 Michael A. Mandell
Michael A. Mandell Bhaskar Saha1
Bhaskar Saha1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 11 March 2020
Sec. Pharmacology of Anti-Cancer Drugs
Volume 11 - 2020 | https://doi.org/10.3389/fphar.2020.00308
This article is part of the Research TopicNovel Cancer Treatments based on Autophagy ModulationView all 17 articles
Autophagy is a cellular degradative process that has multiple important actions in cancer. Autophagy modulation is under consideration as a promising new approach to cancer therapy. However, complete autophagy dysregulation is likely to have substantial undesirable side effects. Thus, more targeted approaches to autophagy modulation may prove clinically beneficial. One potential avenue to achieving this goal is to focus on the actions of tripartite motif-containing protein family members (TRIMs). TRIMs have key roles in an array of cellular processes, and their dysregulation has been extensively linked to cancer risk and prognosis. As detailed here, emerging data shows that TRIMs can play important yet context-dependent roles in controlling autophagy and in the selective targeting of autophagic substrates. This review covers how the autophagy-related actions of TRIM proteins contribute to cancer and the possibility of targeting TRIM-directed autophagy in cancer therapy.
Macroautophagy is a promising new target for cancer treatment as this cellular pathway has both cancer-suppressing and cancer-promoting mechanisms. Macroautophagy (autophagy hereafter) is a process of cellular self-digestion that involves the sequestration of cytoplasmic contents into a vesicle (the autophagosome) that fuses with the lysosome where it is degraded. The “core” molecular machinery that is required for autophagy consists of more than 30 proteins. These proteins were mostly identified in yeast and their functions are conserved in human cells (Klionsky et al., 2011). However, as the physiological roles of autophagy have been expanded in higher organisms, the number of proteins involved in mammalian autophagy is increased relative to what is seen in single-celled organisms. Autophagy has been classified as being either “bulk” or “selective”, the latter indicating the ability of the autophagy machinery to identify and selectively degrade substrates. Selective autophagy is further classified into “–phagies”, denoting the particular substrates degraded: for example, mitophagy is the autophagic degradation of mitochondria, ERphagy involves autophagy of endoplasmic reticulum, ferritinophagy refers to autophagic degradation of ferritin, and so forth. While the different “phagies” all require the same core autophagy machinery, they can vary in terms of their upstream regulators and in the factors required for specific cargo identification (Kirkin and Rogov, 2019). This variability in mechanism opens the possibility to the selective pharmacological modulation of certain autophagic activities.
Autophagic degradation of cytoplasmic contents can generate molecules for biosynthesis or energy during times of cellular starvation. Additionally, autophagy plays an important cytoplasmic quality control function that can eliminate specific proteins, toxic protein aggregates, unnecessary or non-functional organelles, and intracellular pathogens from cells. These pro-survival functions of autophagy have been of interest as potential targets of cancer therapy (Levy et al., 2017), and work in experimental rodent cancer models has demonstrated that a variety of tumors require functional autophagy in the tumor cells themselves and in healthy tissue (Poillet-Perez and White, 2019). Furthermore, there are indications that autophagy can render cancer cells more resistant to chemotherapy (Sui et al., 2013; Pan et al., 2019). Together, these findings strongly indicate that pharmacological inhibition of autophagy may be a promising approach to cancer therapy. Nevertheless, there is a problem: complete inhibition of autophagy by inducible whole-body knockout of the core autophagy factor ATG7 is lethal in mice (Karsli-Uzunbas et al., 2014), suggesting the strong likelihood of unacceptably severe side effects if autophagy inhibition were to be tried in human cancer patients. A more tractable approach to modulate autophagy in cancer may be to target specific components of the varieties of autophagic processes.
The challenge of targeting specific elements of autophagy could potentially be alleviated if there was some way to specifically target a cancer-promoting phagy while allowing other varieties of autophagy to proceed normally. Conceptually, this would be done by targeting proteins with phagy-specific functions rather than by targeting the core machinery or lysosomal function to block all autophagy. One possible way of doing this is through the tripartite motif family of proteins (TRIMs). This large protein family has emerged as possessing a wide variety of actions on autophagy regulation and action. Importantly, many TRIMs have very strong connections to oncogenesis or cancer progression. The purpose of this review is to detail how TRIMs intersect with and orchestrate autophagy and to discuss how TRIM-mediated autophagy may affect oncogenesis, cancer progression, and cancer therapy.
The TRIM family of proteins is structurally distinguished by having a cluster of domains starting with an N-terminal RING domain, followed by one or two B box domains, and then a coiled-coil domain (CCD; Figure 1A) (Reymond et al., 2001). The RING domain confers upon TRIMs their catalytic function as E3 ligases, and individual TRIMs have been shown to directly ubiquitylate, SUMOylate, or NEDDylate themselves and/or their interacting partners (Ivanov et al., 2007; Noguchi et al., 2011; Fletcher et al., 2015). While the overwhelming majority of TRIMs possess a RING domain, there are some exceptions (e.g. TRIM16, TRIM20). Interestingly, TRIM16 still has ubiquitin ligase activity due to a cryptic RING-like fold in its B box domain (Bell et al., 2012), emphasizing that the enzymatic activity of TRIMs should be determined empirically. The B box and CCD both mediate protein-protein interactions, with the CCD allowing for TRIM hetero- and homodimerization. At their C terminus, most TRIMs have one or more additional domains with the SPRY domain being the most common variant in human TRIMs. The SPRY domain is important for mediating protein-protein interactions such as the interaction between TRIM5 and retroviral capsids, while other C terminal domains have different interacting specificities (e.g. PHD domain can bind to chromatin) or even have enzymatic activities (the ADP-ribosylation factor/ARF domain of TRIM23).
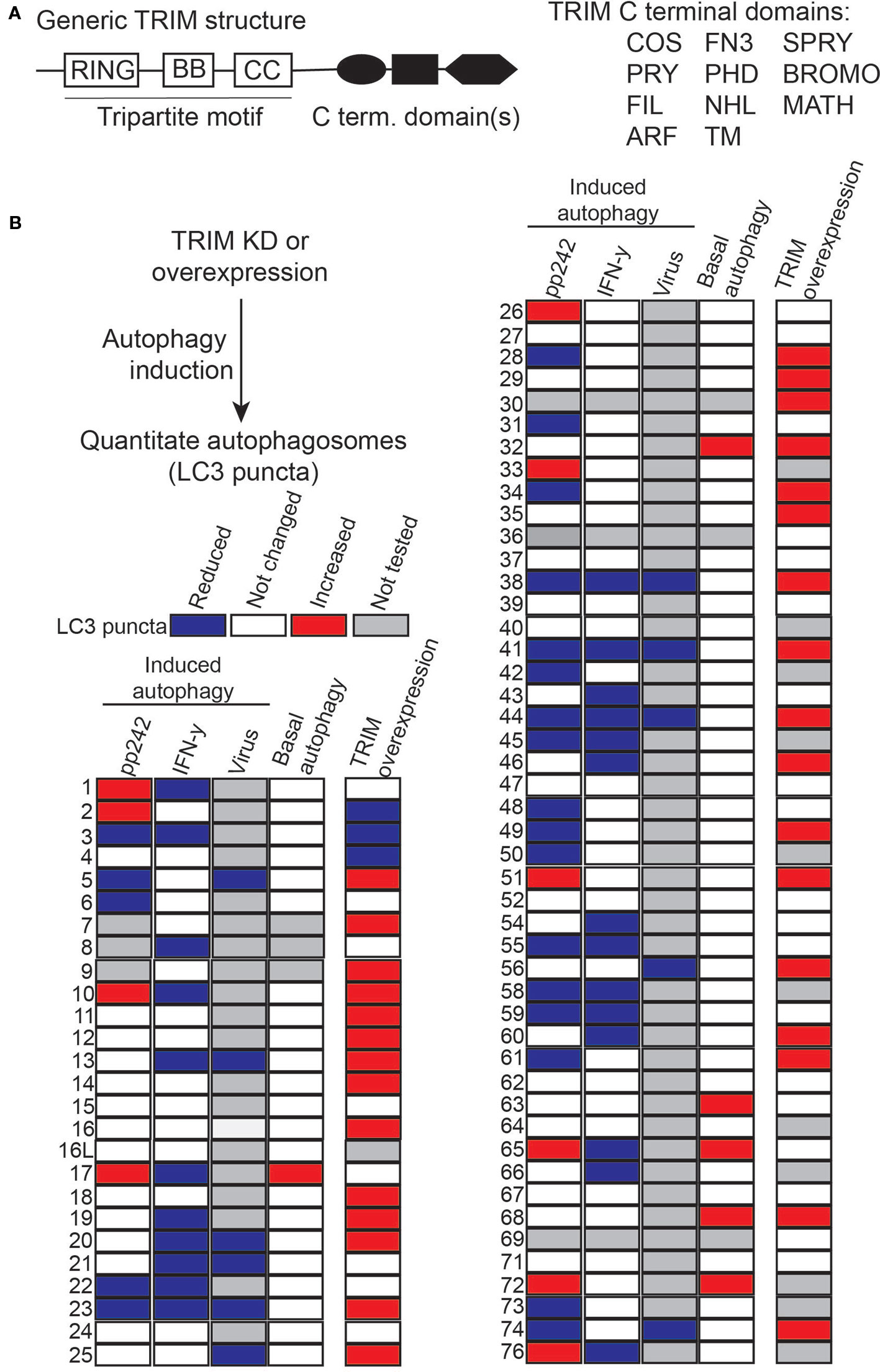
Figure 1 Many tripartite motif-containing protein family members (TRIMs) act as autophagy regulators. (A) Left, schematic of generic TRIM protein domain organization. Typical TRIMs have N-terminal RING domains (RING), one or two B box domains (BB), a coiled-coil (CC) domain and may have one or more C terminal domains. Right, list of C terminal domains present in TRIM family proteins. (B) The results of several previously published TRIM siRNA or over-expression screens are summarized here in heat map format. In all experiments, cells were transfected with TRIM siRNA or expression plasmids and treated or not with a known inducer of autophagy (e.g. pp242) prior to imaging-based quantitation of cytoplasmic LC3B or GFP-LC3B puncta (autophagosomes). TRIMs that significantly increased or decreased autophagosome abundance relative to negative controls are colored red or blue, respectively. Changes in the abundance of autophagosomes can result from either increased autophagy activation or decreased autophagy flux, thus in isolation these data do not indicate mechanisms of individual TRIMs on autophagy but illustrate the broad involvement of TRIMS in autophagy regulation.
TRIMs are a metazoan-specific protein family, with seven TRIMs found in the genome the fruit fly Drosophila melanogaster and 18 TRIMs in the Caenorhabditis elegans genome (Sardiello et al., 2008). The number of TRIM genes is substantially elevated in vertebrates, with more than 200 TRIMs or TRIM-like genes found in the zebrafish (Danio rerio) genome (Sardiello et al., 2008). The human genome includes more than 80 TRIMs which have been assigned into eleven sub-families based on their domain organization (Short and Cox, 2006; Ozato et al., 2008). Many of these genes encode for multiple isoforms, thus further expanding the protein sequence diversity and possibly the functionality of TRIM proteins. At a cellular level, these functions include governing gene expression, regulating signal transduction pathways, contributing to cytoplasmic quality control, direct antiviral action, and effects on cell survival and metabolism. At the organismal level, TRIMs play important roles in development and in immune regulation, and alterations in TRIM protein function/expression are linked to a variety of diseases including cancer (Watanabe and Hatakeyama, 2017; Park et al., 2020).
Many TRIM proteins are found as relevant biomarkers of cancer, where they may show decreased or increased levels of expression (Table 1). A significant decrease in TRIM expression associated with cancers is suggestive of a tumor suppressive role. In contrast, a significant overexpression of TRIM proteins may reflect a contribution to cancer development and/or cancer progression. TRIMs with the greatest association with cancer include 11, 14, 24, 25, 27, 28, 29, 33, 37, 44, and 59, each associated with at least five different cancers. It is likely the expression of TRIMs in cancers is relevant to the development and/or progression of the disease and TRIM expression may have prognostic value for cancer. Furthermore, TRIMs associated with specific cancers may provide insight into the development of novel TRIM targeted cancer therapies. Importantly, associations between individual TRIMs and different cancers are regularly being discovered, strongly predicting that the list presented in Table 1 will grow with further study. The following paragraphs details a few of the connections between TRIMs and individual cancers.
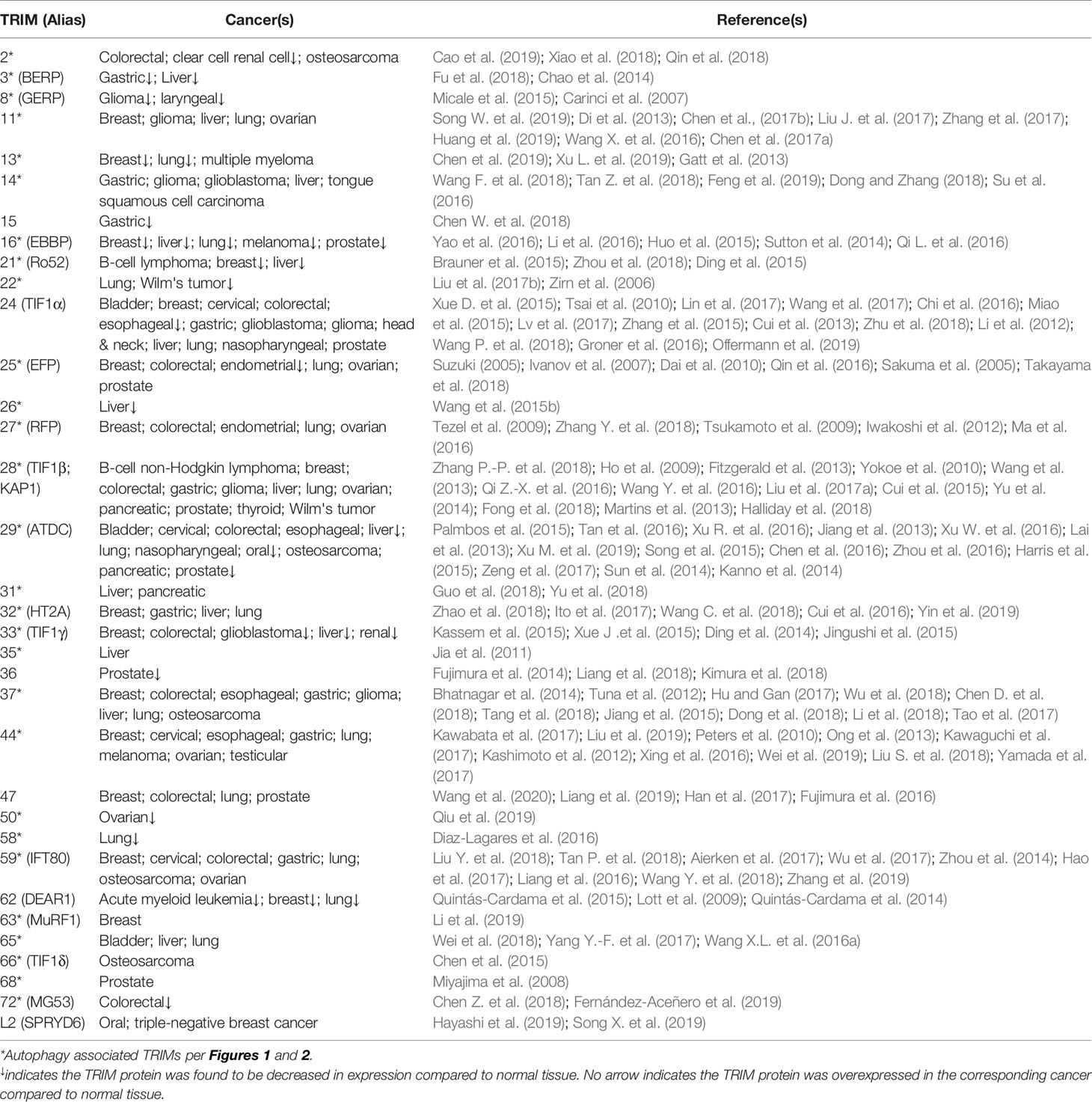
Table 1 TRIM expression changes found in cancers.
Lung cancer has the highest mortality among cancers in the U.S. (Siegel et al., 2020), in part due to the poor response of current cancer chemotherapeutic regimens for lung cancer. In addition to the importance of identifying new lung cancer biomarkers, the identification of novel therapeutic targets or approaches to increase the efficacy of lung cancer chemotherapies is critically needed. Many TRIMs are altered in expression in lung cancer (Table 1). Of these, TRIMs 13, 16, 58, and 62 have reduced levels of expression, whereas the majority of TRIMs associated with lung cancer are increased in expression. TRIM13 expression is reduced in non-small-cell lung cancer (NSCLC), where its overexpression was found to inactivate NF-κB (Xu L. et al., 2019). TRIM16 was also found decreased in NSCLC with concurrent upregulation of the sonic hedgehog pathway, suggesting a role for TRIM16 in epithelial-mesenchymal-transition in NSCLC (Huo et al., 2015). Interestingly, hypermethylation of TRIM58 in lung cancer may account for its down-regulation (Diaz-Lagares et al., 2016). Overexpression of TRIMs 11, 22, 44, 47, 59, and 65 were correlated with poor prognosis in lung cancers (Liu et al., 2013; Wang X.-L. et al., 2016; Xing et al., 2016; Han et al., 2017; Hao et al., 2017; Liu et al., 2017b; Huang et al., 2019; Yin et al., 2019). Overexpression of TRIM44 induced mTOR signaling, epithelial-mesenchymal transition, and cyclin/CDK upregulation in lung cancer cells (Xing et al., 2016). Poor prognosis of lung cancer patients was observed in those overexpressing TRIM27 (RFP) and possessing epidermal growth factor receptor mutations (Iwakoshi et al., 2012). The multitude of TRIMs found increased in lung cancers may serve as promising targets for improved lung cancer therapy.
