 Qianqian Li
Qianqian Li Jian Gao
Jian Gao Xiaohan Pang1
Xiaohan Pang1 Yi Wang
Yi Wang- 1School of Traditional Chinese Medicine, Beijing University of Chinese Medicine, Beijing, China
- 2College of Pharmaceutical Sciences, Pharmaceutical Informatics Institute, Zhejiang University, Hangzhou, China
Emodin is a natural occurring anthraquinone derivative isolated from roots and barks of numerous plants, molds, and lichens. It is found to be an active ingredient in different Chinese herbs including Rheum palmatum and Polygonam multiflorum, and it is a pleiotropic molecule with diuretic, vasorelaxant, anti-bacterial, anti-viral, anti-ulcerogenic, anti-inflammatory, and anti-cancer effects. Moreover, emodin has also been shown to have a wide activity of anti-cardiovascular diseases. It is mainly involved in multiple molecular targets such as inflammatory, anti-apoptosis, anti-hypertrophy, anti-fibrosis, anti-oxidative damage, abnormal, and excessive proliferation of smooth muscle cells in cardiovascular diseases. As a new type of cardiovascular disease treatment drug, emodin has broad application prospects. However, a large amount of evidences detailing the effect of emodin on many signaling pathways and cellular functions in cardiovascular disease, the overall understanding of its mechanisms of action remains elusive. In addition, by describing the evidence of the effects of emodin in detail, the toxicity and poor oral bioavailability of mice have been continuously discovered. This review aims to describe a timely overview of emodin related to the treatment of cardiovascular disease. The emphasis is to summarize the pharmacological effects of emodin as an anti-cardiovascular drug, as well as the targets and its potential mechanisms. Furthermore, the treatment of emodin compared with conventional cardiovascular drugs or target inhibitors, the toxicity, pharmacokinetics and derivatives of emodin were discussed.
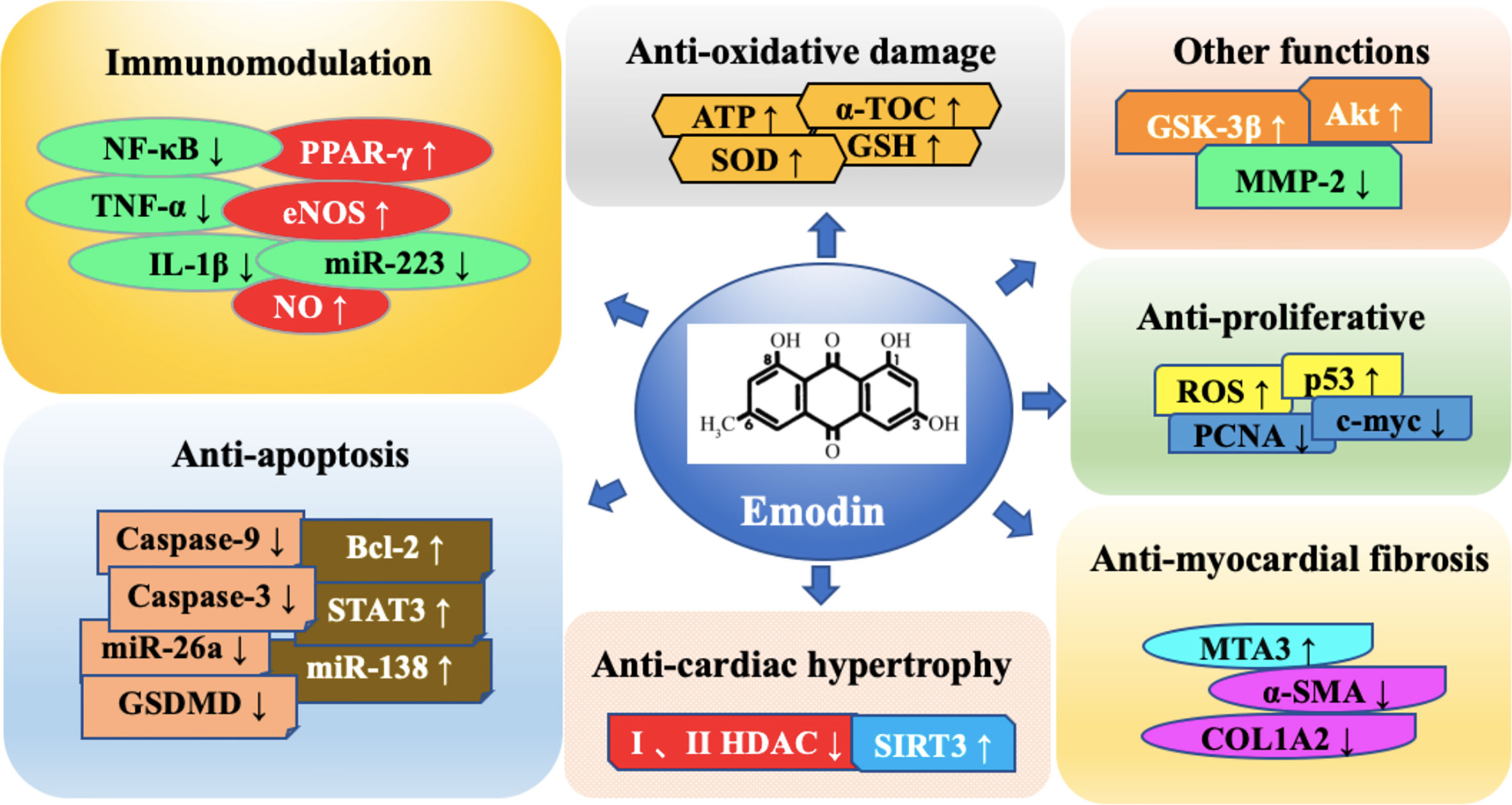
Graphical Abstract
Introduction
Emodin (1, 3, 8-trihydroxy-6-methylanthraquinone) is a natural anthraquinone derivative, and is the main ingredient of many herbs including rhubarb, polygonum multiflorum, and polygonum cuspidatum (Dong et al., 2016; Monisha et al., 2016). Pharmacological studies have shown that emodin, as a polyvalent molecule, has a variety of biological functions, such as anti-inflammatory (Li X. et al., 2020; Xu et al., 2020), anti-bacterial (Ji et al., 2020), anti-cancer (Wu C. et al., 2019; Ma et al., 2020; Rossi et al., 2020) and inhibition of oxidative stress(Lee et al., 2020). Emodin has also been shown to have potent anti-cardiovascular activities in vitro and in vivo, including anti-inflammatory (Wu et al., 2007), antioxidant damage (Du and Ko, 2005), inhibition of myocardial cell fibrosis (Evans et al., 2020) and other pharmacological effects. Based on these pharmacological effects, emodin has been shown to be functional in myocarditis, myocardial hypertrophy, ischemia-reperfusion injury, and myocardial infarction. This indicates that emodin plays an important role in the treatment of cardiovascular diseases (Shrimali et al., 2013). Numerous studies have also made huge strides in revealing the mechanisms behind these effects. In addition, comparable therapeutic effect and better assisting effect were also shown in the comparative study between emodin and conventional therapeutic drugs or target inhibitors (Du and Ko, 2005; Du and Ko, 2006; Liu et al., 2013; Evans et al., 2020). However, whether emodin has a specific therapeutic effect in cardiovascular diseases is still unclear, which limits its further research and application. In addition, although there is abundant evidence detailing the effects of emodin on various signaling pathways and cellular functions, the overall understanding of its mechanisms of action remains elusive. Therefore, the purpose of this paper is to provide an overview of emodin’s role in the fight against cardiovascular disease timely. The emphasis is to summarize the pharmacological effects of emodin as a possible anti-cardiovascular drug, as well as its targets and mechanisms of action.
Targets and Studies of Emodin Against Anti-Cardiovascular Disease
Emodin has been shown to be active in a variety of cardiovascular diseases in vivo and in vitro. And numerous studies have explored the underlying mechanisms of these effects (Table 1).
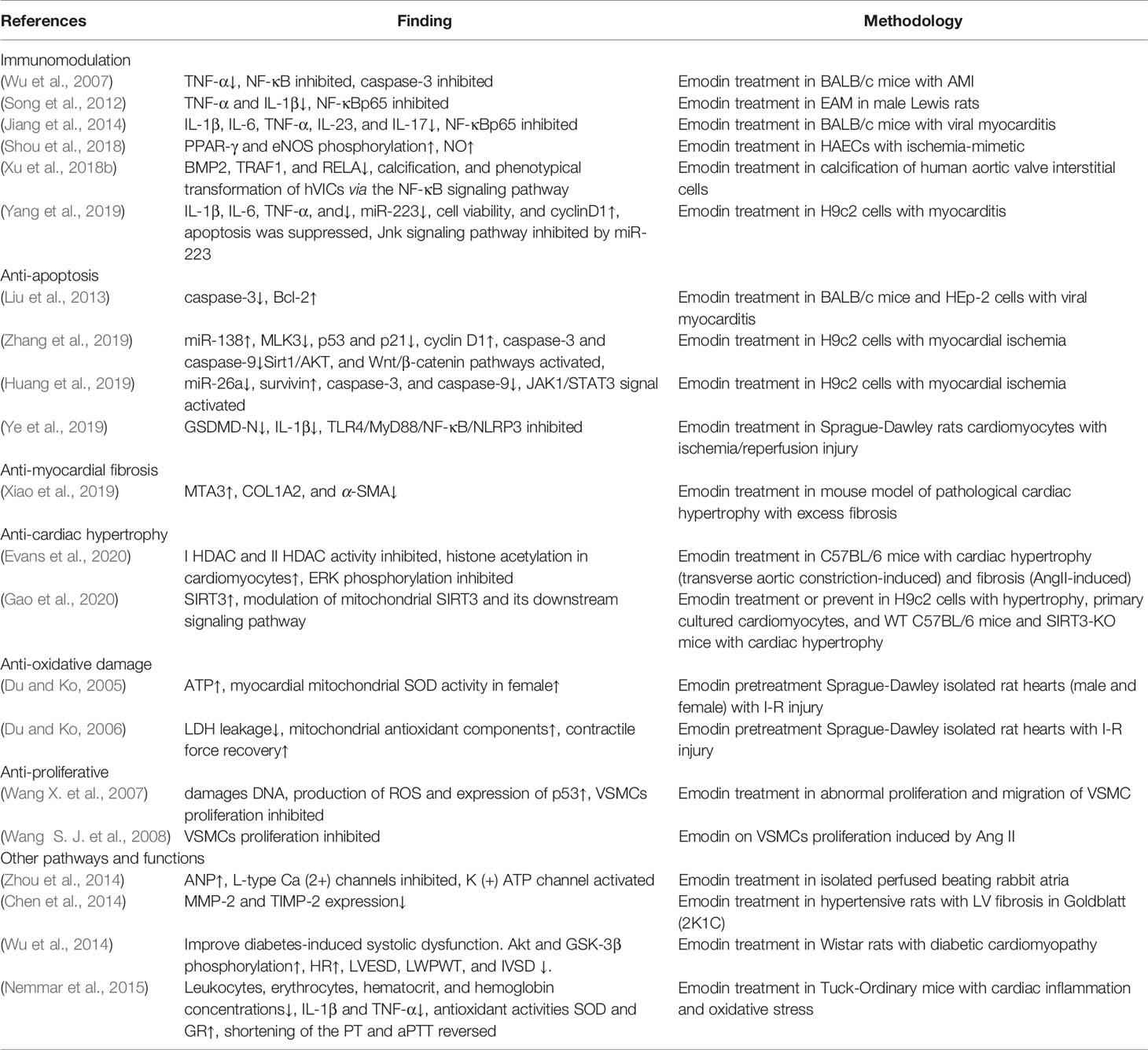
Table 1 Pharmacological effects of emodin in anti-cardiovascular diseases.
