 Kenny M. Van Theemsche
Kenny M. Van Theemsche Dieter V. Van de Sande†
Dieter V. Van de Sande† Alain J. Labro
Alain J. Labro- Laboratory of Molecular, Cellular, and Network Excitability, University of Antwerp, Antwerp, Belgium
In the Nav channel family the lipophilic drugs/toxins binding sites and the presence of fenestrations in the channel pore wall are well defined and categorized. No such classification exists in the much larger Kv channel family, although certain lipophilic compounds seem to deviate from binding to well-known hydrophilic binding sites. By mapping different compound binding sites onto 3D structures of Kv channels, there appear to be three distinct lipid-exposed binding sites preserved in Kv channels: the front and back side of the pore domain, and S2-S3/S3-S4 clefts. One or a combination of these sites is most likely the orthologous equivalent of neurotoxin site 5 in Nav channels. This review describes the different lipophilic binding sites and location of pore wall fenestrations within the Kv channel family and compares it to the knowledge of Nav channels.
Introduction
Voltage-gated ion channels are transmembrane proteins that are selectively permeable to physiological important ions such as Na+, K+, Ca2+, and Cl-. Under influence of the membrane potential (Vm) these channels change their conductance. In the conductive open (or activated) state, ions flow down their electrochemical gradient through the channel pore. The flux of these ions elicits an electrical current that directly influences the Vm. For voltage-gated sodium and potassium channels (Nav and Kv), the main focus of this review, the Vm will shift towards the ion's equilibrium potential, which under normal conditions is depolarizing and repolarizing, respectively (Hille, 2001). Although Nav and Kv channels differ in selectivity from one another, their structure is quite similar. However, a main difference is that Nav channels are characterized by one large α-subunit containing four recognizable domains (DI–IV), whereas Kv channels are formed by tetramerization of four individual α-subunits. In both cases, these four entities comprise six transmembrane segments (S1–S6), which are divided into a voltage sensing domain (VSD, S1–S4) and a pore-forming domain (PD, S5–S6) that are connected by the S4–S5 linker. The four PDs assemble into the ion permeation pathway (or pore) that is surrounded by four VSDs. In the non-conductive closed (or deactivated) state, ion permeation is prevented by the intracellular activation gate, located at the point where the four S6 helices cross. The aperture-like opening and closure of this gate is controlled by the VSD (Doyle et al., 1998; Bavro et al., 2012; Labro and Snyders, 2012; Lenaeus et al., 2017). The main component of the VSD is the S4 segment that physically moves in response to a change in Vm, due to the presence of positively charged residues (arginine and lysine) that detect changes in the membrane electric field (Bezanilla, 2008). The S4–S5 linker is a component of the electro-mechanical coupling that translates S4 movements into opening or closing of the activation gate (Bezanilla, 2008; Blunck and Batulan, 2012). After opening, fast inactivation occurs in Nav and in some Kv channels, which is caused by the physical occlusion of the pore by an inactivation particle. For Nav channels this inactivation particle is the linker between DIII and DIV, while in Kv channels it is located at the N-terminus of each subunit, hence termed N-type inactivation (Hoshi et al., 1990; West et al., 1992; Armstrong and Hollingworth, 2018). Alternatively, Kv channel inactivation can occur via the slower C- or U-type inactivation mechanism that makes the channels non or less conductive (Hoshi et al., 1991; Klemic et al., 1998; Cuello et al., 2010).
Binding of drugs/toxins to Nav and Kv channels may alter the activation, deactivation, and/or inactivation process(es), which may cause or alleviate aberrant electrical excitability. Therefore, knowledge about the different binding sites is key for drug development and pharmacovigilance. The binding sites for these drugs/toxins are well defined and categorized within the Nav channel family, as opposed to the much larger Kv channel family. Most binding sites are enveloped by water, locating either inside or outside the channel's pore. However, some compounds bind to a site(s) that does not fit any of the hydrophilic binding sites. For instance, brevetoxins and ciguatoxins bind to a conserved hydrophobic site within the Nav channel family, termed neurotoxin site 5 (Catterall and Risk, 1981; Cestele and Catterall, 2000). For the Kv channel family no such site has been described, but certain compounds have been shown to deviate from binding to hydrophilic binding sites like; retigabine, gambierol, psora-4, polyunsaturated fatty acids (PUFAs), ICA-compounds (Vennekamp et al., 2004; Kopljar et al., 2009; Lange et al., 2009; Borjesson and Elinder, 2011). It is notable that these are rather lipophilic compounds and there has been, and still is, a growing interest in such compounds for their use in treating neurological disorders (e.g., as anti-convulsant).
So, is there a unifying picture of the lipid exposed/accessible drug/toxin binding sites within the large Kv channel family and even between Kv and Nav channels? Several lipophilic binding sites have been described in different Kv channels, while in fact some may converge to just one binding region preserved between Kv channel (sub)families. In this review, the well-documented Nav lipophilic binding sites, neurotoxin site 2, site 5, and the access to the local anaesthetic (LA) binding site within the pore through fenestrations is compared to what has been reported for Kv channels.
Voltage-Gated Sodium Channels
The Nav channel family contains nine isoforms (Nav1.1 to Nav1.9) that display a high sequence homology, especially within the transmembrane segments (Marban et al., 1998; Ahern et al., 2016). This facilitated the categorization of drug/toxin binding sites within the Nav channel family. Over the past decades a detailed picture emerged on where compounds bind within these channels and resulted in a well-documented classification of seven different sites (site 1 to 7) and a LA binding site (Stevens et al., 2011; De Lera Ruiz and Kraus, 2015). As the focus of this review is on the binding sites that involve lipid soluble and/or transmembrane binding compounds only binding site 5 and site 2 will be briefly discussed. The LA binding site is also mentioned as some compounds can reach their binding site via hydrophobic fenestrations in the pore wall of the channel protein. To maintain an orderly overview, all Nav residues are numbered according to the Nav1.4 channel when possible. In case the sequence could not be aligned, as for bacterial Nav structures, it will be noted and the original numbering is maintained.
The Closed State Accessible LA Binding Site: Pore-Accessibility Through Channel Fenestrations
LA compounds and anti-arrhythmic drugs inhibit Nav channels by occlusion of the pore. Most LA compounds have a similar structure consisting of a tertiary hydrophilic amine domain (head) linked with an aromatic hydrophobic ring domain, with a total length of 10–15Å (Courtney, 1988). Three types of block can be observed. First type is the use dependent open state block, or high affinity block, which occurs after channel opening and LA compounds enter the pore via the intracellular side (Grant et al., 1989; Benz and Kohlhardt, 1991; Gingrich et al., 1993). The second type is flicker block, or fast block, which is only observed when the channel's inactivation process is modulated. For example, by other compounds such as batrachotoxin (BTX) (CAS No.:23509-16-2) that binds at site 2 (Cahalan, 1978; Uehara and Moczydlowski, 1986; Zamponi et al., 1993). The third and least common type is the resting or hydrophilic block that establishes when the channel is closed. LA compounds are thought to enter the ion conductive pore and find their binding site trough fenestrations in the lipid exposed part of the PD (Gamal El-Din et al., 2018). LA compounds are protonated but for crossing the cell membrane they need to be deprotonated. This is possible when their pKA value is close to the physiological pH values in the extracellular environment. After traversing the cell membrane the compounds are protonated again to exert their pharmacological effect (Hille, 1977). As the binding site is located within the pore, block occurs with the highest affinity when the channel is opened or used, while applying frequent channel activating stimuli. Two residues within the S6 of domain four (DIV-S6) have been identified to be important for LA binding/modulation, namely, F1586 and Y1593, (Nav1.4 numbering). F1586 probably binds to the alkylamino head, while Y1593 interacts with the aromatic ring structure (Ragsdale et al., 1994; Yarov-Yarovoy et al., 2002) (Figures 1A, B). As these residues reside within the pore and certain compounds display closed state block, this requires a hydrophobic pathway to the binding site when the channel gate is closed (Hille, 1977).
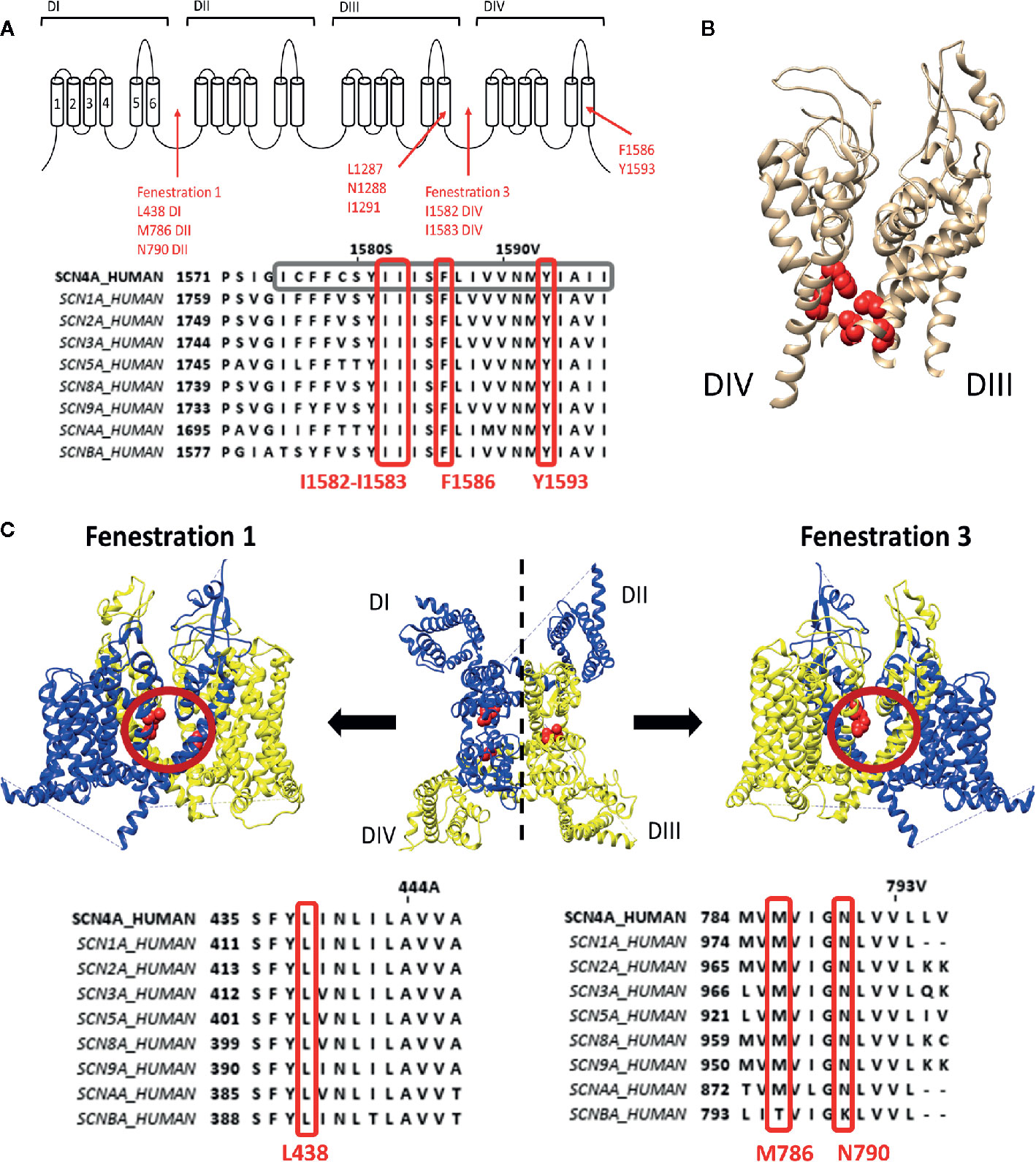
Figure 1 Representation of the local anaesthetic (LA) binding site and the hydrophobic access paths or fenestrations. (A) Schematic representation of the Nav channel topology which contains four domains (DI to DIV) each consisting out of six transmembrane segments (S1–S6). Regions and location of residues important for LA binding are indicated with red arrows. Fenestrations that are sufficiently large to allow passage of LA compound are between DI–DII (fenestration 1) and DIII–DIV (fenestration 3) with the bottleneck residues listed below. This schematic representation is then followed by an alignment of Nav1.4 DIV with the other human isoforms, with marked in grey S6 and marked in red well conserved residues for LA binding and the fenestration bottleneck (I1582–I1583). Mutation of I1582 can also create a pathway connecting the inner pore with the extracellular environment. (B) 3D structure of the Nav1.4 channel representing the S5–S6 segments of DIII and DIV. In red are the residues, forming the LA binding site, visualized which are listed in panel A, clearly marking the inner pore LA binding site. (C) In the middle a top view of the 3D structure of the Nav1.4 channel is shown with in blue the DI–DII domains and in yellow the DIII-DIV domains. Fenestration 1 locates between DI and DII a side view of it shown on the left. A side view of fenestration 3 is shown on the right. Both fenestrations are highlighted by a red circle and the residues responsible for creating the bottleneck of the fenestration are represented in red. Below are the alignments of Nav1.4 with the other isoforms. On the left the bottleneck residue of DI is marked in red, on the right the bottleneck residues of DII. Amino acid sequence alignment and 3D structures are visualized using Jalview and chimera software, respectively (Pettersen et al., 2004; Waterhouse et al., 2009).