As listed in Table 1, breast cancer has been associated with at least 15 different TRIM proteins. Decreased levels of TRIM expression in breast cancer were observed with TRIMs 13, 21, and 62. Chen et al. (2019) found that decreased TRIM13 expression was associated with worse distant metastasis free survival, disease specific survival, metastatic relapse free survival, and relapse free survival. Zhou et al. (2018) found decreased TRIM21 expression correlated with poor overall survival in breast cancer patients. Reduced TRIM62 (DEAR1) expression was found in many breast cancer tissues and was found to strongly correlate with early-onset breast cancer (Lott et al., 2009). In a 3D cell culture model, restoration of TRIM62 expression inhibited uncontrolled cell growth and directed the cells to form organoids reminiscent of health breast tissue (Lott et al., 2009). In contrast, TRIM11 levels were found increased in breast cancer tissues, where TRIM11 may act through the AKT/GLUT1 signaling pathway in breast cancer (Song W. et al., 2019). Elevated TRIM24 and TRIM37 in breast cancer may act through modifications of histone proteins, H2A and H3, respectively (Tsai et al., 2010; Bhatnagar et al., 2014). Elevated levels of TRIM32 and TRIM44 were associated with actions on NF-κB pathways in breast cancer (Kawabata et al., 2017; Zhao et al., 2018). Interestingly, TRIM59 was found upregulated in metastatic breast cancer, where it was observed to suppress the selective autophagic degradation of a tumor suppressor (Tan P. et al., 2018), underscoring an autophagic role for TRIM proteins in cancer.
Though the incidence of liver cancer is relatively low in the U.S., the mortality rate for this cancer is high (Siegel et al., 2020). TRIMs 3, 16, 21, and 29 were found at reduced levels in liver cancers (Table 1), where this reduced expression was consistently found associated with poorer prognosis among liver cancer patients. In contrast, TRIMs 11, 14, 24, 28, 31, 32, 37, and 65 have been found elevated in human liver cancers.
Colorectal cancer is the third most common cancer in both incidence and mortality among men and women in the U.S. (Siegel et al., 2020). TRIM72 expression is reduced in the serum of colon cancer patients and in colon cancer tumors (Chen Z. et al., 2018; Fernández-Aceñero et al., 2019). In contrast, most TRIMs associated with colorectal cancers have been found up-regulated, including TRIMs 2, 24, 25, 27, 29, 33, 37, 47, and 59 (Table 1). Up-regulation of TRIM47 in colorectal cancers was associated with SMAD4 degradation, enhancing growth and invasion of colorectal cancer cells (Liang et al., 2019).
The androgen receptor possesses a key function in prostate cancer progression serving as the main target for the treatment of advanced, hormone-responsive disease (Yuan et al., 2014). TRIM36 expression is increased in response to androgen and has a prostate cancer suppressive role that includes inhibiting prostate cancer cell proliferation and migration while promoting prostate cancer cell death (Kimura et al., 2018; Liang et al., 2018). Reduced levels of TRIM36 are associated with advanced stages of prostate cancer (Fujimura et al., 2014; Kimura et al., 2018; Liang et al., 2018) and TRIM36 was reported to be an independent predictor of survival in prostate cancer patients (Kimura et al., 2018). Like TRIM36, expression TRIMs 16 and 29 is decreased in prostate cancer, suggesting that these proteins may act as tumor suppressors in normal prostate tissue. In contrast, high expression levels of TRIMs 24 and 28 are associated with more advanced prostate cancer disease, particularly in androgen non-responsive, castration resistant cancer. TRIM24 can augment androgen receptor signaling (Groner et al., 2016), apparently downstream of the actions of TRIM28 (Fong et al., 2018). Increased levels of TRIMs 25, 47, and 68 are also associated with poorer prognosis of prostate cancer (Table 1).
A number of TRIM genes are associated with chromosomal translocations that likely contribute to oncogenesis. One of the most investigated involves a translocation between the TRIM19 gene (also known as PML) on chromosome 15 and the retinoic acid receptor alpha (RARα) gene located on chromosome 17, which is associated with acute promyelocytic leukemia (Cambiaghi et al., 2012). This fusion protein acts by repressing genes associated with retinoic acid signaling. The RET gene on chromosome 10 has been found in translocations with a number of TRIM genes including TRIM24, TRIM27, and TRIM33 associated with papillary thyroid cancer (Klugbauer and Rabes, 1999), lymphoma (Takahashi et al., 1985), and non-small cell lung carcinoma (Lin et al., 2015), respectively. Similarly, the BRAF gene has been found translocated with TRIM4 in lung cancer (Shim et al., 2015) and TRIM24 in both melanoma (Hutchinson et al., 2013) and lung cancer (Nakaoku et al., 2014) and the FGFR1 gene is translocated with TRIM24 in myeloproliferative syndrome (Belloni et al., 2005). These fusion proteins lead to the unregulated activity of the RET, BRAF, or FGFR1 kinases, resulting in the activation of multiple pro-survival signaling pathways. In summary, a number of TRIM-containing oncogenic gain-of-function fusion genes have been found with profound effects in oncogenesis.
A small sub-population of cancer cells have properties reminiscent of embryonic stem cells including a high degree of cellular plasticity, resistance to cell death, and the capacity of self-renewal. These cancer stem cells are important contributors to metastasis, drug resistance, and cancer recurrence, leading to an interest in targeting them as part of cancer therapy (Yang et al., 2020). Several TRIMs can regulate pathways seminal to cancer stemness including STAT signaling, AKT signaling, NANOG-Sox2-Oct-3/4 networks, and pathways related to epithelial-mesenchymal transition (EMT). In this section we cite a few examples of how TRIMs can regulate stemness, a topic that was recently reviewed by Jaworska et al. (2020). TRIM28 is reported to maintain of Oct-3/4-Sox2-NANOG expression in breast cancer cells (Czerwinska et al., 2017). TRIM24 is also reported to promote cancer stemness in glioblastoma by enhancing STAT3-mediate transcriptional activation (Lv et al., 2017). Additionally, TRIMs 14 and 24 have been shown to enhance EMT through promoting AKT signaling in gastric and colorectal cancer, respectively (Wang F. et al., 2018; Zhang Y. et al., 2018). In contrast to TRIMs that up-regulate stemness pathways, TRIM16 has been associated as a negative regulator of stemness in breast and ovarian cancer cells (Yao et al., 2016; Tan et al., 2017). In both cases, TRIM16 acted to reduce expression of the Hh signaling-activated transcription factor Gli-1, a positive regulator of cancer stem cell self-renewal. Importantly, autophagy is another key contributor to cancer stem cells promoting their longevity as quiescent cells (Vera-Ramirez et al., 2018) and its downregulation is associated with cancer stem cell reactivation (La Belle Flynn et al., 2019). TRIMs are major regulators of autophagic processes, and their autophagic actions may constitute another TRIM-dependent contribution to cancer stemness.
The tumor suppressor protein p53 promotes genomic stability and can induce cell cycle arrest and apoptosis resulting from extensive cellular DNA damage. Interactions between TRIM proteins and p53 are well-established and have recently been reviewed in detail (Valletti et al., 2019). TRIMs 11, 21, 24, 25, 28, 29, 31, 32, 39, and 59 all negatively regulate p53. Mechanistically, TRIMs can directly ubiquitylate p53, leading to its consequent degradation or sequestration in the cytoplasm where it cannot impact gene expression or carry out its pro-apoptotic or cell-cycle controlling functions.
In addition to being subject to proteasomal degradation, multiple studies have demonstrated that p53 can also be regulated by delivery to the lysosome for degradation by the autophagy pathway (White, 2016). Autophagy regulation is a very prominent feature of many TRIMs. Collectively, recent studies have identified individual TRIMs that impact stages of the autophagy pathway. The following sections will focus on the mechanisms whereby TRIMs impact autophagy and highlight examples of how TRIM-directed autophagy contributes to cancer.
In addition to these activities in cancer, TRIMs also have been shown to impact oncogenesis and tumor progression through their actions on autophagy. A panel of published siRNA screens have demonstrated that a surprisingly high percentage of human TRIMs appear to regulate autophagy in cells under basal autophagy conditions (Mandell et al., 2014) or in response to autophagy induction by mTOR inhibition (Mandell et al., 2014; Sparrer et al., 2017), interferon γ stimulation (Kimura et al., 2015), lysosomal damage (Chauhan et al., 2016), or viral infection (Sparrer et al., 2017). Collectively, these screens identified 49 TRIMs whose knockdown either decreased or increased the number of autophagosomes in cells (Figure 1B). Additional sets of TRIMs have been identified as having autophagy regulatory roles under other experimental conditions. The fact that so many TRIMs were identified in these screens illustrated that some of the actions of TRIMs in autophagy are context dependent with some TRIMs contributing to defined subsets of autophagic outputs. For example, TRIM16 was uniquely required among TRIMs for an autophagic response to lysosomal damage but was dispensable for autophagy induced by mTOR inhibition, interferon γ, or viruses. The context-specific action of TRIMs in autophagy is important because it suggests that the modulation of a specific TRIM may affect only the subset of autophagic activities governed by that TRIM. The ability to precisely alter the cancer-related subset of autophagic activities may be therapeutically beneficial. Additionally, the broad requirement for TRIMs in autophagy suggested that they may act via non-redundant mechanisms. This notion is supported by subsequent studies, which have demonstrated that some TRIMs affect the cellular abundance of autophagy-related proteins whereas other TRIMs appear to affect the activation status of autophagy regulators and/or alter their protein-protein interactions (Figure 2).
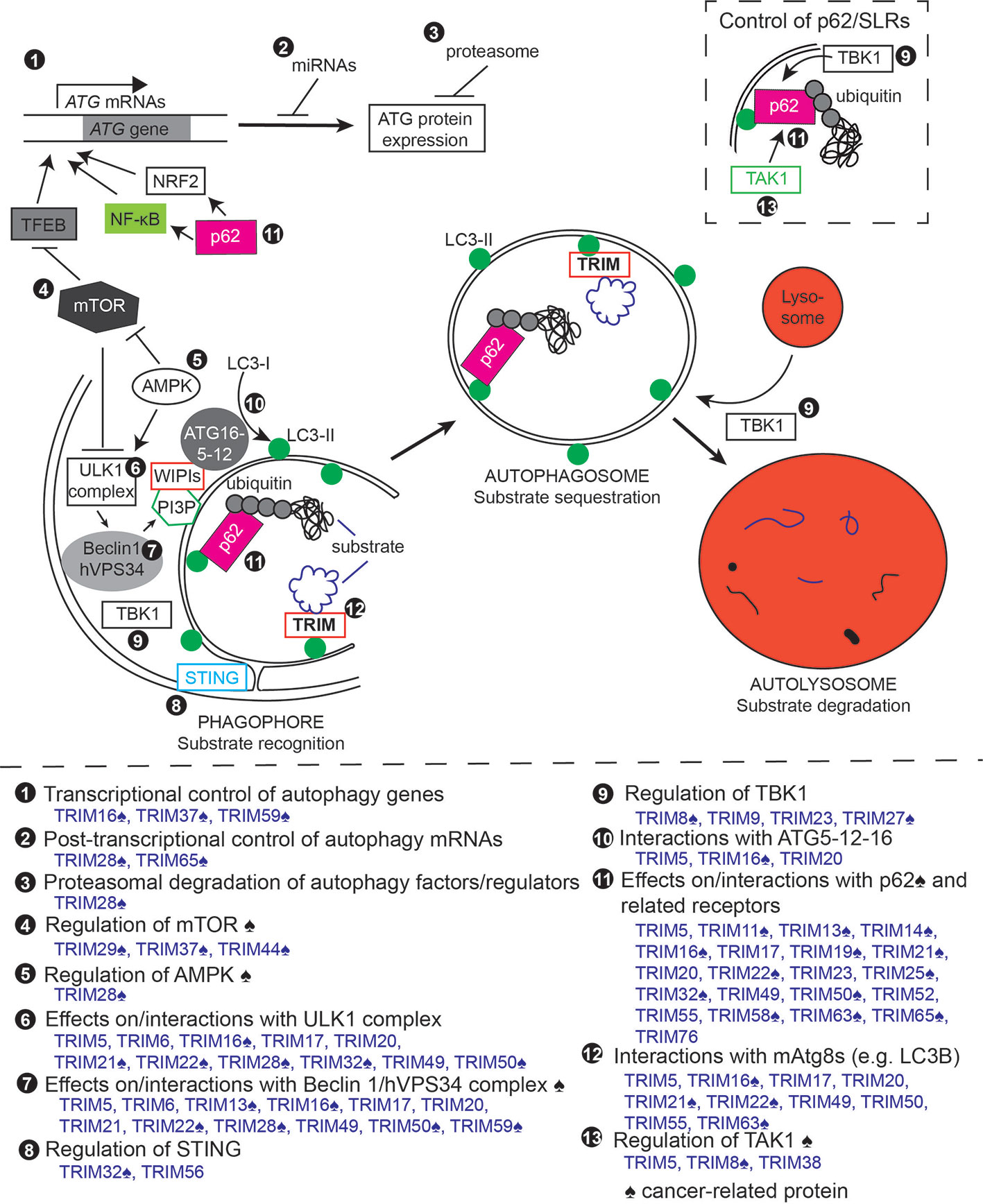
Figure 2 Tripartite motif-containing protein family members (TRIMs) regulate the autophagy pathway at multiple points. Top, schematic of different steps/stages of the autophagy pathway. Circled numbers indicate steps of the autophagy pathway or autophagy regulators and factors that are impacted by individual TRIMs. Bottom, summary of TRIM actions in autophagy. Circled numbers correspond with those on the schematic. ♠ symbol indicates proteins with reported cancer relevance.
Several TRIMs have been shown to affect the transcription of autophagy genes. In some cases, this is through TRIM actions on transcription factors that activate expression of autophagy-related genes. For example, the expression of TRIM59 in the lung carcinoma cell line H1299 inhibits autophagy by negatively regulating the expression of Becn1 mRNA (Han et al., 2018), an effect that was connected to TRIM59's observed inhibitory action on NF-κB activation. TRIM37, a known oncogene (Bhatnagar et al., 2014), suppresses autophagic flux and inhibits the activation and nuclear translocation of the pro-autophagy transcription factor TFEB (Wang W. et al., 2018). Conversely, TRIM16 promotes its own expression along with that of the autophagy receptor p62 by driving Nrf2 activation under conditions of oxidative stress (Jena et al., 2018). TRIM16 is also found in protein complexes with TFEB (Chauhan et al., 2016), but how this interaction shapes TFEB activation separately from the role of TRIM16 in maintaining lysosomal health has not been fully explored. It is likely that additional TRIMs will be identified that can regulate autophagy by effecting diverse signal transduction pathways that result in the activation of transcription factors (e.g. IRF3, AP1, Nf-κB).
In addition to regulating transcription factor activity, some TRIMs localize to the nucleus and can directly act as transcriptional regulators or co-regulators. While a subset of TRIMs have a C terminal domain (plant homeodomain, PHD) that mediates chromatin binding, some TRIMs lacking these domains can localize to the nucleus and affect gene expression: an example being TRIM22 which was shown to reduce retroviral gene expression (Kajaste-Rudnitski et al., 2011). The transcriptional regulatory activities of a TRIM on autophagy was first demonstrated for the PHD domain-containing TRIM28 (also known as KAP1, Table 1) (Barde et al., 2013). Hematopoietic-specific knockout of TRIM28 resulted in abnormal erythroblasts that contained elevated numbers of mitochondria. Accordingly, TRIM28 knockout erythroblasts expressed substantially lower levels of mRNAs coding for core autophagy factors (e.g. Ulk1, Becn1, Atg12) and for proteins with mitophagy-specific functions (Nix and Bnip3L). Mechanistically, TRIM28 was found to repress the expression of miRNAs that target autophagy factors. Similarly, TRIM65 promotes autophagy by preventing miRNA-based down-regulation of ATG7 in a non-small-cell lung cancer cell line (Pan et al., 2019). In this study, the TRIM65 knockdown potentiated the cytotoxic effects of cisplatin.
The autophagy pathway is central to many cellular functions under both homeostatic and stress conditions, and hence a large number of signal transduction pathways have roles in positively or negatively regulating autophagy. The mTOR and AMPK signaling pathways, both of which are involved in sensing a cell's nutritional status and are of critical importance to cancer, are the best-known autophagy regulating pathways. mTOR (mammalian target of rapamycin) is activated under amino acid replete conditions and in turn activates signaling that promotes anabolic processes and cellular growth. Active mTOR attenuates the expression of autophagy genes by inhibiting the activation and nuclear localization of TFEB and other MiTF transcription factors through direct phosphorylation. mTOR also directly inhibits the activation of autophagosome initiation by phosphorylation of the most upstream autophagy factor ULK1 at serine 757. AMPK (5' AMP-activated protein kinase) is activated by low glucose conditions in cells and directly opposes the actions of mTOR. When activated, AMPK phosphorylates mTOR, leading to the disassembly and inactivation of mTOR complexes. AMPK also phosphorylates ULK1 in an activating manner at Ser317, Ser555, and Ser777, resulting in autophagy activation (Galluzzi et al., 2014).
Several TRIMs are now known to regulate autophagy through actions on mTOR or AMPK signaling. For instance, TRIM37 (Table 1) was recently reported to physically interact with mTOR complex components and to promote the assembly of active mTOR complexes at the lysosome (Wang W. et al., 2018). TRIM37-deficient cells carryout unregulated autophagy. Interestingly, TRIM37-deficient cells become “autophagy addicted”, and so inhibition of autophagy flux in these cells leads to pronounced cell death (Wang W. et al., 2018). TRIM29 and TRIM44 (Table 1) are also reported to affect mTOR signaling (Xing et al., 2016; Zhou et al., 2016).
