 Élise Pépin
Élise Pépin Tim Jalinier
Tim Jalinier Guy Massicotte
Guy Massicotte Michel Cyr
Michel Cyr- Groupe de recherche en signalisation cellulaire, Département de biologie médicale, Université du Québec à Trois-Rivières, Trois-Rivières, QC, Canada
Sphingosine-1-phosphate (S1P) is a potent bioactive lipid mediator that acts as a natural ligand upon binding to five different receptors that are located in astrocytes, oligodendrocytes, microglial and neuronal cells. Recently, global activation of these receptors by FTY720 (fingolimod) has been suggested to provide neuroprotection in animal model of Parkinson’s disease (PD). Among S1P receptors, the subtype 1 (S1P1R) has been linked to features of neuroprotection and, using the selective agonist SEW2871, the present investigation assessed potential benefits (and mechanisms) of this receptor subtype in an established animal model of PD. We demonstrated that oral treatments with SEW2871 are able to provide protection to the same levels as FTY720 against loss of dopaminergic neurons and motor deficits in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (30 mg/kg, i.p., 5 days) mouse model of PD. At the molecular level, we observed that the beneficial effects of both S1PR agonists were not associated with alterations in ERK and Akt levels, two markers of molecular adaptations in the striatum neurons. However, these compounds have the capacity to prevent signs of neuroinflammation such as the activation of astrocytes and glial cells, as well as MPTP-induced reduction of BDNF levels in key regions of the brain implicated in motor functions. These findings suggest that selective S1P1R modulation has the ability to provide neuroprotection in response to MPTP neurotoxicity. Targeting S1P1R in PD therapy may represent a prominent candidate for treatment of this neurodegenerative conditions.
Introduction
Parkinson’s disease (PD) is a common age-related neurodegenerative condition, characterized by progressive loss of the nigrostriatal dopaminergic pathway and erosion of several neurological functions. Biochemical studies performed on postmorterm brains suggest that pathogenic factors most likely contributing to PD include a progressive neuroinflammatory reaction involving microglial activation and subsequent formation of pro-inflammatory cytokines such as the tumor necrosis factor (TNF-α) (Nagatsu and Sawada, 2007). Reduction in neurotrophins synthesis such as brain-derived nerve growth factor (BDNF) is another important feature associated with PD pathology (Shen et al., 2018). In support to this theory, several in vitro and in vivo studies propose that BDNF depletion occurring in various neuropathological conditions is mediated by the release of pro-inflammatory cytokines (Calabrese et al., 2014).
Although symptomatic improvement can be achieved by pharmacologically restoring dopaminergic transmission, the development of neuroprotective treatments that prevent or halt PD pathogenic processes is still awaiting. Emerging evidence have established that Fingolimod (FTY720), a non-selective sphingosine-1-phosphate receptors (S1PRs) modulator approved for the treatment of multiple sclerosis, can provide significant protection in mouse models of neurodegenerative conditions including two recent studies on PD (Aytan et al., 2016; Zhao et al., 2017). These later studies suggest that neuroprotective properties of FTY720 in a murine model of PD require direct effects of the drug on neuronal cells, which are presumably dependent on ERK activity. However, which specific S1PRs is responsible for these beneficial effects and whether non-neuronal mechanisms such as neuroinflammation are associated with brain damages is still unknown.
The present study will examine the efficacy of an oral treatment with the non-selective agonist FTY720 to subtype 1, 3, and 5 of S1PRs, or the selective agonist SEW2871 to subtype 1 of S1PRs (S1P1R), to prevent the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induced nigrostriatal loss and motor deficits in mice. In addition, potential mechanisms of action that include neuronal signaling, BDNF synthesis, and neuroinflammatory pathways will be investigated.
Materials and Methods
Animals
Twelve week-old male C57BL/6j mice (Charles River Laboratories, QC, CAN) were individually housed in a controlled room under a 14 h light/10 h dark cycle. Food and water were available ad libitum. All experiments were approved and carried out with the recommendations of the UQTR Institutional Animal Care and Use Committee (protocol #2016-MiC.24) in accordance with the Canadian Council on Animal Care.
