 Aarti Singh
Aarti Singh Sharon L. Kendall
Sharon L. Kendall Michelangelo Campanella
Michelangelo Campanella- 1Department of Comparative Biomedical Sciences, Royal Veterinary College, London, United Kingdom
- 2Department of Pathology and Pathogen Biology, Royal Veterinary College, Hertfordshire, United Kingdom
- 3UCL Consortium for Mitochondrial Research, London, United Kingdom
Selective autophagy contributes to the wellbeing of eukaryotic cells by recycling cellular components, disposing damaged organelles, and removing pathogens, amongst others. Both the quality control process of selective mitochondrial autophagy (Mitophagy) and the defensive process of intracellular pathogen-engulfment (Xenophagy) are facilitated via protein assemblies which have shared molecules, a prime example being the Tank-Binding Kinase 1 (TBK1). TBK1 plays a central role in the immunity response driven by Xenophagy and was recently shown to be an amplifying mechanism in Mitophagy, bring to attention the potential cross talk between the two processes. Here we draw parallels between Xenophagy and Mitophagy, speculating on the inhibitory mechanisms of specific proteins (e.g., the 18 kDa protein TSPO), how the preferential sequestering toward one of the two pathways may undermine the other, and in this way impair cellular response to pathogens and cellular immunity. We believe that an in depth understanding of the commonalities may present an opportunity to design novel therapeutic strategies targeted at both the autonomous and non-autonomous processes of selective autophagy.
Introduction
Running comparative investigations on species-specific processes allows the comprehension of the underlying biological phenomena; thus, better framing their general value and devising accurate strategies of intervention. Studies on Xenophagy and Mitophagy are steadily bringing to light shared elements between these two evolutionary divergent selective types of autophagy detailing their molecular biology and inspiring novel approaches of exploitation.
Autophagy patrols the intracellular environment and can do so selectively by targeting either mitochondria (mitophagy) (Lemasters, 2005), protein aggregates (aggrephagy) (Lamark and Johansen, 2012), lipids (lipophagy) (Weidberg et al., 2009) or pathogens (xenophagy) (Levine, 2005) with new selective autophagy mechanisms being discovered continuously. These means of cellular quality control rely on molecular mechanism, which may be common between them and therefore account for a subtle interplay to which little attention has been devoted.
The recent advancements on the molecular function of Tank-binding kinase 1 (TBK1) unveiled a role in mitophagy thus complementing the established one in Xenophagy (Thurston et al., 2009; Wild et al., 2011; Pilli et al., 2012) This has provided us with an opportunity to discuss values and dangers of a similar molecular co-sharing besides posing novel questions on core regulatory aspects of mammalian cells homeostasis in health and disease.
In this short contribution, we shall snapshot the fundamentals of Mitophagy and Xenophagy with the aim of highlighting the relevance of common elements and in this way pave a path forward to learn how to enhance or inhibit their unfolding for potential therapeutic benefit.
How Selective Autophagy Protects Against Extra- and Intracellular Toxic Elements
Autophagy is the conserved and genetically programmed homeostatic process which traps and degrades intracellular components that are no longer necessary or have become dysfunctional or damaged (Mizushima and Klionsky, 2007; Yang and Klionsky, 2010a). It targets damaged or excessive organelles by engulfing them into a double-membraned autophagosome which ultimately fuses with lysosomes for degradation (Levine and Klionsky, 2004; Mizushima, 2007; Glick et al., 2010). Examples of autophagy regulators include: autophagy-related genes (ATGs) (Itakura and Mizushima, 2010), mechanistic target of rapamycin complexes (mTORC1) (Wong et al., 2015), beclin-1 (mammalian ortholog of Atg6) (Kang et al., 2011), unc-51 like autophagy activating kinase 1 (ULK1) (Russell et al., 2013) and Microtubule-associated proteins 1A/1B light chain 3B (LC3) (Glick et al., 2010; Yang and Klionsky, 2010b).
The main process of macroautophagy, considered to be the main form of autophagy, a double-membraned phagophore is formed around ubiquitinated proteins or organelles, which matures into an autophagosome that ultimately fuses with a lysosome (Feng et al., 2014). Whereas, during the process of microautophagy the substrates are directed into the lysosome through invagination resulting in their degradation (Li et al., 2012). The chaperone Mediated Autophagy (CMA) occurs instead through the recognition of a specific motif to which the chaperone complex binds and forms a substrate/chaperone complex that fuses with the lysosome upon recognition of the CMA receptor (Kaushik et al., 2011; Kaushik and Cuervo, 2012).
Core elements of the autophagy machinery are retained in the selective versions of the process of which the most extensively characterized versions are: (i) Mitophagy which consists of the degradation of dysfunctional or damaged mitochondria (Youle and Narendra, 2011; Jin and Youle, 2012) and (ii) Xenophagy which is instead the removal of invading pathogens such as bacteria and viruses (Knodler and Celli, 2011; Mao and Klionsky, 2017) (Figure 1). Mitophagy and Xenophagy are finely tuned processes, which share key steps such as the ubiquitination of the unwanted elements prior their disposal via the autophagy-lysosomal pathway (Alomairi et al., 2015). Both processes depend on three key steps: flagging the problem (ubiquitination), fusing with degradative machinery (autophagosome and lysosomal fusion) and breakdown (acidic and enzymatic degradation). These common elements may represent co-regulatory framework.
