 Daniel M. Sussman
Daniel M. Sussman Daniel A. Beller
Daniel A. Beller- 1Department of Physics, Syracuse University, Syracuse, NY, United States
- 2Department of Physics, Emory University, Atlanta, GA, United States
- 3Department of Physics, University of California, Merced, Merced, CA, United States
Numerical modeling of nematic liquid crystals using the tensorial Landau-de Gennes (LdG) theory provides detailed insights into the structure and energetics of the enormous variety of possible topological defect configurations that may arise when the liquid crystal is in contact with colloidal inclusions or structured boundaries. However, these methods can be computationally expensive, making it challenging to predict (meta)stable configurations involving several colloidal particles, and they are often restricted to system sizes well below the experimental scale. Here we present an open-source software package that exploits the embarrassingly parallel structure of the lattice discretization of the LdG approach. Our implementation, combining CUDA/C++ and OpenMPI, allows users to accelerate simulations using both CPU and GPU resources in either single- or multiple-core configurations. We make use of an efficient minimization algorithm, the Fast Inertial Relaxation Engine (FIRE) method, that is well-suited to large-scale parallelization, requiring little additional memory or computational cost while offering performance competitive with other commonly used methods. In multi-core operation we are able to scale simulations up to supra-micron length scales of experimental relevance, and in single-core operation the simulation package includes a user-friendly GUI environment for rapid prototyping of interfacial features and the multifarious defect states they can promote. To demonstrate this software package, we examine in detail the competition between curvilinear disclinations and point-like hedgehog defects as size scale, material properties, and geometric features are varied. We also study the effects of an interface patterned with an array of topological point-defects.
1. Introduction
Nematic liquid crystals' combination of fluidity and orientational order both underlies nematics' widespread technological applications and endows them with topological defects, localized breakdowns in the orientational order stabilized by the medium's broken symmetries. The topological defects of nematics have been integral to the study of liquid crystals since the field's infancy [1].
Besides their role as tabletop physical realizations of profound topological ideas, nematic topological defects—including disclination lines, point-like hedgehogs, and surface-bound boojums—are of great interest for their importance in nematic colloidal suspensions [2]. These composite materials, formed by suspensions of colloidal particles or nanoparticles in nematics, promise new routes to directed self-assembly and self-organization. Nanoparticles in nematics are pushed by elastic forces to assemble in pre-existing defect lines, meaning that sculpted disclinations provide a path to controlled nanoparticle assembly. Applications include plasmonic properties for metamaterials [3, 4], molecular self-assembly [5], and quantum-dot assembly in microshells [6, 7]. Even greater complexity arises in the cases of colloidal particles in the size range of tens of nanometers to several microns, which often have companion topological defects and which interact through forces mediated by nematic elasticity. Self-assembled structures of colloidal particles with companion defects include bound pairs, chains [2, 8] and triclinic 3D crystals [9]; with the aid of laser tweezers, other configurations such as 3D crystals with tetragonal symmetry [10] and sophisticated disclination knots [11–14] can be stabilized. Tailored self-assembled colloidal structures hold promise as optical metamaterials for photonics applications, such as photonic bandgap crystals and microlasers [15–18].
Nematic defect configurations can also be controlled by non-trivial boundary surfaces [19]. Substrate patterning strategies include topographic variations such as “lock-and-key” docking sites for colloidal particles [20–22] and chemical patterning where the boundary condition shifts abruptly [23, 24]. Complex director fields, including disclinations, can be prescribed on a substrate by photoalignment [25] or by scribing with an atomic force microscope [26]. Confinement in geometries such as capillaries [27], droplets [28], shells [29], and thin films [30] produces a wealth of point- and line-defect behaviors stabilized by topology and energetics.
The rapidly expanding variety of experimentally created nematic defect configurations has benefited greatly from the understanding provided by robust modeling approaches. One set of approaches is based on the Frank-Oseen elastic free energy, which penalizes deformations of the nematic director , and which in its simplest form reads
The superscript (1) refers to the approximation of a single elastic constant K in this expression. However, the symmetry of nematics presents challenges for this model in the presence of disclinations with half-integer winding number, especially if their locations are not known beforehand.
This difficulty is resolved by the Landau-de Gennes (LdG) model, the theoretical approach which is the focus of this work and which we review in section 2. The LdG framework takes as its order parameter the second-rank traceless nematic order tensor Qij(x), and is well-suited to modeling arbitrary disclination configurations, as well as biaxial nematics and the blue phases [15, 31]. While little is known analytically about free energy minimizers in LdG theory in any but the simplest geometries [32, 33], numerical minimization of the LdG free energy has been fruitfully applied over a wide range of systems [10–12, 15, 21, 34–48]. Additionally, flow dynamics of nematics, including active nematic systems, can be modeled by supplementing the LdG free energy with hydrodynamical equations as formulated by Beris and Edwards [49] or by Qian and Sheng [50] and solved by methods such as lattice Boltzmann [51–55], multiparticle collision dynamics and related off-lattice methods [56–58], or finite difference and finite element approaches [59, 60]. Some methods incorporate a fast relaxation of the momentum compared to the director, to account for the separation in time scales for these relaxations in typical molecular liquid crystals [52, 61].
The broad usefulness of the LdG theory goes hand in hand with a significant limitation of scale: Resolving defects at a priori unknown locations requires the simulation lattice spacing to be comparable to or smaller than the size of the defect core, the region in which nematic order breaks down, which in thermotropic nematics is typically a few nanometers. This is often thousands of times smaller than the individual micron-scale colloidal particles of interest. Therefore, a faithful rescaling of the experimental system in numerics would require of order at least 109 lattice sites even for configurations involving only a small number of such colloids.
Accessing such experimentally relevant lattice sizes presents computational challenges not often seen in the simulations of glassy and polymeric soft matter systems. The demands on system memory quickly become prohibitive: simply maintaining the five independent degrees of freedom at each lattice site and storing the necessary change in those variables from one minimization step to the next at 109 lattice sites requires 80 GB—more than on most current commodity desktops and larger than the memory capacity of any CUDA-capable GPU1. Additionally, there is a large direct computational cost of even simple manipulations acting on so many degrees of freedom; this contributes to the significant wall-time required for most numerical energy minimizations and presents challenges for efficient exploration of parameter spaces and colloidal particle positions.
Consequently, LdG numerical modeling is typically applied to systems significantly scaled down, with respect to a fixed defect core size, as compared with the experiments that they are intended to model. While important qualitative insights about defects and director fields can often be obtained by scaling down the experimental dimensions, the change in size ratios makes quantitative prediction challenging. There can also be major qualitative differences. The most well-known of these is the form of the companion defect to a particle with homeotropic (normal) anchoring: Micron-scale particles typically have hyperbolic hedgehog companions (in the absence of confinement or external fields) [2], whereas particles in the few hundred nm or smaller size range have disclination loops in the “Saturn ring” configuration [62, 63]. This constitutes a major challenge in modeling systems with multiple colloid-hedgehog pairs. Experimental work on high-aspect ratio colloidal particle shapes observes both hedgehogs and disclination loops, but numerical modeling has been limited to the line defect case [35, 38–43, 64, 65]. Adaptive mesh refinement in finite-element simulations can help to avoid computational and memory expense in regions not near defects [66] but typically does not remove the need to scale down.
In this work we present an open-source finite difference-based implementation of LdG free energy minimization with non-trivial boundary conditions, “openQmin” [67], using a combination of approaches designed to address the challenges described above. It is written for heterogeneous CPU and GPU operation to target two complementary research goals. First, it offers a user-friendly GUI environment for rapid prototyping of topological defect configurations as a function of liquid crystal parameters, boundary geometry, and the presence of colloidal inclusions. Simultaneously, it targets large-scale systems using OpenMPI [68] to support parallelization across both CPU and GPU resources to scale up to the supra-micron length scales of experimental relevance. We employ efficient minimization algorithms, such as the Fast Inertial Relaxation Engine (FIRE) method, to maintain reasonable convergence times even for large-scale parallelized calculations.
The remainder of the paper is structured as follows. We begin with a review of the LdG theory in section 2. Section 3 lays out our numerical approach, first discretizing the LdG theory for a finite-difference method, and then outlining our use of minimization algorithms and OpenMPI parallelization. In section 4 we present two sample studies demonstrating the effectiveness of this approach. We first perform a classic analysis of the companion defects to homeotropic spherical particles at varying system sizes, and then examine the effects of a boundary patterned with surface disclinations in a supra-micron-scale system. Section 5 briefly describes the GUI version of openQmin with an example of the rapid prototyping workflow it enables. Finally, in section 6, we discuss both the range of use we foresee for openQmin and some future directions for additional physics that could be studied in this framework.
2. Landau de-Gennes Theory for Nematic Liquid Crystals
Here we give a brief overview of those aspects of the LdG theory used in our numerical approach. The theory is of course well-established [69] and its use in a finite difference numerical free energy minimization scheme is described in several sources; the reader is directed to Ravnik and Žumer [47] for a thorough explanation.