Pharmacological Treatments
The experimental design is detailed in Figure 1. FTY720 and SEW2871 [5-(4-phenyl-5-trifluoromethylthiophen-2-yl)-3-(3-trifluoromethylphenyl)-1,2,4-oxadiazole] were purchased from Cayman Chemical (Ann Arbor, MI, USA) and MPTP hydrochloride from Toronto Research Chemicals (North York, ON, CAN). All mice (n = 24) were orally treated with either vehicle [10% dimethyl sulfoxide (DMSO) and 25% Tween 20 v/v dissolved in saline 0.9% sodium chloride], 1 mg/kg FTY720 (dissolved in vehicle) (Yazdi et al., 2015), or 20 mg/kg SEW2871 (dissolved in vehicle) (Dong et al., 2014) daily for 14 days. Mice were also intraperitoneally (i.p.) injected with saline or 30 mg/kg MPTP (dissolved in saline) (Xiao-Feng et al., 2016) once a day for five consecutive days. Our experimental groups were defined as follow: 1) vehicle + saline, n = 3 mice; 2) FTY720 + saline, n = 3; 3) SEW2871 + saline, n = 3; 4) Vehicle + MPTP, n = 5; 5) FTY720 + MPTP, n = 5; and 6) SEW2871 + MPTP, n = 5.

Figure 1 Experimental design. Three days before 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) injections, mice were pre-trained at the pole and beam tests by completing three trials within a day. Two days before MPTP injections, mice received an oral treatment with either vehicle [10% dimethyl sulfoxide (DMSO) and 25% Tween 20 v/v dissolved in saline 0.9% sodium chloride], FTY720 (1 mg/kg), or SEW2871 (20 mg/kg) once a day for a total of 14 days. At day 0, saline or MPTP (30 mg/kg/day) treatments were administered i.p. during 5 days. Motor abilities at the pole and beam tests were evaluated 8 days after the first MPTP injections. On day 11, all mice were sacrificed. The experimental setting involved six groups: 1) vehicle with saline treatments, n = 3; 2) FTY720 with saline treatments, n = 3; 3) SEW2871 with saline treatments, n = 3; 4) vehicle with MPTP treatments, n = 5; 5) FTY720 with MPTP treatments, n = 5; (6) SEW2871 with MPTP treatments, n = 5.
Behavioral Assessments
Motor behavior was assessed using the pole and beam tests, as previously described (Chagniel et al., 2016; Bergeron et al., 2017). Briefly, at the pole test, mice were placed at the upper end of a vertical pole (diameter: 1.5 cm; length: 50 cm). The time required for each mouse to turn down and reach the base of the pole was recorded for three trials. The balance beam assessed two mouse abilities: the first one is the time taken to go across a narrow beam (width: 6 mm; length: 100 cm) to reach a dark goal box whereas the second one measured the stepping errors, i.e., foot-faults, occurring during the same trial. Mouse performances were recorded for three trials. A foot-fault was considered each time a paw fell under the beam midline. In all tests, mice were pre-trained three consecutive periods to remove the learning variables and a maximum time of 60 s was allowed to execute the tasks.
Protein Levels Quantification
Mice were sacrificed by decapitation and brains were divided in half. The posterior part of the brain was immediately processed for immunofluorescence analyses. In the anterior part of the brain, we rapidly dissected out the striatum structure in both hemispheres and immediately freeze them on powdered dry ice. The striatum were pooled together and stored at −80°C until we performed protein extractions for Western blot analysis. Samples were homogenized in ice-cold radioimmunoprecipitation assay (RIPA) lysis buffer containing a cocktail of protease and phosphatase inhibitors (Roche, Indianapolis, IN, USA). Protein concentrations were quantified by Bradford assay (Bio-Rad, Hercules, CA, USA). Equal amounts of proteins (30–40 μg) were separated on sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS/PAGE) gels and transferred onto nitrocellulose membranes. Immunoblots were performed overnight using the following primary antibodies: mouse monoclonal antibody against tyrosine hydroxylase (TH; 1:2,000; Millipore, Billerica, MA, USA), rabbit polyclonal antibody against dopamine transporter (DAT; 1:1,000; Millipore), rabbit polyclonal antibody against sphingosine 1-phosphate receptor 1 (EDG-1; 1:10,000; Abcam, Cambridge, MA, USA), rabbit polyclonal antibody against phospho-p44/42 MAPK (ERK1/2; Thr202/Tyr204; 1:1,000; Cell Signaling Technology, Whitby, ON, CAN), mouse monoclonal antibody against p44/42 MAPK (ERK1/2; 1:2,000; Cell Signaling Technology), rabbit monoclonal antibody against protein kinase B (p-Akt-Thr308; 1:1,000; Cell Signaling Technology), rabbit monoclonal antibody against total protein kinase B (Akt; 1:1,000; Cell Signaling Technology), rabbit polyclonal antibody against BDNF (1:400; Abcam), mouse monoclonal antibody against tumor necrosis factor alpha (TNF-α; 1:2,000; Abcam), mouse monoclonal antibody against glial fibrillary acidic protein (GFAP; 1:1,000; Cell Signaling Technology), and mouse monoclonal antibody against GAPDH (1:10,000; Abcam). Membranes were washed in tris-buffered saline (TBS)-Tween 0.1% and incubated with appropriate horseradish peroxidase-conjugated secondary antibody (1:5,000; Thermo Scientific, Ottawa, ON, CAN). To visualize protein bands, chemiluminescence reactions were performed (SuperSignal West Femto Chemiluminescence Kit, Pierce Chemical Co, IL, USA). Densitometry analysis were achieved using the VisionWorks LS software (UVP Bioimaging Upland, Upland, CA, USA) and expressed as relative optical density.