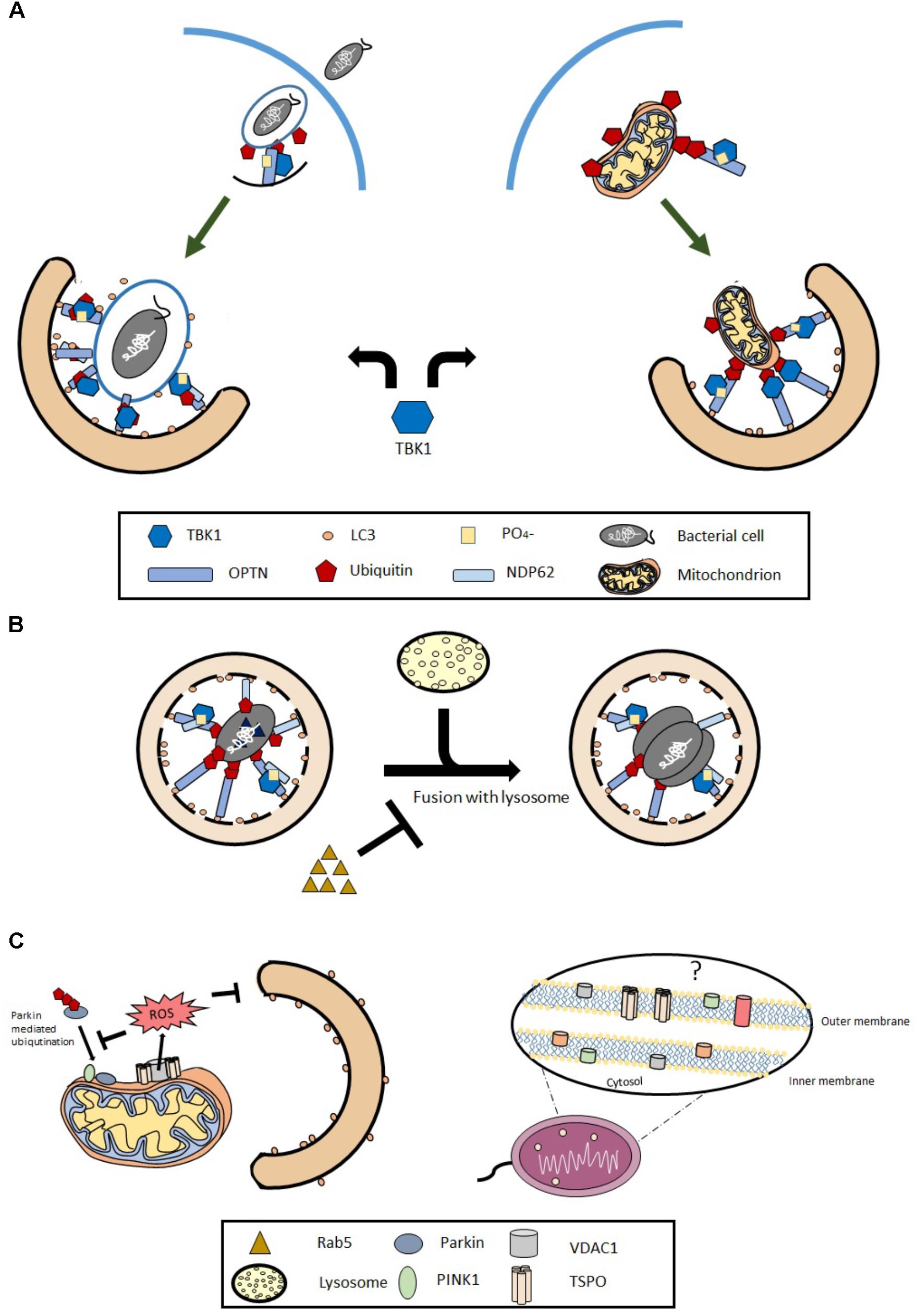
FIGURE 1. Tank-binding kinase 1 (TBK1) as a common functional element between Xenophagy and Mitophagy. Panel (A) depicts pivotal steps in the two processes of selective autophagy against pathogens and mitochondria in which TBK1 plays an equally important role. Panel (B) highlights instead that the similarity may embrace also inhibitory mechanisms among which the TSPO pathway is proposed (C).
The innate immune system is the frontline defense against pathogens, which also acts as a bridge for the adaptive immune response to further control and prevent the invasion (Iwasaki and Medzhitov, 2015). Innate immunity functions through a multitude of signaling pathways, which are conserved across species and grant organisms the fundamental ability to make a distinction between self and non-self (Mogensen, 2009) with autophagy playing part in this (Deretic, 2011). Xenophagy is the activation of a selective breakdown specifically in the context of invading microbial organisms by contributing the prominent processes of phagocytosis and recognition (Figure 1). Xenophagy is distinct from the biological process of phagocytosis as the former acts as a specialized protective mechanism for cells which have already been targeted and breached by pathogens (Flannagan et al., 2009) while phagocytosis is not specific to pathogens alone, and is often utilized to engulf other cells or debris as well.
When pathogens undergo recognition through pattern recognition receptors (PRRs) (Levine and Klionsky, 2004) whereby PRRs identify the pathogen associated molecular patterns (PAMPs). This then initiates the immune signaling preceding the internalization of the pathogen and the activation of the autophagy machinery resulting in entrapment in autophagosomes once within the cytosol and subsequent autolysosomal degradation (Delgado et al., 2009; Oh and Lee, 2014). This is particularly relevant in mammalian cells, which adopt cytosolic or cell surface bound PRRs [such as Toll-like receptors (TLRs) or NOD-like receptors (NLRs)] to detect invading pathogens and signal the upregulation of targeted Autophagy via Xenophagy (Sanjuan et al., 2007).
Xenophagy relies on components of the immunity pathways such as Stimulator of interferon genes (STING) and galectin-8 (Thurston et al., 2012; Watson et al., 2012) which act as cytosolic sensors of the pathogen and recruit downstream effectors (Crotzer and Blum, 2010). Studies have now shown that when the MHC class I protein surface expression is diminished (Li et al., 2010; Oh and Lee, 2014) a reduction in the levels of Xenophagy occurs thus implying that a response could not be successful without selective autophagy embedded and functional therein.
Xenophagy and Mitophagy are both mediating selective disposal of unfit elements and therefore considered to be part of an immune-like response key to maintain cellular homeostasis (Figure 1).
