 Izumi V. Hinkson
Izumi V. Hinkson Benjamin Madej
Benjamin Madej
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Pharmacol. , 30 June 2020
Sec. Translational Pharmacology
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.00770
This article is part of the Research Topic Artificial intelligence for Drug Discovery and Development View all 18 articles
Conventional drug discovery is long and costly, and suffers from high attrition rates, often leaving patients with limited or expensive treatment options. Recognizing the overwhelming need to accelerate this process and increase success, the ATOM consortium was formed by government, industry, and academic partners in October 2017. ATOM applies a team science and open-source approach to foster a paradigm shift in drug discovery. ATOM is developing and validating a precompetitive, preclinical, small molecule drug discovery platform that simultaneously optimizes pharmacokinetics, toxicity, protein-ligand interactions, systems-level models, molecular design, and novel compound generation. To achieve this, the ATOM Modeling Pipeline (AMPL) has been developed to enable advanced and emerging machine learning (ML) approaches to build models from diverse historical drug discovery data. This modular pipeline has been designed to couple with a generative algorithm that optimizes multiple parameters necessary for drug discovery. ATOM's approach is to consider the full pharmacology and therapeutic window of the drug concurrently, through computationally-driven design, thereby reducing the number of molecules that are selected for experimental validation. Here, we discuss the role of collaborative efforts such as consortia and public-private partnerships in accelerating cross disciplinary innovation and the development of open-source tools for drug discovery.
Preclinical drug discovery typically takes five and a half years and accounts for about one third of the cost of drug development (Paul et al., 2010). The process is largely empirical with a sequential, iterative approach to optimizing key drug discovery parameters—efficacy, pharmacokinetics (PK), safety, and developability. Millions of molecules are tested, thousands are produced, and most fail to progress in preclinical or clinical settings (Shannon Decker and Atkinson, 2007; Mohs and Greig, 2017). Furthermore, translation from R&D to the clinic is insufficient with a success rate of less than 10%, and safety liabilities and poor efficacy cited as the main causes of attrition (Miller et al., 2017; Lowe, 2019).
Patients are waiting for the field of drug discovery to innovate new processes that will help improve the success rate of pharmaceutical development, lower drug costs, and get medicines to the clinic more quickly. With the average cost of developing a new molecular entity at over $2 billion, in large part due to the costs of failures, researchers are challenged to work outside the conventional slow, sequential, and costly drug development paradigm to better meet the urgent needs of patients (Kramer et al., 2007; Munos, 2009; Mullin, 2014; DiMasi et al., 2016). To increase the generation of successful new molecular entities, a number of groups have called for more innovation around the culture of and approach to drug discovery (Munos, 2006; Papadaki and Hirsch, 2013; Parekh et al., 2015). In particular, because so much of the cost of development stems from the cost of failures, approaches that improve our ability to distinguish early which molecules will ultimately succeed can have a disproportionate impact on improving the output of new medicines illustrate the potential for accelerating drug discovery through artificial intelligence (AI)-driven approaches (Ringel et al., 2013).
The demonstrations of ML for polypharmacological drug design, deep neural nets for predicting quantitative structure-activity relationships (QSAR), and generative molecular design through the use of variational autoencoders and generative adversarial networks (Besnard et al., 2012; Ma et al., 2015; Blaschke et al., 2018) hold great promise. To this end, significant interest has been raised in the application of approaches that combine AI, simulation, and experimentation to drug discovery (Vamathevan et al., 2019). Recognizing the compelling need for a paradigm shift in drug development, the ATOM consortium was established in October 20171. ATOM's founders, the Frederick National Laboratory for Cancer Research (FNLCR, on behalf of the National Cancer Institute), Lawrence Livermore National Laboratory (LLNL, on behalf of the Department of Energy), GSK (GlaxoSmithKline), and the University of California, San Francisco (UCSF), have joined forces to leverage resources toward the common goal of benefiting patients. ATOM is applying an integrated approach to combine capabilities such as high-performance computing, human-relevant in vitro experimentation, data-driven and mechanistic modeling, and curation of pharmacological data toward the development of a novel preclinical drug discovery and development platform.