These pathways, or fenestrations, in the lipid exposed part of the PD were first observed in the crystal structure of bacterial Nav channels such as NavAb (Payandeh et al., 2011; Payandeh et al., 2012), NavMs (Mccusker et al., 2012) and NavRh (Zhang et al., 2012). It should be noted that bacterial Nav channels are constructed out of four separate α-subunits instead of one large subunit. For the bacterial Nav channels, four fenestrations are observed between the different domains where the radius of the fenestration varies from 0.8Å minimally to 2.59-2.83Å (Kaczmarski and Corry, 2014). Nonetheless, these fenestrations are wide enough for small LA compounds and anti-arrhythmic drugs to pass. The narrowest point in the fenestration, termed “bottleneck”, is created by the amino acid residues M174, T175, F203, T206, and M209 (NavAb numbering), with F203 being the most important residue (Figure 1C). These amino acids will sterically hinder the passage of compounds through the fenestration. Mutation of F203 to an alanine increased the size of the fenestration allowing easier access of flecainide (CAS No.:54143-55-4, polar surface area=59.6Å) to its binding site within the pore, with as result an increased tonic, closed state, block (Gamal El-Din et al., 2018). The mutation did not affect lidocaine block as this compound is smaller (polar surface area=32.3Å) and can easily traverse the wild type fenestration (Courtney, 1988). The F203W mutation on the other hand reduces fenestration size and consequently the access of both lidocaine and flecainide is reduced, decreasing tonic block (Gamal El-Din et al., 2018). Computational modelling using the Nav1.4 3D structure resulted in the observation of four fenestrations just as in bacterial Nav channels, with the exception that the size of two out of the four fenestrations seemed inadequate for compound access, namely the fenestration constructed by DII–DIII and DIV–DI (Kaczmarski and Corry, 2014). Fenestrations in between DI-DII and DIII-DIV seemed sufficiently wide for compounds to cross (Figure 1C). The bottleneck of these fenestrations is formed by residues N790, L438, and M786 for the fenestration between DI-DII and by I1582, I1583, and F1586 (if rotated) for the fenestration between DIII-DIV. These fenestrations lining residues, are well conserved in the different Nav channel isoforms. Mutating DIV-S6 residue I1582 appeared to create an extra pore access pathway (Ragsdale et al., 1994) and allowed the external blocker QX314 (CAS No.:24003-58-5, polar surface area: 29.1Å), which is a charged LA compound at physiological pH and therefore not able to traverse the membrane, to access the LA binding site when added in the extracellular environment (Sunami et al., 2001).
Apart from LAs that have a distinct binding site, sevoflurane (CAS No.: 28523-86-6), an inhalational anaesthetic, has a more complex binding profile. Sevoflurane binding results in a decrease of the peak sodium current, a hyperpolarised shift in the voltage dependence of inactivation and a slowing of the recovery from inactivation (Horishita et al., 2008; Ouyang et al., 2009). Within the bacterial sodium channel NaChBac, binding regions have been located at the pore region, selectivity filter, and the S4–S5 linker/S6 interface (Ouyang et al., 2007; Raju et al., 2013). MD simulations suggested that the binding of sevoflurane to the selectivity filter and the S4–S5 linker occurs mainly when the channel is in the activated/open state, while in the closed state the channel gate and the VSD are targeted (Barber et al., 2014). A state independent binding site is possibly the central cavity that is accessed by the fenestrations. Residues T220 and F227 (NavChBac numbering) are proposed to be responsible for sevoflurane binding in the central cavity, which are the homologue residues for LA binding in Nav1.4 (F1764, Y1771).
Binding Site 2 Compounds, Though Being Lipophilic, Bind to the Inner Pore
Compounds binding at site 2 of the Nav channel include batrachotoxin (Daly et al., 1965; Huang et al., 1982), grayanotoxin (Yuki et al., 2001; Jansen et al., 2012), CAS No.: 54781-61-2), and alkaloids from plant such as veratridine (Ulbricht, 1969; Sutro, 1986), CAS No.:71-62-5), aconitine and mesaconitine (Herzog et al., 1964; Friese et al., 1997) (CAS No.: 302-27-2). All these compounds are lipid soluble and need to traverse the cell membrane before influencing the channel in a use-dependent manner, comparable to the action of LA compounds (Herzog et al., 1964; Catterall, 1980; Huang et al., 1982; Dubois et al., 1983; Sutro, 1986; Barnes and Hille, 1988; Ameri et al., 1996; Yuki et al., 2001; Wang and Wang, 2003). Their effects can be a combination of: (1) a hyperpolarised shift of the voltage dependence of activation, (2) inhibition of the fast inactivation process, leading to persistent sodium currents, (3) decrease in ion conductance, and/or (4) decrease of the Na+ selectivity. The location of binding site 2 is thought to be at the S6 of all four domains and to overlap with the LA binding site or at least allosterically hinder LA binding.
To describe neurotoxin site 2 in more detail, we focus on the most potent site 2 toxin reported to date, batrachotoxin (BTX) a steroidal alkaloid indirectly produced by the South-American poison dart frogs of the genus Phyllobates (Daly et al., 1965). Binding of BTX is highly irreversible and only possible when the channel is in its activated open state (Huang et al., 1982; Dubois et al., 1983). BTX shifts the voltage dependence of channel activation by approximately −40 mV toward more hyperpolarized potentials (Linford et al., 1998), reducing the channel's ion conductance and selectivity. BTX also reduces the affinity for LA, which is thought to be caused by non-competitive antagonism (i.e., allosteric effect) because of overlapping binding sites as BTX and LA share the binding residue F1586 (Linford et al., 1998). Residue mutations affecting BTX in Nav1.4 were first described in DI-S6 and DIV-S6: I439K, N440K, L443K, F1586K, and N1591K, respectively (Wang and Wang, 1998; Wang and Wang, 1999). Afterwards the DIII-S6 residues Ser1283 and leu1287 were identified (Wang et al., 2000) and finally DII-S6 residues N790 and L794 (Wang et al., 2001). All these residues line the inside of the pore (Figure 2). Non-pore lining residues involved in BTX sensitivity locate at the putative hinge region of the channel gate, Gly1282 and Phe1284, respectively (Du et al., 2011). While Phe1284 is important for the stabilisation of the ammonium group of BTX, Gly1282 is not a direct binding receptor for BTX and mutations of this residue affect binding allosterically by changing the channels gating properties. Modeling studies suggest that BTX binds to the pore but does not completely prevent ion conduction because of its “horseshoe-like” structure (Du et al., 2011). Only upon mutation of DII-S6 N790 BTX becomes a full blocker (Wang et al., 2007).
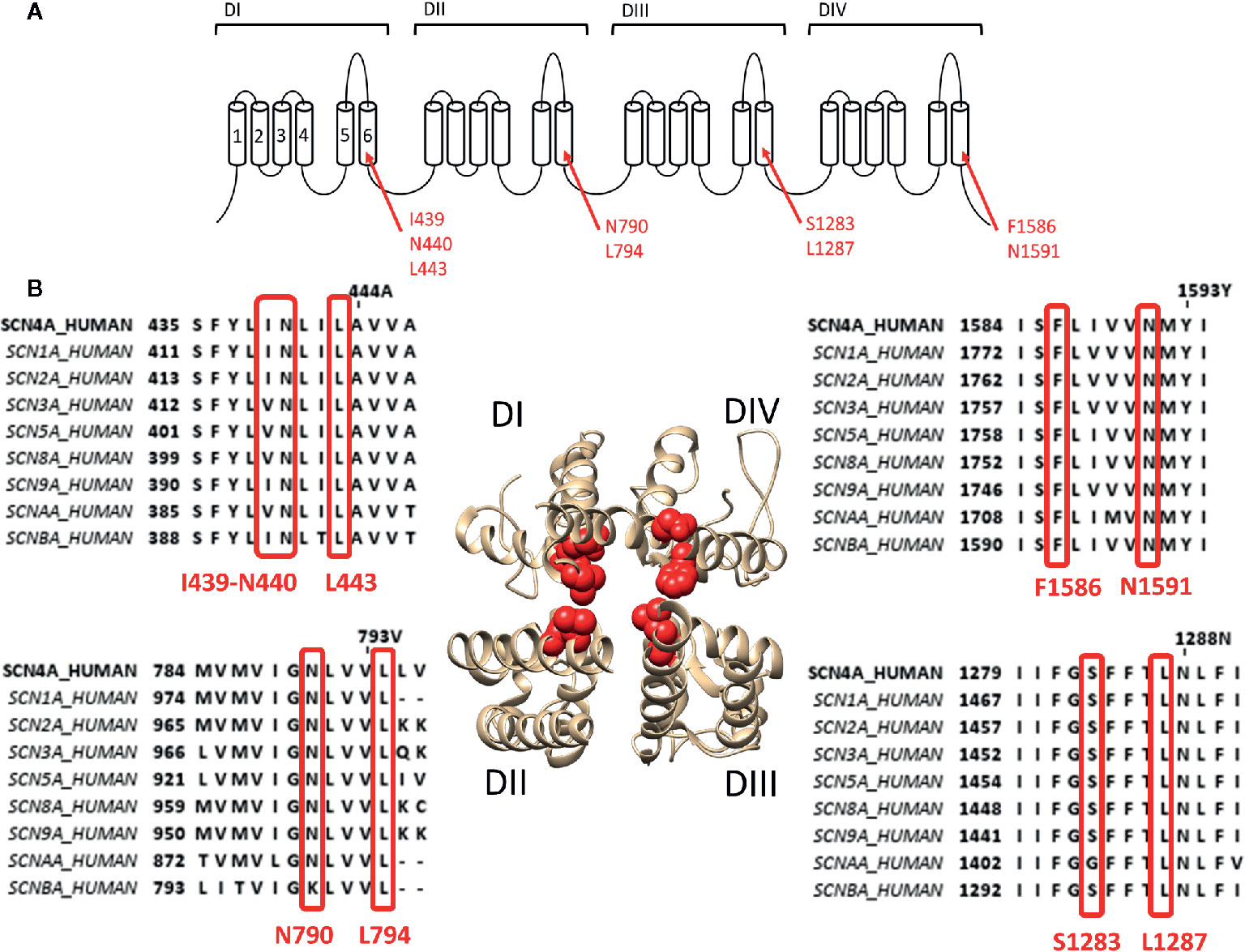
Figure 2 Location of binding site 2 within the Nav1.4 channel. (A) Schematic visualisation of the Nav ion channel structure which is constructed out of four domains (DI to DIV) each consisting out of six transmembrane segments (S1–S6). The red arrows indicate the probable residues responsible for constructing the site 2 receptor, residues are noted in their Nav1.4 annotation. (B) The bottom view of a 3D structure of the Nav1.4 channel with in red marking the residues constructing the binding site 2. Only the S5–S6 segments of each domain is shown for a clear view on the inner pore binding site. Around the 3D structure are the alignments of the known human sodium channels with the Nav1.4 channel (SCN4A) as reference, all positioned at the corresponding domain of the 3D structure. Marked in red are the residues which are necessary for site 2 toxin binding. While some variability is observed between some isoform, most of the residues are well conserved. Amino acid sequence alignment and 3D structure are visualized using Jalview and chimera software, respectively (Pettersen et al., 2004; Waterhouse et al., 2009).
Binding Site 5 Locates Between DIS6 and DIVS5
Some of the toxins that bind to site 5 of Nav channels are brevetoxins (Catterall and Gainer, 1985; Poli et al., 1986) and ciguatoxins (Murata et al., 1990; Lewis et al., 1998), both originate from marine dinoflagellates (Gambierdiscus toxicus) and are structurally comparable. Brevetoxins are produced by unarmoured marine dinoflagellates (e.g., Karenia brevis, Gymnodinium breve, or Ptychodiscus brevis). Ingestion of these toxins can lead to poisoning and death of marine animals and cause the disease neurotoxic shellfish poisoning in humans (Baden, 1983; Flewelling et al., 2005). Eleven different brevetoxins have been discovered today with brevetoxin A (PbTX1, CAS No.:98112-41-5) and brevetoxin B (PbTX2 and 3, CAS No.:79580-28-2 and 85079-48-7) being the most investigated ones. These toxins are about 30Å in length, 6Å in width, and 6Å high. They are composed of four recognizable parts: a lactone ring (the head) linked with a linker to a multiple carbon ring (the tail) ending in a rest-group (Lin et al., 1981). While the head is responsible for the effect, without the linker and the tail the toxin cannot modulate the channel as it allows the head region to reach its binding site (Rein et al., 1994). PbTX binds to the Nav channel at the DI-S6, DIV-S5, and DIV-S6 segments (Trainer et al., 1991; Trainer et al., 1994; Konoki et al., 2019). The region spans within DI-S6 from Ala418/Thr422 to Lys465/Lys477 and within DIV-S5 from Glu1510 to lys1557 with possible extension to Lys1565 (Figure 3). The tail of the molecule will be situated at the S5–S6 extracellular loop while the head is able to reach, due to the long linker and tail region, the inactivation gate at the intracellular side of the channel (Trainer et al., 1991; Trainer et al., 1994). Interaction of the head with the inactivation gate reduces channel inactivation, leading to a persistent sodium current (Sheridan and Adler, 1989; Schreibmayer and Jeglitsch, 1992).