Mitochondria are pivotal to cellular function as producers of the majority of cellular adenosine triphosphate (ATP), intracellular signaling decoders and docking base for cyclic adenosine monophosphate (cAMP) effectors (Tarasov et al., 2012; Finkel et al., 2015; Zhang et al., 2016). Mitochondria are not originally part of the ancestral cell as they are likely of bacterial origin which make of them “hosted elements” despite the successful co-habitation (Embley and Martin, 2006; Gray, 2012). There are several theories describing how mitochondria ended up in mammalian cells; the most prominent of which is the endosymbiosis (Martin et al., 2001; Archibald, 2015), whereby the mitochondrion was originally an extracellular organism [likely α-proteobacterial (Andersson et al., 1998)] capable of oxidative phosphorylation and therefore engulfed in eukaryotic cell to improve the energetic capacity (Taanman, 1999; Thiergart et al., 2012; Martin et al., 2015). This evolved into a successful symbiotic relationship, which crossed evolution. Mitophagy may have evolved as a response to this, thus acting as a controller for these foreign organelles. Mitophagy recognizes and clears the cell of damaged mitochondria preventing the accumulation of dysfunctional mitochondria harmful to the intracellular environment. Even though it exploits the same upstream initiators to general autophagy, the overall mitophagic response is regulated by process-specific proteins to distinguish damaged self from integer self within the mitochondrial network (Ding and Yin, 2012).
Given their origin, the adaptation of the mitochondrion to the early ancestral eukaryotic cell would require a unique subset of proteins to sense and regulate the organelle. Examples include the PTEN-induced kinase 1 (PINK1) which is capable of recognizing dysfunctional mitochondria (Jin and Youle, 2012). It is expressed at very low levels in healthy mitochondria due to successful cleaving of the protein into smaller products after its import into the inner mitochondrial membrane. If a mitochondrion is damaged, however, full-length PINK1 will accumulate on the outer membrane. This leads to the recruitment of the E3 ubiquitin ligase Parkin which ubiquinates the mitochondria, tagging them for lysosomal degradation (Jin and Youle, 2013; Kane et al., 2014; Hamacher-Brady and Brady, 2016).
Notably, various conditions exploit this pathway leading to its impairment spanning from metabolic diseases (e.g., Fanconi anemia) to neurodegeneration, all leading to persistent cellular and tissue damage. Dysfunctional mitochondria can lead to cytotoxicity (Nicholls, 2002; Akbar et al., 2016), hyperactivation of the NLRP3 inflammasome (Lopez-Armada et al., 2013) and cell death via uncontrolled release of the Cytochrome c (Kubli and Gustafsson, 2012). Mitophagy therefore maintains the balance of multiple cellular signaling pathways, downregulating ROS production and helping to maintain a healthy population of mitochondria in the cell (Lazarou, 2014).
Another common element between Mitophagy and Xenophagy is that may remain functional in absence of ubiquitin. In ubiquitin-independent mitophagy mitochondrial receptors like Nip3-like protein X (Nix) (Koentjoro et al., 2017) and FUNDC1 (Liu et al., 2012) interact directly with LC3 (and hence with the autophagosome) leading to lysosomal degradation. Ubiquitin-independent Xenophagy sees galectin-8 capable of recognizing the glycans of the vacuole within which the pathogen resides: this recruits the cargo receptor NDP52 (CALCOCO2) to complete degradation via autophagy (Thurston et al., 2012). In addition, the LC3-associated phagocytosis (LAP), a novel form of non-canonical autophagy, can also be considered an ubiquitin-independent type of Xenophagy hijacking components of the autophagy machinery to aid phagocytosis of extracellular particles and pathogens (Martinez et al., 2015). In LAP LC3 is quickly covering the phagosome for a rapid fusion with lysosome resulting in degradation without pro-inflammatory immune response (Heckmann et al., 2017; Schille et al., 2018).
Whether Mitophagy should be considered equal to Xenophagy in defining the immune response process is debatable. Undeniable though is that operates as an adaptor via mechanisms (memory based) resembling features of immunity exploited against mitochondria (Krysko et al., 2011). Can Mitophagy inform Xenophagy and vice-versa? Are there common functional elements between the two, which could dictate their mutual influence and dictate their efficiency according to the physiopathology of the cell? The recent advances on TBK1 imply this may be highly plausible.
The Uncovering of Another Common Conduit
The Serine/threonine-protein kinase, TBK1, is known to be required in Xenophagy to maintain structural integrity of the pathogen-containing vacuoles. Studies have convincingly shown that knocking down of TBK1 as well as of NDP52, with which it complexes, results in defective clearance of bacteria allowing their escape into the cytosol (Radtke et al., 2007; Thurston et al., 2009; Pilli et al., 2012). The cargo-associated “eat-me” signals as well as the receptors mediating selective autophagy to bridge cargo and phagosomes have been previously unveiled for both processes as reviewed by Randow and Youle (2014).
In 2016, Dikic and colleagues showed that TBK1 integrates the ubiquitin dependent signaling events in Mitophagy upstream of the process (Heo et al., 2015; Moore and Holzbaur, 2016; Richter et al., 2016). They have convincingly shown that TBK1 phosphorylates the Mitophagy receptor Optineurin (OPTN), on the ubiquitin-binding domain (UBD) and the LC3-binding ones. Via this processing TBK1 control and regulates the degradation of dysfunctional mitochondria to which it is selectively recruited. TBK1 mediates phosphorylation of OPTN on the S473 thus expanding the binding capacity of OPTN to multiple ubiquitin chains necessary for both TBK1 recruitment and OPTN targeting to ubiquitinated mitochondria. Mutated TBK1 instead fails to phosphorylate OPTN and therefore stalls the downstream signaling cascade for the activation of Mitophagy. This process of phosphorylation is also implicated in the Parkin independent and PINK1 mediated Mitophagy, highlighting an important molecular partnership in the regulation of homeostatic Mitophagy. Dikic and colleagues were also able to show that multiple Mitophagy receptors beyond OPTN are targeted by TBK1 such as NDP52 (CALCOCO2), TAX1BP1, and p62 (SQSTM1).
This TBK1-mediated phosphorylation establishes therefore an amplification loop that activates the molecular pathway driving the selective degradation of mitochondria.