As the complexity of biomedical research questions has increased, so too has the need to bring together expertise and resources from multiple disciplines and organizations (Cooke et al., 2015). Consequently, several articles by thought leaders have called for more collaboration in the drug development process (Altshuler et al., 2010; Dahlin et al., 2015; Alteri and Guizzaro, 2018; Takebe et al., 2018; Chaturvedula et al., 2019). Open innovation and open-source research strategies which emphasize the value of collaboration and use of both internal and external information, are creating the opportunity for the drug research and development industry to leverage know-how from across organizations (Munos, 2006; Hunter and Stephens, 2010; Owens, 2016). Cross-industry collaboration is particularly important in the application of computational approaches to drug discovery, where for instance, most companies have one or fewer drugs approved per year, far too small a sample size to support these approaches (Munos, 2009). The advantages of bringing together organizations into public-private partnerships (PPP) and consortia include not just scale, but also new-found agility and increased creativity alongside risk reduction and cost sharing (Papadaki and Hirsch, 2013; Slusher et al., 2013; Rosenberg, 2017; Kuchler, 2019). In fact, the US Food and Drug Administration (FDA), acknowledges the critical role of PPPs and consortia with respect to the innovation and modernization of medical product development (Maxfield et al., 2017).
One notable example of cross-sector collaboration is the Merk Molecular Activity Challenge2 where the pharmaceutical company provided contestants with a training set of molecular descriptors and activities and a test set of descriptors only, and spurred the development of innovative ML methods for QSAR (Ma et al., 2015). In the last 2 years, new academic-industry consortia projects have emerged, focusing on applications of ML in drug discovery. The Machine Learning for Pharmaceutical Discovery and Synthesis Consortium, with membership from three Massachusetts Institute of Technology departments and several leading pharmaceutical companies, focuses on the application of ML to automate drug discovery and synthesis3. Summer 2019 saw the start of a new Innovative Medicines Initiative collaborative project led by Janssen, dubbed Machine Learning Ledger Orchestration for Drug Discovery (MELLODDY)4 (Kuchler, 2019). With a 3-year timeframe, the MELLODDY project focuses on employing federated ML to foster sharing data insights while preserving organizational intellectual property. Pharmaceutical industry participants will train models on their own proprietary data and share those models to increase the impact of AI and ML in the industry.
As an open consortium backed by major public entities, the Department of Energy, the National Cancer Institute, and the University of California Office of the President, as well as pharmaceutical leader GSK, the Accelerating Therapeutics for Opportunities in Medicine consortium (ATOM) is committed to creating new tools for drug discovery that can be shared broadly and benefit the public good. Computational approaches to drug design hold the potential to drastically improve the field's ability to generate novel drugs for patients in need. Harnessing advances in computational power and AI, ATOM is building a new, comprehensive, integrated platform for efficient molecular property prediction, optimization, and design. Drawing from team science, open innovation, and open-source concepts, the ATOM platform combines ML, simulation, and experimentation to generate novel drug candidates more rapidly than traditional approaches. ATOM's current scope focuses within the area of preclinical drug discovery, but its outcomes aim to benefit not only the member organizations and their immediate stakeholders, but the biomedical community at large including academicians, start-ups, private industry, clinicians, and patients.
Drug discovery is relying increasingly on computational and AI-driven methods. Collaborative efforts that combine scientific know-how and computational power are being stood up to incubate innovative methods while sharing risk and accelerating progress. In the past decade significant advances have been made to accelerate the drug discovery process such as the development of computational and AI-based methods for virtual screening and in silico drug design. Moving beyond structure-based approaches and virtual screens, several seminal publications have demonstrated the use of generative adversarial networks and variational autoencoders for de novo drug design (Kadurin et al., 2017; Olivecrona et al., 2017; Gómez-Bombarelli et al., 2018; Merk et al., 2018; Polykovskiy et al., 2018; Putin et al., 2018; Segler et al., 2018; Ståhl et al., 2019; Hong et al., 2020). For example, a recently published deep generative model demonstrated the design of small-molecule drug candidates for discoidin domain receptor 1 prioritizing synthetic feasibility, efficacy, and uniqueness with respect to known small molecules, showcasing the ability to rapidly discover drugs at low cost (Zhavoronkov et al., 2019).
The promise of AI-driven drug design carries with it, several challenges—the need for appropriate datasets, ability to generate and test evolving biological hypotheses, multi-parameter optimization, reduction in design-make-test-analyze cycle times, and adaptability of research culture (Schneider et al., 2020). ATOM is tackling these challenges through the collaborative development of a preclinical, open-source, small-molecule drug discovery platform (Chaturvedula et al., 2019). The initial stages have focused on building computational infrastructure, curating preclinical data from both GSK and public sources, and creating and testing data-driven modeling capabilities.