The results and potential mechanisms of cardiovascular protection are shown below.
Immunomodulation
NF-κB
Inflammation is closely related to the occurrence of cardiovascular events (Della Corte et al., 2016; Del Pinto and Ferri, 2018). Abnormal secretion and expression of inflammatory factors are essential for the injury and necrosis of cardiovascular cells (Ait-Oufella et al., 2019). Emodin has been found to have effective anti-inflammatory effects (Shrimali et al., 2013; Ding et al., 2020), and it also has the activity in cardiovascular diseases. It is reported that emodin can control the expression and secretion of inflammatory factors by inhibiting the transcription factor nuclear factor kappa-B (NF‐κB) in a variety of inflammatory diseases. For example, emodin (30 mg/kg) significantly inhibited the expression of tumor necrosis factor-α (TNF-α) and the activation of NF-κB in the local myocardial infarction area of acute myocardial infarction (AMI) model mice, protected myocardial cell injury by inhibiting local inflammation, reduced the range of myocardial infarction induced by occluding the left anterior descending coronary artery in BALB/c mice in a dose-dependent manner (Wu et al., 2007). Similarly, emodin can regulate the inflammatory process in experimental autoimmune myocarditis (EAM) (50 mg/kg) and viral myocarditis (VMC) (0.3 mg) model mice by inhibiting the transcription factor NF‐κB, reducing the production of pro-inflammatory cytokines, such as interleukin-1β (IL-1β), interleukin-6 (IL-6), TNF-α, and decreasing the severity of myocarditis (Song et al., 2012; Jiang et al., 2014). Moreover, in lipopolysaccharide (LPS)-induced H9c2 cells (5, 10, and 15 μM of emodin), it has a similar effect to myocarditis (Yang et al., 2019). Emodin (10 μM) can also inhibit TNF-α-induced calcification and phenotypical transformation of human aortic valve interstitial cells (hVICs) via the NF-κB signaling pathway, thereby preventing calcification events stimulated in acute inflammatory responses (Xu et al., 2018b).
PPAR-γ and eNOS
Abnormal expression of endothelial inflammatory cytokines and mediators is a critical factor leading to myocardial ischemia-reperfusion (I-R) injury (Sager and Nahrendorf, 2016). Peroxisome proliferator-activated receptor γ (PPAR-γ) is identified as a cardio-protective factor (Agrawal et al., 2014). I-R can inhibit the activation of PPAR-γ signaling pathway by reducing the phosphorylation of PPAR-protein (Xiong D. et al., 2016). PPAR-γ agonists can reduce I-R cell damage in multiple organs, including brain, liver, heart, and lung, and further activate endothelial nitric oxide synthase (eNOS), which plays a key role in maintaining and improving endothelial function (Kobayashi et al., 2008). In addition, eNOS inhibited the production of inflammatory cytokines by NO dependent mechanisms (Wang et al., 2017). Shou et al. (2018) established an in vitro hypoxia-reoxygenation model of human aortic endothelial cells (HAECs). The inactivation of PPAR-γ and eNOS signaling pathways is essential for the expression of inflammatory cytokines in endothelial cells induced by hypoxia-reoxygenation. The intervention of emodin reversed the levels of endothelial inflammatory cytokines and the expression of mediators in a concentration-dependent manner. Emodin recovered the PPAR-γ activity of hypoxia-reoxygenated endothelial cells by the molecular interaction between Heat Shock Proteins90 (HSP90) and eNOS. Therefore, it advances the production of NO, inhibits the inflammatory response, and has a protective effect on hypoxia-reoxygenation injury.
MiR-223
Studies have reported that MicroRNA (miR)-223 can be invoked as a new potential target for the diagnosis and treatment of inflammatory diseases, such as thrombophlebitis (Li H. et al., 2020), acute peritonitis (Brook et al., 2019), viral myocarditis (Gou et al., 2018), and acute lung injury (Feng et al., 2017). It can inhibit the inflammatory response and prevent the indirect damage in the process of infection (Yuan et al., 2018; Cavallari et al., 2019; Kim et al., 2019). Jun kinase enzyme (JNK) signaling regulates a wide range of cellular processes, including cell proliferation, differentiation, survival, apoptosis, and inflammation (Hammouda et al., 2020). (Yang et al. (2019) explored the effects of miR-223 and JNK signaling pathways on LPS-induced H9c2 cells, and found that LPS exposure significantly up-regulated the expression levels of miR-223 and JNK-related proteins (p-JNK and p-c-Jun), promoted apoptosis and the release of inflammatory factors IL-1β, IL-6, TNF-α. Furthermore, 20 μM of emodin intervention eliminated the upregulation. Emodin alleviated the LPS-induced myocarditis damage by inhibiting the over expression of miR-223. And by down-regulating miR-223 to inactivate the JNK signaling pathway. Therefore, miR-223 is considered as a possible target of emodin.
Anti-Apoptosis
Caspase-3 and Caspase-9
Effector caspase-3 and the initiator caspase-9 have been recorded as important agents of apoptosis (Cao et al., 2002; Yao et al., 2007). Emodin has been repeatedly reported to down-regulate caspase-3 and caspase-9 due to myocardial cell injury in vivo and in vitro (Wu et al., 2007; Yang et al., 2019). Liu et al. (2013) first reported that the apoptosis rate of hep-2 cells treated with emodin was significantly lower than that of the control group infected with the virus. The 50% effective concentration (EC50) of the pro-apoptotic gene caspase-3 in the heart tissue of mice with coxsackievirus B4 (CVB4)-induced viral myocarditis down-regulated by emodin was 12.06 μM, and the selectivity index (SI) was 5.08. At the same time, the apoptosis inhibitor gene bcl-2 was up-regulated to play an anti-apoptotic role. Furthermore, survival rate of mice or cells was improved, MTD was prolonged, and HW/BW, viral titer and myocardial pathological score caused by viral infection were decreased. Studies (Huang et al., 2019; Zhang et al., 2019) also showed that H9c2 cells with myocardial ischemia (hypoxia-induced) after treatment with emodin (15 μM or 20 μM) had the effect of down-regulating caspase-3 and caspase-9. Regarding the role of emodin, Leung et al. (2020) also reported that emodin can regulate the kinase (ERK)-1/2 signaling pathway in vivo and in vitro by activating extracellular signaling, increasing the expression of Bcl-2 and glutamate transporter-1 (GLT-l), and inhibiting the activated level of caspase-3.
MiR-138
MicroRNA (miRNA) is essential in a variety of biological processes, including cell differentiation and proliferation, apoptosis, and metabolism (Brennecke et al., 2003; Chen et al., 2004). MiRNA deregulation often disrupts critical cellular processes, leading to the onset and progression of various human diseases. The identified miRNA can target various target genes, and the target miRNA can also contain multiple miRNA binding sequences. Currently, it have shown hope of miRNA-based therapies in treating human diseases (Zhang Y. et al., 2018; Cheng et al., 2019; Wang et al., 2019; Pottoo et al., 2020; Wu et al., 2020). Among them, miR-138 has been reported to protect cardiomyocytes from hypoxia-induced apoptosis (He et al., 2013; Xiong H. et al., 2016). Emodin (10 μM) has been demonstrated to inhibit hypoxia-induced injury. And it can significantly reduce the decline in cell viability and apoptosis caused by hypoxia in a dose-dependent manner (Lei et al., 2014; Fan et al., 2018). Subsequently, it was reported that emodin can protect cardiomyocytes by regulating miRNA expression (Hua et al., 2015; Xiang et al., 2017). Overexpression of miR-138 enhanced the protective effect of emodin, while miR-138 silencing weakened the protective effect of emodin on hypoxia-induced injury, suggesting that emodin played a protective effect on cardiomyocytes by upregulation of miR-138 (Rukov and Shomron, 2011; Zhang et al., 2019). In addition, in hypoxia-induced damaged cells, the overexpression of Mixed Lineage Kinase 3 (MLK3), which is synergistic with miR-138, inactivates the Sirt1/AKT and Wnt/wp-catenin pathways (He et al., 2013). The activation of Sirt1/AKT and Wnt/w1-catenin pathways plays an important role in anti-apoptosis (Lehwald et al., 2011). Zhang et al. (2019) showed that emodin (15 and 20 μM) inhibited the hypoxia-induced damage of H9c2 cells by up-regulated miR-138 and activating Sirt1/AKT and Wnt/wp-catenin pathways. It suggested that emodin may promote hypoxia-induced cell viability and inhibit cell apoptosis by de-regulating the expression of miR-138.
MiR-26a
MiR-26a plays a crucial role in the regulation of cardiomyocytes (Liu et al., 2016). Huang et al. found that the up-regulation of miR-26a promoted the apoptosis of hypoxia-treated H9c2 cells, while emodin (15 and 20 μM) could negatively regulate the expression of miR-26a and reduce the apoptosis by regulating miR-26a (Huang et al., 2019). Meanwhile, survivin is a member of the inhibitor of apoptosis protein (IAPs) family. It promotes cell survival by interfering with various cycle-related proteins, such as inner centromere protein and Aurora B kinase (Cheung et al., 2011). In addition, the relationship between hypoxic treatment and survivin expression has also been found in previous studies (Zhu et al., 2001; Zhang et al., 2007). Huang et al. (2019) revealed that hypoxia decreased the accumulated level of survivin while emodin enhanced the expression of survivin, which provided a reason for emodin to inhibit cell apoptosis due to the anti-apoptosis effects of survival. It is observed that MiR-26a achieves its functions via targeting survivin.
STAT3
It has been reported that signal transducer and activator of transcription (STAT)3 is an anti-apoptotic gene (Gu et al., 2020; Turovskaya et al., 2020). The activation of STAT3 may prevent cardiomyocyte apoptosis and enhance regulatory cardiovascular protective genes in vivo and in vitro during hypoxia-reoxygenation and I-R therapy (Mascareno et al., 2005; Ke et al., 2019). In a study (Huang et al., 2019), emodin can activate the inactivated JAK1/STAT3 pathway after hypoxia. Further results indicated that emodin (at the concentration of 15 and 20 μM) increased the phosphorylation of pathway-related proteins and that the JNK/STAT3 pathway inhibitor AG490 could weaken the protective effect of emodin on hypoxia-induced damage. This indicates that emodin alleviates hypoxia-induced cell damage through activation of the JAK1/STAT3 signaling pathway.
