaxations, probably utilizing an endothelium-dependent mechanism distinct from ACh. Since relaxin-2 was the strongest vasodilator, we continued the following mechanistic studies using this relaxin peptide.
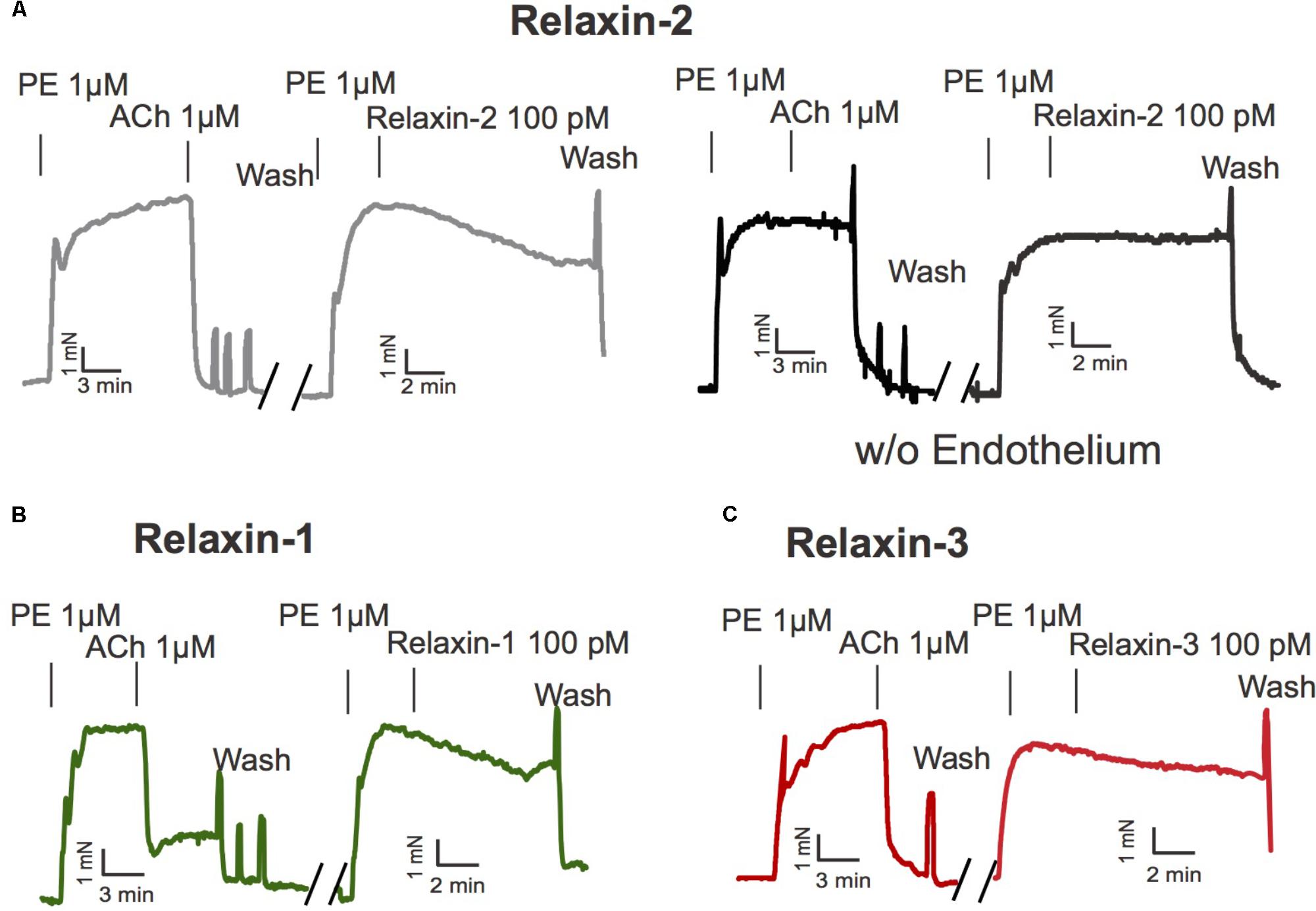
FIGURE 1. Effects of relaxins on mesenteric arteries. (A) Original representative recording for relaxin-2 induced relaxation with (left) and without endothelium (right). (B) Original representative recording for relaxin-1 induced relaxation. (C) Original representative recording for relaxin-3 induced relaxation. PE, phenylephrine. ACh, acetylcholine. For numbers of experiments, see Figure 2.
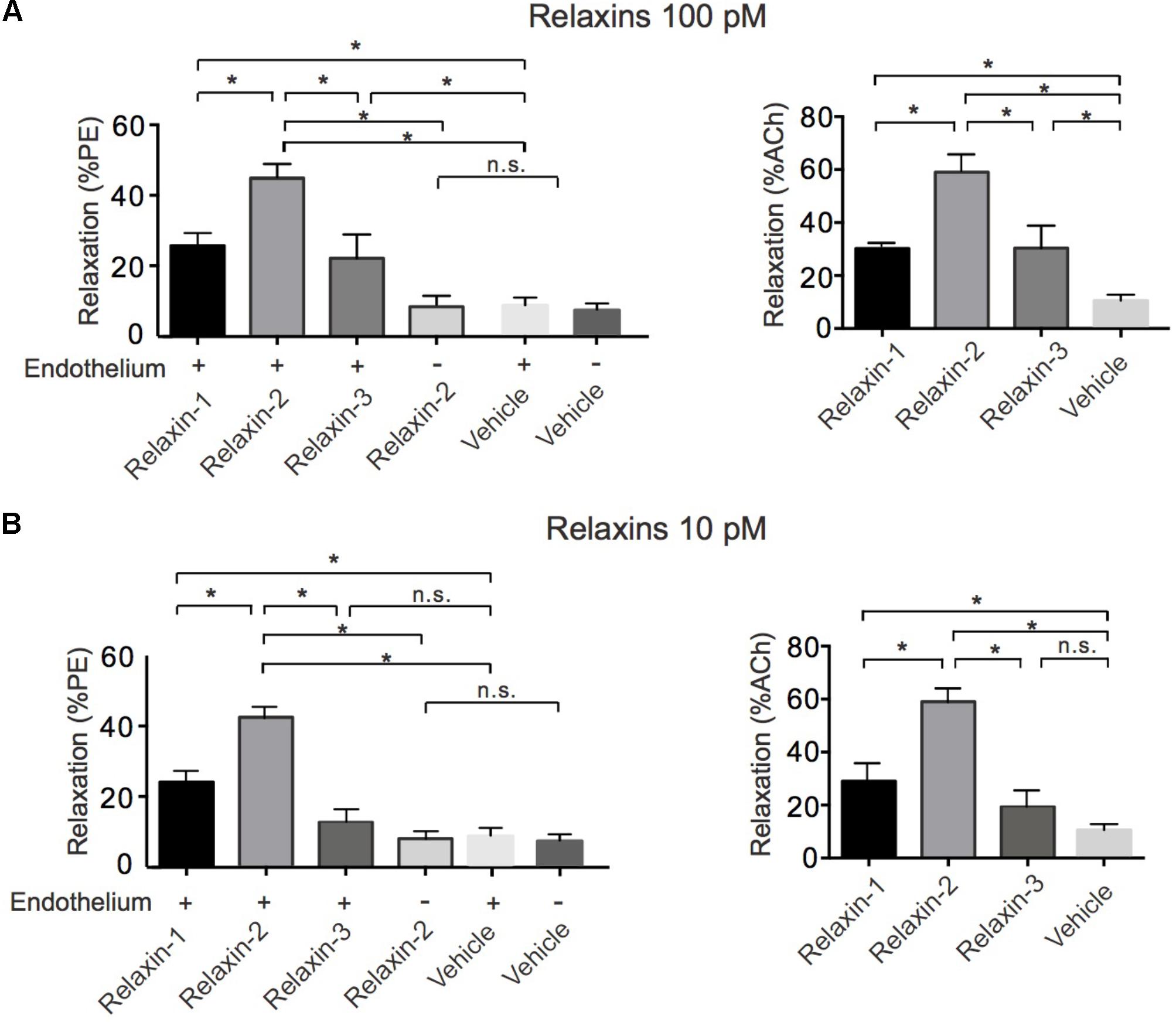
FIGURE 2. Summary data for relaxations induced by relaxins. (A) Summary data for % relaxation (PE) by 100 pM relaxin-1 (n = 6 rings out of 5 mice), 100 pM relaxin-2 (n = 8 out of six mice), 100 pM relaxin-3 (n = 6 out of five mice), vehicle control in vessels with endothelium (n = 6 out of four mice), and vehicle control in vessels without endothelium (n = 6 out of four mice) and for % relaxation relative to ACh (100%) response. Relaxin-1 (n = 6 out of five mice), relaxin-2 (n = 8 out of six mice), relaxin-3 (n = 6 out of five mice), and vehicle control in vessels with endothelium (n = 6 out of four mice). (B) Summary data for % relaxation (PE) by 10 pM relaxin-1 (n = 6 rings out of four mice), 10 pM relaxin-2 (n = 7 out of four mice), 10 pM relaxin-3 (n = 6 out of four mice), vehicle control in vessels with endothelium (n = 6 out of four mice), and vehicle control in vessels without endothelium (n = 6 out of four mice) and for % relaxation relative to ACh (100%) response. Relaxin-1 (n = 6 out of four mice), relaxin-2 (n = 7 out of four mice), relaxin-3 (n = 6 out of four mice), and vehicle control in vessels with endothelium (n = 6 out of four mice). ∗P < 0.05 using one-way ANOVA followed by Bonferroni multiple comparisons test; n.s., not significant.