ATOM has developed a data-driven modeling pipeline capable of rapidly building and optimizing ML models for bioassay activity and molecular property predictions. This modeling pipeline is important for developing predictive models for public and private pharmaceutical assay datasets. While ML-based techniques to predict drug properties from structures are regularly used in the field of computational drug design, there remains a need for an automated modular pipeline for common modeling tasks. Some key features for such a software package are to enable reproducibility, incorporate new models, support a variety of chemical representations, allow for hyperparameter optimization, and validate predictive performance (Dahl et al., 2014; Gilmer et al., 2017; Feinberg et al., 2018; Yang et al., 2019).
Existing commercial pipeline tools such as BIOVIA Pipeline Pilot are limited in their customizability and can be cost prohibitive to small academic research groups and start-up companies5. On the other end of the spectrum, open-source pipeline tools such as KNIME are useful as GUI-based platforms for data processing, model fitting, and analysis, (Berthold, 2008) but have yet to demonstrate the suitability for large scale model generation.
AMPL6, or the ATOM Modeling Pipeline, extends the popular DeepChem7 library and supports ML and molecular featurization tools (Minnich et al., 2020). AMPL is implemented as a Python library that integrates with existing data science ecosystems and utilities. AMPL automates and optimizes many common ML model fitting tasks that are performed for pharmaceutical datasets including model fitting, validation, and prediction. AMPL allows researchers to reproducibly train and test models, incorporate new models, and provide utilities for automated dataset characterization, model validation, and uncertainty quantification. AMPL is designed to be a versatile library that can interface with many services and tools.
AMPL allows users to build in silico models based on molecular properties to aid in drug discovery. With an initial focus on safety and pharmacokinetic modeling, AMPL has been extensively tested on activity and property assay datasets. In preparation for the initial release of the pipeline, 11,552 regression and classification models were built to evaluate data splitting algorithms, model types, and feature types (Minnich et al., 2020). AMPL supports a wide variety of dataset splitting algorithms for validation and testing, including random splits, Butina clustering, scaffold splits, and temporal splits. AMPL uses models from scikit-learn and DeepChem including random forest, XGBoost, fully connected neural network, and graph convolution neural network models. Small molecules were represented as SMILES strings using the RDKit cheminformatics library and the molecule validation and standardization tool, MolVS. AMPL's data curation module was applied to datasets to filter out compound assay values with wide variability, and to characterize the datasets with Tanimoto distances between chemical fingerprints or Euclidean distances between descriptor feature vectors. Several featurization approaches were compared including Extended Connectivity Fingerprints (ECFP), DeepChem graph convolution latent vectors, Mordred chemical descriptors, and Molecular Operating Environment (MOE) descriptors. Due to the modular nature of AMPL's implementation, extensions to the pipeline are available for additional splitting algorithms, model types, and feature types.
Hyperparameter optimization is an important task for cheminformatics ML model fitting that may improve model predictive performance. AMPL supports basic hyperparameter optimization functions including searches using basic linear grids, logistic grids, random searches, and user-specified searches. Model fitting for safety and pharmacokinetic parameters used AMPL's hyperparameter optimization module to explore model parameter combinations. Generally, hyperparameter optimization improved predictive performance on properties of external test sets except for certain cases with limited data or ECFP featurization.
AMPL automatically calculates standard model performance metrics for regression and classification models. The regression performance statistics include R2, mean absolute error, and mean square error to evaluate the level of agreement between the model predicted values and actual experimental ground truth values. AMPL also includes classification performance metrics such as precision and recall, area under the precision-recall curve (PRC-AUC), negative predictive value, cross entropy, and accuracy metrics. As previously described, model prediction uncertainty was calculated for several of PK datasets for comparison with model prediction error (Minnich et al., 2020). AMPL enables this type of uncertainty quantification analysis toward better understanding model predictions, uncertainty, and error.
AMPL is open-source, modular, and flexible, allowing for additions or extensions as needed. This makes data-driven modeling using modern ML libraries accessible to the wider scientific community including academic or government laboratories and small companies. AMPL is now available for download on Github8. The website includes detailed library documentation as well as example Jupyter notebooks to learn to use the pipeline.
Bioassay data, specifically the half-maximal effective drug concentration (EC50), and the half-maximal inhibitory drug concentration (IC50), of known hepatic, central nervous system, cardiovascular, and cellular toxicity safety liabilities were used to benchmark safety models. Models were fit for assays such as BSEP, β2 adrenoceptor, muscarinic acetylcholine receptor, dopamine D2, voltage-gated potassium channels, and phospholipidosis induction. For each assay type, model hyperparameters were optimized resulting in 2,130 classification models with thresholds appropriate set for each assay. As described by Minnich et al, the predictive performance of the classification models was evaluated using common validation statistics including receiver operating characteristic area under the curve (ROC AUCs) built on safety datasets. Predictive performance varied based on assay type, dataset size, dataset split type, feature type, and model type, but overall produced many useful models for pharmaceutical safety properties (Minnich et al., 2020).
