
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol., 17 June 2020
Sec. Translational Pharmacology
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.00905
This article is part of the Research TopicNatural Product Epigenetic Modulators and InhibitorsView all 7 articles
 Donya Naz Divsalar1†
Donya Naz Divsalar1† Conrad Veranso Simoben2,3†Cole Schonhofer1Khumoekae Richard1
Conrad Veranso Simoben2,3†Cole Schonhofer1Khumoekae Richard1 Wolfgang Sippl3
Wolfgang Sippl3 Fidele Ntie-Kang2,3*
Fidele Ntie-Kang2,3* Ian Tietjen1,4*
Ian Tietjen1,4*Current antiretroviral therapies used for HIV management do not target latent viral reservoirs in humans. The experimental “shock-and-kill” therapeutic approach involves use of latency-reversal agents (LRAs) that reactivate HIV expression in reservoir-containing cells, followed by infected cell elimination through viral or host immune cytopathic effects. Several LRAs that function as histone deacetylase (HDAC) inhibitors are reported to reverse HIV latency in cells and in clinical trials; however, none to date have consistently reduced viral reservoirs in humans, prompting a need to identify new LRAs. Toward this goal, we describe here a virtual screening (VS) approach which uses 14 reported HDAC inhibitors to probe PubChem and identifies 60 LRA candidates. We then show that four screening “hits” including (S)-N-Hydroxy-4-(3-methyl-2-phenylbutanamido)benzamide (compound 15), N-(4-Aminophenyl)heptanamide (16), N-[4-(Heptanoylamino)phenyl]heptanamide (17), and 4-(1,3-Dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-N-(2-hydroxyethyl)butanamide (18) inhibit HDAC activity and/or reverse HIV latency in vitro. This study demonstrates and supports that VS-based approaches can readily identify novel HDAC inhibitors and LRAs, which in turn may help toward inhibitor design and chemical optimization efforts for improved HIV shock-and-kill-based efforts.
While combination antiretroviral therapy (cART) can durably suppress HIV replication, it does not act on resting CD4+ T cells containing latent proviral reservoirs. As these cells can reactivate at any time to produce infectious virus, they preclude a readily accessible HIV cure (Finzi et al., 1997; Siliciano et al., 2003; Cary et al., 2016). As a result, cART currently must be taken for life.
One proposed therapeutic-based method toward identifying and eliminating HIV reservoir-containing cells, frequently termed “shock-and-kill,” involves the use of latency reversal agents (LRAs) to initially stimulate virus production (i.e., “shock”). Following LRA administration, cells expressing reactivated HIV are then eliminated through apoptosis or immune-enhancing mechanisms (“kill”), while the concurrent use of cART prevents reservoir reseeding (Deeks, 2012). Among numerous LRAs identified to date, one of the most common functional classes is comprised of inhibitors of class I histone deacetylases (HDAC; Margolis, 2011; Zaikos et al., 2018). During proviral latency, HDACs are recruited to the HIV promoter resulting in transcriptional repression (Archin and Margolis, 2014; Darcis et al., 2017). In the presence of HDAC inhibitors, lysine acetylation within histone tails results in euchromatin formation and subsequent binding of transcription factors that drive provirus expression. Several HDAC inhibitors, including both natural products (e.g., romidepsin) and synthetic derivatives of natural product leads (e.g., vorinostat), have been investigated in clinical trials for their ability to reactivate provirus expression and reduce HIV reservoir levels in cART-treated, virally suppressed, HIV-infected individuals (Rasmussen and Lewin, 2016; Andersen et al., 2018). However, while a subset of these studies reports transient increases of virus or viral RNA following HDAC inhibitor administration, no trial to date has shown a significant reduction of HIV reservoir size in humans (Abner and Jordan, 2019; Zerbato et al., 2019). These results suggest that additional HDAC inhibitors with improved efficacy and/or selectivity for proviral integration sites may be needed to achieve sufficient HIV latency reversal and viral reservoir clearance in vivo.
One approach toward identifying new HDAC inhibitor prototypes, to serve as the basis for developing potentially improved efficacy and selectivity over existing agents, involves virtual screening (VS) of compound library databases. This method frequently entails searching for potential “hit” molecules which are stored in an electronic format and are able to interact favorably with a drug target site or otherwise “fit” into the structural, electronic, and steric features of known bioactive molecules in silico. Common VS approaches involve molecular docking of an electronic three dimensional molecular library toward a drug target site, searching for common pharmacophores, and calculating binding free energies (Chaput et al., 2016; Chen et al., 2017). Previous efforts by us and others have shown that “hits” identified from VS are frequently biologically active, for example as novel inhibitors of HIV replication (Brigo et al., 2005; Shityakov and Dandekar, 2010; Tietjen et al., 2015; Panwar and Singh, 2017; Berinyuy and Soliman, 2017). We therefore hypothesized that similar efforts could be used to identify novel LRA candidates.
Toward this goal, we describe here the results of a VS beginning with 5,687 unique compound structures obtained from ~100 million compounds present in PubChem (Kim et al., 2016). We describe a designed procedure that combines similarity searching, docking, and scoring and which resulted in the identification of 60 hit compounds as potential HDAC inhibitors. We then confirm that four commercially available compounds from this list function as HDAC inhibitors and/or HIV LRAs in vitro.
Jurkat T cells (Clone E6-1) were obtained from the American Tissue Culture Collection. J-Lat 10.6 cells were obtained from the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH (contributed by Dr. Eric Verdin; Jordan et al., 2003). Cells were cultured in R10+ media [RPMI 1640 with HEPES and L-Glutamine, 10% fetal bovine serum, 100 U of penicillin/ml, and 100 µg of streptomycin/ml (Sigma)] and incubated at 37°C and 5% CO2.
HDAC-Glo I/II Assay kits were obtained from Promega. Panobinostat was obtained from Sigma-Aldrich. Compounds 15, 16 and 17, and 18 were purchased from Sellick Chemical, Enamine, and Molport, respectively. Serpulanine A was gratefully obtained as a gift from Dr. Raymond J. Andersen (University of British Columbia). Compounds stocks were diluted in DMSO and stored at minus 20°C until use.
The VS dataset was collected from selected molecules in PubChem (Kim et al., 2016). We then searched for compounds from PubChem having close similarity with 14 known class I HDAC inhibitors including belinostat, entinostat, givinostat, mocetinostat, oxamflatin, panobinostat, psammaplin A, romidepsin, scriptaid, serpulanine A, thiophenyl benzamide, trichostatin A, valproic acid, and vorinostat (Kiernan et al., 1999; Contreras et al., 2009; Matalon et al., 2010; Ying et al., 2010; Matalon et al., 2011; Yin et al., 2011; Archin et al., 2012; Rasmussen et al., 2014; Wei et al., 2014; Huang et al., 2018; Richard et al., 2018; Williams et al., 2018; Zaikos et al., 2018; Table 1). A two dimensional (2D) similarity search was conducted on the PubChem website, with similarity between chemical structures being quantified by use of the Tanimoto equation (Chen and Reynolds, 2002; Holliday et al., 2002; Holliday et al., 2003) in combination with the PubChem substructure fingerprint (ftp://ftp.ncbi.nlm.nih.gov/pubchem/specifications/pubchem_fingerprints.pdf). For 12 of 14 compounds which were listed in PubChem at the time of study [all except the thiophenylbenzamide (compound 1) and serpulanine A (compound 11)], the canonical SMILES were used to conduct similarity searches. For the remaining two compounds (1 and 11), 2D structure files built with ChemDraw (.cdx) were converted to their respective.mol files, uploaded to PubChem, and used for the similarity search. Initially, a random search was carried out for 80%, 90%, and 95% Tanimoto similarities. The hit lists for similarity searches with less than 95% similarity cut offs yielded several hundred thousand entries for some of the 14 HDAC inhibitors; as a result, we selected a cut off of 95% similarity for all 14 input compounds. Resulting outputs from the search were collected into a combined dataset and duplicates were removed, leading to an initial library of 5,867 unique compound entries.