Immunofluorescence Analysis
Posterior part of the brain was post-fixed overnight at 4°C in 4% paraformaldehyde in phosphate buffered saline (PBS), pH 7.5, and a few hours in 10% sucrose/4% paraformaldehyde (wt/vol). They were frozen in isopentane and stored at −80°C. Coronal brain sections (60 μm) containing the subtantia nigra pars compacta (SNc) and the ventral tegmental area (VTA) (−2.92 to −3.64 mm from the Bregma; Paxinos and Franklin, 2001) were sliced using a Leica CM3050S Cryostat (Leica, Richmond Hill, ON, Canada) and kept at 4°C in PBS. For free-floating immunofluorescence, sections were incubated in permeabilizing solution containing 1.2% Triton X-100 in PBS followed by blocking solution containing 10% normal goat serum in PBS to avoid non-specific binding. They were then incubated with primary antibodies: rabbit polyclonal antibody or mouse monoclonal antibody against TH (1:500; Millipore), mouse monoclonal antibody against neuronal nuclear antigen (NeuN; 1:200; Millipore), mouse monoclonal antibody against GFAP (1:300; Cell Signaling Technology), and rabbit monoclonal antibody against ionized calcium binding adaptor molecule 1 (Iba-1; 1:100; Abcam). The sections were rinsed in PBS and incubated in appropriate secondary antibody: goat anti-mouse conjugated with fluorescein isothiocyanate (FITC) or DyLight 594-conjugated goat anti-rabbit (1:500; Cell Signaling Technology), diluted in PBS containing 0.3% Triton X-100 and 2% next-generation sequencing (NGS) for 1 h at room temperature. After several washes in PBS, they were incubated with Hoescht 33342 (1:10,000/PBS, Invitrogen, Burlington, ON, CAN) for 15 min. Finally, the sections were rinsed several times in PBS and mounted in VECTASHIELD medium on Superfrost slides for visualization under a confocal spinning disk microscope (MBF Bioscience, Williston, VT, USA).
TH and NeuN positive cells were determined by unbiased stereological quantification using the optical fractionator of Stereo Investigator software (MBF Bioscience, Vermont, USA). Three coronal sections containing the SNc and VTA of both hemispheres were considered per animal: −3.16, −3.28, and −3.40 mm from the bregma. Borders of the SNc and VTA were defined using TH-immunostaining from a random starting point with 2X objective. Inside these borders, positive cells were counted with a 60X PlanApo oil-immersion objective and 1.4 numerical aperture attached to an Olympus BX51 microscope. A systematic sampling of the outlined area was made from a random starting point. Counts were recorded at predetermined intervals (x = 250, y = 150) and a counting frame (50x50 μm) was superimposed on the live image of each tissue section. Section thickness was measured by focusing on the top of the section, zeroing the z-axis, and focusing on bottom of the section (average section thickness was 60 μm with a range of 58.9–61.1 μm). The dissector height was set at 50 μm. Immunolabeled neurons were counted only if the first recognizable profile came into focus within the counting frame. This method certified a uniform, random, and systematic cell count. Focusing through the z-axis revealed that NeuN and TH antibodies penetrated the full depth of tissue sections. Positive cell counts were expressed as total number/mm3.
Semiquantitative optical densitometry measurements in the SNc and VTA structures were also conducted to evaluate Iba-1 and GFAP immunofluorescences. Three sections of both hemispheres were considered per animal: −3.16, −3.28, and −3.40 mm from Bregma. To delineate SNc and VTA, coronal midbrain sections labeled with Iba-1 or GFAP were co-immunostained with TH. Images of Iba-1 and GFAP immunolabeling were captured bilaterally with 2X objective using Olympus BX51 microscope and optical densitometry measurements were obtained using ImageJ software (NIH, USA).
