 Theodora Markati
Theodora Markati Liesbeth De Waele
Liesbeth De Waele Urlike Schara-Schmidt4
Urlike Schara-Schmidt4 Laurent Servais
Laurent Servais- 1MDUK Oxford Neuromuscular Center, Department of Paediatrics, University of Oxford, Oxford, United Kingdom
- 2KU Leuven Department of Development and Regeneration, Leuven, Belgium
- 3Department of Paediatric Neurology, University Hospitals Leuven, Leuven, Belgium
- 4Department of Pediatric Neurology, Center for Neuromuscular Diseases, Center for Translational Neuro- and Behavioral Sciences, University Duisburg-Essen, Essen, Germany
- 5Division of Child Neurology, Reference Center for Neuromuscular Disease, Centre Hospitalier Régional de Références des Maladies Neuromusculaires, Department of Pediatrics, University Hospital Liège, Liège, Belgium
Duchenne muscular dystrophy (DMD) is an X-linked condition caused by a deficiency of functional dystrophin protein. Patients experience progressive muscle weakness, cardiomyopathy and have a decreased life expectancy. Standards of care, including treatment with steroids, and multidisciplinary approaches have extended the life expectancy and improved the quality of life of patients. In the last 30 years, several compounds have been assessed in preclinical and clinical studies for their ability to restore functional dystrophin levels or to modify pathways involved in DMD pathophysiology. However, there is still an unmet need with regards to a disease-modifying treatment for DMD and the attrition rate between early-phase and late-phase clinical development remains high. Currently, there are 40 compounds in clinical development for DMD, including gene therapy and antisense oligonucleotides for exon skipping. Only five of them have received conditional approval in one jurisdiction subject to further proof of efficacy. In this review, we present data of another 16 compounds that failed to complete clinical development, despite positive results in early phases of development in some cases. We examine the reasons for the high attrition rate and we suggest solutions to avoid similar mistakes in the future.
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked condition caused by deficiency of functional dystrophin protein. Dystrophin deficiency causes increased susceptibility of the sarcolemma to mechanical stress and results in high intracellular calcium levels, mitochondrial dysfunction, and increased production of reactive oxygen species that lead to a cycle of loss of muscle fibres, chronic inflammation, and decreased muscle regenerative capacity. Ultimately, muscle fibres are replaced by connective and adipose tissue resulting in muscle dysfunction. Patients experience progressive weakness in skeletal muscles and cardiomyopathy. Several isoforms of dystrophins are expressed in the central nervous system, and a large proportion of patients have cognitive involvement ranging from mild attention deficit to severe autism (Doorenweerd, 2020). DMD patients lose ambulation and have a shortened life expectancy. For a detailed review of DMD pathophysiology, we refer readers to the following reviews (Duan et al., 2021; Starosta and Konieczny, 2021). Standards of care includes steroids, multidisciplinary approaches and depending on the country, ataluren for nonsense mutation or exon skipping therapy for patients with eligible mutations. Standards of care and steroids extend life expectancy and the quality of life for patients with DMD (Birnkrant et al., 2018a; Birnkrant et al., 2018b; Birnkrant et al., 2018c); however, there is an unmet need with regards to treatment.
In the past decades, several compounds have been tested in preclinical and clinical studies for their abilities to restore functional dystrophin levels in the muscle cells or to alter the fate of downstream molecular pathways involved in DMD pathophysiology. Breakthroughs in biomedical research have led to the development of ASOs and gene therapy treatments that increase the levels of dystrophin. Except for the US Food and Drug Administration (FDA)-approved steroid for use in DMD (deflazacort), four other antisense oligonucleotides (ASOs) for exon skipping (eteplirsen, golodirsen, casimersen, viltolarsen) have received approval by the FDA through the accelerated approval pathway; viltolarsen has additionally received approval in Japan [1 (Aartsma-Rus and Krieg, 2017; Aartsma-Rus and Corey, 2020; Dhillon, 2020; Shirley, 2021)]. One more compound, the small molecule for stop codon readthrough namely ataluren, has received conditional approval by the European Medicines Association (EMA), but not the FDA (Ryan, 2014). However, the clinical development of several compounds with promising early-phase results has been halted due to disappointing late-phase results, tempering expectations in the DMD community.
The difficulty in translating results from animal models to humans, the heterogeneity of the DMD genotypes and symptoms such as cognitive involvement, the rarity of the disease, the lack of efficacious outcome measures or fully validated biomarkers are some of the most important reasons for the high attrition rate between early- and late-phase clinical development. In this article, we review data on the compounds that have failed in clinical development with published results within the last 5 years. Additionally, we present our perspective on the reasons for the high attrition rate in DMD clinical development and propose solutions.
Methods
To identify compounds for which clinical development in the DMD population has been terminated, we performed a comprehensive review of four databases (Ovid MEDLINE, Embase, Scopus, and Cochrane) in English and French, restricted for the last 5 years (2016–2021). For a full list of the keywords used for the different databases, please refer to Supplementary Material. We selected full-text peer-reviewed publications of clinical studies of DMD (+/− Becker muscular dystrophy) patients of pharmacological interventions only. We made a list of the different compounds identified in the selected papers of our research strategy and we checked the status of their development online and on ClinicalTrials.gov.
Additionally, we searched ClinicalTrials.gov using the term “Duchenne muscular dystrophy” and filters “terminated” OR “suspended” OR “withdrawn” with no time limitation. Studies with unknown status were not included and we selected only studies of a pharmacological intervention intended to treat DMD (+/− Becker muscular dystrophy). In total, we identified 10 compounds from the literature review and another six from the ClinicalTrials.gov search whose development has been discontinued.
Results
Identification of Compounds with Terminated Clinical Development
By searches of relevant databases, we identified 16 compounds that had been in clinical development in the DMD population (Table 1). Overall, compounds aiming to treat DMD can be classified into two broad categories: upstream treatments and downstream treatments. Upstream treatments aim to treat the cause of the disease, namely the deficiency of functional dystrophin. This category includes the therapeutic approaches of gene transfer therapy as well as exon skipping and stop codon readthrough. The two latter are highly mutation-specific. Downstream treatments target different levels of DMD pathophysiology. This therapeutic category applies to all DMD mutations and potentially to other conditions with shared pathophysiology such as Becker muscular dystrophy or limb-girdle muscular dystrophies. The Figure 1 is a schematic presentation of DMD pathophysiology on which the different levels of action of the discontinued clinical developments are demonstrated.
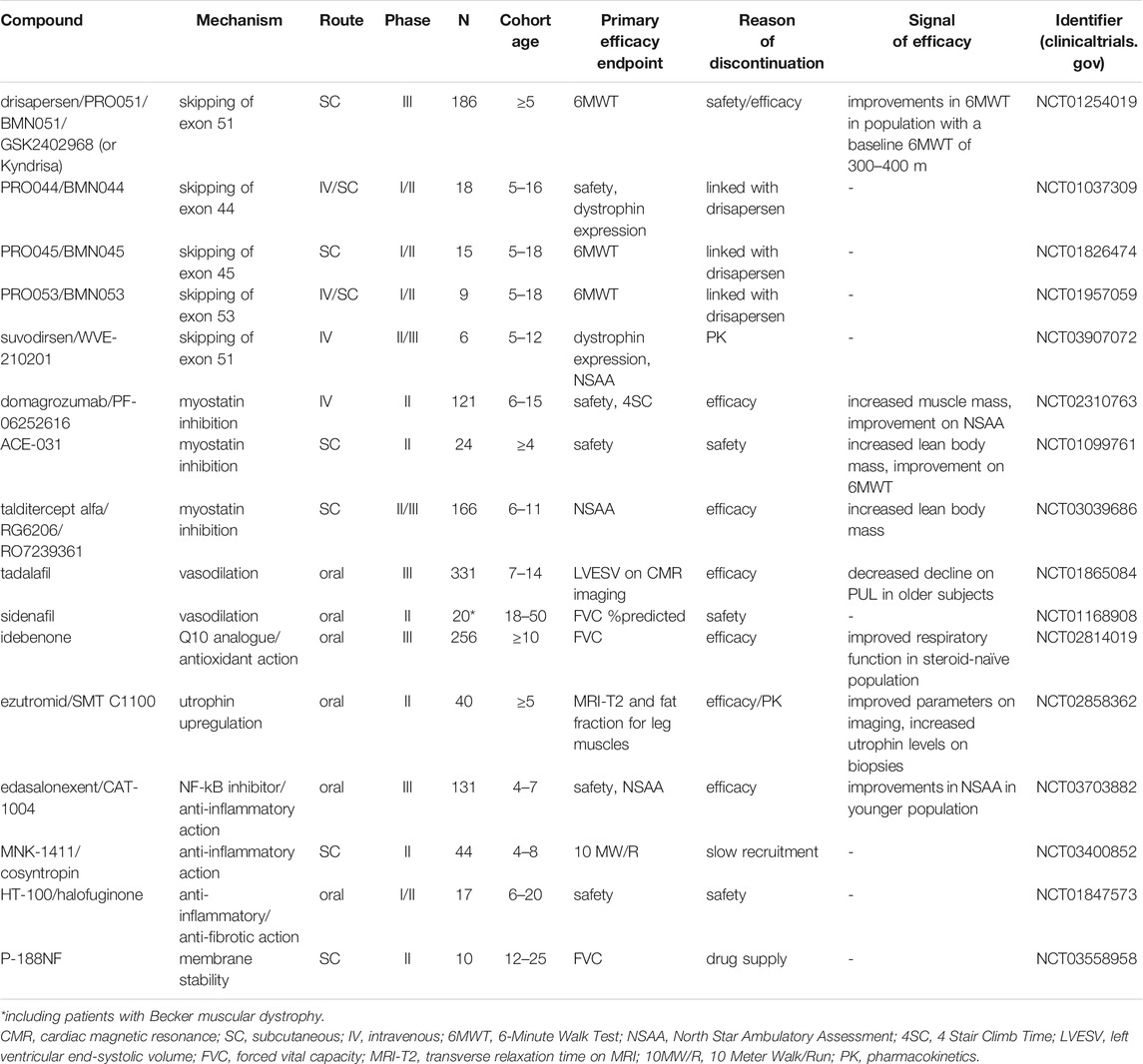
TABLE 1. Compounds discontinued during clinical development for DMD.