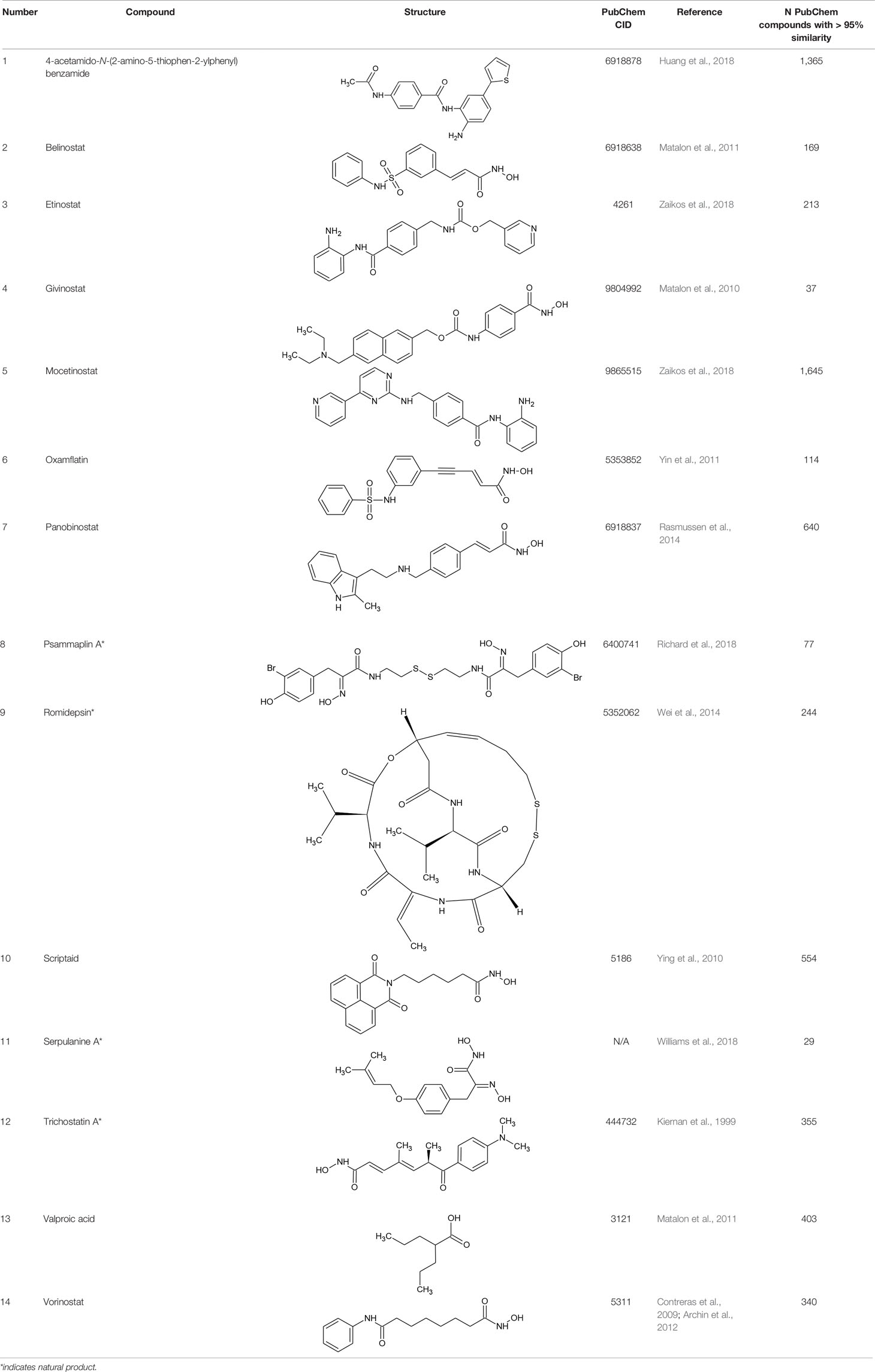
Table 1 Structures of reported histone deacetylase (HDAC) inhibitors used for virtual screening (VS).
The high-resolution crystal structure of the human histone deacetylase 1 (hmHDAC1, class I HDAC family), with PDB ID: 5ICN, chain B, (Resolution: 3.3 Å) was downloaded from the Protein Databank (PDB; www.rcsb.org; Burley et al., 2018). Preparation of the protein structure was performed using protocols similar to what we have previously reported (Heimburg et al., 2016; Simoben et al., 2018). MOE software (v. 2016.08) was used to delete all water molecules. Further preparation steps on the protein were applied using the default settings of the Protein Preparation Wizard of Schrödinger software (Sastry et al., 2013). Assignment of the bond orders and hydrogen atoms as well as protonation of the heteroatom states were added using Epik-tool (with the pH set at biologically relevant values, i.e., at 7.0 ± 2.0). Optimization of the H-bond network was done, and the structure was finally subjected to a restrained energy minimization step using the Optimized Potentials for Liquid Simulations (OPLS) 2005 force field. The root-mean-square deviation (RMSD) of the atom displacement for terminating the minimization was 0.3 Å (Banks et al., 2005).
Ligands for docking were prepared using similarly reported protocols (Heimburg et al., 2016; Simoben et al., 2018). Preparation of ligands for docking was done using the LigPrep tool (Schroedinger, 2017-u1), as implemented in Schrödinger's software (version 2017-1). All possible tautomers, as well as possible combinations of stereoisomers for molecules without well-defined stereochemistries, were generated for pH 7.0 ± 2.0 using the Epik ionization method. Additionally, the optimized integrated OPLS-2005 force field (Banks et al., 2005) was used to minimize all ligands. Pan-Assay Interference (PAIN) molecules were subsequently discarded after the application of PAIN filters implemented in the Schroedinger's Canvas tool. Finally, the generation of 30 conformers for each of the prepared ligand molecules, followed by the minimization of each conformer output, was performed using the settings of ConfGen (Watts et al., 2010).
Prepared protein and ligands were docked using the standardized Glide program procedure of Schrödinger's software (version 2017-1; Friesner et al., 2004; Halgren et al., 2004). The receptor grid preparation for the docking procedure was carried out by assigning the nonpolymer fraction of the cocrystallized ligand (6A0) as the centroid of the grid box. The generated three-dimensional conformers of the prepared ligand were subsequently docked into the receptor model using the Glide program. An additional metal constraint on the catalytic Zn2+ ion was used to optimize our docking results. A total of five docking poses per ligand conformer were included in the postdocking minimization step, and a maximum of two docking poses was generated for each ligand conformer. The GlideScore Standard Precision (SP) score mode was used as the scoring function to rank the resulting binding poses (Friesner et al., 2004; Halgren et al., 2004). The Schroedinger clustering package was subsequently used to cluster the resulting poses using an RMSD value of 1.5 Å and the Glide SP score as properties of interest. Further reduction in the number of molecules was done by choosing only the top 200,000 docking poses each from the clustered outcomes after ranking by Glide SP score and Glide Ligand efficiency (LE) scores. The resulting sets of molecules were merged and duplicates were removed. The final set of hits was proposed after manual inspection of each docking pose and interaction(s) with the conserved amino acid residues His140, His141, and Tyr303 in the HDAC active site.
HDAC activity in the presence of compounds was performed using the HDAC-Glo I/II assay (Promega) according to the manufacturer's instructions. HDAC reactions were performed in white 384-well plates with a final volume of 20 µl per well. Stock compounds and DMSO were diluted in manufacturer buffer to desired concentrations and added to wells. Jurkat cells were then resuspended in phenol red-free and fetal bovine serum-free RPMI 1640 and seeded into wells at 3*103 cells/well. Wells containing no cells were included as negative controls for the signal background. Following incubation at 37°C and 5% CO2 for 90 min, 20 µl of HDAC-Glo I/II Reagent plus 1% Triton-X100 (prepared as per manufacturer's instructions) was added to each well. Plates were gently mixed for 30 s and then incubated at room temperature for an additional 30 min. Luminescence was detected using an Infinity M200 multimode plate reader (Tecan Life Sciences). Data were normalized between no-inhibitor (100%) and no-cell (0%) controls and presented as the mean ± s.e.m. from three independent experiments.
J-Lat 10.6 cells were seeded in 96-well plates at 2*105 cells/well and coincubated with LRAs at defined concentrations or 0.1 to 1.0% DMSO vehicle control for 24 or 72 h. Cells were then examined for GFP expression by flow cytometry (Guava EasyCyte 8HT, Millipore). Culture viability was estimated by flow cytometry and based on the relative percentage of compound-treated J-Lat cells displaying the characteristic forward- and side-scatter parameters of vehicle-treated control cells (Tietjen et al., 2015). Flow cytometry data were analyzed using FlowJo v. 10.5.3 software (FlowJo LLC, Ashland, OR), where background GFP signals in live J-Lat cells treated with respective DMSO concentrations were set to 0.05%-positive cells. For all results, data are presented as the mean ± s.e.m. from at least three independent experiments.
Table 1 lists the structures of 14 compounds (compounds 1–14) that were used for similarity searching in PubChem. Table 2 lists published activities of these 14 compounds to inhibit recombinant HDAC1. All selected compounds were also reported to reverse HIV latency in one or more in vitro and/or primary cell assays with the exception of serpulanine A (Williams et al., 2018), which we confirmed at 10 µM induced a 31.0% increase in HIV provirus expression, as measured by the GFP reporter, in J-Lat 10.6 cells (Jordan et al., 2003 and see below). A similarity search was then performed to reduce the virtual chemical space to a manageable number of molecules for VS (see Materials and Methods), which resulted in 6,175 compounds. After the removal of duplicates and pan assay interference molecules, we obtained 5,867 molecules for VS.
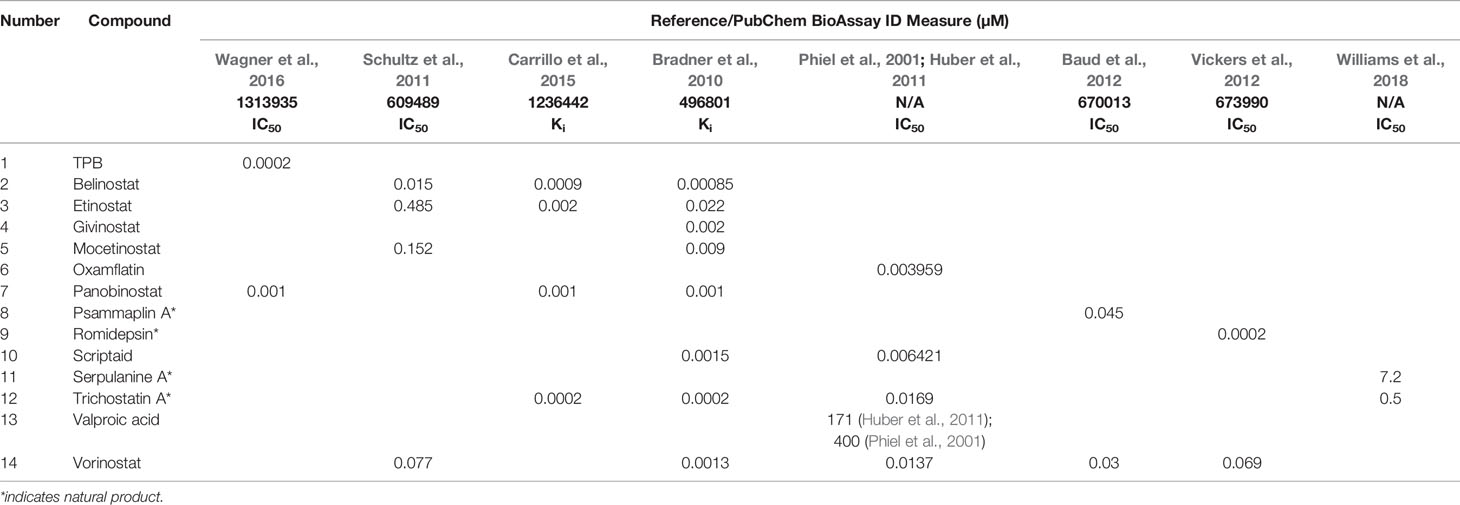
Table 2 Reported inhibition of recombinant HDAC1 by histone deacetylase (HDAC) inhibitors.