Uniaxial nematic liquid crystals are characterized by orientational ordering of nematogens (molecules or suspended anisotropic particles) along a director, , which is a unit vector with the identification = -. To respect that symmetry consistently, which is important at disclinations of half-integer winding number, we take as order parameter not a director but a second-rank tensor. This is the traceless, symmetric tensor field Q(x), whose lattice discretization is the fundamental object of the LdG modeling approach. Q is related to by [70]
Here, S is the degree of uniaxial nematic order, and SB is the degree of biaxial order distinguishing a preferred direction , perpendicular to , from . The nematic director can be recovered as the eigenvector corresponding to the largest eigenvalue of Q, which equals S. Most nematics are unaxial, so the equality SB = 0 is true in the absence of distortions and represents a good approximation away from defects. In this uniaxial limit, Equation (2) reduces to
2.1. Phenomenological Free Energy Density
The LdG theory constructs a phenomenological free energy as a functional of Q(x). We can write this functional schematically as [47, 70]:
The first integral, over the volume of the nematic, has three free energy density terms incorporating respectively the energetic costs arising from deviations away from the thermodynamically preferred degree of nematic order S = S0, from elastic distortions, and from external fields. The second integral, summing over all boundary surfaces Sα in contact with the nematic, incorporates the anchoring energy associated with each interface, including the surfaces of colloidal particles; its form may be different for different surfaces. We address each component in turn.
2.1.1. Bulk Free Energy
The first free energy density term in Equation (4) gives a Landau free energy for the isotropic-nematic phase transition, written in terms of rotational invariants of Q in a Taylor expansion about the isotropic, Q = 0 state [71]:
The parameter , where is the temperature at which the isotropic phase is destabilized. In the uniaxial limit fbulk becomes a polynomial in the degree of order,
which is minimized either by S = 0 or by
In the nematic phase, the absolute value of
provides a free energy penalty per unit volume to the melted cores of defects, where S → 0.
2.1.2. Distortion Free Energy
The distortion free energy density models the elasticity of the nematic phase, and represents the LdG counterpart to the Frank-Oseen free energy density. The latter, in full generality up to second derivatives of , is
The parameters in this expression are the splay (K1), twist (K2), bend (K3), and saddle-splay (K24) elastic constants, and the spontaneous chiral wavenumber q0 which is non-zero in the cholesteric and blue phases. Other common conventions for the saddle-splay energy density replace K24 in Equation (9) by either 2K24 or 2(K2 + K24). Equation (9) reduces to Equation (1) under the “one-constant approximation” K1 = K2 = K3 = K24 ≡ K and q0 = 0. The one-constant approximation is a reasonable simplification for many molecular liquid crystals, where K1, K2, and K3 typically differ by less than a factor of 5 [72, 73].
The most general form of fdistortion that we employ, following Poniewierski and Sluckin [74], Mori et al. [48], and Mottram and Newton [70], includes all gradient terms of quadratic order in Q allowed by symmetry, plus one term at cubic order:
where Einstein summation over repeated indices is implied, and ϵ is the Levi-Civita tensor. Equation (10) corresponds in the uniaxial limit to Equation (9) with the identifications [48]
The one-constant approximation in the absence of spontaneous chiral ordering sets L2 = L3 = L4 = L6 = 0, leaving the much simpler and more computationally efficient form
which corresponds in the uniaxial limit to Equation (1) with .
Taking this simpler form of the distortion energy density, we estimate the defect core size by considering a distorted uniaxial nematic configuration at S = S0 with varying with typical gradient 1/ℓ. Roughly speaking, the energy well depth f0 (Equation 8) gives the threshold value for fdistortion at which distortions become so energetically costly that a local melting of nematic order occurs instead. This length ℓ = ξN, the nematic correlation length (or coherence length), sets the size of the defect core:
2.1.3. External Fields Free Energy
The response of the nematic to an external magnetic field H or an external electric field E is modeled by the free energy density term
where Δχ and Δε are the anisotropic parts (difference in principal values corresponding to and its perpendicular directions) of the magnetic susceptibility tensor and dielectric tensor, respectively [34], and μ0 and ε0 are respectively the magnetic permeability and electric permittivity of free space. (We omit here the terms for the isotropic parts of these tensors, as they do not couple to Q). In the uniaxial limit, the right-hand side becomes (again dropping isotropic terms with no coupling to ). Positive Δχ or Δε will favor alignment of with H or E.
2.1.4. Boundary Free Energy
Boundary surfaces, including the surfaces of embedded colloidal particles, generally impose an anchoring surface energy density fboundary representing the surface tension's dependence on the director at the surface. In terms of the director, a common modeling choice for the anchoring energy is the Rapini-Papoular form where is the surface normal vector and |Wα| is the anchoring strength of surface α [75]. Homeotropic (normal) anchoring follows from WRP > 0, whereas WRP < 0 creates degenerate planar anchoring, which equally favors every direction perpendicular to . The same anchoring functional can favor a different anchoring direction, for example an in-plane direction in the case of oriented planar anchoring, using WRP > 0 with the replacement of by the favored direction.
In LdG theory, for homeotropic or other oriented anchoring, the Rapini-Papoular form is generalized as the Nobili-Durand surface anchoring form [76],
where is the anchoring strength of surface α and the surface-preferred Q-tensor, Qα, is usually taken to be , with or some other surface-preferred director.
For degenerate planar anchoring, the Nobili-Durand form is not suitable, and we use instead the following free energy due to Fournier and Galatola [77]:
where and is the projection onto via for . Assuming Q is uniaxial with S = S0, the Rapini-Papoular anchoring is recovered with .
3. Numerical Approach
3.1. Overview
The primary contribution of this work is the presentation of an open-source numerical implementation that exploits the embarrassingly parallel structure of the lattice discretization of the above phenomenological theory. Our implementation, combining CUDA/C++ [78] and OpenMPI [68], was written with extreme flexibility in mind to allow users to accelerate simulations large and small using combinations of available CPU and GPU resources in either single- or multiple-core configurations.
The foundation of the software package, “dDimensionalSimulation,” is a set of generic classes meant to execute simulations of N interacting units, each consisting of d scalar degrees of freedom, using data structures appropriate for efficient execution on either CPU or GPU resources. These generic classes serve as the template for models which instantiate the dN total degrees of freedom, forces which compute interactions between degrees of freedom, updaters which can change the degrees of freedom (e.g., by implementing equations of motion), and simulations which tie objects of these various types together. The present work focuses on implementing the details of these classes to carry out lattice-based LdG modeling to find energy-minimized configurations of equilibrium nematics in the presence of various boundary conditions. The general structure we have employed was chosen to allow flexibility in future development, for example to derive new model classes which include not only the Q-tensor but also density and velocity degrees of freedom, as would be appropriate for modeling active nematic systems [53–55, 60].
In addition to writing efficient code to carry out the required lattice-based minimizations of the Q-tensor field in a domain, we also advocate the use of the graphical user interface (GUI) we developed to rapidly prototype and explore the effects of particular boundaries, colloidal inclusions, and external fields that may be of experimental interest. The GUI allows a wide variety of user operations—adding boundary objects at any stage of the simulation, starting and stopping minimization, adding or removing external fields at will—all while visualizing the resulting defect structure and recording configurational details. A snapshot of the GUI is shown in Figure 1, and more details of the available features are given in section 5. We envision that this capability will allow for rapid prototyping of experimental geometries in the search for particular controllable defect states; running on a single GPU allows real-time visualization of lattices in the low-millions of total sites. We have also included several example files that use the code in a non-GUI mode; these can then use OpenMPI to parallelize across either CPU or GPU resources to scale up to lattices that represent supra-micron-scale liquid crystal systems.
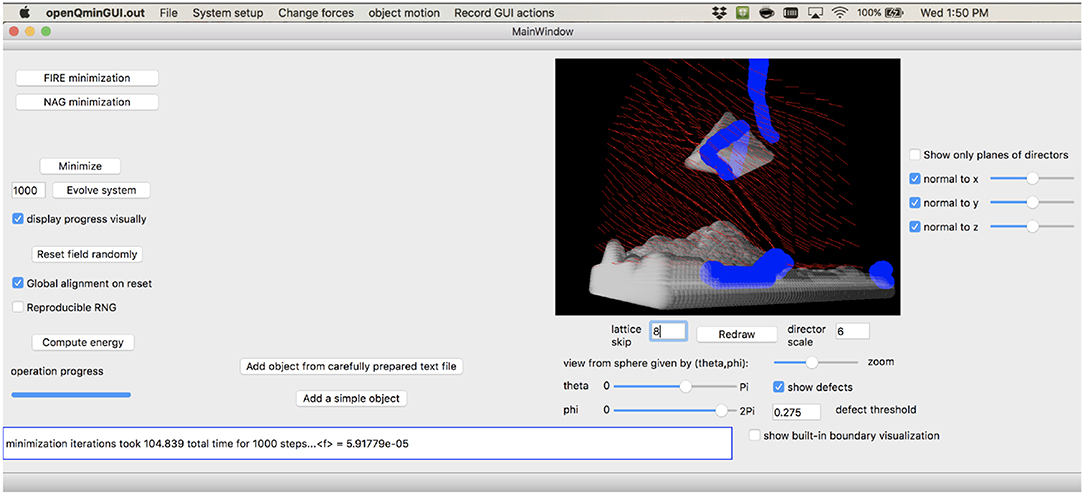
Figure 1. Snapshot of the graphical user interface provided by openQmin, here shown simulating the defect structure near a pyramidal colloid above a topographically non-trivial boundary, all with oriented anchoring along user-specified directions to approximately model homeotropic surfaces.