The ability of AMPK to induce autophagy is also subject to regulation by TRIMs. In a subset of cancers, TRIM28 ubiquitylates the AMPKα1 subunit, resulting in its proteasomal degradation and repression of autophagy (Pineda et al., 2015). This effect is mediated by two proteins whose expression is largely cancer-specific, the melanoma antigen A3 and A6 (MAGE-A3/6), which interact with TRIM28 and recruit it to AMPKα1. AMPK activity has also been linked to the pro-cancer kinase TAK1 (Xie et al., 2006; Herrero-Martin et al., 2009), and TAK1 is in turn activated by TRIMs 5 (Pertel et al., 2011) and 8 (Li et al., 2011) and inhibited by TRIM38 (Hu et al., 2014), likely through autophagic degradation of TAK1 complex components.
The STING-TBK1 signaling axis is another autophagy-regulating pathway whose activity is orchestrated by multiple TRIMs. STING is a crucial component of cytosolic DNA signaling pathways. Under homeostatic conditions, STING, which contains four transmembrane domains, is localized to the endoplasmic reticulum membrane in an inactive state. In response to cytosolic DNA detection, STING undergoes a conformational change that allows for the recruitment of the kinase TBK1. The STING-TBK1 complex re-localizes to the ER-Golgi intermediate compartment, where TBK1 can phosphorylate and activate the transcription factor IRF3 (Chen Q. et al., 2016). TBK1 can also be activated by other pattern recognition receptors (Louis et al., 2018). These two proteins have key roles in autophagy regulation. STING has been shown to be important for autophagy induction in response to various microbial stimuli (Watson et al., 2015; Moretti et al., 2017), and cytosolic DNA-activated STING was recently shown to provide a membrane source for autophagosome formation independently of the “core” autophagy upstream regulator ULK1 and the hVPS34/Beclin 1 complex (Gui et al., 2019). TBK1 also plays important roles in autophagy regulation, at least some of which are independent of STING. For instance, TBK1-mediated phosphorylation of syntaxin 17 is required for the earliest steps of autophagosome formation (Kumar et al., 2019) and TBK1 also has a role in allowing for autophagic maturation (Pilli et al., 2012). These activities may be in addition to or in conjunction with TBK1's roles in promoting autophagic cargo selectivity through actions on autophagy receptors p62, NDP52, and optineurin (Pilli et al., 2012; Richter et al., 2016; Vargas et al., 2019). Both TRIM56 and TRIM32 have been shown to potentiate the STING-TBK1 pathway by carrying out the K63-linked poly-ubiquitylation of STING (Tsuchida et al., 2010; Zhang et al., 2012), while the mouse-specific TRIM30α catalyzes K48-linked ubiquitylation of STING, resulting in its degradation (Wang et al., 2015a). TRIM27 promotes the proteasomal degradation of TBK1 (Zheng et al., 2015), while other TRIMs can affect the activity of TBK1 by modifying its protein-protein interactions (Qin et al., 2016; Ye et al., 2017). Whereas autophagy was not addressed in the studies cited above, TRIM23 has been demonstrated to affect virus-induced autophagy through direct actions on TBK1 (Sparrer et al., 2017). TRIM23 is unique among the human TRIM family in that it is the only TRIM to feature an ADP ribosylation factor-like ARF domain as a C terminal domain. This domain is required for the interaction between TRIM23 and TBK1 and for TRIM23-mediated autophagy. Both genetic and pharmacological inhibition of TBK1 impaired autophagy driven by TRIM23 expression. This finding provided the first evidence that a TRIM could regulate autophagy by acting on TBK1, suggesting the possibility that other TRIMs may act similarly.
The previous sections dealt with mechanisms whereby TRIMs regulate autophagy indirectly at the transcriptional level or by affecting upstream signal transduction pathways that affect autophagy. In this section, we discuss evidence that multiple TRIMs directly interact with and modulate the activity of the conserved core autophagy machinery. The first indication that TRIMs as a family could directly intersect with the autophagy machinery was published in 2014, when TRIM5, TRIM6, TRIM17, TRIM22, and TRIM49 were shown to interact with the autophagy regulators ULK1 and Beclin 1 (Mandell et al., 2014). ULK1 (the mammalian homologue of yeast Atg1) is the most upstream autophagy regulator. One of the roles of ULK1 in autophagy is to activate the Beclin 1/hVPS34 complex, which it does through phosphorylation of Beclin 1 (Russell et al., 2013). This process was potentially enhanced by expression of the TRIMs listed above, which recruited ULK1 into Beclin 1 multi-molecular complexes (Mandell et al., 2014) in co-immunoprecipitation experiments. Further studies with TRIM5 showed that it also interacted with ATG14L1 and AMBRA1, both proteins that interact with the Beclin 1 complex. Additional studies have broadened the list of TRIMs that interact with the ULK1 and/or Beclin 1 complexes to include TRIMs 13, 16, 20, 21, 28, 32, and 50 (Yang et al., 2013; Kimura et al., 2015; Chauhan et al., 2016; Fusco et al., 2018; Di Rienzo et al., 2019; Ji et al., 2019). However, the finding that a TRIM interacts with these upstream regulators of autophagy does not necessarily prove that it acts to promote autophagy, as exemplified by TRIM17 which binds to ULK1 and Beclin 1 yet was found to inhibit autophagosome formation (Mandell et al., 2014; Mandell et al., 2016). Instead, TRIM17 promoted the formation of inhibitory Beclin 1 complexes including the protein Mcl-1 (Mandell et al., 2016).
Active Beclin 1 complexes generate phosphatidylinositol 3-phosphate (PI3P) at autophagosome initiation sites. PI3P recruits proteins including ATG16L1 and ATG5 that carry out the elongation of the autophagosome membrane. A key part of this process involves the lipidation of the mammalian Atg8 orthologues (LC3 and GABARAP proteins; mAtg8s). Lipidated mAtg8 proteins are important for the elongation of the autophagosome membrane, the closure of the autophagosome, and its fusion with lysosomes. TRIM5, TRIM16, and TRIM20 have all been reported to form protein complexes with ATG16L1 and TRIM5 was also shown to co-immunoprecipitate with ATG5 (Kimura et al., 2015; Ribeiro et al., 2016). Whether these interactions specifically modulate mAtg8 lipidation and autophagosome membrane elongation has not yet been demonstrated.
One critical question is whether the enzymatic activity of TRIMs as E3 ligases is important for their actions in autophagy. The answer to this question appears to be “sometimes”. For example, TRIM20 lacks a catalytic RING domain but can still assemble active autophagy initiation complexes (Kimura et al., 2015). On the other hand, TRIM28 has been shown to enhance the PI3 kinase activity of Beclin 1 complexes by directly SUMOylating hVPS34, and TRIM50 attaches K63-linked poly-ubiquitin to Beclin 1 in an autophagy-activating manner (Yang et al., 2013; Fusco et al., 2018). TRIM32 has been reported to promote the activity of the ULK1 complex through the generation of unattached K63-linked poly-ubiquitin chains (Di Rienzo et al., 2019). Whether the enzymatic activity of other autophagy-regulating TRIMs is required for their actions in autophagy remains to be answered.
One of the primary ways that autophagy can impact cellular health and physiology is through the degradative elimination of cytoplasmic contents. Once considered a bulk cellular recycling mechanism, the autophagy pathway is now known to selectively target certain substrates for degradation. This selective autophagy presents an opportunity for the potential deployment of autophagy-modulating therapies. While the wholesale induction or inhibition of autophagy may have deleterious side effects, the still-theoretical ability to activate or inhibit the autophagic degradation of a specific cancer-related target could be considerably safer since only some of autophagy's many physiological roles would be impacted.
The autophagy machinery's ability to selectively recognize substrates is based on proteins that act as autophagy receptors. These receptors are thought to act by bridging autophagic cargoes with mAtg8s associated with the nascent autophagosomal/phagophore membrane. Receptors can interact with cargos directly or indirectly through a protein “eat-me” tag; these tags are often ubiquitin-based (Kirkin et al., 2009). Receptors interact with mAtg8s via two different defined peptide sequences termed LC3-interacting regions (LIRs) (Birgisdottir et al., 2013) or ubiquitin interacting motif-like (UIM) (Marshall et al., 2019). The best recognized autophagy receptors are the sequestosome-like receptors (SLRs), which include the proteins p62/Sequestosome 1, NDP52, NBR1, Optineurin, and TAX1BP1. These proteins all include ubiquitin binding domains for substrate recognition and LIRs, and these domains have been shown to be important for these proteins to carry out the autophagic degradation of specific proteins, organelles, or intracellular pathogens. Autophagy receptors also have autophagy-regulatory roles by linking selective autophagy substrates with upstream autophagy regulators as exemplified by NDP52, which recruits the ULK1/FIP200 complex to depolarized mitochondria during mitophagy (Vargas et al., 2019). In addition to regulating the autophagy pathway, multiple TRIMs impact the autophagic targeting and degradation of select substrates by themselves acting as autophagy receptors or by modulating the actions of SLRs.
Most TRIM family members have N-terminal RING catalytic domains that act as E3 ubiquitin ligases. As such, it may be expected that TRIM-mediated ubiquitination of autophagy substrates leading to their recognition by ubiquitin binding receptors such as the SLRs would be a common mechanism of TRIM-mediated selective autophagy. However, to date this mechanism is not well-established; although there is an indication that TRIM21-mediated ubiquitination of the kinase IKKβ may facilitate IKKβ degradation by autophagy (Niida et al., 2010). On the contrary, TRIM14 and TRIM59 have been shown to prevent the ubiquitination and subsequent p62-mediated autophagic degradation of the DNA sensing enzyme cGAS (Chen M. et al., 2016) and PDCD10 (programmed cell death protein 10) (Tan P. et al., 2018). Interestingly, the TRIM59-mediated protection of PDCD10 from autophagy was shown to promote the survival and growth of breast cancer cells (Tan P. et al., 2018).
Instead of tagging autophagy substrates with ubiquitin “eat-me tags”, TRIMs appear to act as autophagy receptors that directly bind to their substrates (Table 2). This was originally demonstrated for TRIM5 (Mandell et al., 2014), a protein that is known to assemble into a lattice around incoming retroviral cores in a host- and viral-species specific manner (Ganser-Pornillos and Pornillos, 2019). TRIM5 was found to include two LIR motifs and to bind directly to mAtg8 proteins (Mandell et al., 2014; Keown et al., 2018). Rhesus TRIM5 promoted the autophagy-dependent degradation of HIV-1 viral components (Mandell et al., 2014), which can be recognized and bound by the TRIM5 SPRY domain (Stremlau et al., 2006). Binding to mAtg8s appears to be a feature of multiple TRIMs in addition to TRIM5 (Pizon et al., 2013; Mandell et al., 2014; Kimura et al., 2015; Kimura et al., 2016; Overå et al., 2019). This feature puts these mAtg8-binding TRIMs into a position where they can recruit their interacting partners to autophagosomes for degradation. Autophagic degradation of cancer-relevant targets is one possible mechanism explaining how autophagy may impact cancer progression (Table 2). Given the large size of the TRIM family, it is possible that TRIMs provide cells with a breadth of selective autophagy receptors.
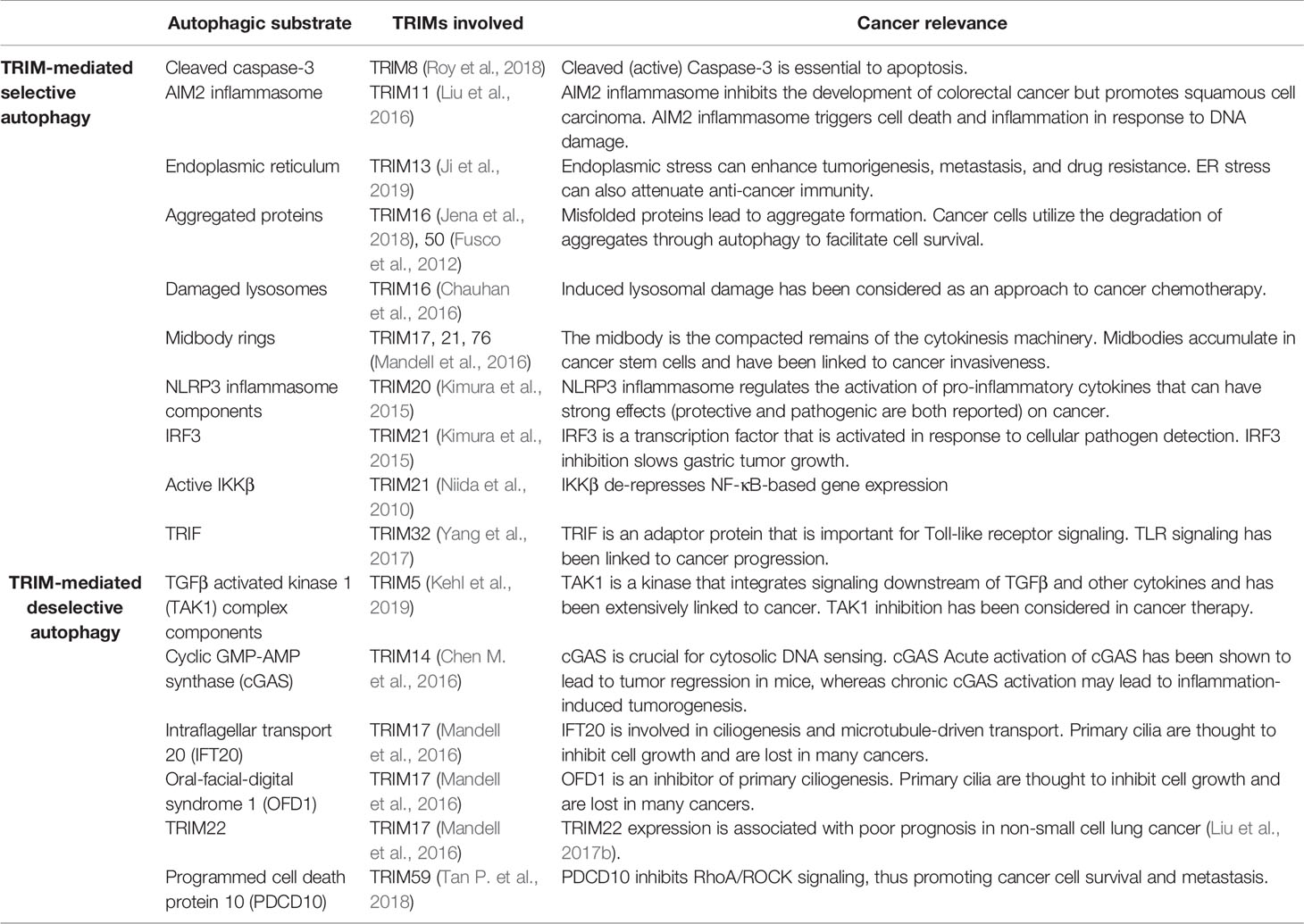
Table 2 Substrates whose autophagic degradation is controlled by TRIMs. Top, in some cases, TRIMs promote the selective autophagic degradation of the listed substrates. In other cases (bottom), TRIMs ‘deselect’ potential autophagic substrates allowing them to accumulate in cells.
While ubiquitin tagging of substrates by TRIMs has not yet been definitively reported, several studies indicate that auto-ubiquitination of TRIMs when bound to their substrates is important for their actions as autophagy receptors. For instance, TRIM11 binds to the DNA sensor AIM2 following activation by cytoplasmic DNA. TRIM11 then auto-ubiquitinates at lysine 458. This modification is required for p62 recruitment and AIM2 degradation by autophagy (Liu et al., 2016). AIM2 has been suggested to play roles in several cancers, and the TRIM11-p62-autophagy axis attenuated AIM2 signaling. Auto-ubiquitination is required for the recently uncovered roles of TRIM13 in ER-phagy (the autophagic targeting of damaged endoplasmic reticulum) (Ji et al., 2019). Analogously, auto-ubiquitinated TRIM32 binds to the signaling adapter TRIF and acts as an autophagic “eat-me” signal that is detected by TAX1BP1 (Yang Q. et al., 2017). This raises the question as to how TRIM auto-ubiquitination is regulated. In the case of TRIM5, the spatial arrangement of TRIM5 dimers scaffolded on a retroviral core allows the RING domain of one TRIM5 molecule to poly-ubiquitylate the RING domain on another TRIM5 molecule (Fletcher et al., 2018). Thus, substrate recognition may be required for TRIM auto-ubiquitination and action as autophagy receptors. This is likely the case for TRIM17. Under normal conditions, TRIM17 assembles Beclin 1 with an inhibitory binding partner to inhibit autophagy. However, TRIM17-Beclin 1 complexes localized to midbody rings lack this inhibitory binding partner, and TRIM17 contributes to the autophagic elimination of midbody rings (Mandell et al., 2016). Whether and how TRIMs coordinate their substrate binding activities as selective autophagy receptors with their enzymatic activities and their actions as autophagy regulators (described above) remains an open question.
By definition, autophagy receptors are co-degraded with their targets in the autolysosome. So far, autophagic degradation has been demonstrated for at least TRIMs 5, 13, 16, 20, 21, 23, 27, 31, 32, 45, 49, 50, and 56 (Imam et al., 2016; Mandell et al., 2016; Ji et al., 2019; Overå et al., 2019), several of which have known connection to cancer (Table 1). This finding raises the autophagic degradation can also regulate the cancer-related activities of these TRIMs. Interestingly, this effect is seen with the oncogenic fusion protein PML-RARα (TRIM19), whose autophagic degradation is induced in following exposure to all-trans retinoic acid (Isakson et al., 2010; Wang et al., 2011), which is a standard treatment for acute promyelocytic leukemia (Wang and Chen, 2008).