In Xenophagy the identification of the Serine/threonine-protein kinase TBK1 was paradigm shifting since it helped to clarify the regulatory and recruitment mechanisms of pathogen ubiquitin regulation. The wealth of subsequent literature better contextualized the significance of TBK1 as amplifying signaling in Xenophagy (Weidberg and Elazar, 2011; Helgason et al., 2013; Yang et al., 2016).
Being now aware that TBK1 is required to amplify the removal of both invading pathogens and damaged mitochondria makes us wonder whether defective Mitophagy may indirectly impact Xenophagy. Explicitly, whether defective Mitophagy may undermine Xenophagy recruiting pools of TBK1 to undertake the process (i) or whether a high degree of Xenophagy could impact the unfolding of Mitophagy (ii). Above all interesting to encrypt would be whether a detectable hierarchy exists between the two processes.
The potential cross talk between these two processes, as well as the mutual hijacking of core molecules here hypothesized, calls for further studies which should begin by considering the inhibitory mechanisms of both these processes which we detail below.
The Implications for Common Conduits
Pathogens have evolved distinct mechanisms to evade Xenophagy, particularly by avoiding autophagic consumption. There are a variety of methods that pathogens exploit to avoid lysosomal degradation. These include: (i) creating a neutral compartment within cells where the pathogen can replicate and then escape as exploited by Brucella abortus (Case and Samuel, 2016); (ii) Hijacking the pathway and persisting within quiescent membrane reservoirs inside the autophagosome to later re-establish recurrent infections as done by the uropathogenic Escherichia coli (UPEC) (Mulvey et al., 2001; Mysorekar and Hultgren, 2006; Lewis et al., 2016); (iii) Mycobacterium Tuberculosis (MT) instead, prevents the maturation of the phagosome into the autolysosome by releasing inhibitory factors of the likes of ESAT-6 and Rab5 (Chandra et al., 2015; López de Armentia et al., 2016; Russell, 2016).
It can therefore be considered that the evolutionary selective pressure exerted by the innate immune response, in the form of Xenophagy, has driven adaptation in pathogens for enhanced virulence.
Concomitantly, the subversion of Xenophagy by pathogens could have evolved to form a symbiotic relationship and underpin the successful co-habitation of mitochondria within the hosting cell. This is particularly relevant for a pathogen like MT and UPEC which can lie in a state of dormancy for many years and cause diseases which are increasingly difficult to treat. Based on this, Mitophagy-inhibiting molecules could exploit the same evasion mechanisms as understood with Xenophagy. Hitherto, there is one prominent molecule described as Mitophagy inhibitor: the mitochondrial Translocator Protein (TSPO) (Gatliff et al., 2014) whose role in Xenophagy remains unaddressed (Figure 1) in spite of its high degree of conservation between mammalian and bacterial genomes (Li et al., 2016). A bacterial homolog of the mammalian TSPO, Tryptophan-rich sensory protein (TspO) was first identified in the photosynthetic bacterium Rhodobacter Sphaeroides (RS), where it is thought to be enrolled in the biosynthetic pathway for photosynthetic pigments acting as a negative regulator of photosynthesis genes in response to light and oxygen availability (Yeliseev and Kaplan, 1999). Since its initial discovery in RS, Tspo homologs have been found in a wide range of bacterial taxonomic groups (Chapalain et al., 2009) and human pathogens such as Bacillus anthracis, Legionella pneumophila, Staphylococcus haemolyticus, and Clostridium perfringens. Expression of tspO in Pseudomonas fluorescens increases adhesion and decreases apoptosis (Chapalain et al., 2009) (Figure 2). These observations suggest a role for bacterial TspOs in virulence, particularly when this is delivered intracellularly. It is therefore arguable that bacterial tspOs might represent a conduit to: a) give further insight on the Xenophagy evasion mechanisms exploited by intracellular parasites and b) Enlighten on the crosstalk with Mitophagy (Figure 1).

FIGURE 2. Sequence alignment of mycobacterial TspOs with the human homolog. Sequences were aligned using Clustal Omega and show approximately 30% amino acid identity with the human homolog. Stars indicate identical amino acid residues and dots indicate semi-conserved (similar residues) in all three sequences.
In this regard, what if mammalian cells overexpressing TSPO (and subsequently bearing impairment in the cellular mitophagic response) undermine xenophagy, enabling the establishment of bacterial infections? We are tempted to speculate that if Mitophagy requires greater commitment by the machinery dedicated to the process (such as in the cases in which TSPO is overexpressed) this is likely to de-potentiate Xenophagy. Intriguingly, we know nothing of the ability of TBK1 to retain its amplificatory role in both Mitophagy and Xenophagy in presence of inhibitory elements such as TSPO thus posing the question whether unfolding of the processes are preserved during pathological conditions, as it is known that TSPO is generally overexpressed in these (Liu et al., 2014; Roncaroli et al., 2016).
The possibility for which defective Mitophagy could undermine the efficiency of Xenophagy has never been properly contemplated nor considered in depth, in spite of some evidences available in the literature. The Streptomyces antibioticus, for example, is capable of producing antimycin, an inhibitor of the respiratory chain complex III (Rehacek et al., 1968) also used in combination to trigger mitophagy. In line with this Francione et al., 2009 reported that patients suffering from mitochondrial diseases show an increased susceptibility for infection by legionella supporting that pathogens could well exploit Mitophagy enhancing factors to repress Xenophagy.
The above evidence, as well as the mechanistic advancements on TBK1 and the parallel characterization of anti-mitophagic stress response elements such as TSPO (Gatliff et al., 2014) make us speculate that if amplificatory mechanisms are required for Mitophagy completion, this is likely impaired and therefore the recruitment of the shared elements, may deprive Xenophagy of core elements for its proper unfolding allowing the spread of invading pathogens (Gatliff and Campanella, 2016; Kimmey and Stallings, 2016) (Figure 3).