3.2. Lattice Discretization and Energy Minimization
The finite difference lattice calculations employed in this work use a regular cubic lattice discretization of space, with a Q-tensor defined at each site . The lattice spacing Δx can be related to physical quantities through a natural non-dimensionalization of the free energy density, , which implies a non-dimensionalization of the elastic constants . In the one-constant approximation, we thus have . In this work we set . To model 5CB, following Ravnik and Žumer [47] we take A = −0.172 × 106 J/m3, B = −2.12 × 106 J/m3, C = 1.73 × 106 J/m3, and where S0 ≈ 0.53. These give a lattice spacing of Δx ≈ 4.5 nm, which is at the few-nm scale of the defect core in 5CB. Note that the non-dimensionalization of all constants by an energy scale |A| and a length Δx is implicitly made for all values in openQmin, including in the GUI.
The symmetry and tracelessness of Q leaves five independent degrees of freedom, which we take to be at each of the N lattice sites in the simulation domain. We write the local free energy density in terms of these five independent variables, so that the symmetry and tracelessness of Q are automatic (rather than being maintained by projection operations following update steps [47]). We also label each site with an integer “type,” indicating whether it is a bulk site, a boundary site, or a site inside an object (for instance, the interior of a colloidal inclusion, or part of a bounding surface), depending on the geometry of the problem. Only bulk and boundary sites are “simulated sites,” meaning Q is defined there.
We discretize the total free energy, , using a finite-difference approach over the 5N independent variables. For the distortion terms we allow the user to select either the more general expression, Equation (10), or the more computationally efficient expression of Equation (11). For the terms in fdistortion which contain spatial first derivatives of Q, we consider first-order forward and backward finite difference approximations,
Here is the unit vector in the xk direction, and is the site where the calculation is taking place. The choice of a regular cubic lattice makes these derivative approximations straightforward to calculate. The forward and backward finite differences are each compatible with the simulation domain only if (), , respectively, are simulated (bulk or boundary) sites. We then take, as the discretized expression of f, Equation (4) averaged over all forward and backward finite difference expressions for each of k = 1, 2, 3 that are allowed by the geometry of the simulation domain. A bulk site, therefore, has a local free energy averaged over 23 such combinations, while a boundary site has fewer. We use these averages over different expressions for the finite differences, rather than using a single centered finite difference formula (, because using the latter form in Equation (11) produces no terms coupling to its nearest neighbors, of the form . This use of the centered first derivative expression would therefore create an artificial (and undesirable) lattice doubling effect in our approach, with even sites and odd sites evolving independently. For curved boundaries such as on spherical colloidal particles, well-known inaccuracies are introduced in the finite difference calculations by the discretization of boundaries as sites in the cubic lattice [79]. Specifically, errors of order O(Δx) in are introduced, leading to truncation errors of O(1) (which do not diverge as the lattice spacing is refined) in the first derivative approximations of Equation (16).
Finally, we minimize as a cost function over the 5N independent variables , i = 1, …, 5, j = 1, …, N. The gradient of in this 5N-dimensional space is calculated by explicitly differentiating the expression for with respect to each degree of freedom. This explicit differentiation of a cost function is an alternative to the approach of analytically deriving local forces (molecular fields) from the Euler-Lagrange equations, projecting to recover symmetry and tracelessness, and then discretizing those expressions. While the Euler-Lagrange equations have separate forms for the bulk and the boundaries, in the approach used here forces are derived from the cost function in formally the same way for bulk and boundary sites.
We emphasize that by discretizing space, we can directly map the problem of solving the LdG partial differential equations to finding the minima of a complex energy landscape (where the Q-tensors on each lattice site are the degrees of freedom). For instance, many PDE solvers implement steepest descent relaxation, which can be directly interpreted as overdamped molecular dynamics at zero temperature. This allows us to turn to the wealth of existing algorithmic approaches from the field of non-linear optimization, including minimization techniques such as quasi-Newton methods (conjugate), gradient descent, and momentum-based techniques such as Nesterov's accelerated gradient [80]. Since our aim is to be able to scale up to large systems, we ignore minimizers which require second-order derivatives of the cost function, and we find that even limited-memory quasi-Newton methods such as L-BFGS impose too-strong a memory requirement for many of our purposes. Additionally, while conjugate gradient is appealing in having only marginal extra memory requirements and being much faster than simple gradient descent, it involves frequent line searches that require expensive repeated evaluations of the free energy density and imposes additional parallelization costs.
Thus, although we have implemented many of the above-named minimizers in openQmin, we focus our attention on the use of the Fast Inertial Relaxation Engine (FIRE) method of energy minimization [81]. FIRE falls into the class of “gradient plus momentum”-style minimization algorithms, and it additionally rescales the “velocity” (fictitious additional variables introduced to make the analogy with molecular dynamics even more complete and corresponding to the velocities at which the Q-tensor components change) of the degrees of freedom and adaptively changes the size of the time step itself based on the behavior of the force and velocity during the most recent update. For convex optimization problems the addition of inertia can be proven to enhance convergence [82], although for more complex energy landscapes in general little can be proven. Thus, while it is a heuristic approach, FIRE has been shown to be competitive with (or even faster than) conjugate gradient minimization [81, 83, 84], all while maintaining an extremely light additional memory footprint and being highly amenable to parallelization across multiple cores or multiple GPU units. Note that FIRE was originally developed with atomistic simulations in mind, but it is increasingly being used more generally, including in the solution of PDEs [85] and in machine learning applications [86]. By the straightforward mapping mentioned above we are able to directly apply the pseudocode presented in Algorithm 1.

Algorithm 1: Pseudocode for FIRE minimization [81].
We first demonstrate this efficient minimization in Figure 2, where we compare the system energy and average norm of the force on the degrees of freedom during the minimization of a lattice of N = 2503 sites in a cubic geometry with periodic boundary conditions. To make a fair comparison, we have performed both a FIRE and a gradient descent (GD) minimization on the same system using separately tuned minimization parameters for each algorithm. We use the same hardware for each simulation, and report the minimization progress in terms of the wall-clock time taken. Although it is sometimes common to report efficiency in such comparisons in units of function calls, for algorithms with very different numbers of arithmetic operations (each FIRE iteration requires more than twice the number of arithmetic operations compared to GD) such comparisons are often misleading.
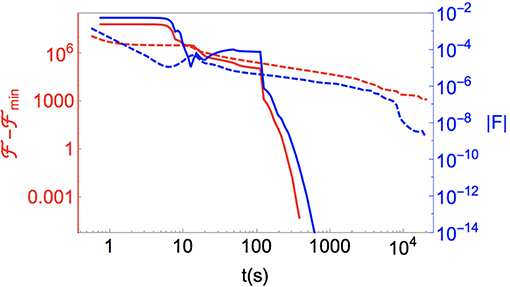
Figure 2. (Red) Energy relative to the uniform texture with preferred nematic order, , and (blue) the norm of the residual force vector, , for bulk nematic (lattice size is N = 2503), starting from a randomly initialized configuration, as a function of wall-clock time. Solid lines are minimizations using FIRE and dashed lines are those using gradient descent. As described in the text, we have tuned the minimization parameters (step size, etc.) for each algorithm separately and use identical hardware to make a one-to-one comparison.
As Figure 2 makes clear, even in the trivial case of finding the uniform nematic ground state for a system with no boundary terms from a system initialized with random Q-tensors at each lattice site, FIRE provides orders of magnitude improvement in the time taken to find minima. This performance of our default minimizer is not restricted to simple, bulk states of the liquid crystals. As we demonstrate in Figure 3 for a handful of simple (and well-studied) arrangements of colloidal inclusions and boundaries, FIRE is very rapidly able to find these more complex minima, too. As with any non-convex optimization solver, though, no guarantees are made by FIRE about avoiding particular local minima in favor of a true global minimum. Where this is a concern, we adopt the standard approach of minimizing from multiple different random initializations. Particularly when coupled with a GPU, the substantial acceleration of FIRE-based minimizations enables the usefulness of the GUI, as the evolution of defect structures in response to user-instigated changes can be seen in real time.
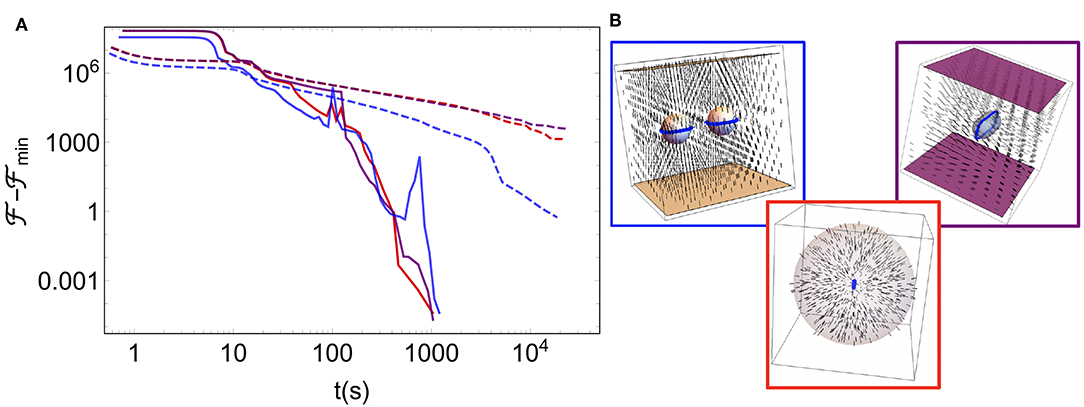
Figure 3. (A) Energy relative to the minimized energy for three different geometries as a function of wall-clock time, in a lattice of size N = 2503 and starting from a randomly initialized configuration. As in Figure 2, solid lines are minimizations using FIRE and dashed lines are those using gradient descent. As depicted in (B) showing the minimized configurations, the three sets of lines correspond to (Blue) two spherical colloids between parallel plates, all with homeotropic anchoring, (Red) the interior of a spherical droplet with homeotropic anchoring, and (Purple) a spherocylinder with homeotropic anchoring between parallel plates with degenerate planar anchoring. These images were created using the “multirankImages.nb” Mathematica file included in the repository for making simple visualizations.
