 Fengping Liu1,2,3,4†
Fengping Liu1,2,3,4† Zongxin Ling1,3†
Zongxin Ling1,3† Yonghong Xiao1,3†Qing Yang3Baohong Wang1,3
Yonghong Xiao1,3†Qing Yang3Baohong Wang1,3 Li Zheng4Ping Jiang4Lanjuan Li1,3*†
Li Zheng4Ping Jiang4Lanjuan Li1,3*† Wei Wang4*†
Wei Wang4*†- 1State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 2Nursing School, Yancheng Medical College, Yancheng, China
- 3Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 4Department of Urology, First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
Evidence shows urine specimens from different women have different populations of bacteria. The co-occurrence of hypertension and hyperlipidemia in those with diabetes may alter the composition of urine and the microenviroment of the bladder in which bacteria live. The aim of this study was to characterize the urinary microbiota in women with type 2 diabetes mellitus only and those with diabetes plus hypertension and/or hyperlipidemia, and to explore whether the composition of the urinary microbiota is affected by fasting blood glucose, blood pressure, and blood lipids. We enrolled 28 individuals with diabetes only, 24 with diabetes plus hypertension, 7 with diabetes plus hyperlipidemia, and 11 with diabetes plus both hypertension and hyperlipidemia. Modified midstream urine collection technique was designed to obtain urine specimens. Bacterial genomic DNA was isolated using magnetic beads and the urinary microbiota was analyzed using the Illumina MiSeq Sequencing System based on the V3-V4 hypervariable regions of the 16S rRNA gene. Among the four cohorts, the diabetes plus hypertension cohort had the highest relative abundance of Proteobacteria. In contrast, the diabetes plus hyperlipidemia cohort had the lowest relative abundance of Proteobacteria. In addition, Escherichia and Gardnerella were not found in the diabetes plus hyperlipidemia cohort but they were found in all of the other cohorts. Cetobacterium was only present in the diabetes plus hypertension cohort. The most abundant bacteria in the diabetes only and diabetes plus hyperlipidemia cohorts was Lactobacillus, while Prevotella was the most abundant bacteria in the diabetes plus hypertension and diabetes plus hypertension and hyperlipidemia cohorts. Moreover, the relative abundance of Lactobacillus was significantly lower in the diabetes plus hypertension cohort than in the diabetes only and diabetes plus hyperlipidemia cohorts. Several bacteria were correlated with the participants' fasting blood glucose, blood pressure, and blood lipids. In conclusion, hypertension and/or hyperlipidemia and other patient factors can affect the composition of the urinary microbiota in those with diabetes. The insights from this study could be used to develop microbiota-based treatment for comorbid conditions, including urinary tract infections, in those with diabetes.
Introduction
The number of people around the world with diabetes has risen from 108 million in 1980 to 422 million in 2014 (World Health Organization, 2016). Type 2 diabetic mellitus (T2DM) accounts for approximately 90–95% of cases of diabetes (Rubino, 2008). Up to 75% of the adult population with diabetes has hypertension and approximately 52.8% of those with diabetes have hyperlipidemia (de Sereday et al., 2004; Long and Dagogo-Jack, 2011).
Recent evidence has shown that urinary microbiota profiles are complex and they vary considerably between individuals, both in women and men (Dong et al., 2011; Siddiqui et al., 2011, 2012; Fouts et al., 2012; Wolfe et al., 2012; Lewis et al., 2013; Fricke et al., 2014; Nienhouse et al., 2014; Pearce et al., 2014, 2015; Santiago-Rodriguez et al., 2015; Shoskes et al., 2016; Thomas-White et al., 2016a,b). The main urinary bacteria in healthy women are Lactobacillus, Prevotella, and Gardnerella (Siddiqui et al., 2011). Although patients with urinary urgency incontinence have been shown to have higher levels of Gardnerella and Lactobacillus gasseri than controls, they have also been shown to have lower levels of Lactobacillus crispatus (Pearce et al., 2014). Women undergoing surgery for stress urinary incontinence have been shown to have increased urinary bacterial diversity, and this diversity has been shown to be affected by hormone status (there were differences between premenopausal women, and postmenopausal women with or without current exogenous hormone use), body mass index, and the symptoms of stress urinary incontinence (Thomas-White et al., 2016b). In addition, the urinary microbiota has been demonstrated to be affected by age. For example, the urinary microbiota of men and women who were 70 years old or over were dominated by Jonquetella, Parvimonas, Proteiniphilum, and Saccharofermentans (Lewis et al., 2013). Elderly healthy females had significantly lower relative abundance of Lactobacillus than non-elderly (Liu et al., 2016).
Studies have demonstrated that individuals with T2DM, and those with comorbid conditions such as hypertension and hyperlipidemia, have alterations in their intestinal microbiota profiles. The risk of developing T2DM and hyperlipidemia has been reported to be correlated with an alteration in the composition and the function of the intestinal microbiota (Suez et al., 2016). For instance, functional analysis demonstrated that the microbiota of T2DM patients had decreased pathways of bacterial chemotaxis, flagellar assembly, butyrate biosynthesis and metabolism of cofactors and vitamins (Qin et al., 2012).
The occurrence of T2DM may also be associated with changes in urinary microbiota profiles, as having diabetes can change the microenvironment of the urinary tract in which bacterium live. For example, those with diabetes can have increased levels of glucose in their urine as a result of their high levels of blood glucose. These high levels of blood glucose can progressively damage the glomeruli, reducing their ability to filtrate blood and causing protein to leak out of the kidneys into the urine.
Hypertension may also be associated with changes in urinary microbiota profiles as it is correlated with an increase in the activity of the renin-angiotensin-aldosterone system, which affects the amount of water excreted by the kidneys (Basi et al., 2008). In addition, higher sodium and lower potassium in the urine are more prevalent in those with hypertension than in those without hypertension (Kieneker et al., 2014; Mente et al., 2016). Furthermore, impaired kidney function is associated with hyperlipidemia (Chen et al., 2016), which can cause high levels of proteinuria (Gordillo and Spitzer, 2009). Thus, the co-occurrence of hypertension and/or hyperlipidemia with T2DM can affect the composition of urine, which can then affect the microenvironment of the urinary tract and consequently alter bacterial growth.
So far, no study has explored the relationship between the urinary microbiota and comorbidities in those with T2DM. We speculate that the composition changes in urine caused by comorbid hypertension, hyperlipidemia and urinary tract infections in female T2DM patients can alter the urinary microbiota profile. Thus, the aim of this study was to investigate the changes in the urinary microbiota profiles of women with T2DM only and those with T2DM plus hypertension and/or hyperlipidemia, and the correlation between the profiles and patient characteristics, i.e., fasting blood glucose (FBG), blood pressure (BP), blood lipids (BL), asymptomatic bacteriuria (according to urine cultures) and nitrite-positive samples (which are indicative of urinary tract infections).
Materials and Methods
Recruitment of Subjects
Seventy individuals with T2DM were divided into four cohorts: 28 were allocated to the T2DM only (DM) cohort, 24 to the T2DM plus hypertension (DM+HT) cohort, 7 to the T2DM plus hyperlipidemia (DM+HLP) cohort, and 11 to the T2DM plus both hypertension and hyperlipidemia (DM+HT+HLP) cohort. The participants had been previously diagnosed (with T2DM only or with T2DM plus hypertension and/or hyperlipidemia) by clinicians in local provincial or municipal hospitals in China. They were enrolled at the Endocrinology Department of the First Affiliated Hospital, School of Medicine, Zhejiang University from June 2015 to January 2016. Written informed consent was obtained from the participants prior to enrollment, and approval for the study was obtained from the Ethics Committee of the First Affiliated Hospital (reference number: 295). The following exclusion criteria were applied: a urinary tract infection (UTI) in the previous month, antibiotic use in the previous 3 months, inability to complete the questionnaire, menstruation, urinary incontinence, known anatomical abnormalities of the urinary tract (e.g., cystoceles, hydronephrosis, renal atrophy, or neurogenic bladder), and use of a urinary catheter. Each participant's most recent FBG, BP, and BL values were obtained from their medical records. Water intake was assessed using the Chinese Food Frequency Questionnaire (Zhao et al., 2010).