Figure 3 shows that the RXFP1 blocker simazine (100 nM) inhibited relaxations induced by relaxin-2. Figure 4 shows that the Gαq protein inhibitor FR900359 (100 nM) (Schrage et al., 2015) inhibited concentration-dependent relaxations by ACh (Figure 4B), but had no effect on relaxin-2-dependent relaxations (Figure 4A). In these experiments, pre-tension was induced by KCl-PSS, which causes membrane depolarization to cause Ca2+ influx into vascular smooth muscle cells and hence vasocontraction independently of Gαq protein activation (Wirth et al., 2008). In contrast, the eNOS inhibitor L-NAME (100 μM) inhibited both relaxin-2- and ACh-induced relaxations (Figures 4A,B, respectively). Together, the data indicate that both relaxin-2 and ACh produce relaxation by an endothelial-dependent mechanism involving eNOS/NO release. However, whereas ACh utilizes an eNOS/NO signaling pathway involving muscarinic receptors coupled to Gq proteins (Kruse et al., 2012), relaxin-2 stimulates RXFP1 receptors coupled to G proteins other than Gq to produce eNOS/NO-dependent relaxation.
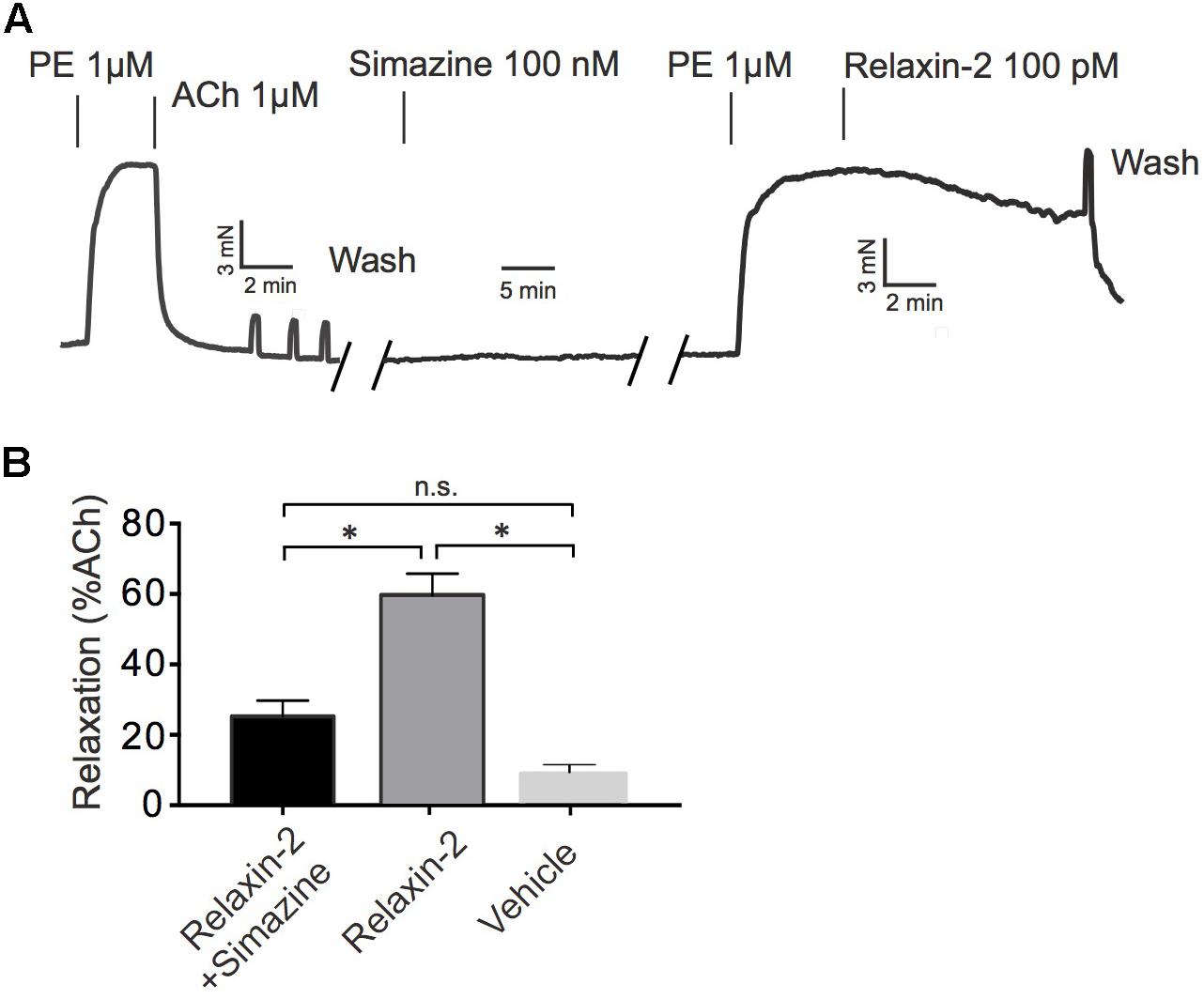
FIGURE 3. Effects of simazine on relaxin-2 induced relaxations. (A) Original representative recording on relaxin-2 induced relaxation in the presence of 100 nM simazine. (B) Summary data for relaxin-2 induced relaxation in the presence of 100 nM simazine (n = 9 out of six mice). ∗P < 0.05 using one-way ANOVA followed by Bonferroni multiple comparisons test; n.s., not significant.
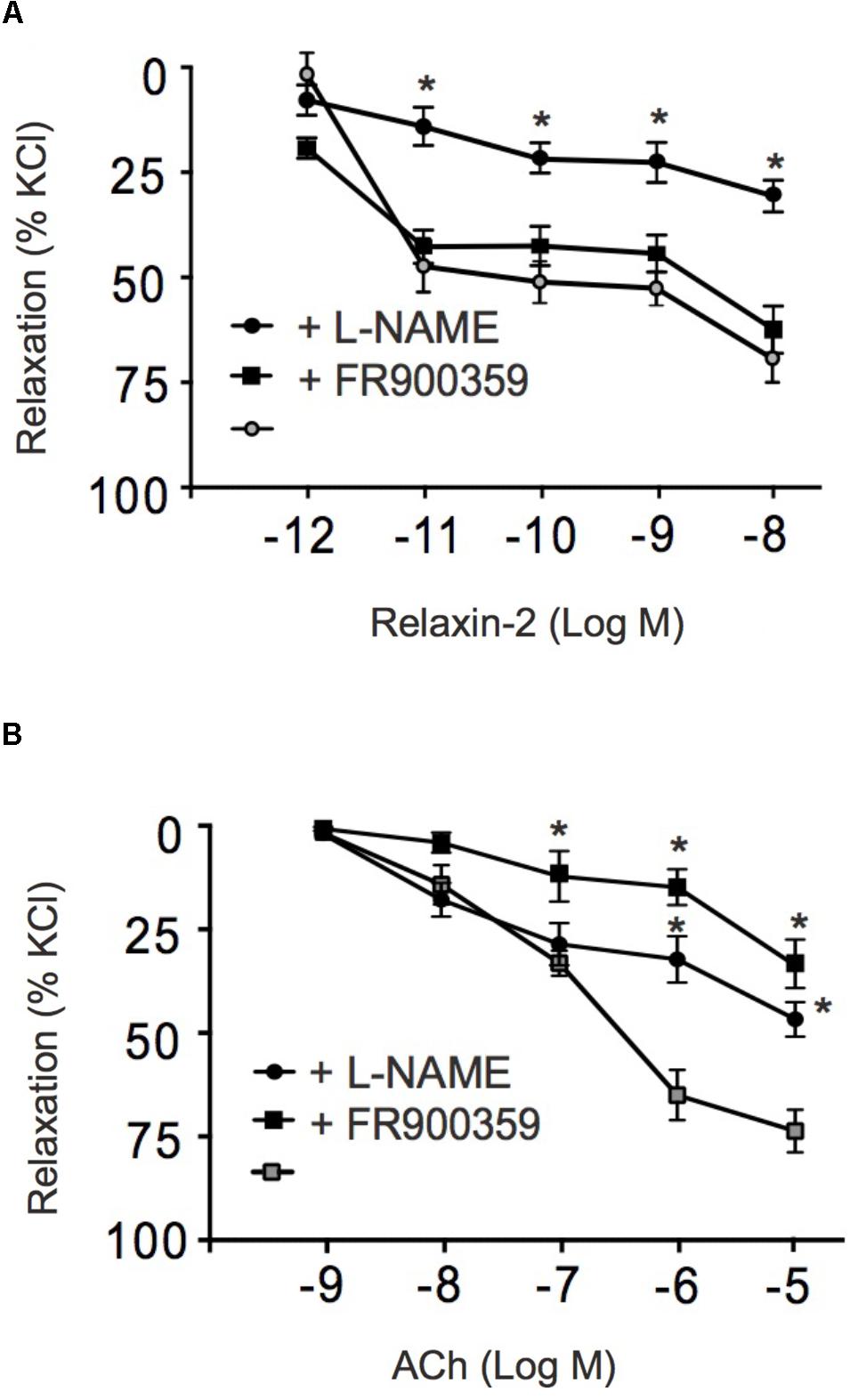
FIGURE 4. Effects of FR900359 and L-NAME on acetylcholine and relaxin-2 induced relaxations. (A) Summary data for relaxin-2 induced relaxationsin arteries in the presence of L-NAME (100 μM, 30 min) ( , n = 6 out of four mice), in the presence of FR900359 (100 nM, 30 min) (
, n = 6 out of four mice), in the presence of FR900359 (100 nM, 30 min) ( , n = 6 out of four mice), and in non-treated arteries (control group) (∙, n = 6 out of four mice). (B) Summary data for ACh-induced relaxations in arteries in the presence of L-NAME (100 μM) (
, n = 6 out of four mice), and in non-treated arteries (control group) (∙, n = 6 out of four mice). (B) Summary data for ACh-induced relaxations in arteries in the presence of L-NAME (100 μM) ( , n = 6 out of four mice), in the presence of FR900359 (100 nM) (, n = 6 out of four mice), and non-treated arteries (control group) (
, n = 6 out of four mice), in the presence of FR900359 (100 nM) (, n = 6 out of four mice), and non-treated arteries (control group) ( , n = 6 out of four mice). ∗P < 0.05 for relaxin-2 + L-NAME vs. control or ACh + L-NAME vs. non-treated vessels or ACh + FR900359 vs. non-treated vessels; repeated-measures two-way ANOVA, followed by Bonferroni post hoc test.