Although numerical simulations of this size have been commonly used to make contact with experiments, in single-core operation it is impractical to simulate lattices much larger than N ~ 3003, with the limiting factor being the wall-clock time required for CPUs and memory constraints for GPUs. Given a simulation with degrees of freedom and spreading the work across P processing units (either GPUs or CPUs), achieving ideal scaling requires both low-latency communication between processors and algorithms that are themselves linear in . Fortunately, lattice-based models with only nearest- and next-nearest-neighbor interactions are trivial to parallelize using a pattern common to, e.g., spin glasses [87]. We use a standard spatial decomposition of the total number of lattice sites into rectilinear sub-regions (typically cubes, although other spatial partitions are easily implemented and may be preferable for some simulation geometries). Each processing unit is assigned to one of these subregions, and is responsible for controlling and updating the lattice sites in that subregion. It also maintains information about the state of the “halo” of lattice sites that are neighbors, nearest-neighbors, and next-nearest neighbors of lattice sites at the boundary of the subregion it controls. Standard OpenMPI protocols [68] are used during each simulation step to communicate information about the state of these halo sites to and from each processing unit in optimized sequences of uni-directional transfers.
We now assess how our method's efficiency scales as the problem size is increased. Although strong scaling (Amdahl's law)—in which the total problem size is kept fixed and P is increased—is often important, it is well-established that the structure of the near-neighbor lattice interactions we simulate is embarrassingly parallel. Our real aim is to scale up the problem size itself and use many processors to simulate lattices that approach experimental scales. As such, weak scaling (Gustafson's law)—in which the amount of work per processing unit is kept constant—is the relevant test.
One challenge to mention here is that when targeting energy minima—as opposed to simply advancing a molecular dynamics simulation for a fixed number of time steps—the number of minimization steps itself grows with the total system size. In general the convergence properties of different minimizers in non-convex settings are highly non-trivial. For simple geometries we are able to numerically probe this scaling—for instance, we find that in the absence of any boundary the number of minimization steps to achieve a target small force tolerance scales with the linear size of the system, whereas in the presence of a spherical colloid it scales roughly with L3/2. In general, though, the approximate scaling may be hard to ascertain (and may depend on the target threshold for declaring a configuration to be in a minimum).
Turning instead, then, to the per-minimization-step timings, we present the weak scaling performance of openQmin in Figure 4, where we compute the total number of lattice-site updates (i.e., N times the number of simulated time steps) during a minimization in which we fix Np, the number of lattice sites per processing unit, at several values and vary P. Consistent with a globally cubic simulation, we parallelized across P = 13, 23, 33, 43, 53, 63, 73, 83, 93, 103 processors on the Comet XSEDE cluster, and studied computational performance for . As expected, there are systematic drops due to increased communication costs as one goes from 1 core to multiple cores to multiple nodes, but openQmin recovers ideal linear scaling of lattice updates with P as P grows very large. Additionally, there is a systematic degradation of performance for small Np, since in that case there is a more unfavorable ratio of halo sites to controlled sites for each processor.
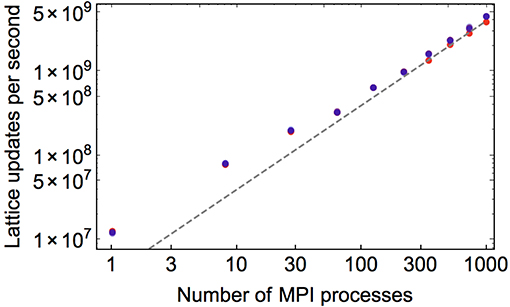
Figure 4. Weak scaling performance of openQmin on Comet, in total number of lattice site updates [i.e., (time steps)×(ranks)×(Np)] per second vs. the number of CPU processes, P, for a constant number of lattice sites per process. The points from dark red to light blue correspond to lattice sites per rank. The dashed gray line corresponds to ideal ∝P scaling.
Note that when we set the characteristic lattice spacing to correspond to 4.5 nm, the largest system simulated in this study, , corresponds to a simulation domain of volume 1,424 μm3.
4. Sample Studies
4.1. Companion Defects to Homeotropic Spherical Colloids
In this section we apply openQmin to the question of whether a hyperbolic hedgehog or a Saturn ring disclination loop provides the minimum-energy form of the topological companion defect to a homeotropic spherical colloid. As mentioned above, a larger colloid radius a favors the dipolar configuration with a hedgehog, whereas smaller a favors the quadrupolar configuration with a Saturn ring. As a result, the common rescaling of experimental dimensions to smaller a/ξN in numerical modeling risks obtaining qualitatively different topological defect configurations. Besides increasing the simulation box size, altering the modeled material constants can restore qualitative agreement between experiment and simulation. Here we explore the issue in detail, using openQmin to systematically investigate the stability of hedgehogs relative to Saturn rings over a range of sizes and material parameters.
The dipolar configuration with a hyperbolic hedgehog is the ground state for homeotropic colloidal particles near or above the micron scale [2]. Terentjev's prediction of the alternative quadrupolar director field configuration with a Saturn ring disclination loop [62] can be stabilized for large particles by confinement or external fields [88, 89]. Stark [63] demonstrated numerically using the Frank-Oseen free energy that the Saturn ring becomes metastable relative to the dipole for a ≲ 720 nm, with a defect core size rc = 10 nm. For a ≲ 270 nm, the Saturn ring becomes the global ground state.
While the elastic energies of the two configurations are complicated to express, the Saturn ring is additionally penalized by a simple core energy per unit length, or line tension, γ = πK/8 [63, 69]. Because the Saturn ring maintains a radius rd just slightly larger than that of the colloidal particle, rd ≈ 1.1a [63], the total defect core energy penalty Ec = 2πrdγ ∝ Krd of the Saturn ring grows linearly with the colloid radius. In contrast, the hyperbolic hedgehog has no defect core dimension growing in size with the colloidal particle, helping to stabilize the dipole over the Saturn ring at larger colloid sizes.
In order to numerically model multi-particle configurations in the dipolar size regime—if we cannot exploit crystal symmetries to obtain a small unit cell [10, 45]—we must either scale up the simulation volume to larger lattices, or stabilize the dipole at smaller particle sizes. We can achieve the latter by altering the materials-constant ratios , in Equation (5). Together, these two ratios determine S0 via Equation (7), as well as the non-dimensionalized free energy density of the nematic ground state with the energy well depth f0 defined as in Equation (8).
By varying and such that S0 remains fixed, we alter the energetic cost per unit volume of melted nematic order in defect cores, |f0|. The defect core size rc varies with the nematic correlation length ξN, which, from Equation (12), scales as . Thus, an increase in |f0| implies a decrease in the defect core size, which means effectively that the ratio a/rc of the particle size to the defect core size is increased without changing the size of the simulation lattice. The dipolar configuration is therefore expected to remain stable at smaller particle sizes. This technique was used in Luo et al. [37] to model a dynamical transition from Saturn ring to dipole as a colloidal particle approaches an undulated boundary, at simulation box sizes up to 50 times smaller than the experimental dimensions.
The results of this study are shown in Figure 5, which we parameterize by varying at fixed S0 = 0.53 [i.e., setting ], along with the size of the spherical colloid and the lattice size. We test the stability of hyperbolic hedgehogs by initializing the surrounding lattice sites in the dipolar defect configuration suggested by Lubensky et al. [90], performing an energy minimization, and testing whether the resulting configuration has remained in the hedgehog state or transitioned to a Saturn ring configuration (thus, testing the meta-stability of the dipolar defect state as a function of system parameterization). At the values , commonly used in modeling of 5CB [47], we find that the lower limit of hedgehog meta-stability is a ≈ 74 lattice spacings, or about 330 nm. In this sample study we have imposed a large but finite anchoring strength at the colloid's surface. Weaker anchoring strength will affect the results, with a “surface ring” configuration replacing the dipole at low anchoring strength [63].