Collection and Processing of Urinary Samples
The samples collected contained the first urine of the day. The participants were taught to use a modified midstream urine (MMSU) collection technique that involved disinfection and a four-tube collection method. The MMSU collection technique involved the following steps: (a) A polyvinylpyrrolidone-iodine antiseptic (Dian'erkang, Shanghai, China) was poured onto sterile cotton balls, which were then placed into a 40 mL sterile sputum cup. (b) Four 50-mL sterile centrifuge tubes (which were labeled tube 1, 2, 3, and 4) were opened and the lids were placed upwards so that the participant would not touch the interior or top edge of either the tubes or the lids. (c) The participant pulled her underwear down to her knees and squatted on a squat toilet, with legs spread apart. (d) The participant disinfected the thumb, middle finger, and index finger of each hand twice with the polyvinylpyrrolidone-iodine antiseptic. (e) The participant used her dominant hand to pick up one of the cotton balls covered with antiseptic, cleaned the far labial fold, starting from above the urinary meatus down toward the rectum. The cotton ball was then discarded without crossing the sterile field that had previously been disinfected. The near labial fold was then disinfected by cleaning down the center of the urinary meatus. The labia were held apart to prevent the labia minora from falling back over the urinary meatus. (f) After disinfection, the participant urinated into tube 1 until half the tube was filled. Then, without stopping the flow of urine, the participant urinated into tubes 2, 3, and 4 in that order. The redundant urine was voided to the toilet. All the tubes were filled up to the halfway mark except for tube 2, which was filled up to the labeled line (representing 40 mL of urine). After training, we asked the participant demonstrating the MMSU technique to assess their competency to perform the procedure. The collection procedure was supervised by a senior nurse.
Each sample was given an anonymous identification code. The urine in tubes 2 and 3 was separated into three aliquots: 15 mL for urinalysis, 1 mL for urine culture, and 40 mL for bacterial DNA sequencing. The urine in tubes 1 and 4 was discarded, which could guarantee that genuine mid-stream urine was used in the experiment. The urinalysis involved testing the sample for nitrites. The urine cultures were performed by inoculating blood agar plates and incubating them at 37°C for 48 h. Asymptomatic bacteriuria was defined as the presence of ≥105 CFU/mL of the same bacterial strain in two consecutive MMSU specimens (Varli et al., 2012). If asymptomatic bacteriuria was diagnosed, urine from the second MMSU specimen was used for bacterial DNA sequencing. A self-report questionnaire was used to collect demographic characteristics.
DNA Extraction, PCR, and MiSeq Sequencing
Forty mL urine was aspirated from tubes 2 and 3, separated into three sections and injected into three 15 mL sterile centrifuge tubes. Each of them was pelleted by centrifugation at 4,000 × g for 15 min at 4°C. Ten milliliter of the supernatant was decanted and the pellet was obtained by centrifugation for 15 min at 4,000 × g at 4°C. The pellet was injected into a 2 mL sterile centrifugation tube which contained 500 μL of lysis buffer, which was composed of 1 M Tris-HCl (pH 8.03), NaCl, 0.5 M EDTA (pH 7.97), and SDS. The tube was kept at −80°C until DNA extraction. The method of magnetic beads isolation of genomic DNA from bacteria was based on the manufacturer's protocol with minor modifications (Shoskes et al., 2016). The tube was placed in liquid nitrogen for 1 min, and transferred into a water bath at 65°C for 5 min, with vigorous mixing. This last process was repeated three times with a final maintenance in the water bath for 30 min. 50 μL Agencourt AMPure XP (Beckman Coulter, USA) was added to 100 μL of the urine pellet, vortexed for 30 s and incubated for 5 min at room temperature. The tube was placed into a magnetic separator for 5 min, and DNA was bound to magnetic beads which were drawn to the wall of the microcentrifuge tube. The supernatant was carefully removed without disrupting the magnetic beads. The sample was washed twice with 200 μL 80% ethanol for 30 s, being placed on a magnet separator between each washing. The purified DNA was eluted with 50 μL ddH2O for 1 min. The beads, now released from the DNA, were collected with the magnet, The DNA-containing supernatant was transferred to a clean tube. The concentration of extracted DNA was determined by using a NanoDrop ND-1000 spectrophotometer (Thermo Electron Corporation, USA); its integrity and size were checked by 1.0% agarose gel electrophoresis containing 0.5 mg/mL ethidium bromide. The DNA complex was placed at −20°C until PCR amplification. We failed to extract sufficient DNA for sequencing from the sample of one participant, so this participant was replaced by another who had similar demographic characteristics.
The 16S rRNA gene V3-V4 regions were PCR-amplified from microbial genome DNA using primers (forward primer, 5′-ACTCCTACGGGAGGCAGCAG-3′; reverse primer, 5′-GGACTACHVGGGTWTCTAAT-3′; Fadrosh et al., 2014). All crucial steps during sample processing, DNA isolation and the entire PCR set up were performed in a laminar air flow bench, illuminated with a UV lamp prior to use in order to avoid possible contaminants. In addition, negative DNA extraction controls (lysis buffer and kit reagents only) were amplified and sequenced as contamination controls. The amplicons were normalized, pooled and sequenced on the Illumina MiSeq instrument using a 300 × 2 V3 kit.
Bioinformatics Analysis and Statistical Analysis
Sequence reads processing was performed using QIIME (version 1.9.0) (Caporaso et al., 2010) and included additional quality trimming, demultiplexing, and taxonomic assignments. Profiling of predictive urinary microbiota was analyzed by using PICRUSt based on the Greengenes database as of 13 August 2013 (DeSantis et al., 2006) (BioProject number: 329477; Accessing number: SRP087709). Specifically, the processed sequences were subjected to subsampled open-reference operational taxonomic unit (OTU) picking against Greengenes, clustering unmatched reads with 97% identity into OTUs. QIIME was used to calculate the Shannon-Weiner indices, and phylogenetic (alpha) diversity. The diversity and richness of the bacteria in the urine samples were calculated using several estimates. These consisted of the level of OTUs, which provides a measure of bacterial richness), Chao1 (which is also an estimate of bacteria richness) and the Shannon and Simpson indices (which are measures of bacterial diversity).
Statistical analysis was performed using the SPSS data analysis program (version 21.0). For continuous variables, Independent t-tests and Mann-Whitney U-test was applied. For categorical variables between groups, using either the Pearson chi-square or Fisher's exact test, depending on assumption validity. For taxon between cohorts, Mann-Whitney U-test was applied. All tests of significance were two sided, and p < 0.05 was considered statistically significant. FDR was used as a correction approach to control the false discovery rate (Shoskes et al., 2016). The receiver operating characteristic curve was plotted and the area under curve of each predictive model was calculated using SPSS.
Results
Cohort Description
Table 1 displays the baseline characteristics of the participants in each cohort. The mean age in the DM+HT cohort was significantly greater than those in the DM and DM+HLP cohorts, the mean age in the DM+HT+HLP cohort was significantly greater than those in the DM and DM+HLP cohorts. The duration of T2DM was significantly shorter in the DM and DM+HLP cohorts than in the DM+HT cohort. Those in the DM+HT cohort were also less likely to be premenstrual compared to those in the DM cohort.