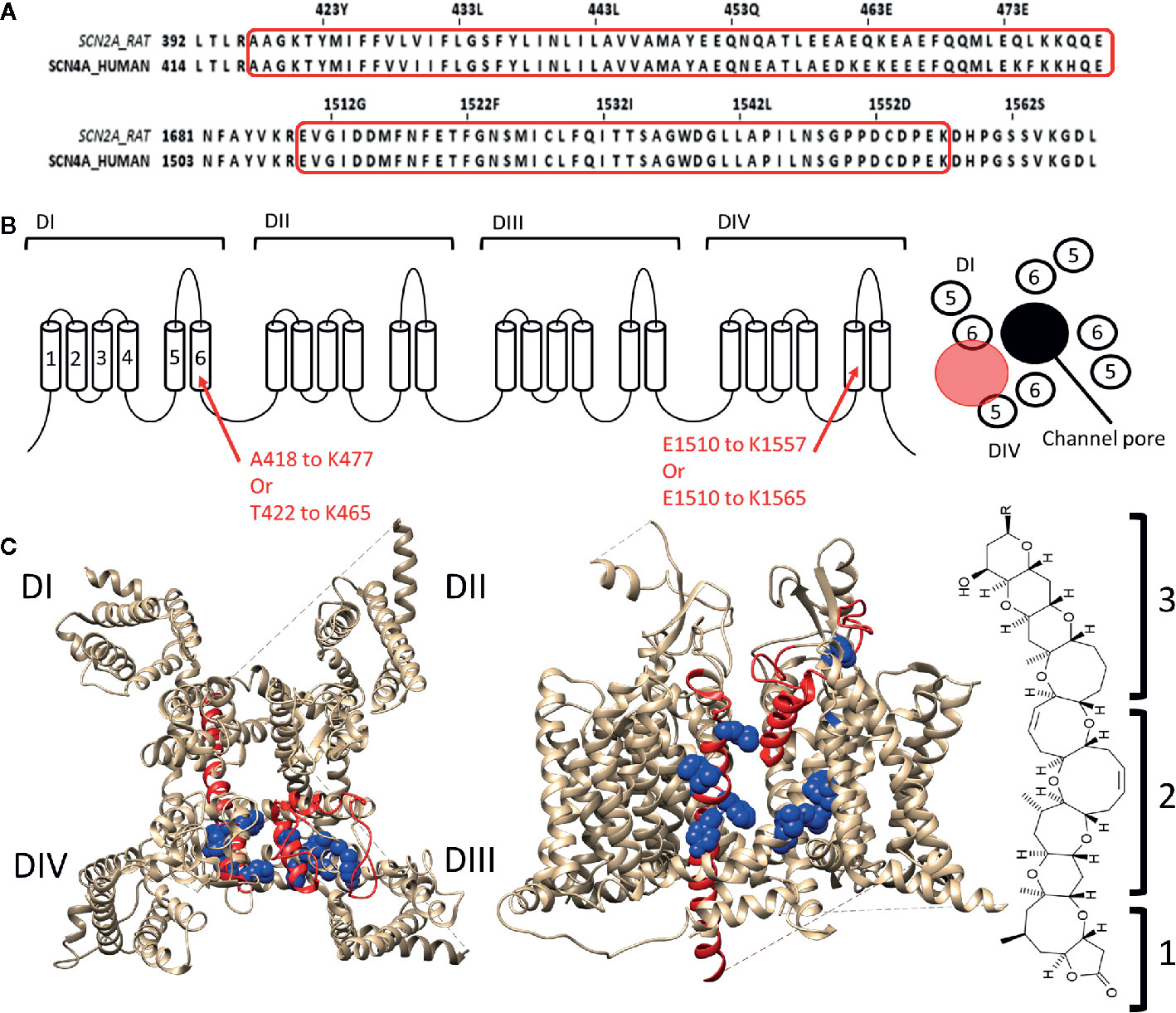
Figure 3 Location of binding site 5 in Nav1.4. (A) Alignment of the rat brain IIa sodium channel (SCN2A) with the human Nav1.4 (SCN4A) as reference. Marked in red are the regions where brevetoxin could bind. (B) Schematic topology of the Nav1.4 channel with the residues of DIS6 and DIVS5 that form the binding site listed below. On the right a schematic representation of the PD (top view) with the ion permeation pore represented in black and the location of binding site 5 indicated by the red circle. (C) 3D structures of the Nav1.4 channel seen from the top (left) and side (center, between DIII and DIV) are represented with in red the regions which are, using radioactive labelling studies, probably the location for brevetoxin binding. In blue residues are shown which had an effect on brevetoxin binding. With for DI M424, V429, I430, G434 decreasing affinity and F436, Y437 increasing affinity when mutated to alanine. In case of DIV mutation of I1485, G1486, L1488, L1489 lead to decrease and L1491, V1492, G1500, Y1506 to increase of affinity when mutated (Nav1.4 numbering) (Konoki et al., 2019). A cleft can be observed in between DIS6 and DIVS5, at the side view, where the toxin could bind. This cleft is also located around fenestration four of the ion channel. At the left the structure of brevetoxin with the head region (1) attached with a linker (2) to the tail structure (3). The structure is orientated as it would bind at the channel compared to the side view. Amino acid sequence alignment and 3D structures are visualized using Jalview and chimera software, respectively (Pettersen et al., 2004; Waterhouse et al., 2009).
Like brevetoxins, ciguatoxins (CTX) bind at site 5 and their effects are consequently similar. Ciguatoxins are classified based on their geographic origin, with P-CTX (Murata et al., 1990) and C-CTX (Lewis et al., 1998) standing for Pacific and Caribbean, respectively. In all Nav channel isoforms P-CTX1 (CAS No.:11050 21 8), the most potent CTX discovered to date, induces a hyperpolarizing shift in the voltage dependence of channel activation (Bidard et al., 1984; Benoit et al., 1986; Strachan et al., 1999) concomitantly with a shift in the voltage dependence of inactivation in some (Inserra et al., 2017). As CTX and brevetoxin share the same binding site, they are logically going into competition with each other (Lombet et al., 1987). Gambierol (CAS No.: 146763-62-4, origin Gambierdiscus Toxicus) has a similar structure as brevetoxins and CTXs, being a lipophilic multi-ring polyether toxin, but has no effect on the sodium currents (Lepage et al., 2007). However, when administered simultaneously it decreases the effect of CTX and brevetoxin, suggesting that gambierol acts as a competitive antagonist that binds to site 5 or at least exerts a negative allosteric effect.
Voltage-Gated Potassium Channels
The Kv channel family is impressively large compared to this of Nav channels, due to an extensive library of genes encoding different α-subunits that in some subfamilies can “mix-and-match” to form functional Kv channels. Additionally, alternative splicing, RNA editing, and post-translational modification further expand on the Kv channel family (Jan and Jan, 2012). This explains why the binding sites of the much larger Kv channel family are less well categorized than those of the Nav channel family. The next part will highlight different binding sites and pore wall fenestrations within several, but not all, members of the Kv channel family. After a short overview of the well-documented extracellular and intracellular exposed “hydrophilic” binding sites, we will discuss in detail the lesser-known lipid embedded “hydrophobic” binding site(s) in different Kv channels.
Extracellular and Intracellular Exposed “Hydrophilic” Binding Sites
The well-documented Kv channel binding sites can be topologically located on the intracellular or extracellular side of the channels. Extracellularly the most well-known conserved binding sites are those of the pore blockers and VSD targeting gating modifiers, while intracellularly the inner pore block is the most notable one (Wulff et al., 2009). Certain toxins from a variety of venomous animals target the extracellular binding sites. For example, scorpion toxins like charybdotoxin (CTX, CAS No.: 95751-30-7) and agitoxin (AgTx, CAS No.: 168147-41-9) target the water enveloped extracellular mouth of Kv channels, thereby physically occluding the permeation pore (Eriksson and Roux, 2002; Banerjee et al., 2013). Other scorpion toxins, like ergotoxin (ErgTx1, CAS No.: 8006-25-5) and BeKm-1 (CAS No.: 524962-01-4), cause an incomplete block of Kv current. This is because they only partially occlude the permeation pathway, which is known as “turret-block” (Xu et al., 2003; Zhang et al., 2003; Hill et al., 2007). Gating modifier toxins, like tarantula toxins (Hanatoxin, SGTx1, and VSTx1), bind to the paddle motif (S3b helices, S3–S4 linkers, and S4 helices) at the extracellular protein-lipid interface of the VSD. This binding site cannot be characterized as strictly hydrophilic as the hydrophobic residues of these amphipathic peptide toxins allow it to partially partition into the membrane, and interact with the VSD (Lee et al., 2004; Jung et al., 2005; Alabi et al., 2007; Swartz, 2007; Jung et al., 2010; Mihailescu et al., 2014). Intracellularly, compounds can bind in the inner cavity, either physically blocking ion permeation (e.g., quaternary ammonium ions) or allosterically modulating the gating machinery of Kv channels (e.g., 4-aminopyridine, CAS No.: 504-24-5) (Armstrong, 1971; Armstrong and Loboda, 2001; Del Camino et al., 2005). Also, the quaternary ammonium ion TEA can occlude the external Kv pore, similar to the scorpion toxins mentioned above (Luzhkov and Aqvist, 2001). Apart from the gating modifier binding site, which is partially enveloped by water, the binding sites mentioned here are exposed to an aqueous environment. Hence, referring them here as “hydrophilic” binding sites.
Lipid Embedded “Hydrophobic” Binding Site(s) and Pore Wall Fenestrations in the Shaker-Type Kv Family, Kv7 Family, and Kv10.1/Kv11.1 Channel
The next part reviews the potential lipophilic binding sites and pore wall fenestrations in different Kv channel types. To define the binding site(s) of certain compounds, the PD is divided into a “front side” and “back side”. Residues of the front side point towards the VSD of the same α-subunit, while residues of the “back side” point in the opposite direction, thus towards the VSD of a neighboring α-subunit (Figures 4D, 5D and 6C).
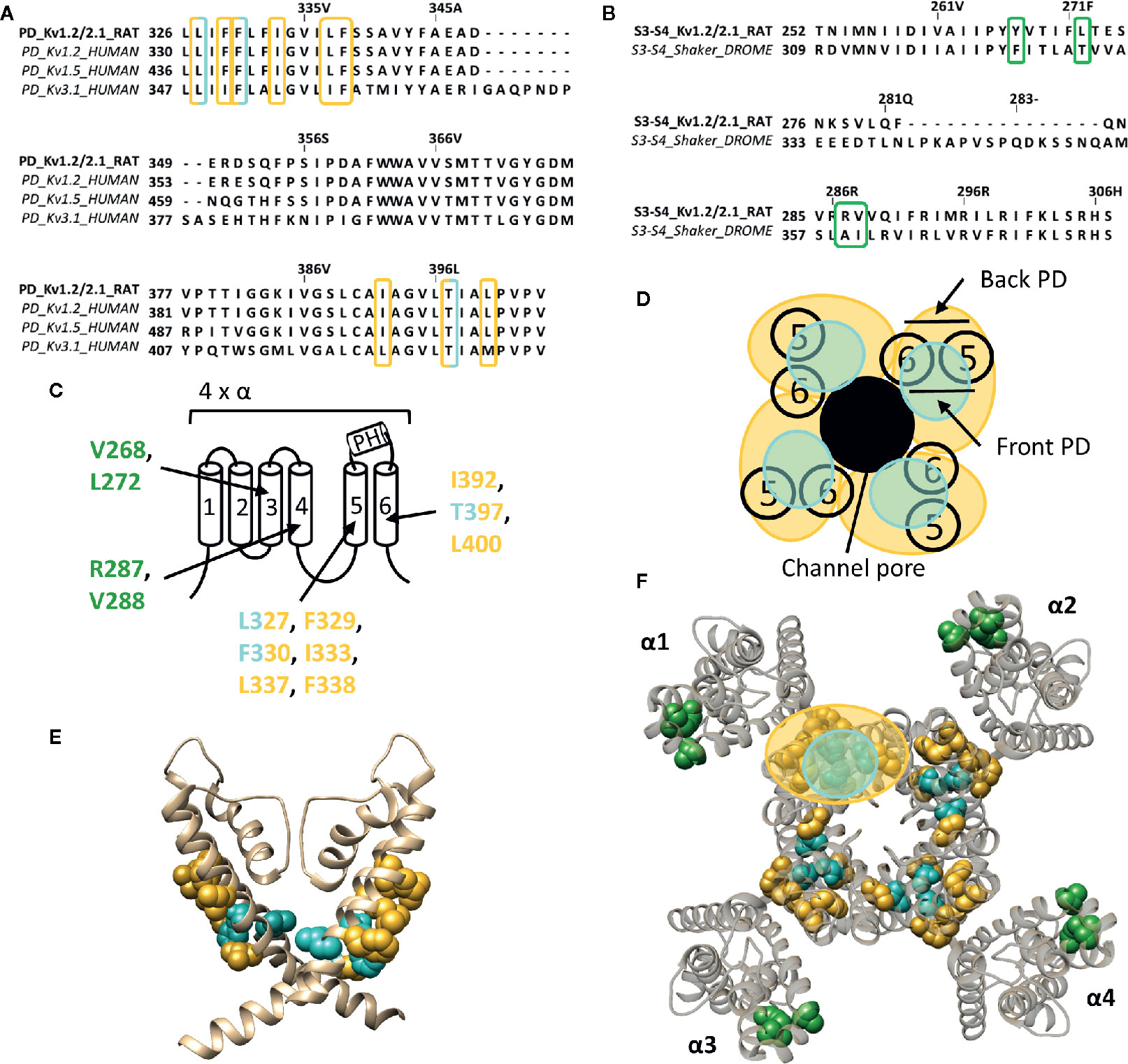
Figure 4 Lipid-exposed binding sites within the crystal structure of a Kv1.2–Kv2.1 paddle chimera channel (PDB: 2R9R). (A) Sequence alignment of part of the PD (S5-S6 segment) of Kv1.2-2.1, Kv1.2, Kv1.5, and Kv3.1, with Kv1.2-2.1 as reference. Residues important for gambierol and psora-4 action are highlighted in blue and yellow respectively. (B) Sequence alignment of the S3–S4 segments of Kv1.2–2.1 and shaker, with Kv1.2–2.1 as reference. Highlighted in green are the residues important for PUFA action. (C) Schematic visualization of one Kv channel α-subunit consisting out of six transmembrane segments (1–6) and a pore helix (PH). In blue the residues important for gambierol binding (L327, F330, and T397 according to Kv1.2–2.1 numbering) and in yellow those for psora-4 binding (L327, F329, F330, I333, L337, F338, I392, T397, and L400). Residues important for PUFA interaction are shown in green (V268, R287, V288, and L272). (D) Schematic visualization of the pore domain of the Kv1.2–Kv2.1 channel. Four pore-forming domains tetramerize to form the channel pore. The blue and yellow circle highlights the proposed gambierol/psora-4 binding site regions on the front- and/or backside of the pore-forming domain, respectively. (E) Side view of the Kv1.2–Kv2.1 channel with the front and back subunit omitted for clarity. Residues involved in gambierol and psora-4 interaction are shown in blue (L327, F330, and T397) and yellow (I392, T397, and L400), respectively. The PUFA action site is visualized in green (V268, R287, V288, and L272). (F) Top view of the Kv1.2–Kv2.1 channel, with each α subunit named α1–α4. Residues involved in gambierol and psora-4 interaction are shown in blue (L327, F330, and T397) and yellow (I392, T397, and L400), respectively. In green the PUFA action site comprising residuesV268, R287, V288, and L272. Kv1.2–Kv2.1 crystal structure (PDB: 2R9R) (Long et al., 2007) was visualized with chimera software (Pettersen et al., 2004) and amino acid sequence alignment with Jalview (Waterhouse et al., 2009).