These 5,867 molecules were next docked into the active site of the hmHDAC1 protein (PDB ID: 5ICN, chain B; Resolution: 3.3 Å). The proposed docking procedure was able to redock the cocrystallized inhibitor within the receptor-binding pocket with RMSD < 1 Å. The redocked inhibitor pose was observed to reproduce the bidentate coordination to the catalytic Zn-ion as well as H-bond interaction with the conserved Tyr303 (Figure 1). Further reduction of the compound set was then performed via clustering of the docking results (generated after sorting by docking score as well as ligand efficiency) using an average RMSD of 1.5 Å base on the docking score. The final selection and proposal of hits were based on visual inspection of the selected docked poses for conserved interactions (such as coordination to the conserved catalytic Zn2+ ion and hydrogen bond interactions) and based on published works (Figure 2).
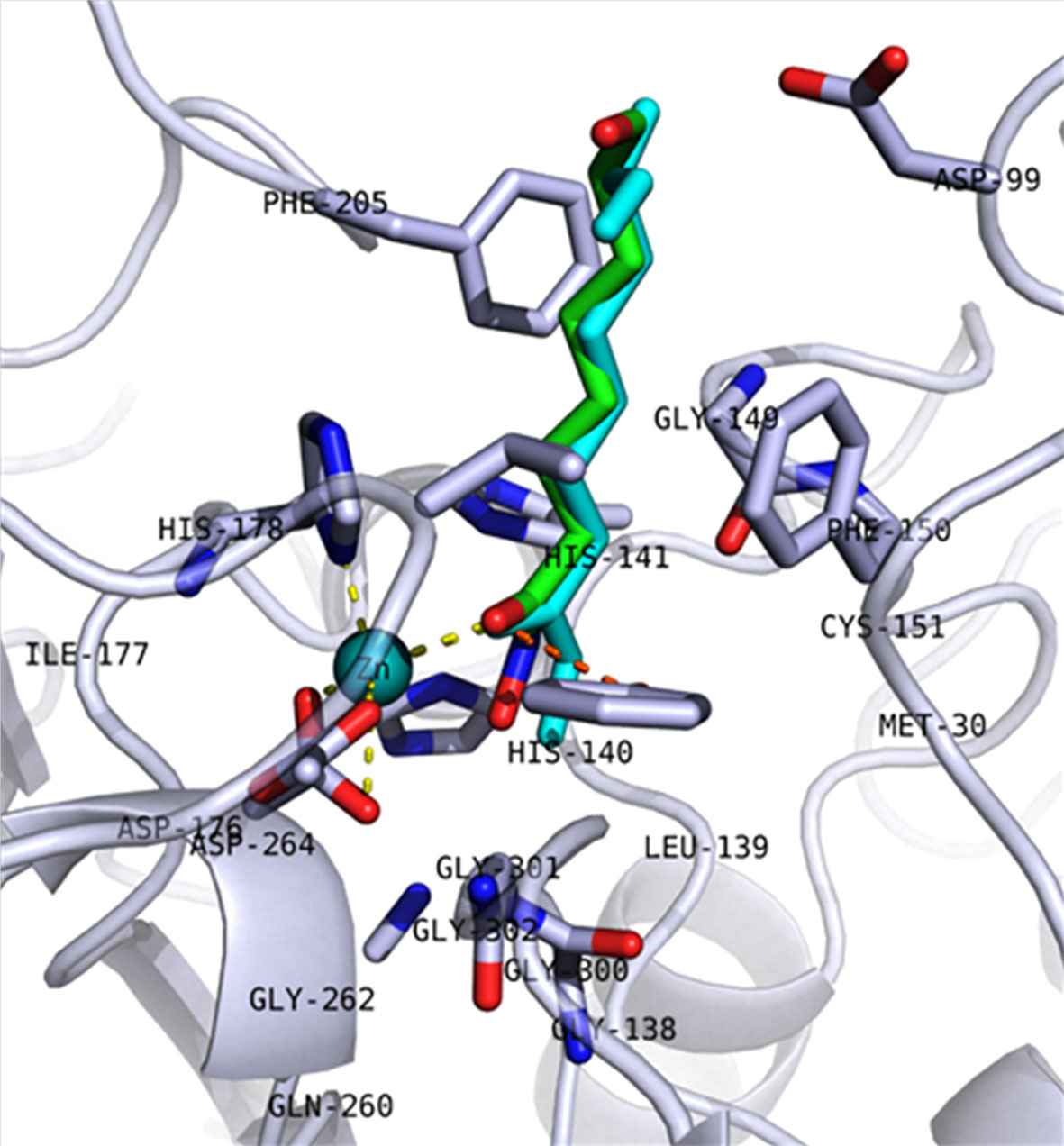
Figure 1 Redocking pose of 5ICN_B native ligand (6A0). The backbone of the protein is shown as a cartoon, and key amino acid residues in the active site are shown in stick representation. The redocked molecule is shown in cyan, while the crystalized pose is shown in green. Coordination of Zn2+ ion (cyan sphere) and hydrogen bond interactions with Tyr303 are shown as yellow and orange dashed lines, respectively.
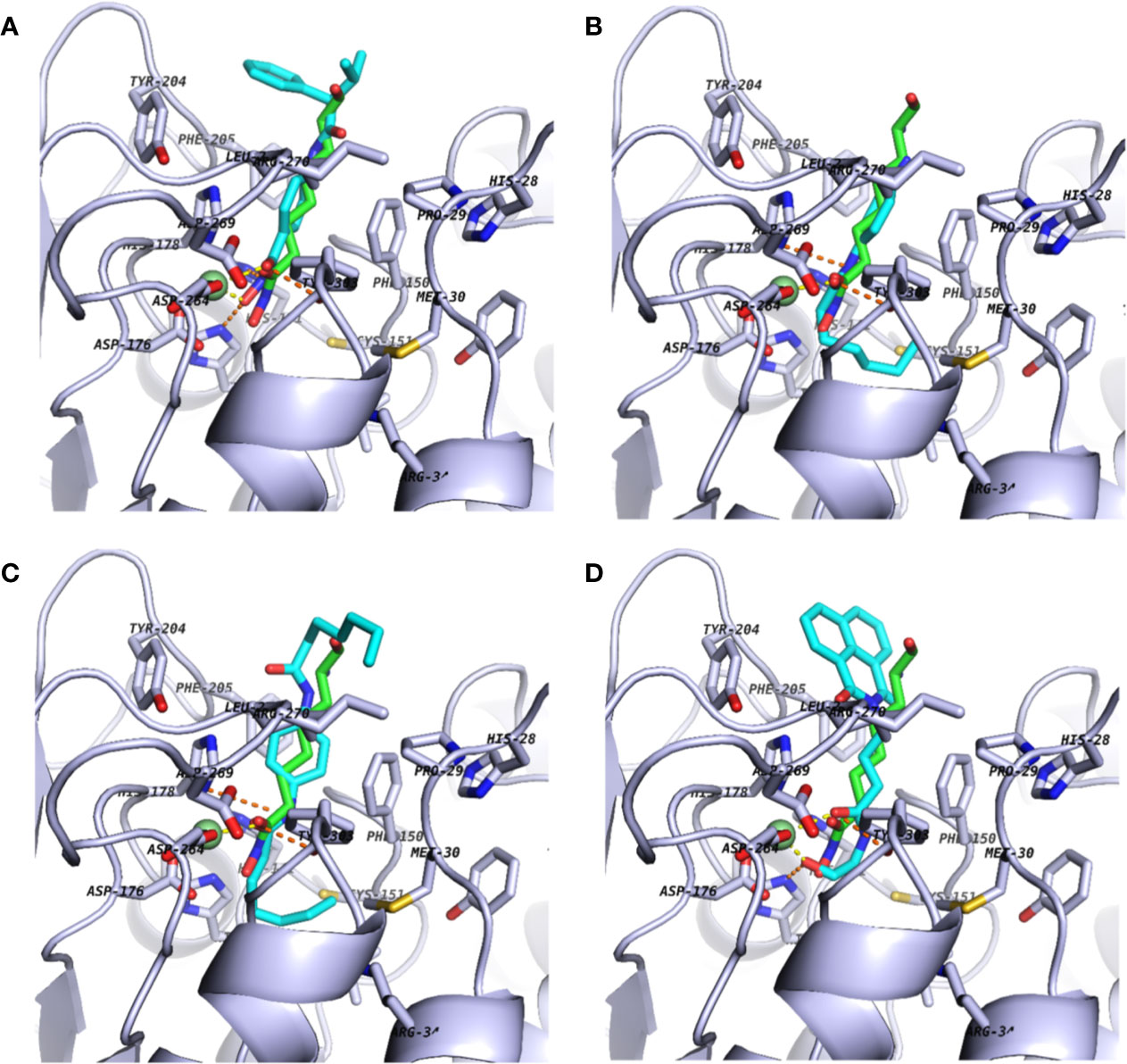
Figure 2 Docking poses of commercially available screening hits: (A) (S)-N-Hydroxy-4-(3-methyl-2-phenylbutanamido)benzamide (compound 15); (B) N-(4-Aminophenyl)heptanamide (16); (C) N-[4-(Heptanoylamino)phenyl]heptanamide (17); and (D) 4-(1,3-Dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-N-(2-hydroxyethyl)butanamide (18). Schematics are shown as described in Figure 1.