The protein p62/Sequestosome 1 (p62) has multiple known roles in a variety of cancers (Moscat et al., 2016; Sanchez-Martin et al., 2019). The best known cellular function of p62 is as a selective autophagy receptor (Pankiv et al., 2007; Deretic, 2012). Separate from its actions in autophagy, p62 also plays a key role in a number of cellular signaling pathways that can profoundly affect cellular survival and growth (Sanz et al., 2000; Jin et al., 2009; Jain et al., 2010; Komatsu et al., 2010; Duran et al., 2011; Linares et al., 2013; Paul et al., 2014; Moscat et al., 2016; Goodall et al., 2016; Sanchez-Martin and Komatsu, 2018). Numerous studies have reported p62 over-expression in tumors and have associated elevated p62 expression with poor prognosis (Duran et al., 2008; Inui et al., 2013; Li et al., 2013; Adams et al., 2016; Saito et al., 2016; Umemura et al., 2016; Karras et al., 2019; Nguyen et al., 2019; Polonen et al., 2019). Furthermore, p62 has been shown to limit the efficacy of sorafenib treatment against liver cancer (Sun et al., 2016). Given its multifunctional nature, it is unsurprising that dysregulation of p62 in cancer cells can promote their growth by several mechanisms including through selective autophagy (Nguyen et al., 2019), activation of pro-survival signaling and gene expression (Duran et al., 2011; Linares et al., 2013; Umemura et al., 2016; Lam et al., 2017; Polonen et al., 2019), or stabilization of a set of pro-metastatic mRNAs (Karras et al., 2019). In contrast, p62 expression in non-transformed cells can reduce cancer progression (Valencia et al., 2014; Huang et al., 2018). In light of all of p62's possible pathogenic or protective effects in cancer, a key question is how p62 coordinates its various cellular activities and what factors govern its behavior.
The control of p62 action is emerging as a conserved feature of TRIM family members. As discussed above, TRIMs can regulate the levels of p62 indirectly through their actions as autophagy regulators. However, TRIMs employ additional mechanisms for affecting p62 abundance and activity. TRIMs 5,11,13, 17, 21, 22, 23, 32, 49, 50, 55, and 63 have been demonstrated to biochemically interact with p62 (Witt et al., 2008; O'Connor et al., 2010; Fusco et al., 2012; Tomar et al., 2012; Mandell et al., 2014; Kimura et al., 2015; Liu et al., 2016; Pan et al., 2016; Sparrer et al., 2017; Overå et al., 2019), and additional TRIMs such as TRIM19 colocalize with p62 in cellular structures (Clausen et al., 2010). A primary cellular function of p62 is to organize and sequester ubiquitylated proteins into cytoplasmic punctate structures termed p62 bodies which have liquid droplet-like properties (Zaffagnini et al., 2018) and that may function as platforms for p62-mediated signaling while also concentrating cellular wastes destined for autophagic degradation. At least 14 TRIMs have roles in regulating the formation and/or clearance of these structures, with TRIMs 5, 16, 17, 32, 50, 52, and 58 increasing their abundance in cells while TRIMs 14, 19, 21, 22, 25, 65, and 76 having the opposite effect (Mandell et al., 2016; Pan et al., 2016; Jena et al., 2018; Kehl et al., 2019; Overå et al., 2019). The relevance of TRIM-regulated p62 localization to cancer is illustrated by TRIM21 and TRIM16, which act in opposing manners on p62 condensation into cytoplasmic bodies and on activation of the transcription factor Nrf2.
As a master transcriptional regulator of antioxidant stress resistance genes, Nrf2 is a prominent actor in cancers (Kitamura and Motohashi, 2018). Under homeostatic conditions, Nrf2 is targeted for proteasomal degradation through its interaction with Keap1. In response to oxidative stress, p62 binds to Keap1, leading to Keap1's sequestration and eventual autophagic degradation. This liberates Nrf2 from Keap1, allowing Nrf2 to enter the nucleus and to activate its target genes (Komatsu et al., 2010; Taguchi et al., 2012; Ichimura et al., 2013). TRIM21 directly catalyzes the K63-linked poly-ubiquitylation of p62 at lysine 7 in the p62 PB1 domain, a region required for p62 dimerization and cytoplasmic body formation. Consequently, K7-ubiquitylated p62 loses its ability to sequester Keap1, establishing TRIM21 as a negative regulator of Nrf2-directed cytoprotective antioxidant responses and providing a possible mechanism explaining the observation that reduced TRIM21 expression is associated with poor prognosis in hepatocellular carcinoma and B cell lymphoma (Brauner et al., 2015; Ding et al., 2015). In contrast, TRIM16 is required for the formation of p62 bodies in response to proteotoxic or oxidative stress, and its expression decouples Nrf2 from its inhibitor Keap1 (Jena et al., 2018). Knockout of TRIM16 reduces cellular survival and growth under stress conditions in vitro and in a mouse xenograft tumor model (Jin et al., 2009). While the precise molecular mechanism of TRIM16 action on the p62-Nrf2 system has not been completely defined, TRIM16 expression is associated with increased p62 phosphorylation (Ichimura et al., 2013), suggesting that TRIM16 may be involved in the activation of a kinase that controls p62 action. A likely candidate for this role is TAK1, a kinase activated downstream of TRIM5 (Pertel et al., 2011) and other TRIMs (Versteeg Gijs et al., 2013), that phosphorylates p62 (Hashimoto et al., 2016; Kehl et al., 2019) and is required for p62 body formation in response to multiple cellular stresses (Kehl et al., 2019). How other TRIMs affect p62 localization and Nrf2 activation, and what role(s) these activities play in cancer has not been fully elucidated.
Can the connections between TRIMs and autophagy be leveraged for the therapeutic benefit of cancer patients? While this concept has not yet been tested, there is reason to believe that TRIMs could be druggable targets. TRIMs are a heterogeneous group of proteins organized into subclasses that possess a defining cluster of domains (e.g. RING, B-box, coiled-coil, FN3, SPRY, bromobox/bromodomain, etc.) (Gushchina et al., 2018). One approach to drug design would be to target the activity of specific domains critical to TRIM function in cancer. In fact, efforts to identify inhibitors of TRIM bromodomains are underway, with the bromodomains present in TRIM24, 28, and 33 being of particular interest to cancer therapy. These three TRIMs are transcriptional modulators associated with multiple cancers (Table 1). Bromodomains are involved in the recognition of acetylated lysines on histones and can recruit chromatin remodeling enzymes, resulting in transcriptional activation or repression including of autophagy-related genes (Sakamaki et al., 2017). Small-molecule bromodomain inhibitors have been identified that display robust target specificity, including against TRIM24 (Zhan et al., 2015; Bennett et al., 2016; Romero et al., 2016). Such agents may serve as useful epigenetic based anti-cancer therapies. As discussed above, bromodomain-containing TRIM28 has multiple connections to autophagy, and TRIM33 was among TRIM “hits” as regulators of autophagy. The role of the bromodomain of these TRIMs in their autophagic function(s) has not yet been tested, but one could imagine that bromodomain-targeted therapies could also impact autophagy directed by bromodomain-containing TRIMs.
The RING domain found in most TRIMs may also present an opportunity for therapeutic targeting. Most TRIM RINGs possess E3 ubiquitin ligase activity that is often crucial to TRIM functionality. While inhibitors specific to TRIM RING domains have not yet been reported, the fact that small molecule inhibitors of the RING domains from other protein families exist (Bulatov et al., 2018) suggests that TRIM RING inhibition may be feasible. The search for E3 ligase inhibitors in general has been sparked by the desire to identify molecules that could modulate ubiquitination-dependent proteasomal degradation of selected proteins. This process has been targeted therapeutically using drugs such as bortezomib, a 26S proteasome inhibitor, which is used in the treatment of cancers including multiple myeloma. Bortezomib's utility in cancer therapy is limited by toxicity, possibly resulting from the general, non-specific nature of proteasomal inhibition. However, because E3 ubiquitin ligases have some level of specificity in their action, their pharmacological targeting may provide greater therapeutic utility. A prime example of this approach that is currently under investigation is to target interactions between TRIMs and p53, a notable tumor suppressor (Valletti et al., 2019). Inhibiting the E3 ligase activity of these TRIMs to improve p53 stability may represent a selective therapeutic target for cancer. Inhibition of the E3 ligase activity of TRIMs would also impact their ubiquitination-dependent but proteasome independent activities, including those in autophagy regulation and in autophagic substrate selection.
A third possible modality for TRIM-directed cancer therapy would be to interfere with or enhance the interactions between a TRIM and its cancer-relevant binding partners. Depending on the TRIM and its mechanism of action in cancer, these interacting partners could include autophagy factors (e.g. Beclin 1, p62, mAtg8s). Alternatively, compounds capable of disrupting the oligomerization of cancer-relevant TRIMs would be expected to block their action, since the higher-order assembly of TRIMs is thought to be essential for their function. In total, there is strong support for targeting TRIMs in an effort to develop effective therapies for a wide-array of cancers.
TRIM proteins are positioned as hubs connecting cellular signaling, metabolism, and autophagy. As such, it is unsurprising that so many of them have prominent roles in cancer. To date, no efforts to effectively target TRIMs as a strategy to therapeutically modulate autophagy in cancer have been reported. However, the strong connections between TRIMs and a variety of diseases in addition to cancer suggest that TRIM targeting may hold promise pending future mechanistic studies. These studies could be initiated by two complementary approaches. First, the requirement for autophagy should be investigated in cancers showing TRIM dysregulation. Second, TRIMs should be assessed for whether they promote the survival of autophagy-addicted cancers. Either approach might identify cancers in which TRIM-directed autophagy plays a significant role in tumor survival and/or resistance to chemotherapeutic agents. This could justify further cellular, biochemical, and structural studies aimed at identifying TRIM structures or activities to target in the development of more effective cancer therapies.
MM and BS wrote the sections on autophagy regulation by TRIMs. TT wrote the sections on the connections between TRIMs and cancer and on TRIMs as therapeutic targets. Other sections were written collaboratively by all three authors.
MM is supported by P20GM121176 and R21AI131964 from NIGMS and NIAID, respectively.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Graham S. Timmins for critically reading the manuscript and for the suggestions.
Adams, O., Dislich, B., Berezowska, S., Schlafli, A. M., Seiler, C. A., Kroll, D., et al. (2016). Prognostic relevance of autophagy markers LC3B and p62 in esophageal adenocarcinomas. Oncotarget 7 (26), 39241–39255. doi: 10.18632/oncotarget.9649
Aierken, G., Seyiti, A., Alifu, M., Kuerban, G. (2017). Knockdown of tripartite-59 (TRIM59) inhibits cellular proliferation and migration in human cervical cancer cells. Oncol. Res. 25 (3), 381–388.
Barde, I., Rauwel, B., Marin-Florez, R. M., Corsinotti, A., Laurenti, E., Verp, S., et al. (2013). A KRAB/KAP1-miRNA cascade regulates erythropoiesis through stage-specific control of mitophagy. Science 340 (6130), 350–353. doi: 10.1126/science.1232398
Bell, J. L., Malyukova, A., Holien, J. K., Koach, J., Parker, M. W., Kavallaris, M., et al. (2012). TRIM16 acts as an E3 ubiquitin ligase and can heterodimerize with other TRIM family members. PloS One 7 (5), e37470. doi: 10.1371/journal.pone.0037470
Belloni, E., Trubia, M., Gasparini, P., Micucci, C., Tapinassi, C., Confalonieri, S., et al. (2005). 8p11 myeloproliferative syndrome with a novel t(7;8) translocation leading to fusion of the FGFR1 and TIF1 genes. Genes Chromosomes Cancer 42 (3), 320–325. doi: 10.1002/gcc.20144
Bennett, J., Fedorov, O., Tallant, C., Monteiro, O., Meier, J., Gamble, V., et al. (2016). Discovery of a Chemical Tool Inhibitor Targeting the Bromodomains of TRIM24 and BRPF. J. Med. Chem. 59 (4), 1642–1647. doi: 10.1021/acs.jmedchem.5b00458
Bhatnagar, S., Gazin, C., Chamberlain, L., Ou, J., Zhu, X., Tushir, J. S., et al. (2014). TRIM37 is a new histone H2A ubiquitin ligase and breast cancer oncoprotein. Nature 516 (7529), 116–120. doi: 10.1038/nature13955
Birgisdottir, A. B., Lamark, T., Johansen, T. (2013). The LIR motif - crucial for selective autophagy. J. Cell Sci. 126 (Pt 15), 3237–3247. doi: 10.1242/jcs.126128
Brauner, S., Zhou, W., Backlin, C., Green, T. M., Folkersen, L., Ivanchenko, M., et al. (2015). Reduced expression of TRIM21/Ro52 predicts poor prognosis in diffuse large B-cell lymphoma patients with and without rheumatic disease. J. Intern. Med. 278 (3), 323–332. doi: 10.1111/joim.12375
Bulatov, E., Zagidullin, A., Valiullina, A., Sayarova, R., Rizvanov, A. (2018). Small Molecule Modulators of RING-Type E3 Ligases: MDM and Cullin Families as Targets. Front. Pharmacol. 9, 450. doi: 10.3389/fphar.2018.00450
Cambiaghi, V., Giuliani, V., Lombardi, S., Marinelli, C., Toffalorio, F., Pelicci, P. G. (2012). TRIM proteins in cancer. Adv. Exp. Med. Biol. 770, 77–91. doi: 10.1007/978-1-4614-5398-7_6
Cao, H., Fang, Y., Liang, Q., Wang, J., Luo, B., Zeng, G., et al. (2019). TRIM2 is a novel promoter of human colorectal cancer. Scand. J. Gastroenterol. 54 (2), 210–218.
Carinci, F., Arcelli, D., Lo Muzio, L., Francioso, F., Valentini, D., Evangelisti, R., et al. (2007). Molecular classification of nodal metastasis in primary larynx squamous cell carcinoma. Trans. Res. 150 (4), 233–245.
Chao, J., Zhang, X.-F., Pan, Q.-Z., Zhao, J.-J., Jiang, S.-S., Wang, Y., et al. (2014). Decreased expression of TRIM3 is associated with poor prognosis in patients with primary hepatocellular carcinoma. Med. Oncol. 31 (8), 102 (7p).
Chauhan, S., Kumar, S., Jain, A., Ponpuak, M., Mudd, M. H., Kimura, T., et al. (2016). TRIMs and Galectins Globally Cooperate and TRIM16 and Galectin-3 Co-direct Autophagy in Endomembrane Damage Homeostasis. Dev. Cell 39 (1), 13–27. doi: 10.1016/j.devcel.2016.08.003
Chen, D., You, X., Pan, Y., Liu, Q., Cao, G. (2018). TRIM37 promotes cell invasion and metastasis by regulating SIP1-mediated epithelial-mesenchymal transition in gastric cancer. OncoTargets Ther. 11, 8803–8813.
Chen, M., Meng, Q., Qin, Y., Liang, P., Tan, P., He, L., et al. (2016). TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor p62 to Promote Innate Immune Responses. Mol. Cell 64 (1), 105–119. doi: 10.1016/j.molcel.2016.08.025
Chen, Q., Sun, L., Chen, Z. J. (2016). Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17 (10), 1142–1149. doi: 10.1038/ni.3558
Chen, W., Lu, C., Hong, J. (2018). TRIM15 exerts anti-tumor effects through suppressing cancer cell invasion in gastric adenocarcinoma. Med. Sci. Monit. 24, 8033–8041.
Chen, W. X., Cheng, L., Xu, L. Y., Qian, Q., Zhu, Y. L. (2019). Bioinformatics analysis of prognostic value of TRIM13 gene in breast cancer. Biosci. Rep. 39 (3), BSR20190285–BSR20190294. doi: 10.1042/BSR20190285
Chen, Y., Guo, Y., Yang, H., Shi, G., Xu, G., Shi, J., et al. (2015). TRIM66 overexpresssion contributes to osteosarcoma carcinogenesis and indicates poor survival outcome. Oncotarget 6 (27), 23708–23719.
Chen, Y., Li, L., Qian, X., Ge, Y., Xu, G. (2017b). High expression of TRIM11 correlates with poor prognosis in patients with hepatocellular carcinoma. Clinics Res. Hepatol. Gastroenterol. 41 (2), 190–196.
Chen, Y., Sun, J., Ma, J. (2017a). Proliferation and invasion of ovarian cancer cells are suppressed by knockdown of TRIM11. Oncol. Lett. 14 (2), 2125–2130.
Chen, Z., Long, L., Wang, K., Cui, F., Zhu, L., Tao, Y., et al. (2016). Identification of nasopharyngeal carcinoma metastasis-related biomarkers by iTRAQ combined with 2D-LC-MS/MS. Oncotarget 7 (23), 34022–34037.
Chen, Z., Yin, X., Li, K., Chen, S., Li, H., Li, Y., et al. (2018). Serum Levels of TRIM72 Are Lower among Patients with Colon Cancer: Identification of a Potential Diagnostic Marker. Tohoku J. Exp. Med. 245 (1), 61–68. doi: 10.1620/tjem.245.61
Cui, Z., Cao, W., Li, J., Song, X., Mao, L., Chen, W. (2013). TRIM24 overexpression is common in locally advanced head and neck squamous cell carcinoma and correlates with aggressive malignant phenotypes. PloS One 8 (5), e63887 (10p).
Chi, J., Yang, Q., Xie, X.-F., Yang, X.-Z., Zhang, M.-Y., Wang, H.-Y., et al. (2016). Clinical significance and prognostic value of TRIM24 expression in esophageal squamous cell carcinoma. Aging 8 (9), 2204–2221.
Clausen, T. H., Lamark, T., Isakson, P., Finley, K., Larsen, K. B., Brech, A., et al. (2010). p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 6 (3), 330–344. doi: 10.4161/auto.6.3.11226
Cui, X., Lin, Z., Chen, Y., Mao, X., Ni, W., Liu, J., et al. (2016). Upregulated TRIM32 correlates with enhanced cell proliferation and poor prognosis in hepatocellular carcinoma. Mol. Cell Biochem. 421 (1–2), 127–137.
Cui, Y., Yang, S., Fu, X., Feng, J., Xu, S., Ying, G. (2015). High levels of KAP1 expression are associated with aggressive clinical features in ovarian cancer. Int. J. Mol. Sci. 16 (1), 363–377.