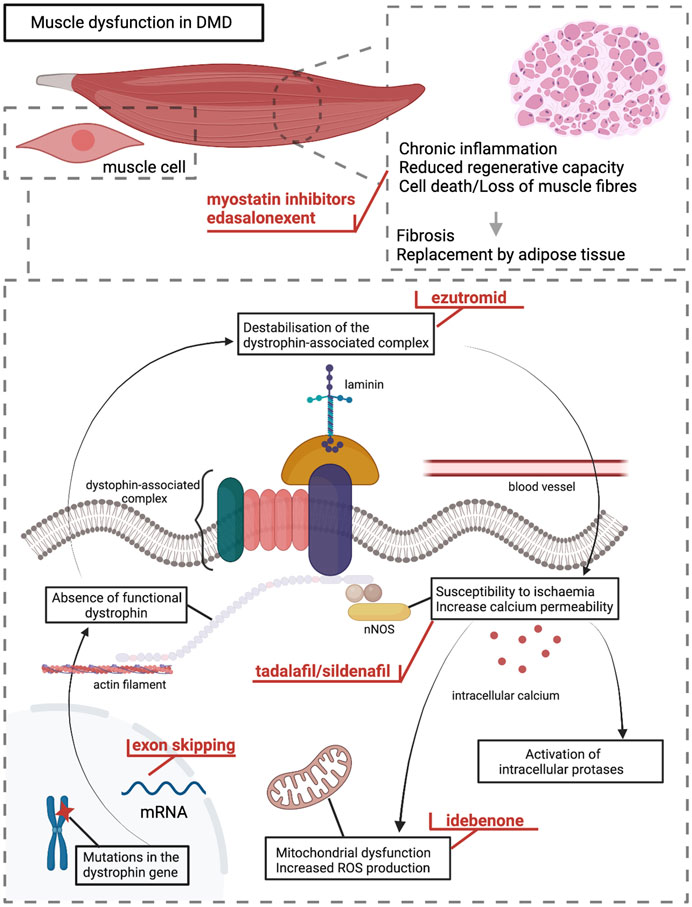
FIGURE 1. DMD pathophysiology and levels of action for the different compounds discussed in the paper. Mutations in the dystrophin gene lead to absence of functional dystrophin protein in muscle cells and destabilisation of the dystrophin-associated complex at the sarcolemma. This leads to increased susceptibility of the membrane to mechanical stress and ischaemia leading to increased membrane permeability, disturbed cellular signalling, and higher intracellular calcium levels causing activation of intracellular proteases. Mitochondrial dysfunction and increased production of reactive oxygen species contribute to DMD pathophysiology. These trigger a cycle of cell necrosis, chronic inflammation, and decreased muscle regenerative capacity. Ultimately, muscle fibres are lost and replaced by connective and adipose tissue resulting in muscle dysfunction. The different levels of action for the compounds discussed in the paper are indicated in red colour.
Compounds Targeting Specific DMD Mutations
The goal of exon-skipping approaches, which are mutation-specific, is modulation of splicing of the dystrophin pre-mRNA to restore the reading frame in cases of frameshift mutations (deletions or duplications) which result in truncated dystrophin protein. Two ASOs that induce exon 51 skipping (applicable to approximately 14% of patients) have failed in late-phase trials. One of these is drisapersen, an ASO modified with 2′-O-methyl and phosphorothioate that showed promising preclinical results (van Deutekom et al., 2001; Lu et al., 2005; Alter et al., 2006; McClorey et al., 2006). Drisapersen was assessed in an open-label extension of a dose-finding phase I trial that included 12 DMD patients. Participants who were able to complete 330 m or more in the 6-Minute Walking Test (6MWT), a functional assessment of mobility, at baseline showed remarkable improvement after 177 weeks. However, two patients who had a baseline 6MWT of less than 330 m lost ambulation (Goemans et al., 2016). Drisapersen was tested in two phase II and a phase III, placebo-controlled trials of approximately 300 DMD patients. In the first phase II (DMD114117; NCT01153932) trial 53 participants (≥5 years) received continuously (weekly) or intermittently (combination of twice weekly and weekly administration, followed by no administration periods) placebo or drisapersen at 6 mg/kg for 48 weeks. A key inclusion criterion was the ability to raise from the floor in less than 7 s, which is a strong positive prognostic factor for maintaining ambulation over the next 2 years (Mazzone et al., 2016). There was a statistically significant improvement on the 6MWT (primary outcome) for continuously treated patients when compared with the placebo group at week 25 (study midpoint) but not for the intermittently treated group. Analysis of the population of the two subgroups suggested that drisapersen might benefit younger patients at an earlier stage of the disease more than older subjects. In the second phase II (DMD114876; NCT01462292) trial, the 51 participants (≥5 years) were randomised to placebo, 3 mg/kg/week drisapersen or 6 mg/kg/week drisapersen. The placebo and the low-dose group showed a decline in the 6MWT at 24 weeks (primary endpoint), whereas the high-dose group improved; however, this difference was not statistically significant (Voit et al., 2014; McDonald et al., 2018a). The randomised, placebo-controlled phase III (DMD114044; NCT01254019) trial involved 186 participants (≥5 years) with a later stage disease and who were on background treatment with steroids. Lack of inclusion criteria on the time-to-rise from floor test translated into the inclusion of patients who lost ambulation during the trial period. As the number of patients was much larger than in the phase II studies, broader inclusion criteria were needed as were a larger number of investigation sites. Ultimately, the difference between drisapersen-treated (with 6 mg/kg/week) and placebo-treated patients did not reach statistical significance on the 6MWT at 48 weeks or on key secondary endpoints. Only the post-hoc-defined subgroup of patients scoring between 300 and 400 m on the 6MWT at baseline significantly benefited from therapy. The occurrence of adverse events including injection site reactions, thrombocytopenia, and proteinuria in combination with the lack of efficacy led to the termination of the clinical development of this compound (Goemans et al., 2018).
The company developing drisapersen was also developing ASOs PRO044, PRO045, and PRO053 designed to induce exon 44, exon 45 and exon 53 skipping, respectively. Development of these compounds was discontinued at the same time drisapersen development was halted. The development of drisapersen demonstrated that it is crucial to recruit a large, representative sample of patients because of the heterogeneity in the disease trajectory. Because of the rarity of the disease, this is very challenging, leading to broad inclusion criteria that further increases heterogeneity. Moreover, the need to include a big number of sites considerably increases the variability in standards of care and data collection.
Suvodirsen was another ASO designed to induce exon 51 skipping. Suvodirsen is a stereopure ASO, which was expected to result in a better pharmacodynamic and pharmacokinetic profile, that would translate into improved safety and efficacy. Suvodirsen has shown very promising results in vitro; however, in a phase II/III (DYSTANCE 51; NCT03907072) trial, suvodirsen failed to increase dystrophin expression as assessed by western blot in muscle biopsies; its development was stopped due to inability to show target engagement (Sun et al., 2020).