Figure 3 lists 60 compounds that were identified as putative hits of the VS. From these 60 compounds, four (6.6%) including (S)-N-Hydroxy-4-(3-methyl-2-phenylbutanamido)benzamide (compound 15), N-(4-Aminophenyl)heptanamide (16), N-[4-(Heptanoylamino)phenyl]heptanamide (17), and 4-(1,3-Dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-N-(2-hydroxyethyl)butanamide (18), were readily available through commercial sources and therefore selected for further validation in functional studies (Table 3). Compound 15 contains a hydroxamic acid and the same core pharmacophore as compound 1. Also called AR-42, 15 was previously reported to reverse HIV latency in vitro (Mates et al., 2015) but was not included in our original list of HDAC inhibitors for VS, providing initial validation of this experimental approach. Compounds 16-17 are both N-phenylheptamides, with 17 almost being the dimeric form of 16 (except for an additional amine group). 16 and 17 structurally resemble vorinostat (14), however missing the crucial hydroxamic acid moiety. Finally, compound 18 resembles scriptaid (compound 10; Ying et al., 2010) with the difference being that 18 has an amide and a hydroxyl group, separated by two methylene groups, in place of the Zinc-binding hydroxamic acid moiety in scriptaid and which is often found in HDAC inhibitors. The abilities of 16–18 to inhibit HDAC and reverse HIV latency have not been reported.
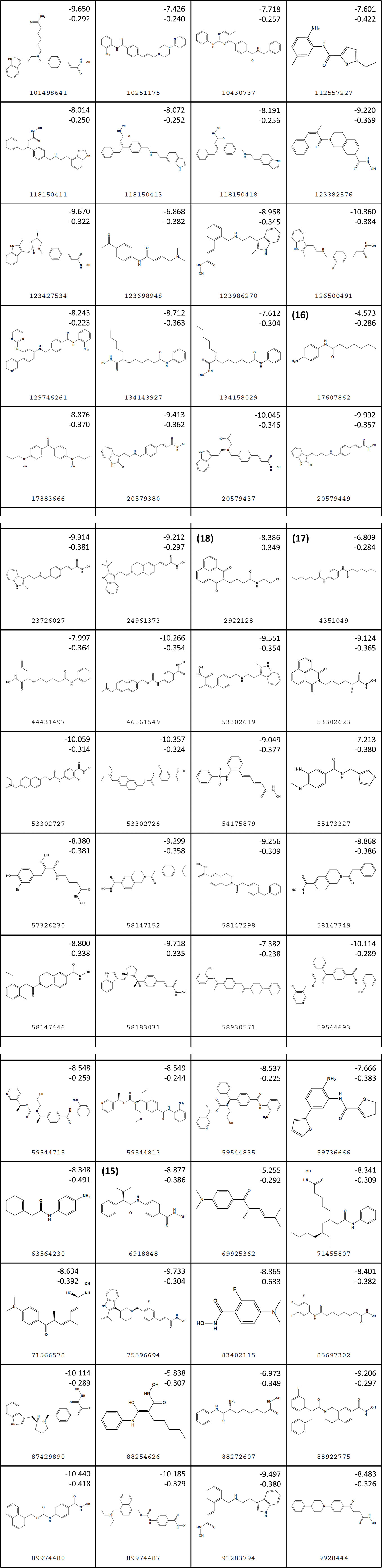
Figure 3 Structures of virtual screening (VS) hits. Compounds 15–18 are highlighted. For each structure, PubChem ID numbers are located at bottom center. Values at top right indicate Glide Standard Precision (SP) (top) and Glide Ligand efficiency (LE) (bottom) scores.
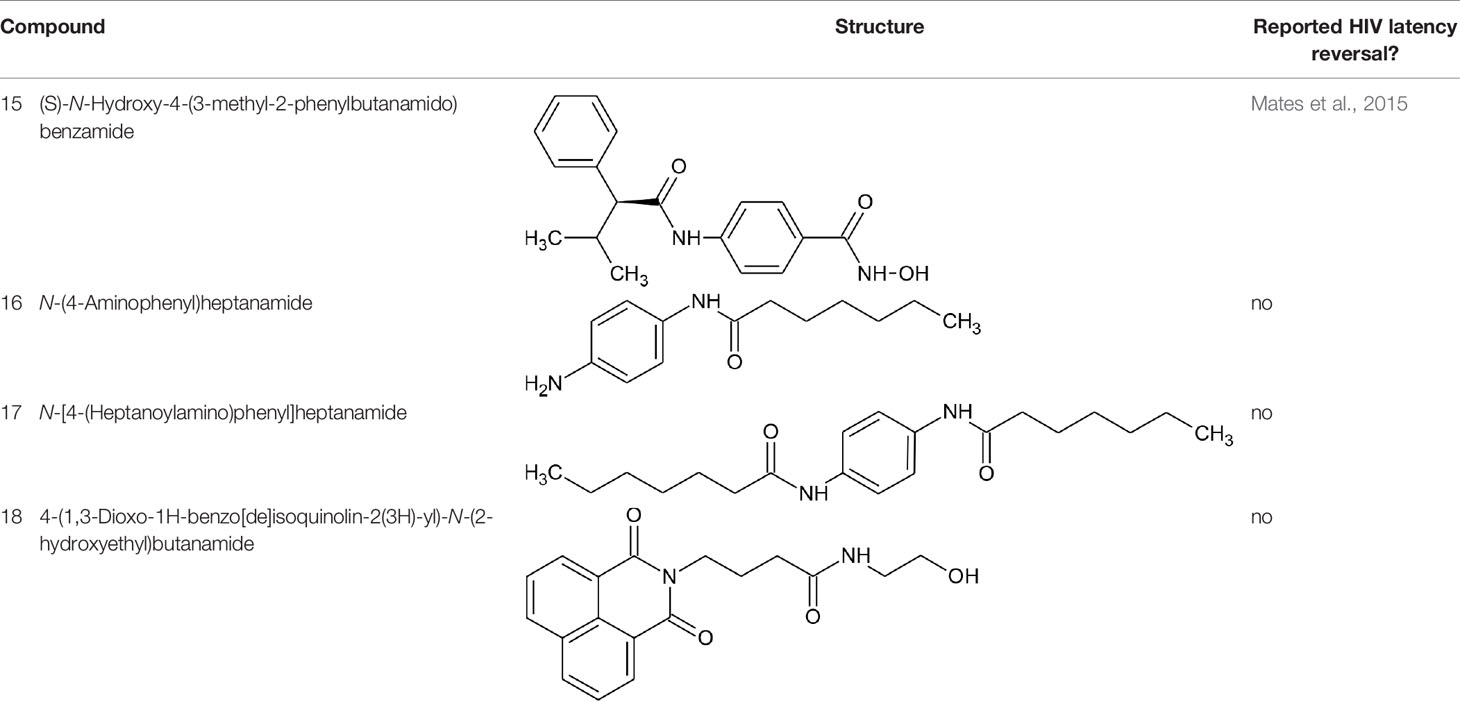
Table 3 Compounds hits from virtual screening (VS) selected for biological validation.
To examine whether compounds 15–18 inhibit HDAC activity in vitro, we used the HDAC-Glo I/II assay (Promega). This assay allows quantification of HDAC I/II activity in live cells via a cell-permeable, acetylated, luminogenic peptide substrate. The deacetylated peptide is then cleaved by the addition of a developer reagent, which releases aminoluciferin from the peptide. Finally, aminoluciferin release is quantified in a firefly luciferase reaction. As a result, increased deacetylation of the substrate by cellular HDACs results in increased firefly luciferase production, while suppression of HDAC activity with inhibitors results in decreased luminescence production.
Using this assay, the treatment of Jurkat cells with the control HDAC inhibitor panobinostat (compound 7) resulted, as expected, in substantial inhibition of in vitro HDAC activity (Figure 4). For example, as little as 300 pM of panobinostat inhibited 55.9% ± 3.5% of HDAC activity (mean ± s.e.m.), with > 99% HDAC inhibition observed at 0.3 µM, which is consistent with other results from in vitro assays (Scuto et al., 2008). Also consistent with its known activity as an HDAC inhibitor (Mates et al., 2015), compound 15 additionally inhibited HDAC activity, albeit at much higher concentrations (e.g., 52.5% ± 11.2% inhibition at 0.1 µM and > 99% inhibition at 100 µM; Figure 4). In contrast, no more than 32.7% ± 9.1% and 27.1% ± 7.2% inhibition at 300 µM were observed for compounds 16 and 17, respectively, indicating weak but detectable HDAC inhibition in vitro. No inhibitory activity was detected for compound 18 at any concentration up to 300 µM. Thus three of the four assessed VS hits detectibly inhibit HDAC activity in Jurkat cells, although none approach the activity of the control HDAC inhibitor panobinostat.