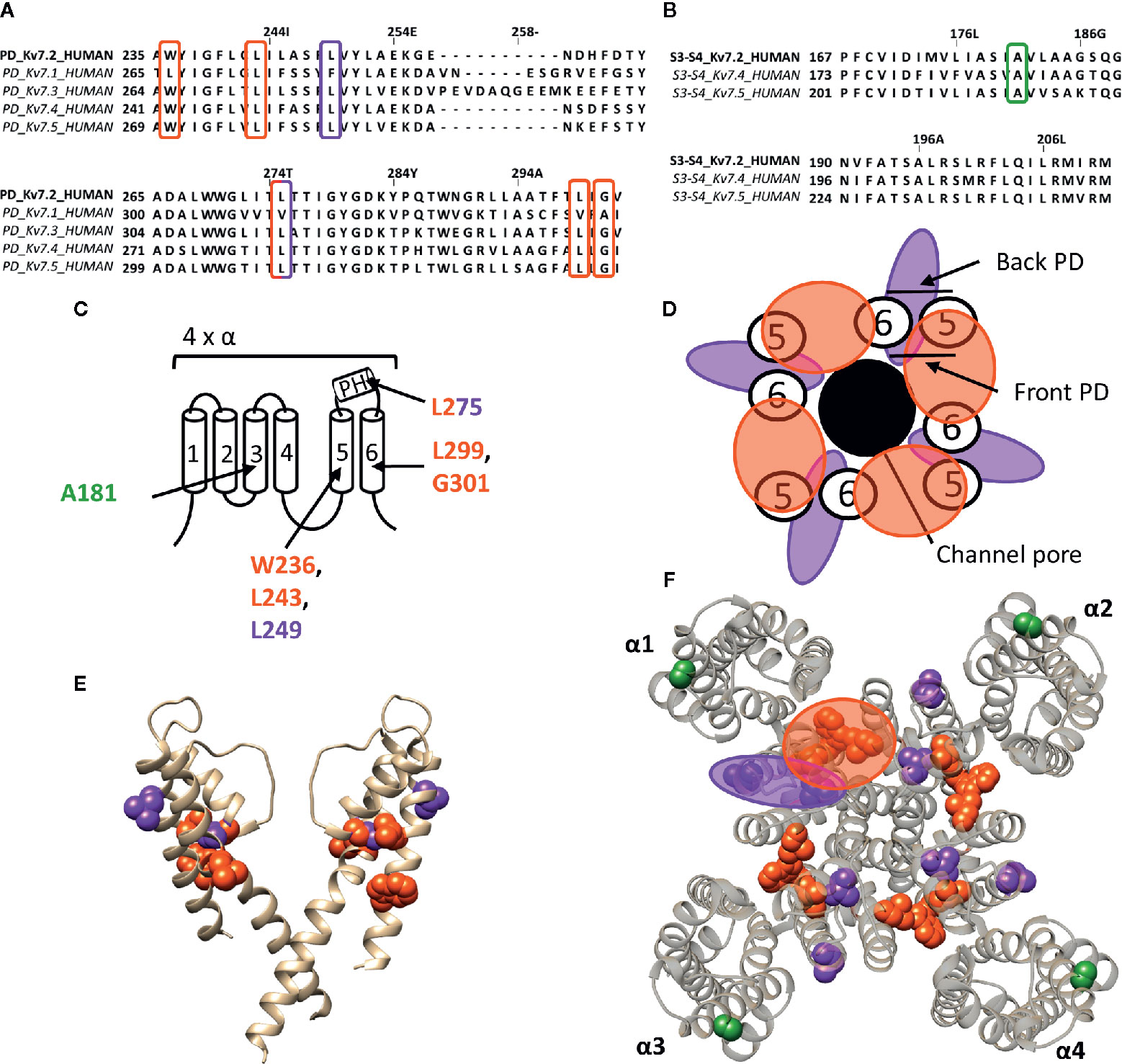
Figure 5 Lipid-exposed binding sites within a homology model of the Kv7.2 channel based on the 3D structure of Kv7.1 (PDB: 5VMS). (A) Sequence alignment of part of the PD of Kv7.1–Kv7.5, with Kv7.2 residue numbering. Highlighted in red and purple are the residues important for RTG and zinc pyrithione action, respectively. (B) Sequence alignment of the S3-S4 segment of Kv7.2, Kv7.4, and Kv7.5, with Kv7.2 as reference. Highlighted in green is the residue important for ICA73 action. (C) Schematic visualization of one Kv7 channel α-subunit consisting out of 6 transmembrane segments (1–6) and a pore helix (PH). Location of the residues involved in the interaction of Kv7.2 with retigabine are represented in red. Represented in purple are residues important for zinc pyrithione action). The residue important for ICA73 interaction is shown in green. (D) Schematic visualization of the PD of Kv7.2. The red and purple circle highlight the proposed retigabine/zinc pyrithione binding site on either the front- or back-side of the PD pore-forming domain, respectively. (E) Side view of Kv7.2 with the front and back subunit omitted for clarity. Residues involved in retigabine and zinc pyrithione interaction are shown in red (W236, L243, L299, and G301) and purple (L249 and L275), respectively. In green the ICA73 action site is visualized (A181). (F) Top view of Kv7.2, with each α subunit named α1–α4. Residues important for retigabine, zinc pyrithione, and ICA73 action are shown in red (W236, L243, L299, and G301), purple (L249 and L275), and green (A181), respectively. Shown Kv7.2 structure is a homology model of the 3D structure of Kv7.1 (PDB: 5VMS) (Sun and Mackinnon, 2017), generated with SWISS-MODEL (Waterhouse et al., 2018) and visualized using chimera software (Pettersen et al., 2004). The amino acid sequences are aligned using Jalview (Waterhouse et al., 2009).
The Shaker-Type Kv Channel Family
Whereas gambierol does not modulate Nav channels, it is capable of inhibiting Kv1 and Kv3 channels (Nicholson and Lewis, 2006; Cuypers et al., 2008; Kopljar et al., 2009). Gambierol's inhibitory mechanism has been extensively studied, whereby a threonine on S6, T427 in Kv3.1, is an important determinant (Kopljar et al., 2009). Substitution of the polar threonine by a hydrophobic valine abolishes the high gambierol affinity (Kopljar et al., 2009). Additional determinants are a leucine and phenylalanine on S5: L348 and F351 in Kv3.1, respectively. Kv1 channels possess a threonine residue equivalent to T427 (T401 in Kv1.2), explaining their similar gambierol sensitivity (Cuypers et al., 2008; Kopljar et al., 2009; Martinez-Morales et al., 2016). These residues are mostly positioned on the front side of the PD (Figure 4).
On the other hand, psora-4 (CAS No.: 724709-68-6), a potent inhibitor of Kv1.3, has been shown to predominantly bind to the back side of the PD (Figure 4) (Vennekamp et al., 2004; Marzian et al., 2013). A single psora-4 molecule acts as a central pore blocker of Kv1 channels (Kv1.1-Kv1.5, and Kv1.7), thereby preventing ions from permeating. However, four additional drug molecules can bind the lipid-exposed pocket on the back side of the PD, thereby causing the selectivity filter to narrow. Thus, the binding of five psora-4 molecules leads to a stable non-conducting state. The residues identified in Kv1.5 as playing a key role in psora-4 action map on the back side of S5–S6 (Figure 4). Additionally, some residues in S4 and the S4–S5 linker also seem involved (Marzian et al., 2013).
The inhalation anaesthetic, sevoflurane, seems to bind within the central cavity and to a similar hydrophobic pocket as psora-4 (Stock et al., 2018). Apart from these binding regions, the polar lipophilic molecule has been shown to interact with the S4–S5 linker, pore helix, segment S6, and even the VSD, likewise to the many binding regions of sevoflurane on the Nav channel (Barber et al., 2011; Barber et al., 2014). These sites are primarily dehydrated and lipid accessible, which is highly favourable for the polar lipophilic sevoflurane molecule (Stock et al., 2018). The specific residues involved in sevoflurane binding are not known, but one residue within the S4–S5 linker (G329 according to Kv1.2 numbering) has been identified to play an important role (Liang et al., 2015).
PUFAs are charged lipophilic compounds that position at the lipid membrane interface, although in small quantities (Yazdi et al., 2016). Nonetheless, PUFAs may play an important role in the treatment of arrhythmias and epilepsy, due to their modulating effect on, but not limited to, voltage-gated ion channels (Lefevre and Aronson, 2000; Leaf, 2007; Boland and Drzewiecki, 2008; Borjesson and Elinder, 2011). The Shaker Kv channel has been extensively studied as one of the targets of PUFAs. They interact with the channel at several sites, but the major one seems located within the lipid-facing cleft between S3 and S4 (S3–S4 cleft) of the VSD (Borjesson and Elinder, 2011). This hydrophobic cleft is perfectly shaped to accommodate the lipophilic carbon tail of the PUFAs, causing the negatively charged carboxyl head group to be positioned close to the S4 segment. In this way, PUFAs electrostatically affect the VSD by trapping S4 toward the extracellular position, stabilizing the open state of Shaker Kv channels (Borjesson et al., 2008; Yazdi et al., 2016). Accordingly, a series of point mutations on the lipid facing side of S3-S4 (I325C, T329C, A359C, and I360C, Shaker numbering) had a significant impact on the PUFA-induced hyperpolarizing shift in the channel's voltage dependence of activation (Figure 4) (Borjesson and Elinder, 2011). Although the binding site for gating modifier toxins is in close proximity to that of PUFAs, the action sites do most likely not overlap as the residues important for PUFA action are more deeply embedded into the lipid bilayer (Borjesson and Elinder, 2011). Interestingly, dehydroabietic acid (DHAA) and some of its derivatives, the most potent being Wu32and Wu122, have a similar effect on Shaker Kv channels. The carboxyl group of DHAA is positioned at roughly the same site, namely the S3–S4 cleft (Ottosson et al., 2017). Wu32 possibly interacts with residues between the S2–S3 and/or S3–S4 cleft as five cysteine mutations in S3, which have been shown to alter its affinity and/or efficacy, point towards S2 (I320C and F324C, shaker numbering) and S4 (I318C, P322C, and T326C), respectively (Ottosson et al., 2017).
The Kv7 Family
The well characterized retigabine (RTG, CAS No.: 150812-12-7) binding site is localized between the front side of one PD and the back side of an adjacent PD of Kv7.2–Kv7.5 (KCNQ2-5) channels (Figure 5) (Schenzer et al., 2005; Lange et al., 2009). These channels are predominantly expressed in neurons where they underlie the native M-current that plays a major role in regulating neuronal excitability (Wang et al., 1998; Kubisch et al., 1999; Schroeder et al., 2000). RTG amplifies the Kv7.2–Kv7.5 currents by stabilizing the open-channel conformation, by which it acts as a brake on neuronal excitability in vivo (Lange et al., 2009). Hence, the potential use of RTG and RTG-derived compounds as anticonvulsants (Rundfeldt, 1997; Wang et al., 2018). RTG binding has been attributed to several conserved residues on S5–S6, lining a hydrophobic pocket near the channel gate of Kv7.2–Kv7.5. According to Kv7.2 numbering these residues are: W236, L243, L275, L299, and G301 (Figure 5). Kv7.1 channels lack these amino acids (apart from L243), explaining their RTG-insensitivity (Schenzer et al., 2005; Lange et al., 2009). Because of the role of Kv7 in diseases of neuronal hyperexcitability, the search for positive allosteric modulators such as RTG, for which the clinical use is currently discontinued (Wang et al., 2018), is pursued. For example, BMS-204352, ML-213, and the acrylamide compound (S)-1 act on the canonical RTG binding site (Bentzen et al., 2006; Kim et al., 2015; Wang et al., 2018).
The hydrophobic pocket of the RTG binding site also seems to be able to accommodate endogenous hydrophilic neurotransmitters like γ-aminobutyric acid (GABA), which directly activates Kv7.3 and Kv7.5 via W236. In contrast to RTG, GABA does not readily cross the plasma membrane to reach its site of action. Based on the Kv1.2–Kv2.1 paddle chimera structure the tryptophan also seems to be accessible from the extracellular side (Manville et al., 2018). It is possible that this accessibility is dependent of the state of the channel, such that in certain conformations the mostly hydrophobic binding pocket can be reached by hydrophilic compounds like GABA. Additionally, not all Kv7 channel openers interact with residue W236. Zinc pyrithione for instance interacts with two residues at the back side of S5 and the pore helix (L249 and L275) (Figure 5) (Schenzer et al., 2005; Xiong et al., 2007; Lange et al., 2009).
ICA-compounds (ICAgen, Durham, NC, US), which are benzanilide Kv7 channel openers, were developed as RTG alternatives (Mcnaughton-Smith et al., 2002). Of these, the most well-documented is ICA-27243 (ICA43), which has been shown to be more selective than RTG (Wickenden et al., 2008). The individual residues that determine ICA43 binding have not been identified, but the C-terminal end of S2 and the N-terminal part of S3 (S2–S3 cleft) are proposed to be involved (Padilla et al., 2009). Later, Wang AW, et al. continued the investigation of the mechanism of action of ICA-compounds on Kv7 channels, but focussed on ICA-069673 (ICA73) (Wang et al., 2017). They identified two key residues in S3 of Kv7.2: A181 and F168, respectively. Mutation of these residues did not affect RTG-mediated gating, but did alter the action of ICA73 (Wang et al., 2017). Furthermore, it has been shown that ICA43 and ICA73 are resistant to mutation of the RTG binding site, supporting that not all Kv7 channel openers bind to the PD, but also can interact with a VSD site (Padilla et al., 2009; Wang et al., 2017). However, it remains debated whether residues A181 and F168 are involved in ICA binding directly or allosterically (Wang et al., 2018). In case these residues are binding ICA73, the position of residue A181 toward the lipid-exposed surface of the VSD suggests the presence of a drug binding site at the lipid-exposed cleft of S2–S3 and/or S3–S4 in Kv7 channels, similar to the interaction of Wu32 with Shaker Kv channels (Figure 5).
PUFAs have also been described to electrostatically affect Kv7.1 channels, resulting in a negative shift of the conductance-voltage curve. This modulation of Kv7.1 channels by PUFAs is similar to what has been described for shaker Kv channels (Borjesson et al., 2008; Borjesson and Elinder, 2011). Hence, the binding site and mechanism of action of PUFAs are most likely similar for both channels (Liin et al., 2015).