GSDMD
Myocardial I-R injury activates apoptosis and is accompanied by an inflammatory response (Toldo et al., 2018; Wu M. Y. et al., 2018). The TLR4/MyD88/NF-κB/NLRP3 inflammasome pathway is essential for activation of inflammation. Toll-like receptors recruit myeloiddifferentiationfactor88 (MyD88), leading to activation of NF-κB (Ve et al., 2017). NF-κB increases synthesis of NLRP3, pro-IL-1β, and IL-1β (Afonina et al., 2017). Activated NLRP3 inflammasome cleaves pro-caspase-1 to produce activated caspase-1 (Ives et al., 2015). GSDMD-FL could be cleaved by activated caspase-1 (canonical inflammasomes) or activated caspase-11 (non-canonical inflammasomes) (Jorgensen and Miao, 2015). Then, the GSDMD-N forms a large pore in the plasma membrane (Sborgi et al., 2016) and controls IL-1β release (Shi et al., 2015). Subsequently, water enters into the cell, causing cell swelling and eventual lysis (Shi et al., 2017). Therefore, GSDMD-N is the executioner of pyroptotic cell death. It has been reported that I-R injury could activate pyroptosis, which were at least partially mediated by the TLR4/MyD88/NF-κB/NLRP3 inflammasome pathway by Ye et al. (2019). Emodin (10 µM) inhibits the expression of TLR4 by decreasing ROS production and reduces the expression of GSDMD-N by decreasing the expression of TLR4, MyD88, NF-κB, and the NLRP3 inflammasome.
Anti-Myocardial Fibrosis
MTA3
Cardiac fibrosis has been identified as the cause of heart failure (Jin et al., 2018). Emodin can improve multi-tissue fibrosis (Guan et al., 2016; Wang et al., 2016). Therefore, Xiao et al. (2019) evaluated the beneficial effects and potential mechanisms of emodin on cardiac fibrosis. Firstly, it was observed that emodin can reduce cardiac fibrosis by TAC-induced and cardiac fibroblast activation by AngII-induced. Then, it was found that the emodin/MTA3 axis play a crucial role in regulating cardiac fibrosis. This is consistent with the recognized role of MTA3 in regulating a variety of cellular events (Jiao et al., 2017). As a transcription factor, MTA3 becomes a cancer suppressor by inhibiting the proliferation, invasion, and migration of cancer cells (Ma et al., 2016). In addition, MTA3 also has anti-fibrosis effect (Qin et al., 2015). Xiao et al. (2019) further demonstrated that the upregulation of MTA3 may be molecular mechanisms of emodin inhibiting cardiac fibrosis.
Anti-Cardiac Hypertrophy
HDAC
Lysine acetylation on histone tails is a reversible epigenetic modification that is regulated by two enzymes: histone deacetylases (HDACs) and histone acetyltransferases (HATs) (Yang and Seto, 2007). It has been reported that inhibition of class I and class II HDAC can effectively treat pathological myocardial hypertrophy in animal models (Antos et al., 2003). The pan-HDAC inhibitor TSA increases the acetylation of histones and attenuates cardiac hypertrophy (Yoshimura et al., 1993; Nakagawa et al., 1995). Recently, it has been reported that emodin can inhibit HDAC activity in a test tube (Godoy et al., 2017). Levi W. Evans et al. (Evans et al., 2020) has reported that emodin (10 μM) normalized cardiac gene expression with changes similar to the well-known HDAC inhibitor TSA. Emodin has been shown to block pathological cardiac hypertrophy in vivo and in vitro, which is consistent with its role as an HDAC inhibitor.
SIRT3
SIRT3 is a member of the Class III HDACs family (Zheng et al., 2019). It has been shown to play a key role in maintaining normal mitochondrial biological function through reversible deacetylation of protein lysine. Inhibition of SIRT3 can lead to defects in mitochondrial function, leading to the development of various cardiovascular diseases. The activation of SIRT3 may represent a promising therapeutic strategy to improve mitochondrial function and treat related cardiovascular diseases (Mihanfar et al., 2018; Wu J. et al., 2019). Gao et al. (2020) extracted rhubarb ingredients and cardiac hypertrophy targets based on machine learning and the network pharmacology method, and the optimal component-target combination (emodin-SIRT3) was obtained and further functional analysis was carried out on the combination. The results showed that emodin could treat or prevent agonist-induced and pressure-overload mediated cardiac hypertrophy. This suggested that activation of SIRT3 may be potential mechanisms for emodin to prevent mitochondrial dysfunction.
Anti-Oxidative Damage
Emodin can act on the antioxidant system (Huang et al., 1991; Yi et al., 2004). Due to the ability of single electron transfer, emodin is considered as a generator of reactive oxygen species (Shi et al., 1994). Studies have showed that the formation of myocardial oxygen free radicals (oxygen free radicals) increased during I-R, and mitochondria were the main source of oxygen free radicals. Myocardial I-R injury was associated with decreased mitochondrial ATP production and antioxidant level/activity (Munro and Treberg, 2017; Ryoo and Kwak, 2018). The decrease of mitochondrial ATP production might be due to the oxidative damage of the protein complexes involved in the electron transfer process, while the consumption of non-enzymatic antioxidants, such as GSH and a-toc and the inhibition of antioxidant enzymes such as GRD and SOD will aggravate the oxidative damage of mitochondria (Yim et al., 1998). Du and Ko (2005) studied the sensitivity of emodin in a single dose treatment to mitochondrial ATP production and antioxidant composition in male and female rats and ischemia reperfusion (IR) injury. The results showed that a single dose of emodin can enhance the antioxidant components of mitochondria and prevent IR damage. Early experimental results have demonstrated the increased formation of oxygen-derived free radicals (oxy-radicals) in the myocardium during reperfusion after ischemia, wherein mitochondria are the major source of oxy-radical production. Pharmacological pretreatment of myocardium with low dose emodin chronic therapy can significantly enhance the antioxidant components of mitochondria. Du and Ko (2006) demonstrated that the combination of emodin in a low dose (25 μM/kg/day ×15) and IPC pretreatment produced a protective effect on the heart in a semi-additive manner. It protects the heart muscle from I-R damage.
Anti-Proliferation
Atherosclerosis is the main cause of ischemic heart disease, and percutaneous coronary intervention is the main method to treat coronary artery occlusion (Fischman et al., 1994; Serruys et al., 1994). However, progressive arterial restenosis caused by abnormal proliferation and migration of vascular smooth muscle cells (VSMC) impedes effective treatment (Hao et al., 2003). Therefore, the concern is inhibition of migration and proliferation of vascular smooth muscle cells. It has been reported that emodin can inhibit the proliferation of VSMC, thereby inhibiting the hyperplasia of the intima (Xu et al., 2018a). It is reported by Wang X. et al. (2007) that emodin in a dose-dependent induced apoptosis of hVSMC and two additional forms of injury: autophagy and senescence. It may be regulated by ROS and p53 pathways. Subsequently, Wang S. J. et al. (2008) also proved that emodin directly inhibited VSMCs proliferation induced by Ang II in a dose-dependent manner. And inhibiting the expression of PCNA and c-myc may contribute to its anti-proliferative activity. Further evidence suggested that the proto-oncogene c-myc plays a key role in the cardiac protective properties of emodin.
Other Functions
The cardiovascular protective effects of emodin are still under further exploration. Zhou et al. (2014) demonstrated that emodin (30 μM) increased ANP secretion via inhibition of L-type Ca(2+) channels through an activation of K(+)ATP channel in isolated beating rabbit atria. The results also provided role of emodin in the treatment of cardiovascular homeostasis. Wu et al. (2014) has reported that emodin may have great therapeutic potential for DCM in the treatment of diabetes-induced cardiomyopathy by Akt/gsk-3 signaling pathway. A study has demonstrated that emodin significantly prevented DEP induced cardiac inflammation and oxidative stress, and thrombotic complications in vivo and in vitro (Nemmar et al., 2015). This suggested that the use of functional food such as emodin can be considered a useful agent and may have the potential to protect people of all ages against the cardiovascular events in cities with high concentrations of particulate air pollution which was pending further studies.
Potential Applications in Cardiovascular Diseases Therapy
Emodin targets multiple features of cardiovascular diseases through a series of pathways and has a variety of targets. Antioxidant, anti-viral, and anti-fibrotic effects are similar to conventional therapeutic methods or target protein inhibitors. In addition, it also showed good synergistic therapeutic effect. All these make emodin have broad application prospects in the treatment of cardiovascular diseases (Table 2). Among them, emodin has antioxidant properties. Pretreatment in Sprague-Dawley isolated mouse hearts (male and female) with I-R injury improved their antioxidant capacity, which was similar to the antioxidant a-tocopherol (a-TOC) and a-lipoic acid (a-LA) (Du and Ko, 2005). Emodin antiviral, reported by Liu et al. (2013) in BALB/c mice with viral myocarditis (CVB4), 15 mg/kg/d emodin treatment was equivalent to 10 mg/kg/d ribavirin can reduce viral cardiomyopathy. Antifibrosis role in hypertensive rats with LV fibrosis in Goldblatt (2K1C), Emodin, irbesartan or two drugs together can potentially inhibit the ventricular fibrosis in Goldblatt hypertensive rats. Furthermore, the combination of these two drugs may provide a better anti-fibrosis effect than the single application (Chen et al., 2014). In neonatal rat ventricular myocytes (NRVMs) with myocardial hypertrophy (phenylephrine induced), emodin blocks pathological cardiac hypertrophy and reverses stress-induced transcriptome changes in cardiomyocytes, consistent with HDAC inhibitor TSA (Evans et al., 2020). Emodin has a protective effect on myocardial I-R injury. In Sprague-Dawley isolated rat hearts with I-R injury, the protective effect of emodin pretreatment on myocardial I-R injury is consistent with that of ischemic preconditioning (IPC), and the protective effect of emodin and IPC combined pretreatment on myocardium is semi-additive (Du and Ko, 2006).