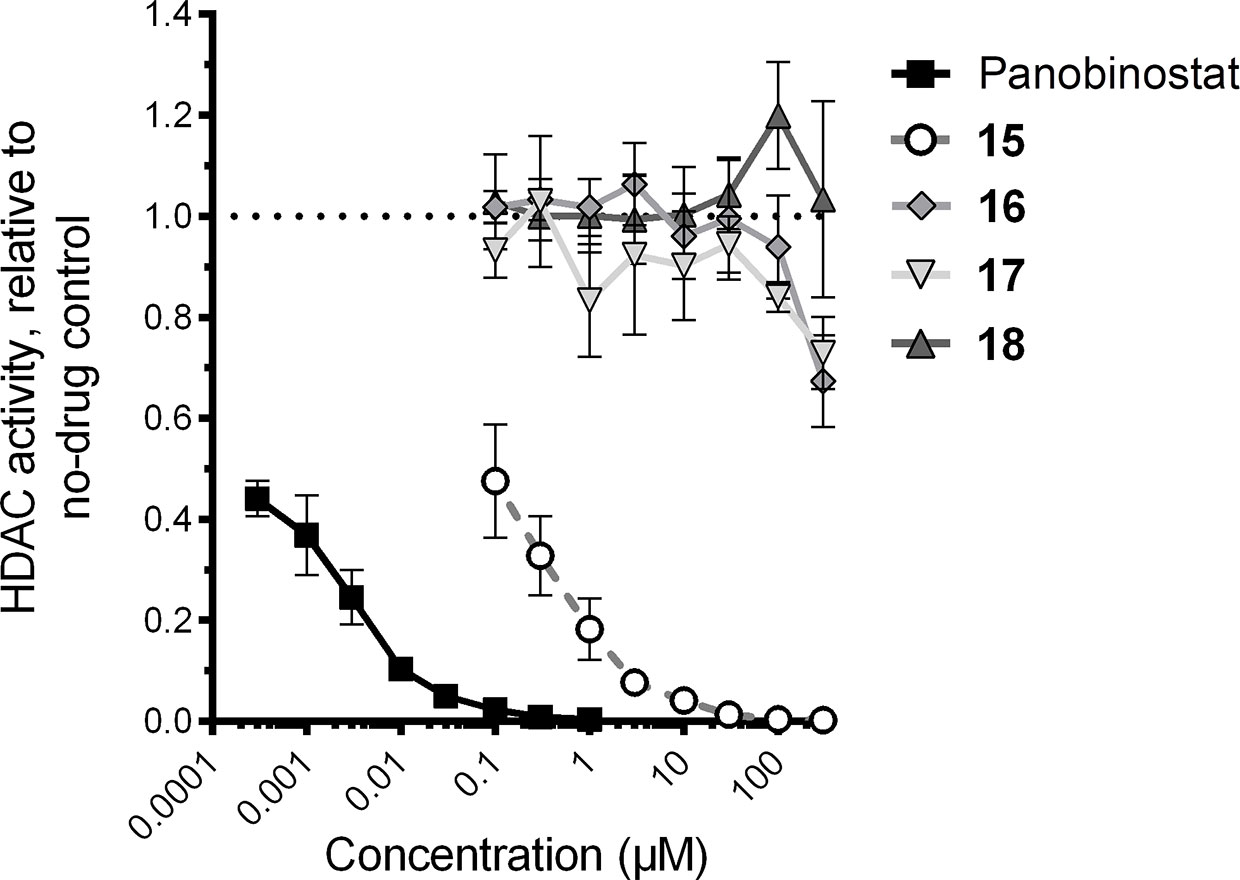
Figure 4 Effects of four virtual screening (VS) hits on cellular histone deacetylase (HDAC) activity, as recorded by HDAC-Glo assay. HDAC activity in the presence of each compound is presented relative to HDAC activity in cells treated with 0.1% DMSO vehicle control.
To investigate whether compounds 15–18 reverse HIV latency in vitro, we used the J-Lat 10.6 cell line. These cells are derived from Jurkat T cells and contain an inducible latent provirus with a frameshift mutation in Env, rendering the virus noninfectious, in addition to a GFP reporter expressed from the deleted viral Nef locus (Jordan et al., 2003). As such, HIV latency reversal in these cells can be measured by an increase in GFP expression, as measured by flow cytometry. Resting J-Lat 10.6 cells feature a proportion of spontaneously GFP-expressing cells that are not fully latent; however, they are also highly sensitive to stimulation by LRAs (Williams et al., 2004; Cummins et al., 2017). In our hands, we observed an average of 8.0% ± 1.7% GFP-positive cells in resting conditions. J-Lat 10.6 cells were therefore treated with panobinostat, compounds 15–18, or DMSO vehicle control for 24 h. Figure 5A shows representative examples of GFP expression in J-Lat 10.6 cells plus LRAs.
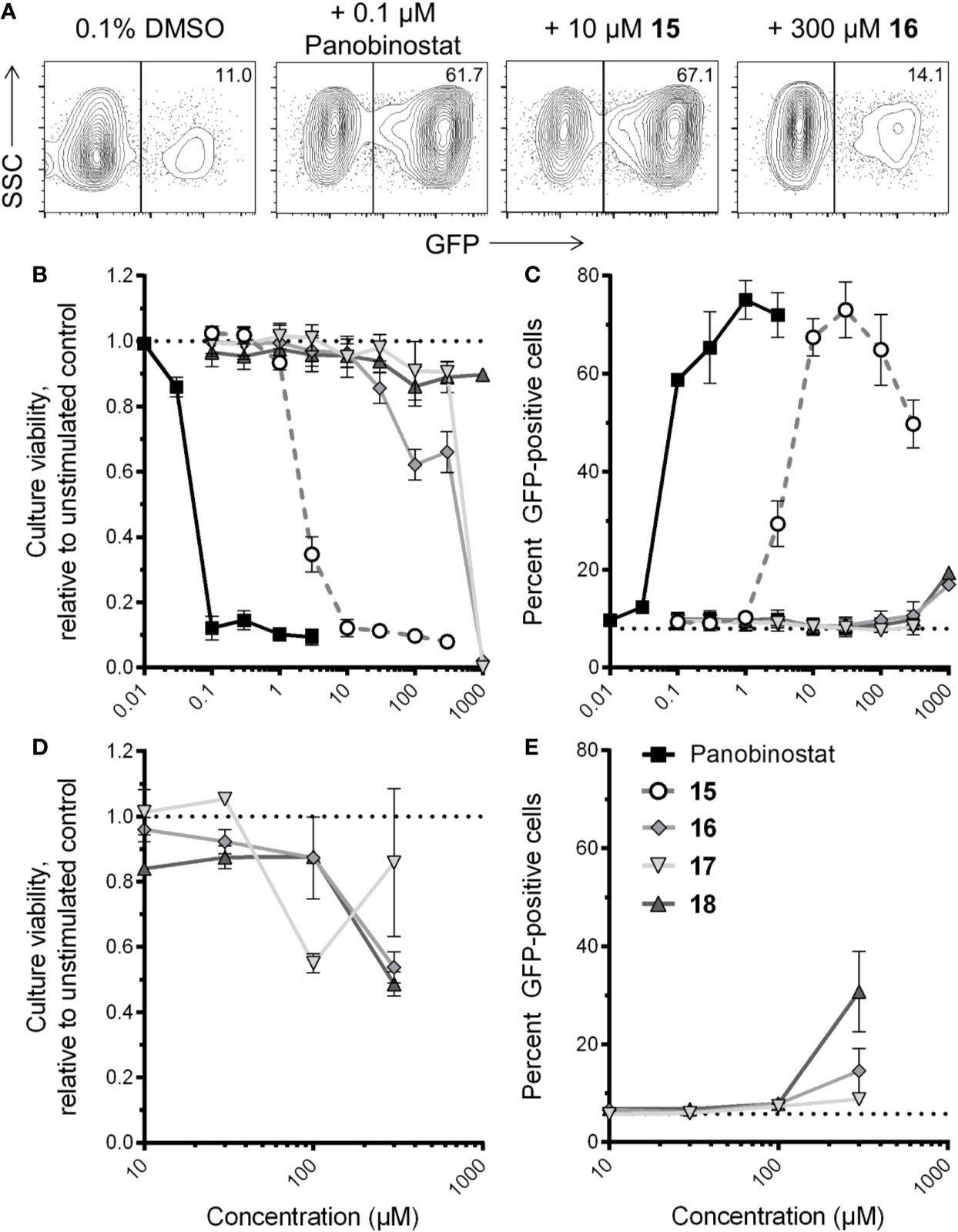
Figure 5 Effects of four virtual screening (VS) compound hits on HIV latency reversal. (A) Representative flow cytometry data of HIV latency reversal, as measured by GFP expression, in J-Lat 10.6 cells in the presence of 0.1% DMSO, panobinostat, or select VS hits following 24-h incubation. For each panel, values indicate percent GFP-positive cells. (B) Dose-response profiles of panobinostat and compounds 15–18 on J-Lat 10.6 estimated cell culture viability following 24-h incubation. Data are presented relative to culture viability of cells treated with 0.1% DMSO vehicle control. (C) Dose-response profiles of compounds on HIV-1 latency reversal, as measured by percent GFP-positive cells, following 24-h incubation. Dotted line indicates baseline GFP expression in untreated cells. (D) Effects of compounds 16–18 on J-Lat 10.6 estimated culture viability after 72-h incubation. Data are presented as described in (B). (E) Effects of 16–18 on HIV-1 latency reversal after 72-h incubation. Data are presented as described in (C).
In parallel, we also estimated cell culture viability by measuring the percentage of LRA-treated cells displaying the characteristic forward and side-scatter parameters of control cultures treated with 0.1% DMSO (Tietjen et al., 2015; Figure 5B). After 24-h incubation, the control HDAC inhibitor panobinostat was observed to induce substantial cytotoxicity, consistent with our previous observations (Richard et al., 2018): for example, treatment with 0.1 µM panobinostat resulted in an estimated 12.1% ± 3.6% culture viability relative to cells treated with 0.1% DMSO, and a 50% cytotoxicity concentration (CC50) of 35.3 ± 4.1 nM (Figure 5B). Compound 15 exhibited a similar level of toxicity at 30 µM (11.3% ± 2.0% estimated culture viability), with a CC50 of 1.6 ± 0.3 µM, thereby indicating a comparable therapeutic window to panobinostat in this assay. Substantial cellular toxicity (i.e. > 50%) for compounds 16 and 17 were not observed except at 1 mM, where estimated culture viabilities for both relative to DMSO-treated cells were 2.0% ± 0.0% and 0.0% ± 0.0%, respectively, while no substantial toxicity was observed for compound 18 at any concentration up to 1 mM (i.e. minimum 86.1% ± 6.0% estimated culture viability at 100 µM; Figure 5B). Thus, when panobinostat and compounds 15–18 are rank-ordered, these activities match their respective in vitro HDAC inhibition efficacies (Figure 4).