, n = 6 out of four mice). ∗P < 0.05 for relaxin-2 + L-NAME vs. control or ACh + L-NAME vs. non-treated vessels or ACh + FR900359 vs. non-treated vessels; repeated-measures two-way ANOVA, followed by Bonferroni post hoc test.
To determine a possible role of Gi proteins in relaxin-2-dependent relaxation, we treated mice with pertussis toxin (PTX), which is used as pan-Gαi-inhibitor in vivo (Devanathan et al., 2015). Control mice were treated with 0.9% NaCl only. Figure 5A shows that Gq-dependent relaxations by ACh were preserved in mesenteric arteries treated with PTX. However, relaxation in response to relaxin-2 were abolished in mesenteric arteries from mice pre-treated with PTX (Figures 5A,C) compared to control arteries (Figures 5B,C). In contrast, relaxations in response to ACh were not changed by PTX treatment (Figure 5D). The Gi-family comprises three closely related Gα members, Gαi1-3, with Gαi2 and Gαi3 abundantly expressed in the cardiovascular system (Hippe et al., 2015). We therefore used Gαi2-deficient (Gnai2-/-) and Gαi3-deficient (Gnai3-/-) mice to determine which Gαi isoforms are involved in relaxin-2 mediated relaxations. Figure 6 shows that relaxin-2 induced relaxations were reduced in Gαi2-deficient arteries (Figures 6A,C) but not in Gαi3-deficient arteries (Figures 6B,D). These data indicate that Gαi2 plays an important role in RXFP1 mediated relaxations.
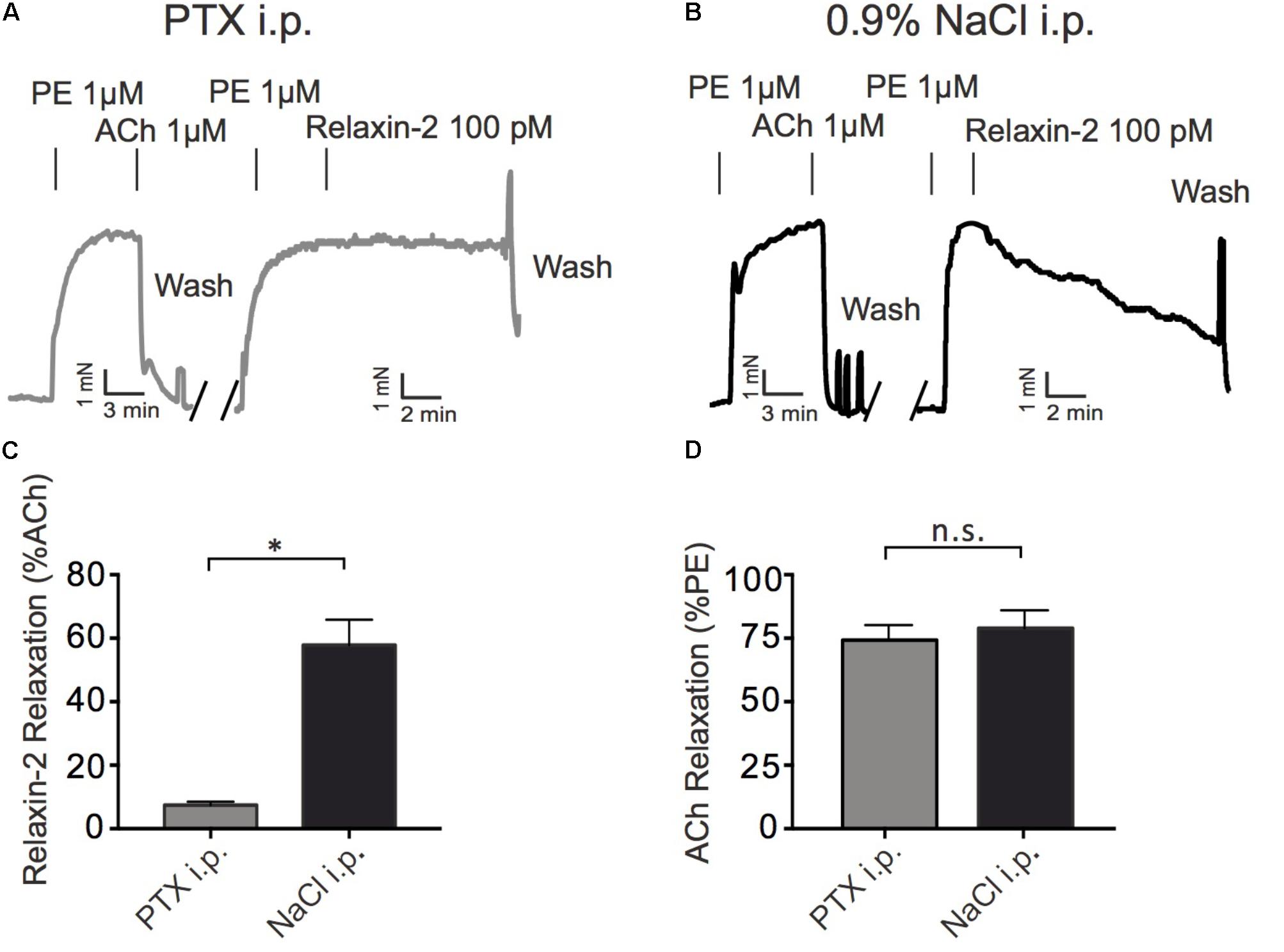
FIGURE 5. Effects of treatment of mice with pertussis toxin (PTX) or sham (0.9% NaCl) on relaxin-2 relaxations. (A) Original representative recording of relaxin-2 induced relaxations in arteries of PTX treated mice. (B) Original representative recording of relaxin-2 induced relaxations in arteries of 0.9% NaCl treated (control) mice. (C) Summary data of relaxin-2 induced relaxation. (D) Summary data of ACh induced relaxation. PTX group; n = 11 out of eight mice. 0.9% NaCl group; n = 7 out of four mice. ∗P < 0.05 using unpaired t-test; n.s., not significant.
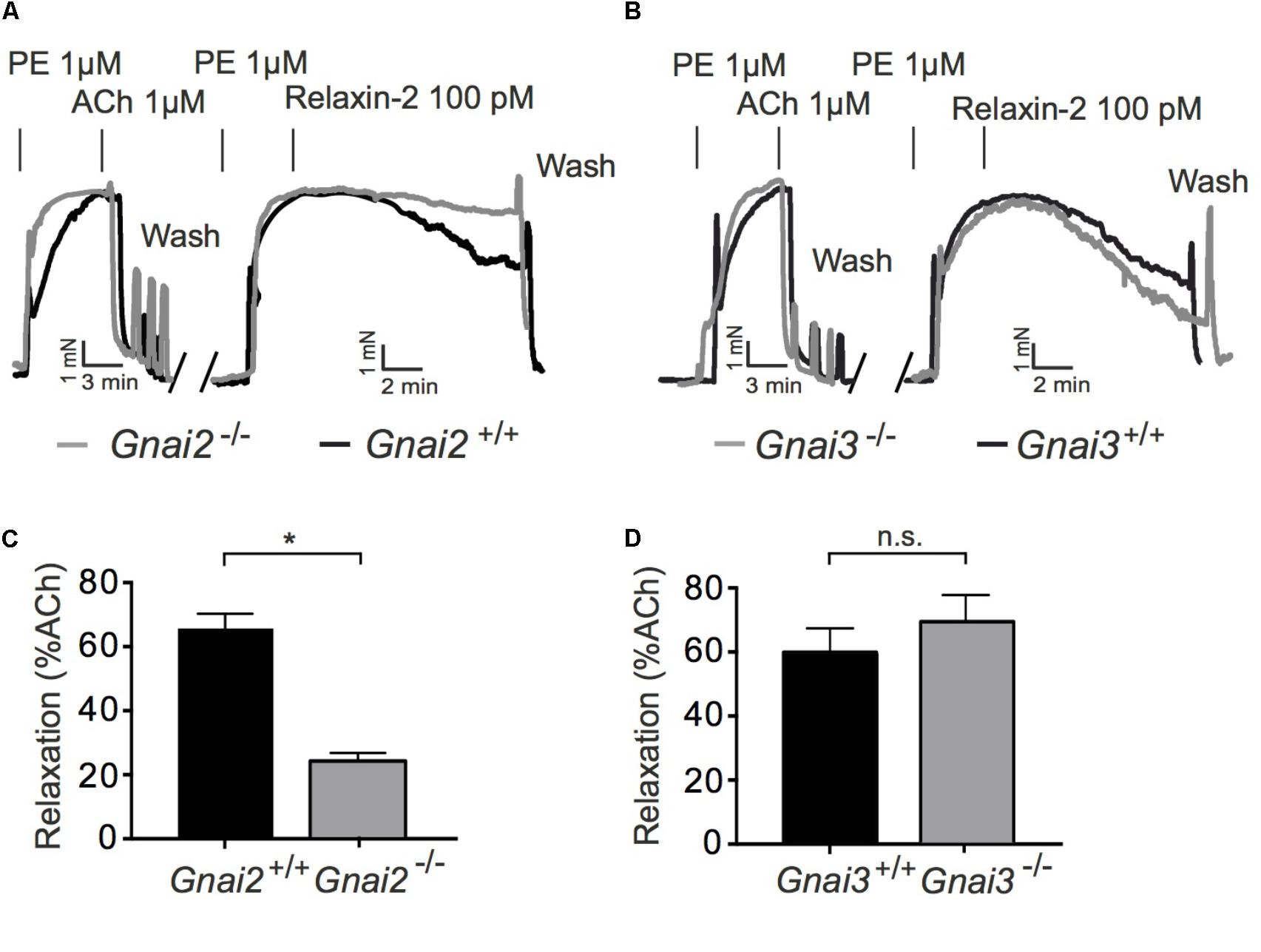
FIGURE 6. Vasorelaxant effects of relaxin-2 in Gαi2 deficient (Gnai2-/-), Gαi3 deficient (Gnai3-/-), and control arteries. (A,B) Original representative recordings. (C) Summary of data. Gαi2 deficient arteries; n = 9 out of five mice. Control; n = 9 out of four mice. (D) Summary of data. Gαi3 deficient arteries; n = 6 out of five mice. Control arteries; n = 7 out of four mice. ∗P < 0.05 using unpaired t-test.; n.s., not significant.