Compounds Applicable to all DMD Mutations
Myostatin Inhibitors
Myostatin is a member of the transforming growth factor-beta family, and its levels are negatively associated with muscle growth. Myostatin inhibition has long been considered a promising intervention to reverse pathology in muscle-wasting diseases, and in preclinical studies decreased myostatin levels lead to increased muscle mass and strength as well as decreased fibrosis (Wagner et al., 2002; McCroskery et al., 2005; Wagner et al., 2005). Three myostatin inhibitors have been tested in DMD patients in randomised, placebo-controlled trials including the monoclonal neutralising antibody domagrozumab, the soluble ActRIIB ligand ACE-031, and talditercept alfa, which is an adnectin anti-myostatin protein. In a randomised, placebo-controlled phase II (NCT02310763) trial of 121 patients (6–15 years), domagrozumab-treated patients at three different doses did not differ significantly from placebo-treated patients in the 4-Stair Climb (4SC) assessment at 49 weeks (primary endpoint). Development of domagrozumab was discontinued despite the fact that treated participants showed a trend of increasing muscle mass volume, assessed by MRI, and improvement on the North Star Ambulatory Assessment (NSAA) score (Wagner et al., 2020). ACE-031 injection in a randomised, placebo-controlled phase II (NCT01099761) trial of 24 patients (≥4 years) caused dose-dependent increases in lean body mass and improvement in 6MWT; however, spontaneous bleeding associated with angiogenetic action of ACE-031 was observed. Due to safety reasons, the development was terminated (Campbell et al., 2017). In a phase I trial, talditercept alfa had a good safety profile and induced an increase in the lean body mass of participants. A phase II (NCT03039686) trial in 166 ambulant participants (6–11 years) was discontinued based on the prediction that a significant change in NSAA score (primary endpoint) at 48 weeks would not be achieved (Wagner et al., 2019).
There has been a failure to translate positive results of myostatin inhibition in animal models to humans. Myostatin is downregulated in muscle-wasting conditions in humans and dystrophin-deficient mdx mice. However, the myostatin levels in mdx mice remain much higher (25% of wild-type levels) than in humans with DMD (8% of normal values). Thus, further inhibition of myostatin in humans may not have the therapeutic effect seen in mice. In addition, myostatin is naturally downregulated in many neuromuscular diseases. Of note, myostatin inhibitors showed signals of efficacy as assessed by the lean/muscle mass with non-invasive imaging, but failed to show effect on functional outcomes (domagrozumab, talditercept alfa) or raised safety concerns (ACE-031) (Mariot et al., 2017; Koch et al., 2020; Rybalka et al., 2020; Wagner, 2020).
Tadalafil and Sildenafil
Repurposing compounds with a known safety and tolerability profile for rare diseases can accelerate approval processes. A case of repurposing was that of the phosphodiesterase type 5 inhibitors, tadalafil and sildenafil, which are approved for use in erectile dysfunction and pulmonary arterial hypertension. Dystrophin deficiency leads to decreased levels of the muscle-specific neuronal nitric oxide (NO) synthase at the sarcolemma and subsequently decreased production of NO. The NO-cyclic guanosine monophosphate pathway is involved in protective functional sympatholysis, a mechanism by which muscle blood flow increases with activity, to provide the muscle with oxygen and nutrients. Disruption of this protective mechanism exacerbates DMD pathology by making muscles prone to activity-induced ischemia and by increasing production of reactive oxygen species (Timpani et al., 2017; Timpani et al., 2020). The hypothesis that phosphodiesterase inhibition would increase muscle blood flow was supported by preclinical data in mdx mice (Percival et al., 2012; Nio et al., 2017). However, neither compound showed efficacy during clinical assessment (Timpani et al., 2020). The development of sildenafil was terminated earlier than that of tadalafil due to cardiomyopathy worsening observed in some of the treated participants in a placebo-controlled phase II (REVERSE DBMD; NCT01168908) trial, which also included patients affected by Becker muscular dystrophy (Leung et al., 2014). Assessment of tadalafil progressed to a randomised, placebo-controlled phase III (NCT01865084) trial in 331 participants (7–14 years) with a background treatment of steroids in which treated participants received either 0.3 mg/kg/d or 0.6 mg/kg/d tadalafil for 48 weeks. Tadalafil failed to show efficacy, as assessed by the 6MWT, which declined faster in the treated groups (Victor et al., 2017). Of note, these trials included one of the largest numbers of participants.
Idebenone
Another disappointing story of drug repurposing is that of the antioxidant Q10 analogue idebenone, which showed promising preclinical results in the mdx mouse model (Buyse et al., 2009). In a proof-of-concept randomised, placebo-controlled phase II trial, which included 21 patients (8–16 years) and lasted 12 months, it was demonstrated that idebenone slowed the loss of respiratory function in steroid-naïve subjects (Buyse et al., 2013). A randomised, placebo-controlled phase III (DELOS; NCT01027884) trial that included 64 steroid-naïve patients (10–18 years) with change in peak expiratory flow (PEF) from baseline to week 52 of treatment as primary endpoint reported significant effect in favour of the patients treated with idebenone (900 mg/d) (Buyse et al., 2015), which was sustained when patients were followed for up to 6 years through the Expanded Access Program (Servais et al., 2019). However, the primary outcome of the study was not an approved endpoint at that time (Buyse et al., 2017; Mayer et al., 2017). Idebenone (900 mg/d) was further assessed in a randomised, placebo-controlled, phase III (SIDEROS; NCT02814019) trial in 256 participants (≥10 years), who were not steroid-naïve and who were in the respiratory decline phase of the disease. Early interim analysis suggested that the study was unlikely to meet its primary endpoint (FVC% predicted) at 18 months. The development of idebenone was terminated despite earlier signs of efficacy in improving respiratory function in steroid-naïve patients.
Ezutromid
Ezutromid is an aryl hydrocarbon receptor antagonist that upregulates the production of utrophin, a dystrophin paralogue, and reverses DMD pathology in mdx mice (Tinsley et al., 1998). Ezutromid was overall safe and well-tolerated in early clinical development (Tejura et al., 2017; Muntoni et al., 2019a). A randomised, placebo-controlled phase II (PhaseOut DMD; NCT02858362) trial of two different ezutromid formulations (F3: microfluidised aqueous suspension, F6: powder suspension) included 40 participants (≥5 years), who were on stable steroid treatment. The primary endpoints were the fat fraction and the transverse relaxation time T2 measured by magnetic resonance imaging (MRI-T2) to assess muscle structure and inflammation levels at 12-weeks intervals. As secondary endpoints, the levels of utrophin and heavy chain myosin on muscle biopsies, as well as other functional outcomes were used (Muntoni et al., 2017). An interim analysis at 24 weeks based on the results from imaging and the muscle biopsies was positive, but at 48 weeks improvement was not seen. The lack of clinical efficacy signs combined with the challenging pharmacokinetics that ezutromid showed in earlier phases of development led to the cessation of its development (Wilkinson et al., 2019).
Edasalonexent
In DMD patients, the NF-kB pathway is activated, which is associated with inflammation and muscle degradation. The potent anti-inflammatory action of NF-kB inhibitors justified clinical testing as an alternative to the long-term use of steroids, which is linked to serious adverse effects (McDonald et al.,2018b). One of these NF-kB inhibitors, edasalonexent was safe, well-tolerated, and had good target engagement, as assessed by its ability to decrease NF-kB downstream pathways after 1 week of treatment in a phase I trial (Finanger et al., 2019). In a three-part phase I/II (MoveDMD; NCT02439216) trial, 31 participants (4–7 years), not on treatment with steroids were enrolled and the primary endpoint was the MRI-T2 of 5 lower leg muscles at 12 weeks. The primary endpoint was not met in the pooled analysis of the two edasalonexent doses (67 mg/kg/d, 100 mg/kg/d). This was attributed to the short observation period and the small sample size combined with the phenotypic variability. However, for the highest dose (100 mg/kg) MRI-T2 was improved compared to the off-treatment period after 12-weeks of treatment, with a good safety profile (Finkel et al., 2021a). Therefore, edasalonexent progressed in the subsequent randomised, placebo-controlled phase III (PolarisDMD; NCT03703882) trial, which enrolled 131 boys (4–7 years), who had been on steroids for at least 6 months, in which the primary outcome was not met. More specifically, there was no significant difference between the treated and the placebo group in the NSAA score (primary outcome) after 12 months of treatment with 100 mg/kg/d edasalonexent. Additionally, none of the secondary outcomes was met. Younger patients showed a non-significant one-point difference in the NSAA score compared to the placebo group, and a significant difference on secondary endpoints, including the 4SC assessment and timed function tests. However, when considering the overall population, the one-year change was similar between the two groups for all endpoints. This led to the discontinuation of the clinical development (Finkel et al., 2021b). Other NF-kB inhibitors (e.g., vamorolone, flavocoxid) are still in clinical development.