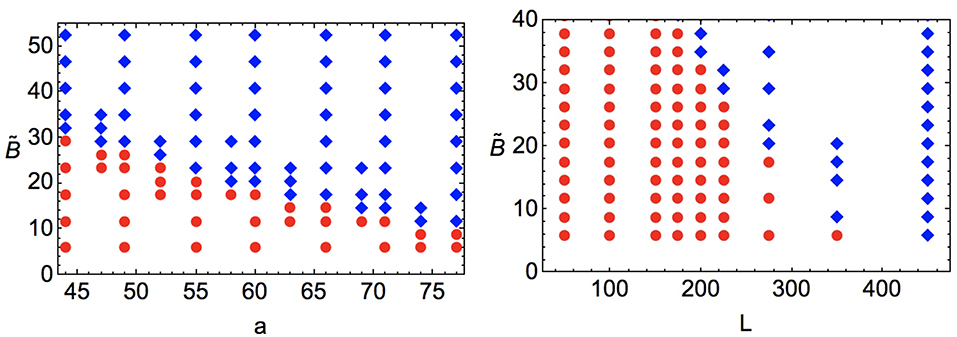
Figure 5. Stability of dipolar defects around a spherical colloidal inclusion at fixed S0 = 0.53 as a function of dimensionless bulk free energy density constant , colloid size a, and linear system size L, with a and L in units of the lattice spacing. Regions of parameter space with (meta-)stable dipolar configurations are shown with blue diamonds, stable quadrupolar configurations are shown with red circles. (Left) For fixed lattice size of N = 2753, we vary the dimensionless bulk free energy density constant and the colloidal radius a. (Right) For fixed ratio of colloidal radius to linear system size, a = 0.22 × L = 0.22 × N1/3, we vary and L. Particularly for the larger values of a, one can see dependence of hedgehog meta-stability on L, indicating the importance of far-field distortions and boundary conditions (here, periodic).
We have also tested the meta-stability of the quadrupolar defect configuration by initializing the system in a Saturn ring configuration and minimizing, but we have not observed the spontaneous appearance of hedgehog defects from such simulations, indicating at least the meta-stability (if not absolute stability) of Saturn rings over the entire parameter range studied here. In addition to the effect of defect core size mentioned above, slight deviations in hedgehog meta-stability as a function of lattice size at fixed and a seen in Figure 5 indicate the importance of far-field distortion terms on the (meta-) stability of defect configurations.
4.2. Patterned Boundary Conditions
To demonstrate the modeling of patterned boundaries in openQmin, we examine a square array of alternating ±1 disclinations imprinted as a spatially varying anchoring direction on a planar substrate. Such an array was created experimentally by the authors of Murray et al. [26], by scribing lines into a polyimide surface with an atomic force microscope. As in that experiment, we give the opposing surface degenerate planar anchoring. In openQmin, these boundary conditions are specified at each boundary lattice site through a user-prepared text file (see section 5 below). We employ periodic boundary conditions in the horizontal directions, and the anchoring strength W at both surfaces is set to make the extrapolation length K/W roughly equal to the lattice spacing.
Figure 6A shows the result of minimizing a cell of thickness h = 224 lattice spacings, corresponding to ≈1 μm for 5CB, and a spacing d between defects equal to h. We create an 8 × 8 array of defects, so the total volume modeled is 64 μm3, larger than the maximum size achievable with single core minimizations on a typical CPU (≈10 − 20 μm3). Simulating several unit cells of the substrate patterning in this way allows us to observe a labyrinthine configuration of half-integer disclination lines near the plane of the substrate, connecting neighboring surface-defects. Meanwhile, some disclination lines are vertical, traveling between the two surfaces and imprinting a or defect profile on the top surface. The stopping condition for the minimization here was a somewhat modest force tolerance, allowing these large-system-size studies to be completed in less than 24 hours. While clearly not completely equilibrated, the horizontal disclination labyrinth is similar to a domain wall texture observed experimentally in Murray et al. [26], which may also be kinetically trapped. Absent from the texture in Figure 6A is the ±1 non-singular escaped configuration, which did appear in the experiments.
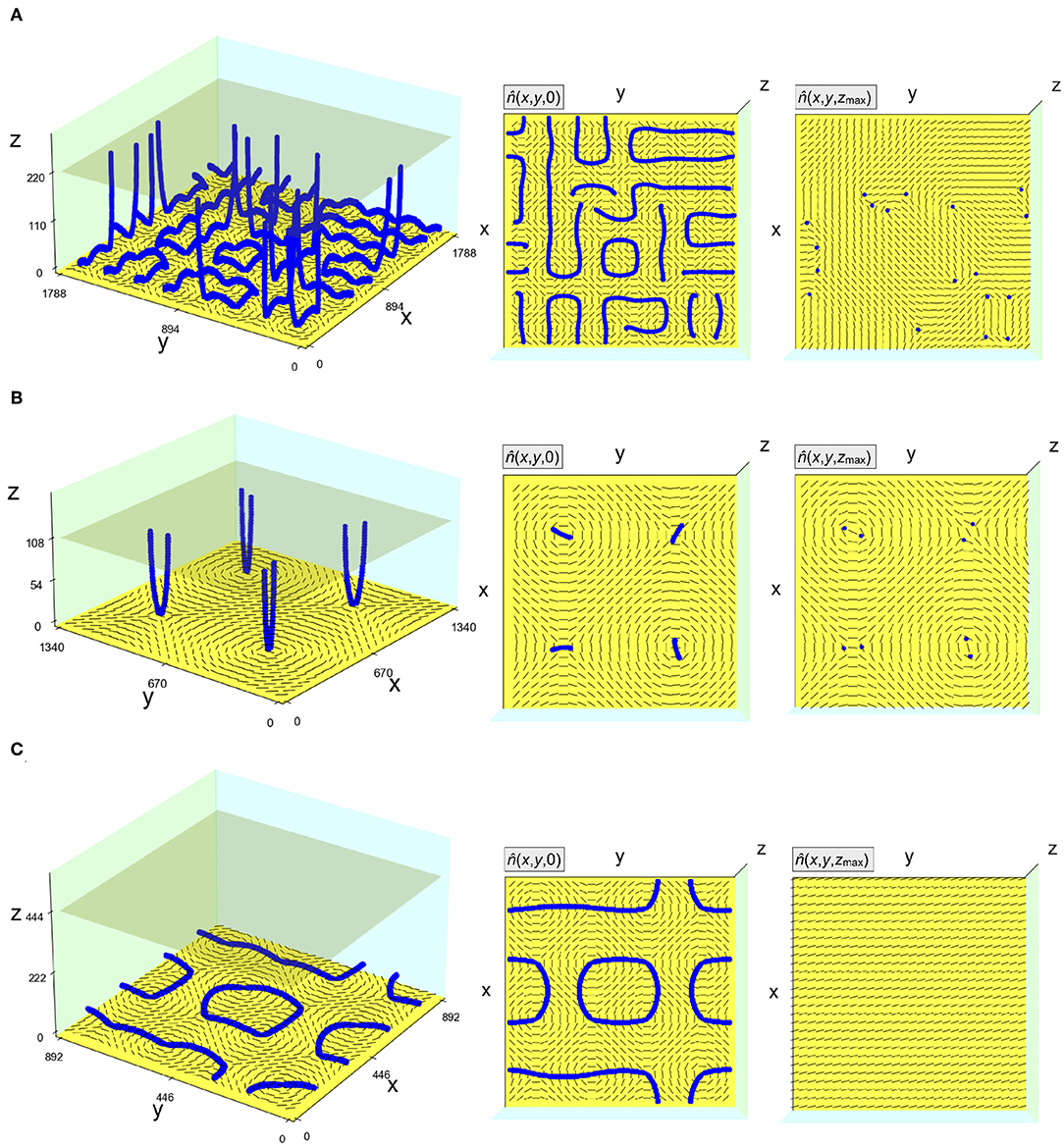
Figure 6. Numerically computed disclination configurations near planar substrates patterned with square arrays of alternating ±1 surface disclinations. The opposite planar boundary (transparent square) has degenerate planar anchoring. (A) An 8 × 8 array of surface disclinations with spacing equal to the cell thickness. (B) A 2 × 2 array of surface disclinations with spacing equal to six times the cell thickness. (C) A 4 × 4 array of surface disclinations with spacing equal to half the cell thickness. Configurations in (A–C) are partially energy-minimized. Disclinations are colored blue. Axes values are given in units of the lattice spacing. In each row, the second panel shows a top view of the disclinations in bulk and the director field in the plane of the patterned substrate; the third panel shows a top view of the director field on the opposite surface, along with the half-integer disclination points (if any) in that surface. These images were made using the “visualize.py” Python script included in the repository for taking saved configurations and making simple visualizations from the command line.
The energetic cost per unit length of disclination lines implies that the vertical configuration is favored by smaller cell thickness h. Indeed, as shown in Figure 6B, when we decrease h/d from 1 to , only vertical disclinations appear, in pairs of or disclinations from the “splitting” of the ±1 surface defects. This defect splitting was sometimes observed in Murray et al. [26] in place of the escaped configuration. Conversely, as shown in Figure 6C, only horizontal disclinations appear when h/d is increased to 2. Extensions to even larger defect arrays, to curved boundaries, and to spatially non-uniform anchoring types can be explored in the same manner in openQmin.
5. Rapid Prototyping With GUI Interface
Figure 1 shows a screenshot of the graphical user interface in action; the Supplemental Video and accompanying narrative transcript of the video in Supplementary Data Sheet 1 shows a representative demonstration of its use. Here we discuss some of its current functionality. Initialization dialog boxes allow the user to set the simulation size, the computational resource to use (CPU or GPU, auto-detecting whether CUDA-capable resources are available for use), and parameters for the bulk and distortion free energy density. This generates a random bulk configuration of Q-tensor lattice sites with periodic boundary conditions. For the visualization pane the user can specify the density and magnitude of directors to draw (taken to be the direction of the largest eigenvector of Q at each site), and can freely rotate and zoom in on the configuration, as well as highlight in blue defects defined locally by regions where the largest eigenvalue of Q falls below some threshold.