Data Analysis
The data were analyzed using the GraphPad Prism software (version 5.0, Graph Pad Software, San Diego, CA, USA) to perform one-way ANOVA followed by the Newman-Keuls post hoc tests. Data were reported as mean ± SEM. and statistical significance was set at P < 0.05.
Results
S1PRs Modulators Reduced MPTP-Induced Inflammation and BDNF Depletion Without Interfering With S1P1R in Striatum Homogenates
We first examined the manifestation of neuroinflammation in the striatum by the determination of GFAP and TNF-α levels using Western blot analyses. While the SEW2871 and FTY720 treatments alone have no effect, they were able to prevent the surge of striatal GFAP [Figure 2A; F(5,23) = 5.31, P < 0.01] and TNF-α [Figure 2B; F(5,23) = 8.20, P < 0.001] levels induced by MPTP. In parallel, expression levels of BDNF proteins have also been investigated and we observed a significant decrease of its protein levels after MPTP administrations, an effect that was totally prevented by SEW2871 and FTY720 treatments [Figure 2C; F(5,23) = 10.77, P < 0.01].
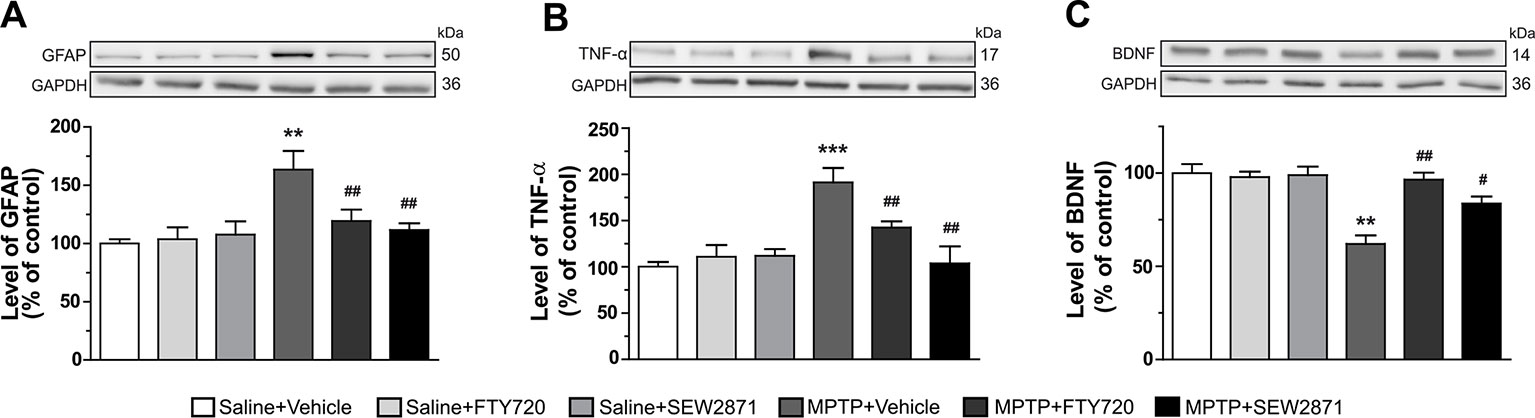
Figure 2 Levels of inflammatory markers are affected in the striatum. Levels of GFAP (A), TNF-α (B), and BDNF (C) were assessed by Western blot in the striatum of mice. The data, expressed relative to GAPDH, represent the mean of relative optical density in triplicate experiments of GFAP, TNF-α, and BDNF (expressed as a percentage of control values) ± S.E.M., n = 3–5 mice/group. **p < 0.01, ***p < 0.001 vs. vehicle + saline; #p < 0.05, ##p < 0.01 vs. vehicle + MPTP.
The levels of S1P1R were estimated by Western blot analysis in the striatum of MPTP mice treated with the SEW2871 or FTY720. Statistical analyses did not revealed any difference between all groups [Figure 3A; F(5,23) = 0.20, P > 0.05]. Levels of phosphorylated ERK1/2 and Akt proteins were also investigated by Western blot in order to assess adaptive response to neuronal activation. We observed that in addition to total ERK1/2 and total Akt, levels of phospho-Thr202-ERK1 [Figure 3B; F(5,23) = 1.62, P > 0.05] and phospho-Thr308-Akt [Figure 3C; F(5,23) = 1.69, P > 0.05] were not altered by the different treatments.