A diverse set of pharmacokinetic data including blood-to-plasma ratio, plasma protein binding, in vivo clearance, volume of distribution, hepatocyte clearance, and microsomal clearance, logD was used to fit predictive models with AMPL (Minnich et al., 2020). Nine thousand four hundred twenty-two regression models were fit for all the assay types and corresponding model parameters were evaluated for improvements to predictive performance as described by Minnich et al. General trends between different training and test splits, feature types, and model types were examined. When using neural network models with calculated descriptors for many of these PK datasets, model predictions with MOE descriptors were slightly better than predictions with open-source Mordred descriptors. Several PK datasets with larger numbers of measurements (10,000 or more) benefitted from DeepChem's graph convolutional neural network models with better predictions compared to experiment than ECFP or calculated descriptors. For smaller PK datasets, random forest models with MOE descriptors had slightly better performance than other feature and model combinations (Minnich et al., 2020).
AMPL is designed to automatically and rapidly build and evaluate cheminformatics models. Automation of deep learning model training, parallelized hyperparameter search, performance benchmarking, and data and model storage are essential for reproducible ML predictions in drug discovery. Given the wide range of activity and property assay types, the validation performed by Minnich et al. demonstrate there is no single best model fitting approach for every dataset. This underscores the need to rapidly search and fit predictive models for new datasets enabled by the AMPL software suite.
Two examples of model fitting on publicly accessible datasets are available with the AMPL repository. Each example describes a general method of curating datasets, fitting a ML model, and using the created model for new predictions. Example code is included to download the datasets from their original source, perform basic curation on the datasets, train a model on the curated datasets, and then load the fitted model for prediction on a withheld test set. In the first example, AMPL mimicked a DeepChem example model by fitting a model to a public aqueous solubility dataset using DeepChem's graph convolutional neural network model (Delaney, 2004). In a second example, AMPL was used to fit a predictive neural network model using Mordred descriptors for human liver microsomal clearance from a public PK dataset (Wenzel et al., 2019). The entire process of data curation to analysis and visualization for these sample datasets is automated and reproducible with the AMPL library and tools.
AMPL models can be applied toward related compounds to rapidly predict bioassay activity or safety and pharmacokinetic properties. In the context of ATOM, AMPL is a key component in the overall mission to accelerate the drug discovery process.
Given heavy reliance on expensive and lengthy experimentation, the field of drug discovery is increasingly integrating both computational and AI-driven methods for virtual screening and in silico drug design. Further, the application of deep neural network architectures in generative design in conjunction with data-driven and mechanistic modeling for functional property prediction and an in silico framework for rapid lead optimization will drastically change how drug discovery is done.
Collaborative efforts have been employed in recent efforts to develop new capabilities where risks and required investment have been high. ATOM provides an avenue for collaborative AI-driven drug discovery that results in an open-source framework that broadens availability and an opportunity to raise the level of collaborative drug discovery efforts.
The AMPL serves as the initial step toward the development of an open-source preclinical drug design platform that will accelerate the process of getting more effective therapies to patients. Future efforts involve extending the modeling capability of AMPL toward the development of an open-source pre-clinical drug discovery platform (Figure 1).