In the top left are buttons allowing the user to specify parameters from one of two energy minimization techniques to use (FIRE and Nesterov's Accelerated Gradient Descent); the resulting dialog boxes are populated with values that we typically find to be efficient for default parameter choices in the bulk and distortion energies, although some amount of tuning may be quite beneficial (particularly when changing the distortion terms L2 through L6). The “Minimize” button performs the requested energy minimization (either until a target force tolerance is attained or the maximum number of iterations is reached), with the option to visualize the results only at the end or to watch the minimization proceed. The “File” dialog box allows the currently visualized state of the system to be saved for separate analysis or processing.
Note that menu items allow any of the terms in the energy functional governing the simulation, Equation (4), to be changed on the fly. This allows, for example, the user to first minimize a system with some values of the distortion constants and then perform repeated minimizations as those values are changed, observing the stability or meta-stability of defect structures as this is done.
Two buttons allow the user to introduce boundaries and colloidal inclusions into the system. “Simple” objects are spheres and flat walls with either normal homeotropic or degenerate planar anchoring conditions. Arbitrarily complex boundary conditions (taking any shape, with degenerate planar and homeotropic anchoring conditions not restricted by the direction of the surface normal) can be added by preparing a simple text file that the program can read in – an example script that generates the custom boundary file used in Figure 1 is included in the “/tools” directory of the project's GitHub repository [67].
With boundaries and colloids (“objects”) in place, some manipulations of these objects are accessible via drop-down menus. The positions of these objects within the simulation can be directly modified, so the user could place an ellipsoidal particle, perform a minimization, change the position, re-minimize the system, and record the different energy minima attained. We include an option to automate this type of operation (which can be used to build up the potential of mean force from the liquid crystal and colloid interactions) for convenience. A near-term addition will be allowing objects to move according to the integrated stresses at their surface (or according to the energetic results of various trial moves); the user will then be able to separately “Minimize” just the liquid crystal sites or “Evolve [the] system” by allowing both liquid crystalline and colloidal degrees of freedom to change simultaneously.
Finally, to facilitate moving from GUI prototyping to larger-scale MPI studies, we have included the ability to record system initialization and sequences of commands entered in the graphical user interface, and then save this sequence of commands as a new file that can be separately compiled and executed in non-GUI operation. This file has its own set of command line options, primarily so that it can be made to work as an MPI executable and so that the system size of the simulation it represents can be rescaled to a larger value. We highlight this GUI-prototyping approach as a visual alternative to the scripting-language approaches of molecular-dynamics packages like LAMMPS [91] or HOOMD-blue [92] for specifying complex sequences of system initialization, energy minimizations, and the introduction of objects, fields, and boundary conditions. We believe that this seamless visual-prototyping-to-MPI-scalable pipeline will be beneficial to researchers interested in accessing experimental-scale simulations.
6. Discussion
As demonstrated in our sample study, openQmin utilizes MPI to enable LdG modeling at typical size scales of experimental relevance, at the ~ 10 μm range, with fast convergence enabled by the FIRE algorithm. Besides the colloidal defect configurations and patterned boundaries discussed here, another immediate use is for the study of cholesterics, where typically fewer than ten pitches can fit inside a simulation box using a single processor, but using openQmin tens of pitches can be modeled. While it may not be realistic at present to frequently conduct simulations with 103 processors, using openQmin on computer clusters will facilitate demonstration of how numerical results scale with system size, allowing reasonable extrapolations to experimental scales.
For modeling at the ~ 1 μm range or smaller, openQmin's combination of FIRE with GPU computing offers a substantial speedup, enabling users to manipulate the simulated conditions in a GUI environment and observe the change in energy-minimized configurations. The GUI is useful for running “real-time” tests of proposed configurations which can then be modeled at larger scales with MPI.
Likewise, the GUI will also be useful to experimentalists in quickly identifying more optimal properties of nematics, colloidal particles, boundaries, etc. in order to achieve targeted topological or self-assembled configurations. In general, numerical modeling can aid experimental studies not only in developing theoretical understanding of nematic structures and energy landscapes, but also in performing high-throughput searches through these design spaces. For example, geometric compatibility conditions favoring lock-and-key assembly of particles and patterned walls [21, 37], or particle design promoting assembly into photonic crystals, can be optimized more efficiently in numerics, to help guide the increasingly sophisticated uses of fabrication techniques such as photolithography and two-photon polymerization [93]. An ambitious but important direction for future development is therefore to efficiently explore design parameter spaces in numerical modeling, possibly employing genetic algorithms and techniques from machine learning.
There are some near-term directions for future development of openQmin that we anticipate will increase the usefulness of this open-source software to the liquid crystals research community. An expanded library of Q-initialization options will facilitate investigations of chiral liquid crystals, topologically entangled or knotted defect configurations [11–14], and periodic defect arrays [26, 94], for example. A major advance would be adding a flow field coupled to Q by Beris-Edwards nematodynamics, for investigations of microfluidic geometries and active nematics.
Incorporating motion of colloidal particles into the modeling is another area for useful developments. In the experimental system, energy is minimized not only over Q but also over the positions and (if applicable) orientations of colloidal particles. At present, openQmin takes these latter degrees of freedom as input parameters, and a free energy landscape can be mapped either informally using the GUI or more systematically on a computer cluster. Thus one desired future improvement is to allow overdamped translation and rotation of colloidal particles within the program, downhill in the energy landscape, based on trial moves or on estimated nematic elastic stresses felt by the particle [46]. The trial move approach, requiring several re-minimizations of Q at each time step, is made less cumbersome by improved convergence speed of the FIRE algorithm.
Finally, we hope that openQmin's GUI interface will be useful in physics education. Interacting with a fast and “hands-on” version of the numerical modeling, students at the undergraduate or beginning graduate level can quickly gain experience and intuition for liquid crystals. This will help to capitalize on the position of liquid crystals as one of the most accessible, and visualizable, physical realizations of abstract topological ideas relevant to many areas of physics.
Data Availability Statement
The open source code described in this work can be found at Reference [67] and used to reproduce all data in the manuscript. Documentation for the software is maintained at Reference [95], and can also be generated with doxygen from the source code.
Author Contributions
DS and DB designed the study, developed the open-source software, and wrote the manuscript.
Funding
DS was supported by NSF-POLS-1607416 as well as Simons Foundation Grant Number 454947. The Tesla K40s used for this research were donated by the NVIDIA Corporation. We acknowledge computing support via an XSEDE allocation on Comet through Grant No. NSF-TG-PHY190027, and from the Multi-Environment Computer for Exploration and Discovery (MERCED) cluster at UC Merced, which was funded by National Science Foundation Grant No. ACI-1429783.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
DB thanks Gareth Alexander for introducing him to LdG numerical modeling, and gratefully acknowledges illuminating discussions with Simon Čopar, Miha Ravnik, and Slobodan Žumer.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphy.2019.00204/full#supplementary-material
Footnote
1. ^As of July, 2019.