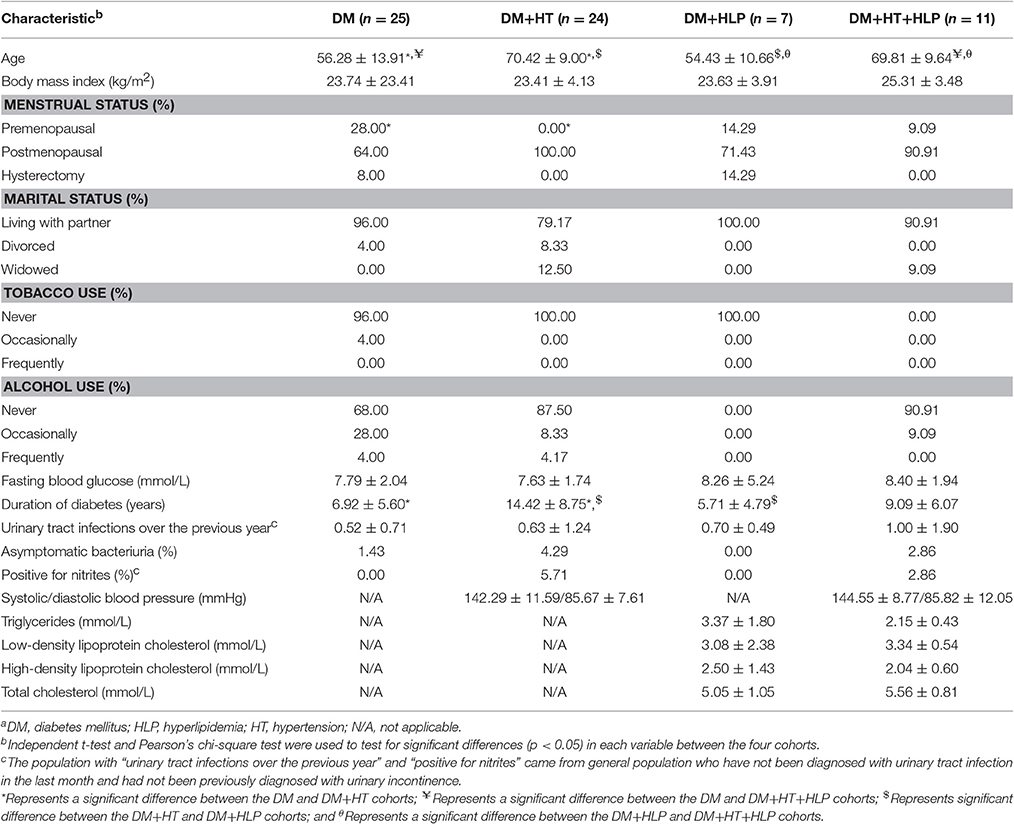
Table 1. Characteristics of the DM, DM+HT, DM+HLP, and DM+HT+HLP cohortsa.
Sequencing Data
We obtained 5,175,509 raw sequences from the 70 samples, with a median read length of 2 × 300 and 443 base pairs. After filtering and removing the chimeric sequences, we obtained 3,981,519 reads for further analysis, which accounted for 76.93% of the valid reads, with a mean of 56,878.84 reads (ranging from 13,419 to 146,528) per sample. The mean read length was 438 bp (ranging from 423 to 436). The value of Good's coverage estimator was 98%.
As shown in Table 2, the numbers of reads and OTUs were significantly higher in the DM+HT cohort compared to in the DM and DM+HLP cohorts. In addition, the number of OTUs was dramatically higher in the DM+HT+HLP cohort compared to in the DM+HLP cohort.
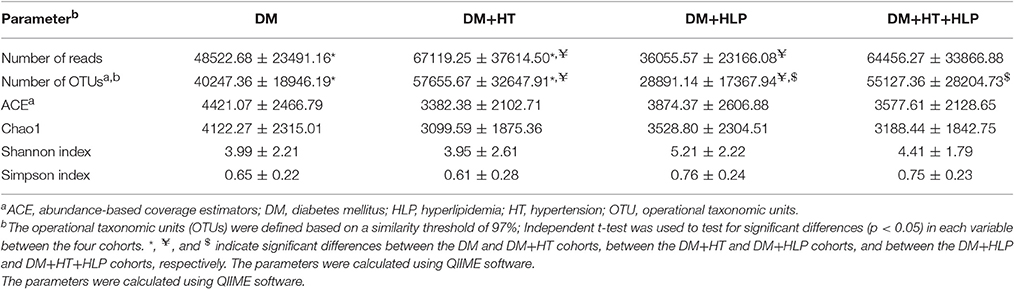
Table 2. Richness and diversity estimators in the DM, DM+HT, DM+HLP, and DM+HT+HLP cohortsa.
Comorbid Conditions with T2DM were Associated with Alterations in the Urinary Microbiota Profiles
The numbers of bacterial phyla detected in the DM, DM+HT, DM+HLP, and DM+HT+HLP cohorts were 40, 37, 30, and 26, respectively. The predominant bacterial phyla in the DM cohort were Proteobacteria (58.27%), Firmicutes (20.83%), Bacteroidetes (10.14%), Actinobacteria (7.52%), and Fusobacteria (1.06%). The DM+HT cohort was dominated by Proteobacteria (60.18%), Firmicutes (20.60%), Bacteroidetes (11.20%), Actinobacteria (5.36%), and Synergistetes (0.54%). The DM+HLP cohort was dominated by Proteobacteria (50.26%), Firmicutes (24.43%), Bacteroidetes (13.36%), Actinobacteria (9.21%), and Acidobacteria (1.18%). The DM+HT+HLP cohort was dominated by Proteobacteria (37.29%), Firmicutes (22.78%), Bacteroidetes (22.47%), Actinobacteria (15.33%), and Fusobacteria (0.82%).
The numbers of bacterial genera in the DM, DM+HT, DM+HLP, and DM+HT+HLP cohorts were 320, 303, 236, and 225, respectively. The genera detected in each of the cohorts are shown in Table S1. Of the genera in the DM cohort, 61, 103, and 108 were not detected in the DM+HT, DM+HLP, and DM+HT+HLP cohorts, respectively. Of the genera in the DM+HT cohort, 41, 86, and 102 were not found in the DM, DM+HLP, and DM+HT+HLP cohorts, respectively. Of the genera in the DM+HLP cohort, 26, 6, and 60 were absent from the DM, DM+HT, and DM+HT+HLP cohorts, respectively. Finally, of the genera in the DM+HT+HLP cohort, 23, 26, and 53 were not present in the DM, DM+HT, and DM+HT+HLP cohorts.
The predominant genera in the DM cohort were Lactobacillus, Prevotella, and Acinetobacter (Figure 1A). The DM+HT cohort was dominated by Prevotella, Streptococcus, and Bacteroides, and Lactobacillus only accounted for 5% of the DNA sequences (Figure 1B). The samples from the DM+HLP cohort primarily consisted of Lactobacillus, Prevotella, and Halomonas (Figure 1C). The most common genera in the DM+HT+HLP cohort were Prevotella, Lactobacillus, and Bacillus (Figure 1D).