Most compounds were also observed to reverse HIV latency in live cells after 24 h, as measured by GFP expression (Figure 5C). For example, in the presence of panobinostat, GFP was detected in a maximum of 75.1% ± 3.9% of cells at 1 µM, while a maximum of 73.0% ± 5.7% of cells were GFP-positive in the presence of 30 µM of compound 15. In contrast, no more than 16.9% ± 0.2% GFP expression in live cells was observed in the presence of compound 16, which occurred at 1 mM, reflecting only a 2.1-fold induction of GFP over spontaneous expression in DMSO-treated cells (Figure 5C). For compound 17, no GFP expression was observed at any concentration, although no data could be obtained at 1 mM due to complete cytotoxicity. Interestingly, despite exhibiting no in vitro HDAC inhibition or toxicity, compound 18 did induce weak but consistent GFP expression in 19.4% ± 0.2% of cells at 1 mM, or 2.4-fold over spontaneous expression. These results indicate that compound 15 is a robust LRA, although with efficacy approximately one to two orders of magnitude lower than control panobinostat, while compounds 18 and 16 may exhibit latency reversal at very high concentrations.
To confirm whether compounds 16–18 could consistently induce latency reversal at levels above spontaneous GFP expression in DMSO-treated J-Lat 10.6 cells, we repeated the assays described above but incubated cells for a total of 72 h. In these conditions, no more than 50% loss of estimated culture viability was observed for any compound at up to 300 µM. (Figure 5D). Notably, at 300 µM, compounds 16 and 17 consistently induced 15.5% ± 4.6% and 8.8% ± 1.4% at 300 µM, respectively, or 2.7 and 1.5-fold increased GFP over spontaneous expression (5.8% ± 0.2%; Figure 5E). Interestingly, 300 µM of compound 18 induced 30.8% ± 8.2% GFP expression, a 5.3-fold increase over spontaneous expression, indicating consistent HIV latency reversal despite no activity in HDAC inhibition assays and no obvious cytotoxicity.
Taken together, these results indicate that compounds 15–18 all induce latency reversal and/or cytotoxicity, albeit at concentrations that are orders of magnitude higher than panobinostat.
Current LRAs used in shock-and-kill therapeutic strategies to date have yet to consistently clear viral reservoirs in humans, prompting the need to identify new chemical leads to inform ongoing efforts to improve existing LRA strategies. Toward this goal, we described here a VS-based approach to probe 5,867 nonredundant molecules from PubChem to identify 60 which structurally resemble LRAs of the HDAC inhibitor functional class. The four compounds that were readily commercially available were further tested and confirmed to inhibit HDAC activity and/or reverse HIV latency in vitro. In a related approach, Gallastegui et al. (2012) used the J-Lat A2 cell line to screen 6,000 small molecules in vitro, and a VS-based approach was then used to probe 7.5 million compounds to identify those with similarity to the eight biologically identified hits. This strategy led to discovery of the novel LRA 8-methoxy-6-methylquinolin-4-ol (Gallastegui et al., 2012). Our study, which used VS to discover new LRAs of the HDAC inhibitor class, further demonstrates that new LRAs can be readily identified using similarity-based VS approachesWhile none of the compounds reached the efficacy of the control HDAC inhibitor panobinostat in our study, this we demonstrate that this proof-of-concept approach represents a simple, efficient, and cost-effective method to identify new LRA leads. The VS approaches described here are readily transferrable to resource-constrained researchers and also allow improved access and ability toward informing worldwide HIV eradication efforts for even remote and very small research groups. However, while VS can be faster and more cost-effective than biological laboratory-based compound screens, care must be taken as several methods for scoring active and inactive compounds are often unsuccessful (Chen, 2015). Another caveat is compound availability for biological validation; for example, a recent study showed that only ~10% of compounds contained in electronic libraries potentially identifiable by VS are accessible as samples through commercial sources and academic collaborations (Chen et al., 2017). This clearly impacted our ability to validate all hits identified in the VS, as only four compounds (6.7%) were available to us from commercial sources. The low commercial availability of identified hits is therefore a significant limitation of this study and precludes determining statistically whether identifying hits by VS is likely to translate into functional effects. However, the published compounds available in PubChem, even if not commercially available, can still inform further design studies such as chemical optimization from a starting point. Future synthetic chemistry efforts beyond the scope of this work will likely be required to address this question. Limited availability becomes even more heightened when rare compounds such as those obtained in natural product-based isolation efforts are considered, and this highlights the continuing need for the building and efficient management and accessibility of consortiums of publicly available synthetic and natural compound repositories (Ntie-Kang et al., 2014; Tietjen et al., 2015; Ntie-Kang et al., 2017). Furthermore, as biological activities for compounds 16–8 were only found at concentrations > 100 µM, synthetic chemistry efforts are needed to identify derivatives with improved efficacies before additional studies are pursued, for example to assess ability to reverse viral latency in primary cells from donors with HIV. Future VS efforts should also focus on identifying compounds with structural similarities to only the most efficacious HDAC inhibitors.
Of the four VS hits validated here, (S)-N-Hydroxy-4-(3-methyl-2-phenylbutanamido)benzamide (15), also called AR-42, was previously described as an HDAC inhibitor and HIV LRA with cell toxicity at single micromolar to high nanomolar concentrations (Mates et al., 2015). Our results are consistent with this previous study. As 15 was not included in our initial set of screening probes, the rediscovery of 15 here supports both the previously reported results of Mates et al. but also serves as a positive control for the VS approach described here. In contrast, compounds 16 and 17 exhibited limited HDAC1 inhibition at 300 µM (32.7 ± 9.1 and 27.1% ± 7.2% respectively) which, although clearly weak functional hits, do place them within the range of efficacies observed for the established HDAC inhibitor valproic acid in similarly designed assays (i.e., reported IC50s = 171–400 µM; Table 2; Phiel et al., 2001; Huber et al., 2011). Furthermore, while 16 and 17 respectively induced only 2.7 or 1.5-fold increased GFP over spontaneous expression at 300 µM in J-Lat 10.6 cells, these results are consistent with the ability of valproic acid to induce latency reversal in the comparable Jurkat-LAT-GFP cell line, where 1 and 2.5 mM resulted in 1.6- and 2.2-fold increases in GFP expression, respectively (Pérez et al., 2010). Notably, 4-(1,3-Dioxo-1H-benzo[de]isoquinolin-2(3H)-yl)-N-(2-hydroxyethyl)butanamide (compound 18) was also able to weakly reverse HIV latency at high concentrations (> 100 µM) but did not inhibit in vitro HDAC activity at up to 300 µM. This result was unexpected from a biological perspective, although not from a chemical perspective as 18 has no zinc-chelating moiety. The reason for the discrepancy in biological assays for 18 is not immediately clear but could reflect exceptionally slow binding of HDACs in vitro which is not captured within the time-course of the HDAC-Glo assay performed here. However, the parent compound of 18 (scriptaid) has also been reported to act on other biological pathways which drive HIV latency reversal including inhibition of histone methyltransferase at H3K9 moieties, induction of PI3K/AKT signaling, and perhaps other putative activities which allow scriptaid to synergize with other HDAC inhibitors in vitro (Chen et al., 2013; Wang et al., 2013; Liang et al., 2015; Andersen et al., 2018). Thus 18 could function as an LRA through one of these alternative proviral signaling pathways. However, further studies with more potent analogs of 18 and detailing of structure-activity relations based on the scriptaid scaffold are desired to test this hypothesis.
In summary, we demonstrate that VS-based approaches can readily identify novel LRA candidates of the HDAC inhibitor functional class. This approach can be used, in principle, to identify additional, novel LRAs representing other functional classes such as PKC activators, BET bromodomain inhibitors, and others (Andersen et al., 2018). It also identifies new chemical scaffolds which can serve as starting points to design more potent LRAs and which may be useful toward improving shock-and-kill based HIV eradication therapies in humans.
All datasets generated for this study are included in the article/supplementary material.
DD, CVS, CS, and KR performed experiments and wrote and reviewed the manuscript. WS, FN-K, and IT conceived the study, planned and supervised experiments, and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding was provided by the Canadian Institutes for Health Research (CIHR PJT-153057) (IT) and the New Frontiers in Research Fund – Explorations (NFRFE-2018-01386) (IT). This work was also supported through the Sub-Saharan African Network for TB/HIV Research Excellence (SANTHE) (KR, IT), a DELTAS African Initiative [grant no. DEL-15-006]. The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)'s Alliance for Accelerating Excellence in Science in Africa (AESA) and supported by the New Partnership for Africa's Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust [grant no. 107752/Z/15/Z] and the UK government. DD was supported by a Simon Fraser University – Social Sciences and Humanities Research Council of Canada (SSHRC) Undergraduate Research Student Award. CVS acknowledges financial support from the German Academic Exchange Services (DAAD), Germany, awarded to Doctoral Candidates. CS was supported by a CIHR Frederick Banting and Charles Best MSc Award. KR was a recipient of a Canadian Queen Elizabeth II Diamond Jubilee Scholarship, a partnership between the Rideau Hall Foundation, Community Foundations of Canada and Universities Canada, in addition to a SANTHE Ph.D. Fellowship. FN-K acknowledges a return fellowship and equipment subsidy from the Alexander von Humboldt Foundation, Germany.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank David E. Williams and Raymond J. Andersen for serpulanine A, Silven Read for technical assistance and training, and Luis J. Montaner for critical review of the manuscript.
cART, combination antiretroviral therapy; CC50, 50% cytotoxicity concentration; HDAC, histone deacetylase; LE, ligand efficiency; LRA, latency reversal agent; OPLS, optimized potentials for liquid simulations; PAIN, pan-assay interference; PDB, protein data bank; RMSD, root-mean-square deviation; SP, standard precision; VS, virtual screening; 2D, two dimensional.