PI3K signaling has been proposed to mediate slow and sustained activation of eNOS and subsequent NO release in cell culture experiments (Dimmeler et al., 1999). We tested the contribution of different PI3K isoforms in eNOS/NO-dependent RXFP1 receptor-mediated relaxations in comparison to ACh-induced relaxations. Vessels were pre-contracted with PE, incubated with L-NAME or various PI3K inhibitors and subsequently exposed to relaxin-2 (Figure 7). Figures 7A,B shows that relaxin-2-induced relaxations were inhibited by L-NAME. The PI3Kβ inhibitor TGX-221 (Figure 7B) and the PI3Kγ inhibitor AS-252424 (Figure 7B) had similar inhibitory effects on stimulation by relaxin-2. However, relaxin-2-induced relaxations were not affected by the PI3Kα inhibitor PI-103 (Figure 7B). Together, the data suggest that PI3K isoforms β and γ are essential intermediate signaling components in eNOS/NO-dependent relaxation by relaxin-2, which are controlled by relaxin-2 that act on RXFP1 receptors coupled to Gi2 proteins.
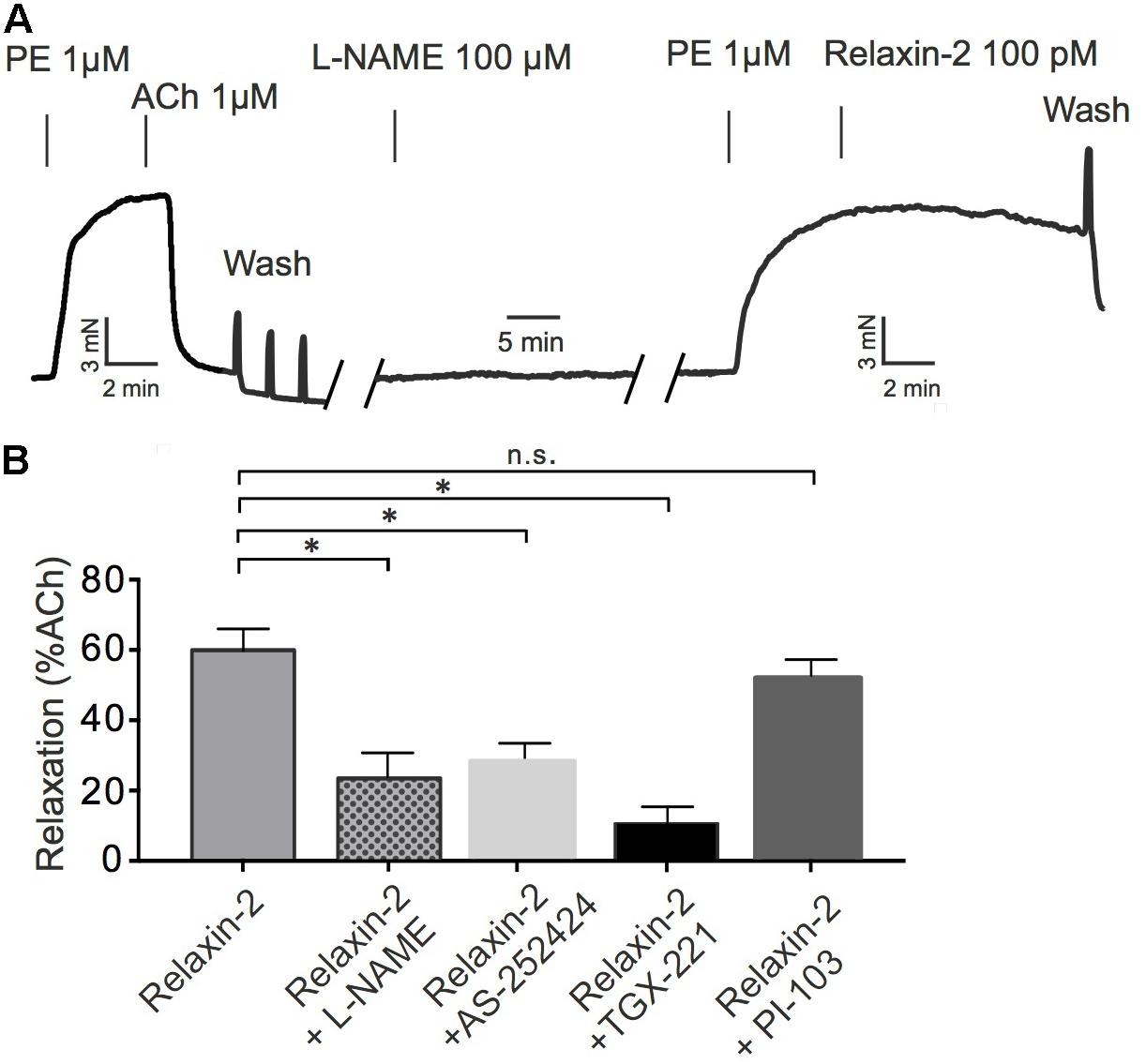
FIGURE 7. Effects of PI3K inhibitors on relaxin-2 induced relaxation. (A) Original representative recording. (B) Summary of data. Relaxin-2-induced relaxation in the presence of 100 μM L-NAME (n = 6 out of four mice), 100 nM AS-252424 (n = 7 out of four mice), 100 nM TGX-221 (n = 7 out of four mice), or 100 nM PI-103 (n = 6 out of four mice). ∗P < 0.05 using one-way ANOVA followed by Bonferroni multiple comparisons test; n.s., not significant.
Perivascular adipose tissue (PVAT) plays a functional role in regulating the contractile state of arteries by production of numerous vasodilatory substances (Gollasch, 2017). Since relaxin(s) are expressed in adipose tissue (Hausman et al., 2006), these polypeptide hormones might represent an adipose-derived relaxing factor released by PVAT to contribute to anti-contractile effects on arterial tone. Therefore, we studied vascular contractions in response to PE in arterial ring in the presence and absence of PVAT. Figure 8 shows that in the absence of PVAT phenylephrine was more potent in causing effective contractions of arterial rings than in the presence of PVAT. This anti-contractile effect of PVAT was not influenced by the incubation of the arteries with the RXFP1-receptor antagonist simazine (100 nM) (Figure 8). We also performed control experiments on whether simazine or the other drugs used in this study affect contractions caused by PE. The results in Supplementary Figure S1 show that contractions caused by PE in the absence (first PE application) and presence of the respective drugs (second PE application) were not different. The data demonstrate that neither simazine nor the other drugs used in this study affected PE contractions.
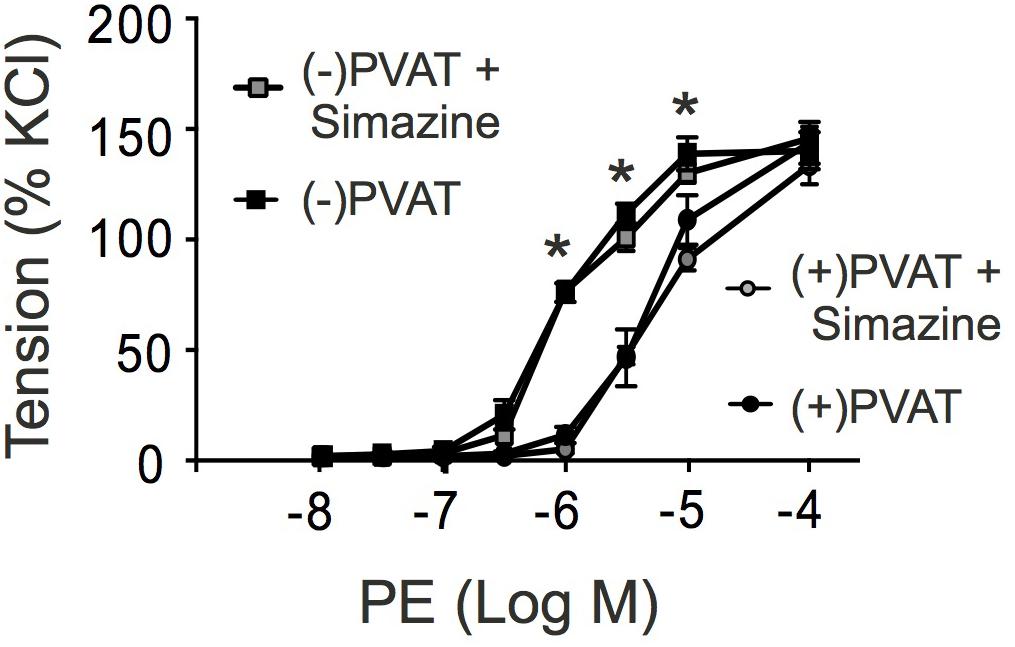
FIGURE 8. Effects of simazine on phenylephrine (PE)-dependent contractions in the presence (+) or absence (-) of perivascular adipose tissue (PVAT). Summary data for PE induced contractions in (+) PVAT (∙, n = 8 out of four mice) or (-) PVAT (, n = 8 out of four mice) rings. Incubation of the rings with simazine (100 nM, 30 min) had no effects on contractions caused by PE in (+) PVAT (∙, n = 8 out of four mice) and (-) PVAT (, n = 9 out of 4 mice) arterial rings (P > 0.05 each). ∗P < 0.05 for (-) PVAT vs. (+) PVAT or (-) PVAT + Simazine vs. (+) PVAT + Simazine; repeated-measures two-way ANOVA, followed by Bonferroni post hoc test.