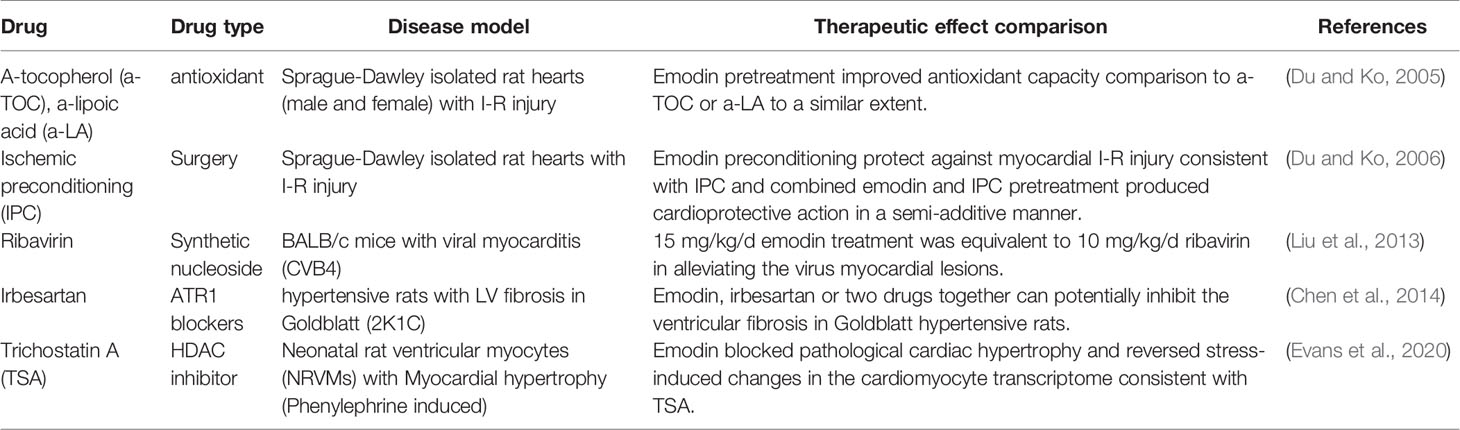
Table 2 Emodin and conventional therapeutic methods against cardiovascular disease.
Toxicity
Although emodin has beneficial pharmacology activities, it has been found that hepatotoxicity, nephrotoxicity, and reproductive toxicity limit its further study. First, it was found in studies that emodin has hepatotoxic effects (Zhang M. et al., 2018). Chen et al. (2020b) found that long-term or high-dose emodin showed a cytotoxic effect in hepatocytes and inhibited hepatocyte nuclear factor 4α expression, thereby reducing UDP-glucuronosyltransferase 2B7 (UGT2B7) expression and causing hepatotoxicity. Wu L. et al. (2018) also showed that there were gender differences in the hepatotoxicity and toxicology of emodin, which may be mediated by the coupling of UGT2B7 and multidrug-resistant-protein 2 in vivo. Second, with regard to the nephrotoxicity of emodin, the results of the United States National Toxicology Program showed that mice taking emodin at 22 mg·kg−1 were associated with tubular injury (increased incidence of tubular pigmentation) and increased incidence of kidney disease in female mice (National Toxicology Program, 2001). After 24 h of emodin was applied (40 and 80 µm), cell viability was significantly reduced by approximately 29% and 45%, respectively, which was mediated by the induction of apoptosis in the caspase 3‐dependent pathway and the mitochondrial pathway (Wang C. et al., 2007, Wang et al., 2015). Wang C. et al. (2008) have shown that emodin has cytotoxic effects on HK-2 cells, partially via induction of cathepsin B (CB) protein and activation of CB protease. And then, the CK2α’ knockout mice were given emodin orally for 5 days (1000 mg/kg/day), and it was found that emodin had testosterone toxicity due to disruption of testicular gene expression (Oshida et al., 2011). The different doses of emodin (25–400 μM) were applied to ejaculated human sperm, showed that sperm functions were inhibited by reducing sperm [Ca(2+)]i and prohibiting tyrosine phosphorylation in vitro (Luo et al., 2015). In addition, it was found that 24 h emodin (100 μM) interrupted several metabolites and biological processes including amino acid metabolism, purine metabolism, and Krebs cycle disruption (Chen et al., 2018). Since the ROS scavenging agent NAC can counteract the toxic effect of emodin, the increase in mitochondrial reactive oxygen production was also considered as an aspect of emodin toxicity (Kolitsida and Abeliovich, 2017).
Pharmacokinetics
Emodin has a wide range of biological activities, but its bioavailability is low when taken orally (Zhang M. et al. (2018). Shia et al. (2010) reported that after intragastric administration at doses of 20 or 40 mg/kg, emodin rapidly underwent phase II metabolism to form its glucuronide, and the parent form of emodin was almost undetectable in vivo. Even at higher doses (82.4 mg/kg), the levels of emodin detected in vivo remained very low. The appearance of glucuronidation metabolism and oxidative conversion into rhein may be the main reasons for the extremely poor oral bioavailability of emodin (Liu et al., 2012; Li et al., 2017; Cheng et al., 2020). Li et al. (2019) revealed that the absorption of emodin was faster, and the effect was rapider when used in combination with aloe-emodin, rhein, chrysophanol, or physcion. Trans-2,3,5,4′-tetrahydroxystilbene-2-O-β-D-glucopyranoside (TSG) can promote the absorption of emodin and increase its plasma concentration to enhance the efficacy of emodin (Xing et al., 2019). A similar study revealed that piperine significantly increased the Cmax and area under concentration‐time curve (AUC) of emodin and decreased the Cmax and AUC of emodin glucuronide (Di et al., 2015). Therefore, the application of emodin is greatly limited, because of its poor intestinal absorption, rapid elimination and low bioavailability in vivo after oral administration. In future studies, it is important and urgent to improve oral bioavailability in clinical practice.
Emodin Derivatives
In order to apply emodin safely and effectively, the research on emodin derivatives has been carried out continuously. Chen et al. (2016) reported that the emodin derivatives, madagascine, has been ameliorated vasospasm related cardiovascular diseases. It exerted vasodilatation effect through activating adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK), leading to the activation of eNOS in endothelium and inhibition of rho-associated protein kinase/myosine phosphatase target subunit 1(ROCK/MYPT1) in vascular smooth muscle (VSM). Furthermore, under the same experimental conditions, compared with emodin, it has higher biological activity and safety. Huang et al. (2014) provided evidence that PPemd26, an anthraquinone derivative, suppressed angiogenesis through inhibiting the vascular Endothelial Growth Factor Receptor 2 (VEGFR2) signaling pathway, suggesting that it is a potential drug candidate for the development of anti-angiogenic agents for angiogenesis-related diseases, including coronary heart disease. Ji et al. (2020) reported that haloemodin had strong antibacterial activity against Gram-positive bacterial strains. Compared with emodin, haloemodin more rapidly increases the bacterial plasma membrane permeability for potassium ions. More importantly, compared with emodin, halomodin binds to protein tighter and more stable. The results partially explain the stronger antibacterial activities of haloemodin over their parent nucleus, emodin. Chen et al. (2020a) disclosed that Aloe‐Emodin (AE) could alleviate high-fat diet/palmitic acid-induced cardiac inflammation via inhibition of the TLR4/NF-κB signaling pathway. Thus, AE may be a promising therapeutic strategy to prevent myocardial injury κB and p-P65l caused by hyperlipidemia. Bai’s findings suggested a novel pharmacological role of AE in high-fat diet-induced cardiac electrical remodelling (Bai et al., 2017). Yu et al. (2019) findings indicated that AE can protect against myocardial infarction via the upregulation of miR-133, inhibition of ROS production and suppression of caspase-3 apoptotic signaling pathway. Zhang et al. (2020) results revealed that inhibiting the activation of NLRP3 inflammasome and reducing the release of HMGB1 by promoting NLRP3 ubiquitination, thereby restoring the endothelial tight junction proteins and permeability, which indicated that AE exhibited immense potential therapeutic value in hypertension-related cardiovascular disease and the development of innovative drugs (Table 3).
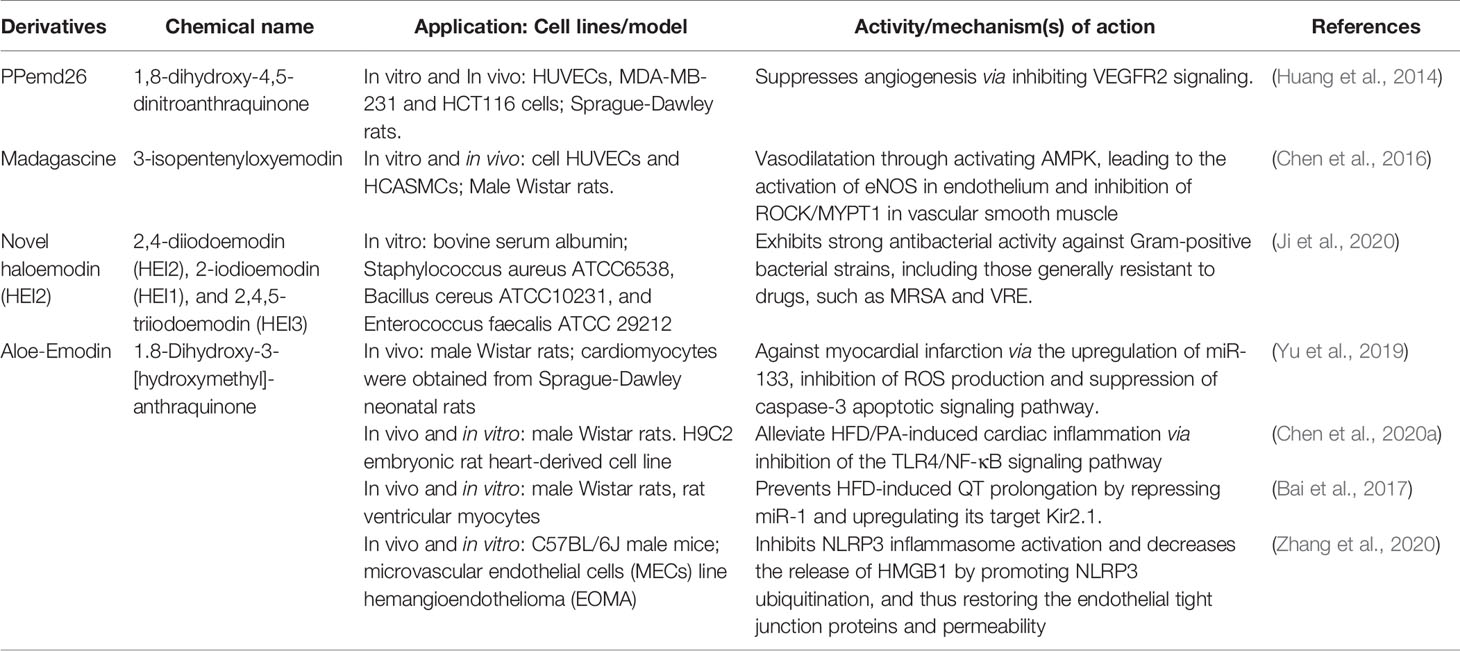
Table 3 Anti-cardiovascular effects of emodin derivatives.