Abner, E., Jordan, A. (2019). HIV “shock and kill” therapy: In need of revision. Antiviral Res. 166, 19–34. doi: 10.1016/j.antiviral.2019.03.008
Andersen, R. J., Ntie-Kang, F., Tietjen, I. (2018). Natural product-derived compounds in HIV suppression, remission, and eradication strategies. Antiviral Res. 158, 63–77. doi: 10.1016/j.antiviral.2018.07.016
Archin, N. M., Margolis, D. M. (2014). Emerging strategies to deplete the HIV reservoir. Curr. Opin. Infect. Dis. 27, 29–35. doi: 10.1097/QCO.0000000000000026
Archin, N. M., Liberty, A. L., Kashuba, A. D., Choudhary, S. K., Kuruc, J. D., Crooks, A. M., et al. (2012). Administration of vorinostat disrupts HIV-1 latency in patents on antiretroviral therapy. Nature. 487, 482–485. doi: 10.1038/nature11286
Banks, J. L., Beard, H. S., Cao, Y., Cho, A. E., Damm, W., Farid, R., et al. (2005). Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 26, 1752–1780. doi: 10.1002/jcc.20292
Baud, M. G. J., Leiser, T., Haus, P., Samlal, S., Wong, A. C., Wood, R. J., et al. (2012). Defining the mechanism of action and enzymatic selectivity of psammaplin A against its epigenetic targets. J. Med. Chem. 55, 1731–1750. doi: 10.1021/jm2016182
Berinyuy, E., Soliman, M. E. S. (2017). Identification of novel potential gp120 of HIV-1 antagonist using per-residue energy contribution-based pharmacophore modelling. Interdiscip. Sci. 9, 406–418. doi: 10.1007/s12539-016-0174-7
Bradner, J. E., West, N., Grachan, M. L., Greenberg, E. F., Haggarty, S. J., Warnow, T., et al. (2010). Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 6, 238–243. doi: 10.1038/nchembio.313
Brigo, A., Mustata, G. I., Briggs, J. M., Moro, S. (2005). Discovery of HIV-1 integrase inhibitors through a novel combination of ligand and structure-based drug design. Med. Chem. 1, 263–275. doi: 10.2174/1573406053765440
Burley, S. K., Berman, H. M., Christie, C., Duarte, J. M., Feng, Z., Westbrook, J., et al. (2018). RCSB Protein Data Bank: Sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education.Protein. Sci. 27, 316–330. doi: 10.1002/pro.3331
Carrillo, A. K., Guiguemde, W. A., Guy, R. K. (2015). Evaluation of histone deacetylase inhibitors (HDACi) as therapeutic leads for human African trypanosomiasis (HAT). Bioorg. Med. Chem. 23, 5151–5155. doi: 10.1016/j.bmc.2014.12.066
Cary, D. C., Fujinaga, K., Peterlin, B. M. (2016). Molecular mechanisms of HIV latency. J. Clin. Investig. 126, 448–454. doi: 10.1172/JCI80565
Chaput, L., Martinez-Sanz, J., Saettel, N., Mouawad, L. (2016). Benchmark of four popular virtual screening programs: construction of the active/decoy dataset remains a major determinant of measured performance. J. Cheminf. 8, 56. doi: 10.1186/s13321-016-0167-x
Chen, X., Reynolds, C. H. (2002). Performance of similarity measures in 2D fragment-based similarity searching: Comparison of structural descriptors and similarity coefficients. J. Chem. Inf. Comput. Sci. 42, 1407–1414. doi: 10.1021/ci025531g
Chen, C. H., Du, F., Xu, J., Chang, W. F., Liu, C. C., Su, H. Y., et al. (2013). Synergistic effect of trichostatin A and scriptaid on the development of cloned rabbit embryos. Theriogenology. 79, 1284–1293. doi: 10.1016/j.theriogenology.2013.03.003
Chen, Y., de Bruyn Kops, C., Kirchmair, J. (2017). Data resources for the computer-guided discovery of bioactive natural products. J. Chem. Inf. Model. 57, 2099–2111. doi: 10.1021/acs.jcim.7b00341
Chen, Y. C. (2015). Beware of docking! Trends Pharmacol. Sci. 36, 78–95. doi: 10.1016/j.tips.2014.12.001
Contreras, X., Schweneker, M., Chen, C. S., McCune, J. M., Deeks, S. G., Martin, J., et al. (2009). Suberoylanilidehydroxamic acid reactivates HIV from latently infected cells. J. Biol. Chem. 284, 6782–6789. doi: 10.1074/jbc.M807898200
Cummins, N. W., Sainski-Nguyen, A. M., Natesampillai, S., Aboulnasr, F., Kaufmann, S., Badley, A. D. (2017). Maintenance of the HIV reservoir is antagonized by selective BCL2 inhibition. J. Virol. 91, e00012–e00017. doi: 10.1128/JVI.00012-17
Darcis, G., Van Driessche, B., Van Lint, C. (2017). HIV latency: Should we shock or lock? Trends Immunol. 38, 217–228. doi: 10.1016/j.it.2016.12.003
Finzi, D., Hermankova, M., Pierson, T., Carruth, L. M., Buck, C., Chaisson, R. E., et al. (1997). Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 278, 1295–1300. doi: 10.1126/science.278.5341.1295
Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Klicic, J. J., Mainz, D. T., et al. (2004). Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749. doi: 10.1021/jm0306430
Gallastegui, E., Marshall, B., Vidal, D., Sanchez-Duffhues, G., Collado, J. A., Alvarez-Fernández, C., et al. (2012). Combination of biological screening in a cellular model of viral latency and virtual screening identifies novel compounds that reactivate HIV-1. J. Virol. 86, 3795–3808. doi: 10.1128/JVI.05972-11
Halgren, T. A., Murphy, R. B., Friesner, R. A., Beard, H. S., Frye, L. L., Pollard, W. T., et al. (2004). Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 47, 1750–1759. doi: 10.1021/jm030644s
Heimburg, T., Chakrabarti, A., Lancelot, J., Marek, M., Melesina, J., Hauser, A. T., et al. (2016). Structure-Based Design and Synthesis of Novel Inhibitors Targeting HDAC8 from Schistosoma mansoni for the Treatment of Schistosomiasis. J. Med. Chem. 59, 2423–2435. doi: 10.1021/acs.jmedchem.5b01478
Holliday, J. D., Hu, C. Y., Willett, P. (2002). Grouping of coefficients for the calculation of inter-molecular similarity and dissimilarity using 2D fragment bit-strings. Comb. Chem. High Throughput Screen. 5, 155–166. doi: 10.2174/1386207024607338
Holliday, J. D., Salim, N., Whittle, M., Willett, P. (2003). Analysis and display of the size dependence of chemical similarity coefficients. J. Chem. Inf. Comput. Sci. 43, 819–828. doi: 10.1021/ci034001x
Huang, L., Lai, W. H., Zhu, L., Li, W., Wei, L., Lee, K. H., et al. (2018). Elimination of HIV-1 latently infected cells by gnidimacrin and a selective HDAC inhibitor. ACS Med. Chem. Lett. 9, 268–273. doi: 10.1021/acsmedchemlett.8b00012
Huber, K., Doyon, G., Plaks, J., Fyne, E., Mellors, J. W., Sluis-Cremer, N. (2011). Inhibitors of histone deacetylases: Correlation between isoform specificity and reactivation of HIV type 1 (HIV-1) from latently infected cells. J. Biol. Chem. 286, 22211–22218. doi: 10.1074/jbc.M110.180224
Jordan, A., Bisgrove, D., Verdin, E. (2003). HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 22, 1868–1877. doi: 10.1093/emboj/cdg188
Kiernan, R. E., Vanhulle, C., Schiltz, L., Adam, E., Xiao, H., Maudoux, F., et al. (1999). HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J. 18, 6106–6118. doi: 10.1093/emboj/18.21.6106
Kim, S., Thiessen, P. A., Bolton, E. E., Chen, J., Fu, G., Gindulyte, A., et al. (2016). PubChem substance and compound databases. Nucleic Acids Res. 44, D1202–D1213. doi: 10.1093/nar/gkv951
Kim, Y., Anderson, J. L., Lewin, S. R. (2018). Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 23, 14–26. doi: 10.1016/j.chom.2017.12.004
Liang, S., Zhao, M. H., Choi, J. W., Kim, N. H., Cui, X. S. (2015). Scriptaid treatment decreases DNA methyltransferase 1 expression by induction of MicroRNA-152 expression in procine somatic cell nuclear transfer embryos. PloS One 10, e0134567. doi: 10.1371/journal.pone.0134567
Margolis, D. M. (2011). Histone deacetylase inhibitors and HIV latency. Current. Opin. HIV AIDS. 6, 25–29. doi: 10.1097/COH.0b013e328341242d
Matalon, S., Palmer, B. E., Nold, M. F., Furlan, A., Kassu, A., Fossati, G., et al. (2010). The histone deacetylase inhibitor ITF2357 decreases surface CXCR5 and CCR5 expression on CD4(+) T-cells and monocytes and is superior to valproic acid for latent HIV-1 expression in vitro. J. Acquir. Immune Defic. Syndr. 54, 1–9. doi: 10.1097/QAI.0b013e3181d3dca3