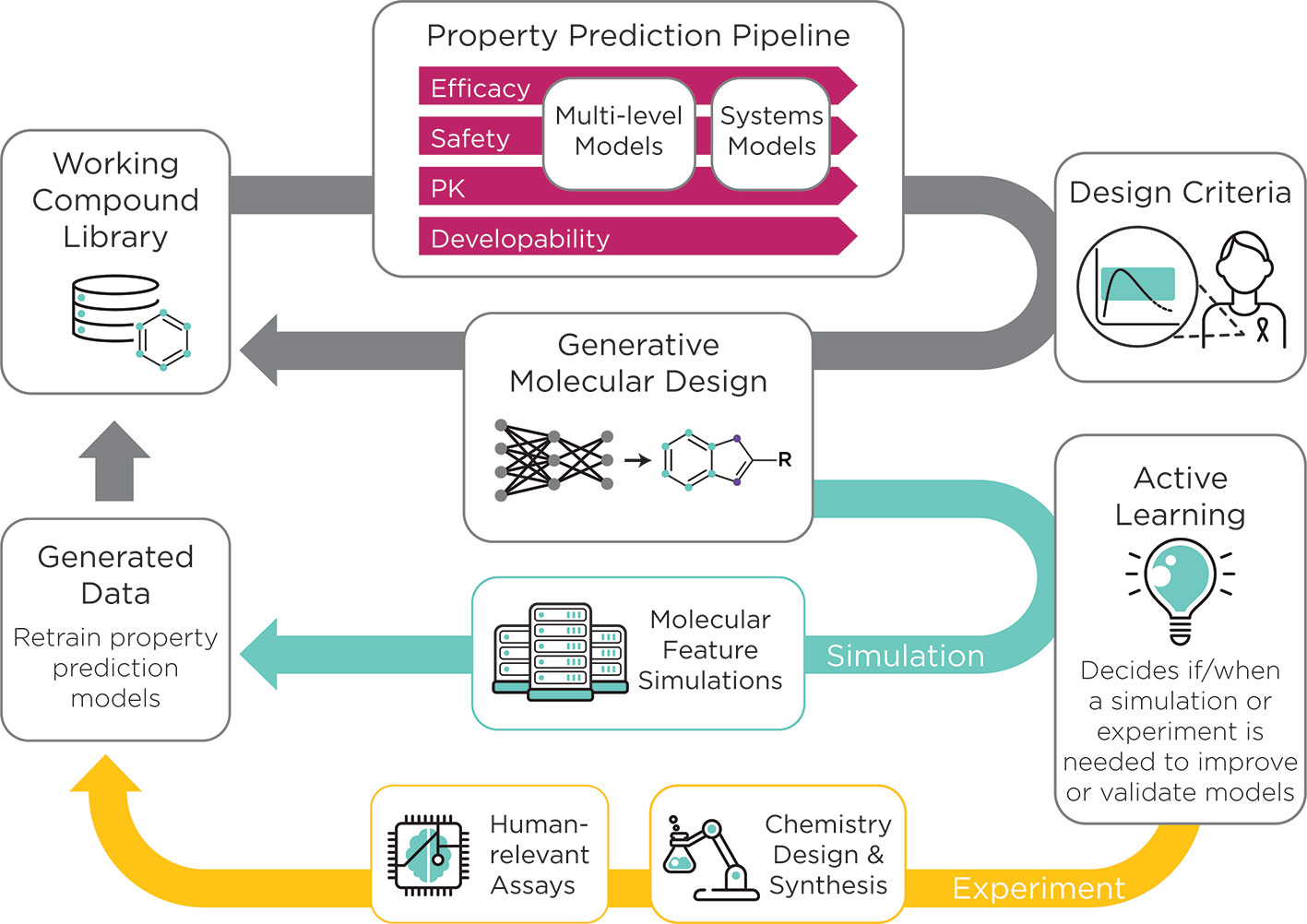
Figure 1 The ATOM preclinical drug discovery workflow. ATOM is developing an active learning drug discovery framework that uses a compound library as input to a property prediction pipeline. The pipeline begins with historic data collected on a working compound library to train machine learning-based models for property prediction. Next, multi-level and systems-level models of efficacy, safety, and pharmacokinetics as well as developability are integrated to generate a set of drug design criteria. These parameters are simultaneously optimized for the generation of novel molecules by the generative molecular design framework. The multi-parameter optimization loop, in grey, can be run for numerous cycles. An active learning approach is used to decide whether a molecular simulation or experiment is needed to improve or validate the models. Data that result from these simulations and experiments are then used to re-train the property prediction models. The result of this workflow is a set of optimized drug candidates.
At ATOM, efforts are underway to integrate current and emerging computational capabilities with active learning in an AI-driven platform. ATOM is creating a generative molecular design framework that integrates predictive models from AMPL and initiates cycles of generative molecular design and multiparameter optimization. The goal of ATOM's generative molecular design framework is to propose novel small-molecule drug candidates with optimized properties based on design criteria such as potency, selectivity, cardiotoxicity, hepatoxicity, solubility, clearance, and synthetic accessibility9. New experimental and molecular simulation data will be selectively acquired to support the ML-based approach and will be integrated into the computational pipeline to kick start additional cycles of the molecular design and optimization. The integration of active learning will streamline time-consuming and costly experimentation and will guide the design of novel drug candidates (Figure 1). Collectively, these efforts usher in a paradigm shift in drug discovery that emphasizes collaboration, innovation, and the development of open-source tools.
IH: manuscript writing and figure design. BM: manuscript writing. ES and ATOM consortium: manuscript and figure revision, approval of final manuscript.