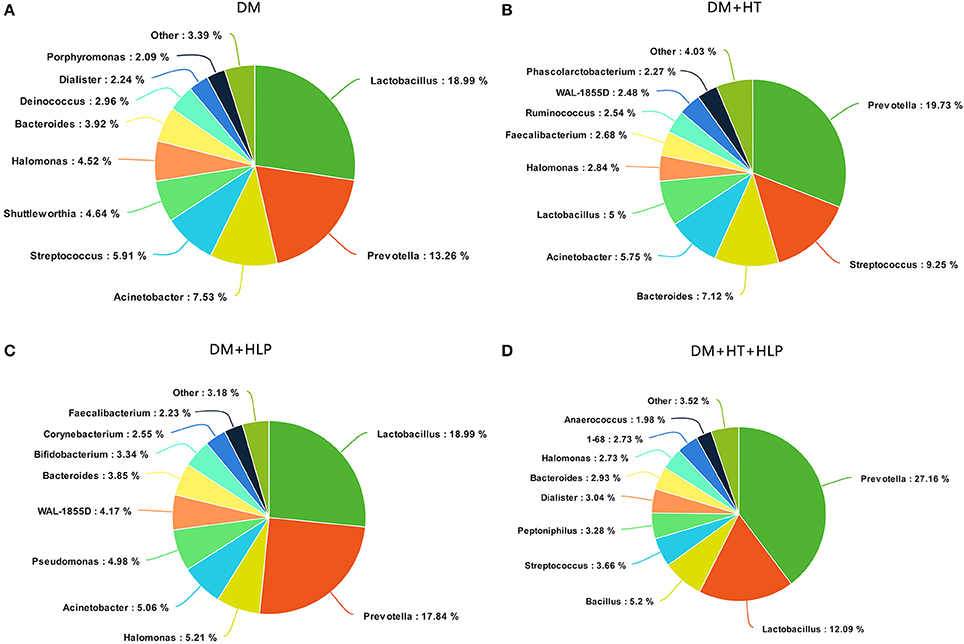
Figure 1. Summary of bacterial genera detected in the four cohorts. (A–D) indicate the top 10 most abundant genera detected in the DM, DM+HT, DM+HLP, and DM+HT+HLP cohorts, respectively. DM, diabetes mellitus; HLP, hyperlipidemia; HT, hypertension.
The levels of Aeromonas, Roseburia, and Ruminococcus were significantly lower in the DM cohort compared to in the DM+HT cohort, while Lactobacillus, Enterobacter, Klebsiella, etc. were dramatically higher in the DM cohort compared to in the DM+HT cohort (Figure 2A). Compared to the DM cohort, the relative abundances of Faecalibacterium, Oscillospira, and Collinsella were significantly higher in the DM+HLP cohort (Figure 2B). In addition, compared to in the DM cohort, the relative abundance of Prevotella was significantly higher in the DM+HT+HLP cohort, while those of Shuttleworthia, Acinetobacter, and Gemella were dramatically lower in the DM+HT+HLP cohort (Figure 2C). The levels of Klebsiella, Novosphingobium, Lactobacillus, and Enterobacter were significantly lower in the DM+HT cohort compared to in the DM+HLP cohort (Figure 2D). The relative abundance of Faecalibacterium was significantly higher in the DM+HLP cohort compared to in the DM+HT+HLP cohort (0.02 ± 0.02 vs. 0.01 ± 0.01, p = 0.040). In addition, as shown in Figure 3, the receiver operating characteristic curve analysis demonstrated that Oscillospira was capable of being used as a diagnostic factor for distinguishing those in the DM+HLP cohort from those in the DM cohort.
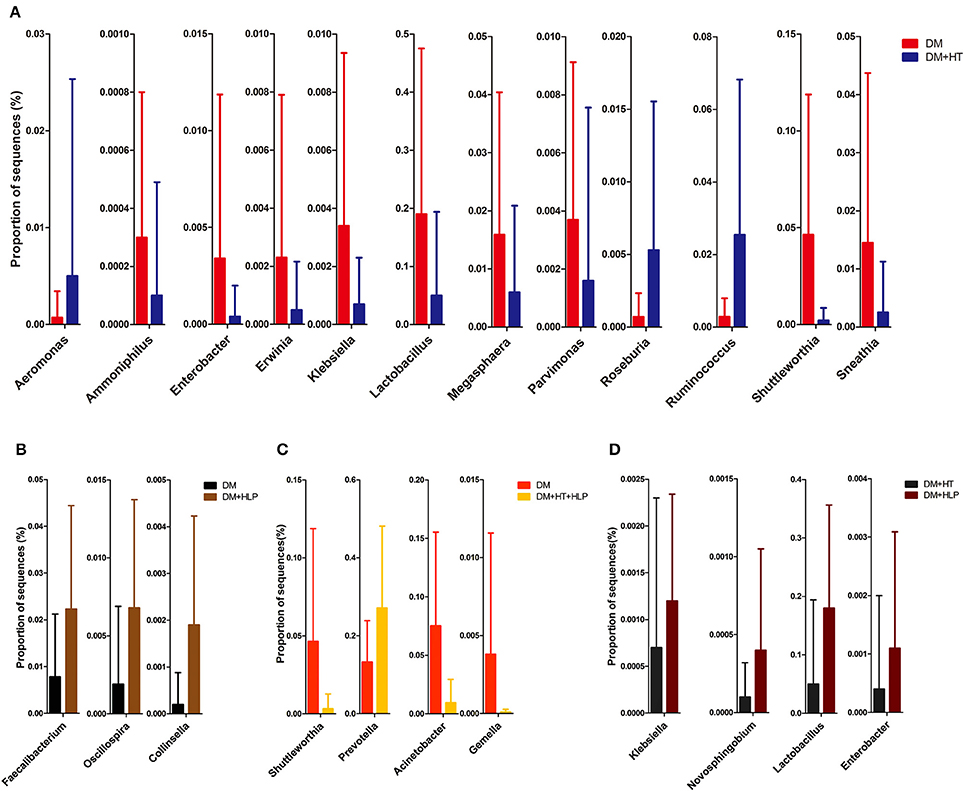
Figure 2. Genus-level operational taxonomic units that were significantly different between the four cohorts. Mann-Whitney U-tests were used to compare the differences in the relative abundance of bacterial genera between pairs of cohorts. (A–D) indicate the significant differences (p < 0.05) between the DM and DM+HT cohorts, between the DM and DM+HLP cohorts, between the DM and DM+HT+HLP cohorts, and between the DM+HT and DM+HLP cohorts, respectively. DM, diabetes mellitus; HLP, hyperlipidemia; HT, hypertension.
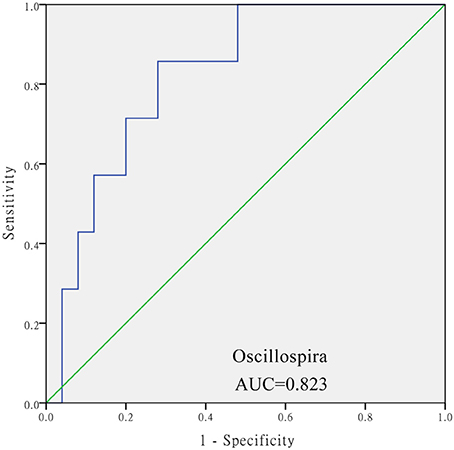
Figure 3. Receiver operating characteristic curve analysis of sensitivity and specificity of the taxon sequence. The analysis evaluated the use of the relative abundance of Oscillospira as a diagnostic factor for distinguishing between those with diabetes only from those with diabetes plus hyperlipidemia. SPSS software was used for the ROC curve analysis. The diabetes only was labeled with “0” and the diabetes plus hypertenstion was labeled with “1,” and the value of state variable was set “1.”
To better understand the relationships between urinary microbiota and individual characteristics such as FBG, BP, and BL, correlation analyses were carried out. The bacteria that were significantly correlated with the three factors are listed in Tables S2–S5. In the DM cohort, the levels of 4 genera were positively correlated with FBG (Table S2). In the DM+HT cohort, the relative abundance of 8 and 14 genera was positively and negatively correlated with systolic BP, respectively. In addition, 15 genera were negatively correlated with diastolic BP in the DM+HT cohort (Table S3). In the DM+HLP cohort, the levels of 15 and 29 genera were positively correlated with triglycerides and high-density lipoprotein cholesterol (HDL-C), respectively. In the DM+HLP cohort, 8 and 10 genera were negatively correlated with low-density lipoprotein cholesterol (LDL-C) and total cholesterol, respectively (Table S4). In the DM+HT+HLP cohort, the levels of 11 and 1 genera were positively and negatively correlated with systolic and diastolic BP, respectively. Thirteen genera were negatively correlated with triglycerides, 37 were positively correlated with LDL-C, 2 were correlated with HDL-C (one was positively correlated and the other was negatively correlated), and 5 and 10 genera were positively and negatively correlated with total cholesterol (Table S5). No genera were correlated with FBG in any of the cohorts.