Summary and Outlooks
In summary, it is reported that emodin has a wide range of anti-cardiovascular activities. It is involved in multiple molecular targets, such as inflammatory regulation, anti-apoptosis, anti-hypertrophy, anti-myocardial fibrosis, inhibition of oxidative damage, and abnormal and excessive proliferation of smooth muscle cells. It showed a good therapeutic prospect in the treatment of myocardial hypertrophy, coronary heart disease, and myocardial ischemia/reperfusion injury. Numerous studies have also made huge strides in revealing the mechanisms behind these effects. In addition, similar therapeutic effects and better synergistic effects were also shown in studies comparing emodin with conventional therapeutic methods or target protein inhibitors. However, despite the encouraging results in this research field, the current research only exists in the animal research stage, and a systematic and overall understanding of its mechanisms of action has not yet been formed. More importantly, attentions should be paid to the toxicity of emodin due to the limitation of its use caused by its increased content and low bioavailability. They are the most important factor limiting in the future research. With the deepening of research on emodin, its toxicity has been continuously discovered, and it is necessary to search for more bioavailability and toxicity neutralization solutions.
Author Contributions
Authors’ contributions were as follows: conception and design of the study (QL, JG, YW); combing the literature (QL, XP); drafting the article (QL); revisions for content (QL, AC); and critical revisions for important intellectual content (QL, YW). All authors contributed to the article and approved the submitted version.
Funding
We acknowledge the financial supports from the National Natural Science Foundation of China (81573832), National Key Research and Development Program of China [2017YFC1700106], and Double First-Class Personnel Office-High Level Scientific Research Team Research Fund (1000061020100, 1000061223245).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AMI, acute myocardial infarction; BMP, bone morphogenetic protein; AngII, angiotensin II; ANP, atrial natriuretic peptide; CVB3, coxsackievirus B3; CVB4, coxsackievirus B4; EAM, experimental autoimmune myocarditis; eNOS, endothelial nitric oxide synthase; I-R, ischemia/reperfusion; IL-1β, interleukin-1β; IL-6, interleukin-6; IL-23, interleukin-23; IL-17, interleukin-17; ISO, isoproterenol; GSDMD, gasdermin D; GLT-l, glutamate transporter-1; HATs, histone acetyltransferases; HAECs, human aortic endothelial cells; HDACs, histone deacetylases; hVICs, human aortic valve interstitial cells; HSP, heat shock protein; JNK, jun kinase enzyme; MTA3, metastasis-associated protein 3; miR-223, microRNA-223; miR-138, microRNA-138; miR-26a, microRNA-26a; NO, nitric oxide; NF-kB, nuclear factor kappa-B; PPAR-γ, peroxisome proliferator-activated receptor-γ; MyD88, myeloiddifferentiationfactor88; TNF-α, tumor necrosis factor-a; TAC, transaortic constriction; VMC, viral myocarditis; VSMC, vascular smooth muscle cells; α-SMA, alpha-smooth muscle actin; PPAR-γ, peroxisome proliferator-activated receptor γ; SOD, superoxide dismutase; STZ, streptozotocin; STAT, signal transducer and activator of transcription; HSP90, heat shock proteins90; MLK3, mixed lineage kinase 3; AMP, adenosine 5′-monophosphate; AMPK, activated protein kinase; VEGFR2, vascular endothelial growth factor receptor 2; AE, aloe–emodin.
References
Afonina, I., Zhong, Z., Karin, M., Beyaert, R. (2017). Limiting inflammation-the negative regulation of NF-κB and the NLRP3 inflammasome. Nat. Immunol. 18 (8), 861–869. doi: 10.1038/ni.3772
Agrawal, Y. O., Sharma, P. K., Shrivastava, B., Ojha, S., Upadhya, H. M., Arya, D. S., et al. (2014). Hesperidin produces cardioprotective activity via PPAR-γ pathway in ischemic heart disease model in diabetic rats. PloS One 9 (11), e111212–e111212. doi: 10.1371/journal.pone.0111212
Ait-Oufella, H., Libby, P., Tedgui, A. (2019). Anticytokine Immune Therapy and Atherothrombotic Cardiovascular Risk. Arterioscler. Thromb. Vasc. Biol. 39 (8), 1510–1519. doi: 10.1161/atvbaha.119.311998
Antos, C., McKinsey, T., Dreitz, M., Hollingsworth, L., Zhang, C., Schreiber, K., et al. (2003). Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J. Biol. Chem. 278 (31), 28930–28937. doi: 10.1074/jbc.M303113200
Bai, Y., Su, Z., Sun, H., Zhao, W., Chen, X., Hang, P., et al. (2017). Aloe-Emodin Relieves High-Fat Diet Induced QT Prolongation via MiR-1 Inhibition and IK1 Up-Regulation in Rats. Cell Physiol. Biochem. 43 (5), 1961–1973. doi: 10.1159/000484120
Brennecke, J., Hipfner, D., Stark, A., Russell, R., Cohen, S. (2003). bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 113 (1), 25–36. doi: 10.1016/s0092-8674(03)00231-9
Brook, A., Jenkins, R., Clayton, A., Kift-Morgan, A., Raby, A., Shephard, A., et al. (2019). Neutrophil-derived miR-223 as local biomarker of bacterial peritonitis. Sci. Rep. 9 (1), 10136. doi: 10.1038/s41598-019-46585-y
Cao, G., Luo, Y., Nagayama, T., Pei, W., Stetler, R., Graham, S., et al. (2002). Cloning and characterization of rat caspase-9: implications for a role in mediating caspase-3 activation and hippocampal cell death after transient cerebral ischemia. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 22 (5), 534–546. doi: 10.1097/00004647-200205000-00005
Cavallari, C., Dellepiane, S., Fonsato, V., Medica, D., Marengo, M., Migliori, M., et al. (2019). Online Hemodiafiltration Inhibits Inflammation-Related Endothelial Dysfunction and Vascular Calcification of Uremic Patients Modulating miR-223 Expression in Plasma Extracellular Vesicles. J. Immunol. (Balt. Md. 1950) 202 (8), 2372–2383. doi: 10.4049/jimmunol.1800747
Chen, C. Z., Li, L., Lodish, H. F., Bartel, D. P. (2004). MicroRNAs modulate hematopoietic lineage differentiation. Science 303 (5654), 83–86. doi: 10.1126/science.1091903
Chen, Q., Pang, L., Huang, S., Lei, W., Huang, D. (2014). Effects of emodin and irbesartan on ventricular fibrosis in Goldblatt hypertensive rats. Die Pharm. 69 (5), 374–378. doi: 10.1691/ph.2014.3816
Chen, D., Lv, B., Kobayashi, S., Xiong, Y., Sun, P., Lin, Y., et al. (2016). Madagascine Induces Vasodilatation via Activation of AMPK. Front. Pharmacol. 7, 435. doi: 10.3389/fphar.2016.00435
Chen, C., Gao, J., Wang, T.-S., Guo, C., Yan, Y.-J., Mao, C.-Y., et al. (2018). NMR-based Metabolomic Techniques Identify the Toxicity of Emodin in HepG2 Cells. Sci. Rep. 8 (1), 9379–9379. doi: 10.1038/s41598-018-27359-4
Chen, Y., Feng, B., Yuan, Y., Hu, J., Zhao, W., Jiang, H., et al. (2020a). Aloe Emodin Reduces Cardiac Inflammation Induced by a High-Fat Diet through the TLR4 Signaling Pathway. Mediators Inflam. 2020, 6318520–6318520. doi: 10.1155/2020/6318520
Chen, Y., Zhang, T., Wu, L., Huang, Y., Mao, Z., Zhan, Z., et al. (2020b). Metabolism and Toxicity of Emodin: Genome-Wide Association Studies Reveal Hepatocyte Nuclear Factor 4α Regulates UGT2B7 and Emodin Glucuronidation. Chem. Res. Toxicol. doi: 10.1021/acs.chemrestox.0c00047.
Cheng, X., Wang, Y., Du, L. (2019). Epigenetic Modulation in the Initiation and Progression of Pulmonary Hypertension. Hypertension (Dallas Tex. 1979) 74 (4), 733–739. doi: 10.1161/hypertensionaha.119.13458
Cheng, W., Li, Y., Yang, W., Wu, S., Wei, M., Gao, Y., et al. (2020). Simultaneous Determination of 13 Constituents of Radix Polygoni Multiflori in Rat Plasma and Its Application in a Pharmacokinetic Study. Int. J. Analyt Chem. 2020, 4508374. doi: 10.1155/2020/4508374
Cheung, C., Cheng, L., Chang, K., Chen, H., Chang, J. (2011). Investigations of survivin: the past, present and future. Front. Biosci. (Landmark Ed) 16, 952–961. doi: 10.2741/3728
Del Pinto, R., Ferri, C. (2018). Inflammation-Accelerated Senescence and the Cardiovascular System: Mechanisms and Perspectives. Int. J. Mol. Sci. 19 (12), 3701. doi: 10.3390/ijms19123701
Della Corte, V., Tuttolomondo, A., Pecoraro, R., Di Raimondo, D., Vassallo, V., Pinto, A. (2016). Inflammation, Endothelial Dysfunction and Arterial Stiffness as Therapeutic Targets in Cardiovascular Medicine. Curr. Pharm. Des. 22 (30), 4658–4668. doi: 10.2174/1381612822666160510124801
Di, X., Wang, X., Di, X., Liu, Y. (2015). Effect of piperine on the bioavailability and pharmacokinetics of emodin in rats. J. Pharm. BioMed. Anal. 115, 144–149. doi: 10.1016/j.jpba.2015.06.027
Ding, Y., Xu, J., Cheng, L., Huang, Y., Wang, Y., Li, H., et al. (2020). Effect of Emodin on Coxsackievirus B3m mediated-Encephalitis in Hand, Foot and Mouth Disease by inhibiting Toll-Like Receptor 3 pathway in vitro and in vivo. J. Infect. Dis. 222 (3), 443–455. doi: 10.1093/infdis/jiaa093.