Czerwinska, P., Shah, P. K., Tomczak, K., Klimczak, M., Mazurek, S., Sozanska, B., et al. (2017). TRIM28 multi-domain protein regulates cancer stem cell population in breast tumor development. Oncotarget 8 (1), 863–882. doi: 10.18632/oncotarget.13273
Dai, H., Zhao, S., Xu, L., Chen, A., Dai, S. (2010). Expression of Efp, VEGF and bFGF in normal, hyperplastic and malignant endometrial tissue. Oncol. Rep. 23, 795–799.
Di, K., Linskey, M. E., Bota, D. A. (2013). TRIM11 is overexpressed in high-grade gliomas and promotes proliferation, invasion, migration and glial tumor growth. Oncogene 32 (42), 5038–5047.
Deretic, V. (2012). Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Curr. Opin. Immunol. 24 (1), 21–31. doi: 10.1016/j.coi.2011.10.006
Di Rienzo, M., Antonioli, M., Fusco, C., Liu, Y., Mari, M., Orhon, I., et al. (2019). Autophagy induction in atrophic muscle cells requires ULK1 activation by TRIM32 through unanchored K63-linked polyubiquitin chains. Sci. Adv. 5 (5), eaau8857. doi: 10.1126/sciadv.aau8857
Diaz-Lagares, A., Mendez-Gonzalez, J., Hervas, D., Saigi, M., Pajares, M. J., Garcia, D., et al. (2016). A Novel Epigenetic Signature for Early Diagnosis in Lung Cancer. Clin. Cancer Res. : an Off. J. Am. Assoc. Cancer Res. 22 (13), 3361–3371. doi: 10.1158/1078-0432.CCR-15-2346
Ding, Q., He, D., He, K., Zhang, Q., Tang, M., Dai, J., et al. (2015). Downregulation of TRIM21 contributes to hepatocellular carcinoma carcinogenesis and indicates poor prognosis of cancers. Tumour Biol. 36 (11), 8761–8772. doi: 10.1007/s13277-015-3572-2
Ding, Z., Jin, G., Wang, W., Chen, W., Wu, Y., Ai, X., et al. (2014). Reduced expression of transcriptional intermediary factor 1 gamma promotes metastasis and indicates poor prognosis of hepatocellular carcinoma. Hepatology 60 (5), 1620–1636.
Dong, B., Zhang, W. (2018). High Levels of TRIM14 Are Associated with poor prognosis in hepatocellular carcinoma. Oncol. Res. Treat. 41 (3), 129–134.
Dong, S., Pang, X., Sun, H., Yuan, C., Mu, C., Zheng, S. (2018). TRIM37 targets AKT in the growth of lung cancer cells. OncoTargets Ther. 11, 7935–7945.
Duran, A., Amanchy, R., Linares, J. F., Joshi, J., Abu-Baker, S., Porollo, A., et al. (2011). p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol. Cell 44 (1), 134–146. doi: 10.1016/j.molcel.2011.06.038
Duran, A., Linares, J. F., Galvez, A. S., Wikenheiser, K., Flores, J. M., Diaz-Meco, M. T., et al. (2008). The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 13 (4), 343–354. doi: 10.1016/j.ccr.2008.02.001
Feng, S., Cai, X., Li, Y., Jian, X., Zhang, L., Li, B. (2019). Tripartite motif-containing 14 (TRIM14) promotes epithelial-mesenchymal transition via ZEB2 in glioblastoma cells. J. Exp. Clin. Cancer Res. 38 (1), 57 (13p).
Fitzgerald, S., Sheehan, K. M., O'Grady, A., Kenny, D., O'Kennedy, R., Kay, E. W., et al. (2013). Relationship between epithelial and stromal TRIM28 expression predicts survival in colorectal cancer patients: TRIM28 predicts survival in colorectal cancer. J. Gastroenterol. Hepatol. 28 (6), 967–974.
Fernández-Aceñero, M. J., Cruz, M., Sastre-Varela, J., Casal, J. I., Nieto, M. A. C., Del Puerto-Nevado, L., et al. (2019). TRIM72 Immunohistochemical Expression Can Predict Relapse in Colorectal Carcinoma. Pathol. Oncol. Res. doi: 10.1007/s12253-019-00629-w
Fletcher, A. J., Christensen, D. E., Nelson, C., Tan, C. P., Schaller, T., Lehner, P. J., et al. (2015). TRIM5alpha requires Ube2W to anchor Lys63-linked ubiquitin chains and restrict reverse transcription. EMBO J. 34 (15), 2078–2095. doi: 10.15252/embj.201490361
Fletcher, A. J., Vaysburd, M., Maslen, S., Zeng, J., Skehel, J. M., Towers, G. J., et al. (2018). Trivalent RING Assembly on Retroviral Capsids Activates TRIM5 Ubiquitination and Innate Immune Signaling. Cell Host Microbe 24 (6), 761–75 e6. doi: 10.1016/j.chom.2018.10.007
Fong, K. W., Zhao, J. C., Song, B., Zheng, B., Yu, J. (2018). TRIM28 protects TRIM24 from SPOP-mediated degradation and promotes prostate cancer progression. Nat. Commun. 9 (1), 5007. doi: 10.1038/s41467-018-07475-5
Fu, H., Yang, H., Zhang, X., Wang, B., Mao, J., Li, X., et al. (2018). Exosomal TRIM3 is a novel marker and therapy target for gastric cancer. J. Exp. Clin. Cancer Res. 37 (1), 162 (16p).
Fujimura, T., Inoue, S., Urano, T., Takayama, K., Yamada, Y., Ikeda, K., et al. (2016). Increased expression of tripartite motif (TRIM) 47 is a negative prognostic predictor in human prostate cancer. Clin. Genitourinary Cancer 14 (4), 298–303.
Fujimura, T., Takahashi, S., Urano, T., Takayama, K., Sugihara, T., Obinata, D., et al. (2014). Expression of androgen and estrogen signaling components and stem cell markers to predict cancer progression and cancer-specific survival in patients with metastatic prostate cancer. Clin. Cancer Res. : Off. J. Am. Assoc. Cancer Res. 20 (17), 4625–4635. doi: 10.1158/1078-0432.CCR-13-1105
Fusco, C., Mandriani, B., Di Rienzo, M., Micale, L., Malerba, N., Cocciadiferro, D., et al. (2018). TRIM50 regulates Beclin 1 proautophagic activity. Biochim. Biophys. Acta Mol. Cell Res. 1865 (6), 908–919. doi: 10.1016/j.bbamcr.2018.03.011
Fusco, C., Micale, L., Egorov, M., Monti, M., D'Addetta, E. V., Augello, B., et al. (2012). The E3-ubiquitin ligase TRIM50 interacts with HDAC6 and p62, and promotes the sequestration and clearance of ubiquitinated proteins into the aggresome. PloS One 7 (7), e40440. doi: 10.1371/journal.pone.0040440
Galluzzi, L., Pietrocola, F., Levine, B., Kroemer, G. (2014). Metabolic control of autophagy. Cell 159 (6), 1263–1276. doi: 10.1016/j.cell.2014.11.006
Ganser-Pornillos, B. K., Pornillos, O. (2019). Restriction of HIV-1 and other retroviruses by TRIM5. Nat. Rev. Microbiol. 17 (9), 546–556. doi: 10.1038/s41579-019-0225-2
Gatt, M. E., Takada, K., Mani, M., Lerner, M., Pick, M., Hideshima, T., et al. (2013). TRIM13 (RFP2) downregulation decreases tumour cell growth in multiple myeloma through inhibition of NF Kappa B pathway and proteasome activity. Br. J. Haematol. 162 (2), 210–220.
Goodall, M. L., Fitzwalter, B. E., Zahedi, S., Wu, M., Rodriguez, D., Mulcahy-Levy, J. M., et al. (2016). The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Dev. Cell 37 (4), 337–349. doi: 10.1016/j.devcel.2016.04.018
Groner, A. C., Cato, L., de Tribolet-Hardy, J., Bernasocchi, T., Janouskova, H., Melchers, D., et al. (2016). TRIM24 Is an Oncogenic Transcriptional Activator in Prostate Cancer. Cancer Cell 29 (6), 846–858. doi: 10.1016/j.ccell.2016.04.012
Gui, X., Yang, H., Li, T., Tan, X., Shi, P., Li, M., et al. (2019). Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567 (7747), 262–266. doi: 10.1038/s41586-019-1006-9
Guo, P., Ma, X., Zhao, W., Huai, W., Li, T., Qiu, Y., et al. (2018). TRIM31 is upregulated in hepatocellular carcinoma and promotes disease progression by inducing ubiquitination of TSC1–TSC2 complex. Oncogene 37 (4), 478–488.
Gushchina, L. V., Kwiatkowski, T. A., Bhattacharya, S., Weisleder, N. L. (2018). Conserved structural and functional aspects of the tripartite motif gene family point towards therapeutic applications in multiple diseases. Pharmacol. Ther. 185, 12–25. doi: 10.1016/j.pharmthera.2017.10.020
Halliday, B. J., Fukuzawa, R., Markie, D. M., Grundy, R. G., Ludgate, J. L., Black, M. A., et al. (2018). Germline mutations and somatic inactivation of TRIM28 in Wilms tumour. PloS Genet. 14 (6), e1007399 (17p).
Han, T., Guo, M., Gan, M., Yu, B., Tian, X., Wang, J. B. (2018). TRIM59 regulates autophagy through modulating both the transcription and the ubiquitination of BECN1. Autophagy 14 (12), 2035–2048. doi: 10.1080/15548627.2018.1491493
Han, Y., Tian, H., Chen, P., Lin, Q. (2017). TRIM47 overexpression is a poor prognostic factor and contributes to carcinogenesis in non-small cell lung carcinoma. Oncotarget 8 (14), 22730–22740. doi: 10.18632/oncotarget.15188
Hao, L., Du, B., Xi, X. (2017). TRIM59 is a novel potential prognostic biomarker in patients with non-small cell lung cancer: A research based on bioinformatics analysis. Oncol. Lett. 14 (2), 2153–2164. doi: 10.3892/ol.2017.6467
Harris, T. M., Du, P., Kawachi, N., Belbin, T. J., Wang, Y., Schlecht, N. F., et al. (2015). Proteomic analysis of oral cavity squamous cell carcinoma specimens identifies patient outcome–associated proteins. Arch. Pathol. Lab. Med. 139 (4), 494–507.
Hashimoto, K., Simmons, A. N., Kajino-Sakamoto, R., Tsuji, Y., Ninomiya-Tsuji, J. (2016). TAK1 Regulates the Nrf2 Antioxidant System Through Modulating p62/SQSTM1. Antioxidants Redox Signaling 25 (17), 953–964. doi: 10.1089/ars.2016.6663
Hayashi, F., Kasamatsu, A., Endo-Sakamoto, Y., Eizuka, K., Hiroshima, K., Kita, A., et al. (2019). Increased expression of tripartite motif (TRIM) like 2 promotes tumoral growth in human oral cancer. Biochem. Biophys. Res. Commun. 508 (4), 1133–1138.
Herrero-Martin, G., Hoyer-Hansen, M., Garcia-Garcia, C., Fumarola, C., Farkas, T., Lopez-Rivas, A., et al. (2009). TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J. 28 (6), 677–685. doi: 10.1038/emboj.2009.8
Ho, J., Kong, J-W-F, Choong, L.-Y., Loh, M-C-S, Toy, W., Chong, P.-K., et al. (2009). Novel Breast Cancer Metastasis-Associated Proteins. J. Proteome Res. 8 (2), 583–594.
Hu, C.-E., Gan, J. (2017). TRIM37 promotes epithelial-mesenchymal transition in colorectal cancer. Mol. Med. Rep. 15 (3), 1057–1062.
Hu, M. M., Yang, Q., Zhang, J., Liu, S. M., Zhang, Y., Lin, H., et al. (2014). TRIM38 inhibits TNFalpha- and IL-1beta-triggered NF-kappaB activation by mediating lysosome-dependent degradation of TAB2/3. Proc. Natl. Acad. Sci. U. S. A. 111 (4), 1509–1514. doi: 10.1073/pnas.1318227111
Huang, J., Duran, A., Reina-Campos, M., Valencia, T., Castilla, E. A., Muller, T. D., et al. (2018). Adipocyte p62/SQSTM1 Suppresses Tumorigenesis through Opposite Regulations of Metabolism in Adipose Tissue and Tumor. Cancer Cell. 33 (4), 770–84 e6. doi: 10.1016/j.ccell.2018.03.001
Huang, J., Tang, L., Zhao, Y., Ding, W. (2019). TRIM11 promotes tumor angiogenesis via activation of STAT3/VEGFA signaling in lung adenocarcinoma. Am. J. Cancer Res. 9 (9), 2019–2027.
Huo, X., Li, S., Shi, T., Suo, A., Ruan, Z., Yao, Y. (2015). Tripartite motif 16 inhibits epithelial-mesenchymal transition and metastasis by down-regulating sonic hedgehog pathway in non-small cell lung cancer cells. Biochem. Biophys. Res. Commun. 460 (4), 1021–1028. doi: 10.1016/j.bbrc.2015.03.144
Hutchinson, K. E., Lipson, D., Stephens, P. J., Otto, G., Lehmann, B. D., Lyle, P. L., et al. (2013). BRAF fusions define a distinct molecular subset of melanomas with potential sensitivity to MEK inhibition. Clin. Cancer Res. : Off. J. Am. Assoc. Cancer Res. 19 (24), 6696–6702. doi: 10.1158/1078-0432.CCR-13-1746
Ichimura, Y., Waguri, S., Sou, Y. S., Kageyama, S., Hasegawa, J., Ishimura, R., et al. (2013). Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol. Cell. 51 (5), 618–631. doi: 10.1016/j.molcel.2013.08.003
Imam, S., Talley, S., Nelson, R. S., Dharan, A., O'Connor, C., Hope, T. J., et al. (2016). TRIM5alpha degradation via autophagy is not required for retroviral restriction. J. Virol. 90 (7), 3400–3410. doi: 10.1128/jvi.03033-15
Inui, T., Chano, T., Takikita-Suzuki, M., Nishikawa, M., Yamamoto, G., Okabe, H. (2013). Association of p62/SQSTM1 excess and oral carcinogenesis. PloS One 8 (9), e74398. doi: 10.1371/journal.pone.0074398
Isakson, P., Bjoras, M., Boe, S. O., Simonsen, A. (2010). Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 116 (13), 2324–2331. doi: 10.1182/blood-2010-01-261040
Ito, M., Migita, K., Matsumoto, S., Wakatsuki, K., Tanaka, T., Kunishige, T., et al. (2017). Overexpression of E3 ubiquitin ligase tripartite motif 32 correlates with a poor prognosis in patients with gastric cancer. Oncol. Lett. 13 (5), 3131–3138.
Ivanov, A. V., Peng, H., Yurchenko, V., Yap, K. L., Negorev, D. G., Schultz, D. C., et al. (2007). PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 28 (5), 823–837. doi: 10.1016/j.molcel.2007.11.012
Iwakoshi, A., Murakumo, Y., Kato, T., Kitamura, A., Mii, S., Saito, S., et al. (2012). RET finger protein expression is associated with prognosis in lung cancer with epidermal growth factor receptor mutations. Pathol. Int. 62 (5), 324–330. doi: 10.1111/j.1440-1827.2012.02797.x
Jain, S. (2011). Association of overexpression of TIF1γ with colorectal carcinogenesis and advanced colorectal adenocarcinoma. World J. Gastroenterol. 17 (35), 3994–4000.
Jain, A., Lamark, T., Sjottem, E., Larsen, K. B., Awuh, J. A., Overvatn, A., et al. (2010). p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 285 (29), 22576–22591. doi: 10.1074/jbc.M110.118976
Jaworska, A. M., Wlodarczyk, N. A., Mackiewicz, A., Czerwinska, P. (2020). The role of TRIM family proteins in the regulation of cancer stem cell self-renewal. Stem Cells 38 (2), 165–173. doi: 10.1002/stem.3109
Jena, K. K., Kolapalli, S. P., Mehto, S., Nath, P., Das, B., Sahoo, P. K., et al. (2018). TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. EMBO J. 37 (18), e98358–e98381. doi: 10.15252/embj.201798358
Ji, C. H., Kim, H. Y., Heo, A. J., Lee, S. H., Lee, M. J., Kim, S. B., et al. (2019). The N-Degron Pathway Mediates ER-phagy. Mol. Cell 75 (5), 1058–72 e9. doi: 10.1016/j.molcel.2019.06.028
Jia, D., Wei, L., Guo, W., Zha, R., Bao, M., Chen, Z., et al. (2011). Genome-wide copy number analyses identified novel cancer genes in hepatocellular carcinoma. Hepatology 54 (4), 1227–1236.
Jiang, J., Yu, C., Chen, M., Tian, S., Sun, C. (2015). Over-expression of TRIM37 promotes cell migration and metastasis in hepatocellular carcinoma by activating Wnt/β-catenin signaling. Biochem. Biophys. Res. Commun. 464 (4), 1120–1127.
Jiang, T., Tang, H., Lu, S., Yan, D., Yang, Y., Peng, Z. (2013). Up-regulation of tripartite motif-containing 29 promotes cancer cell proliferation and predicts poor survival in colorectal cancer. Med. Oncol. 30 (4), 715.
Jin, Z., Li, Y., Pitti, R., Lawrence, D., Pham, V. C., Lill, J. R., et al. (2009). Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell 137 (4), 721–735. doi: 10.1016/j.cell.2009.03.015
Jingushi, K., Ueda, Y., Kitae, K., Hase, H., Egawa, H., Ohshio, I., et al. (2015). miR-629 targets TRIM33 to promote TGFβ/Smad signaling and metastatic phenotypes in ccRCC. Mol. Cancer Res. 13 (3), 565–574.
Kajaste-Rudnitski, A., Marelli, S. S., Pultrone, C., Pertel, T., Uchil, P. D., Mechti, N., et al. (2011). TRIM22 inhibits HIV-1 transcription independently of its E3 ubiquitin ligase activity, Tat, and NF-kappaB-responsive long terminal repeat elements. J. Virol. 85 (10), 5183–5196. doi: 10.1128/JVI.02302-10
Kanno, Y., Watanabe, M., Kimura, T., Nonomura, K., Tanaka, S., Hatakeyama, S. (2014). TRIM29 as a novel prostate basal cell marker for diagnosis of prostate cancer. Acta Histochem. 116 (5), 708–712.