At the species level, those in the DM+HT cohort had significantly lower relative abundances of Lactobacillus iners, Acinetobacter rhizosphaerae, and Acinetobacter schindleri than those in the DM cohort (2.14 ± 10.23 vs. 9.15 ± 22.36, p = 0.001; 0.03 ± 0.08 vs. 0.16 ± 0.19, p = 0.001; 0.00 ± 0.00 vs. 0.01 ± 0.03, p = 0.019, respectively). In contrast, they also had a higher relative abundance of Kocuria palustris compared to those in the DM cohort (0.01 ± 0.03 vs. 0.00 ± 0.0, p = 0.042). Interestingly, Eggerthella lenta, which was not detected in the DM cohort, was found in the DM+HT cohort. In addition, Deinococcus aquatilis, which was detected in the DM cohort, was absent in the DM+HT cohort. Additionally, A. rhizosphaerae was significantly lower in the DM+HT+HLP cohort compared to in the DM cohort (0.01 ± 0.03 vs. 0.16 ± 0.19, p = 0.003). A. rhizosphaerae was also significantly lower in the DM+HT+HLP cohort compared to in the DM+HLP cohort (0.00 ± 0.00 vs. 0.06 ± 0.07, p = 0.001).
To explore the association between nutrients intake and the relative abundance of bacterial genus in urinary microbiota and between nutrients intake and medicine intake, we compared the difference of nutrients intake among DM, DM+HT, DM+HLP, and DM+HT+HLP group, and there were difference in energy, protein, fat, carbohydrate, vitamin B1, vitamin B2, vitamin C, vitamin E, potassium, sodium, magnesium, selenium, copper, manganese, monounsaturated fatty acid, polyunsaturated fatty acid and aspirin intake among groups. We have found that some nutrients intake were weak or mild positively or negatively associated with urinary microbiota (r < 0.7, p < 0.05; Tables S6, S7).
Discussion
Hypertension appears to increase urinary bacterial diversity, as those in the DM+HT and DM+HT+HLP cohorts had larger numbers of reads and/or OTUs compared to those in the DM and DM+HLP cohorts. Hyperlipidemia may lower the diversity of the microbiota, as the DM+HLP cohort had lower numbers of reads and OTUs compared to the DM+HT cohort. The results were similar at both the phylum and genus levels. Among the four cohorts, subjects with hyperlipidemia had a lower presence of bacteria than those without hyperlipidemia.
The top four most abundant bacteria in the four cohorts were (in various orders) Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria. This is somewhat similar to the findings of several previous studies on urinary microbiota profiles, as in these previous studies, one of the most abundant bacteria was Proteobacteria, which were generally ranked as the second or third most abundant bacteria, though they sometimes ranked lower (Siddiqui et al., 2011, 2012; Pearce et al., 2014; Karstens et al., 2016; Thomas-White et al., 2016a).
Among the four cohorts, the rates of participants with asymptomatic bacteriuria and nitrite-positive samples were the highest in the DM+HT cohort. Interestingly, among the four cohorts, the relative abundance of Proteobacteria was also the highest in the DM+HT cohort. Lewis et al. (2013) reported that Proteobacteria was the predominant phylum in nitrite-positive samples in their study. The findings in our study and the study by Lewis et al. indicate that the relative abundance of Proteobacteria may be correlated with UTIs. Evidence has shown that hypertension is a risk factor for UTIs in those with diabetes (Al-Rubeaan et al., 2013). This suggests that Proteobacteria may play a role in the high prevalence of UTIs in those with diabetes and comorbid hypertension.
Interestingly, asymptomatic bacteriuria and nitrite-positive samples were not found in the DM+HLP cohort and, among the four cohorts, the DM+HLP cohort had the lowest relative abundance of Proteobacteria. In addition, Escherichia which is one of the most frequent uropathogens (Reygaert, 2014), a genus belonging to Proteobacteria, was not detected in the DM+HLP cohort (Table S1). This suggests that the absence of asymptomatic bacteriuria and nitrite-positive samples in the DM+HLP cohort may be due to the low relative abundance of Proteobacteria and the absence of Escherichia in the urine. This may indicate that there is a microbiota-related regulating system that prevented those in the DM+HLP cohort from developing UTIs. This is consistent with the finding that, although the co-occurrence of hyperlipidemia and diabetes can increase the body's inflammatory response (Casqueiro et al., 2012; Chen S. et al., 2014), there were no indications of UTIs (by either asymptomatic bacteriuria or nitrite-positive samples) in the DM+HLP cohort.
Several bacteria were not present in the DM+HLP cohort, but were detected in the three other cohorts such as Gardnerella, which has been reported to be a major urotype in previous studies (Thomas-White et al., 2016a,b). Price et al. found that Gardnerella was present more often in non-UTI samples than in UTI samples (Price et al., 2016), but in the present study, there were no participants with either asymptomatic bacteriuria or nitrite-positive samples in the DM+HLP cohort. Notably, Akkermansia and Bifidobacterium, which are considered probiotic bacteria, were detected in all four cohorts. In a previous intestinal microbiota study, the relative abundance of Akkermansia was lower in patients with inflammatory diseases compared to healthy humans, and was also lower in diabetic mice (Dingemanse et al., 2015). Bifidobacterium has been shown to be able to reduce levels of intestinal endotoxins and improve the mucosal barrier, which results in reduced systemic inflammation (Cani et al., 2008). Although T2DM, hypertension, and hyperlipidemia can increase an individual's inflammatory response (Casqueiro et al., 2012; Mangin, 2014; Chen S. et al., 2014), the subjects in the DM, DM+HT, and DM+HLP cohorts did not have a higher incidence of UTIs (in the previous year) compared to the incidence of UTIs (0.5–0.7 per year) reported by Hooten et al. in healthy women (Hooton et al., 1996). This may be due to the presence of bacteria in urine (such as Akkermansia and Bifidobacterium) that help to combat the inflammatory responses caused by diabetes, hypertension, and hyperlipidemia in order to maintain healthy urinary conditions.
The most abundant bacteria in the DM and DM+HLP cohorts was Lactobacillus, which is similar to the findings of previous studies (Siddiqui et al., 2011, 2012; Pearce et al., 2014, 2015; Thomas-White et al., 2016b). However, Lactobacillus was the fifth most abundant bacteria and only accounted for 5% of the DNA sequences in the DM+HT cohort, and the level of Lactobacillus was significantly lower in the DM+HT cohort compared to in the DM cohort. The relative abundance of Lactobacillus may have been affected by the participants' ages, as the participants in the DM+HT and DM+HT+HLP cohorts were significantly older than those in the DM and DM+HLP cohorts. Thomas-White et al. reported a similar finding in their study of urgency incontinence patients, i.e., the urgency incontinence patients were significantly older than the healthy controls, and they had a significantly lower proportion of Lactobacillus in their urine (Thomas-White et al., 2016a). Moreover, the DM+HT cohort included fewer premenopausal women than the DM cohort. A recent study demonstrated that premenopausal women and postmenopausal women taking exogenous hormones had higher levels of Lactobacillus in their urine compared to postmenopausal women who were not taking exogenous hormones (Thomas-White et al., 2016b). In addition, another study reported that Lactobacillus was a dominant genus in urine samples from premenopausal women (Karstens et al., 2016). Although age and menopausal status play a role in the relative abundance of Lactobacillus, hypertension may also be a risk factor for reduced levels of Lactobacillus. The DM+HT cohort did not include a significantly greater number of postmenopausal women than the DM+HLP cohort, but the relative abundance of Lactobacillus was still significantly lower in the DM+HT compared to in the DM+HLP cohort. Furthermore, it has been reported that oral administration of recombinant Lactobacillus can significantly decrease systolic BP (Yang et al., 2015). Future studies should explore whether a deficiency in Lactobacillus is responsible for the co-occurrence of hypertension in those with diabetes or whether the co-occurrence of hypertension causes the low relative abundance of Lactobacillus in urine. Notably, among the four cohorts, the DM+HT subjects had the highest rates of asymptomatic bacteriuria and nitrite-positive samples and lowest relative abundance of Lactobacillus. Hypertension is a risk factor for UTIs in diabetic patients (Al-Rubeaan et al., 2013) and Lactobacillus can reduce UTIs (Marelli et al., 2004).