Discussion
Despite almost three decades of preclinical and clinical research, no treatment other than steroids has formally demonstrated clinical efficacy in DMD. The attrition rate between early-phase and late-phase development is high (Dovari et al., 2021). Several developments have been discontinued due to inefficacy despite early-phase positive results or signals of efficacy in secondary outcomes. The pressure by investors and by stakeholders to present and publish positive results has probably contributed to an inflation of confirmatory trials with compounds that are unlikely to gain approval. Currently, several compounds are in clinical development for DMD, with five of them having received approval subject to further proof of efficacy. There is a clear need for better clinical trial design and more reliable outcome measures. Reasons for failures and suggested solutions are summarised in Table 2.
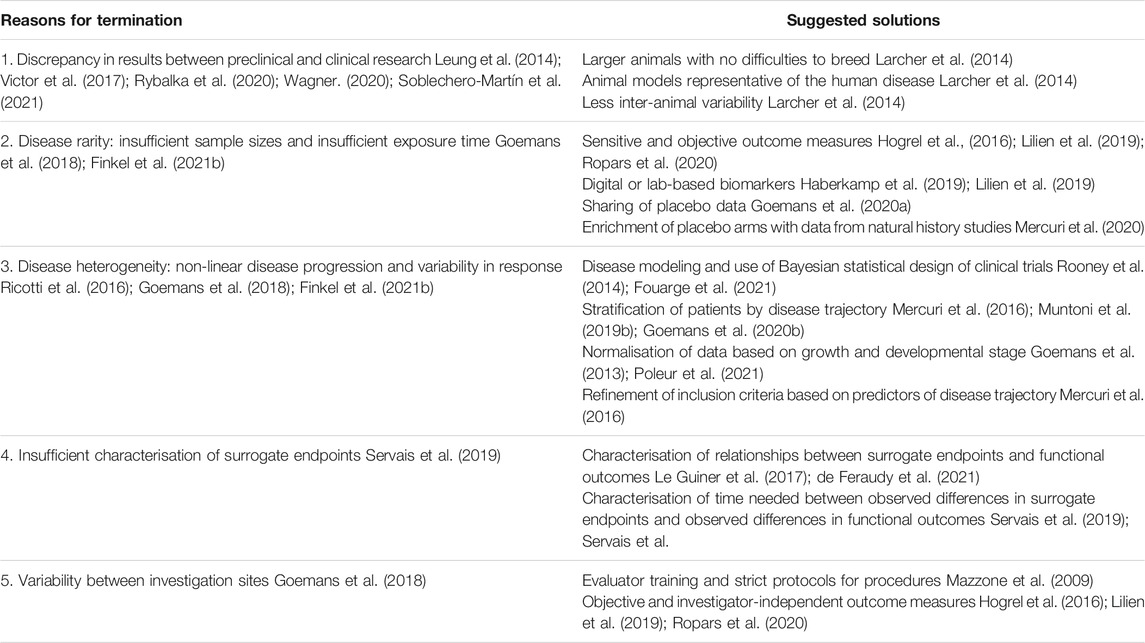
TABLE 2. Reasons for termination and suggested solutions.
More extensive preclinical studies could contribute to more effective clinical trials; however, current preclinical evaluation modes are not optimal. The commonly used mdx mouse model has significant differences from patients affected by DMD, mainly because an accelerated muscle regeneration rate in mice results in milder symptoms (Egorova et al., 2021). The pathophysiology of the disease may also differ. For example, myostatin levels are higher in mdx mice. This likely resulted in differences in the efficacy of myostatin inhibitors in mdx mice and DMD patients. Additionally, confounding factors encountered in clinical trials such as the use of steroids are not an issue in preclinical studies. The recently reported dystrophin-deficient rat could allow more rigorous preclinical studies in a more relevant model and a larger number of animals, which would help tackle the issue of inter-animal variability (Larcher et al., 2014; Nakamura et al., 2015).
The variability of DMD genotypes results in a wide range of phenotypes. For example, in patients with certain point mutations, alternate splicing can lead to some production of functional dystrophin and consequently less severe symptoms. Other genetic modifiers, such as LTBP4, may also contribute to phenotypic variability (Barp et al., 2015; Hightower and Alexander, 2018; Bello et al., 2020). Furthermore, differences in standards of care may result in significant differences in the disease trajectory. The age at which steroids are started, the type of steroids used (deflazacort vs. prednisone) as well as the steroid regimen (e.g., daily vs. alternate days vs. 10 days on -10 days off) also influence disease trajectory (Schreiber et al., 2016; de la Peña et al., 2017; Pradhan and Das, 2018). Additionally, the disease follows a non-linear trajectory. Loss of ambulation occurs between 6 and 18 years of age, which leads to significant variability in phenotype around the age of 7, the typical age of a patient included in a clinical trial. In children between 4 and 7 years, motor function outcomes used in clinical trials, such as the NSAA score, might show development-associated improvement before declining due to the natural course of the disease (Goemans et al., 2013; Mcdonald et al., 2013; Ciafaloni et al., 2016; Ricotti et al., 2016). For another example, respiratory outcomes may improve due to the normal growth of patients leading to increased lung volumes. Furthermore, it is highly important to normalise data for both age and height in this population, as some DMD patients have significantly stunted growth, mainly due to chronic steroid treatment (Hogrel et al., 2020). With regards to clinical trial design, the need for larger samples and longer duration is hampered by the rarity of the disease. Additionally, the regulatory pressure for the use of placebo arms contradicts the wishes of the community (Peay et al., 2018). The stratification of patients by disease trajectory (Muntoni et al., 2019b) to better define inclusion criteria, the use of natural history data to enrich placebo arms (Bello et al., 2016; Goemans et al., 2020a; Mercuri et al., 2020; Osorio et al., 2020), the sharing of placebo data via platforms (Bello et al., 2016; Goemans et al., 2020a), the use of innovative and sensitive outcome measures (Seferian et al., 2015; Lilien et al., 2019), or the use of Bayesian modelling could all result in more efficient and informative clinical trials (Fouarge et al., 2021).
There is a significant unmet need with regards to outcome measures used in clinical trials; different endpoints are meaningful depending on the age and disease stage of DMD patients. The use of surrogate endpoints, such as dystrophin expression for the compounds which induce dystrophin production, require further investigation, as the extent to which these measures predict efficacy is unknown (Aartsma-Rus and Arechavala-Gomeza, 2018). The optimisation of dystrophin quantification methods with the aim to improve accuracy and reproducibility will provide an undeniable pharmacodynamic endpoint for clinical trials. Towards this step, the harmonisation of protocols of the most used assays (e.g., immunochemistry, western blot) for the quantification and localisation of dystrophin will help minimise intra- and inter-lab variability. Several steps are currently being taken towards the optimisation of current assays and the use of new technologies which will help the reliable and accurate dystrophin quantification (Wells, 2019). However, there are different arguments for the clinical relevance of low levels of dystrophin; some patients who produce low dystrophin levels from birth show a milder phenotype, but it is still under investigation if low levels can have the same result after years of disease evolution (de Feraudy et al., 2021). Additionally, dystrophin levels can be variable in healthy individuals (Aartsma-Rus et al., 2019).
The FDA has granted accelerated approval based on truncated dystrophin expression as a surrogate endpoint, whereas the EMA has been consistently requiring functional data (Aartsma-Rus and Arechavala-Gomeza, 2018). This more conservative attitude translates into a clear controversy between the two agencies when it comes to drug approvals. Given the burden of the intravenous infusion, the risk of infections associated with the use of port-a-cath, the potential side effects as well as the cost of such drugs, the EMA position seems reasonable. Nevertheless, the difference in the position of the two agencies complicates clinical development, which must integrate both the possibility of accelerated approval and the need to prove efficacy by strong clinical data -sometimes even when this drug is commercially distributed in the US-. This is currently the case for exon skipping ASOs, like casimersen, golodirsen and viltolarsen, which are commercially distributed in the US, and still in large scale placebo-controlled trials. The qualification of dystrophin levels as a validated surrogate endpoint will be key in the next years; the translation of a limited re-expression of dystrophin into a sustainable clinical benefit could potentially not only support early approval but also help to compare compounds with a mechanism of action based on dystrophin re-expression. The minimum amount of dystrophin that is needed, the critical age or tissue quality at which re-expression must be initiated to be efficient and the time needed to observe clinically significant benefit is currently unknown. On the other side, functional outcome measures are very much dependent on both the observer’s opinion and the patient’s fatigue and motivation. The use of endpoints based on continuous monitoring with wearable technologies, such as the 95th percentile stride velocity, has the potential to dramatically decrease the number of patients needed as well as the study duration (Haberkamp et al., 2019; Poleur et al., 2021).