The hERG (Kv11.1) and EAG (Kv10.1) Channel
The human ether-a-go-go related gene (hERG) type 1 encodes for the Kv11.1 channel, which functions as the rapid component of the delayed rectifier K+ current contributing to the repolarization of cardiac action potentials (Vandenberg et al., 2012). Alteration of the native functioning of Kv11.1 channels, either genetically or pharmacologically, can disrupt this repolarization, leading to various cardiac rhythm disorders (Thomas et al., 2006). Furthermore, the role of Kv11.1 is not limited to the heart, as it also seems to play a role in the central nervous system, digestive, secretory, and reproductive system, and even cancer (Babcock and Li, 2013). Hence, it has become common practice to screen compounds on hERG channel activity during the early stages of drug development, as unintentional side-effects may lead to disease and sudden-death (Mitcheson et al., 2000; Thomas et al., 2006; Babcock and Li, 2013). Most hERG inhibitors interact with residues inside the channel's permeation pathway, either located on the pore helix (T623,S624, and V625, according to hERG numbering) or on segment S6 (G648, Y652, and F656) (Lees-Miller et al., 2000; Mitcheson et al., 2000; Kamiya et al., 2001; Sanchez-Chapula et al., 2002; Sanchez-Chapula et al., 2003; Fernandez et al., 2004; Kamiya et al., 2008). On the other hand, some small molecule hERG activators have been discovered who deviate from this binding site, like ICA-105574 (ICA74) and PD-118057 (PD57). The most critical binding determinants for ICA74 are F557 and L622 (Garg et al., 2011), which topologically would situate the binding site at the front side of the PD (Figure 6). Apart from these critical binding determinants several other residues have been proposed to line the hydrophobic ICA binding pocket of Kv11.1 (F619, T623, M645, L646, M651, Y652, and F656), situating the ICA74 binding site between the front side of one α-subunit and the back side of an adjacent α-subunit (Garg et al., 2013). PD57 seems to bind the same hydrophobic pocket, with key residues being L646 on segment S6 and F619 on the pore helix of an adjacent subunit (Figure 6) (Perry et al., 2009).
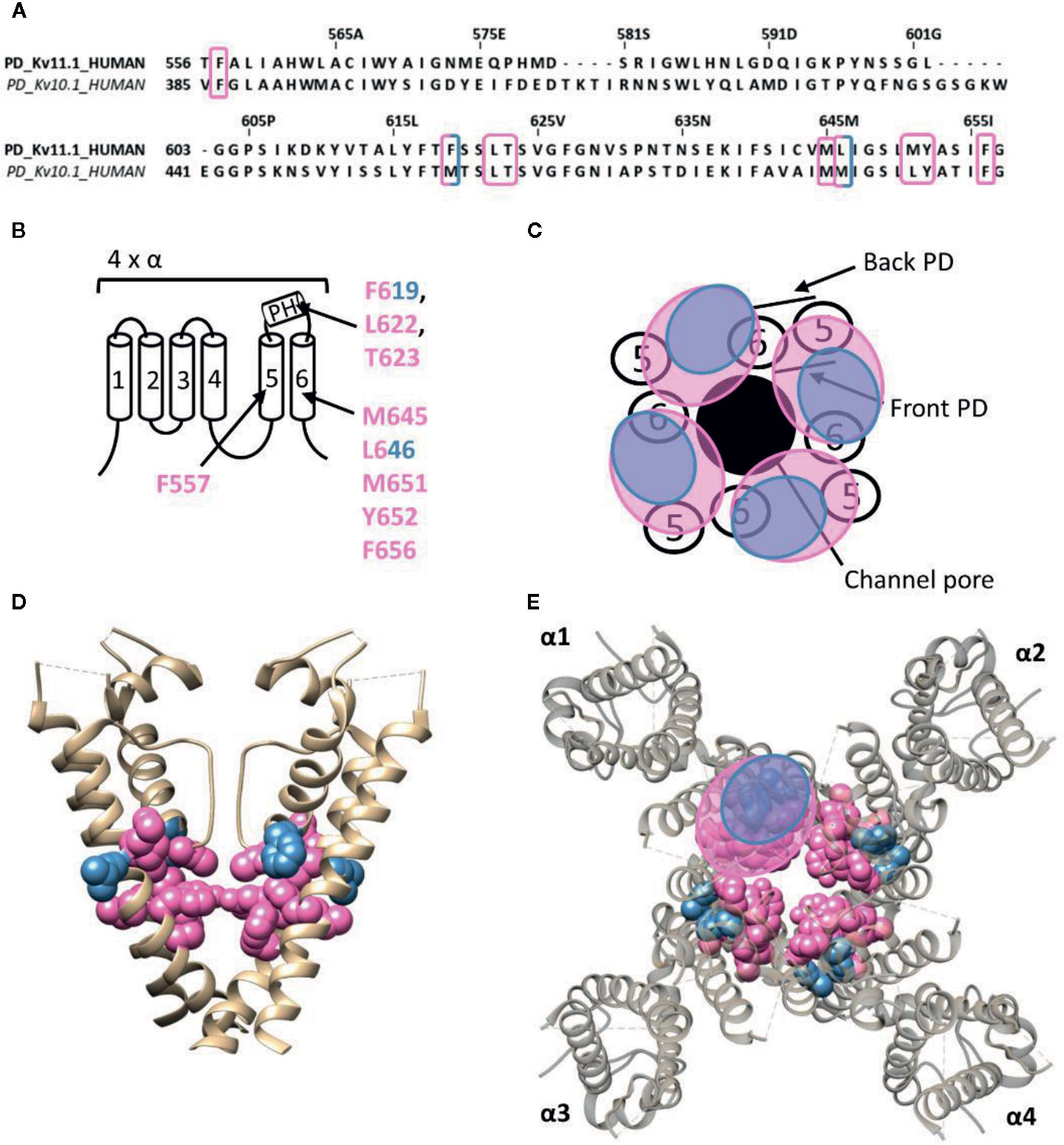
Figure 6 Lipid-exposed binding site(s) within the cryo-EM structure of the Kv11.1 (hERG) channel (PDB: 5VA1). (A) Sequence alignment of part of the PD of Kv11.1 and Kv10.1, with Kv11.1 residue numbering. Highlighted in pink and blue are the residues important for ICA74 and PD57 action, respectively. (B) Schematic visualization of one Kv channel α-subunit consisting out of six transmembrane segments (1–6) and a pore helix (PH). In blue the critical residues of PD57 (F619 and L646 according to Kv11.1 numbering) and in pink the residues lining the proposed hydrophobic ICA74 binding pocket (F619, F557, L622, T623, M645, L646, M651, Y652, and F656). (C) Schematic visualization of the pore domain of the Kv11.1 channel. Four pore-forming domains tetramerize to form the channel pore. The blue and pink circle highlights the proposed PD57 and ICA74 binding site regions on the front- and/or backside of the pore-forming domain. (D) Side view of the Kv11.1 channel with the front and back subunit omitted for clarity. Residues involved in PD57 and ICA74 interaction are shown in blue (F619 and L646) and pink (F619, F557, L622, T623, M645, L646, M651, Y652, and F656), respectively. The residues F619 and L646 are both critical residues for PD57 and ICA74, but are shown in blue. (E) Top view of the Kv11.1 channel, with each α subunit named α1–α4. Residues involved in PD57 and ICA74 interaction are shown in blue (F619 and L646) and pink (F619, F557, L622, T623, M645, L646 M651, Y652, and F656), respectively. Kv11.1 cryo-EM structure (PDB: 5VA1) (Wang and Mackinnon, 2017) was visualized with chimera software (Pettersen et al., 2004) and amino acid sequence alignment with Jalview (Waterhouse et al., 2009).
Interestingly, ICA74 binds to a similar hydrophobic pocket in the related ether-a-go-go (EAG) type 1 channel (Kv10.1), although eliciting an opposite effect as in Kv11.1. ICA74 inhibits Kv10.1 currents by enhancing channel inactivation (Garg et al., 2012). The key residues for ICA74 binding in Kv11.1 (F557 and L622) are indeed conserved in Kv10.1 (F359 and L434). Furthermore, only three residues (F619, L646, and M651) of the proposed ICA binding pocket in Kv11.1 appeared not to be conserved in the Kv10.1 channel. This strongly suggests that ICA74 binds to the same hydrophobic site in Kv10.1 and Kv11.1 channels (Garg et al., 2013).
Lateral Pore Wall Fenestrations
The reports of compounds that can access the inner cavity from lipid exposed side-pocket through lateral pore wall fenestrations in Kv channels is still very limited. In Kv7.1 a pore wall fenestration is formed upon interaction with the β-subunit KCNE1 such that adamantane compounds, AC-1 (CAS No.: 878489-28-2) and its analogs (ACs), can reach their binding site (Jaraskova et al., 2005; Wrobel et al., 2016). Interestingly, AC-1 does not affect currents generated by homomeric Kv7.1, channels, nor Kv7.1 co-expressed with other KCNE isoforms (KCNE2-5). Thus, the “β-subunit-induced fenestrations” seem to be required for AC binding. Within these fenestrations many residues have been identified as important for AC-1 activity (V334, F335, I337, F340, and A344, according Kv7.1 numbering), but its position relative to the central cavity and lipophilic side-pocket could not be elucidated (Wrobel et al., 2016).
In Kv11.1, a lateral pore wall fenestration is possibly formed upon mutation of residue F557 to leucine (F557L), explaining the decrease in current inhibition of six known hERG blockers (dofetilide, haloperidol, terfenadine, astemizole, cisapride, and amiodarone) (Saxena et al., 2016). For Kv1.5 it has been proposed that psora-4 molecules can move in the I502A mutant between the central cavity and the lipophilic side-pockets through fenestration between segments S5–S6 (Marzian et al., 2013).Hence, the presence of pore wall fenestrations has thus far only been observed upon β-subunit interaction with Kv7.1 and to be induced by mutations in Kv1.5 and Kv11.1 (Marzian et al., 2013; Saxena et al., 2016; Wrobel et al., 2016). Although the presence of fenestrations has not been reported yet for wild-type channels, the likelihood that some Kv channel types express lateral pore wall fenestrations increases. If present, these fenestrations may be similar to those characterized in Nav channels, which allow LA's to pass between the central cavity and lipophilic side-pockets (Payandeh et al., 2011; Mccusker et al., 2012; Payandeh et al., 2012; Zhang et al., 2012; Kaczmarski and Corry, 2014; Wrobel et al., 2016).
Unifying the Lipid Exposed/Accessible Drug/Toxin Binding Sites of Kv and Nav Channels
Whereas a classification exists for the different drug/toxin binding sites in Nav channels, such a categorization is currently lacking for the Kv channel family. In this review we take a first step and describe the hydrophobic binding sites reported in different Kv channel families. A compound like gambierol (Figures 4 and 7) has been shown to mostly bind to the front side of the PD, while RTG (Figures 5 and 7) PD57, and ICA74 (Figure 6) also interact with the back side of the PD of an adjacent α-subunit. When mapping all the sites it appears that gambierol, ICA74, RTG, and PD57 bind to an analogues binding site present in different Kv channel types (Figure 7) (Schenzer et al., 2005; Kopljar et al., 2009; Lange et al., 2009; Perry et al., 2009; Garg et al., 2011; Martinez-Morales et al., 2016). This indicates that an analogues lipophilic binding site is conserved between the different Kv channel types.
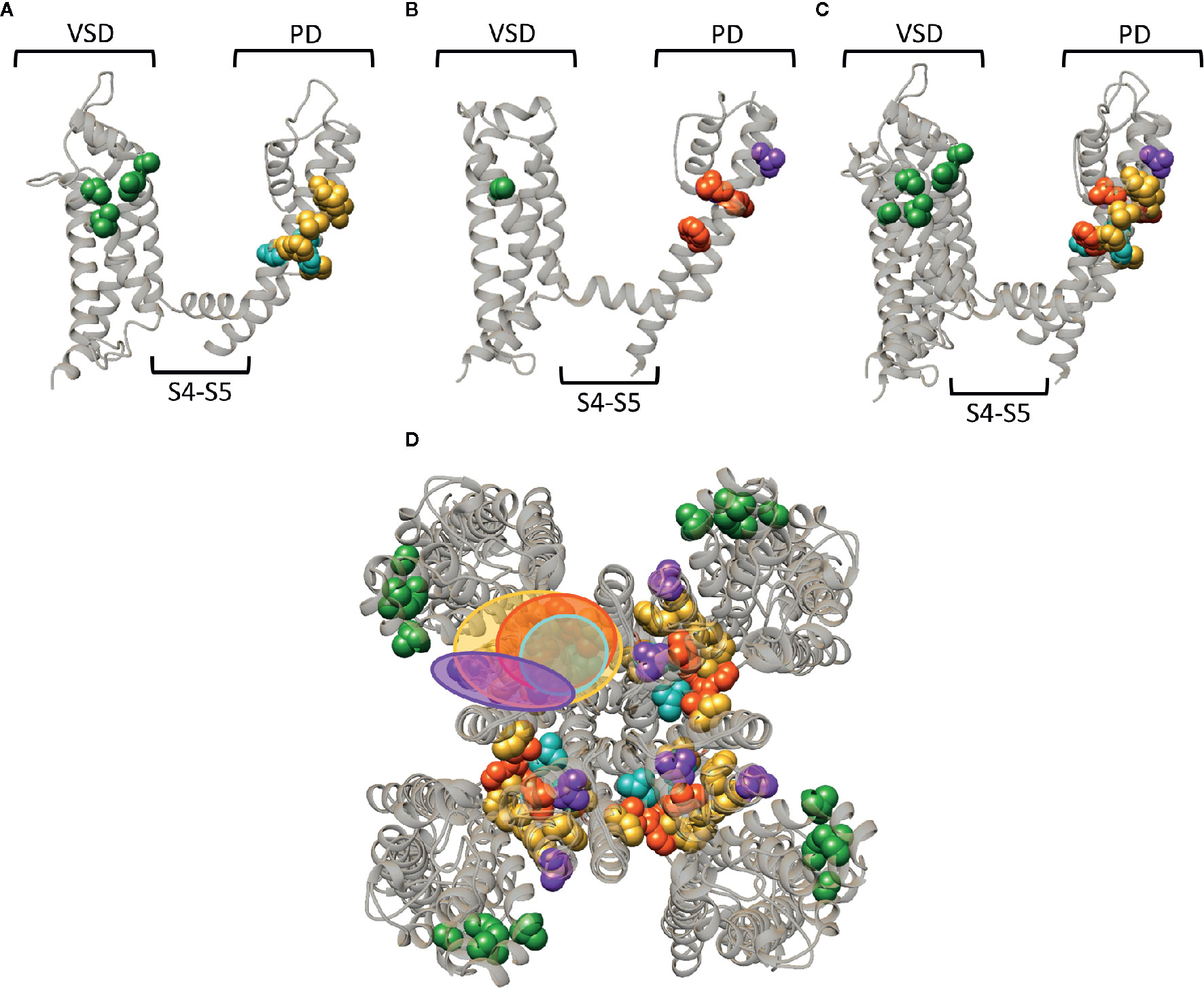
Figure 7 Superposition of the lipid-exposed binding sites within the Kv1.2–2.1 paddle chimera channel (PDB: 2R9R) and Kv7.2. (A) Side view of one α-subunit of the Kv1.2–2.1 channel with the residues important for gambierol, psora-4, and PUFA action represented by spheres and highlighted in blue, yellow, and green, respectively. The VSD, PD, and S4–S5 linker are also indicated. (B) Side view of one Kv7.2 α-subunit with the residues important for retigabine, zinc pyrithione, and ICA73 action represented by red, purple, and green spheres, respectively. (C) Superposition of the structures shown in panel (A, B). (D) Superposition of the Kv1.2–2.1 and Kv7.2 channel structure shown as top view. The red and blue circle highlight the proposed retigabine/gambierol binding site on the “front side” of the PD, while the binding sites for zinc pyrithione and psora-4 on the “back side” of the PD are highlighted with the purple and yellow circles, respectively. Structures are visualized using chimera software (Pettersen et al., 2004).