Karras, P., Riveiro-Falkenbach, E., Canon, E., Tejedo, C., Calvo, T. G., Martinez-Herranz, R., et al. (2019). p62/SQSTM1 Fuels Melanoma Progression by Opposing mRNA Decay of a Selective Set of Pro-metastatic Factors. Cancer Cell 35 (1), 46–63 e10. doi: 10.1016/j.ccell.2018.11.008
Karsli-Uzunbas, G., Guo, J. Y., Price, S., Teng, X., Laddha, S. V., Khor, S., et al. (2014). Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 4 (8), 914–927. doi: 10.1158/2159-8290.CD-14-0363
Kashimoto, K., Komatsu, S., Ichikawa, D., Arita, T., Konishi, H., Nagata, H., et al. (2012). Overexpression of TRIM44 contributes to malignant outcome in gastric carcinoma. Cancer Sci. 103 (11), 2021–2026.
Kassem, L., Deygas, M., Fattet, L., Lopez, J., Goulvent, T., Lavergne, E., et al. (2015). TIF1γ interferes with TGFβ1/SMAD4 signaling to promote poor outcome in operable breast cancer patients. BMC Cancer 15 (1), 453 (13p).
Kawabata, H., Azuma, K., Ikeda, K., Sugitani, I., Kinowaki, K., Fujii, T., et al. (2017). TRIM44 Is a Poor Prognostic Factor for Breast Cancer Patients as a Modulator of NF-kappaB Signaling. Int. J. Mol. Sci. 18 (9). doi: 10.3390/ijms18091931
Kawaguchi, T., Komatsu, S., Ichikawa, D., Hirajima, S., Nishimura, Y., Konishi, H., et al. (2017). Overexpression of TRIM44 is related to invasive potential and malignant outcomes in esophageal squamous cell carcinoma. Tumor Biol. 39 (6). doi: 10.1177/1010428317700409 (9p)
Kehl, S. R., Soos, B. A., Saha, B., Choi, S. W., Herren, A. W., Johansen, T., et al. (2019). TAK1 converts Sequestosome 1/p62 from an autophagy receptor to a signaling platform. EMBO Rep. 20 (9), e46238. doi: 10.15252/embr.201846238
Keown, J. R., Black, M. M., Ferron, A., Yap, M., Barnett, M. J., Pearce, F. G., et al. (2018). A helical LC3-interacting region mediates the interaction between the retroviral restriction factor Trim5alpha and mammalian autophagy-related ATG8 proteins. J. Biol. Chem. 293 (47), 18378–18386. doi: 10.1074/jbc.RA118.004202
Kimura, T., Jain, A., Choi, S. W., Mandell, M. A., Schroder, K., Johansen, T., et al. (2015). TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity. J. Cell Biol. 210 (6), 973–989. doi: 10.1083/jcb.201503023
Kimura, T., Mandell, M., Deretic, V. (2016). Precision autophagy directed by receptor regulators - emerging examples within the TRIM family. J. Cell Sci. 129 (5), 881–891. doi: 10.1242/jcs.163758
Kimura, N., Yamada, Y., Takayama, K. I., Fujimura, T., Takahashi, S., Kume, H., et al. (2018). Androgen-responsive tripartite motif 36 enhances tumor-suppressive effect by regulating apoptosis-related pathway in prostate cancer. Cancer Sci. 109 (12), 3840–3852. doi: 10.1111/cas.13803
Kirkin, V., Rogov, V. V. (2019). A Diversity of Selective Autophagy Receptors Determines the Specificity of the Autophagy Pathway. Mol. Cell 76 (2), 268–285. doi: 10.1016/j.molcel.2019.09.005
Kirkin, V., McEwan, D. G., Novak, I., Dikic, I. (2009). A role for ubiquitin in selective autophagy. Mol. Cell 34 (3), 259–269. doi: 10.1016/j.molcel.2009.04.026
Kitamura, H., Motohashi, H. (2018). NRF2 addiction in cancer cells. Cancer Sci. 109 (4), 900–911. doi: 10.1111/cas.13537
Klionsky, D. J., Baehrecke, E. H., Brumell, J. H., Chu, C. T., Codogno, P., Cuervo, A. M., et al. (2011). A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 7 (11), 1273–1294. doi: 10.4161/auto.7.11.17661
Klugbauer, S., Rabes, H. M. (1999). The transcription coactivator HTIF1 and a related protein are fused to the RET receptor tyrosine kinase in childhood papillary thyroid carcinomas. Oncogene 18 (30), 4388–4393. doi: 10.1038/sj.onc.1202824
Komatsu, M., Kurokawa, H., Waguri, S., Taguchi, K., Kobayashi, A., Ichimura, Y., et al. (2010). The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12 (3), 213–223. doi: 10.1038/ncb2021
Kumar, S., Gu, Y., Abudu, Y. P., Bruun, J. A., Jain, A., Farzam, F., et al. (2019). Phosphorylation of Syntaxin 17 by TBK1 Controls Autophagy Initiation. Dev. Cell 49 (1), 130–44 e6. doi: 10.1016/j.devcel.2019.01.027
La Belle Flynn, A., Calhoun, B. C., Sharma, A., Chang, J. C., Almasan, A., Schiemann, W. P. (2019). Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat. Commun. 10 (1), 3668. doi: 10.1038/s41467-019-11640-9
Lai, W., Zhao, J., Zhang, C., Cui, D., Lin, J., He, Y., et al. (2013). Upregulated ataxia-telangiectasia group D complementing gene correlates with poor prognosis in patients with esophageal squamous cell carcinoma: ATDC and ESCC. Dis. Esophagus 26 (8), 817–822.
Lam, H. C., Baglini, C. V., Lope, A. L., Parkhitko, A. A., Liu, H. J., Alesi, N., et al. (2017). p62/SQSTM1 Cooperates with Hyperactive mTORC1 to Regulate Glutathione Production, Maintain Mitochondrial Integrity, and Promote Tumorigenesis. Cancer Res. 77 (12), 3255–3267. doi: 10.1158/0008-5472.CAN-16-2458
Levy, J. M. M., Towers, C. G., Thorburn, A. (2017). Targeting autophagy in cancer. Nat. Rev. Cancer 17 (9), 528–542. doi: 10.1038/nrc.2017.53
Li, H., Sun, L., Tang, Z., Fu, L., Xu, Y., Li, Z., et al. (2012). Overexpression of TRIM24 Correlates with Tumor Progression in Non-Small Cell Lung Cancer. PloS One 7 (5), e37657 (12p).
Li, K., Pan, W., Ma, Y., Xu, X., Gao, Y., He, Y., et al. (2019). A novel oncogene TRIM63 promotes cell proliferation and migration via activating Wnt/β-catenin signaling pathway in breast cancer. Pathol. - Res. Pract. 215 (10), 152573 (7p).
Li, L., Dong, L., Qu, X., Jin, S., Lv, X., Tan, G. (2016). Tripartite motif 16 inhibits hepatocellular carcinoma cell migration and invasion. Int. J. Oncol. 48 (4), 1639–1649.
Li, L., Shen, C., Nakamura, E., Ando, K., Signoretti, S., Beroukhim, R., et al. (2013). SQSTM1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer Cell 24 (6), 738–750. doi: 10.1016/j.ccr.2013.10.025
Li, Q., Yan, J., Mao, A. P., Li, C., Ran, Y., Shu, H. B., et al. (2011). Tripartite motif 8 (TRIM8) modulates TNFalpha- and IL-1beta-triggered NF-kappaB activation by targeting TAK1 for K63-linked polyubiquitination. Proc. Natl. Acad. Sci. U. S. A. 108 (48), 19341–19346. doi: 10.1073/pnas.1110946108
Li, Y., Deng, L., Zhao, X., Li, B., Ren, D., Yu, L., et al. (2018). Tripartite motif-containing 37 (TRIM37) promotes the aggressiveness of non-small-cell lung cancer cells by activating the NF-κB pathway: TRIM37 promotes the aggressiveness of NSCLC. J. Pathol. 246 (3), 366–378.
Liang, C., Wang, S., Qin, C., Bao, M., Cheng, G., Liu, B., et al. (2018). TRIM36, a novel androgen-responsive gene, enhances anti-androgen efficacy against prostate cancer by inhibiting MAPK/ERK signaling pathways. Cell Death Dis. 9 (2), 155. doi: 10.1038/s41419-017-0197-y
Liang, J., Xing, D., Li, Z., Shen, J., Zhao, H., Li, S. (2016). TRIM59 is upregulated and promotes cell proliferation and migration in human osteosarcoma. Mol. Med. Rep. 13 (6), 5200–5206.
Liang, Q., Tang, C., Tang, M., Zhang, Q., Gao, Y., Ge, Z. (2019). TRIM47 is up-regulated in colorectal cancer, promoting ubiquitination and degradation of SMAD4. J. Exp. Clin. Cancer Res. 38 (1), 159. doi: 10.1186/s13046-019-1143-x
Lin, C., Wang, S., Xie, W., Chang, J., Gan, Y. (2015). The RET fusion gene and its correlation with demographic and clinicopathological features of non-small cell lung cancer: a meta-analysis. Cancer Biol. Ther. 16 (7), 1019–1028. doi: 10.1080/15384047.2015.1046649
Lin, L., Zhao, W., Sun, B., Wang, X., Liu, Q. (2017). Overexpression of TRIM24 is correlated with the progression of human cervical cancer. Am. J. Transl. Res. 9 (2), 620–628.
Linares, J. F., Duran, A., Yajima, T., Pasparakis, M., Moscat, J., Diaz-Meco, M. T. (2013). K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol. Cell. 51 (3), 283–296. doi: 10.1016/j.molcel.2013.06.020
Liu, L., Zhao, E., Li, C., Huang, L., Xiao, L., Cheng, L., et al. (2013). TRIM28, a new molecular marker predicting metastasis and survival in early-stage non-small cell lung cancer. Cancer Epidemiol. 37 (1), 71–78. doi: 10.1016/j.canep.2012.08.005
Liu, J., Rao, J., Lou, X., Zhai, J., Ni, Z., Wang, X. (2017). Upregulated TRIM11 exerts its oncogenic effects in hepatocellular carcinoma through inhibition of p53. Cell Physiol. Biochem. 44 (1), 255–266.
Liu, L., Xiao, L., Liang, X., Chen, L., Cheng, L., Zhang, L., et al. (2017a). TRIM28 knockdown increases sensitivity to etoposide by upregulating E2F1 in non-small cell lung cancer. Oncol. Rep. 37 (6), 3597–3605.
Liu, L., Zhou, X. M., Yang, F. F., Miao, Y., Yin, Y., Hu, X. J., et al. (2017b). TRIM22 confers poor prognosis and promotes epithelial-mesenchymal transition through regulation of AKT/GSK3beta/beta-catenin signaling in non-small cell lung cancer. Oncotarget 8 (37), 62069–62080. doi: 10.18632/oncotarget.18911
Liu, S., Meng, F., Ding, J., Ji, H., Lin, M., Zhu, J., et al. (2019). High TRIM44 expression as a valuable biomarker for diagnosis and prognosis in cervical cancer. Biosci. Rep. 39 (3), BSR20181639 (9p).
Liu, S., Yin, H., Ji, H., Zhu, J., Ma, R. (2018). Overexpression of TRIM44 is an independent marker for predicting poor prognosis in epithelial ovarian cancer. Exp. Ther. Med. 16, 3034–3040.
Liu, T., Tang, Q., Liu, K., Xie, W., Liu, X., Wang, H., et al. (2016). TRIM11 Suppresses AIM2 Inflammasome by Degrading AIM2 via p62-Dependent Selective Autophagy. Cell Rep. doi: 10.1016/j.celrep.2016.07.019
Liu, Y., Dong, Y., Zhao, L., Su, L., Diao, K., Mi, X. (2018). TRIM59 overexpression correlates with poor prognosis and contributes to breast cancer progression through AKT signaling pathway. Mol. Carcinog. 57 (12), 1792–1802.
Lott, S. T., Chen, N., Chandler, D. S., Yang, Q., Wang, L., Rodriguez, M., et al. (2009). DEAR1 is a dominant regulator of acinar morphogenesis and an independent predictor of local recurrence-free survival in early-onset breast cancer. PloS Med. 6 (5), e1000068. doi: 10.1371/journal.pmed.1000068
Louis, C., Burns, C., Wicks, I. (2018). TANK-Binding Kinase 1-Dependent Responses in Health and Autoimmunity. Front. Immunol. 9, 434. doi: 10.3389/fimmu.2018.00434
Lv, D., Li, Y., Zhang, W., Alvarez, A. A., Song, L., Tang, J., et al. (2017). TRIM24 is an oncogenic transcriptional co-activator of STAT3 in glioblastoma. Nat. Commun. 8 (1), 1454. doi: 10.1038/s41467-017-01731-w
Ma, Y., Wei, Z., Bast, R. C., Wang, Z., Li, Y., Gao, M., et al. (2016). Downregulation of TRIM27 expression inhibits the proliferation of ovarian cancer cells in vitro and in vivo. Lab. Invest. 96 (1), 37–48.
Mandell, M. A., Jain, A., Arko-Mensah, J., Chauhan, S., Kimura, T., Dinkins, C., et al. (2014). TRIM Proteins Regulate Autophagy and Can Target Autophagic Substrates by Direct Recognition. Dev. Cell 30 (4), 394–409. doi: 10.1016/j.devcel.2014.06.013
Mandell, M. A., Jain, A., Kumar, S., Castleman, M. J., Anwar, T., Eskelinen, E. L., et al. (2016). TRIM17 contributes to autophagy of midbodies while actively sparing other targets from degradation. J. Cell Sci. 129 (19), 3562–3573. doi: 10.1242/jcs.190017
Marshall, R. S., Hua, Z., Mali, S., McLoughlin, F., Vierstra, R. D. (2019). ATG8-Binding UIM Proteins Define a New Class of Autophagy Adaptors and Receptors. Cell 177 (3), 766–81 e24. doi: 10.1016/j.cell.2019.02.009
Martins, M. B., Marcello, M. A., Morari, E. C., Cunha, L. L., Soares, F. A., Vassallo, J., et al. (2013). Clinical utility of KAP-1 expression in thyroid lesions. Endocr. Pathol. 24 (2), 77–82.
Miao, Z.-F., Wang, Z.-N., Zhao, T.-T., Xu, Y.-Y., Wu, J.-H., Liu, X.-Y., et al. (2015). TRIM24 is upregulated in human gastric cancer and promotes gastric cancer cell growth and chemoresistance. Virchows Arch. May466 (5), 525–532.
Micale, L., Fusco, C., Fontana, A., Barbano, R., Augello, B., De Nittis, P., et al. (2015). TRIM8 downregulation in glioma affects cell proliferation and it is associated with patients survival. BMC Cancer 15 (1), 470 (10p).
Miyajima, N., Maruyama, S., Bohgaki, M., Kano, S., Shigemura, M., Shinohara, N., et al. (2008). TRIM68 regulates ligand-dependent transcription of androgen receptor in prostate cancer cells. Cancer Res. 68 (9), 3486–3494.
Moretti, J., Roy, S., Bozec, D., Martinez, J., Chapman, J. R., Ueberheide, B., et al. (2017). STING Senses Microbial Viability to Orchestrate Stress-Mediated Autophagy of the Endoplasmic Reticulum. Cell 171 (4), 809–823 e13. doi: 10.1016/j.cell.2017.09.034
Moscat, J., Karin, M., Diaz-Meco, M. T. (2016). p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell 167 (3), 606–609. doi: 10.1016/j.cell.2016.09.030
Nakaoku, T., Tsuta, K., Ichikawa, H., Shiraishi, K., Sakamoto, H., Enari, M., et al. (2014). Druggable oncogene fusions in invasive mucinous lung adenocarcinoma. Clin. Cancer Res. : Off. J. Am. Assoc. Cancer Res. 20 (12), 3087–3093. doi: 10.1158/1078-0432.CCR-14-0107
Nguyen, T. D., Shaid, S., Vakhrusheva, O., Koschade, S. E., Klann, K., Tholken, M., et al. (2019). Loss of the selective autophagy receptor p62 impairs murine myeloid leukemia progression and mitophagy. Blood 133 (2), 168–179. doi: 10.1182/blood-2018-02-833475
Niida, M., Tanaka, M., Kamitani, T. (2010). Downregulation of active IKK beta by Ro52-mediated autophagy. Mol. Immunol. 47 (14), 2378–2387. doi: 10.1016/j.molimm.2010.05.004
Noguchi, K., Okumura, F., Takahashi, N., Kataoka, A., Kamiyama, T., Todo, S., et al. (2011). TRIM40 promotes neddylation of IKKgamma and is downregulated in gastrointestinal cancers. Carcinogenesis 32 (7), 995–1004. doi: 10.1093/carcin/bgr068
O'Connor, C., Pertel, T., Gray, S., Robia, S. L., Bakowska, J. C., Luban, J., et al. (2010). p62/sequestosome-1 associates with and sustains the expression of retroviral restriction factor TRIM5alpha. J. Virol. 84 (12), 5997–6006. doi: 10.1128/JVI.02412-09
Offermann, A., Roth, D., Hupe, M. C., Hohensteiner, S., Becker, F., Joerg, V., et al. (2019). TRIM24 as an independent prognostic biomarker for prostate cancer. Urol. Oncol.: Semin. Original Invest. 37 (9), 576.e1–576.e10 (10p).
Ong, C.-A. J., Shapiro, J., Nason, K. S., Davison, J. M., Liu, X., Ross-Innes, C., et al. (2013). Three-gene immunohistochemical panel adds to clinical staging algorithms to predict prognosis for patients with esophageal adenocarcinoma. J. Clin. Oncol. 31 (12), 1576–1582.