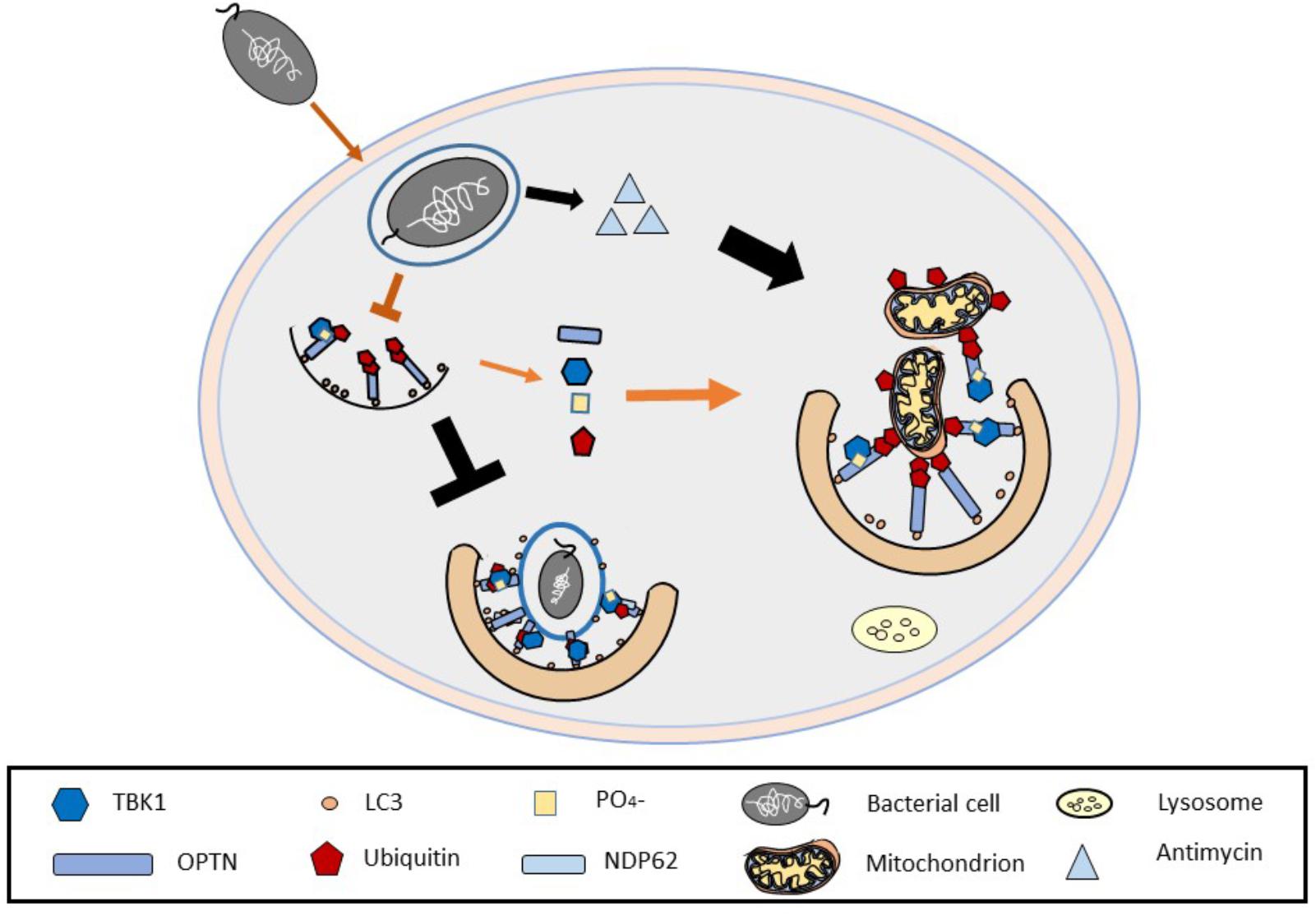
FIGURE 3. Undermining mitochondria to establish bacterial infections. The figure depicts the ability of certain bacterial species to release mitochondria impairing toxins (i.e., antimycin to impair mitochondria) which upregulate mitophagy, perhaps as a way to hijack the common molecules used in both autophagy processes and as such allow the bacteria to propagate within the host cell.
We conclude that the unveiling of common elements may represent a viable approach to succeed in manipulating cellular fate and with it the ability to combat diseases and disorders caused by deficient or abnormally upregulated Mitophagy and Xenophagy.
Below we summarize the unanswered questions in the field of selective autophagy hoping for greater attention and ad hoc investigation:
1. How many common traits between mitophagy and xenophagy remain unaddressed?
2. Which are the consequences on cellular pathophysiology of the shared amplificatory role of TBK1 in mitophagy and xenophagy when both these pathways are activated?
3. Could the high degree of conservation between bacterial and mammalian genomes of the antimitophagy protein TSPO represent an exploitable target to maximize the therapeutic value of xenophagy to co-adjuvate antimicrobial therapy?