Our study shows that relaxins are extremely potent (in the low picomolar range) endothelium-dependent and L-NAME-sensitive vasodilators in mouse mesenteric arteries. Similar vasodilatory potencies of relaxins have been observed renal arteries of rats (Novak et al., 2002) and human small gluteal and subcutaneous arteries (Fisher et al., 2002). Among the three human relaxins studied, we identified relaxin-2 as the most effective vasodilator, which produces eNOS/NO-dependent relaxation most likely due to activation of RXFP1 coupled to a Gi2-PI3kβ/PI3kγ pathway. Although, we probed human relaxins in a non-human vascular preparation, that is, in isolated arteries from mice, this is the first study suggesting that RXFP1 coupled to a Gi2-PI3kβ/PI3kγ pathway is capable of producing vascular relaxation. Furthermore, our data indicate that this pathway does not contribute to PVAT control of arterial tone.
Relaxin-encoding genes are present in all mammals and responsible for the production of the relaxin peptides that have been initially found in circulating blood during pregnancy. However, more recent studies have observed that relaxins are produced in many tissues in mammals as paracrine or autocrine factors to exert a number of different physiological roles in the vasculature, which may exhibit protective effects in cardiovascular disease (Samuel et al., 2006). The injection of recombinant human relaxins to normotensive rats for 1 to 6 h induced a systemic vasodilatory response (Debrah et al., 2005). This finding suggests that certain vascular beds, for example, in the kidney or mesentery, are able to respond by a vasodilatory response caused by relaxins. Relaxin-2 has been identified as the most important member of the relaxin family and major circulating form of relaxin peptides in humans (Grossman and Frishman, 2010). Relaxin-1 is also believed to exist as circulatory peptide in the circulation (Bathgate et al., 2013), but the function of the relaxin-1 in humans and higher primates is mostly unclear. Relaxin-3 is the most recently identified member of the relaxin family and is primarily expressed in the brain of mammals (Heidari et al., 2018). In our study, we first tested the vasoactive function of all three relaxins in mouse mesenteric arteries. We found that all three relaxins can produce relaxations, with relaxin-2 being the most effective member of the relaxin family. In contrast to ACh, which produces a rapid relaxation, relaxin-1, relaxin-2, and relaxin-3 induced slow and delayed relaxations, indicating different underlying signaling mechanisms between ACh and relaxins to cause endothelium-dependent relaxations.
Studies on isolated vessels have shown that the endothelium is necessary for vasodilation by relaxin in renal and human subcutaneous arteries (McGuane et al., 2011). In our study on mouse mesenteric arteries, we also found that removal of the endothelium or treatment with L-NAME inhibited relaxation by relaxin-2. Our data support previous findings indicating that relaxins cause vasodilation primarily by an eNOS/NO-dependent mechanism (McGuane et al., 2011; Ng et al., 2015; Leo et al., 2016). Relaxin produces its major effects via specific G-protein-coupled receptors (GPCRs), that is, RXFP 1-4 (Hsu et al., 2002; Bathgate et al., 2006). Among them, RXFP1 was the first to be identified and remains in focus of interest because of its crucial role in the cardiovascular system (Bathgate et al., 2013). RXFP2 receptors are mainly activated by insulin-like (INSL) 3 in the gubernaculum to facilitate testicular descent into the scrotum, and RXFP3 and RXFP4 receptors are activated by relaxin-3 and INSL5, respectively (van der Westhuizen et al., 2008). RXFP1 has the highest binding potency for relaxin-2 within the several members of the relaxin peptide family (Samuel et al., 2006; Nistri et al., 2007). RXFP1 is predominantly expressed in endothelial cells of mesenteric arteries and veins, but also expressed in the aorta and vena cava (Novak et al., 2006; Jelinic et al., 2014). In our experiments, the RXFP1 antagonist simazine (Park et al., 2016) largely inhibited the endothelium-dependent relaxation induced by relaxin-2, which supports the idea that relaxin-2 produces relaxation primarily through binding to and activation of RXFP1 in the endothelium (Figure 9). The remaining relaxation of about 25% in simazine-treated vessels may represent spontaneous relaxation or caused by RXFP activation distinct from RXFP1 in the vasculature or both of them.
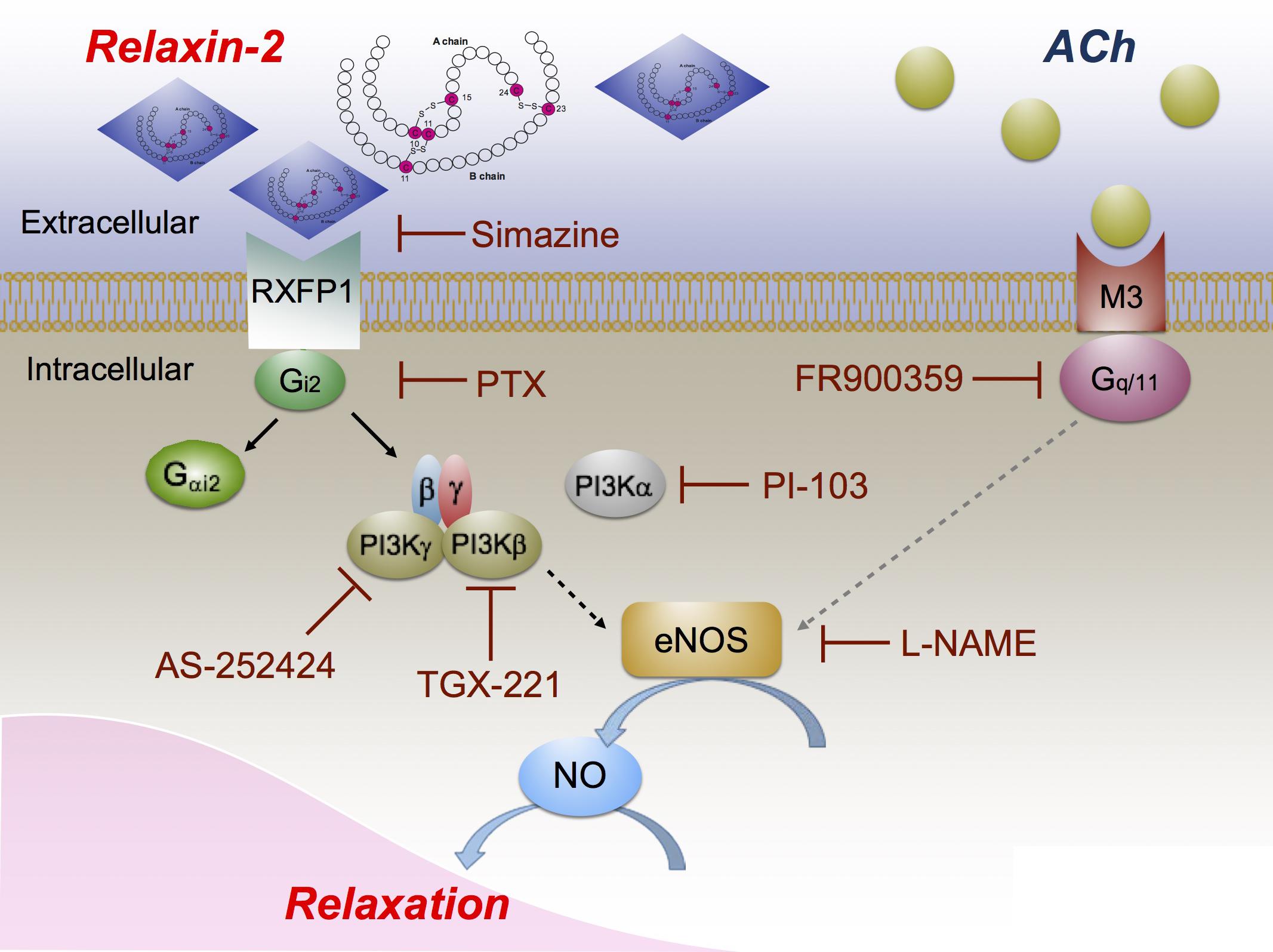
FIGURE 9. Proposed vasodilatory pathways caused by relaxin-2. Relaxin-2 activates RXFP1 (blocked by simazine), which leads to Gi2 activation and dissociation of Gαi2 and βγ subunits (blocked by PTX). Gβγ subunits in turn activate PI3Kβ (blocked by TGX-221) and PI3Kγ (blocked by AS-252424) to initiate eNOS activation and NO release (blocked by L-NAME) to cause relaxation. PI3Kα (blocked by PI-103) seems not to be involved in this pathway. On the other hand, ACh binds to muscarinic M3 receptors coupled to Gq/11 (blocked by FR900359) to produce eNOS/NO dependent arterial relaxation.