Overå, K. S., Garcia Garcia, J., Bhujabal, Z., Jain, A., Overvatn, A., Larsen, K. B., et al. (2019). TRIM32, but not its muscular dystrophy-associated mutant, positively regulates and is targeted to autophagic degradation by p62/SQSTM1. J. Cell Sci. 132 (23), jcs236596. doi: 10.1242/jcs.236596
Ozato, K., Shin, D. M., Chang, T. H., Morse, H. C., 3rd (2008). TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 8 (11), 849–860. doi: 10.1038/nri2413
Palmbos, P. L., Wang, L., Yang, H., Wang, Y., Leflein, J., Ahmet, M. L., et al. (2015). ATDC/TRIM29 drives invasive bladder cancer formation through miRNA-mediated and epigenetic mechanisms. Cancer Res. 75 (23), 5155–5166.
Pan, J. A., Sun, Y., Jiang, Y. P., Bott, A. J., Jaber, N., Dou, Z., et al. (2016). TRIM21 Ubiquitylates SQSTM1/p62 and Suppresses Protein Sequestration to Regulate Redox Homeostasis. Mol. Cell. 61 (5), 720–733. doi: 10.1016/j.molcel.2016.02.007
Pan, X., Chen, Y., Shen, Y., Tantai, J. (2019). Knockdown of TRIM65 inhibits autophagy and cisplatin resistance in A549/DDP cells by regulating miR-138-5p/ATG7. Cell Death Dis. 10 (6), 429. doi: 10.1038/s41419-019-1660-8
Pankiv, S., Clausen, T. H., Lamark, T., Brech, A., Bruun, J. A., Outzen, H., et al. (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282 (33), 24131–24145. doi: 10.1074/jbc.M702824200
Park, J. S., Burckhardt, C. J., Lazcano, R., Solis, L. M., Isogai, T., Li, L., et al. (2020). Mechanical regulation of glycolysis via cytoskeleton architecture. Nature. doi: 10.1038/s41586-020-1998-1
Paul, S., Traver, M. K., Kashyap, A. K., Washington, M. A., Latoche, J. R., Schaefer, B. C. (2014). T cell receptor signals to NF-kappaB are transmitted by a cytosolic p62-Bcl10-Malt1-IKK signalosome. Sci. Signaling 7 (325), ra45. doi: 10.1126/scisignal.2004882
Pertel, T., Hausmann, S., Morger, D., Zuger, S., Guerra, J., Lascano, J., et al. (2011). TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472 (7343), 361–365. doi: 10.1038/nature09976
Peters, C. J., Rees, J. R. E., Hardwick, R. H., Hardwick, J. S., Vowler, S. L., Ong, C. J., et al. (2010). A 4-gene signature predicts survival of patients with resected adenocarcinoma of the esophagus, junction, and gastric cardia. Gastroenterology 139 (6), 1995–2004.
Pilli, M., Arko-Mensah, J., Ponpuak, M., Roberts, E., Master, S., Mandell, M. A., et al. (2012). TBK-1 Promotes Autophagy-Mediated Antimicrobial Defense by Controlling Autophagosome Maturation. Immunity 37 (2), 223–234. doi: 10.1016/j.immuni.2012.04.015
Pineda, C. T., Ramanathan, S., Fon Tacer, K., Weon, J. L., Potts, M. B., Ou, Y. H., et al. (2015). Degradation of AMPK by a cancer-specific ubiquitin ligase. Cell 160 (4), 715–728. doi: 10.1016/j.cell.2015.01.034
Pizon, V., Rybina, S., Gerbal, F., Delort, F., Vicart, P., Baldacci, G., et al. (2013). MURF2B, a Novel LC3-Binding Protein, Participates with MURF2A in the Switch between Autophagy and Ubiquitin Proteasome System during Differentiation of C2C12 Muscle Cells. PloS One 8 (10), e76140. doi: 10.1371/journal.pone.0076140
Poillet-Perez, L., White, E. (2019). Role of tumor and host autophagy in cancer metabolism. Genes Dev. 33 (11-12), 610–619. doi: 10.1101/gad.325514.119
Polonen, P., Jawahar Deen, A., Leinonen, H. M., Jyrkkanen, H. K., Kuosmanen, S., Mononen, M., et al. (2019). Nrf2 and SQSTM1/p62 jointly contribute to mesenchymal transition and invasion in glioblastoma. Oncogene. doi: 10.1038/s41388-019-0956-6
Qi, L., Lu, Z., Sun, Y.-H., Song, H.-T., Xu, W.-K. (2016). TRIM16 suppresses the progression of prostate tumors by inhibiting the Snail signaling pathway. Int. J. Mol. Med. 38 (6), 1734–1742.
Qi, Z.-X., Cai, J.-J., Chen, L.-C., Yue, Q., Gong, Y., Yao, Y., et al. (2016). TRIM28 as an independent prognostic marker plays critical roles in glioma progression. J. Neurooncol. 126 (1), 19–26.
Qin, Y., Liu, Q., Tian, S., Xie, W., Cui, J., Wang, R. F. (2016). TRIM9 short isoform preferentially promotes DNA and RNA virus-induced production of type I interferon by recruiting GSK3beta to TBK1. Cell Res. 26 (5), 613–628. doi: 10.1038/cr.2016.27
Qin, Y., Ye, J., Zhao, F., Hu, S., Wang, S. (2018). TRIM2 regulates the development and metastasis of tumorous cells of osteosarcoma. Int. J. Oncol. 53, 1643–1656.
Qiu, Y., Liu, P., Ma, X., Ma, X., Zhu, L., Lin, Y., et al. (2019). TRIM50 acts as a novel Src suppressor and inhibits ovarian cancer progression. Biochim. Biophys. Acta - Mol. Cell Res. 1866 (9), 1412–1420.
Quintás-Cardama, A., Post, S. M., Solis, L. M., Xiong, S., Yang, P., Chen, N., et al. (2014). Loss of the novel tumour suppressor and polarity gene Trim62 (Dear1) synergizes with oncogenic Ras in invasive lung cancer. J. Pathol. 234 (1), 108–119.
Quintás-Cardama, A., Zhang, N., Qiu, Y. H., Post, S., Creighton, C. J., Cortes, J., et al. (2015). Loss of TRIM62 expression is an independent adverse prognostic factor in acute myeloid leukemia. Clin. Lymphoma Myeloma Leukemia 15 (2), 115–127.
Reymond, A., Meroni, G., Fantozzi, A., Merla, G., Cairo, S., Luzi, L., et al. (2001). The tripartite motif family identifies cell compartments. EMBO J. 20 (9), 2140–2151. doi: 10.1093/emboj/20.9.2140
Ribeiro, C. M., Sarrami-Forooshani, R., Setiawan, L. C., Zijlstra-Willems, E. M., van Hamme, J. L., Tigchelaar, W., et al. (2016). Receptor usage dictates HIV-1 restriction by human TRIM5alpha in dendritic cell subsets. Nature 540 (7633), 448–452. doi: 10.1038/nature20567
Richter, B., Sliter, D. A., Herhaus, L., Stolz, A., Wang, C., Beli, P., et al. (2016). Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. U. S. A. 113 (15), 4039–4044. doi: 10.1073/pnas.1523926113
Romero, F. A., Taylor, A. M., Crawford, T. D., Tsui, V., Cote, A., Magnuson, S. (2016). Disrupting Acetyl-Lysine Recognition: Progress in the Development of Bromodomain Inhibitors. J. Med. Chem. 59 (4), 1271–1298. doi: 10.1021/acs.jmedchem.5b01514
Roy, M., Tomar, D., Singh, K., Lakshmi, S., Prajapati, P., Bhatelia, K., et al. (2018). TRIM8 regulated autophagy modulates the level of cleaved Caspase-3 subunit to inhibit genotoxic stress induced cell death. Cell Signal 48, 1–12. doi: 10.1016/j.cellsig.2018.04.003
Russell, R. C., Tian, Y., Yuan, H., Park, H. W., Chang, Y. Y., Kim, J., et al. (2013). ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 15 (7), 741–750. doi: 10.1038/ncb2757
Saito, T., Ichimura, Y., Taguchi, K., Suzuki, T., Mizushima, T., Takagi, K., et al. (2016). p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 7, 12030. doi: 10.1038/ncomms12030
Sakamaki, J. I., Wilkinson, S., Hahn, M., Tasdemir, N., O'Prey, J., Clark, W., et al. (2017). Bromodomain Protein BRD4 Is a Transcriptional Repressor of Autophagy and Lysosomal Function. Mol. Cell 66 (4), 517–32 e9. doi: 10.1016/j.molcel.2017.04.027
Sakuma, M., Akahira, J., Suzuki, T., Inoue, S., Ito, K., Moriya, T., et al. (2005). Expression of estrogen-responsive finger protein (Efp) is associated with advanced disease in human epithelial ovarian cancer. Gynecologic Oncol. 99 (3), 664–670.
Sanchez-Martin, P., Komatsu, M. (2018). p62/SQSTM1 - steering the cell through health and disease. J. Cell Sci. 131 (21), jcs222836–jcs222849. doi: 10.1242/jcs.222836
Sanchez-Martin, P., Saito, T., Komatsu, M. (2019). p62/SQSTM1: ‘Jack of all trades’ in health and cancer. FEBS J. 286 (1), 8–23. doi: 10.1111/febs.14712
Sanz, L., Diaz-Meco, M. T., Nakano, H., Moscat, J. (2000). The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 19 (7), 1576–1586. doi: 10.1093/emboj/19.7.1576
Sardiello, M., Cairo, S., Fontanella, B., Ballabio, A., Meroni, G. (2008). Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol. Biol. 8, 225. doi: 10.1186/1471-2148-8-225
Shim, H. S., Kenudson, M., Zheng, Z., Liebers, M., Cha, Y. J., Hoang Ho, Q., et al. (2015). Unique Genetic and Survival Characteristics of Invasive Mucinous Adenocarcinoma of the Lung. J. Thorac. Oncol. : Off. Publ. Int. Assoc. Study Lung Cancer 10 (8), 1156–1162. doi: 10.1097/JTO.0000000000000579
Short, K. M., Cox, T. C. (2006). Subclassification of the RBCC/TRIM superfamily reveals a novel motif necessary for microtubule binding. J. Biol. Chem. 281 (13), 8970–8980. doi: 10.1074/jbc.M512755200
Siegel, R. L., Miller, K. D., Jemal, A. (2020). Cancer statistics, 2020. CA: Cancer J. Clin. 70 (1), 7–30. doi: 10.3322/caac.21590
Song, W., Wang, Z., Gu, X., Wang, A., Chen, X., Miao, H., et al. (2019). TRIM11 promotes proliferation and glycolysis of breast cancer cells via targeting AKT/GLUT1 pathway. OncoTargets Ther. 12, 4975–4984. doi: 10.2147/OTT.S207723
Song, X., Fu, C., Yang, X., Sun, D., Zhang, X., Zhang, J. (2015). Tripartite motif-containing 29 as a novel biomarker in non-small cell lung cancer. Oncol. Lett. 10 (4), 2283–2288.
Song, X., Zhang, C., Liu, Z., Liu, Q., He, K., Yu, Z. (2019). Characterization of ceRNA network to reveal potential prognostic biomarkers in triple-negative breast cancer. PeerJ 7, e7522 (17p).
Sparrer, K. M. J., Gableske, S., Zurenski, M. A., Parker, Z. M., Full, F., Baumgart, G. J., et al. (2017). TRIM23 mediates virus-induced autophagy via activation of TBK1. Nat. Microbiol. 2 (11), 1543–1557. doi: 10.1038/s41564-017-0017-2
Stremlau, M., Perron, M., Lee, M., Li, Y., Song, B., Javanbakht, H., et al. (2006). Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. U. S. A. 103 (14), 5514–5519. doi: 10.1073/pnas.0509996103
Su, X., Wang, J., Chen, W., Li, Z., Fu, X., Yang, A. (2016). Overexpression of TRIM14 promotes tongue squamous cell carcinoma aggressiveness by activating the NF-κB signaling pathway. Oncotarget 7 (9), 9939–9950.
Sui, X., Chen, R., Wang, Z., Huang, Z., Kong, N., Zhang, M., et al. (2013). Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 4, e838. doi: 10.1038/cddis.2013.350
Sun, H., Dai, X., Han, B. (2014). TRIM29 as a novel biomarker in pancreatic adenocarcinoma. Dis. Markers 2014, 1–7.
Sun, N., Xue, Y., Dai, T., Li, X., Zheng, N. (2017). Tripartite motif containing 25 promotes proliferation and invasion of colorectal cancer cells through TGF-β signaling. Biosci. Rep. 37 (4), BSR20170805 (9p).
Sun, X., Ou, Z., Chen, R., Niu, X., Chen, D., Kang, R., et al. (2016). Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63 (1), 173–184. doi: 10.1002/hep.28251
Sutton, S. K., Koach, J., Tan, O., Liu, B., Carter, D. R., Wilmott, J. S., et al. (2014). TRIM16 inhibits proliferation and migration through regulation of interferon beta 1 in melanoma cells. Oncotarget 5 (20), 10127–10139.
Suzuki, T. (2005). Estrogen-responsive finger protein as a new potential biomarker for breast cancer. Clin. Cancer Res. 11 (17), 6148–6154.
Taguchi, K., Fujikawa, N., Komatsu, M., Ishii, T., Unno, M., Akaike, T., et al. (2012). Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. U. S. A. 109 (34), 13561–13566. doi: 10.1073/pnas.1121572109
Takahashi, M., Ritz, J., Cooper, G. M. (1985). Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell 42 (2), 581–588. doi: 10.1016/0092-8674(85)90115-1
Takayama, K., Suzuki, T., Tanaka, T., Fujimura, T., Takahashi, S., Urano, T., et al. (2018). TRIM25 enhances cell growth and cell survival by modulating p53 signals via interaction with G3BP2 in prostate cancer. Oncogene 37 (16), 2165–2180.
Tan, H., Qi, J., Chu, G., Liu, Z. (2017). Tripartite Motif 16 Inhibits the Migration and Invasion in Ovarian Cancer Cells. Oncol. Res. 25 (4), 551–558. doi: 10.3727/096504016X14758370595285
Tan, P., Ye, Y., He, L., Xie, J., Jing, J., Ma, G., et al. (2018). TRIM59 promotes breast cancer motility by suppressing p62-selective autophagic degradation of PDCD10. PloS Biol. 16 (11), e3000051. doi: 10.1371/journal.pbio.3000051
Tan, S.-T., Liu, S.-Y., Wu, B. (2016). TRIM29 Overexpression Promotes Proliferation and Survival of Bladder Cancer Cells through NF-κB Signaling. Cancer Res. Treat. 48 (4), 1302–1312.
Tan, Z., Song, L., Wu, W., Zhou, Y., Zhu, J., Wu, G., et al. (2018). TRIM14 promotes chemoresistance in gliomas by activating Wnt/β-catenin signaling via stabilizing Dvl2. Oncogene 37 (40), 5403–5415.
Tang, S., Gao, Y., Wen-zhong, H. (2018). Knockdown of TRIM37 suppresses the proliferation, migration and invasion of glioma cells through the inactivation of PI3K/Akt signaling pathway. Biomed. Pharmacother. 99, 59–64.
Tao, Y., Xin, M., Cheng, H., Huang, Z., Hu, T., Zhang, T., et al. (2017). TRIM37 promotes tumor cell proliferation and drug resistance in pediatric osteosarcoma. Oncol. Lett. 14, 6365–6372.
Tezel, G. G., Uner, A., Yildiz, I., Guler, G., Takahashi, M. (2009). RET finger protein expression in invasive breast carcinoma: Relationship between RFP and ErbB2 expression. Pathol. - Res. Pract. 205 (6), 403–408.
Tomar, D., Singh, R., Singh, A. K., Pandya, C. D. (2012). TRIM13 regulates ER stress induced autophagy and clonogenic ability of the cells. Biochim. Biophys. Acta 1823 (2), 316–326. doi: 10.1016/j.bbamcr.2011.11.015
Tsai, W. W., Wang, Z., Yiu, T. T., Akdemir, K. C., Xia, W., Winter, S., et al. (2010). TRIM24 links a non-canonical histone signature to breast cancer. Nature 468 (7326), 927–932. doi: 10.1038/nature09542
Tsuchida, T., Zou, J., Saitoh, T., Kumar, H., Abe, T., Matsuura, Y., et al. (2010). The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 33 (5), 765–776. doi: 10.1016/j.immuni.2010.10.013
Tsukamoto, H., Kato, T., Enomoto, A., Nakamura, N., Shimono, Y., Jijiwa, M., et al. (2009). Expression of Ret finger protein correlates with outcomes in endometrial cancer. Cancer Sci. 100 (10), 1895–1901.
Tuna, M., Smid, M., Martens, J. W. M., Foekens, J. A. (2012). Prognostic value of acquired uniparental disomy (aUPD) in primary breast cancer. Breast Cancer Res. Treat. 132 (1), 189–196.
Umemura, A., He, F., Taniguchi, K., Nakagawa, H., Yamachika, S., Font-Burgada, J., et al. (2016). p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 29 (6), 935–948. doi: 10.1016/j.ccell.2016.04.006
Valencia, T., Kim, J. Y., Abu-Baker, S., Moscat-Pardos, J., Ahn, C. S., Reina-Campos, M., et al. (2014). Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell 26 (1), 121–135. doi: 10.1016/j.ccr.2014.05.004
Valletti, A., Marzano, F., Pesole, G., Sbisa, E., Tullo, A. (2019). Targeting Chemoresistant Tumors: Could TRIM Proteins-p53 Axis Be a Possible Answer? Int. J. Mol. Sci. 20 (7), 1776–1798. doi: 10.3390/ijms20071776
Vargas, J. N. S., Wang, C., Bunker, E., Hao, L., Maric, D., Schiavo, G., et al. (2019). Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol. Cell 74 (2), 347–62 e6. doi: 10.1016/j.molcel.2019.02.010
Vera-Ramirez, L., Vodnala, S. K., Nini, R., Hunter, K. W., Green, J. E. (2018). Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 9 (1), 1944. doi: 10.1038/s41467-018-04070-6
Versteeg Gijs, A., Rajsbaum, R., Sánchez-Aparicio Maria, T., Maestre Ana, M., Valdiviezo, J., Shi, M., et al. (2013). The E3-Ligase TRIM Family of Proteins Regulates Signaling Pathways Triggered by Innate Immune Pattern-Recognition Receptors. Immunity 38 (2), 384–398. doi: 10.1016/j.immuni.2012.11.013
Wang, C., Xu, J., Fu, H., Zhang, Y., Zhang, X., Yang, D., et al. (2018). TRIM32 promotes cell proliferation and invasion by activating β-catenin signaling in gastric cancer. J. Cell Mol. Med. 22 (10), 5020–5028.