Dong, X., Fu, J., Yin, X., Cao, S., Li, X., Lin, L., et al. (2016). Emodin: A Review of its Pharmacology, Toxicity and Pharmacokinetics. Phytother. Res. 30 (8), 1207–1218. doi: 10.1002/ptr.5631
Du, Y., Ko, K. (2005). Effects of emodin treatment on mitochondrial ATP generation capacity and antioxidant components as well as susceptibility to ischemia-reperfusion injury in rat hearts: single versus multiple doses and gender difference. Life Sci. 77 (22), 2770–2782. doi: 10.1016/j.lfs.2005.03.027
Du, Y., Ko, K. M. (2006). Effects of pharmacological preconditioning by emodin/oleanolic acid treatment and/or ischemic preconditioning on mitochondrial antioxidant components as well as the susceptibility to ischemia-reperfusion injury in rat hearts. Mol. Cell. Biochem. 288 (1-2), 135–142. doi: 10.1007/s11010-006-9129-3
Evans, L., Bender, A., Burnett, L., Godoy, L., Shen, Y., Staten, D., et al. (2020). Emodin and emodin-rich rhubarb inhibits histone deacetylase (HDAC) activity and cardiac myocyte hypertrophy. J. Nutr. Biochem. 79, 108339. doi: 10.1016/j.jnutbio.2019.108339
Fan, L., Zhang, H., Li, X., Yang, G., Ru, J., Liu, T. (2018). Emodin protects hyperglycemia-induced injury in PC-12cells by up-regulation of miR-9. Mol. Cell Endocrinol. 474, 194–200. doi: 10.1016/j.mce.2018.03.009
Feng, Z., Qi, S., Zhang, Y., Qi, Z., Yan, L., Zhou, J., et al. (2017). Ly6G+ neutrophil-derived miR-223 inhibits the NLRP3 inflammasome in mitochondrial DAMP-induced acute lung injury. Cell Death Dis. 8 (11), e3170. doi: 10.1038/cddis.2017.549
Fischman, D. L., Leon, M. B., Baim, D. S., Schatz, R. A., Savage, M. P., Penn, I., et al. (1994). A randomized comparison of coronary-stent placement and balloon angioplasty in the treatment of coronary artery disease. Stent Restenosis Study Investigators. N. Engl. J. Med. 331 (8), 496–501. doi: 10.1056/nejm199408253310802
Gao, J., Zhang, K., Wang, Y., Guo, R., Liu, H., Jia, C., et al. (2020). A machine learning-driven study indicates emodin improves cardiac hypertrophy by modulation of mitochondrial SIRT3 signaling. Pharmacol. Res. 155, 104739. doi: 10.1016/j.phrs.2020.104739
Godoy, L., Lucas, J., Bender, A., Romanick, S., Ferguson, B. (2017). Targeting the epigenome: Screening bioactive compounds that regulate histone deacetylase activity. Mol. Nutr. Food Res. 61 (4). doi: 10.1002/mnfr.201600744
Gou, W., Zhang, Z., Yang, C., Li, Y. (2018). MiR-223/Pknox1 axis protects mice from CVB3-induced viral myocarditis by modulating macrophage polarization. Exp. Cell Res. 366 (1), 41–48. doi: 10.1016/j.yexcr.2018.03.004
Gu, M., Liu, J., Shi, N. N., Li, X. D., Huang, Z. D., Wu, J. K., et al. (2020). [Analysis of property and efficacy of traditional Chinese medicine in staging revention and treatment of coronavirus disease 2019]. China J. Chin. Mater. Med. 45 (6), 1253–1258. doi: 10.19540/j.cnki.cjcmm.20200225.501
Guan, R., Wang, X., Zhao, X., Song, N., Zhu, J., Wang, J., et al. (2016). Emodin ameliorates bleomycin-induced pulmonary fibrosis in rats by suppressing epithelial-mesenchymal transition and fibroblast activation. Sci. Rep. 6, 35696. doi: 10.1038/srep35696
Hammouda, M., Ford, A., Liu, Y., Zhang, J. (2020). The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer. Cells 9 (4), 857. doi: 10.3390/cells9040857
Hao, H., Gabbiani, G., Bochaton-Piallat, M. L. (2003). Arterial smooth muscle cell heterogeneity: implications for atherosclerosis and restenosis development. Arterioscler. Thromb. Vasc. Biol. 23 (9), 1510–1520. doi: 10.1161/01.Atv.0000090130.85752.Ed
He, S., Liu, P., Jian, Z., Li, J., Zhu, Y., Feng, Z., et al. (2013). miR-138 protects cardiomyocytes from hypoxia-induced apoptosis via MLK3/JNK/c-jun pathway. Biochem. Biophys. Res. Commun. 441 (4), 763–769. doi: 10.1016/j.bbrc.2013.10.151
Hua, J.-y., He, Y.-z., Xu, Y., Jiang, X.-h., Ye, W., Pan, Z.-m. (2015). Emodin prevents intima thickness via Wnt4/Dvl-1/β-catenin signaling pathway mediated by miR-126 in balloon-injured carotid artery rats. Exp. Mol. Med. 47 (6), e170–e170. doi: 10.1038/emm.2015.36
Huang, H. C., Chu, S. H., Chao, P. D. (1991). Vasorelaxants from Chinese herbs, emodin and scoparone, possess immunosuppressive properties. Eur. J. Pharmacol. 198 (2-3), 211–213. doi: 10.1016/0014-2999(91)90624-y
Huang, S. W., Lien, J. C., Kuo, S. C., Huang, T. F. (2014). PPemd26, an anthraquinone derivative, suppresses angiogenesis via inhibiting VEGFR2 signalling. Br. J. Pharmacol. 171 (24), 5728–5742. doi: 10.1111/bph.12872
Huang, J. C., Li, X. B., Liu, P. J., Wang, J., Li, H. Y. (2019). Emodin protects H9c2 cells against hypoxia-induced injury via regulation of miR-26a/survivin and the JAK1/STAT3 pathway. J. Cell. Biochem. 120 (7), 11081–11090. doi: 10.1002/jcb.28385
Ives, A., Nomura, J., Martinon, F., Roger, T., LeRoy, D., Miner, J., et al. (2015). Xanthine oxidoreductase regulates macrophage IL1β secretion upon NLRP3 inflammasome activation. Nat. Commun. 6, 6555. doi: 10.1038/ncomms7555
Ji, C., Xin, G., Duan, F., Huang, W., Tan, T. (2020). Study on the antibacterial activities of emodin derivatives against clinical drug-resistant bacterial strains and their interaction with proteins. Ann. Trans. Med. 8 (4), 92–92. doi: 10.21037/atm.2019.12.100
Jiang, N., Liao, W., Kuang, X. (2014). Effects of emodin on IL-23/IL-17 inflammatory axis, Th17 cells and viral replication in mice with viral myocarditis. Nan Fang Yi Ke Da Xue Xue Bao J. South. Med. Univ. 34 (3), 373–378.
Jiao, T., Li, Y., Gao, T., Zhang, Y., Feng, M., Liu, M., et al. (2017). MTA3 regulates malignant progression of colorectal cancer through Wnt signaling pathway. Tumour Biol. J. Int. Soc. Oncodevelop. Biol. Med. 39 (3):1010428317695027. doi: 10.1177/1010428317695027
Jin, L., Sun, S., Ryu, Y., Piao, Z., Liu, B., Choi, S., et al. (2018). Gallic acid improves cardiac dysfunction and fibrosis in pressure overload-induced heart failure. Sci. Rep. 8 (1), 9302. doi: 10.1038/s41598-018-27599-4
Jorgensen, I., Miao, E. (2015). Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 265 (1), 130–142. doi: 10.1111/imr.12287
Ke, M., Tang, Q., Pan, Z., Yin, Y., Zhang, L., Wen, K. (2019). Sphingosine-1-phosphate attenuates hypoxia/reoxygenation-induced cardiomyocyte injury via a mitochondrial pathway. Biochem. Biophys. Res. Commun. 510 (1), 142–148. doi: 10.1016/j.bbrc.2019.01.067
Kim, G., Ng, H., Patel, N., Mahabeleshwar, G. (2019). Kruppel-like factor 6 and miR-223 signaling axis regulates macrophage-mediated inflammation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 33 (10), 10902–10915. doi: 10.1096/fj.201900867RR
Kobayashi, N., Ohno, T., Yoshida, K., Fukushima, H., Mamada, Y., Nomura, M., et al. (2008). Cardioprotective mechanism of telmisartan via PPAR-gamma-eNOS pathway in dahl salt-sensitive hypertensive rats. Am. J. Hypertens. 21 (5), 576–581. doi: 10.1038/ajh.2008.27
Kolitsida, P., Abeliovich, H. (2017). Selective emodin toxicity in cancer cells. Oncotarget 8 (23), 36932–36933. doi: 10.18632/oncotarget.16611
Lee, E., Baek, S., Park, J., Kim, Y. (2020). Rheum undulatumEmodin in inhibits oxidative stress in the liver via AMPK with Hippo/Yap signalling pathway. Pharm. Biol. 58 (1), 333–341. doi: 10.1080/13880209.2020.1750658
Lehwald, N., Tao, G., Jang, K., Sorkin, M., Knoefel, W., Sylvester, K. (2011). Wnt-β-catenin signaling protects against hepatic ischemia and reperfusion injury in mice. Gastroenterology 141 (2), 718.e701–705. doi: 10.1053/j.gastro.2011.04.051
Lei, Q., Qiang, F., Chao, D., Di, W., Guoqian, Z., Bo, Y., et al. (2014). Amelioration of hypoxia and LPS-induced intestinal epithelial barrier dysfunction by emodin through the suppression of the NF-kappaB and HIF-1alpha signaling pathways. Int. J. Mol. Med. 34 (6), 1629–1639. doi: 10.3892/ijmm.2014.1965
Leung, S., Lai, J., Wu, J., Tsai, Y., Chen, Y., Kang, S., et al. (2020). Neuroprotective Effects of Emodin against Ischemia/Reperfusion Injury through Activating ERK-1/2 Signaling Pathway. Int. J. Mol. Sci. 21 (8), 2899. doi: 10.3390/ijms21082899
Li, P., Lu, Q., Jiang, W., Pei, X., Sun, Y., Hao, H., et al. (2017). Pharmacokinetics and pharmacodynamics of rhubarb anthraquinones extract in normal and disease rats. Biomed. Pharmacother. 91, 425–435. doi: 10.1016/j.biopha.2017.04.109
Li, R.-R., Liu, X.-F., Feng, S.-X., Shu, S.-N., Wang, P.-Y., Zhang, N., et al. (2019). Pharmacodynamics of Five Anthraquinones (Aloe-emodin, Emodin, Rhein, Chysophanol, and Physcion) and Reciprocal Pharmacokinetic Interaction in Rats with Cerebral Ischemia. Mol. (Basel Switzerland) 24 (10), 1898. doi: 10.3390/molecules24101898
Li, H., Liu, Y., Shang, X., Li, Y., Feng, L., Qi, D. (2020). Effect of miR-223 on thrombophlebitis rats through regulating Toll-like receptor signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 24 (4), 2020–2027. doi: 10.26355/eurrev_202002_20380
Li, X., Shan, C., Wu, Z., Yu, H., Yang, A., Tan, B. (2020). Correction to: Emodin alleviated pulmonary inflammation in rats with LPS-induced acute lung injury through inhibiting the mTOR/HIF-1α/VEGF signaling pathway. Inflammation Res. 69 (7), 711. doi: 10.1007/s00011-020-01344-y
Liu, W., Feng, Q., Li, Y., Ye, L., Hu, M., Liu, Z. (2012). Coupling of UDP-glucuronosyltransferases and multidrug resistance-associated proteins is responsible for the intestinal disposition and poor bioavailability of emodin. Toxicol. Appl. Pharmacol. 265 (3), 316–324. doi: 10.1016/j.taap.2012.08.032
Liu, Z., Wei, F., Chen, L., Xiong, H., Liu, Y., Luo, F., et al. (2013). In vitro and in vivo studies of the inhibitory effects of emodin isolated from Polygonum cuspidatum on Coxsakievirus B4. Mol. (Basel Switzerland) 18 (10), 11842–11858. doi: 10.3390/molecules181011842
Liu, Y., Wang, Z., Xiao, W. (2016). MicroRNA-26a protects against cardiac hypertrophy via inhibiting GATA4 in rat model and cultured cardiomyocytes. Mol. Med. Rep. 14 (3), 2860–2866. doi: 10.3892/mmr.2016.5574
Luo, T., Li, N., He, Y. Q., Weng, S. Q., Wang, T., Zou, Q. X., et al. (2015). Emodin inhibits human sperm functions by reducing sperm [Ca(2+)]i and tyrosine phosphorylation. Reprod. Toxicol. 51, 14–21. doi: 10.1016/j.reprotox.2014.11.007
Ma, L., Yao, Z., Deng, W., Zhang, D., Zhang, H. (2016). The Many Faces of MTA3 Protein in Normal Development and Cancers. Curr. Protein Pept. Sci. 17 (8), 726–734. doi: 10.2174/1389203717666160401150122
Ma, W., Liu, F., Yuan, L., Zhao, C., Chen, C. (2020). Emodin and AZT synergistically inhibit the proliferation and induce the apoptosis of leukemia K562 cells through the EGR1 and the Wnt/β−catenin pathway. Oncol. Rep. 43 (1), 260–269. doi: 10.3892/or.2019.7408
Mascareno, E., Beckles, D., Siddiqui, M. (2005). Janus kinase-2 signaling mediates apoptosis in rat cardiomyocytes. Vasc. Pharmacol. 43 (5), 327–335. doi: 10.1016/j.vph.2005.08.023
Mihanfar, A., Nejabati, H., Fattahi, A., Latifi, Z., Faridvand, Y., Pezeshkian, M., et al. (2018). SIRT3-mediated cardiac remodeling/repair following myocardial infarction. Biomed. Pharmacother. 108, 367–373. doi: 10.1016/j.biopha.2018.09.079
Monisha, B. A., Kumar, N., Tiku, A. B. (2016). Emodin and Its Role in Chronic Diseases. Adv. Exp. Med. Biol. 928, 47–73. doi: 10.1007/978-3-319-41334-1_3
Munro, D., Treberg, J. (2017). A radical shift in perspective: mitochondria as regulators of reactive oxygen species. J. Exp. Biol. 220, 1170–1180. doi: 10.1242/jeb.132142
Nakagawa, O., Itoh, H., Harada, M., Komatsu, Y., Yoshimasa, T., Nakao, K. (1995). Gene regulation of brain natriuretic peptide in cardiocyte hypertrophy by alpha1-adrenergic stimulation. Clin. Exp. Pharmacol. Physiol. Supplement. 22 (1), S183–S185. doi: 10.1111/j.1440-1681.1995.tb02873.x
National Toxicology Program (2001). NTP Toxicology and Carcinogenesis Studies of EMODIN (CAS NO. 518-82-1) Feed Studies in F344/N Rats and B6C3F1 Mice. Natl. Toxicol. Program Tech. Rep. Ser. 493, 1–278.