References
1. Friedel G. Les états mésomorphes de la matière. Ann Phys. (1922) 18:273–474. doi: 10.1051/anphys/192209180273
2. Poulin P, Stark H, Lubensky TC, Weitz DA. Novel colloidal interactions in anisotropic fluids. Science. (1997) 275:1770–3. doi: 10.1126/science.275.5307.1770
3. Dickson W, Wurtz GA, Evans PR, Pollard RJ, Zayats AV. Electronically controlled surface plasmon dispersion and optical transmission through metallic hole arrays using liquid crystal. Nano Lett. (2008) 8:281–6. doi: 10.1021/nl072613g
4. Liu Q, Cui Y, Gardner D, Li X, He S, Smalyukh II. Self-alignment of plasmonic gold nanorods in reconfigurable anisotropic fluids for tunable bulk metamaterial applications. Nano Lett. (2010) 10:1347–53. doi: 10.1021/nl9042104
5. Wang X, Miller DS, Bukusoglu E, De Pablo JJ, Abbott NL. Topological defects in liquid crystals as templates for molecular self-assembly. Nat Mater. (2016) 15:106–12. doi: 10.1038/nmat4421
6. Rodarte AL, Pandolfi RJ, Ghosh S, Hirst LS. Quantum dot/liquid crystal composite materials: self-assembly driven by liquid crystal phase transition templating. J Mater Chem C. (2013) 1:5527–32. doi: 10.1039/c3tc31043d
7. Rodarte A, Cisneros F, Hein J, Ghosh S, Hirst L. Quantum dot/liquid crystal nanocomposites in photonic devices. Photonics. (2015) 2:855–64. doi: 10.3390/photonics2030855
8. Muševič I, Škarabot M, Tkalec U, Ravnik M, Žumer S. Two-dimensional nematic colloidal crystals self-assembled by topological defects. Science. (2006) 313:954–8. doi: 10.1126/science.1129660
9. Mundoor H, Senyuk B, Smalyukh II. Triclinic nematic colloidal crystals from competing elastic and electrostatic interactions. Science. (2016) 352:69–73. doi: 10.1126/science.aaf0801
10. Nych A, Ognysta U, Škarabot M, Ravnik M, Žumer S, Muševič I. Assembly and control of 3D nematic dipolar colloidal crystals. Nat Commun. (2013) 4:1489. doi: 10.1038/ncomms2486
11. Ravnik M, Škarabot M, Žumer S, Tkalec U, Poberaj I, Babič D, et al. Entangled nematic colloidal dimers and wires. Phys Rev Lett. (2007) 99:247801. doi: 10.1103/PhysRevLett.99.247801
12. Tkalec U, Ravnik M, Čopar S, Žumer S, Muševič I. Reconfigurable knots and links in chiral nematic colloids. Science. (2011) 333:62–5. doi: 10.1126/science.1205705
13. Tasinkevych M, Campbell MG, Smalyukh II. Splitting, linking, knotting, and solitonic escape of topological defects in nematic drops with handles. Proc Natl Acad Sci USA. (2014) 111:16268–73. doi: 10.1073/pnas.1405928111
14. Machon T, Alexander GP. Knotted defects in nematic liquid crystals. Phys Rev Lett. (2014) 113:027801. doi: 10.1103/PhysRevLett.113.027801
15. Ravnik M, Alexander GP, Yeomans JM, Žumer S. Three-dimensional colloidal crystals in liquid crystalline blue phases. Proc Natl Acad Sci USA. (2011) 108:5188–92. doi: 10.1073/pnas.1015831108
16. Lavrentovich OD. Liquid crystals, photonic crystals, metamaterials, and transformation optics. Proc Natl Acad Sci USA. (2011) 108:5143–4. doi: 10.1073/pnas.1102130108
17. Humar M, Muševič I. 3D microlasers from self-assembled cholesteric liquid-crystal microdroplets. Opt Express. (2010) 18:26995–7003. doi: 10.1364/OE.18.026995
18. Muševič I. Interactions, topology and photonic properties of liquid crystal colloids and dispersions. Eur Phys J Spec Top. (2019) 227:2455–85. doi: 10.1140/epjst/e2019-800107-y
19. Serra F. Curvature and defects in nematic liquid crystals. Liq Cryst. (2016) 43:1920–36. doi: 10.1080/02678292.2016.1209698
20. Hung FR, Gettelfinger BT, Koenig GM Jr, Abbott NL, de Pablo JJ. Nanoparticles in nematic liquid crystals: interactions with nanochannels. J Chem Phys. (2007) 127:124702. doi: 10.1063/1.2770724
21. Eskandari Z, Silvestre N, da Gama MT, Ejtehadi M. Particle selection through topographic templates in nematic colloids. Soft Matter. (2014) 10:9681–7. doi: 10.1039/C4SM02231A
22. Luo Y, Serra F, Stebe KJ. Experimental realization of the “lock-and-key” mechanism in liquid crystals. Soft Matter. (2016) 12:6027–32. doi: 10.1039/C6SM00401F
23. Guillamat P, Sagués F, Ignés-Mullol J. Electric-field modulation of liquid crystal structures in contact with structured surfactant monolayers. Phys Rev E. (2014) 89:052510. doi: 10.1103/PhysRevE.89.052510
24. Kos Ž, Ravnik M. Relevance of saddle-splay elasticity in complex nematic geometries. Soft Matter. (2016) 12:1313–23. doi: 10.1039/C5SM02417J
25. Peng C, Guo Y, Conklin C, Viñals J, Shiyanovskii SV, Wei QH, et al. Liquid crystals with patterned molecular orientation as an electrolytic active medium. Phys Rev E. (2015) 92:052502. doi: 10.1103/PhysRevE.92.052502
26. Murray BS, Pelcovits RA, Rosenblatt C. Creating arbitrary arrays of two-dimensional topological defects. Phys Rev E. (2014) 90:052501. doi: 10.1103/PhysRevE.90.052501
27. Williams C, Cladis P, Kleman M. Screw disclinations in nematic samples with cylindrical symmetry. Mol Cryst Liquid Cryst. (1973) 21:355–73. doi: 10.1080/15421407308083329
28. Volovik G, Lavrentovich O. Topological dynamics of defects: boojums in nematic drops. Zh Eksp Teor Fiz. (1983) 85:1997–2010.
29. Nelson DR. Toward a tetravalent chemistry of colloids. Nano Lett. (2002) 2:1125–9. doi: 10.1021/nl0202096
30. Lavrentovich O, Nastishin YA. Defects in degenerate hybrid aligned nematic liquid crystals. Europhys Lett. (1990) 12:135. doi: 10.1209/0295-5075/12/2/008
31. Wright DC, Mermin ND. Crystalline liquids: the blue phases. Rev Modern Phys. (1989) 61:385. doi: 10.1103/RevModPhys.61.385
32. Alama S, Bronsard L, Lamy X. Minimizers of the Landau–de Gennes energy around a spherical colloid particle. Arch Ration Mech Anal. (2016) 222:427–50. doi: 10.1007/s00205-016-1005-z
33. Alama S, Bronsard L, Lamy X. Analytical description of the Saturn-ring defect in nematic colloids. Phys Rev E. (2016) 93:012705. doi: 10.1103/PhysRevE.93.012705
34. Kralj S, Žumer S. Fréedericksz transitions in supra-um nematic droplets. Phys Rev A. (1992) 45:2461–70. doi: 10.1103/PhysRevA.45.2461
35. Tkalec U, Škarabot M, Muševič I. Interactions of micro-rods in a thin layer of a nematic liquid crystal. Soft Matter. (2008) 4:2402–9. doi: 10.1039/b807979j
36. Emeršič T, Zhang R, Kos Ž, Čopar S, Osterman N, de Pablo JJ, et al. Sculpting stable structures in pure liquids. Sci Adv. (2019) 5:eaav4283. doi: 10.1126/sciadv.aav4283
37. Luo Y, Beller DA, Boniello G, Serra F, Stebe KJ. Tunable colloid trajectories in nematic liquid crystals near wavy walls. Nat Commun. (2018) 9:3841. doi: 10.1038/s41467-018-06054-y
38. Tasinkevych M, Mondiot F, Mondain-Monval O, Loudet JC. Dispersions of ellipsoidal particles in a nematic liquid crystal. Soft Matter. (2014) 10:2047–58. doi: 10.1039/c3sm52708e
39. Hashemi SM, Ejtehadi MR. Equilibrium state of a cylindrical particle with flat ends in nematic liquid crystals. Phys Rev E. (2015) 91:012503. doi: 10.1103/PhysRevE.91.012503
40. Hung FR, Bale S. Faceted nanoparticles in a nematic liquid crystal: defect structures and potentials of mean force. Mol Simul. (2009) 35:822–34. doi: 10.1080/08927020902801563
41. Hung FR. Quadrupolar particles in a nematic liquid crystal: effects of particle size and shape. Phys Rev E. (2009) 79:021705. doi: 10.1103/PhysRevE.79.021705
42. Hung FR, Guzmán O, Gettelfinger BT, Abbott NL, de Pablo JJ. Anisotropic nanoparticles immersed in a nematic liquid crystal: defect structures and potentials of mean force. Phys Rev E. (2006) 74:011711. doi: 10.1103/PhysRevE.74.011711
43. Beller DA, Gharbi MA, Liu IB. Shape-controlled orientation and assembly of colloids with sharp edges in nematic liquid crystals. Soft Matter. (2015) 11:1078–86. doi: 10.1039/C4SM01910E
44. Araki T, Tanaka H. Colloidal aggregation in a nematic liquid crystal: topological arrest of particles by a single-stroke disclination line. Phys Rev Lett. (2006) 97:127801. doi: 10.1103/PhysRevLett.97.127801
45. Škarabot M, Ravnik M, Žumer S, Tkalec U, Poberaj I, Babič D, et al. Interactions of quadrupolar nematic colloids. Phys Rev E. (2008) 77:031705. doi: 10.1103/PhysRevE.77.031705
46. Škarabot M, Ravnik M, Žumer S, Tkalec U, Poberaj I, Babič D, et al. Hierarchical self-assembly of nematic colloidal superstructures. Phys Rev E. (2008) 77:061706. doi: 10.1103/PhysRevE.77.061706
47. Ravnik M, Žumer S. Landau–de Gennes modelling of nematic liquid crystal colloids. Liq Cryst. (2009) 36:1201–14. doi: 10.1080/02678290903056095
48. Mori H, Gartland EC, Kelly JR, Box PJ. Multidimensional director modeling using the Q tensor repre- sentation in a liquid crystal cell and its application to the π cell with patterned electrodes. Jpn J App Phys. (1999) 38:135. doi: 10.1143/JJAP.38.135
49. Beris AN, Edwards BJ. Thermodynamics of Flowing Systems: With Internal Microstructure. New York, NY; Oxford: Oxford University Press (1994).