Matalon, S., Rasmussen, T. A., Dinarello, C. A. (2011). Histone deacetylase inhibitors for purging HIV-1 from the latent reservoir. Mol. Med. 17, 466–472. doi: 10.2119/molmed.2011.00076
Mates, J. M., de Silva, S., Lustberg, M., Van Deusen, K., Baiocchi, R. A., Wu, L., et al. (2015). A Novel Histone Deacetylase Inhibitor, AR-42, Reactivates HIV-1 from Chronically and Latently Infected CD4+ T-cells. Retrovirol. (Auckl). 7, 1–5. doi: 10.4137/RRT.S31632
Ntie-Kang, F., Amoa Onguéné, P., Fotso, G. W., Andrae-Marobela, K., Bezabih, M., Ndom, J. C., et al. (2014). Virtualizing the p-ANAPL Library: A Step towards Drug Discovery from African Medicinal Plants. PloS One 9, e90655. doi: 10.1371/journal.pone.0090655
Ntie-Kang, F., Telukunta, K. K., Döring, K., Simoben, C. V., Moumbock, A. F. A., Malange, Y. I., et al. (2017). NANPDB: a resource for natural products from Northern African sources. J. Nat. Prod. 80, 2067–2076. doi: 10.1021/acs.jnatprod.7b00283
Panwar, U., Singh, S. K. (2017). Structure-based virtual screening toward the discovery of novel inhibitors for impeding the protein-protein interaction between HIV-1 integrase and human lens epithelium-derived growth factor (LEDGF/p75). J. Biomol. Struct. Dyn. 23, 1–19. doi: 10.1080/07391102.2017.1384400
Pérez, M., de Vinuesa, A. G., Sanchez-Duffhues, G., Marquez, N., Bellido, M. N., Muñoz-Fernandez, M. Á., et al. (2010). Bryostatin-1 synergizes with histone deacetylase inhibitors to reactivate HIV-1 from latency? Curr. HIV Res. 8, 418–429. doi: 10.2174/157016210793499312
Phiel, C. J., Zhang, F., Huang, E. Y., Guenther, M. G., Lazar, M. A., Klein, P. S. (2001). Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stablilizer, and teratogen. J. Biol. Chem. 276, 36734–36741. doi: 10.1074/jbc.M101287200
Rasmussen, T. A., Lewin, S. R. (2016). Shocking HIV out of hiding: Where are we with clinical trials of latency reversing agents? Curr. Opin. HIV AIDS. 11, 394–401. doi: 10.1097/COH.0000000000000279
Rasmussen, T. A., Tolstrup, M., Brinkmann, C. R., Olesen, R., Erikstrup, C., Solomon, A., et al. (2014). Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV. 1, e13–e21. doi: 10.1016/S2352-3018(14)70014-1
Richard, K., Williams, D. E., de Silva, E. D., Brockmann, M. A., Brumme, Z. L., Andersen, R. J., et al. (2018). Identification of novel HIV-1 latency-reversing agents from a library of marine natural products. Viruses 10 (7), 348. doi: 10.3390/v10070348
Sastry, G. M., Adzhigirey, M., Day, T., Annabhimoju, R., Sherman, W. (2013). Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 27, 221–234. doi: 10.1007/s10822-013-9644-8
Schultz, M., Fan, J., Chen, C., Cho, Y. S., Davis, N., Bickford, S., et al. (2011). The design, synthesis, and structure-activity relationships of novel isoindoline-based histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 21, 4909–4912. doi: 10.1016/j.bmcl.2011.06.015
Scuto, A., Kirschbaum, M., Kowolik, C., Kretzner, L., Juhasz, A., Atadja, P., et al. (2008). The novel histone deacetylase inhibitor, LBH589, induces expression of DNA damage response genes and apoptosis in Ph- acute lymphoblastic leukemia cells. Blood. 111, 5093–5100. doi: 10.1182/blood-2007-10-117762
Shityakov, S., Dandekar, T. (2010). Lead expansion and virtual screening of Indinavir derivate HIV-1 protease inhibitors using pharmacophoric-shape similarity scoring function. Bioinformation. 4, 295–299. doi: 10.6026/97320630004295
Siliciano, J. D., Kajdas, J., Finzi, D., Quinn, T. C., Chadwick, K., Margolick, J. B., et al. (2003). Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 9, 727–728. doi: 10.1038/nm880
Simoben, C. V., Robaa, D., Chakrabarti, A., Schmidtkunz, D., Marek, M., Lancelot, J., et al. (2018). A Novel Class of Schistosoma mansoni Histone Deacetylase 8 (HDAC8) Inhibitors Identified by Structure-Based Virtual Screening and In Vitro Testing. Molecules 23 (3), 566. doi: 10.3390/molecules23030566
Tietjen, I., Ntie-Kang, F., Mwimanzi, P., Onguéné, P. A., Scull, M. A., Idowu, T. O., et al. (2015). Screening of the Pan-African natural product library identifies ixoratannin A-2 and boldine and novel HIV-1 inhibitors. PloS One 10, e0121099. doi: 10.1371/journal.pone.0121099
Vickers, C. J., Olsen, C. A., Leman, L. J., Ghadiri, M. R. (2012). Discovery of HDAC inhibitors that lack an active site Zn(2+)-binding functional group. ACS Med. Chem. Lett. 3, 505–508. doi: 10.1021/ml300081u
Wagner, F. F., Weïmer, M., Steinbacher, S., Schomburg, A., Reinemer, P., Gale, J. P., et al. (2016). Kinetic and structural insights into the binding of histone deacetylase 1 and 2 (HDAC1, 2) inhibitors. Bioorg. Med. Chem. 24, 4008–4015. doi: 10.1016/j.bmc.2016.06.040
Wang, G., Jiang, X., Pu, H., Zhang, W., An, C., Hu, X., et al. (2013). Scriptaid, a novel histone deacetylase inhibitor, protects against traumatic brain injury via modulation of PTEN and AKT pathway: Scriptaid protects against TBI via AKT. Neurotherapeutics. 10, 124–142. doi: 10.1007/s13311-012-0157-2
Watts, K. S., Dalal, P., Murphy, R. B., Sherman, W., Friesner, R. A., Shelley, J. C. (2010). ConfGen: a conformational search method for efficient generation of bioactive conformers. J. Chem. Inf. Model. 50, 534–546. doi: 10.1021/ci100015j
Wei, D. G., Chiang, V., Fyne, E., Balakrishnan, M., Barnes, T., Grapue, M., et al. (2014). Histone deacetylase inhibitor romidepsin induces HIV expression in CD4 T cells from patients on suppressive antiretroviral therapy at concentrations achieved by clinical dosing. PloS Pathog. 10, e1004071. doi: 10.1371/journal.ppat.1004071
Williams, S. A., Chen, L. F., Kwon, H., Fenard, D., Bisgrove, D., Verdin, E., et al. (2004). Prostratin antagonizes HIV latency by activating NF-κB. J. Biol. Chem. 279, 42008–42017. doi: 10.1074/jbc.M402124200
Williams, D. E., Gunasekara, N. W., Ratnaweera, P. B., Zheng, Z., Ellis, S., Dada, S., et al. (2018). Serpulanines A to C, N-oxidized tyrosine derivatives isolated from the Sri Lankan fungus Serpula sp.: Structure elucidation, synthesis, and histone deacetylase inhibition. J. Nat. Prod. 81, 78–84. doi: 10.1021/acs.jnatprod.7b00680
Yin, H., Zhang, Y., Zhou, X., Zhu, H. (2011). Histone deacetylase inhibitor Oxamflatin increase HIV-1 transcription by including histone modification in latently infected cells. Mol. Biol. Rep. 38, 5071–5078. doi: 10.1007/s11033-010-0653-6
Ying, H., Zhang, Y., Lin, S., Han, Y., Zhu, H. Z. (2010). Histone deacetylase inhibitor scriptaid reactivates latent HIV-1 promoter by inducing histone modification in in vitro latency cell lines. Int. J. Mol. Med. 26, 265–272. doi: 10.3892/ijmm_00000461
Zaikos, T. D., Painter, M. M., Sebastian Kettinger, N. T., Terry, V. H., Collins, K. L. (2018). Class 1-selective histone deacetylase (HDAC) inhibitors enhance HIV latency reversal while preserving the activity of HDAC isoforms necessary for maximal HIV gene expression. J. Virol. 92, e02110–e02117. doi: 10.1128/JVI.02110-17
Keywords: virtual screening, histone deacetylase, histone deacetylase inhibitor, HIV, latency reversal, drug discovery
Citation: Divsalar DN, Simoben CV, Schonhofer C, Richard K, Sippl W, Ntie-Kang F and Tietjen I (2020) Novel Histone Deacetylase Inhibitors and HIV-1 Latency-Reversing Agents Identified by Large-Scale Virtual Screening. Front. Pharmacol. 11:905. doi: 10.3389/fphar.2020.00905
Received: 23 January 2020; Accepted: 03 June 2020;
Published: 17 June 2020.
Edited by:
Roberto Paganelli, University of Studies G. d'Annunzio Chieti and Pescara, ItalyReviewed by:
Stevan Pecic, California State University, United StatesCopyright © 2020 Divsalar, Simoben, Schonhofer, Richard, Sippl, Ntie-Kang and Tietjen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fidele Ntie-Kang, ZmlkZWxlLm50aWUta2FuZ0B1YnVlYS5jbQ==; Ian Tietjen, aXRpZXRqZW5Ad2lzdGFyLm9yZw==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.