Now, breakthroughs in the fields of genetic therapies bring several opportunities in the preclinical and clinical pipeline of DMD, which need to be efficiently assessed in clinical trials. The rarity and variability of DMD render the design of clinical trials challenging. Undoubtedly, the DMD community needs to act to avoid repeating the mistakes of the past. The development of representative disease models for preclinical studies, the identification of sensitive biomarkers, the use of innovative technology, and the application of statistics will assist the roadmap to a more efficacious clinical trial design.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
TM and LS drafted and conducted the study research. TM wrote the initial draft including figure and tables. All authors have critically contributed to the different versions of the manuscript and have approved the final version. Figure was created using the tool BioRender.com.
Funding
TM is a scholar of the Onassis Foundation (Scholarship ID: F ZQ 040-1/2020-2021).
Conflict of Interest
LS has participated on Data Safety Monitoring or Advisory Boards of FibroGen, Santhera Pharmaceuticals, and Catabasis Pharmaceuticals. LS has received consulting fees by Sarepta, Pfizer and F. Hoffmann La Roche Ltd. US-S has received honoraria for attending boards, invited talks and consultancy from Santhera, Sarepta, PTC Therapeutics. LD has received honoraria for attending an advisory board of Santhera.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, orclaim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphar.2021.735912/full#supplementary-material
Footnotes
1https://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation.
References
Aartsma-Rus, A., and Arechavala-Gomeza, V. (2018). Why Dystrophin Quantification Is Key in the Eteplirsen Saga. Nat. Rev. Neurol. 14 (8), 454–456. doi:10.1038/s41582-018-0033-8
Aartsma-Rus, A., and Corey, D. R. (2020). The 10th Oligonucleotide Therapy Approved: Golodirsen for Duchenne Muscular Dystrophy. Nucleic Acid Ther. 30 (2), 67–70. doi:10.1089/nat.2020.0845
Aartsma-Rus, A., and Krieg, A. M. (2017). FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid Ther. 27 (1), 1–3. doi:10.1089/nat.2016.0657
Aartsma-Rus, A., Morgan, J., Lonkar, P., Neubert, H., Owens, J., Binks, M., et al. (2019). Report of a TREAT-NMD/World Duchenne Organisation Meeting on Dystrophin Quantification Methodology. J. Neuromuscul. Dis. 6 (1), 147–159. doi:10.3233/JND-180357
Alter, J., Lou, F., Rabinowitz, A., Yin, H., Rosenfeld, J., Wilton, S. D., et al. (2006). Systemic Delivery of Morpholino Oligonucleotide Restores Dystrophin Expression Bodywide and Improves Dystrophic Pathology. Nat. Med. 12 (2), 175–177. doi:10.1038/nm1345
Barp, A., Bello, L., Politano, L., Melacini, P., Calore, C., Polo, A., et al. (2015). “Genetic Modifiers of Duchenne Muscular Dystrophy and Dilated Cardiomyopathy, Editor A. Kumar, PLoS One 10, e0141240. doi:10.1371/journal.pone.0141240
Barthélémy, I., Calmels, N., Weiss, R. B., Tiret, L., Vulin, A., Wein, N., et al. (2020). X-linked Muscular Dystrophy in a Labrador Retriever Strain: Phenotypic and Molecular Characterisation. Skelet Muscle 10 (1), 23. doi:10.1186/s13395-020-00239-0
Bello, L., D'Angelo, G., Villa, M., Fusto, A., Vianello, S., Merlo, B., et al. (2020). Genetic Modifiers of Respiratory Function in Duchenne Muscular Dystrophy. Ann. Clin. Transl Neurol. 7 (5), 786–798. doi:10.1002/acn3.51046
Bello, L., Morgenroth, L. P., Gordish-Dressman, H., Hoffman, E. P., McDonald, C. M., Cirak, S., et al. (2016). DMD Genotypes and Loss of Ambulation in the CINRG Duchenne Natural History Study. Neurology 87 (4), 401–409. doi:10.1212/WNL.0000000000002891
Birnkrant, D. J., Bushby, K., Bann, C. M., Alman, B. A., Apkon, S. D., Blackwell, A., et al. (2018a). Diagnosis and Management of Duchenne Muscular Dystrophy, Part 2: Respiratory, Cardiac, Bone Health, and Orthopaedic Management. Lancet Neurol. 17 (4), 347–361. doi:10.1016/S1474-4422(18)30025-5
Birnkrant, D. J., Bushby, K., Bann, C. M., Apkon, S. D., Blackwell, A., Brumbaugh, D., et al. (2018b). Diagnosis and Management of Duchenne Muscular Dystrophy, Part 1: Diagnosis, and Neuromuscular, Rehabilitation, Endocrine, and Gastrointestinal and Nutritional Management. Lancet Neurol. 17 (3), 251–267. doi:10.1016/S1474-4422(18)30024-3
Birnkrant, D. J., Bushby, K., Bann, C. M., Apkon, S. D., Blackwell, A., Colvin, M. K., et al. (2018c). Diagnosis and Management of Duchenne Muscular Dystrophy, Part 3: Primary Care, Emergency Management, Psychosocial Care, and Transitions of Care across the Lifespan. Lancet Neurol. 17 (5), 445–455. doi:10.1016/S1474-4422(18)30026-7
Buyse, G. M., Goemans, N., van den Hauwe, M., and Meier, T. (2013). Effects of Glucocorticoids and Idebenone on Respiratory Function in Patients with Duchenne Muscular Dystrophy. Pediatr. Pulmonol 48 (9), 912–920. doi:10.1002/ppul.22688
Buyse, G. M., Van der Mieren, G., Erb, M., D'hooge, J., Herijgers, P., Verbeken, E., et al. (2009). Long-term Blinded Placebo-Controlled Study of SNT-MC17/idebenone in the Dystrophin Deficient Mdx Mouse: Cardiac protection and Improved Exercise Performance. Eur. Heart J. 30 (1), 116–124. doi:10.1093/eurheartj/ehn406
Buyse, G. M., Voit, T., Schara, U., Straathof, C. S., D'Angelo, M. G., Bernert, G., et al. (2017). Treatment Effect of Idebenone on Inspiratory Function in Patients with Duchenne Muscular Dystrophy. Pediatr. Pulmonol 52 (4), 508–515. doi:10.1002/ppul.23547
Buyse, G. M., Voit, T., Schara, U., Straathof, C. S. M., D'Angelo, M. G., Bernert, G., et al. (2015). Efficacy of Idebenone on Respiratory Function in Patients with Duchenne Muscular Dystrophy Not Using Glucocorticoids (DELOS): a Double-Blind Randomised Placebo-Controlled Phase 3 Trial. Lancet 385 (9979), 1748–1757. doi:10.1016/S0140-6736(15)60025-3
Campbell, C., McMillan, H. J., Mah, J. K., Tarnopolsky, M., Selby, K., McClure, T., et al. (2017). Myostatin Inhibitor ACE-031 Treatment of Ambulatory Boys with Duchenne Muscular Dystrophy: Results of a Randomized, Placebo-Controlled Clinical Trial. Muscle Nerve 55 (4), 458–464. doi:10.1002/mus.25268
Ciafaloni, E., Kumar, A., Liu, K., Pandya, S., Westfield, C., Fox, D. J., et al. (2016). “Age at Onset of First Signs or Symptoms Predicts Age at Loss of Ambulation in Duchenne and Becker Muscular Dystrophy: Data from the MD STARnet,” in Age at Onset of First Signs or Symptoms Predicts Age at Loss of Ambulation in Duchenne and Becker Muscular Dystrophy: Data from the MD STARnet. Editor D. J. Matthews (Amsterdam, Netherlands: J. Pediatr. Rehabil. Med, IOS Press) 9, 5–11. doi:10.3233/prm-160361
de Feraudy, Y., Ben Yaou, R., Wahbi, K., Stalens, C., Stantzou, A., Laugel, V., et al. (2021). Very Low Residual Dystrophin Quantity Is Associated with Milder Dystrophinopathy. Ann. Neurol. 89 (2), 280–292. doi:10.1002/ana.25951
de la Peña, N., Luna, E., and Castellanos, A. (2017). Influencia de los esteroides en los parametros de la marcha en distrofia de Duchenne. Rehabilitación 51 (1), 5–10. doi:10.1016/j.rh.2016.09.006