This project has been funded in whole or in part with funds from University of California, San Francisco, funds from GlaxoSmithKline, federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E, and federal funds from U.S. Department of Energy, National Nuclear Security Administration under contract DE-AC52-07NA27344. The federal funds include the Cancer Moonshot funds that Congress passed the 21st Century Cures Act in December 2016, authorizing $1.8 billion in funding for the Cancer Moonshot over 7 years. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services nor the Department of Energy; nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. Frederick National Laboratory is a Federally Funded Research and Development Center operated by Leidos Biomedical Research, Inc., for the National Cancer Institute. Lawrence Livermore National Laboratory is operated by Lawrence Livermore National Security, LLC, for the Department of Energy, National Nuclear Security Administration.
The authors declare that this study received funding from GSK. The funder had the following involvement with the study: GSK is a founding member of ATOM and has contributed to R&D and consortium activities at ATOM.
We express thanks to ATOM team members John Baldoni, Frank Blanchard, Stacie Calad-Thomson, Ethan Dmitrovsky, Leonard Freedman, Mary Ellen Hackett, Deirdre Olynick, Michael Ringel, Tom Rush, and Jeremy Thomas for their advice on the manuscript. We thank the ATOM team for their research, technical, and partnership contributions to the projects described within. We also thank Frederick National Laboratory for Cancer Research, GlaxoSmithKline, Lawrence Livermore National Laboratory, University of California San Francisco, the National Cancer Institute, the Department of Energy, and the University of California Office of the President for guidance and consortium support.
Alteri, E., Guizzaro, L. (2018). Be open about drug failures to speed up research. Nature 563 (7731), 317–319. doi: 10.1038/d41586-018-07352-7
Altshuler, J. S., Balogh, E., Barker, A. D., Eck, S. L., Friend, S. H., Ginsburg, G. S., et al. (2010). Opening up to precompetitive collaboration. Sci. Transl. Med. 2 (52), 52cm26. doi: 10.1126/scitranslmed.3001515
Berthold, M. R. E. A. (2008). “KNIME: The Konstanz Information Miner,” in Data Analysis, Machine Learning and Applications. Studies in Classification, Data Analysis, and Knowledge Organization. Eds. Preisach, C. B. H., Schmidt-Thieme, L., Decker, R. (Berlin, Heidelberg: Springer).
Besnard, J., Ruda, G. F., Setola, V., Abecassis, K., Rodriguiz, R. M., Huang, X.-P., et al. (2012). Automated design of ligands to polypharmacological profiles. Nature 492 (7428), 215–220. doi: 10.1038/nature11691
Blaschke, T., Olivecrona, M., Engkvist, O., Bajorath, J., Chen, H. (2018). Application of Generative Autoencoder in De Novo Molecular Design. Mol. Inf. 37 (1-2), 1700123. doi: 10.1002/minf.201700123
Chaturvedula, A., Calad-Thomson, S., Liu, C., Sale, M., Gattu, N., Goyal, N. (2019). Artificial Intelligence and Pharmacometrics: Time to Embrace, Capitalize, and Advance? CPT Pharmacometr. Syst. Pharmacol. 8 (7), 440–443. doi: 10.1002/psp4.12418
Cooke, N. J., Hilton, M. L., National Research Council (U.S.), Committee on the Science of Team Science (2015). “Enhancing the effectiveness of team science” (Washington, D.C: The National Academies Press).
Dahl, G. E., Jaitly, N., Salakhutdinov, R. (2014). Multi-task Neural Networks for QSAR Predictions. ArXiv E-prints. [Online]. Available: https://ui.adsabs.harvard.edu/abs/2014arXiv1406.1231D [Accessed June 01, 2014].
Dahlin, J. L., Inglese, J., Walters, M. A. (2015). Mitigating risk in academic preclinical drug discovery. Nat. Rev. Drug Discovery 14 (4), 279–294. doi: 10.1038/nrd4578
Delaney, J. S. (2004). ESOL: Estimating Aqueous Solubility Directly from Molecular Structure. J. Chem. Inf. Comput. Sci. 44 (3), 1000–1005. doi: 10.1021/ci034243x
DiMasi, J. A., Grabowski, H. G., Hansen, R. W. (2016). Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ. 47, 20–33. doi: 10.1016/j.jhealeco.2016.01.012
Feinberg, E. N., Sur, D., Wu, Z., Husic, B. E., Mai, H., Li, Y., et al. (2018). PotentialNet for Molecular Property Prediction. ACS Cent. Sci. 4 (11), 1520–1530. doi: 10.1021/acscentsci.8b00507
Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O., Dahl, G. E. (2017). Neural message passing for Quantum chemistry (Sydney, NSW, Australia: JMLR.org).