Zinc pyrithione, on the other hand, seems to solely bind to the back side of the PD, implying that the front and back side of the PD could serve as distinct binding sites (Figures 5 and 7) (Xiong et al., 2007). Psora-4 and sevoflurane are less specific regarding their binding site, as they both bind to the front and back side of the PD, among others (Marzian et al., 2013; Liang et al., 2015; Stock et al., 2018). Although certain compounds solely bind to the front or back side of the PD it seems that all residues point toward a similar lipophilic region, leading to the speculation that these seemingly distinct binding sites may converge to just one conserved lipophilic binding region in Kv channels. This binding site is then most likely similar to neurotoxin site 5 in Nav channels (Figure 3) (Catterall and Risk, 1981; Cestele and Catterall, 2000), located between DIS6-DIVS5 (Figures 3, 4, 5) (Konoki et al., 2019). The idea that these binding sites are orthologous equivalents is because gambierol presumably binds to site 5 in Nav channels (Lepage et al., 2007). In the case of Kv channels four such binding sites are present due to its tetrameric nature, as opposed to Nav channels who only have one neurotoxin site 5 (Schenzer et al., 2005; Kopljar et al., 2009; Lange et al., 2009; Marzian et al., 2013; Stock et al., 2018).
Another lipid facing binding site is located on the VSD, in particular the cleft between segments S2–S3 and S3–S4. PUFAs, DHAA and its derivatives, and ICA-compounds allegedly bind to these clefts in Shaker and Kv7 channels, respectively (Figures 4D, 5D, and 7) (Padilla et al., 2009; Borjesson and Elinder, 2011; Ottosson et al., 2017; Wang et al., 2017). This leads to the assumption that also this lipophilic binding site is conserved between different Kv channel types. Additionally, certain compounds of Nav (LAs and sevoflurane) and Kv (AC-1, psora-4, and several hERG blockers) channels have been proposed to use hydrophobic lateral pore wall fenestrations to reach their binding sites. The location of these fenestrations in Nav channels are situated between DI–DII and DIII–DIV (Figure 1), while in Kv channels they are most likely present between segments S5–S6, allowing lipid soluble compounds to reach their binding site even when the channel is in its closed state (Payandeh et al., 2011; Mccusker et al., 2012; Payandeh et al., 2012; Zhang et al., 2012; Marzian et al., 2013; Barber et al., 2014; Kaczmarski and Corry, 2014; Saxena et al., 2016; Wrobel et al., 2016).
Of note, the residues reported to affect drug/toxin affinity were in this review mapped on available 3D structures that are snapshots of the channel in a certain state, which should not be the high affinity state for the respective drug/toxin. As mentioned, several drugs/toxins are state dependent and bind with highest affinity to a certain conformation of the channel (e.g., the closed or open channel configuration). Consequently, the residues reported to be important for drug/toxin effect might orient differently when the conformation of the channel changes. Thus, when the channel is in its high affinity drug/toxin state, the orientation of the residues might be slightly different resulting in possibly broader binding regions than highlighted in the figures. Furthermore, several of the residues reported to be important for drug/toxin effect are likely not the binding partners of the drugs/toxins but alter affinity in an allosteric way. Nonetheless, there seem to be three distinct lipid-exposed binding sites preserved in Kv channels: the front and back side of the PD, and S2–S3/S3–S4 clefts. Future experiments will determine if the front and back PD binding sites are two distinct entities or if they converge to just one larger lipophilic binding site region.
Author Contributions
All authors contributed in writing this review.
Funding
This review was supported by a grant of the FWO (Fonds voor Wetenschappelijk onderzoek) G0C6220N (DJS) and by a GOA grant of the University of Antwerp.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahern, C. A., Payandeh, J., Bosmans, F., Chanda, B. (2016). The hitchhiker's guide to the voltage-gated sodium channel galaxy. J. Gen. Physiol. 147, 1–24. doi: 10.1085/jgp.201511492
Alabi, A. A., Bahamonde, M. I., Jung, H. J., Kim, J. I., Swartz, K. J. (2007). Portability of paddle motif function and pharmacology in voltage sensors. Nature 450, 370–375. doi: 10.1038/nature06266
Ameri, A., Shi, Q., Aschoff, J., Peters, T. (1996). Electrophysiological effects of aconitine in rat hippocampal slices. Neuropharmacology 35, 13–22. doi: 10.1016/0028-3908(95)00153-0
Armstrong, C. M., Hollingworth, S. (2018). A perspective on Na and K channel inactivation. J. Gen. Physiol. 150, 7–18. doi: 10.1085/jgp.201711835
Armstrong, C. M., Loboda, A. (2001). A model for 4-aminopyridine action on K channels: similarities to tetraethylammonium ion action. Biophys. J. 81, 895–904. doi: 10.1016/S0006-3495(01)75749-9
Armstrong, C. M. (1971). Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J. Gen. Physiol. 58, 413–437. doi: 10.1085/jgp.58.4.413
Babcock, J. J., Li, M. (2013). hERG channel function: beyond long QT. Acta Pharmacol. Sin. 34, 329–335. doi: 10.1038/aps.2013.6
Baden, D. G. (1983). Marine food-borne dinoflagellate toxins. Int. Rev. Cytol. 82, 99–150. doi: 10.1016/S0074-7696(08)60824-4
Banerjee, A., Lee, A., Campbell, E., Mackinnon, R. (2013). Structure of a pore-blocking toxin in complex with a eukaryotic voltage-dependent K(+) channel. Elife 2, e00594. doi: 10.7554/eLife.00594
Barber, A. F., Liang, Q., Amaral, C., Treptow, W., Covarrubias, M. (2011). Molecular mapping of general anesthetic sites in a voltage-gated ion channel. Biophys. J. 101, 1613–1622. doi: 10.1016/j.bpj.2011.08.026
Barber, A. F., Carnevale, V., Klein, M. L., Eckenhoff, R. G., Covarrubias, M. (2014). Modulation of a voltage-gated Na+ channel by sevoflurane involves multiple sites and distinct mechanisms. Proc. Natl. Acad. Sci. United States America 111, 6726–6731. doi: 10.1073/pnas.1405768111
Barnes, S., Hille, B. (1988). Veratridine modifies open sodium channels. J. Gen. Physiol. 91, 421–443. doi: 10.1085/jgp.91.3.421
Bavro, V. N., De Zorzi, R., Schmidt, M. R., Muniz, J. R., Zubcevic, L., Sansom, M. S., et al. (2012). Structure of a KirBac potassium channel with an open bundle crossing indicates a mechanism of channel gating. Nat. Struct. Mol. Biol. 19, 158–163. doi: 10.1038/nsmb.2208
Benoit, E., Legrand, A. M., Dubois, J. M. (1986). Effects of ciguatoxin on current and voltage clamped frog myelinated nerve fibre. Toxicon 24, 357–364. doi: 10.1016/0041-0101(86)90195-9
Bentzen, B. H., Schmitt, N., Calloe, K., Dalby Brown, W., Grunnet, M., Olesen, S. P. (2006). The acrylamide (S)-1 differentially affects Kv7 (KCNQ) potassium channels. Neuropharmacology 51, 1068–1077. doi: 10.1016/j.neuropharm.2006.07.001
Benz, I., Kohlhardt, M. (1991). Responsiveness of cardiac Na+ channels to antiarrhythmic drugs: the role of inactivation. J. Membr. Biol. 122, 267–278. doi: 10.1007/BF01871427
Bezanilla, F. (2008). How membrane proteins sense voltage. Nat. Rev. Mol. Cell Biol. 9, 323–332. doi: 10.1038/nrm2376
Bidard, J. N., Vijverberg, H. P., Frelin, C., Chungue, E., Legrand, A. M., Bagnis, R., et al. (1984). Ciguatoxin is a novel type of Na+ channel toxin. J. Biol. Chem. 259, 8353–8357.
Blunck, R., Batulan, Z. (2012). Mechanism of electromechanical coupling in voltage-gated potassium channels. Front. Pharmacol. 3, 166. doi: 10.3389/fphar.2012.00166
Boland, L. M., Drzewiecki, M. M. (2008). Polyunsaturated fatty acid modulation of voltage-gated ion channels. Cell Biochem. Biophys. 52, 59–84. doi: 10.1007/s12013-008-9027-2
Borjesson, S. I., Elinder, F. (2011). An electrostatic potassium channel opener targeting the final voltage sensor transition. J. Gen. Physiol. 137, 563–577. doi: 10.1085/jgp.201110599
Borjesson, S. I., Hammarstrom, S., Elinder, F. (2008). Lipoelectric modification of ion channel voltage gating by polyunsaturated fatty acids. Biophys. J. 95, 2242–2253. doi: 10.1529/biophysj.108.130757
Cahalan, M. D. (1978). Local anesthetic block of sodium channels in normal and pronase-treated squid giant axons. Biophys. J. 23, 285–311. doi: 10.1016/S0006-3495(78)85449-6
Catterall, W. A., Gainer, M. (1985). Interaction of brevetoxin A with a new receptor site on the sodium channel. Toxicon 23, 497–504. doi: 10.1016/0041-0101(85)90034-0
Catterall, W. A., Risk, M. (1981). Toxin T4(6) from Ptychodiscus brevis (formerly Gymnodinium breve) enhances activation of voltage-sensitive sodium channels by veratridine. Mol. Pharmacol. 19, 345–348.
Catterall, W. A. (1980). Neurotoxins that act on voltage-sensitive sodium channels in excitable membranes. Annu. Rev. Pharmacol. Toxicol. 20, 15–43. doi: 10.1146/annurev.pa.20.040180.000311
Cestele, S., Catterall, W. A. (2000). Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 82, 883–892. doi: 10.1016/S0300-9084(00)01174-3
Courtney, K. R. (1988). Why do some drugs preferentially block open sodium channels? J. Mol. Cell Cardiol. 20, 461–464. doi: 10.1016/S0022-2828(88)80073-7
Cuello, L. G., Jogini, V., Cortes, D. M., Perozo, E. (2010). Structural mechanism of C-type inactivation in K(+) channels. Nature 466, 203–208. doi: 10.1038/nature09153
Cuypers, E., Abdel-Mottaleb, Y., Kopljar, I., Rainier, J. D., Raes, A. L., Snyders, D. J., et al. (2008). Gambierol, a toxin produced by the dinoflagellate Gambierdiscus toxicus, is a potent blocker of voltage-gated potassium channels. Toxicon : Off. J. Int. Soc. Toxinol. 51, 974–983. doi: 10.1016/j.toxicon.2008.01.004
Daly, J. W., Witkop, B., Bommer, P., Biemann, K. (1965). Batrachotoxin. The active principle of the Colombian arrow poison frog, Phyllobates bicolor. J. Am. Chem. Soc. 87, 124–126. doi: 10.1021/ja01079a026
De Lera Ruiz, M., Kraus, R. L. (2015). Voltage-Gated Sodium Channels: Structure, Function, Pharmacology, and Clinical Indications. J. Med. Chem. 58, 7093–7118. doi: 10.1021/jm501981g
Del Camino, D., Kanevsky, M., Yellen, G. (2005). Status of the intracellular gate in the activated-not-open state of shaker K+ channels. J. Gen. Physiol. 126, 419–428. doi: 10.1085/jgp.200509385
Doyle, D. A., Morais Cabral, J., Pfuetzner, R. A., Kuo, A., Gulbis, J. M., Cohen, S. L., et al. (1998). The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280, 69–77. doi: 10.1126/science.280.5360.69
Du, Y., Garden, D. P., Wang, L., Zhorov, B. S., Dong, K. (2011). Identification of new batrachotoxin-sensing residues in segment IIIS6 of the sodium channel. J. Biol. Chem. 286, 13151–13160. doi: 10.1074/jbc.M110.208496
Dubois, J. M., Schneider, M. F., Khodorov, B. I. (1983). Voltage dependence of intramembrane charge movement and conductance activation of batrachotoxin-modified sodium channels in frog node of Ranvier. J. Gen. Physiol. 81, 829–844. doi: 10.1085/jgp.81.6.829
Eriksson, M. A., Roux, B. (2002). Modeling the structure of agitoxin in complex with the Shaker K+ channel: a computational approach based on experimental distance restraints extracted from thermodynamic mutant cycles. Biophys. J. 83, 2595–2609. doi: 10.1016/S0006-3495(02)75270-3
Fernandez, D., Ghanta, A., Kauffman, G. W., Sanguinetti, M. C. (2004). Physicochemical features of the HERG channel drug binding site. J. Biol. Chem. 279, 10120–10127. doi: 10.1074/jbc.M310683200
Flewelling, L. J., Naar, J. P., Abbott, J. P., Baden, D. G., Barros, N. B., Bossart, G. D., et al. (2005). Brevetoxicosis: red tides and marine mammal mortalities. Nature 435, 755–756. doi: 10.1038/nature435755a
Friese, J., Gleitz, J., Gutser, U. T., Heubach, J. F., Matthiesen, T., Wilffert, B., et al. (1997). Aconitum sp. alkaloids: the modulation of voltage-dependent Na+ channels, toxicity and antinociceptive properties. Eur. J. Pharmacol. 337, 165–174. doi: 10.1016/S0014-2999(97)01268-5
Gamal El-Din, T. M., Lenaeus, M. J., Zheng, N., Catterall, W. A. (2018). Fenestrations control resting-state block of a voltage-gated sodium channel. Proc. Natl. Acad. Sci. U. S. A 115, 13111–13116. doi: 10.1073/pnas.1814928115
Garg, V., Stary-Weinzinger, A., Sachse, F., Sanguinetti, M. C. (2011). Molecular determinants for activation of human ether-a-go-go-related gene 1 potassium channels by 3-nitro-n-(4-phenoxyphenyl) benzamide. Mol. Pharmacol. 80, 630–637. doi: 10.1124/mol.111.073809
Garg, V., Sachse, F. B., Sanguinetti, M. C. (2012). Tuning of EAG K(+) channel inactivation: molecular determinants of amplification by mutations and a small molecule. J. Gen. Physiol. 140, 307–324. doi: 10.1085/jgp.201210826
Garg, V., Stary-Weinzinger, A., Sanguinetti, M. C. (2013). ICA-105574 interacts with a common binding site to elicit opposite effects on inactivation gating of EAG and ERG potassium channels. Mol. Pharmacol. 83, 805–813. doi: 10.1124/mol.112.084384
Gingrich, K. J., Beardsley, D., Yue, D. T. (1993). Ultra-deep blockade of Na+ channels by a quaternary ammonium ion: catalysis by a transition-intermediate state? J. Physiol. 471, 319–341. doi: 10.1113/jphysiol.1993.sp019903
Grant, A. O., Dietz, M. A., Gilliam, F. R., Starmer, C. F. (1989). Blockade of cardiac sodium channels by lidocaine. Single-channel analysis. Circ. Res. 65, 1247–1262. doi: 10.1161/01.res.65.5.1247
Herzog, W. H., Feibel, R. M., Bryant, S. H. (1964). The Effect of Aconitine on the Giant Axon of the Squid. J. Gen. Physiol. 47, 719–733. doi: 10.1085/jgp.47.4.719
Hill, A. P., Sunde, M., Campbell, T. J., Vandenberg, J. I. (2007). Mechanism of block of the hERG K+ channel by the scorpion toxin CnErg1. Biophys. J. 92, 3915–3929. doi: 10.1529/biophysj.106.101956
Hille, B. (1977). Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 69, 497–515. doi: 10.1085/jgp.69.4.497
Horishita, T., Eger, E. I., 2nd, Harris, R. A. (2008). The effects of volatile aromatic anesthetics on voltage-gated Na+ channels expressed in Xenopus oocytes. Anesth Analg. 107, 1579–1586. doi: 10.1213/ane.0b013e318184b966
Hoshi, T., Zagotta, W. N., Aldrich, R. W. (1990). Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 250, 533–538. doi: 10.1126/science.2122519
Hoshi, T., Zagotta, W. N., Aldrich, R. W. (1991). Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron 7, 547–556. doi: 10.1016/0896-6273(91)90367-9
Huang, L. Y., Moran, N., Ehrenstein, G. (1982). Batrachotoxin modifies the gating kinetics of sodium channels in internally perfused neuroblastoma cells. Proc. Natl. Acad. Sci. U. S. A.. 79, 2082–2085. doi: 10.1073/pnas.79.6.2082
Inserra, M. C., Israel, M. R., Caldwell, A., Castro, J., Deuis, J. R., Harrington, A. M., et al. (2017). Multiple sodium channel isoforms mediate the pathological effects of Pacific ciguatoxin-1. Sci. Rep. 7, 42810. doi: 10.1038/srep42810
Jan, L. Y., Jan, Y. N. (2012). Voltage-gated potassium channels and the diversity of electrical signalling. J. Physiol. 590, 2591–2599. doi: 10.1113/jphysiol.2011.224212
Jansen, S. A., Kleerekooper, I., Hofman, Z. L., Kappen, I. F., Stary-Weinzinger, A., Van Der Heyden, M. A. (2012). Grayanotoxin poisoning: ‘mad honey disease' and beyond. Cardiovasc. Toxicol. 12, 208–215. doi: 10.1007/s12012-012-9162-2
Jaraskova, L., Linders, J. T. M., Van Der Veken, L. J. E., Willemsens, G. H. M., Bischoff, F. P. (2005). N-2 Adamantanyl-2-Phenoxy-Acetamide Derivatives as 11-Beta Hydroxysteroid Dehydrogenase Inhibitors.