50. Qian T, Sheng P. Generalized hydrodynamic equations for nematic liquid crystals. Phys Rev E. (1998) 58:7475. doi: 10.1103/PhysRevE.58.7475
51. Denniston C, Orlandini E, Yeomans J. Lattice Boltzmann simulations of liquid crystal hydrodynamics. Phys Rev E. (2001) 63:056702. doi: 10.1103/PhysRevE.63.056702
52. Spencer T, Care C. Lattice Boltzmann scheme for modeling liquid-crystal dynamics: zenithal bistable device in the presence of defect motion. Phys Rev E. (2006) 74:061708. doi: 10.1103/PhysRevE.74.061708
53. Carenza LN, Gonnella G, Lamura A, Negro G, Tiribocchi A. Lattice Boltzmann methods and active fluids. Eur Phys J E. (2019) 42:81. doi: 10.1140/epje/i2019-11843-6
54. Marenduzzo D, Orlandini E, Cates M, Yeomans J. Steady-state hydrodynamic instabilities of active liquid crystals: hybrid lattice Boltzmann simulations. Phys Rev E. (2007) 76:031921. doi: 10.1103/PhysRevE.76.031921
55. Cates M, Henrich O, Marenduzzo D, Stratford K. Lattice Boltzmann simulations of liquid crystalline fluids: active gels and blue phases. Soft Matter. (2009) 5:3791–800. doi: 10.1039/b908659p
56. Lee KW, Mazza MG. Stochastic rotation dynamics for nematic liquid crystals. J Chem Phys. (2015) 142:164110. doi: 10.1063/1.4919310
57. Shendruk TN, Yeomans JM. Multi-particle collision dynamics algorithm for nematic fluids. Soft Matter. (2015) 11:5101–10. doi: 10.1039/C5SM00839E
58. Mandal S, Mazza MG. Multiparticle collision dynamics for tensorial nematodynamics. Phys Rev E. (2019) 99:063319. doi: 10.1103/PhysRevE.99.063319
59. James R, Willman E, Fernandez FA, Day SE. Computer modeling of liquid crystal hydrodynamics. IEEE Trans Magn. (2008) 44:814–7. doi: 10.1109/TMAG.2007.916029
60. Giomi L, Mahadevan L, Chakraborty B, Hagan M. Excitable patterns in active nematics. Phys Rev Lett. (2011) 106:218101. doi: 10.1103/PhysRevLett.106.218101
61. Svenšek D, Žumer S. Hydrodynamics of pair-annihilating disclination lines in nematic liquid crystals. Phys Rev E. (2002) 66:021712. doi: 10.1103/PhysRevE.66.021712
62. Terentjev EM. Disclination loops, standing alone and around solid particles, in nematic liquid crystals. Phys Rev E. (1995) 51:1330. doi: 10.1103/PhysRevE.51.1330
63. Stark H. Physics of colloidal dispersions in nematic liquid crystals. Phys Rep. (2001) 351:387–474. doi: 10.1016/S0370-1573(00)00144-7
64. Gharbi MA, Cavallaro M Jr, Wu G, Beller DA, Kamien RD, Yang S, et al. Microbullet assembly: interactions of oriented dipoles in confined nematic liquid crystal. Liq Cryst. (2013) 40:1619–27. doi: 10.1080/02678292.2012.755223
65. Luo Y, Yao T, Beller DA, Serra F, Stebe KJ. Deck the walls with anisotropic colloids in nematic liquid crystals. Langmuir. (2019) 35:9274–85. doi: 10.1021/acs.langmuir.9b01811
66. Bajc I, Hecht F, Žumer S. A mesh adaptivity scheme on the Landau–de Gennes functional minimization case in 3D, and its driving efficiency. J Comput Phys. (2016) 321:981–96. doi: 10.1016/j.jcp.2016.02.072
67. Sussman DM, Beller DA. Available online at: https://github.com/sussmanLab/open-Qmin (2019).
68. Gabriel E, Fagg GE, Bosilca G, Angskun T, Dongarra JJ, Squyres JM, et al. Open MPI: goals, concept, and design of a next generation MPI implementation. In: European Parallel Virtual Machine/Message Passing Interface Users' Group Meeting. Berlin: Springer (2004). p. 97–104. doi: 10.1007/978-3-540-30218-6_19
70. Mottram NJ, Newton CJ. Introduction to Q-tensor theory. arXiv [Preprint]. arXiv:14093542 (2014).
71. Schophol N, Sluckin TJ. Defect core structure in nematic liquid crystals. Phys Rev Lett. (1987) 59:2582. doi: 10.1103/PhysRevLett.59.2582
72. Jeu WHD, Claassen WAP, Spruijt AMJ. The determination of the elastic constants of nematic liquid crystals. Mol Cryst Liq Cryst. (1976) 37:269–80. doi: 10.1080/15421407608084362
73. Madhusudana NV, Pratibha R. Elasticity and orientational order in some cyanobiphenyls: part IV. Reanalysis of the data. Mol Cryst Liq Cryst. (1982) 89:249–57. doi: 10.1080/00268948208074481
74. Poniewierski A, Sluckin T. On the free energy density of non-uniform nematics. Mol Phys. (1985) 55:1113–27. doi: 10.1080/00268978500101931
75. Rapini A, Papoular M. Distorsion d'une lamelle nématique sous champ magnétique conditions d'ancrage aux parois. Le J Phys Coll. (1969) 30:C4–54. doi: 10.1051/jphyscol:1969413
76. Nobili M, Durand G. Disorientation-induced disordering at a nematic-liquid-crystal–solid interface. Phys Rev A. (1992) 46:R6174. doi: 10.1103/PhysRevA.46.R6174
77. Fournier JB, Galatola P. Modeling planar degenerate wetting and anchoring in nematic liquid crystals. Europhys Lett. (2005) 72:403. doi: 10.1209/epl/i2005-10253-5
79. Noye B, Arnold R. Accurate finite difference approximations for the Neumann condition on a curved boundary. Appl Math Modell. (1990) 14:2–13. doi: 10.1016/0307-904X(90)90157-Z
80. Nocedal J, Wright S. Numerical Optimization. New York, NY: Springer Science & Business Media (2006).
81. Bitzek E, Koskinen P, Gähler F, Moseler M, Gumbsch P. Structural relaxation made simple. Phys Rev Lett. (2006) 97:170201. doi: 10.1103/PhysRevLett.97.170201
82. Polyak BT. Some methods of speeding up the convergence of iteration methods. USSR Comput Math Math Phys. (1964) 4:1–17. doi: 10.1016/0041-5553(64)90137-5
83. Sheppard D, Terrell R, Henkelman G. Optimization methods for finding minimum energy paths. J Chem Phys. (2008) 128:134106. doi: 10.1063/1.2841941
84. Herbol HC, Stevenson J, Clancy P. Computational implementation of nudged elastic band, rigid rotation, and corresponding force optimization. J Chem Theor Comput. (2017) 13:3250–9. doi: 10.1021/acs.jctc.7b00360
85. Zhou Y, Moseler M, Müser MH. Solution of boundary-element problems using the fast-inertial-relaxation-engine method. Phys Rev B. (2019) 99:144103. doi: 10.1103/PhysRevB.99.144103
86. Wang Y, Jia Z, Wen Z. The Search direction Correction makes first-order methods faster. arXiv [Preprint]. arXiv:190506507 (2019).
87. Lulli M, Bernaschi M, Parisi G. Highly optimized simulations on single-and multi-GPU systems of the 3D Ising spin glass model. Comput Phys Commun. (2015) 196:290–303. doi: 10.1016/j.cpc.2015.06.019
88. Gu Y, Abbott NL. Observation of saturn-ring defects around solid microspheres in nematic liquid crystals. Phys Rev Lett. (2000) 85:4719. doi: 10.1103/PhysRevLett.85.4719
89. Loudet J, Poulin P. Application of an electric field to colloidal particles suspended in a liquid-crystal solvent. Phys Rev Lett. (2001) 87:165503. doi: 10.1103/PhysRevLett.87.165503
90. Lubensky TC, Pettey D, Currier N, Stark H. Topological defects and interactions in nematic emulsions. Phys Rev E. (1998) 57:610. doi: 10.1103/PhysRevE.57.610
91. Plimpton S. Fast parallel algorithms for short-range molecular dynamics. J Comput Phys. (1995) 117:1–19. doi: 10.1006/jcph.1995.1039
92. Anderson JA, Lorenz CD, Travesset A. General purpose molecular dynamics simulations fully implemented on graphics processing units. J Comput Phys. (2008) 227:5342–59. doi: 10.1016/j.jcp.2008.01.047
93. Martinez A, Ravnik M, Lucero B, Visvanathan R, Žumer S, Smalyukh II. Mutually tangled colloidal knots and induced defect loops in nematic fields. Nat Mater. (2014) 13:258–63. doi: 10.1038/nmat3840
94. Suh A, Gim MJ, Beller D, Yoon DK. Topological defects and geometric memory across the nematic-smectic A liquid crystal phase transition. Soft Matter. (2019) 15:5835–41. doi: 10.1039/C9SM00781D
95. Sussman DM, Beller DA. Available online at: https://dmsussman.gitlab.io/landaudegui/ (2019).
Keywords: Landau-de Gennes modeling, nematic, topological defect, numerical modeling, GPGPU
Citation: Sussman DM and Beller DA (2019) Fast, Scalable, and Interactive Software for Landau-de Gennes Numerical Modeling of Nematic Topological Defects. Front. Phys. 7:204. doi: 10.3389/fphy.2019.00204
Received: 15 July 2019; Accepted: 14 November 2019;
Published: 03 December 2019.
Edited by:
Uroš Tkalec, University of Ljubljana, SloveniaReviewed by:
Douglas John Cleaver, Sheffield Hallam University, United KingdomSimon Čopar, University of Ljubljana, Slovenia
Marco G. Mazza, Max-Planck-Institute for Dynamics and Self-Organization, Max Planck Society (MPG), Germany
Copyright © 2019 Sussman and Beller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel M. Sussman, ZGFuaWVsLm0uc3Vzc21hbkBlbW9yeS5lZHU=; Daniel A. Beller, ZGJlbGxlckB1Y21lcmNlZC5lZHU=