We aimed to identify the G proteins coupled with RXFP1 to cause eNOS/NO-dependent vasodilation by relaxin-2. Heterotrimeric G proteins, which mediate signals from cell surface receptors to cellular effectors, are composed of α, β, and γ subunits, of which Gα defines the class of G proteins (Simon et al., 1991). The α subunits that define the basic properties of a heterotrimeric G protein can be divided into four families, namely Gs, Gq/G11, Gi/Go, and G12/13 (Kaziro et al., 1988; Wettschureck and Offermanns, 2005). ACh produces relaxation mainly via the Gq/G11-coupled M3 receptor subtype (Figure 9) (Jaiswal et al., 1991; Kruse et al., 2012). In our study, we found that ACh-induced relaxations were abolished by FR900359, a selective mammalian Gq/G11 signaling inhibitor (Schrage et al., 2015), while the relaxin-2-induced relaxations were not blocked by this drug. We conclude that vasodilatory RXFP1-coupled G proteins are distinct from Gq/G11 (Figure 9). Our data using the pan Gi/Go inhibitor PTX (Simon et al., 1991) indicate that Gi/Go proteins could play a major role in relaxin-2 mediated vasodilation. G0 is particularly abundant in the neuronal and the neuroendocrine system and the Gi-family includes three closely related Gα members, Gαi1-3, which display overlapping expression patterns with Gαi2 and Gαi3, abundantly expressed in the cardiovascular system (Hippe et al., 2015). According to previous studies on cultured cells, RXFP1 has been suggested to couple to Gαi3 to release Gβγ dimers to activate the PI3K pathway via Akt phosphorylation and subsequently initiate NOS (Halls et al., 2006; McGuane et al., 2011). Our experiments using Gαi2-deficient (Gnai2-/-) and Gαi3-deficient (Gnai3-/-) mice failed to implicate an important role of Gαi3, but revealed a key role of Gαi2 in RXFP1-eNOS/NO relaxation. We found that relaxin-2 induced relaxation was impaired in arteries from Gnai2-/-, but not from Gnai3-/- mice. Although structural similarity between the three Gi subforms suggests that they may exhibit overlapping functions, Gαi2/Gαi3-double-deficient mice cannot be used for myography experiments because they die in utero at early embryonic stages. Nevertheless, present data obtained in mice lacking Gαi2 or Gαi3 indicate distinct biological key roles of these two Gαi-isoforms (Köhler et al., 2014). Thus, we believe that relaxin-2 relaxes mouse mesenteric arteries primarily via RXFP1 activation and coupling to Gi2 but not Gi3 (Figure 9). Moreover, since the pan Gi/Go inhibitor PTX shows a stronger inhibitory effect than the absence of Gαi2, we should also consider that relaxin-2 could partly act through other Gi proteins.
Following the release of Gβγ from Gαi, it was recently suggested that the class I PI3K represents a target for Giβγ signaling by relaxins (McGuane et al., 2011). Accordingly, we found that eNOS activation by relaxin was inhibited by the pan PI3K inhibitors Wortmannin or LY294002 in cultured endothelial cells (Dessauer and Nguyen, 2005). Based on the association with non-catalytic binding proteins, catalytic subunits of class I PI3Ks are subdivided into class IA-isoforms (p110α, -β, and -δ) or class IB p110γ (Hennessy et al., 2005). In this study, we found that relaxin-2-RXFP1 relaxation was inhibited by the PI3Kγ and PI3Kβ inhibitors AS-252424 and TGX-221, respectively. The PI3Kα inhibitor PI-103 had no effects. These data indicate that class I PI3Kγ and PI3Kβ represents likely a target for Gi2 signaling by relaxins to cause eNOS/NO dependent relaxation. We are not aware of selective PI3kδ inhibitors to determine a possible additional role of PI3Kδ in RXFP1-mediated relaxation. Nevertheless, our data indicate that class I PI3K activation by a target for RXFP/Gi-βγ signaling to cause eNOS activation is not only a cell culture phenomenon (Dimmeler et al., 1999; Dessauer and Nguyen, 2005), but is important for relaxins to produce vascular relaxation (Figure 9). Of note, the relaxin-2-induced relaxations were not fully abolished by TGX-221 or AS-252424. This may indicate that both PI3K isoforms or another PI3K isoform are involved in this vasoregulatory pathway. Also, in addition to the slow activation process of the eNOS/NO by relaxins via Gi-PI3K (McGuane et al., 2011), there is also an ultra-slow mechanism of eNOS/NO stimulation via κ upon exposure of cultured endothelial cells to relaxin (Dschietzig et al., 2003). Although this putative mechanism cannot be examined by the methodological approach used in our study, such non-RXFP-mediated effects may contribute to the effects of relaxins in the vasculature in vivo.
Microarray studies showed expression of relaxin in pig adipose tissue (Hausman et al., 2006). According to our previous studies, PVAT inhibits vessel contraction and produces endothelium-independent relaxation by releasing adipocyte-derived relaxing factor (ADRF) (Löhn et al., 2002; Dubrovska et al., 2004; Verlohren et al., 2004; Tsvetkov et al., 2016b). PVAT dysfunction is characterized by disturbed secretion of various adipokines, which, together with endothelial dysfunction, contribute to hypertension and cardiovascular risk (Lian and Gollasch, 2016). With our interest, we aimed to unravel whether relaxin(s) acting through RXFP1 receptors may represent an ADRF. Our data showed that the RXFP1 antagonist simazine does not influence the anti-contractile effects of PVAT, which indicates that it is unlikely that relaxin(s) acting though RXFP1 receptors contributes to PVAT control of arterial tone, at least in mouse mesenteric arteries.
There are a number of limitations in the present study. First, we studied human relaxins rather than mouse relaxins to identify Gi protein-dependent vasodilatory pathways in the murine vasculature. Utilizing the murine vasculature enabled us to use advance of transgenic mouse models. Since serelaxin (RLX030) represents the recombinant form of human relaxin-2, which shows quite reliable therapies in cardiovascular diseases (Papadopoulos et al., 2013; Parikh et al., 2013; Teerlink et al., 2013), we are aimed to identify the function and mechanisms of RXFP1signaling pathways utilized by human relaxins in the vasculature. Of note, human relaxin-2 (H2 relaxin) is the counterpart of mouse relaxin-1 (M1 relaxin) within the structurally related insulin/relaxin superfamily, and mouse RXFP1 shows 89% identity to human RXFP1 (Sherwood, 2004; Kong et al., 2010). Binding of relaxins to RXFP1 is mediated via high-affinity binding to extracellular domain of RXFP1 and an additional binding site in the transmembrane (TM) exoloops (Sherwood, 2004). A recent study has shown that specific residues in the center of the H2 relaxin A-chain are necessary for ligand activity at RXFP1 (Park et al., 2008). Importantly, modeling of the ligand–receptor interaction for different RXFP receptors suggests that once the B-chains of the ligands are bound to the primary binding site in a large ectodomain with 10 leucine-rich repeats (LRRs) that the A-chain is presented in a favorable orientation for interaction with the TM exoloops (Hartley et al., 2009). Hence, it is likely that human and murine relaxins utilize similar common mechanisms to activate RXFP1 receptors, although there might be species-dependent differences in the mode of interaction between the individual relaxins, extracellular RXFP1 domains and the TM exoloops of the individual receptors. Second, we used female instead of male Gnai2-/- and Gnai3-/- mice and respective littermate controls. Because of the estrous cycle in females, there could be gender-related differences of the vascular reactivity and endothelial function (Sader and Celermajer, 2002), which should be considered. Nevertheless, our data clearly demonstrate a genotype-dependent inhibition of relaxin-2 relaxations, that is, lack of relaxin-2 relaxation in arteries from Gnai2-/-, but not from Gnai3-/- mice. Lastly, considering that inhibition of endothelium may have influence on PE pre-contraction levels, for example, by mitogen-activated protein kinase kinase/extracellular signal-regulated kinase-dependent mechanisms (Molnar et al., 2008), we compared relaxin relaxations with ACh relaxations also in KCl-precontracted vessels. Our data show relaxin-2 relaxations involve an RXFP1-Gi protein pathway in both conditions. The Gq inhibitor FR900359 did not inhibit relaxin-2 relaxations and PE contractions were not affected the drugs used in our study (Supplementary Figure S1). Nevertheless, removal of the endothelium may stabilize the level of pre-contraction to play an additional role in eliminating endothelium-dependent RXFP1 relaxations.
In summary, we provide evidence that all three relaxins, that is, relaxin-1, relaxin-2, and relaxin-3, are potent vasodilators in mesenteric arteries of mice. Among them, relaxin-2 is the strongest vasodilator, which produces relaxation via activation of endothelial RXFP1 coupled to a Gi2-PI3Kγ/β-eNOS/NO pathway. Based on the fact that long- and intermediate-distance conduction of vasodilation is common in the circulation, localized releases of relaxins within a tissue might be able to produce remote vasodilations in regions of reduced blood flow distribution. As a potent vasodilatory Gαi2-coupled receptor, targeting RXFP1 may represent a promising avenue to study Gi-coupled receptor based drugs in cardiovascular disease that may allow clarifying specific roles for Gαi2 and Gαi3 in response to GPCR activation directly in the vasculature.
XL performed the wire myography experiments. XL and MG drafted the article. All authors planned and designed the experimental studies and contributed to its completion.
This study was supported by Deutsche Forschungsgemeinschaft (DFG), Deutsche Akademische Austauschdienst (DAAD), and Shanghai Tongji Hospital. EK and GK gratefully acknowledge support by DFG-funded research unit FOR2372 to isolate FR900359.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2018.01234/full#supplementary-material
FIGURE S1 | Contractions induced by phenylephrine (PE). Summary data of contractions induced by PE in the absence (first application) and presence (second) application of vehicle (control), simazine, L-NAME, AS252424, TGX-221, or PI-103. For concentrations and number of rings, see other figure legends. w/o endothelium; effects of vehicle in the absence of endothelium. In all other experiments, the endothelium was intact. n.s., not significant.
Ali, K., Camps, M., Pearce, W. P., Ji, H., Ruckle, T., Kuehn, N., et al. (2008). Isoform-specific functions of phosphoinositide 3-kinases: p110 delta but not p110 gamma promotes optimal allergic responses in vivo. J. Immunol. 180, 2538–2544. doi: 10.4049/jimmunol.180.4.2538
Bani-Sacchi, T., Bigazzi, M., Bani, D., Mannaioni, P. F., and Masini, E. (1995). Relaxin-induced increased coronary flow through stimulation of nitric oxide production. Br. J. Pharmacol. 116, 1589–1594. doi: 10.1111/j.1476-5381.1995.tb16377.x
Bathgate, R. A., Halls, M. L., van der Westhuizen, E. T., Callander, G. E., Kocan, M., and Summers, R. J. (2013). Relaxin family peptides and their receptors. Physiol. Rev. 93, 405–480. doi: 10.1152/physrev.00001.2012
Bathgate, R. A., Ivell, R., Sanborn, B. M., Sherwood, O. D., and Summers, R. J. (2006). International union of pharmacology LVII: recommendations for the nomenclature of receptors for relaxin family peptides. Pharmacol. Rev. 58, 7–31. doi: 10.1124/pr.58.1.9
Bell, R. J., Eddie, L. W., Lester, A. R., Wood, E. C., Johnston, P. D., and Niall, H. D. (1987). Relaxin in human pregnancy serum measured with an homologous radioimmunoassay. Obstet. Gynecol. 69, 585–589.