Jung, H. J., Lee, J. Y., Kim, S. H., Eu, Y. J., Shin, S. Y., Milescu, M., et al. (2005). Solution structure and lipid membrane partitioning of VSTx1, an inhibitor of the KvAP potassium channel. Biochemistry 44, 6015–6023. doi: 10.1021/bi0477034
Jung, H. H., Jung, H. J., Milescu, M., Lee, C. W., Lee, S., Lee, J. Y., et al. (2010). Structure and orientation of a voltage-sensor toxin in lipid membranes. Biophys. J. 99, 638–646. doi: 10.1016/j.bpj.2010.04.061
Kaczmarski, J. A., Corry, B. (2014). Investigating the size and dynamics of voltage-gated sodium channel fenestrations. Channels (Austin) 8, 264–277. doi: 10.4161/chan.28136
Kamiya, K., Mitcheson, J. S., Yasui, K., Kodama, I., Sanguinetti, M. C. (2001). Open channel block of HERG K(+) channels by vesnarinone. Mol. Pharmacol. 60, 244–253. doi: 10.1124/mol.60.2.244
Kamiya, K., Niwa, R., Morishima, M., Honjo, H., Sanguinetti, M. C. (2008). Molecular determinants of hERG channel block by terfenadine and cisapride. J. Pharmacol. Sci. 108, 301–307. doi: 10.1254/jphs.08102FP
Kim, R. Y., Yau, M. C., Galpin, J. D., Seebohm, G., Ahern, C. A., Pless, S. A., et al. (2015). Atomic basis for therapeutic activation of neuronal potassium channels. Nat. Commun. 6, 8116. doi: 10.1038/ncomms9116
Klemic, K. G., Shieh, C. C., Kirsch, G. E., Jones, S. W. (1998). Inactivation of Kv2.1 potassium channels. Biophys. J. 74, 1779–1789. doi: 10.1016/S0006-3495(98)77888-9
Konoki, K., Baden, D. G., Scheuer, T., Catterall, W. A. (2019). Molecular Determinants of Brevetoxin Binding to Voltage-Gated Sodium Channels. Toxins 11, 513. doi: 10.3390/toxins11090513
Kopljar, I., Labro, A. J., Cuypers, E., Johnson, H. W., Rainier, J. D., Tytgat, J., et al. (2009). A polyether biotoxin binding site on the lipid-exposed face of the pore domain of Kv channels revealed by the marine toxin gambierol. Proc. Natl. Acad. Sci. U. S. A. 106, 9896–9901. doi: 10.1073/pnas.0812471106
Kubisch, C., Schroeder, B. C., Friedrich, T., Lutjohann, B., El-Amraoui, A., Marlin, S., et al. (1999). KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 96, 437–446. doi: 10.1016/S0092-8674(00)80556-5
Labro, A. J., Snyders, D. J. (2012). Being flexible: the voltage-controllable activation gate of kv channels. Front. Pharmacol. 3, 168. doi: 10.3389/fphar.2012.00168
Lange, W., Geissendorfer, J., Schenzer, A., Grotzinger, J., Seebohm, G., Friedrich, T., et al. (2009). Refinement of the binding site and mode of action of the anticonvulsant Retigabine on KCNQ K+ channels. Mol. Pharmacol. 75, 272–280. doi: 10.1124/mol.108.052282
Leaf, A. (2007). Prevention of sudden cardiac death by n-3 polyunsaturated fatty acids. J. Cardiovasc. Med. (Hagerstown) 8 Suppl 1, S27–S29. doi: 10.2459/01.JCM.0000289270.98105.b3
Lee, C. W., Kim, S., Roh, S. H., Endoh, H., Kodera, Y., Maeda, T., et al. (2004). Solution structure and functional characterization of SGTx1, a modifier of Kv2.1 channel gating. Biochemistry 43, 890–897. doi: 10.1021/bi0353373
Lees-Miller, J. P., Duan, Y., Teng, G. Q., Duff, H. J. (2000). Molecular determinant of high-affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: involvement of S6 sites. Mol. Pharmacol. 57, 367–374.
Lefevre, F., Aronson, N. (2000). Ketogenic diet for the treatment of refractory epilepsy in children: A systematic review of efficacy. Pediatrics 105, E46. doi: 10.1542/peds.105.4.e46
Lenaeus, M. J., Gamal El-Din, T. M., Ing, C., Ramanadane, K., Pomes, R., Zheng, N., et al. (2017). Structures of closed and open states of a voltage-gated sodium channel. Proc. Natl. Acad. Sci. U. S. A 114, E3051–E3060. doi: 10.1073/pnas.1700761114
Lepage, K., Rainier, J., Johnson, H., Baden, D., Murray, T. (2007). Gambierol Acts as a Functional Antagonist of Neurotoxin Site 5 on Voltage-Gated Sodium Channels in Cerebellar Granule Neurons. J. Pharmacol. Exp. Ther. 323, 174–179. doi: 10.1124/jpet.107.124271
Lewis, R. J., Vernoux, J.-P., Brereton, I. M. (1998). Structure of Caribbean Ciguatoxin Isolated from Caranx latus. J. Am. Chem. Soc. 120, 5914–5920. doi: 10.1021/ja980389e
Liang, Q., Anderson, W. D., Jones, S. T., Souza, C. S., Hosoume, J. M., Treptow, W., et al. (2015). Positive Allosteric Modulation of Kv Channels by Sevoflurane: Insights into the Structural Basis of Inhaled Anesthetic Action. PloS One 10, e0143363. doi: 10.1371/journal.pone.0143363
Liin, S. I., Silvera Ejneby, M., Barro-Soria, R., Skarsfeldt, M. A., Larsson, J. E., Starck Harlin, F., et al. (2015). Polyunsaturated fatty acid analogs act antiarrhythmically on the cardiac IKs channel. Proc. Natl. Acad. Sci. U. S. A. 112, 5714–5719. doi: 10.1073/pnas.1503488112
Lin, Y.-Y., Risk, M., Ray, S. M., Van Engen, D., Clardy, J., Golik, J., et al. (1981). Isolation and structure of brevetoxin B from the “red tide” dinoflagellate Ptychodiscus brevis (Gymnodinium breve). J. Am. Chem. Soc. 103, 6773–6775. doi: 10.1021/ja00412a053
Linford, N. J., Cantrell, A. R., Qu, Y., Scheuer, T., Catterall, W. A. (1998). Interaction of batrachotoxin with the local anesthetic receptor site in transmembrane segment IVS6 of the voltage-gated sodium channel. Proc. Natl. Acad. Sci. U. S. A. 95, 13947–13952. doi: 10.1073/pnas.95.23.13947
Lombet, A., Bidard, J. N., Lazdunski, M. (1987). Ciguatoxin and brevetoxins share a common receptor site on the neuronal voltage-dependent Na+ channel. FEBS Lett. 219, 355–359. doi: 10.1016/0014-5793(87)80252-1
Long, S. B., Tao, X., Campbell, E. B., Mackinnon, R. (2007). Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450, 376–382. doi: 10.1038/nature06265
Luzhkov, V. B., Aqvist, J. (2001). Mechanisms of tetraethylammonium ion block in the KcsA potassium channel. FEBS Lett. 495, 191–196. doi: 10.1016/S0014-5793(01)02381-X
Manville, R. W., Papanikolaou, M., Abbott, G. W. (2018). Direct neurotransmitter activation of voltage-gated potassium channels. Nat. Commun. 9, 1847. doi: 10.1038/s41467-018-04266-w
Marban, E., Yamagishi, T., Tomaselli, G. F. (1998). Structure and function of voltage-gated sodium channels. J. Physiol. 508 ( Pt 3), 647–657. doi: 10.1111/j.1469-7793.1998.647bp.x
Martinez-Morales, E., Kopljar, I., Rainier, J. D., Tytgat, J., Snyders, D. J., Labro, A. J. (2016). Gambierol and n-alkanols inhibit Shaker Kv channel via distinct binding sites outside the K(+) pore. Toxicon 120, 57–60. doi: 10.1016/j.toxicon.2016.07.017
Marzian, S., Stansfeld, P. J., Rapedius, M., Rinne, S., Nematian-Ardestani, E., Abbruzzese, J. L., et al. (2013). Side pockets provide the basis for a new mechanism of Kv channel-specific inhibition. Nat. Chem. Biol. 9, 507–513. doi: 10.1038/nchembio.1271
Mccusker, E. C., Bagneris, C., Naylor, C. E., Cole, A. R., D'avanzo, N., Nichols, C. G., et al. (2012). Structure of a bacterial voltage-gated sodium channel pore reveals mechanisms of opening and closing. Nat. Commun. 3, 1102. doi: 10.1038/ncomms2077
Mcnaughton-Smith, G. A., Gross, M. F., Wickenden, A. D. (2002). Benzanilides as potassium channel openers, United States patent application 6372767.
Mihailescu, M., Krepkiy, D., Milescu, M., Gawrisch, K., Swartz, K. J., White, S. (2014). Structural interactions of a voltage sensor toxin with lipid membranes. Proc. Natl. Acad. Sci. U. S. A 111, E5463–E5470. doi: 10.1073/pnas.1415324111
Mitcheson, J. S., Chen, J., Lin, M., Culberson, C., Sanguinetti, M. C. (2000). A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. U. S. A. 97, 12329–12333. doi: 10.1073/pnas.210244497
Murata, M., Legrand, A. M., Ishibashi, Y., Fukui, M., Yasumoto, T. (1990). Structures and configurations of ciguatoxin from the moray eel Gymnothorax javanicus and its likely precursor from the dinoflagellate Gambierdiscus toxicus. J. Am. Chem. Soc. 112, 4380–4386. doi: 10.1021/ja00167a040
Nicholson, G., Lewis, R. (2006). Ciguatoxins: Cyclic Polyether Modulators of Voltage-gated Ion Channel Function. Marine Drugs 4, 82–118. doi: 10.3390/md403082
Ottosson, N. E., Silvera Ejneby, M., Wu, X., Yazdi, S., Konradsson, P., Lindahl, E., et al. (2017). A drug pocket at the lipid bilayer-potassium channel interface. Sci. Adv. 3, e1701099. doi: 10.1126/sciadv.1701099
Ouyang, W., Jih, T. Y., Zhang, T. T., Correa, A. M., Hemmings, H. C., Jr. (2007). Isoflurane inhibits NaChBac, a prokaryotic voltage-gated sodium channel. J. Pharmacol. Exp. Ther. 322, 1076–1083. doi: 10.1124/jpet.107.122929
Ouyang, W., Herold, K. F., Hemmings, H. C., Jr. (2009). Comparative effects of halogenated inhaled anesthetics on voltage-gated Na+ channel function. Anesthesiology 110, 582–590. doi: 10.1097/ALN.0b013e318197941e
Padilla, K., Wickenden, A. D., Gerlach, A. C., Mccormack, K. (2009). The KCNQ2/3 selective channel opener ICA-27243 binds to a novel voltage-sensor domain site. Neurosci. Lett. 465, 138–142. doi: 10.1016/j.neulet.2009.08.071
Payandeh, J., Scheuer, T., Zheng, N., Catterall, W. A. (2011). The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358. doi: 10.1038/nature10238
Payandeh, J., Gamal El-Din, T. M., Scheuer, T., Zheng, N., Catterall, W. A. (2012). Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486, 135–139. doi: 10.1038/nature11077
Perry, M., Sachse, F. B., Abbruzzese, J., Sanguinetti, M. C. (2009). PD-118057 contacts the pore helix of hERG1 channels to attenuate inactivation and enhance K+ conductance. Proc. Natl. Acad. Sci. U. S. A 106, 20075–20080. doi: 10.1073/pnas.0906597106
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Poli, M. A., Mende, T. J., Baden, D. G. (1986). Brevetoxins, unique activators of voltage-sensitive sodium channels, bind to specific sites in rat brain synaptosomes. Mol. Pharmacol. 30, 129–135.