Dhillon, S. (2020). Viltolarsen: First Approval. Drugs 80 (10), 1027–1031. doi:10.1007/s40265-020-01339-3
Doorenweerd, N. (2020). Combining Genetics, Neuropsychology and Neuroimaging to Improve Understanding of Brain Involvement in Duchenne Muscular Dystrophy - a Narrative Review. Neuromuscul. Disord. 30 (6), 437–442. doi:10.1016/j.nmd.2020.05.001
Dovari, A., Inuganti, B., Nadimpally, J., Vatturi, S. M., Hyderboini, R., and Goyal, R. (2021). PRO38 Twenty Years of Clinical Trials in Duchenne Muscular Dystrophy: A Low Clinical Drug Development Success. Value in Health 24, S204. doi:10.1016/j.jval.2021.04.1023
Duan, D., Goemans, N., Takeda, S., Mercuri, E., and Aartsma-Rus, A. (2021). Duchenne Muscular Dystrophy. Nat. Rev. Dis. Primers 7 (1), 13. doi:10.1038/s41572-021-00248-3
Egorova, T. V., Galkin, I. I., Ivanova, Y. V., and Polikarpova, A. V. (2021). “Duchenne Muscular Dystrophy Animal Models,” in Animal Models in Medicine and Biology (London, United Kingdom: IntechOpen). (Online ahead of printing). doi:10.5772/intechopen.96738
Finanger, E., Vandenborne, K., Finkel, R. S., Lee Sweeney, H., Tennekoon, G., Yum, S., et al. (2019). Phase 1 Study of Edasalonexent (CAT-1004), an Oral NF-Κb Inhibitor, in Pediatric Patients with Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 6 (1), 43–54. doi:10.3233/JND-180341
Finkel, R. S., Finanger, E., Vandenborne, K., Sweeney, H. L., Tennekoon, G., Shieh, P. B., et al. (2021). Disease-modifying Effects of Edasalonexent, an NF-Κb Inhibitor, in Young Boys with Duchenne Muscular Dystrophy: Results of the MoveDMD Phase 2 and Open Label Extension Trial. Neuromuscul. Disord. 31 (5), 385–396. doi:10.1016/j.nmd.2021.02.001
Finkel, R. S., McDonald, C. M., Lee Sweeney, H., Finanger, E., Neil Knierbein, E., Wagner, K. R., et al. (2021). A Randomized, Double-Blind, Placebo-Controlled, Global Phase 3 Study of Edasalonexent in Pediatric Patients with Duchenne Muscular Dystrophy: Results of the PolarisDMD Trial. J. Neuromuscul. Dis. 8 (5), 769–784. doi:10.3233/JND-210689
Fouarge, E., Monseur, A., Boulanger, B., Annoussamy, M., Seferian, A. M., De Lucia, S., et al. (2021). Hierarchical Bayesian Modelling of Disease Progression to Inform Clinical Trial Design in Centronuclear Myopathy. Orphanet J. Rare Dis. 16 (1), 3. doi:10.1186/s13023-020-01663-7
Goemans, N., Klingels, K., van den Hauwe, M., Boons, S., Verstraete, L., Peeters, C., et al. (2013). Six-minute Walk Test: Reference Values and Prediction Equation in Healthy Boys Aged 5 to 12 Years. PLoS One 8 (12), e84120. doi:10.1371/journal.pone.0084120
Goemans, N., Mercuri, E., Belousova, E., Komaki, H., Dubrovsky, A., McDonald, C. M., et al. (2018). A Randomized Placebo-Controlled Phase 3 Trial of an Antisense Oligonucleotide, Drisapersen, in Duchenne Muscular Dystrophy. Neuromuscul. Disord. 28 (1), 4–15. doi:10.1016/j.nmd.2017.10.004
Goemans, N., Signorovitch, J., Sajeev, G., Yao, Z., Gordish-Dressman, H., McDonald, C. M., et al. (2020). Suitability of External Controls for Drug Evaluation in Duchenne Muscular Dystrophy. Neurology 95 (10), e1381–91. doi:10.1212/WNL.0000000000010170
Goemans, N., Wong, B., Van den Hauwe, M., Signorovitch, J., Sajeev, G., Cox, D., et al. (2020). Prognostic Factors for Changes in the Timed 4-stair Climb in Patients with Duchenne Muscular Dystrophy, and Implications for Measuring Drug Efficacy: A Multi-Institutional Collaboration. PLoS One 15 (6), e0232870. doi:10.1371/journal.pone.0232870
Goemans, N. M., Tulinius, M., van den Hauwe, M., Kroksmark, A. K., Buyse, G., Wilson, R. J., et al. (2016). Long-Term Efficacy, Safety, and Pharmacokinetics of Drisapersen in Duchenne Muscular Dystrophy: Results from an Open-Label Extension Study. PLoS One 11 (9), e0161955. doi:10.1371/journal.pone.0161955
Haberkamp, M., Moseley, J., Athanasiou, D., de Andres-Trelles, F., Elferink, A., Rosa, M. M., et al. (2019). European Regulator's Views on a Wearable-Derived Performance Measurement of Ambulation for Duchenne Muscular Dystrophy Regulatory Trials. Neuromuscul. Disord. 29 (7), 514–516. doi:10.1016/j.nmd.2019.06.003
Hightower, R. M., and Alexander, M. S. (2018). Genetic Modifiers of Duchenne and Facioscapulohumeral Muscular Dystrophies. Muscle Nerve 57 (1), 6–15. doi:10.1002/mus.25953
Hogrel, J. Y., Decostre, V., Ledoux, I., de Antonio, M., Niks, E. H., de Groot, I., et al. (2020). Normalized Grip Strength Is a Sensitive Outcome Measure through All Stages of Duchenne Muscular Dystrophy. J. Neurol. 267 (7), 2022–2028. doi:10.1007/s00415-020-09800-9
Hogrel, J. Y., Wary, C., Moraux, A., Azzabou, N., Decostre, V., Ollivier, G., et al. (2016). Longitudinal Functional and NMR Assessment of Upper Limbs in Duchenne Muscular Dystrophy. Neurology 86 (11), 1022–1030. doi:10.1212/WNL.0000000000002464
Koch, C., Buono, S., Menuet, A., Robé, A., Djeddi, S., Kretz, C., et al. (2020). Myostatin: a Circulating Biomarker Correlating with Disease in Myotubular Myopathy Mice and Patients. Mol. Ther. Methods Clin. Dev. 17, 1178–1189. doi:10.1016/j.omtm.2020.04.022
Larcher, T., Lafoux, A., Tesson, L., Remy, S., Thepenier, V., François, V., et al. (2014). Characterization of Dystrophin Deficient Rats: a New Model for Duchenne Muscular Dystrophy. PLoS One 9 (10), e110371. doi:10.1371/journal.pone.0110371
Le Guiner, C., Servais, L., Montus, M., Larcher, T., Fraysse, B., Moullec, S., et al. (2017). Long-term Microdystrophin Gene Therapy Is Effective in a Canine Model of Duchenne Muscular Dystrophy. Nat. Commun. 8 (1), 16105. doi:10.1038/ncomms16105
Leung, D. G., Herzka, D. A., Thompson, W. R., He, B., Bibat, G., Tennekoon, G., et al. (2014). Sildenafil Does Not Improve Cardiomyopathy in Duchenne/Becker Muscular Dystrophy. Ann. Neurol. 76 (4), 541–549. doi:10.1002/ana.24214
Lilien, C., Gasnier, E., Gidaro, T., Seferian, A., Grelet, M., Vissière, D., et al. (2019). Home-based Monitor for Gait and Activity Analysis. J. Vis. Exp. (150), 59668. doi:10.3791/59668
Lu, Q. L., Rabinowitz, A., Chen, Y. C., Yokota, T., Yin, H., Alter, J., et al. (2005). Systemic Delivery of Antisense Oligoribonucleotide Restores Dystrophin Expression in Body-wide Skeletal Muscles. Proc. Natl. Acad. Sci. U S A. 102 (1), 198–203. doi:10.1073/pnas.0406700102
Mariot, V., Joubert, R., Hourdé, C., Féasson, L., Hanna, M., Muntoni, F., et al. (2017). Downregulation of Myostatin Pathway in Neuromuscular Diseases May Explain Challenges of Anti-myostatin Therapeutic Approaches. Nat. Commun. 8 (1), 1859. doi:10.1038/s41467-017-01486-4
Mayer, O. H., Leinonen, M., Rummey, C., Meier, T., Buyse, G. M., and Group, D. S. (2017). Efficacy of Idebenone to Preserve Respiratory Function above Clinically Meaningful Thresholds for Forced Vital Capacity (FVC) in Patients with Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 4 (3), 189–198. doi:10.3233/JND-170245
Mazzone, E. S., Coratti, G., Sormani, M. P., Messina, S., Pane, M., D'Amico, A., et al. (2016). Timed Rise from Floor as a Predictor of Disease Progression in Duchenne Muscular Dystrophy: An Observational Study. PLoS One 11 (3), e0151445. doi:10.1371/journal.pone.0151445