Interestingly, not only was the relative abundance of Oscillospira dramatically higher in the DM+HLP cohort compared to in the DM cohort, but it was also capable of being used as a diagnostic factor for differentiating those in the DM+HLP cohort from those in the DM. If this finding is confirmed by a study with a larger sample size, the relative abundance of Oscillospira could be used as a measure for patients with diabetes to self-monitor hyperlipidemia.
The relative abundance of Faecalibacterium was higher in the DM+HLP cohort than in the DM cohort. It has been reported that patients with urgent urinary incontinence also have a higher proportion of Faecalibacterium than healthy controls (Pearce et al., 2014), and hyperlipidemia has been shown to increase urinary frequency and decrease the density of blood vessels and nerves in the bladder (Huang et al., 2010). This suggests that reducing the relative abundance of Faecalibacterium in urine may be useful for controlling hyperlipidemia in patients with urgent urinary incontinence.
There was a significantly higher relative abundance of Prevotella in the DM+HT+HLP cohort compared to in the DM cohort. In addition, among the four cohorts, the DM cohort had the lowest relative abundance of Prevotella. A previous study reported that the relative abundance of Prevotella was higher in patients with urgency urinary incontinence compared to healthy controls (Pearce et al., 2014), and compared to healthy controls, those with urgency urinary incontinence had a significantly higher mean body mass index (which is a good predictor of hyperlipidemia) (Rao et al., 2016), and a greater degree of hypertension (Pearce et al., 2014). Therefore, the high relative abundance of Prevotella may be a causal factor in the development of hypertension and hyperlipidemia.
At the genus level, we found that several bacteria correlated with FBG, BP, and BL. Atopobium was positively correlated with FBG in the DM cohort, which conflicts with the findings of a recent intestinal study in which the relative abundance of Atopobium was lower in those with diabetes compared to in healthy controls (Sato et al., 2014). Allobaculum was negatively correlated with diastolic BP in the DM+HT cohort. An intestinal microbiota study revealed that mice eating a high fat diet (which is associated with high BP) had a lower relative abundance of Allobaculum than controls (Ravussin et al., 2011). Therefore, reducing the relative abundance of Allobaculum in urine may be useful for controlling BP if the association between urinary Allobaculum and BP is similar to that between intestinal Allobaculum and BP. The relative abundance of Rikenella was positively correlated with triglycerides in the DM+HLP cohort. This finding is similar to the findings of a previous study that detected Rikenella in the ceca of diabetic mice with dyslipidemia (Harris et al., 2012). In addition, a recent study of intestinal microbiota revealed that the relative abundance of Odoribacter is negatively correlated with systolic BP (Gomez-Arango et al., 2016), which is somewhat similar to the findings in the present study, as the relative abundance of Odoribacter in the DM+HT+HLP cohort was negatively associated with diastolic BP. Moreover, Chen et al. reported a positive correlation between Dorea and LDL-C in a rat model of hyperlipidemia (Chen D. et al., 2014), and Dorea was also positively associated with LDL-C in the DM+HT+HLP cohort in the present study.
The relative abundance of L. iners, which has been reported to be the most abundant sequence in the urine of patients with sexually transmitted infections (Nelson et al., 2010), was dramatically higher in the DM+HT cohort than in the DM cohort. Several studies have detected L. iners in people with infections, indicating that it does not protect against infections (Saunders et al., 2007; Tamrakar et al., 2007). This corresponds with the fact that higher levels of L. iners might be correlated with asymptomatic bacteriuria and nitrite-positive samples in the DM+HT cohort. In addition, E. lenta, which has been shown to be correlated with the occurrence of bacteremia (Gardiner et al., 2015), was not detected in the DM cohort but was detected in the DM+HT cohort. This suggests that the elevated inflammatory response in those with diabetes plus hypertension may be related to the presence of E. lenta. The relative abundance of A. rhizosphaerae was lower in the DM+HT+HLP cohort compared to in the DM cohort, and the relative abundance of this bacteria was also lower in the DM+HT+HLP cohort compared to in the DM+HLP cohort, which suggests that hypertension plays a role in inhibiting this species.
The main limitation of the study was that the sizes of the four cohorts were not equal, and some were particularly small, which may bias the results as it can make it more difficult to detect differences between the cohorts. Furthermore, in the DM+HT, DM+HLP, and DM+HT+HLP cohorts, the participants could not recall sequence of the occurrences of their conditions, so we could not use information on the onset of these conditions to determine the influences on the urinary microbiota profiles. In addition, although nutrient intakes were not strongly associated with urinary microbiota and no previous study reported that urinay microbiota were affected by food and supplements intake (Nelson et al., 2010; Dong et al., 2011; Siddiqui et al., 2011, 2012, 2014; Fouts et al., 2012; Wolfe et al., 2012; Fricke et al., 2014; Nienhouse et al., 2014; Pearce et al., 2014, 2015; Price et al., 2016; Shoskes et al., 2016; Thomas-White et al., 2016b), it might skew the results. We would enroll participants who has the same amounts of food and supplements intake in our future study to rule out the effects of food, supplements and medicine intake on urinary microbiota.
Conclusions
This is the first study to investigate the association between urinary microbiota profiles and comorbid hypertension and hyperlipidemia in those with T2DM. It demonstrated that the co-occurrence of hypertension and hyperlipidemia in those with T2DM affects the relative abundance of various bacteria in urine. UTIs may be affected by the relative abundances of Proteobacteria, Lactobacillus, and L. iners. FBG, BP, and BL were correlated with the relative abundances of several bacteria. The urinary system is a vital excretory system for metabolic waste and it plays a crucial role in regulating metabolic diseases. The urinary microenvironment (which is produced by the composition of urine) has a large impact on the reproduction of the microflora. Therefore, it is important to further investigate the mechanisms behind the association of urinary microbiota with hypertension, hyperlipidemia, and urinary tract infections, as this may be useful for developing microbiota-based treatment to control hypertension, hyperlipidemia, and bladder infections in those with diabetes.
Author Contributions
LL, WW, YX, and FL conceived and designed the study. ZL generated the sequencing data. FL, LZ, and PJ collected the samples. QY and FL conducted urine cultures and the urinalysis. FL, BW, and LZ extracted the bacterial DNA. FL, ZL, and YX analyzed the data, carried out the computational analysis, interpreted the data, and drafted the manuscript.