Ragsdale, D. S., Mcphee, J. C., Scheuer, T., Catterall, W. A. (1994). Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 265, 1724–1728. doi: 10.1126/science.8085162
Raju, S. G., Barber, A. F., Lebard, D. N., Klein, M. L., Carnevale, V. (2013). Exploring volatile general anesthetic binding to a closed membrane-bound bacterial voltage-gated sodium channel via computation. PloS Comput. Biol. 9, e1003090. doi: 10.1371/journal.pcbi.1003090
Rein, K. S., Baden, D. G., Gawley, R. E. (1994). Conformational Analysis of the Sodium Channel Modulator, Brevetoxin A, Comparison with Brevetoxin B Conformations, and a Hypothesis about the Common Pharmacophore of the “Site 5”. Toxins. J. Organ. Chem. 59, 2101–2106. doi: 10.1021/jo00087a027
Rundfeldt, C. (1997). The new anticonvulsant retigabine (D-23129) acts as an opener of K+ channels in neuronal cells. Eur. J. Pharmacol. 336, 243–249. doi: 10.1016/S0014-2999(97)01249-1
Sanchez-Chapula, J. A., Navarro-Polanco, R. A., Culberson, C., Chen, J., Sanguinetti, M. C. (2002). Molecular determinants of voltage-dependent human ether-a-go-go related gene (HERG) K+ channel block. J. Biol. Chem. 277, 23587–23595. doi: 10.1074/jbc.M200448200
Sanchez-Chapula, J. A., Ferrer, T., Navarro-Polanco, R. A., Sanguinetti, M. C. (2003). Voltage-dependent profile of human ether-a-go-go-related gene channel block is influenced by a single residue in the S6 transmembrane domain. Mol. Pharmacol. 63, 1051–1058. doi: 10.1124/mol.63.5.1051
Saxena, P., Zangerl-Plessl, E. M., Linder, T., Windisch, A., Hohaus, A., Timin, E., et al. (2016). New potential binding determinant for hERG channel inhibitors. Sci. Rep. 6, 24182. doi: 10.1038/srep24182
Schenzer, A., Friedrich, T., Pusch, M., Saftig, P., Jentsch, T. J., Grotzinger, J., et al. (2005). Molecular determinants of KCNQ (Kv7) K+ channel sensitivity to the anticonvulsant retigabine. J. Neurosci. 25, 5051–5060. doi: 10.1523/JNEUROSCI.0128-05.2005
Schreibmayer, W., Jeglitsch, G. (1992). The sodium channel activator Brevetoxin-3 uncovers a multiplicity of different open states of the cardiac sodium channel. Biochim. Biophys. Acta 1104, 233–242. doi: 10.1016/0005-2736(92)90035-K
Schroeder, B. C., Hechenberger, M., Weinreich, F., Kubisch, C., Jentsch, T. J. (2000). KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J. Biol. Chem. 275, 24089–24095. doi: 10.1074/jbc.M003245200
Sheridan, R. E., Adler, M. (1989). The actions of a red tide toxin from Ptychodiscus brevis on single sodium channels in mammalian neuroblastoma cells. FEBS Lett. 247, 448–452. doi: 10.1016/0014-5793(89)81389-4
Stevens, M., Peigneur, S., Tytgat, J. (2011). Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol. 2, 71. doi: 10.3389/fphar.2011.00071
Stock, L., Hosoume, J., Cirqueira, L., Treptow, W. (2018). Binding of the general anesthetic sevoflurane to ion channels. PloS Comput. Biol. 14, e1006605. doi: 10.1371/journal.pcbi.1006605
Strachan, L. C., Lewis, R. J., Nicholson, G. M. (1999). Differential actions of pacific ciguatoxin-1 on sodium channel subtypes in mammalian sensory neurons. J. Pharmacol. Exp. Ther. 288, 379–388.
Sun, J., Mackinnon, R. (2017). Cryo-EM Structure of a KCNQ1/CaM Complex Reveals Insights into Congenital Long QT Syndrome. Cell 169, 1042–1050 e1049. doi: 10.1016/j.cell.2017.05.019
Sunami, A., Glaaser, I. W., Fozzard, H. A. (2001). Structural and gating changes of the sodium channel induced by mutation of a residue in the upper third of IVS6, creating an external access path for local anesthetics. Mol. Pharmacol. 59, 684–691. doi: 10.1124/mol.59.4.684
Sutro, J. B. (1986). Kinetics of veratridine action on Na channels of skeletal muscle. J. Gen. Physiol. 87, 1–24. doi: 10.1085/jgp.87.1.1
Swartz, K. J. (2007). Tarantula toxins interacting with voltage sensors in potassium channels. Toxicon : Off. J. Int. Soc. Toxinol. 49, 213–230. doi: 10.1016/j.toxicon.2006.09.024
Thomas, D., Karle, C. A., Kiehn, J. (2006). The cardiac hERG/IKr potassium channel as pharmacological target: structure, function, regulation, and clinical applications. Curr. Pharm. Des. 12, 2271–2283. doi: 10.2174/138161206777585102
Trainer, V. L., Thomsen, W. J., Catterall, W. A., Baden, D. G. (1991). Photoaffinity labeling of the brevetoxin receptor on sodium channels in rat brain synaptosomes. Mol. Pharmacol. 40, 988–994.
Trainer, V. L., Baden, D. G., Catterall, W. A. (1994). Identification of peptide components of the brevetoxin receptor site of rat brain sodium channels. J. Biol. Chem. 269, 19904–19909.
Uehara, A., Moczydlowski, E. (1986). Blocking mechanisms of batrachotoxin-activated Na channels in artificial bilayers. Membr. Biochem. 6, 111–147. doi: 10.3109/09687688609065446
Ulbricht, W. (1969). The effect of veratridine on excitable membranes of nerve and muscle. Ergeb. Physiol. 61, 18–71. doi: 10.1007/BFb0111446
Vandenberg, J. I., Perry, M. D., Perrin, M. J., Mann, S. A., Ke, Y., Hill, A. P. (2012). hERG K(+) channels: structure, function, and clinical significance. Physiol. Rev. 92, 1393–1478. doi: 10.1152/physrev.00036.2011
Vennekamp, J., Wulff, H., Beeton, C., Calabresi, P. A., Grissmer, S., Hansel, W., et al. (2004). Kv1.3-blocking 5-phenylalkoxypsoralens: a new class of immunomodulators. Mol. Pharmacol. 65, 1364–1374. doi: 10.1124/mol.65.6.1364
Wang, W., Mackinnon, R. (2017). Cryo-EM Structure of the Open Human Ether-a-go-go-Related K(+) Channel hERG. Cell 169, 422–430 e410. doi: 10.1016/j.cell.2017.03.048
Wang, S. Y., Wang, G. K. (1998). Point mutations in segment I-S6 render voltage-gated Na+ channels resistant to batrachotoxin. Proc. Natl. Acad. Sci. U. S. A. 95, 2653–2658. doi: 10.1073/pnas.95.5.2653
Wang, S. Y., Wang, G. K. (1999). Batrachotoxin-resistant Na+ channels derived from point mutations in transmembrane segment D4-S6. Biophys. J. 76, 3141–3149. doi: 10.1016/S0006-3495(99)77465-5
Wang, G. K., Wang, S. Y. (2003). Veratridine block of rat skeletal muscle Nav1.4 sodium channels in the inner vestibule. J. Physiol. 548, 667–675. doi: 10.1113/jphysiol.2002.035469
Wang, H. S., Pan, Z., Shi, W., Brown, B. S., Wymore, R. S., Cohen, I. S., et al. (1998). KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282, 1890–1893. doi: 10.1126/science.282.5395.1890
Wang, S. Y., Nau, C., Wang, G. K. (2000). Residues in Na(+) channel D3-S6 segment modulate both batrachotoxin and local anesthetic affinities. Biophys. J. 79, 1379–1387. doi: 10.1016/S0006-3495(00)76390-9
Wang, S. Y., Barile, M., Wang, G. K. (2001). Disparate role of Na(+) channel D2-S6 residues in batrachotoxin and local anesthetic action. Mol. Pharmacol. 59, 1100–1107. doi: 10.1124/mol.59.5.1100
Wang, S. Y., Tikhonov, D. B., Mitchell, J., Zhorov, B. S., Wang, G. K. (2007). Irreversible block of cardiac mutant Na+ channels by batrachotoxin. Channels (Austin) 1, 179–188. doi: 10.4161/chan.4437
Wang, A. W., Yang, R., Kurata, H. T. (2017). Sequence determinants of subtype-specific actions of KCNQ channel openers. J. Physiol. 595, 663–676. doi: 10.1113/JP272762
Wang, C. K., Lamothe, S. M., Wang, A. W., Yang, R. Y., Kurata, H. T. (2018). Pore- and voltage sensor-targeted KCNQ openers have distinct state-dependent actions. J. Gen. Physiol. 150, 1722–1734. doi: 10.1085/jgp.201812070
Waterhouse, A. M, Procter, J. B., Martin, D. M. A., Clamp, M., Barton, G. J. (2009). Jalview Version 2 - a multiple sequence alignment editor and analysis workbench. Bioinformatics 25 (9), 1189–1191. doi: 10.1093/bioinformatics/btp033
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. doi: 10.1093/nar/gky427
West, J. W., Patton, D. E., Scheuer, T., Wang, Y., Goldin, A. L., Catterall, W. A. (1992). A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc. Natl. Acad. Sci. U. S. A 89, 10910–10914. doi: 10.1073/pnas.89.22.10910
Wickenden, A. D., Krajewski, J. L., London, B., Wagoner, P. K., Wilson, W. A., Clark, S., et al. (2008). N-(6-chloro-pyridin-3-yl)-3,4-difluoro-benzamide (ICA-27243): a novel, selective KCNQ2/Q3 potassium channel activator. Mol. Pharmacol. 73, 977–986. doi: 10.1124/mol.107.043216
Wrobel, E., Rothenberg, I., Krisp, C., Hundt, F., Fraenzel, B., Eckey, K., et al. (2016). KCNE1 induces fenestration in the Kv7.1/KCNE1 channel complex that allows for highly specific pharmacological targeting. Nat. Commun. 7, 1–13. doi: 10.1038/ncomms12795
Wulff, H., Castle, N. A., Pardo, L. A. (2009). Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discovery 8, 982–1001. doi: 10.1038/nrd2983
Xiong, Q., Sun, H., Li, M. (2007). Zinc pyrithione-mediated activation of voltage-gated KCNQ potassium channels rescues epileptogenic mutants. Nat. Chem. Biol. 3, 287–296. doi: 10.1038/nchembio874
Xu, C. Q., Zhu, S. Y., Chi, C. W., Tytgat, J. (2003). Turret and pore block of K+ channels: what is the difference? Trends Pharmacol. Sci. 24, 446–448author reply 448–449. doi: 10.1016/S0165-6147(03)00223-2
Yarov-Yarovoy, V., Mcphee, J. C., Idsvoog, D., Pate, C., Scheuer, T., Catterall, W. A. (2002). Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na+ channel alpha subunit in voltage-dependent gating and drug block. J. Biol. Chem. 277, 35393–35401. doi: 10.1074/jbc.M206126200
Yazdi, S., Stein, E., Elinder, F., Andersson, M., Lindahl, E. (2016). The Molecular Basis of Polyunsaturated Fatty Acid Interactions with the Shaker Voltage-Gated Potassium Channel. PloS Comput. Biol. 12, e1004704. doi: 10.1371/journal.pcbi.1004704
Yuki, T., Yamaoka, K., Yakehiro, M., Seyama, I. (2001). State-dependent action of grayanotoxin I on Na(+) channels in frog ventricular myocytes. J. Physiol. 534, 777–790. doi: 10.1111/j.1469-7793.2001.00777.x
Zamponi, G. W., Doyle, D. D., French, R. J. (1993). Fast lidocaine block of cardiac and skeletal muscle sodium channels: one site with two routes of access. Biophys. J. 65, 80–90. doi: 10.1016/S0006-3495(93)81042-7
Zhang, M., Korolkova, Y. V., Liu, J., Jiang, M., Grishin, E. V., Tseng, G. N. (2003). BeKm-1 is a HERG-specific toxin that shares the structure with ChTx but the mechanism of action with ErgTx1. Biophys. J. 84, 3022–3036. doi: 10.1016/S0006-3495(03)70028-9
Keywords: hydrophobic binding sites, voltage-gated potassium channels, voltage-gated sodium channels, channel fenestrations, lipophilic compounds
Citation: Van Theemsche KM, Van de Sande DV, Snyders DJ and Labro AJ (2020) Hydrophobic Drug/Toxin Binding Sites in Voltage-Dependent K+ and Na+ Channels. Front. Pharmacol. 11:735. doi: 10.3389/fphar.2020.00735
Received: 31 January 2020; Accepted: 04 May 2020;
Published: 15 May 2020.
Edited by:
Mounir Tarek, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Michael E. O'Leary, Cooper Medical School of Rowan University, United StatesRoope Mannikko, University College London, United Kingdom
Copyright © 2020 Van Theemsche, Van de Sande, Snyders and Labro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alain J. Labro, YWxhaW4ubGFicm9AdWFudHdlcnBlbi5iZQ==
†These authors share first authorship