Nemmar, A., Al Dhaheri, R., Alamiri, J., Al Hefeiti, S., Al Saedi, H., Beegam, S., et al. (2015). Diesel Exhaust Particles Induce Impairment of Vascular and Cardiac Homeostasis in Mice: Ameliorative Effect of Emodin. Cell. Physiol. Biochem. 36 (4), 1517–1526. doi: 10.1159/000430315
Oshida, K., Hirakata, M., Maeda, A., Miyoshi, T., Miyamoto, Y. (2011). Toxicological effect of emodin in mouse testicular gene expression profile. J. Appl. Toxicol. 31 (8), 790–800. doi: 10.1002/jat.1637
Pottoo, F., Javed, N., Rahman, J., Abu-Izneid, T., Khan, F. (2020). Targeted delivery of miRNA based therapeuticals in the clinical management of Glioblastoma Multiforme. Semin. Cancer Biol. S1044-579X(20)30084-5. doi: 10.1016/j.semcancer.2020.04.001. 10.1016/j.semcancer.2020.04.001.
Qin, W., Du, N., Zhang, L., Wu, X., Hu, Y., Li, X., et al. (2015). Genistein alleviates pressure overload-induced cardiac dysfunction and interstitial fibrosis in mice. Br. J. Pharmacol. 172 (23), 5559–5572. doi: 10.1111/bph.13002
Rossi, M., Wen, K., Caruso, F., Belli, S. (2020). Emodin Scavenging of Superoxide Radical Includes π-π Interaction. X-Ray Crystal Structure, Hydrodynamic Voltammetry and Theoretical Studies. Antioxid. (Basel) 9 (3), 194. doi: 10.3390/antiox9030194
Rukov, J., Shomron, N. (2011). MicroRNA pharmacogenomics: post-transcriptional regulation of drug response. Trends Mol. Med. 17 (8), 412–423. doi: 10.1016/j.molmed.2011.04.003
Ryoo, I., Kwak, M. (2018). Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol. 359, 24–33. doi: 10.1016/j.taap.2018.09.014
Sager, H. B., Nahrendorf, M. (2016). Inflammation: a trigger for acute coronary syndrome. Q. J. Nucl. Med. Mol. Imaging. 60 (3), 185–193.
Sborgi, L., Rühl, S., Mulvihill, E., Pipercevic, J., Heilig, R., Stahlberg, H., et al. (2016). GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 35 (16), 1766–1778. doi: 10.15252/embj.201694696
Serruys, P. W., de Jaegere, P., Kiemeneij, F., Macaya, C., Rutsch, W., Heyndrickx, G., et al. (1994). A comparison of balloon-expandable-stent implantation with balloon angioplasty in patients with coronary artery disease. Benestent Study Group N. Engl. J. Med. 331 (8), 489–495. doi: 10.1056/nejm199408253310801
Shi, M. M., Kugelman, A., Iwamoto, T., Tian, L., Forman, H. J. (1994). Quinone-induced oxidative stress elevates glutathione and induces gamma-glutamylcysteine synthetase activity in rat lung epithelial L2 cells. J. Biol. Chem. 269 (42), 26512–26517. doi: 10.1016/0092-8674(94)90204-6
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526 (7575), 660–665. doi: 10.1038/nature15514
Shi, J., Gao, W., Shao, F. (2017). Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 42 (4), 245–254. doi: 10.1016/j.tibs.2016.10.004
Shia, C. S., Hou, Y. C., Tsai, S. Y., Huieh, P. H., Leu, Y. L., Chao, P. D. (2010). Differences in pharmacokinetics and ex vivo antioxidant activity following intravenous and oral administrations of emodin to rats. J. Pharm. Sci. 99 (4), 2185–2195. doi: 10.1002/jps.21978
Shou, X., Zhou, R., Zhu, L., Ren, A., Wang, L., Wang, Y., et al. (2018). Emodin, A Chinese Herbal Medicine, Inhibits Reoxygenation-Induced Injury in Cultured Human Aortic Endothelial Cells by Regulating the Peroxisome Proliferator-Activated Receptor-γ (PPAR-γ) and Endothelial Nitric Oxide Synthase (eNOS) Signaling Pathway. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 24, 643–651. doi: 10.12659/msm.908237
Shrimali, D., Shanmugam, M., Kumar, A., Zhang, J., Tan, B., Ahn, K., et al. (2013). Targeted abrogation of diverse signal transduction cascades by emodin for the treatment of inflammatory disorders and cancer. Cancer Lett. 341 (2), 139–149. doi: 10.1016/j.canlet.2013.08.023
Song, Z. C., Wang, Z. S., Bai, J. H., Li, Z., Hu, J. (2012). Emodin, a Naturally Occurring Anthraquinone, Ameliorates Experimental Autoimmune Myocarditis in Rats. Tohoku J. Exp. Med. 227 (3), 225–230. doi: 10.1620/tjem.227.225
Toldo, S., Mauro, A. G., Cutter, Z., Abbate, A. (2018). Inflammasome, pyroptosis, and cytokines in myocardial ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 315 (6), H1553–h1568. doi: 10.1152/ajpheart.00158.2018
Turovskaya, M., Gaidin, S., Vedunova, M., Babaev, A., Turovsky, E. (2020). BDNF Overexpression Enhances the Preconditioning Effect of Brief Episodes of Hypoxia, Promoting Survival of GABAergic Neurons. Neurosci. Bull. 36 (7), 733–760. doi: 10.1007/s12264-020-00480-z.
Ve, T., Vajjhala, P. R., Hedger, A., Croll, T., DiMaio, F., Horsefield, S., et al. (2017). Structural basis of TIR-domain-assembly formation in MAL- and MyD88-dependent TLR4 signaling. Nat. Struct. Mol. Biol. 24 (9), 743–751. doi: 10.1038/nsmb.3444
Wang, C., Wu, X., Chen, M., Duan, W., Sun, L., Yan, M., et al. (2007). Emodin induces apoptosis through caspase 3-dependent pathway in HK-2 cells. Toxicology 231 (2-3), 120–128. doi: 10.1016/j.tox.2006.11.064
Wang, X., Zou, Y., Sun, A., Xu, D., Niu, Y., Wang, S., et al. (2007). Emodin induces growth arrest and death of human vascular smooth muscle cells through reactive oxygen species and p53. J. Cardiovasc. Pharmacol. 49 (5), 253–260. doi: 10.1097/FJC.0b013e318033dfb3
Wang, C., Jiang, Z., Yao, J., Wu, X., Sun, L., Liu, C., et al. (2008). Participation of cathepsin B in emodin-induced apoptosis in HK-2 Cells. Toxicol. Lett. 181 (3), 196–204. doi: 10.1016/j.toxlet.2008.05.013
Wang, S. J., Liu, Y. Y., Fan, F. H., Yan, J., Wang, X. X., Chen, J. Z. (2008). Inhibitory effects of emodin on the proliferation of cultured rat vascular smooth muscle cell-induced by angiotensin II. Phytother. Res. 22 (2), 247–251. doi: 10.1002/ptr.2301
Wang, C., Dai, X., Liu, H., Yi, H., Zhou, D., Liu, C., et al. (2015). Involvement of PPARγ in emodin-induced HK-2 cell apoptosis. Toxicol. In Vitro 29 (1), 228–233. doi: 10.1016/j.tiv.2014.10.021
Wang, H. C., Lin, X. H., Fang, X. P., Mu, X. Y., Li, T. J., Liu, J. L. (2016). Emodin ameliorates the peritoneal dialysis-related peritoneal fibrosis via inhibiting the activation of Notch pathway. Sheng Li Xue Bao 68 (6), 747–756.