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
AS is supported by an iCase BBSRC studentship (Industrial Partner GE). The following funding bodies, which are gratefully acknowledged, supported the research activities led by MC: Biotechnology and Biological Sciences Research Council (Grant Nos. BB/M010384/1 and BB/N007042/1); the Medical Research Council (Grant No. G1100809/2); Bloomsbury Colleges Consortium Ph.D. Studentship Scheme; The Petplan Charitable Trust; Umberto Veronesi Foundation; Marie Curie Actions; and LAM-Bighi Grant Initiative.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akbar, M., Essa, M. M., Daradkeh, G., Abdelmegeed, M. A., Choi, Y., Mahmood, L., et al. (2016). Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress. Brain Res. 1637, 34–55. doi: 10.1016/j.brainres.2016.02.016
Alomairi, J., Bonacci, T., Ghigo, E., and Soubeyran, P. (2015). Alterations of host cell ubiquitination machinery by pathogenic bacteria. Front. Cell. Infect. Microbiol. 5:17. doi: 10.3389/fcimb.2015.00017
Andersson, S. G., Zomorodipour, A., Andersson, J. O., Sicheritz-Ponten, T., Alsmark, U. C., Podowski, R. M., et al. (1998). The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 396, 133–140. doi: 10.1038/24094
Archibald, J. M. (2015). Endosymbiosis and eukaryotic cell evolution. Curr. Biol. 25, R911–R921. doi: 10.1016/j.cub.2015.07.055
Case, E. D. R., and Samuel, J. E. (2016). Contrasting lifestyles within the host cell. Microbiol. Spectr. 4. doi: 10.1128/microbiolspec.VMBF-0014-2015
Chandra, P., Ghanwat, S., Matta, S. K., Yadav, S. S., Mehta, M., Siddiqui, Z., et al. (2015). Mycobacterium tuberculosis inhibits RAB7 recruitment to selectively modulate autophagy flux in macrophages. Sci. Rep. 5:16320. doi: 10.1038/srep16320
Chapalain, A., Chevalier, S., Orange, N., Murillo, L., Papadopoulos, V., and Feuilloley, M. G. J. (2009). Bacterial ortholog of mammalian translocator Protein (TSPO) with virulence regulating activity. PLoS One 4:e6096. doi: 10.1371/journal.pone.0006096
Crotzer, V. L., and Blum, J. S. (2010). Autophagy and adaptive immunity. Immunology 131, 9–17. doi: 10.1111/j.1365-2567.2010.03321.x
Delgado, M., Singh, S., De Haro, S., Master, S., Ponpuak, M., Dinkins, C., et al. (2009). Autophagy and pattern recognition receptors in innate immunity. Immunol. Rev. 227, 189–202. doi: 10.1111/j.1600-065X.2008.00725.x
Deretic, V. (2011). Autophagy in immunity and cell-autonomous defense against intracellular microbes. Immunol. Rev. 240, 92–104. doi: 10.1111/j.1600-065X.2010.00995.x
Ding, W. X., and Yin, X. M. (2012). Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol. Chem. 393, 547–564. doi: 10.1515/hsz-2012-0119
Embley, T. M., and Martin, W. (2006). Eukaryotic evolution, changes and challenges. Nature 440, 623–630. doi: 10.1038/nature04546
Feng, Y., He, D., Yao, Z., and Klionsky, D. J. (2014). The machinery of macroautophagy. Cell Res. 24, 24–41. doi: 10.1038/cr.2013.168
Finkel, T., Menazza, S., Holmström, K. M., Parks, R. J., Liu, J., Sun, J., et al. (2015). The ins and outs of mitochondrial calcium. Circ. Res. 116, 1810–1819. doi: 10.1161/CIRCRESAHA.116.305484
Flannagan, R. S., Cosio, G., and Grinstein, S. (2009). Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 7, 355–366. doi: 10.1038/nrmicro2128
Francione, L., Smith, P. K., Accari, S. L., Taylor, P. E., Bokko, P. B., Bozzaro, S., et al. (2009). Legionella pneumophila multiplication is enhanced by chronic AMPK signalling in mitochondrially diseased Dictyostelium cells. Dis. Model. Mech. 2, 479–489. doi: 10.1242/dmm.003319
Gatliff, J., and Campanella, M. (2016). TSPO: kaleidoscopic 18-kDa amid biochemical pharmacology, control and targeting of mitochondria. Biochem. J. 473, 107–121. doi: 10.1042/BJ20150899
Gatliff, J., East, D., Crosby, J., Abeti, R., Harvey, R., Craigen, W., et al. (2014). TSPO interacts with VDAC1 and triggers a ROS-mediated inhibition of mitochondrial quality control. Autophagy 10, 2279–2296. doi: 10.4161/15548627.2014.991665
Glick, D., Barth, S., and Macleod, K. F. (2010). Autophagy: cellular and molecular mechanisms. J. Pathol. 221, 3–12. doi: 10.1002/path.2697
Gray, M. W. (2012). Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 4:a011403. doi: 10.1101/cshperspect.a011403
Hamacher-Brady, A., and Brady, N. R. (2016). Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell. Mol. Life Sci. 73, 775–795. doi: 10.1007/s00018-015-2087-8
Heckmann, B. L., Boada-Romero, E., Cunha, L. D., Magne, J., and Green, D. R. (2017). LC3-associated phagocytosis and inflammation. J. Mol. Biol. 429, 3561–3576. doi: 10.1016/j.jmb.2017.08.012
Helgason, E., Phung, Q. T., and Dueber, E. C. (2013). Recent insights into the complexity of Tank-binding kinase 1 signaling networks: the emerging role of cellular localization in the activation and substrate specificity of TBK1. FEBS Lett. 587, 1230–1237. doi: 10.1016/j.febslet.2013.01.059
Heo, J. M., Ordureau, A., Paulo, J. A., Rinehart, J., and Harper, J. W. (2015). The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 Recruitment and TBK1 activation to promote mitophagy. Mol. Cell. 60, 7–20. doi: 10.1016/j.molcel.2015.08.016
Itakura, E., and Mizushima, N. (2010). Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6, 764–776. doi: 10.4161/auto.6.6.12709
Iwasaki, A., and Medzhitov, R. (2015). Control of adaptive immunity by the innate immune system. Nat. Immunol. 16, 343–353. doi: 10.1038/ni.3123
Jin, S. M., and Youle, R. J. (2012). PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 125, 795. doi: 10.1242/jcs.093849
Jin, S. M., and Youle, R. J. (2013). The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 9, 1750–1757. doi: 10.4161/auto.26122
Kane, L. A., Lazarou, M., Fogel, A. I., Li, Y., Yamano, K., Sarraf, S. A., et al. (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153. doi: 10.1083/jcb.201402104
Kang, R., Zeh, H. J., Lotze, M. T., and Tang, D. (2011). The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18, 571–580. doi: 10.1038/cdd.2010.191
Kaushik, S., Bandyopadhyay, U., Sridhar, S., Kiffin, R., Martinez-Vicente, M., Kon, M., et al. (2011). Chaperone-mediated autophagy at a glance. J. Cell Sci. 124, 495–499. doi: 10.1242/jcs.073874