Bonner, J. S., Lantier, L., Hocking, K. M., Kang, L., Owolabi, M., James, F. D., et al. (2013). Relaxin treatment reverses insulin resistance in mice fed a high-fat diet. Diabetes Metab. Res. Rev. 62, 3251–3260. doi: 10.2337/db13-0033
Ciccarelli, M., Cipolletta, E., Santulli, G., Campanile, A., Pumiglia, K., Cervero, P., et al. (2007). Endothelial beta2 adrenergic signaling to AKT: role of Gi and SRC. Cell. Signal. 19, 1949–1955. doi: 10.1016/j.cellsig.2007.05.007
Conrad, K. P. (2010). Unveiling the vasodilatory actions and mechanisms of relaxin. Hypertension 56, 2–9. doi: 10.1161/HYPERTENSIONAHA.109.133926
Debrah, D. O., Conrad, K. P., Jeyabalan, A., Danielson, L. A., and Shroff, S. G. (2005). Relaxin increases cardiac output and reduces systemic arterial load in hypertensive rats. Hypertension 46, 745–750. doi: 10.1161/01.HYP.0000184230.52059.33
Dessauer, C. W., and Nguyen, B. T. (2005). Relaxin stimulates multiple signaling pathways: activation of cAMP, PI3K, and PKCzeta in THP-1 cells. Ann. N. Y. Acad. Sci. 1041, 272–279. doi: 10.1196/annals.1282.040
Devanathan, V., Hagedorn, I., Kohler, D., Pexa, K., Cherpokova, D., Kraft, P., et al. (2015). Platelet Gi protein Galphai2 is an essential mediator of thrombo-inflammatory organ damage in mice. Proc. Natl. Acad. Sci. U.S.A. 112, 6491–6496. doi: 10.1073/pnas.1505887112
Dimmeler, S., Fleming, I., Fisslthaler, B., Hermann, C., Busse, R., and Zeiher, A. M. (1999). Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399, 601–605. doi: 10.1038/21224
Dschietzig, T., Bartsch, C., Richter, C., Laule, M., Baumann, G., and Stangl, K. (2003). Relaxin, a pregnancy hormone, is a functional endothelin-1 antagonist: attenuation of endothelin-1-mediated vasoconstriction by stimulation of endothelin type-B receptor expression via ERK-1/2 and nuclear factor-kappaB. Circ. Res. 92, 32–40. doi: 10.1161/01.RES.0000051884.27117.7E
Du, X. J., Bathgate, R. A., Samuel, C. S., Dart, A. M., and Summers, R. J. (2010). Cardiovascular effects of relaxin: from basic science to clinical therapy. Nat. Rev. Cardiol. 7, 48–58. doi: 10.1038/nrcardio.2009.198
Dubrovska, G., Verlohren, S., Luft, F. C., and Gollasch, M. (2004). Mechanisms of ADRF release from rat aortic adventitial adipose tissue. Am. J. Physiol. Heart Circ. Physiol. 286, H1107–H1113. doi: 10.1152/ajpheart.00656.2003
Eisen, A., Fisman, E. Z., Rubenfire, M., Freimark, D., McKechnie, R., Tenenbaum, A., et al. (2004). Ischemic preconditioning: nearly two decades of research. A comprehensive review. Atherosclerosis 172, 201–210. doi: 10.1016/S0021-9150(03)00238-7
Ezan, J., Lasvaux, L., Gezer, A., Novakovic, A., May-Simera, H., Belotti, E., et al. (2013). Primary cilium migration depends on G-protein signalling control of subapical cytoskeleton. Nat. Cell Biol. 15, 1107–1115. doi: 10.1038/ncb2819
Fésüs, G., Dubrovska, G., Gorzelniak, K., Kluge, R., Huang, Y., Luft, F. C., et al. (2007). Adiponectin is a novel humoral vasodilator. Cardiovasc. Res. 75, 719–727. doi: 10.1016/j.cardiores.2007.05.025
Fisher, C., MacLean, M., Morecroft, I., Seed, A., Johnston, F., Hillier, C., et al. (2002). Is the pregnancy hormone relaxin also a vasodilator peptide secreted by the heart? Circulation 106, 292–295.
Gohla, A., Klement, K., Piekorz, R. P., Pexa, K., vom Dahl, S., Spicher, K., et al. (2007). An obligatory requirement for the heterotrimeric G protein Gi3 in the antiautophagic action of insulin in the liver. Proc. Natl. Acad. Sci. U.S.A. 104, 3003–3008. doi: 10.1073/pnas.0611434104
Gollasch, M. (2017). Adipose-vascular coupling and potential therapeutics. Annu. Rev. Pharmacol. Toxicol. 57, 417–436. doi: 10.1146/annurev-pharmtox-010716-104542
Goto, M., Swanson, L. W., and Canteras, N. S. (2001). Connections of the nucleus incertus. J. Comp. Neurol. 438, 86–122. doi: 10.1002/cne.1303
Grossman, J., and Frishman, W. H. (2010). Relaxin: a new approach for the treatment of acute congestive heart failure. Cardiol. Rev. 18, 305–312. doi: 10.1097/CRD.0b013e3181f493e3
Halls, M. L., Bathgate, R. A., and Summers, R. J. (2006). Relaxin family peptide receptors RXFP1 and RXFP2 modulate cAMP signaling by distinct mechanisms. Mol. Pharmacol. 70, 214–226.
Halls, M. L., van der Westhuizen, E. T., Bathgate, R. A., and Summers, R. J. (2007). Relaxin family peptide receptors–former orphans reunite with their parent ligands to activate multiple signalling pathways. Br. J. Pharmacol. 150, 677–691. doi: 10.1038/sj.bjp.0707140
Hansell, D. J., Bryant-Greenwood, G. D., and Greenwood, F. C. (1991). Expression of the human relaxin H1 gene in the decidua, trophoblast, and prostate. J. Clin. Endocrinol. Metab. 72, 899–904. doi: 10.1210/jcem-72-4-899
Hartley, B. J., Scott, D. J., Callander, G. E., Wilkinson, T. N., Ganella, D. E., Kong, C. K., et al. (2009). Resolving the unconventional mechanisms underlying RXFP1 and RXFP2 receptor function. Ann. N. Y. Acad. Sci. 1160, 67–73. doi: 10.1111/j.1749-6632.2009.03949.x
Hausman, G. J., Poulos, S. P., Richardson, R. L., Barb, C. R., Andacht, T., Kirk, H. C., et al. (2006). Secreted proteins and genes in fetal and neonatal pig adipose tissue and stromal-vascular cells. J. Anim. Sci. 84, 1666–1681. doi: 10.2527/jas.2005-539
Heidari, S., Taromchi, A. H., Nejatbakhsh, R., and Shokri, S. (2018). Expression and localisation of RXFP3 in human spermatozoa and impact of INSL7 on sperm functions. Andrologia 50:e12928. doi: 10.1111/and.12928
Hennessy, B. T., Smith, D. L., Ram, P. T., Lu, Y., and Mills, G. B. (2005). Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 4, 988–1004. doi: 10.1038/nrd1902
Hippe, H. J., Ludde, M., Schnoes, K., Novakovic, A., Lutz, S., Katus, H. A., et al. (2015). Competition for Gbetagamma dimers mediates a specific cross-talk between stimulatory and inhibitory G protein alpha subunits of the adenylyl cyclase in cardiomyocytes. Naunyn Schmiedebergs Arch. Pharmacol. 386, 459–469. doi: 10.1007/s00210-013-0876-x
Hsu, S. Y., Nakabayashi, K., Nishi, S., Kumagai, J., Kudo, M., Sherwood, O. D., et al. (2002). Activation of orphan receptors by the hormone relaxin. Science 295, 671–674. doi: 10.1126/science.1065654
Jaiswal, N., Lambrecht, G., Mutschler, E., Tacke, R., and Malik, K. U. (1991). Pharmacological characterization of the vascular muscarinic receptors mediating relaxation and contraction in rabbit aorta. J. Pharmacol. Exp. Ther. 258, 842–850.