Gómez-Bombarelli, R., Wei, J. N., Duvenaud, D., Hernández-Lobato, J. M., Sánchez-Lengeling, B., Sheberla, D., et al. (2018). Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 4 (2), 268–276. doi: 10.1021/acscentsci.7b00572
Hong, S. H., Ryu, S., Lim, J., Kim, W. Y. (2020). Molecular Generative Model Based on an Adversarially Regularized Autoencoder. J. Chem. Inf. Model 60 (1), 29–36. doi: 10.1021/acs.jcim.9b00694
Hunter, J., Stephens, S. (2010). Is open innovation the way forward for big pharma? Nat. Rev. Drug Discovery 9 (2), 87–88. doi: 10.1038/nrd3099
Kadurin, A., Aliper, A., Kazennov, A., Mamoshina, P., Vanhaelen, Q., Khrabrov, K., et al. (2017). The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 8 (7), 10883–10890. doi: 10.18632/oncotarget.14073
Kramer, J. A., Sagartz, J. E., Morris, D. L. (2007). The application of discovery toxicology and pathology towards the design of safer pharmaceutical lead candidates. Nat. Rev. Drug Discovery 6 (8), 636–649. doi: 10.1038/nrd2378
Kuchler, H. (2019). Pharma groups combine to promote drug discovery with AI. Financial Times June 4, 2019.
Lowe, D. (2019). The Latest on Drug Failure and Approval Rates. In The Pipeline [Online]. Available from: https://blogs.sciencemag.org/pipeline/archives/2019/05/09/the-latest-on-drug-failure-and-approval-rates 2019].
Ma, J., Sheridan, R. P., Liaw, A., Dahl, G. E., Svetnik, V. (2015). Deep Neural Nets as a Method for Quantitative Structure–Activity Relationships. J. Chem. Inf. Model. 55 (2), 263–274. doi: 10.1021/ci500747n
Maxfield, K. E., Buckman-Garner, S., Parekh, A. (2017). The Role of Public-Private Partnerships in Catalyzing the Critical Path. Clin. Transl. Sci. 10 (6), 431–442. doi: 10.1111/cts.12488
Merk, D., Friedrich, L., Grisoni, F., Schneider, G. (2018). De Novo Design of Bioactive Small Molecules by Artificial Intelligence. Mol. Inf. 37 (1-2), 1700153. doi: 10.1002/minf.201700153
Miller, S. M., Moos, W. H., Munk, B. H., Munk, S. A. (2017). “10 - Drug discovery: Chaos can be your friend or your enemy,” in Managing the Drug Discovery Process. Eds. Moos, W. H., Miller, S. M., Munk, B. H., Munk, S. A. (Woodhead Publishing), 183–279. doi: 10.1016/B978-0-08-100625-2.00010-6
Minnich, A. J., McLoughlin, K., Tse, M., Deng, J., Weber, A., Murad, N., et al. (2020). AMPL: A Data-Driven Modeling Pipeline for Drug Discovery. J. Chem. Inf. Model. 60 (4), 1955–1968. doi: 10.1021/acs.jcim.9b01053
Mohs, R. C., Greig, N. H. (2017). Drug discovery and development: Role of basic biological research. Alzheimers Dement (N. Y.) 3 (4), 651–657. doi: 10.1016/j.trci.2017.10.005
Mullin, R. (2014). Tufts Study Finds Big Rise In Cost Of Drug Development. Chem. Eng. News. Available: https://cen.acs.org/articles/92/web/2014/11/Tufts-Study-Finds-Big-Rise.html [Accessed 2019].