Mazzone, E. S., Messina, S., Vasco, G., Main, M., Eagle, M., D'Amico, A., et al. (2009). Reliability of the North Star Ambulatory Assessment in a Multicentric Setting. Neuromuscul. Disord. 19 (7), 458–461. doi:10.1016/j.nmd.2009.06.368
McClorey, G., Moulton, H. M., Iversen, P. L., Fletcher, S., and Wilton, S. D. (2006). Antisense Oligonucleotide-Induced Exon Skipping Restores Dystrophin Expression In Vitro in a Canine Model of DMD. Gene Ther. 13 (19), 1373–1381. doi:10.1038/sj.gt.3302800
McCroskery, S., Thomas, M., Platt, L., Hennebry, A., Nishimura, T., McLeay, L., et al. (2005). Improved Muscle Healing through Enhanced Regeneration and Reduced Fibrosis in Myostatin-Null Mice. J. Cel Sci 118 (Pt 15), 3531–3541. doi:10.1242/jcs.02482
McDonald, C. M., Henricson, E. K., Abresch, R. T., Duong, T., Joyce, N. C., Hu, F., et al. (2018a). Long-term Effects of Glucocorticoids on Function, Quality of Life, and Survival in Patients with Duchenne Muscular Dystrophy: a Prospective Cohort Study. Lancet 391 (10119), 451–461. doi:10.1016/S0140-6736(17)32160-8
Mcdonald, C. M., Henricson, E. K., Abresch, R. T., Florence, J. M., Eagle, M., Gappmaier, E., et al. (2013). The 6-minute Walk Test and Other Endpoints in Duchenne Muscular Dystrophy: Longitudinal Natural History Observations over 48 Weeks from a Multicenter Study. Muscle Nerve 48 (3), 343–356. doi:10.1002/mus.23902
McDonald, C. M., Wong, B., Flanigan, K. M., Wilson, R., de Kimpe, S., Lourbakos, A., et al. (2018b). Placebo-controlled Phase 2 Trial of Drisapersen for Duchenne Muscular Dystrophy. Ann. Clin. Transl Neurol. 5 (8), 913–926. doi:10.1002/acn3.579
Mercuri, E., Muntoni, F., Osorio, A. N., Tulinius, M., Buccella, F., Morgenroth, L. P., et al. (2020). Safety and Effectiveness of Ataluren: Comparison of Results from the STRIDE Registry and CINRG DMD Natural History Study. J. Comp. Eff. Res. 9 (5), 341–360. doi:10.2217/cer-2019-0171
Mercuri, E., Signorovitch, J. E., Swallow, E., Song, J., Ward, S. J., Group, D. M. D. I., et al. (2016). Categorizing Natural History Trajectories of Ambulatory Function Measured by the 6-minute Walk Distance in Patients with Duchenne Muscular Dystrophy. Neuromuscul. Disord. 26 (9), 576–583. doi:10.1016/j.nmd.2016.05.016
Muntoni, F., Domingos, J., Manzur, A. Y., Mayhew, A., Guglieri, M., Network, U. K. N., et al. (2019b). Categorising Trajectories and Individual Item Changes of the North Star Ambulatory Assessment in Patients with Duchenne Muscular Dystrophy. PLoS One 14 (9), e0221097. doi:10.1371/journal.pone.0221097
Muntoni, F., Tejura, B., Spinty, S., Roper, H., Hughes, I., Layton, G., et al. (2019a). A Phase 1b Trial to Assess the Pharmacokinetics of Ezutromid in Pediatric Duchenne Muscular Dystrophy Patients on a Balanced Diet. Clin. Pharmacol. Drug Dev. 8 (7), 922–933. doi:10.1002/cpdd.642
Muntoni, F., Maresh, K., Davies, K., Harriman, S., Layton, G., Rosskamp, R., et al. (2017). PhaseOut DMD: a Phase 2, Proof of Concept, Clinical Study of Utrophin Modulation with Ezutromid. Neuromuscul. Disord. 27, S217. doi:10.1016/j.nmd.2017.06.443
Nakamura, K., Fujii, W., Tsuboi, M., Tanihata, J., Teramoto, N., Takeuchi, S., et al. (2015). Generation of Muscular Dystrophy Model Rats with a CRISPR/Cas System. Sci. Rep. 4 (1), 5635. doi:10.1038/srep05635
Nio, Y., Tanaka, M., Hirozane, Y., Muraki, Y., Okawara, M., Hazama, M., et al. (2017). Phosphodiesterase 4 Inhibitor and Phosphodiesterase 5 Inhibitor Combination Therapy Has Antifibrotic and Anti-inflammatory Effects in Mdx Mice with Duchenne Muscular Dystrophy. FASEB J. 31 (12), 5307–5320. doi:10.1096/fj.201700249R
Osorio, A. N., Tulinius, M., Buccella, F., Desguerre, I., Kirschner, J., Mercuri, E., et al. (2020). Pulmonary Function in Non-ambulatory Patients with Nonesense Mutation Duchenne Muscular Dystrophy: Stride Registry and Cinrg Dnhs Matched Cohort Analysis. Muscle and Nerve 62 (Suppl. 1), S6–S7. doi:10.1007/s00259-020-04988-4
Peay, H. L., Biesecker, B. B., Wilfond, B. S., Jarecki, J., Umstead, K. L., Escolar, D. M., et al. (2018). Barriers and Facilitators to Clinical Trial Participation Among Parents of Children with Pediatric Neuromuscular Disorders. Clin. Trials 15 (2), 139–148. doi:10.1177/1740774517751118
Percival, J. M., Whitehead, N. P., Adams, M. E., Adamo, C. M., Beavo, J. A., and Froehner, S. C. (2012). Sildenafil Reduces Respiratory Muscle Weakness and Fibrosis in the Mdx Mouse Model of Duchenne Muscular Dystrophy. J. Pathol. 228 (1), 77–87. doi:10.1002/path.4054
Poleur, M., Ulinici, A., Daron, A., Schneider, O., Farra, F. D., Demonceau, M., et al. (2021). Normative Data on Spontaneous Stride Velocity, Stride Length, and Walking Activity in a Non-controlled Environment. Orphanet J. Rare Dis. 16 (1), 318. doi:10.1186/s13023-021-01956-5
Pradhan, S., and Das, A. (2018). Change in the Natural Profile of Duchenne Muscular Dystrophy with the Judicious Use of Steroids. Neurol. India 66 (3), 893–895. doi:10.4103/0028-3886.232304
Ricotti, V., Ridout, D. A., Pane, M., Main, M., Mayhew, A., Mercuri, E., et al. (2016). The NorthStar Ambulatory Assessment in Duchenne Muscular Dystrophy: Considerations for the Design of Clinical Trials. J. Neurol. Neurosurg. Psychiatry 87 (2), 149–155. doi:10.1136/jnnp-2014-309405
Rooney, W. D., Berlow, Y. A., Triplett, W. T., Forbes, S. C., Willcocks, R. J., Wang, D. J., et al. (2014). Modeling Disease Trajectory in Duchenne Muscular Dystrophy. Neurology 94 (15), e1622–33. doi:10.1212/WNL.0000000000009244
Ropars, J., Gravot, F., Ben Salem, D., Rousseau, F., Brochard, S., and Pons, C. (2020). Muscle MRI: A Biomarker of Disease Severity in Duchenne Muscular Dystrophy? A Systematic Review. Neurology 94 (3), 117–133. doi:10.1212/WNL.0000000000008811
Ryan, N. J. (2014). Ataluren: First Global Approval. Drugs 74 (14), 1709–1714. doi:10.1007/s40265-014-0287-4
Rybalka, E., Timpani, C. A., Debruin, D. A., Bagaric, R. M., Campelj, D. G., and Hayes, A. (2020). The Failed Clinical Story of Myostatin Inhibitors against Duchenne Muscular Dystrophy: Exploring the Biology behind the Battle. Cells 9 (12), 10. doi:10.3390/cells9122657
Schreiber, A., Brochard, S., Rippert, P., Fontainecarbonel, S., Peudenier, S., Payan, C., et al. (2016). The Natural History of Duchenne Muscular Dystrophy with Corticosteroids Using the Motor Function Measure. Develop. Med. Child Neurol. 58 (Suppl. 6), 25. doi:10.1111/dmcn.12341
Seferian, A. M., Moraux, A., Annoussamy, M., Canal, A., Decostre, V., Diebate, O., et al. (2015). Upper Limb Strength and Function Changes during a One-Year Follow-Up in Non-ambulant Patients with Duchenne Muscular Dystrophy: an Observational Multicenter Trial. PLoS One 10 (2), e0113999. doi:10.1371/journal.pone.0113999
Servais, L., Mercuri, E., Straub, V., Guglieri, M., Seferian, A. M., Scoto, M., et al. Long-term Safety and Efficacy Data of Golodirsen in Ambulatory Patients with Duchenne Muscular Dystrophy Amenable to Exon 53 Skipping: A First-In-Human, Multicenter, Two-Part, Open-Label, Phase 1/2 Trial.