Jelinic, M., Leo, C. H., Post Uiterweer, E. D., Sandow, S. L., Gooi, J. H., Wlodek, M. E., et al. (2014). Localization of relaxin receptors in arteries and veins, and region-specific increases in compliance and bradykinin-mediated relaxation after in vivo serelaxin treatment. FASEB J. 28, 275–287. doi: 10.1096/fj.13-233429
Kakouris, H., Eddie, L. W., and Summers, R. J. (1992). Cardiac effects of relaxin in rats. Lancet 339, 1076–1078. doi: 10.1016/0140-6736(92)90665-P
Kaziro, Y., Itoh, H., Kozasa, T., Toyama, R., Tsukamoto, T., Matsuoka, M., et al. (1988). Structures of the genes coding for G-protein alpha subunits from mammalian and yeast cells. Cold. Spring Harb. Symp. Quant. Biol. 53(Pt 1), 209–220. doi: 10.1101/SQB.1988.053.01.027
Köhler, D., Devanathan, V., Bernardo de Oliveira Franz, C., Eldh, T., Novakovic, A., Roth, J. M., et al. (2014). Galphai2- and Galphai3-deficient mice display opposite severity of myocardial ischemia reperfusion injury. PLoS One 9:e98325. doi: 10.1371/journal.pone.0098325
Kong, R. C., Shilling, P. J., Lobb, D. K., Gooley, P. R., and Bathgate, R. A. (2010). Membrane receptors: structure and function of the relaxin family peptide receptors. Mol. Cell. Endocrinol. 320, 1–15. doi: 10.1016/j.mce.2010.02.003
Kruse, A. C., Hu, J., Pan, A. C., Arlow, D. H., Rosenbaum, D. M., Rosemond, E., et al. (2012). Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 482, 552–556. doi: 10.1038/nature10867
Leo, C. H., Jelinic, M., Ng, H. H., Tare, M., and Parry, L. J. (2016). Time-dependent activation of prostacyclin and nitric oxide pathways during continuous i.v. infusion of serelaxin (recombinant human H2 relaxin). Br. J. Pharmacol. 173, 1005–1017. doi: 10.1111/bph.13404
Lian, X., and Gollasch, M. (2016). A clinical perspective: contribution of dysfunctional perivascular adipose tissue (PVAT) to cardiovascular risk. Curr. Hypertens. Rep. 18:82. doi: 10.1007/s11906-016-0692-z
Liao, J. K., and Homcy, C. J. (1993). The G proteins of the G alpha I and G alpha q family couple the bradykinin receptor to the release of endothelium-derived relaxing factor. J. Clin. Invest. 92, 2168–2172. doi: 10.1172/JCI116818
Löhn, M., Dubrovska, G., Lauterbach, B., Luft, F. C., Gollasch, M., and Sharma, A. M. (2002). Periadventitial fat releases a vascular relaxing factor. FASEB J. 16, 1057–1063. doi: 10.1096/fj.02-0024com
McGowan, B. M., Minnion, J. S., Murphy, K. G., White, N. E., Roy, D., Stanley, S. A., et al. (2010). Central and peripheral administration of human relaxin-2 to adult male rats inhibits food intake. Diabetes Obes. Metab. 12, 1090–1096. doi: 10.1111/j.1463-1326.2010.01302.x
McGuane, J. T., Debrah, J. E., Sautina, L., Jarajapu, Y. P., Novak, J., Rubin, J. P., et al. (2011). Relaxin induces rapid dilation of rodent small renal and human subcutaneous arteries via PI3 kinase and nitric oxide. Endocrinology 152, 2786–2796. doi: 10.1210/en.2010-1126
Molnar, G. A., Lindschau, C., Dubrovska, G., Mertens, P. R., Kirsch, T., Quinkler, M., et al. (2008). Glucocorticoid-related signaling effects in vascular smooth muscle cells. Hypertension 51, 1372–1378. doi: 10.1161/HYPERTENSIONAHA.107.105718
Ng, H. H., Jelinic, M., Parry, L. J., and Leo, C. H. (2015). Increased superoxide production and altered nitric oxide-mediated relaxation in the aorta of young but not old male relaxin-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 309, H285–H296. doi: 10.1152/ajpheart.00786.2014
Nistri, S., and Bani, D. (2003). Relaxin receptors and nitric oxide synthases: search for the missing link. Reprod. Biol. Endocrinol. 1:5. doi: 10.1186/1477-7827-1-5
Nistri, S., Bigazzi, M., and Bani, D. (2007). Relaxin as a cardiovascular hormone: physiology, pathophysiology and therapeutic promises. Cardiovasc. Hematol. Agents Med. Chem. 5, 101–108. doi: 10.2174/187152507780363179
Novak, J., Parry, L. J., Matthews, J. E., Kerchner, L. J., Indovina, K., Hanley-Yanez, K., et al. (2006). Evidence for local relaxin ligand-receptor expression and function in arteries. FASEB J. 20, 2352–2362. doi: 10.1096/fj.06-6263com
Novak, J., Ramirez, R. J., Gandley, R. E., Sherwood, O. D., and Conrad, K. P. (2002). Myogenic reactivity is reduced in small renal arteries isolated from relaxin-treated rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 283, R349–R355. doi: 10.1152/ajpregu.00635.2001
Papadopoulos, D. P., Mourouzis, I., Faselis, C., Perrea, D., Makris, T., Tsioufis, C., et al. (2013). Masked hypertension and atherogenesis: the impact of apelin and relaxin plasma levels. J. Clin. Hypertens. 15, 333–336. doi: 10.1111/jch.12075
Parikh, A., Patel, D., McTiernan, C. F., Xiang, W., Haney, J., Yang, L., et al. (2013). Relaxin suppresses atrial fibrillation by reversing fibrosis and myocyte hypertrophy and increasing conduction velocity and sodium current in spontaneously hypertensive rat hearts. Circ. Res. 113, 313–321. doi: 10.1161/CIRCRESAHA.113.301646
Park, J. I., Semyonov, J., Yi, W., Chang, C. L., and Hsu, S. Y. (2008). Regulation of receptor signaling by relaxin A chain motifs: derivation of pan-specific and LGR7-specific human relaxin analogs. J. Biol. Chem. 283, 32099–32109. doi: 10.1074/jbc.M806817200
Park, S. E., Lim, S. R., Choi, H. K., and Bae, J. (2016). Triazine herbicides inhibit relaxin signaling and disrupt nitric oxide homeostasis. Toxicol. Appl. Pharmacol. 307, 10–18. doi: 10.1016/j.taap.2016.07.010
Rudolph, U., Finegold, M. J., Rich, S. S., Harriman, G. R., Srinivasan, Y., Brabet, P., et al. (1995). Ulcerative colitis and adenocarcinoma of the colon in G alpha i2-deficient mice. Nat. Genet. 10, 143–150. doi: 10.1038/ng0695-143
Sader, M. A., and Celermajer, D. S. (2002). Endothelial function, vascular reactivity and gender differences in the cardiovascular system. Cardiovasc. Res. 53, 597–604. doi: 10.1016/S0008-6363(01)00473-4
Samuel, C. S., Du, X. J., Bathgate, R. A., and Summers, R. J. (2006). ‘Relaxin’ the stiffened heart and arteries: the therapeutic potential for relaxin in the treatment of cardiovascular disease. Pharmacol. Ther. 112, 529–552. doi: 10.1016/j.pharmthera.2005.05.012
Schrage, R., Schmitz, A. L., Gaffal, E., Annala, S., Kehraus, S., Wenzel, D., et al. (2015). The experimental power of FR900359 to study Gq-regulated biological processes. Nat. Commun. 6:10156. doi: 10.1038/ncomms10156
Sherwood, O. D. (2004). Relaxin’s physiological roles and other diverse actions. Endocr. Rev. 25, 205–234. doi: 10.1210/er.2003-0013
Simon, M. I., Strathmann, M. P., and Gautam, N. (1991). Diversity of G proteins in signal transduction. Science 252, 802–808. doi: 10.1126/science.1902986
Teerlink, J. R., Cotter, G., Davison, B. A., Felker, G. M., Filippatos, G., Greenberg, B. H., et al. (2013). Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet 381, 29–39. doi: 10.1016/S0140-6736(12)61855-8
Tsvetkov, D., Shymanets, A., Huang, Y., Bucher, K., Piekorz, R., Hirsch, E., et al. (2016a). Better understanding of phosphoinositide 3-kinase (PI3K) pathways in vasculature: towards precision therapy targeting angiogenesis and tumor blood supply. Biochemistry 81, 691–699. doi: 10.1134/S0006297916070051
Tsvetkov, D., Tano, J. Y., Kassmann, M., Wang, N., Schubert, R., and Gollasch, M. (2016b). The role of DPO-1 and XE991-sensitive potassium channels in perivascular adipose tissue-mediated regulation of vascular tone. Front. Physiol. 7:335. doi: 10.3389/fphys.2016.00335
van der Westhuizen, E. T., Halls, M. L., Samuel, C. S., Bathgate, R. A., Unemori, E. N., Sutton, S. W., et al. (2008). Relaxin family peptide receptors–from orphans to therapeutic targets. Drug Discov. Today 13, 640–651. doi: 10.1016/j.drudis.2008.04.002
Vanhoutte, P. M., Shimokawa, H., Feletou, M., and Tang, E. H. (2017). Endothelial dysfunction and vascular disease - a 30th anniversary update. Acta Physiol. 219, 22–96. doi: 10.1111/apha.12646
Verlohren, S., Dubrovska, G., Tsang, S. Y., Essin, K., Luft, F. C., Huang, Y., et al. (2004). Visceral periadventitial adipose tissue regulates arterial tone of mesenteric arteries. Hypertension 44, 271–276. doi: 10.1161/01.HYP.0000140058.28994.ec
Wettschureck, N., and Offermanns, S. (2005). Mammalian G proteins and their cell type specific functions. Physiol. Rev. 85, 1159–1204. doi: 10.1152/physrev.00003.2005
Wiege, K., Ali, S. R., Gewecke, B., Novakovic, A., Konrad, F. M., Pexa, K., et al. (2013). Galphai2 is the essential Galphai protein in immune complex-induced lung disease. J. Immunol. 190, 324–333. doi: 10.4049/jimmunol.1201398
Willcox, J. M., Summerlee, A. J., and Murrant, C. L. (2013). Relaxin induces rapid, transient vasodilation in the microcirculation of hamster skeletal muscle. J. Endocrinol. 218, 179–191. doi: 10.1530/JOE-13-0115
Keywords: serelaxin, relaxin-2, endothelial Gαi2, NO, RXFP1 receptor, perivascular-adipose tissue, ADRF
Citation: Lian X, Beer-Hammer S, König GM, Kostenis E, Nürnberg B and Gollasch M (2018) RXFP1 Receptor Activation by Relaxin-2 Induces Vascular Relaxation in Mice via a Gαi2-Protein/PI3Kß/γ/Nitric Oxide-Coupled Pathway. Front. Physiol. 9:1234. doi: 10.3389/fphys.2018.01234
Received: 27 May 2018; Accepted: 15 August 2018;
Published: 04 September 2018.
Edited by:
Camille M. Balarini, Federal University of Paraíba, BrazilReviewed by:
Ulf Simonsen, Aarhus University, DenmarkCopyright © 2018 Lian, Beer-Hammer, König, Kostenis, Nürnberg and Gollasch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maik Gollasch, bWFpay5nb2xsYXNjaEBjaGFyaXRlLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.