Kaushik, S., and Cuervo, A. M. (2012). Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 22, 407–417. doi: 10.1016/j.tcb.2012.05.006
Kimmey, J. M., and Stallings, C. L. (2016). Bacterial pathogens versus autophagy: implications for therapeutic interventions. Trends Mol. Med. 22, 1060–1076. doi: 10.1016/j.molmed.2016.10.008
Knodler, L. A., and Celli, J. (2011). Eating the strangers within: host control of intracellular bacteria via xenophagy. Cell. Microbiol. 13, 1319–1327. doi: 10.1111/j.1462-5822.2011.01632.x
Koentjoro, B., Park, J.-S., and Sue, C. M. (2017). Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 7:44373. doi: 10.1038/srep44373
Krysko, D. V., Agostinis, P., Krysko, O., Garg, A. D., Bachert, C., Lambrecht, B. N., et al. (2011). Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 32, 157–164. doi: 10.1016/j.it.2011.01.005
Kubli, D. A., and Gustafsson, A. B. (2012). Mitochondria and mitophagy: the yin and yang of cell death control. Circ. Res. 111, 1208–1221. doi: 10.1161/CIRCRESAHA.112.265819
Lamark, T., and Johansen, T. (2012). Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012:736905. doi: 10.1155/2012/736905
Lazarou, M. (2014). Keeping the immune system in check: a role for mitophagy. Immunol. Cell Biol. 93, 3–10. doi: 10.1038/icb.2014.75
Lemasters, J. J. (2005). Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 8, 3–5. doi: 10.1089/rej.2005.8.3
Levine, B. (2005). Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 120, 159–162. doi: 10.1016/j.cell.2005.01.005
Levine, B., and Klionsky, D. J. (2004). Development by self-digestion. Dev. Cell 6, 463–477. doi: 10.1016/S1534-5807(04)00099-1
Lewis, A. J., Richards, A. C., and Mulvey, M. A. (2016). Invasion of host cells and tissues by uropathogenic bacteria. Microbiol. Spectr. 4:UTI–0026–2016. doi: 10.1128/microbiolspec.UTI-0026-2016
Li, F., Liu, J., Liu, N., Kuhn, L. A., Garavito, R. M., and Ferguson-Miller, S. (2016). Translocator protein 18 kDa (TSPO): an old protein with new functions? Biochemistry 55, 2821–2831. doi: 10.1021/acs.biochem.6b00142
Li, J. J., Wang, W., Baines, K. J., Bowden, N. A., Hansbro, P. M., Gibson, P. G., et al. (2010). IL-27/IFN-gamma induce MyD88-dependent steroid-resistant airway hyperresponsiveness by inhibiting glucocorticoid signaling in macrophages. J. Immunol. 185, 4401–4409. doi: 10.4049/jimmunol.1001039
Li, W. W., Li, J., and Bao, J. K. (2012). Microautophagy: lesser-known self-eating. Cell. Mol. Life Sci. 69, 1125–1136. doi: 10.1007/s00018-011-0865-5
Liu, G. J., Middleton, R. J., Hatty, C. R., Kam, W. W., Chan, R., Pham, T., et al. (2014). The 18 kDa translocator protein, microglia and neuroinflammation. Brain Pathol. 24, 631–653. doi: 10.1111/bpa.12196
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185. doi: 10.1038/ncb2422
López de Armentia, M. M., Amaya, C., and Colombo, M. I. (2016). Rab GTPases and the autophagy pathway: bacterial targets for a suitable biogenesis and trafficking of their own vacuoles. Cells 5:11. doi: 10.3390/cells5010011
Lopez-Armada, M. J., Riveiro-Naveira, R. R., Vaamonde-Garcia, C., and Valcarcel-Ares, M. N. (2013). Mitochondrial dysfunction and the inflammatory response. Mitochondrion 13, 106–118. doi: 10.1016/j.mito.2013.01.003
Mao, K., and Klionsky, D. J. (2017). Xenophagy: a battlefield between host and microbe, and a possible avenue for cancer treatment. Autophagy 13, 223–224. doi: 10.1080/15548627.2016.1267075
Martin, W., Hoffmeister, M., Rotte, C., and Henze, K. (2001). An overview of endosymbiotic models for the origins of eukaryotes, their ATP-producing organelles (mitochondria and hydrogenosomes), and their heterotrophic lifestyle. Biol. Chem. 382, 1521–1539. doi: 10.1515/BC.2001.187
Martin, W. F., Garg, S., and Zimorski, V. (2015). Endosymbiotic theories for eukaryote origin. Philos. Trans. R. Soc. B Biol. Sci. 370:20140330. doi: 10.1098/rstb.2014.0330
Martinez, J., Malireddi, R. K., Lu, Q., Cunha, L. D., Pelletier, S., Gingras, S., et al. (2015). Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 17, 893–906. doi: 10.1038/ncb3192
Mizushima, N. (2007). Autophagy: process and function. Genes Dev. 21, 2861–2873. doi: 10.1101/gad.1599207
Mizushima, N., and Klionsky, D. J. (2007). Protein turnover via autophagy: implications for metabolism. Annu. Rev. Nutr. 27, 19–40. doi: 10.1146/annurev.nutr.27.061406.093749
Mogensen, T. H. (2009). Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 22, 240–273. doi: 10.1128/CMR.00046-08
Moore, A. S., and Holzbaur, E. L. (2016). Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. U.S.A. 113, E3349–E3358. doi: 10.1073/pnas.1523810113
Mulvey, M. A., Schilling, J. D., and Hultgren, S. J. (2001). Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infect. Immun. 69, 4572–4579. doi: 10.1128/IAI.69.7.4572-4579.2001
Mysorekar, I. U., and Hultgren, S. J. (2006). Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proc. Natl. Acad. Sci. U.S.A. 103, 14170–14175. doi: 10.1073/pnas.0602136103
Nicholls, D. G. (2002). Mitochondrial function and dysfunction in the cell: its relevance to aging and aging-related disease. Int. J. Biochem. Cell Biol. 34, 1372–1381. doi: 10.1016/S1357-2725(02)00077-8
Oh, J. E., and Lee, H. K. (2014). Pattern recognition receptors and autophagy. Front. Immunol. 5:300. doi: 10.3389/fimmu.2014.00300
Pilli, M., Arko-Mensah, J., Ponpuak, M., Roberts, E., Master, S., Mandell, M. A., et al. (2012). TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37, 223–234. doi: 10.1016/j.immuni.2012.04.015
Radtke, A. L., Delbridge, L. M., Balachandran, S., Barber, G. N., and O’Riordan, M. X. (2007). TBK1 protects vacuolar integrity during intracellular bacterial infection. PLoS Pathog. 3:e29. doi: 10.1371/journal.ppat.0030029
Randow, F., and Youle, R. J. (2014). Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe 15, 403–411. doi: 10.1016/j.chom.2014.03.012
Rehacek, Z., Ramankutty, M., and Kozova, J. (1968). Respiratory chain of antimycin A-producing Streptomyces antibioticus. Appl. Microbiol. 16, 29–32.