Funding
This study was funded by the Opening Foundation of China's State Key Laboratory for Diagnosis and Treatment of Infectious Diseases (grant number: 2015KF05).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge the volunteers who participated in our study. We thank The Charlesworth Group for editing this manuscript, and Chunsheng Xiang and Chunmao Han for their valuable comments on the study.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fphys.2017.00126/full#supplementary-material
References
Al-Rubeaan, K. A., Moharram, O., Al-Naqeb, D., Hassan, A., and Rafiullah, M. R. (2013). Prevalence of urinary tract infection and risk factors among Saudi patients with diabetes. World J. Urol. 31, 573–578. doi: 10.1007/s00345-012-0934-x
Basi, S., Fesler, P., Mimran, A., and Lewis, J. B. (2008). Microalbuminuria in Type 2 Diabetes and Hypertension: a marker, treatment target, or innocent bystander? Diab. Care 31, S194–S201. doi: 10.2337/dc08-s249
Cani, P. D., Delzenne, N. M., Amar, J., and Burcelin, R. (2008). Role of gut microflora in the development of obesity and insulin resistance following high-fat diet feeding. Pathol. Biol. 56, 305–309. doi: 10.1016/j.patbio.2007.09.008
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Casqueiro, J., Casqueiro, J., and Alves, C. (2012). Infections in patients with diabetes mellitus: a review of pathogenesis. Ind. J. Endocrinol. Metab. 16 (Suppl. 1), S27–S36. doi: 10.4103/2230-8210.94253
Chen, C. Y., Chiu, W. N., Lin, Y. C., Jane, S. H., and Chiang, H. H. (2016). Impaired Kidney Function and associated factors among rural adults with disabilities in Taiwan. J. Nurs. Res. doi: 10.1097/jnr.0000000000000141. [Epub ahead of print].
Chen, D., Yang, Z., Chen, X., Huang, Y., Yin, B., Guo, F., et al. (2014). The effect of Lactobacillus rhamnosus hsryfm 1301 on the intestinal microbiota of a hyperlipidemic rat model. BMC Complement Altern. Med. 14:386. doi: 10.1186/1472-6882-14-386
Chen, S., Lin, G., You, X., Lei, L., Li, Y., Lin, M., et al. (2014). Hyperlipidemia causes changes in inflammatory responses to periodontal pathogen challenge: implications in acute and chronic infections. Arch. Oral Biol. 59, 1075–1084. doi: 10.1016/j.archoralbio.2014.06.004
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
de Sereday, M. S., Gonzalez, C., Giorgini, D., De Loredo, L., Braguinsky, J., Cobeñas, C., et al. (2004). Prevalence of diabetes, obesity, hypertension and hyperlipidemia in the central area of Argentina. Diab. Metab. 30, 335–339.
Dingemanse, C., Belzer, C., van Hijum, S. A., Günthel, M., Salvatori, D., den Dunnen, J. T., et al. (2015). Akkermansia muciniphila and Helicobacter typhlonius modulate intestinal tumor development in mice. Carcinogenesis 36, 1388–1396. doi: 10.1093/carcin/bgv120
Dong, Q., Nelson, D. E., Toh, E., Diao, L., Gao, X., Fortenberry, J. D., et al. (2011). The microbial communities in male first catch urine are highly similar to those in paired urethral swab specimens. PLoS ONE 6:e19709. doi: 10.1371/journal.pone.0019709
Fadrosh, D. W., Ma, B., Gajer, P., Sengamalay, N., Ott, S., Brotman, R. M., et al. (2014). An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2:6. doi: 10.1186/2049-2618-2-6
Fouts, D. E., Pieper, R., Szpakowski, S., Pohl, H., Knoblach, S., Suh, M. J., et al. (2012). Integrated next-generation sequencing of 16S rDNA and metaproteomics differentiate the healthy urine microbiome from asymptomatic bacteriuria in neuropathic bladder associated with spinal cord injury. J. Transl. Med. 10:174. doi: 10.1186/1479-5876-10-174
Fricke, W. F., Maddox, C., Song, Y., and Bromberg, J. S. (2014). Human microbiota characterization in the course of renal transplantation. Am. J. Transplant. 14, 416–427. doi: 10.1111/ajt.12588
Gardiner, B. J., Tai, A. Y., Kotsanas, D., Francis, M. J., Roberts, S. A., Ballard, S. A., et al. (2015). Clinical and microbiological characteristics of Eggerthella lenta bacteremia. J. Clin. Microbiol. 53, 626–635. doi: 10.1128/JCM.02926-14
Gomez-Arango, L. F., Barrett, H. L., McIntyre, H. D., Callaway, L. K., Morrison, M., and Dekker Nitert, M. (2016). Increased systolic and diastolic blood pressure is associated with altered gut microbiota composition and butyrate production in early pregnancy. Hypertension 68, 974–981. doi: 10.1161/HYPERTENSIONAHA.116.07910
Gordillo, R., and Spitzer, A. (2009). The nephrotic syndrome. Pediatr. Rev. 30, 94–102. doi: 10.1542/pir.30-3-94
Harris, K., Kassis, A., Major, G., and Chou, C. J. (2012). Is the gut microbiota a new factor contributing to obesity and its metabolic disorders? J. Obes. 2012:879151. doi: 10.1155/2012/879151
Hooton, T. M., Scholes, D., Hughes, J. P., Winter, C., Roberts, P. L., Stapleton, A. E., et al. (1996). A prospective study of risk factors for symptomatic urinary tract infection in young women. N. Engl. J. Med. 335, 468–474. doi: 10.1056/NEJM199608153350703
Huang, Y. C., Shindel, A. W., Ning, H., Lin, G., Harraz, A. M., Wang, G., et al. (2010). Adipose derived stem cells ameliorate hyperlipidemia associated detrusor overactivity in a rat model. J. Urol. 183, 1232–1240. doi: 10.1016/j.juro.2009.11.012
Karstens, L., Asquith, M., Davin, S., Stauffer, P., Fair, D., Gregory, W. T., et al. (2016). Does the urinary microbiome play a role in urgency urinary incontinence and its severity? Front. Cell Infect. Microbiol. 6:78. doi: 10.3389/fcimb.2016.00078
Kieneker, L. M., Gansevoort, R. T., Mukamal, K. J., de Boer, R. A., Navis, G., Bakker, S. J., et al. (2014). Urinary potassium excretion and risk of developing hypertension: the prevention of renal and vascular end-stage disease study. Hypertension 64, 769–776. doi: 10.1161/HYPERTENSIONAHA.114.03750
Lewis, D. A., Brown, R., Williams, J., White, P., Jacobson, S. K., Marchesi, J. R., et al. (2013). The human urinary microbiome; bacterial DNA in voided urine of asymptomatic adults. Front. Cell Infect. Microbiol. 3:41. doi: 10.3389/fcimb.2013.00041
Liu, F., Ling, Z., Xiao, Y., Lv, L., Yang, Q., Wang, B., et al. (2016). Dysbiosis of urinary microbiota is positively correlated with Type 2 diabetes mellitus. Oncotarget 8, 3798–3810. doi: 10.18632/oncotarget.14028
Long, A. N., and Dagogo-Jack, S. (2011). Comorbidities of diabetes and hypertension: mechanisms and approach to target organ protection. J. Clin. Hypertens. (Greenwich) 13, 244–251. doi: 10.1111/j.1751-7176.2011.00434.x
Mangin, M. (2014). Hypertension and inflammation: the infection connection. J. Am. Soc. Hypertens. 8, e7–e10. doi: 10.1016/j.jash.2014.07.016
Marelli, G., Papaleo, E., and Ferrari, A. (2004). Lactobacilli for prevention of urogenital infections: a review. Eur. Rev. Med. Pharmacol. Sci. 8, 87–95.