Wang, Y., Cui, L., Xu, H., Liu, S., Zhu, F., Yan, F., et al. (2017). TRPV1 agonism inhibits endothelial cell inflammation via activation of eNOS/NO pathway. Atherosclerosis 260, 13–19. doi: 10.1016/j.atherosclerosis.2017.03.016
Wang, Q., Yu, X., Dou, L., Huang, X., Zhu, K., Guo, J., et al. (2019). miR-154-5p Functions as an Important Regulator of Angiotensin II-Mediated Heart Remodeling. Oxid. Med. Cell. Longev. 2019, 8768164. doi: 10.1155/2019/8768164
Wu, Y. X., Tu, X., Lin, G. S., Xia, H., Huang, H., Wan, J., et al. (2007). Emodin-mediated protection from acute myocardial infarction via inhibition of inflammation and apoptosis in local ischemic myocardium. Life Sci. 81 (17-18), 1332–1338. doi: 10.1016/j.lfs.2007.08.040
Wu, Z. Q., Chen, Q. W., Ke, D. Z., Li, G. Q., Deng, W. (2014). Emodin Protects against Diabetic Cardiomyopathy by Regulating the AKT/GSK-3 beta Signaling Pathway in the Rat Model. Molecules 19 (9), 14782–14793. doi: 10.3390/molecules190914782
Wu, L., Han, W., Chen, Y., Zhang, T., Liu, J., Zhong, S., et al. (2018). Gender Differences in the Hepatotoxicity and Toxicokinetics of Emodin: The Potential Mechanisms Mediated by UGT2B7 and MRP2. Mol. Pharmaceut. 15 (9), 3931–3945. doi: 10.1021/acs.molpharmaceut.8b00387
Wu, M. Y., Yiang, G. T., Liao, W. T., Tsai, A. P., Cheng, Y. L., Cheng, P. W., et al. (2018). Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol. Biochem. 46 (4), 1650–1667. doi: 10.1159/000489241
Wu, C., Chen, M., Cheng, Y., Ko, Y., Lin, S., Chiu, I., et al. (2019). Emodin Inhibits EBV Reactivation and Represses NPC Tumorigenesis. Cancers 11 (11), 1795. doi: 10.3390/cancers11111795
Wu, J., Zeng, Z., Zhang, W., Deng, Z., Wan, Y., Zhang, Y., et al. (2019). Emerging role of SIRT3 in mitochondrial dysfunction and cardiovascular diseases. Free Radical Res. 53 (2), 139–149. doi: 10.1080/10715762.2018.1549732
Wu, A., Peng, Y., Huang, B., Ding, X., Wang, X., Niu, P., et al. (2020). Genome Composition and Divergence of the Novel Coronavirus, (2019-nCoV) Originating in China. Cell Host Microbe 27 (3), 325–328. doi: 10.1016/j.chom.2020.02.001
Xiang, H., Tao, X., Xia, S., Qu, J., Song, H., Liu, J., et al. (2017). Emodin Alleviates Sodium Taurocholate-Induced Pancreatic Acinar Cell Injury via MicroRNA-30a-5p-Mediated Inhibition of High-Temperature Requirement A/Transforming Growth Factor Beta 1 Inflammatory Signaling. Front. Immunol. 8, 1488. doi: 10.3389/fimmu.2017.01488
Xiao, D., Zhang, Y., Wang, R., Fu, Y. J., Zhou, T., Diao, H. T., et al. (2019). Emodin alleviates cardiac fibrosis by suppressing activation of cardiac fibroblasts via upregulating metastasis associated protein 3. Acta Pharm. Sin. B. 9 (4), 724–733. doi: 10.1016/j.apsb.2019.04.003
Xing, Y., Wang, L., Wang, C., Zhang, Y., Zhang, Y., Hu, L., et al. (2019). Pharmacokinetic studies unveiled the drug-drug interaction between trans-2,3,5,4’-tetrahydroxystilbene-2-O-β-d-glucopyranoside and emodin that may contribute to the idiosyncratic hepatotoxicity of Polygoni Multiflori Radix. J. Pharm. Biomed. Analysis 164, 672–680. doi: 10.1016/j.jpba.2018.11.034
Xiong, D., Deng, Y., Huang, B., Yin, C., Liu, B., Shi, J., et al. (2016). Icariin attenuates cerebral ischemia-reperfusion injury through inhibition of inflammatory response mediated by NF-kappaB, PPARalpha and PPARgamma in rats. Int. Immunopharmacol. 30, 157–162. doi: 10.1016/j.intimp.2015.11.035
Xiong, H., Luo, T., He, W., Xi, D., Lu, H., Li, M., et al. (2016). Up-regulation of miR-138 inhibits hypoxia-induced cardiomyocyte apoptosis via down-regulating lipocalin-2 expression. Exp. Biol. Med. (Maywood N.J.) 241 (1), 25–30. doi: 10.1177/1535370215591831
Xu, K., Al-Ani, M., Wang, C., Qiu, X., Chi, Q., Zhu, P., et al. (2018a). Emodin as a selective proliferative inhibitor of vascular smooth muscle cells versus endothelial cells suppress arterial intima formation. Life Sci. 207, 9–14. doi: 10.1016/j.lfs.2018.05.042
Xu, K., Zhou, T. W., Huang, Y. M., Chi, Q. J., Shi, J. W., Zhu, P., et al. (2018b). Anthraquinone Emodin Inhibits Tumor Necrosis Factor Alpha-Induced Calcification of Human Aortic Valve Interstitial Cells via the NF-kappa B Pathway. Front. Pharmacol. 9, 1328:8. doi: 10.3389/fphar.2018.01328
Xu, C., Zhang, J., Liu, J., Li, Z., Liu, Z., Luo, Y., et al. (2020). Proteomic analysis reveals the protective effects of emodin on severe acute pancreatitis induced lung injury by inhibiting neutrophil proteases activity. J. Proteomics 220, 103760. doi: 10.1016/j.jprot.2020.103760
Yang, X. J., Seto, E. (2007). HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26 (37), 5310–5318. doi: 10.1038/sj.onc.1210599
Yang, Y., Jiang, Z., Zhuge, D. (2019). Emodin Attenuates Lipopolysaccharide-Induced Injury via Down-Regulation of miR-223 in H9c2 Cells. Int. Heart J. 60 (2), 436–443. doi: 10.1536/ihj.18-048
Yao, H., Tang, X., Shao, X., Feng, L., Wu, N., Yao, K. (2007). Parthenolide protects human lens epithelial cells from oxidative stress-induced apoptosis via inhibition of activation of caspase-3 and caspase-9. Cell Res. 17 (6), 565–571. doi: 10.1038/cr.2007.6
Ye, B. Z., Chen, X. D., Dai, S. S., Han, J. B., Liang, X. H., Lin, S., et al. (2019). Emodin alleviates myocardial ischemia/reperfusion injury by inhibiting gasdermin D-mediated pyroptosis in cardiomyocytes. Drug Design Dev. Ther. 13, 975–990. doi: 10.2147/dddt.S195412
Yi, J., Yang, J., He, R., Gao, F., Sang, H., Tang, X., et al. (2004). Emodin enhances arsenic trioxide-induced apoptosis via generation of reactive oxygen species and inhibition of survival signaling. Cancer Res. 64 (1), 108–116. doi: 10.1158/0008-5472.can-2820-2
Yim, T. K., Wu, W. K., Mak, D. H., Ko, K. M. (1998). Myocardial protective effect of an anthraquinone-containing extract of Polygonum multiflorum ex vivo. Planta Med. 64 (7), 607–611. doi: 10.1055/s-2006-957531
Yoshimura, M., Yasue, H., Okumura, K., Ogawa, H., Jougasaki, M., Mukoyama, M., et al. (1993). Different secretion patterns of atrial natriuretic peptide and brain natriuretic peptide in patients with congestive heart failure. Circulation 87 (2), 464–469. doi: 10.1161/01.cir.87.2.464
Yu, Y., Liu, H., Yang, D., He, F., Yuan, Y., Guo, J., et al. (2019). Aloe-emodin attenuates myocardial infarction and apoptosis via up-regulating miR-133 expression. Pharmacol. Res. 146, 104315. doi: 10.1016/j.phrs.2019.104315
Yuan, X., Berg, N., Lee, J., Le, T., Neudecker, V., Jing, N., et al. (2018). MicroRNA miR-223 as regulator of innate immunity. J. Leukoc. Biol. 104 (3), 515–524. doi: 10.1002/jlb.3mr0218-079r
Zhang, Y., Park, T., Gidday, J. (2007). Hypoxic preconditioning protects human brain endothelium from ischemic apoptosis by Akt-dependent survivin activation. Am. J. Physiol. Heart Circulatory Physiol. 292 (6), H2573–H2581. doi: 10.1152/ajpheart.01098.2006
Zhang, M., Lin, L., Lin, H., Qu, C., Yan, L., Ni, J. (2018). Interpretation the Hepatotoxicity Based on Pharmacokinetics Investigated Through Oral Administrated Different Extraction Parts of Polygonum multiflorum on Rats. Front. Pharmacol. 9, 505. doi: 10.3389/fphar.2018.00505
Zhang, Y., Xu, H., Yu, F., Wang, M., Li, M., Xu, T., et al. (2018). Crosstalk between MicroRNAs and Peroxisome Proliferator-Activated Receptors and Their Emerging Regulatory Roles in Cardiovascular Pathophysiology. PPAR Res. 2018, 8530371. doi: 10.1155/2018/8530371
Zhang, X. Z., Qin, Q. J., Dai, H. Y., Cai, S. L., Zhou, C. Y., Guan, J. (2019). Emodin protects H9c2 cells from hypoxia-induced injury by up-regulating miR-138 expression. Braz. J. Med. Biol. Res. 52 (3), 12. doi: 10.1590/1414-431x20187994
Zhang, Y., Song, Z., Huang, S., Zhu, L., Liu, T., Shu, H., et al. (2020). Aloe emodin relieves Ang II-induced endothelial junction dysfunction via promoting ubiquitination mediated NLRP3 inflammasome inactivation. J. Leukoc. Biol. 1–12. doi: 10.1002/jlb.3ma0520-582r
Zheng, Y., Shi, B., Ma, M., Wu, X., Lin, X. (2019). The novel relationship between Sirt3 and autophagy in myocardial ischemia-reperfusion. J. Cell. Physiol. 234 (5), 5488–5495. doi: 10.1002/jcp.27329
Zhou, G. H., Zhang, F., Wang, X. N., Kwon, O. J., Kang, D. G., Lee, H. S., et al. (2014). Emodin accentuates atrial natriuretic peptide secretion in cardiac atria. Eur. J. Pharmacol. 735, 44–51. doi: 10.1016/j.ejphar.2014.04.014
Keywords: emodin, polyvalent molecule, mechanisms of action, targets identification, anti-cardiovascular disease
Citation: Li Q, Gao J, Pang X, Chen A and Wang Y (2020) Molecular Mechanisms of Action of Emodin: As an Anti-Cardiovascular Disease Drug. Front. Pharmacol. 11:559607. doi: 10.3389/fphar.2020.559607
Received: 21 May 2020; Accepted: 13 August 2020;
Published: 27 August 2020.
Edited by:
Changhua Wang, Wuhan University, ChinaReviewed by:
Hong Xiang, Dalian Medical University, ChinaYingyu Chen, Union Hospital, Fujian Medical University, China
Copyright © 2020 Li, Gao, Pang, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Wang, emp1d2FuZ3lpQHpqdS5lZHUuY24=