Servais, L., Straathof, C. S. M., Schara, U., Klein, A., Leinonen, M., Hasham, S., et al. (2019). Long-term Data with Idebenone on Respiratory Function Outcomes in Patients with Duchenne Muscular Dystrophy. Neuromuscul. Disord. 30 (1), 5–16. doi:10.1016/j.nmd.2019.10.008
Shirley, M. (2021). Casimersen: First Approval. Drugs 81 (7), 875–879. doi:10.1007/s40265-021-01512-2
Soblechero‐Martín, P., López‐Martínez, A., Puente‐Ovejero, L., Vallejo‐Illarramendi, A., and Arechavala‐Gomeza, V. (2021). Utrophin Modulator Drugs as Potential Therapies for Duchenne and Becker Muscular Dystrophies. Neuropathol. Appl. Neurobiol 47 (6), 711–723. doi:10.1111/nan.12735
Starosta, A., and Konieczny, P. (2021). Therapeutic Aspects of Cell Signaling and Communication in Duchenne Muscular Dystrophy. Cell Mol Life Sci 78 (11), 4867–4891. doi:10.1007/s00018-021-03821-x
Sun, C., Shen, L., Zhang, Z., and Xie, X. (2020). Therapeutic Strategies for Duchenne Muscular Dystrophy: An Update. Genes (Basel) 11 (8), 837. doi:10.3390/genes11080837
Tejura, B., Spinty, S., Roper, H., Hughes, I., Majumdar, A., Harriman, S., et al. (2017). Overcoming Pharmacokinetic Challenges to Drug Administration in Duchenne Muscular Dystrophy: Lessons from Phase 1 Development of Ezutromid. Eur. J. Paediatric Neurol. 21 (Suppl. 1), e93. doi:10.1016/j.ejpn.2017.04.1207
Timpani, C. A., Hayes, A., and Rybalka, E. (2017). Therapeutic Strategies to Address Neuronal Nitric Oxide Synthase Deficiency and the Loss of Nitric Oxide Bioavailability in Duchenne Muscular Dystrophy. Orphanet J. Rare Dis. 12 (1), 100. doi:10.1186/s13023-017-0652-y
Timpani, C. A., Mamchaoui, K., Butler-Browne, G., and Rybalka, E. (2020). Nitric Oxide (NO) and Duchenne Muscular Dystrophy: NO Way to Go? Antioxidants 9 (12), 1268. doi:10.3390/antiox9121268
Tinsley, J., Deconinck, N., Fisher, R., Kahn, D., Phelps, S., Gillis, J. M., et al. (1998). Expression of Full-Length Utrophin Prevents Muscular Dystrophy in Mdx Mice. Nat. Med. 4 (12), 1441–1444. doi:10.1038/4033
van Deutekom, J. C., Bremmer-Bout, M., Janson, A. A., Ginjaar, I. B., Baas, F., den Dunnen, J. T., et al. (2001). Antisense-induced Exon Skipping Restores Dystrophin Expression in DMD Patient Derived Muscle Cells. Hum. Mol. Genet. 10 (15), 1547–1554. doi:10.1093/hmg/10.15.1547
Victor, R. G., Sweeney, H. L., Finkel, R., McDonald, C. M., Byrne, B., Eagle, M., et al. (2017). A Phase 3 Randomized Placebo-Controlled Trial of Tadalafil for Duchenne Muscular Dystrophy. Neurology 89 (17), 1811–1820. doi:10.1212/WNL.0000000000004570
Voit, T., Topaloglu, H., Straub, V., Muntoni, F., Deconinck, N., Campion, G., et al. (2014). Safety and Efficacy of Drisapersen for the Treatment of Duchenne Muscular Dystrophy (DEMAND II): an Exploratory, Randomised, Placebo-Controlled Phase 2 Study. Lancet Neurol. 13 (10), 987–996. doi:10.1016/S1474-4422(14)70195-4
Wagner, K. R., Abdel-Hamid, H. Z., Mah, J. K., Campbell, C., Guglieri, M., Muntoni, F., et al. (2020). Randomized Phase 2 Trial and Open-Label Extension of Domagrozumab in Duchenne Muscular Dystrophy. Neuromuscul. Disord. 30 (6), 492–502. doi:10.1016/j.nmd.2020.05.002
Wagner, K. R., Liu, X., Chang, X., and Allen, R. E. (2005). Muscle Regeneration in the Prolonged Absence of Myostatin. Proc. Natl. Acad. Sci. U S A. 102 (7), 2519–2524. doi:10.1073/pnas.0408729102
Wagner, K. R., McPherron, A. C., Winik, N., and Lee, S. J. (2002). Loss of Myostatin Attenuates Severity of Muscular Dystrophy in Mdx Mice. Ann. Neurol. 52 (6), 832–836. doi:10.1002/ana.10385
Wagner, K. R. (2020). The Elusive Promise of Myostatin Inhibition for Muscular Dystrophy. Curr. Opin. Neurol. 33 (5), 621–628. doi:10.1097/WCO.0000000000000853
Wagner, K. R., Wong, B. L., Byrne, B. J., Tian, C., Jacobsen, L. K., Tirucherai, G. S., et al. (2019). A Phase 1b/2 Study of the Anti-myostatin Adnectin RG6206 (BMS-986089) in Ambulatory Boys with Duchenne Muscular Dystrophy: A 72-week Treatment Update. Neurol. Conf. 71st Annu. Meet. Am. Acad. Neurol. AAN. 92 (15 Suppl. 1), P1.6–062.
Wells, D. J. (2019). What Is the Level of Dystrophin Expression Required for Effective Therapy of Duchenne Muscular Dystrophy? J. Muscle Res. Cel Motil 40 (2), 141–150. doi:10.1007/s10974-019-09535-9
Wilkinson, I. V. L., Perkins, K. J., Dugdale, H., Moir, L., Vuorinen, A., Chatzopoulou, M., et al. (2019). Chemical Proteomics and Phenotypic Profiling Identifies the Aryl Hydrocarbon Receptor as a Molecular Target of the Utrophin Modulator Ezutromid. Angew. Chem. Int. Ed. Engl. 59 (6), 2420–2428. doi:10.1002/anie.201912392
Keywords: antisense oligonucleotides, clinical trials, duchenne muscular dystrophy, dystrophin, exon skipping, randomized controlled clinical trials, myostatin inhibition, utrophin upregulation
Citation: Markati T, De Waele L, Schara-Schmidt U and Servais L (2021) Lessons Learned from Discontinued Clinical Developments in Duchenne Muscular Dystrophy. Front. Pharmacol. 12:735912. doi: 10.3389/fphar.2021.735912
Received: 03 July 2021; Accepted: 12 October 2021;
Published: 01 November 2021.
Edited by:
Tracey Anne Willis, Robert Jones and Agnes Hunt Orthopaedic Hospital NHS Trust, United KingdomReviewed by:
Gregory Thomas Knipp, Purdue University, United StatesSaeed Anwar, University of Alberta, Canada
Copyright © 2021 Markati, De Waele, Schara-Schmidt and Servais. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laurent Servais, laurent.servais@paediatrics.ox.ac.uk