Munos, B. (2006). Can open-source R&D reinvigorate drug research? Nat. Rev. Drug Discovery 5 (9), 723–729. doi: 10.1038/nrd2131
Munos, B. (2009). Lessons from 60 years of pharmaceutical innovation. Nat. Rev. Drug Discovery 8 (12), 959–968. doi: 10.1038/nrd2961
Olivecrona, M., Blaschke, T., Engkvist, O., Chen, H. (2017). Molecular de-novo design through deep reinforcement learning. J. Cheminformat. 9 (1), 48. doi: 10.1186/s13321-017-0235-x
Papadaki, M., Hirsch, G. (2013). Curing consortium fatigue. Sci. Transl. Med. 5 (200), 200fs235. doi: 10.1126/scitranslmed.3006903
Parekh, A., Buckman-Garner, S., McCune, S., R. O. N., Geanacopoulos, M., Amur, S., et al. (2015). Catalyzing the Critical Path Initiative: FDA's progress in drug development activities. Clin. Pharmacol. Ther. 97 (3), 221–233. doi: 10.1002/cpt.42
Paul, S. M., Mytelka, D. S., Dunwiddie, C. T., Persinger, C. C., Munos, B. H., Lindborg, S. R., et al. (2010). How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat. Rev. Drug Discovery 9 (3), 203–214. doi: 10.1038/nrd3078
Polykovskiy, D., Zhebrak, A., Vetrov, D., Ivanenkov, Y., Aladinskiy, V., Mamoshina, P., et al. (2018). Entangled Conditional Adversarial Autoencoder for de Novo Drug Discovery. Mol. Pharmaceut. 15 (10), 4398–4405. doi: 10.1021/acs.molpharmaceut.8b00839
Putin, E., Asadulaev, A., Ivanenkov, Y., Aladinskiy, V., Sanchez-Lengeling, B., Aspuru-Guzik, A., et al. (2018). Reinforced Adversarial Neural Computer for de Novo Molecular Design. J. Chem. Inf. Model. 58 (6), 1194–1204. doi: 10.1021/acs.jcim.7b00690
Ringel, M., Tollman, P., Hersch, G., Schulze, U. (2013). Does size matter in R&D productivity? If not, what does? Nat. Rev. Drug Discovery 12, 901. doi: 10.1038/nrd4164
Rosenberg, A. (2017). “UC launches drug discovery consortium”. (Los Angeles, CA: University of California Newsroom).
Schneider, P., Walters, W. P., Plowright, A. T., Sieroka, N., Listgarten, J., Goodnow, R. A., et al. (2020). Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discovery 19 (5), 353–364. doi: 10.1038/s41573-019-0050-3
Segler, M. H. S., Kogej, T., Tyrchan, C., Waller, M. P. (2018). Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS Cent. Sci. 4 (1), 120–131. doi: 10.1021/acscentsci.7b00512
Shannon Decker, E. A. S., Atkinson, D. R. A. J. (2007). “Chapter 28 - Drug Discovery”, in Principles of Clinical Pharmacology, 2nd ed. Eds. Daniels, C. E., Dedrick, R. L., Markey, S. P. (Academic Press), 439–447. doi: 10.1016/B978-012369417-1/50068-7
Slusher, B. S., Conn, P. J., Frye, S., Glicksman, M., Arkin, M. (2013). Bringing together the academic drug discovery community. Nat. Rev. Drug Discovery 12 (11), 811–812. doi: 10.1038/nrd4155
Ståhl, N., Falkman, G., Karlsson, A., Mathiason, G., Boström, J. (2019). Deep Reinforcement Learning for Multiparameter Optimization in de novo Drug Design. J. Chem. Inf. Model. 59 (7), 3166–3176. doi: 10.1021/acs.jcim.9b00325
Takebe, T., Imai, R., Ono, S. (2018). The Current Status of Drug Discovery and Development as Originated in United States Academia: The Influence of Industrial and Academic Collaboration on Drug Discovery and Development. Clin. Transl. Sci. 11 (6), 597–606. doi: 10.1111/cts.12577
Vamathevan, J., Clark, D., Czodrowski, P., Dunham, I., Ferran, E., Lee, G., et al. (2019). Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discovery 18 (6), 463–477. doi: 10.1038/s41573-019-0024-5
Wenzel, J., Matter, H., Schmidt, F. (2019). Predictive Multitask Deep Neural Network Models for ADME-Tox Properties: Learning from Large Data Sets. J. Chem. Inf. Model. 59 (3), 1253–1268. doi: 10.1021/acs.jcim.8b00785
Yang, K., Swanson, K., Jin, W., Coley, C., Eiden, P., Gao, H., et al. (2019). Analyzing Learned Molecular Representations for Property Prediction. J. Chem. Inf. Model. 59 (8), 3370–3388. doi: 10.1021/acs.jcim.9b00237
Keywords: artificial intelligence, machine learning, drug discovery and development, data science, in silico modeling
Citation: Hinkson IV, Madej B and Stahlberg EA (2020) Accelerating Therapeutics for Opportunities in Medicine: A Paradigm Shift in Drug Discovery. Front. Pharmacol. 11:770. doi: 10.3389/fphar.2020.00770
Received: 08 October 2019; Accepted: 11 May 2020;
Published: 30 June 2020.
Edited by:
Alex Zhavoronkov, Biogerontology Research Foundation, United KingdomReviewed by:
Evgeny Putin, Insilico Medicine, Hong KongCopyright © 2020 Hinkson, Madej and Stahlberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric A. Stahlberg, ZXJpYy5zdGFobGJlcmdAbmloLmdvdg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.