Mente, A., O'Donnell, M., Rangarajan, S., Dagenais, G., Lear, S., McQueen, M., et al. (2016). Associations of urinary sodium excretion with cardiovascular events in individuals with and without hypertension: a pooled analysis of data from four studies. Lancet 388, 465–475. doi: 10.1016/S0140-6736(16)30467-6
Nelson, D. E., Van Der Pol, B., Dong, Q., Revanna, K. V., Fan, B., Easwaran, S., et al. (2010). Characteristic male urine microbiomes associate with asymptomatic sexually transmitted infection. PLoS ONE 5:e14116. doi: 10.1371/journal.pone.0014116
Nienhouse, V., Gao, X., Dong, Q., Nelson, D. E., Toh, E., McKinley, K., et al. (2014). Interplay between Bladder Microbiota and Urinary Antimicrobial Peptides: Mechanisms for Human Urinary Tract Infection Risk and Symptom Severity. PLoS ONE 9:e114185. doi: 10.1371/journal.pone.0114185
Pearce, M. M., Hilt, E. E., Rosenfeld, A. B., Zilliox, M. J., Thomas-White, K., Fok, C., et al. (2014). The female urinary microbiome: a comparison of women with and without urgency urinary incontinence. mBio 5, e01283–e01214. doi: 10.1128/mBio.01283-14
Pearce, M. M., Zilliox, M. J., Rosenfeld, A. B., Thomas-White, K. J., Richter, H. E., Nager, C. W., et al. (2015). The female urinary microbiome in urgency urinary incontinence. Am. J. Obstet. Gynecol. 213, 347.e1–e11. doi: 10.1016/j.ajog.2015.07.009
Price, T. K., Dune, T., Hilt, E. E., Thomas-White, K. J., Kliethermes, S., Brincat, C., et al. (2016). The clinical urine culture: enhanced techniques improve detection of clinically relevant microorganisms. J. Clin. Microbiol. 54, 1216–1222. doi: 10.1128/JCM.00044-16
Qin, J., Li, Y., Cai, Z., Li, S., Zhu, J., Zhang, F., et al. (2012). A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60. doi: 10.1038/nature11450
Rao, W., Su, Y., Yang, G., Ma, Y., Liu, R., Zhang, S., et al. (2016). Cross-sectional associations between body mass index and hyperlipidemia among adults in Northeastern China. Int. J. Environ. Res. Public Health 13:516. doi: 10.3390/ijerph13050516
Ravussin, Y., Koren, O., Spor, A., LeDuc, C., Gutman, R., Stombaugh, J., et al. (2011). Responses of gut microbiota to diet composition and weight loss in lean and obese mice. Obesity 20, 738–747. doi: 10.1038/oby.2011.111
Reygaert, W. C. (2014). Innate immune response to urinary tract infections involving Escherichia coli. J. Clin. Cell Immunol. 5:280. doi: 10.4172/2155-9899.1000280
Rubino, F. (2008). Is type 2 diabetes an operable intestinal disease? A provocative yet reasonable hypothesis. Diab. Care 31 (Suppl. 2), S290–S296. doi: 10.2337/dc08-s271
Santiago-Rodriguez, T. M., Ly, M., Bonilla, N., and Pride, D. T. (2015). The human urine virome in association with urinary tract infections. Front. Microbiol. 6:14. doi: 10.3389/fmicb.2015.00014
Sato, J., Kanazawa, A., Ikeda, F., Yoshihara, T., Goto, H., Abe, H., et al. (2014). Gut dysbiosis and detection of “live gut bacteria” in blood of Japanese patients with type 2 diabetes. Diab. Care 37, 2343–2350. doi: 10.2337/dc13-2817
Saunders, S., Bocking, A., Challis, J., and Reid, G. (2007). Effect of Lactobacillus challenge on Gardnerella vaginalis biofilms. Colloids Surf. B. Biointerf. 55, 138–142. doi: 10.1016/j.colsurfb.2006.11.040
Shoskes, D. A., Altemus, J., Polackwich, A. S., Tucky, B., Wang, H., and Eng, C. (2016). The urinary microbiome differs significantly between patients with chronic prostatitis/chronic pelvic pain syndrome and controls as well as between patients with different clinical phenotypes. Urology 92, 26–32. doi: 10.1016/j.urology.2016.02.043
Siddiqui, H., Lagesen, K., Nederbragt, A. J., Eri, L. M., Jeansson, S. L., and Jakobsen, K. S. (2014). Pathogens in urine from a female patient with overactive bladder syndrome detected by culture-independent high throughput sequencing: a case report. Open Microbiol. J. 8, 148–153. doi: 10.2174/1874285801408010148
Siddiqui, H., Lagesen, K., Nederbragt, A. J., Jeansson, S. L., and Jakobsen, K. S. (2012). Alterations of microbiota in urine from women with interstitial cystitis. BMC Microbiol. 12:205. doi: 10.1186/1471-2180-12-205
Siddiqui, H., Nederbragt, A. J., Lagesen, K., Jeansson, S. L., and Jakobsen, K. S. (2011). Assessing diversity of the female urine microbiota by high throughput sequencing of 16S rDNA amplicons. BMC Microbiol. 11:244. doi: 10.1186/1471-2180-11-244
Suez, J., Shapiro, H., and Elinav, E. (2016). Role of the microbiome in the normal and aberrant glycemic response. Clin. Nutr. Exper. 6, 59–73. doi: 10.1016/j.yclnex.2016.01.001
Tamrakar, R., Yamada, T., Furuta, I., Cho, K., Morikawa, M., Yamada, H., et al. (2007). Association between Lactobacillus species and bacterial vaginosis-related bacteria, and bacterial vaginosis scores in pregnant Japanese women. BMC Infect. Dis. 7:128. doi: 10.1186/1471-2334-7-128
Thomas-White, K. J., Hilt, E. E., Fok, C., Pearce, M. M., Mueller, E. R., Kliethermes, S., et al. (2016a). Incontinence medication response relates to the female urinary microbiota. Int. Urogynecol. J. 27, 723–733. doi: 10.1007/s00192-015-2847-x
Thomas-White, K. J., Kliethermes, S., Rickey, L., Lukacz, E. S., Richter, H. E., Moalli, P., et al. (2016b). Evaluation of the urinary microbiota of women with uncomplicated stress urinary incontinence. Am. J. Obstet. Gynecol. 216, 55.e1–55.e16. doi: 10.1016/j.ajog.2016.07.049
Varli, M., Guruz, H., Aras, S., Yalcin, A., Atli, T., and Turgay, M. (2012). Asymptomatic bacteriuria among the elderly living in the community: prevalence, risk factors and characteristics. Eur. Geriatr. Med. 3, 87–91. doi: 10.1016/j.eurger.2012.01.007
Wolfe, A. J., Toh, E., Shibata, N., Rong, R., Kenton, K., Fitzgerald, M., et al. (2012). Evidence of uncultivated bacteria in the adult female bladder. J. Clin. Microbiol. 50, 1376–1383. doi: 10.1128/JCM.05852-11
Yang, G., Jiang, Y., Yang, W., Du, F., Yao, Y., Shi, C., et al. (2015). Effective treatment of hypertension by recombinant Lactobacillus plantarum expressing angiotensin converting enzyme inhibitory peptide. Microb. Cell Fact. 14:202. doi: 10.1186/s12934-015-0394-2
Keywords: type 2 diabetes mellitus, hypertension, hyperlipidemia, Lactobacillus, Proteobacteria, urinary microbiota
Citation: Liu F, Ling Z, Xiao Y, Yang Q, Wang B, Zheng L, Jiang P, Li L and Wang W (2017) Alterations of Urinary Microbiota in Type 2 Diabetes Mellitus with Hypertension and/or Hyperlipidemia. Front. Physiol. 8:126. doi: 10.3389/fphys.2017.00126
Received: 24 October 2016; Accepted: 15 February 2017;
Published: 03 March 2017.
Edited by:
Gaetano Santulli, Columbia University, USAReviewed by:
Sang-Bing Ong, Duke NUS Graduate Medical School, SingaporeMarkus Niessen, University of Zurich, Switzerland
Copyright © 2017 Liu, Ling, Xiao, Yang, Wang, Zheng, Jiang, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lanjuan Li, bGpsaUB6anUuZWR1LmNu
Wei Wang, d2FuZ3cyMDAyQDE2My5jb20=
†These authors have contributed equally to this work.