Wang, F., Ruan, L., Yang, J., Zhao, Q., Wei, W. (2018). TRIM14 promotes the migration and invasion of gastric cancer by regulating epithelialtomesenchymal transition via activation of AKT signaling regulated by miR1955p. Oncol. Rep. 40 (6), 3273–3284. doi: 10.3892/or.2018.6750
Wang, F.-Q., Han, Y., Yao, W., Yu, J. (2017). Prognostic relevance of tripartite motif containing 24 expression in colorectal cancer. Pathol. - Res. Pract. 213 (10), 1271–1275.
Wang, P., Shen, N., Liu, D., Ning, X., Wu, D., Huang, X. (2018). TRIM24 siRNA induced cell apoptosis and reduced cell viability in human nasopharyngeal carcinoma cells. Mol. Med. Rep. 18, 369–376.
Wang, W., Xia, Z., Farre, J. C., Subramani, S. (2018). TRIM37 deficiency induces autophagy through deregulating the MTORC1-TFEB axis. Autophagy 14 (9), 1574–1585. doi: 10.1080/15548627.2018.1463120
Wang, X., Shi, W., Shi, H., Lu, S., Wang, K., Sun, C., et al. (2016). TRIM11 overexpression promotes proliferation, migration and invasion of lung cancer cells. J. Exp. Clin. Cancer Res. 35 (1), 100 (9p).
Wang, X. L., Shi, W. P., Shi, H. C., Lu, S. C., Wang, K., Sun, C., et al. (2016). Knockdown of TRIM65 inhibits lung cancer cell proliferation, migration and invasion: A therapeutic target in human lung cancer. Oncotarget 7 (49), 81527–81540. doi: 10.18632/oncotarget.13131
Wang, Y., He, D., Yang, L., Wen, B., Dai, J., Zhang, Q., et al (2015b). TRIM26 functions as a novel tumor suppressor of hepatocellular carcinoma and its downregulation contributes to worse prognosis. Biochem. Biophys. Res. Commun. 463 (3), 458–465.
Wang, Y., Jiang, J., Li, Q., Ma, H., Xu, Z., Gao, Y. (2016c). KAP1 is overexpressed in hepatocellular carcinoma and its clinical significance. Int. J. Clin. Oncol. 21 (5), 927–933.
Wang, Y., Lian, Q., Yang, B., Yan, S., Zhou, H., He, L., et al. (2015a). TRIM30alpha Is a Negative-Feedback Regulator of the Intracellular DNA and DNA Virus-Triggered Response by Targeting STING. PloS Pathog. 11 (6), e1005012. doi: 10.1371/journal.ppat.1005012
Wang, Y., Liu, C., Xie, Z., Lu, H. (2020). Knockdown of TRIM47 inhibits breast cancer tumorigenesis and progression through the inactivation of PI3K/Akt pathway. Chem. Biol. Interact. 317, 108960.
Wang, Y., Zhou, Z., Wang, X., Zhang, X., Chen, Y., Bai, J., et al. (2018). TRIM59 is a novel marker of poor prognosis and promotes malignant progression of ovarian cancer by inducing annexin A2 expression. Int. J. Biol. Sci. 14 (14), 2073–2082.
Wang, Y.-Y., Li, L., Zhao, Z.-S., Wang, H.-J. (2013). Clinical utility of measuring expression levels of KAP1, TIMP1 and STC2 in peripheral blood of patients with gastric cancer. World J. Surg. Onc. 11 (1), 81 (8p).
Wang, Z. Y., Chen, Z. (2008). Acute promyelocytic leukemia: from highly fatal to highly curable. Blood 111 (5), 2505–2515. doi: 10.1182/blood-2007-07-102798
Wang, Z., Cao, L., Kang, R., Yang, M., Liu, L., Zhao, Y., et al. (2011). Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RARalpha oncoprotein. Autophagy 7 (4), 401–411. doi: 10.4161/auto.7.4.14397
Watanabe, M., Hatakeyama, S. (2017). TRIM proteins and diseases. J. Biochem. 161 (2), 135–144. doi: 10.1093/jb/mvw087
Watson, R. O., Bell, S. L., MacDuff, D. A., Kimmey, J. M., Diner, E. J., Olivas, J., et al. (2015). The Cytosolic Sensor cGAS Detects Mycobacterium tuberculosis DNA to Induce Type I Interferons and Activate Autophagy. Cell Host Microbe 17 (6), 811–819. doi: 10.1016/j.chom.2015.05.004
Wei, C.-Y., Wang, L., Zhu, M.-X., Deng, X.-Y., Wang, D.-H., Zhang, S.-M., et al. (2019). TRIM44 activates the AKT/mTOR signal pathway to induce melanoma progression by stabilizing TLR4. J. Exp. Clin. Cancer Res. 38 (1), 137 (13p).
Wei, W.-S., Chen, X., Guo, L.-Y., Li, X.-D., Deng, M.-H., Yuan, G.-J., et al. (2018). TRIM65 supports bladder urothelial carcinoma cell aggressiveness by promoting ANXA2 ubiquitination and degradation. Cancer Lett. 435, 10–22.
White, E. (2016). Autophagy and p53. Cold Spring Harbor Perspect. Med. 6 (4), a026120. doi: 10.1101/cshperspect.a026120
Witt, C. C., Witt, S. H., Lerche, S., Labeit, D., Back, W., Labeit, S. (2008). Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. EMBO J. 27 (2), 350–360. doi: 10.1038/sj.emboj.7601952
Wu, G., Song, L., Zhu, J., Hu, Y., Cao, L., Tan, Z., et al. (2018). An ATM/TRIM37/NEMO axis counteracts genotoxicity by activating nuclear-to-cytoplasmic NF-κB signaling. Cancer Res. 78 (22), 6399–6412.
Wu, W., Chen, J., Wu, J., Lin, J., Yang, S., Yu, H. (2017). Knockdown of tripartite motif-59 inhibits the malignant processes in human colorectal cancer cells. Oncol. Rep. 38 (4), 2480–2488.
Xiao, W., Wang, X., Wang, T., Xing, J. (2018). TRIM2 downregulation in clear cell renal cell carcinoma affects cell proliferation, migration, and invasion and predicts poor patients' survival. Cancer Manage. Res. 10, 5951–5964.
Xie, M., Zhang, D., Dyck, J. R., Li, Y., Zhang, H., Morishima, M., et al. (2006). A pivotal role for endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc. Natl. Acad. Sci. U. S. A. 103 (46), 17378–17383. doi: 10.1073/pnas.0604708103
Xing, Y., Meng, Q., Chen, X., Zhao, Y., Liu, W., Hu, J., et al. (2016). TRIM44 promotes proliferation and metastasis in nonsmall cell lung cancer via mTOR signaling pathway. Oncotarget 7 (21), 30479–30491. doi: 10.18632/oncotarget.8586
Xu, L., Wu, Q., Zhou, X., Wu, Q., Fang, M. (2019). TRIM13 inhibited cell proliferation and induced cell apoptosis by regulating NF-kappaB pathway in non-small-cell lung carcinoma cells. Gene 715, 144015. doi: 10.1016/j.gene.2019.144015
Xu, M., Hu, J., Zhou, B., Zhong, Y., Lin, N., Xu, R. (2019). TRIM29 prevents hepatocellular carcinoma progression by inhibiting Wnt/β-catenin signaling pathway. Acta Biochim. Biophys. Sin. 51 (1), 68–77.
Xu, R., Hu, J., Zhang, T., Jiang, C., Wang, H.-Y. (2016a). TRIM29 overexpression is associated with poor prognosis and promotes tumor progression by activating Wnt/β-catenin. Oncotarget 7 (19), 28579–28591.
Xu, W., Xu, B., Yao, Y., Yu, X., Cao, H., Zhang, J., et al. (2016b). RNA interference against TRIM29 inhibits migration and invasion of colorectal cancer cells. Oncol. Rep. 36 (3), 1411–1418.
Xue, D., Zhang, X., Zhang, X., Liu, J., Li, N., Liu, C., et al. (2015a). Clinical significance and biological roles of TRIM24 in human bladder carcinoma. Tumor Biol. 36 (9), 6849–6855.
Xue, J., Chen, Y., Wu, Y., Wang, Z., Zhou, A., Zhang, S., et al. (2015b). Tumour suppressor TRIM33 targets nuclear β-catenin degradation. Nat. Commun. 6 (1), 6156 (16p.
Yamada, Y., Takayama, K., Fujimura, T., Ashikari, D., Obinata, D., Takahashi, S., et al. (2017). A novel prognostic factor TRIM44 promotes cell proliferation and migration, and inhibits apoptosis in testicular germ cell tumor. Cancer Sci. 108 (1), 32–41.
Yang, Y., Fiskus, W., Yong, B., Atadja, P., Takahashi, Y., Pandita, T. K., et al. (2013). Acetylated hsp70 and KAP1-mediated Vps34 SUMOylation is required for autophagosome creation in autophagy. Proc. Natl. Acad. Sci. U. S. A. 110 (17), 6841–6846. doi: 10.1073/pnas.1217692110
Yang, Y.-F., Zhang, M.-F., Tian, Q.-H., Zhang, C. Z. (2017). TRIM65 triggers β-catenin signaling via ubiquitylation of Axin1 to promote hepatocellular carcinoma. J. Cell Sci. 130(18):3108–3115
Yang, Q., Liu, T. T., Lin, H., Zhang, M., Wei, J., Luo, W. W., et al. (2017). TRIM32-TAX1BP1-dependent selective autophagic degradation of TRIF negatively regulates TLR3/4-mediated innate immune responses. PloS Pathog. 13 (9), e1006600. doi: 10.1371/journal.ppat.1006600
Yang, L., Shi, P., Zhao, G., Xu, J., Peng, W., Zhang, J., et al. (2020). Targeting cancer stem cell pathways for cancer therapy. Signal Transduction Targeted Ther. 5, 8. doi: 10.1038/s41392-020-0110-5
Yao, J., Xu, T., Tian, T., Fu, X., Wang, W., Li, S., et al. (2016). Tripartite motif 16 suppresses breast cancer stem cell properties through regulation of Gli-1 degradation via the ubiquitin-proteasome pathway. Oncol. Rep. 35 (2), 1204–1212. doi: 10.3892/or.2015.4437
Ye, W., Hu, M. M., Lei, C. Q., Zhou, Q., Lin, H., Sun, M. S., et al. (2017). TRIM8 Negatively Regulates TLR3/4-Mediated Innate Immune Response by Blocking TRIF-TBK1 Interaction. J. Immunol. 199 (5), 1856–1864. doi: 10.4049/jimmunol.1601647
Yin, H., Li, Z., Chen, J., Hu, X. (2019). Expression and the potential functions of TRIM32 in lung cancer tumorigenesis. J. Cell. Biochem. 120 (4), 5232–5243. doi: 10.1002/jcb.27798
Yokoe, T., Toiyama, Y., Okugawa, Y., Tanaka, K., Ohi, M., Inoue, Y., et al. (2010). KAP1 is associated with peritoneal carcinomatosis in gastric cancer. Ann. Surg. Oncol. 17 (3), 821–828.
Yu, C., Chen, S., Guo, Y., Sun, C. (2018). Oncogenic TRIM31 confers gemcitabine resistance in pancreatic cancer via activating the NF-κB signaling pathway. Theranostics 8 (12), 3224–3236.
Yu, C., Zhan, L., Jiang, J., Pan, Y., Zhang, H., Li, X., et al. (2014). KAP-1 is overexpressed and correlates with increased metastatic ability and tumorigenicity in pancreatic cancer. Med. Oncol. 31 (7), 25 (9p).
Yuan, X., Cai, C., Chen, S., Chen, S., Yu, Z., Balk, S. P. (2014). Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene 33 (22), 2815–2825. doi: 10.1038/onc.2013.235
Zaffagnini, G., Savova, A., Danieli, A., Romanov, J., Tremel, S., Ebner, M., et al. (2018). p62 filaments capture and present ubiquitinated cargos for autophagy. EMBO J. 37 (5), e98308–e98329. doi: 10.15252/embj.201798308
Zeng, S.-X., Cai, Q.-C., Guo, C.-H., Zhi, L.-Q., Dai, X., Zhang, D.-F., et al. (2017). High expression of TRIM29 (ATDC) contributes to poor prognosis and tumor metastasis by inducing epithelial-mesenchymal transition in osteosarcoma. Oncol. Rep. 38 (3), 1645–1654.
Zhan, Y., Kost-Alimova, M., Shi, X., Leo, E., Bardenhagen, J. P., Shepard, H. E., et al. (2015). Development of novel cellular histone-binding and chromatin-displacement assays for bromodomain drug discovery. Epigenet. Chromatin 8, 37. doi: 10.1186/s13072-015-0026-4
Zhang, J., Hu, M. M., Wang, Y. Y., Shu, H. B. (2012). TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 287 (34), 28646–28655. doi: 10.1074/jbc.M112.362608
Zhang, L.-H., Yin, A.-A., Cheng, J.-X., Huang, H.-Y., Li, X.-M., Zhang, Y.-Q., et al. (2015). TRIM24 promotes glioma progression and enhances chemoresistance through activation of the PI3K/Akt signaling pathway. Oncogene 34 (5), 600–610.
Zhang, P., Zhang, H., Wang, Y., Zhang, P., Qi, Y. (2019). Tripartite motif-containing protein 59 (TRIM59) promotes epithelial ovarian Cancer progression via the focal adhesion kinase(FAK)/AKT/matrix metalloproteinase (MMP) pathway. Med. Sci. Monit. 25, 3366–3373.
Zhang, P.-P., Ding, D.-Z., Shi, B., Zhang, S.-Q., Gu, L.-L., Wang, Y.-C., et al. (2018a). Expression of TRIM28 correlates with proliferation and Bortezomib-induced apoptosis in B-cell non-Hodgkin lymphoma. Leukemia Lymphoma 59 (11), 2639–2649.
Zhang, Y., Feng, Y., Ji, D., Wang, Q., Qian, W., Wang, S., et al. (2018b). TRIM27 functions as an oncogene by activating epithelial-mesenchymal transition and p-AKT in colorectal cancer. Int. J. Oncol. 53 (2), 620–632. doi: 10.3892/ijo.2018.4408
Zhang, Z., Xu, C., Zhang, X., Huang, L., Zheng, C., Chen, H., et al. (2017). TRIM11 upregulation contributes to proliferation, invasion, and EMT of hepatocellular carcinoma cells. Oncol. Res. 25 (5), 691–699.
Zhao, T. T., Jin, F., Li, J. G., Xu, Y. Y., Dong, H. T., Liu, Q., et al. (2018). TRIM32 promotes proliferation and confers chemoresistance to breast cancer cells through activation of the NF-kappaB pathway. J. Cancer 9 (8), 1349–1356. doi: 10.7150/jca.22390
Zheng, Q., Hou, J., Zhou, Y., Yang, Y., Xie, B., Cao, X. (2015). Siglec1 suppresses antiviral innate immune response by inducing TBK1 degradation via the ubiquitin ligase TRIM27. Cell Res. 25 (10), 1121–1136. doi: 10.1038/cr.2015.108
Zhou, W., Zhang, Y., Zhong, C., Hu, J., Hu, H., Zhou, D., et al. (2018). Decreased expression of TRIM21 indicates unfavorable outcome and promotes cell growth in breast cancer. Cancer Manage. Res. 10, 3687–3696. doi: 10.2147/CMAR.S175470
Zhou, X. M., Sun, R., Luo, D. H., Sun, J., Zhang, M. Y., Wang, M. H., et al. (2016). Upregulated TRIM29 promotes proliferation and metastasis of nasopharyngeal carcinoma via PTEN/AKT/mTOR signal pathway. Oncotarget 7 (12), 13634–13650. doi: 10.18632/oncotarget.7215
Zhou, Z., Ji, Z., Wang, Y., Li, J., Cao, H., Zhu, H. H., et al. (2014). TRIM59 is up-regulated in gastric tumors, promoting ubiquitination and degradation of p53. Gastroenterology 147 (5), 1043–1054.
Zhu, Y., Zhao, L., Shi, K., Huang, Z., Chen, B. (2018). TRIM24 promotes hepatocellular carcinoma progression via AMPK signaling. Exp. Cell Res. 367 (2), 274–281.
Keywords: autophagy, cancer therapy, tripartite motif (TRIM) family, Sequestosome 1 (p62/SQSTM1), selective autophagy cargo receptor, autophagy regulation, cancer, autophagy modulating drugs
Citation: Mandell MA, Saha B and Thompson TA (2020) The Tripartite Nexus: Autophagy, Cancer, and Tripartite Motif-Containing Protein Family Members. Front. Pharmacol. 11:308. doi: 10.3389/fphar.2020.00308
Received: 19 December 2019; Accepted: 02 March 2020;
Published: 11 March 2020.
Edited by:
Marco Cordani, IMDEA Nanociencia, SpainReviewed by:
Santosh Chauhan, Institute of Life Sciences (ILS), IndiaCopyright © 2020 Mandell, Saha and Thompson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael A. Mandell, bW1hbmRlbGxAc2FsdWQudW5tLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.