Richter, B., Sliter, D. A., Herhaus, L., Stolz, A., Wang, C., Beli, P., et al. (2016). Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. U.S.A. 113, 4039–4044. doi: 10.1073/pnas.1523926113
Roncaroli, F., Su, Z., Herholz, K., Gerhard, A., and Turkheimer, F. E. (2016). TSPO expression in brain tumours: is TSPO a target for brain tumour imaging? Clin. Transl. Imaging 4, 145–156. doi: 10.1007/s40336-016-0168-9
Russell, D. G. (2016). The ins and outs of the Mycobacterium tuberculosis-containing vacuole. Cell. Microbiol. 18, 1065–1069. doi: 10.1111/cmi.12623
Russell, R. C., Tian, Y., Yuan, H., Park, H. W., Chang, Y. Y., Kim, J., et al. (2013). ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 15, 741–750. doi: 10.1038/ncb2757
Sanjuan, M. A., Dillon, C. P., Tait, S. W., Moshiach, S., Dorsey, F., Connell, S., et al. (2007). Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257. doi: 10.1038/nature06421
Schille, S., Crauwels, P., Bohn, R., Bagola, K., Walther, P., and van Zandbergen, G. (2018). LC3-associated phagocytosis in microbial pathogenesis. Int. J. Med. Microbiol. 308, 228–236. doi: 10.1016/j.ijmm.2017.10.014
Taanman, J. W. (1999). The mitochondrial genome: structure, transcription, translation and replication. Biochim. Biophys. Acta 1410, 103–123. doi: 10.1016/S0005-2728(98)00161-3
Tarasov, A. I., Griffiths, E. J., and Rutter, G. A. (2012). Regulation of ATP production by mitochondrial Ca2+. Cell Calcium 52, 28–35. doi: 10.1016/j.ceca.2012.03.003
Thiergart, T., Landan, G., Schenk, M., Dagan, T., and Martin, W. F. (2012). An evolutionary network of genes present in the eukaryote common ancestor polls genomes on eukaryotic and mitochondrial origin. Genome Biol. Evol. 4, 466–485. doi: 10.1093/gbe/evs018
Thurston, T. L., Ryzhakov, G., Bloor, S., von Muhlinen, N., and Randow, F. (2009). The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10, 1215–1221. doi: 10.1038/ni.1800
Thurston, T. L. M., Wandel, M. P., von Muhlinen, N., Foeglein, A., and Randow, F. (2012). Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418. doi: 10.1038/nature10744
Watson, R. O., Manzanillo, P. S., and Cox, J. S. (2012). Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150, 803–815. doi: 10.1016/j.cell.2012.06.040
Weidberg, H., and Elazar, Z. (2011). TBK1 mediates crosstalk between the innate immune response and autophagy. Sci. Signal. 4:pe39. doi: 10.1126/scisignal.2002355
Weidberg, H., Shvets, E., and Elazar, Z. (2009). Lipophagy: selective catabolism designed for lipids. Dev. Cell 16, 628–630. doi: 10.1016/j.devcel.2009.05.001
Wild, P., Farhan, H., McEwan, D. G., Wagner, S., Rogov, V. V., Brady, N. R., et al. (2011). Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333, 228–233. doi: 10.1126/science.1205405
Wong, P. M., Feng, Y., Wang, J., Shi, R., and Jiang, X. (2015). Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat. Commun. 6:8048. doi: 10.1038/ncomms9048
Yang, S., Imamura, Y., Jenkins, R. W., Canadas, I., Kitajima, S., Aref, A., et al. (2016). Autophagy inhibition dysregulates TBK1 signaling and promotes pancreatic inflammation. Cancer Immunol. Res. 4, 520–530. doi: 10.1158/2326-6066.CIR-15-0235
Yang, Z., and Klionsky, D. J. (2010a). Eaten alive: a history of macroautophagy. Nat. Cell Biol. 12, 814–822. doi: 10.1038/ncb0910-814
Yang, Z., and Klionsky, D. J. (2010b). Mammalian autophagy: core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 22, 124–131. doi: 10.1016/j.ceb.2009.11.014
Yeliseev, A. A., and Kaplan, S. (1999). A novel mechanism for the regulation of photosynthesis gene expression by the TspO outer membrane protein of Rhodobacter sphaeroides 2.4.1. J. Biol. Chem. 274, 21234–21243. doi: 10.1074/jbc.274.30.21234
Youle, R. J., and Narendra, D. P. (2011). Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14. doi: 10.1038/nrm3028
Keywords: xenophagy, mitophagy, TBK1, TSPO, mitochondria, bacteria
Citation: Singh A, Kendall SL and Campanella M (2018) Common Traits Spark the Mitophagy/Xenophagy Interplay. Front. Physiol. 9:1172. doi: 10.3389/fphys.2018.01172
Received: 17 April 2018; Accepted: 03 August 2018;
Published: 20 September 2018.
Edited by:
Ildikò Szabò, Università degli Studi di Padova, ItalyReviewed by:
Sonia Cortassa, National Institutes of Health (NIH), United StatesDhanendra Tomar, Temple University, United States
Copyright © 2018 Singh, Kendall and Campanella. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michelangelo Campanella, bWNhbXBhbmVsbGFAcnZjLmFjLnVr