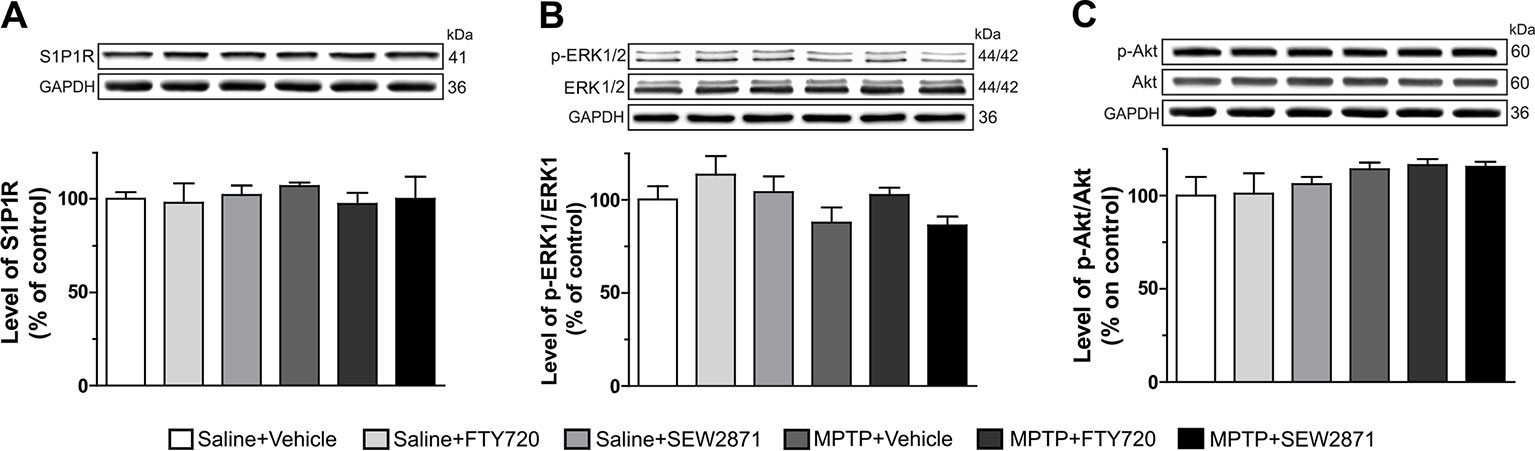
Figure 3 Levels of neuronal activity markers were unaltered in the striatum. Protein levels were evaluated by Western blotting with proteins extracted (30–40 μg of protein) from the mouse striatum. Levels of S1P1R (A), phospho-Thr202-ERK1 (B), and phospho-Thr308-Akt (C) were determined. GAPDH was used as loading control. Data represent the mean of relative optical density in triplicate experiments of S1P1R, phospho-Thr202-ERK1 and phospho-Thr308-Akt (expressed as a percentage of control values) ± S.E.M. Values are respectively expressed relative to GAPDH, total ERK, and total Akt, n = 3–5 mice/group.
S1PRs Modulators Reduced MPTP-Induced Astrogliosis and Microgliosis in the SNc
Because MPTP injections were associated with signs of inflammation in the striatum, we next examined astrocytic and microglial activation in the two major midbrain DAergic centers, the SNc and VTA, using immunofluorescence technique. Astrocytic and microglial activation were revealed by GFAP and Iba-1 immunostaining, respectively. We observed the occurrence of faint immunostaining of Iba-1 positive microglia and GFAP positive astrocytes in the SNc of vehicle-treated mice (Figure 4A). However, in the SNc of MPTP-treated mice, an augmentation in the staining of Iba-1 as well as GFAP were observed, phenotypes known to be reminiscent of reactive microglia and astrocytes, respectively (Figure 4A). FTY720 and SEW2871 were able to prevent the occurrence of microgliosis and astrocytosis as the staining of Iba-1 and GFAP were similar to vehicle-treated mice (Figure 4A). In order to quantify these observations, we performed semi-quantitative optical densitometry measurements in the SNc and VTA of mice treated with MPTP and S1PR modulators. These analyses confirmed our qualitative observations and revealed that FTY720 and SEW2871 treatments prevented the robust increase of Iba-1 [Figure 4B; F(5,23) = 48.66, P < 0.001] and GFAP [Figure 4D; F(5,23) = 11.43, P < 0.001] staining in the SNc of mice after MPTP treatments. No effect of FTY720 and SEW2871 treatments alone were noticed in the SNc and no signs of reactive microglia [Figure 4C; F(5,23) = 0.24, P > 0.05] or astrocytes [Figure 4E; F(5,23) = 0.19, P > 0.05] were observed in the VTA following the different treatments.
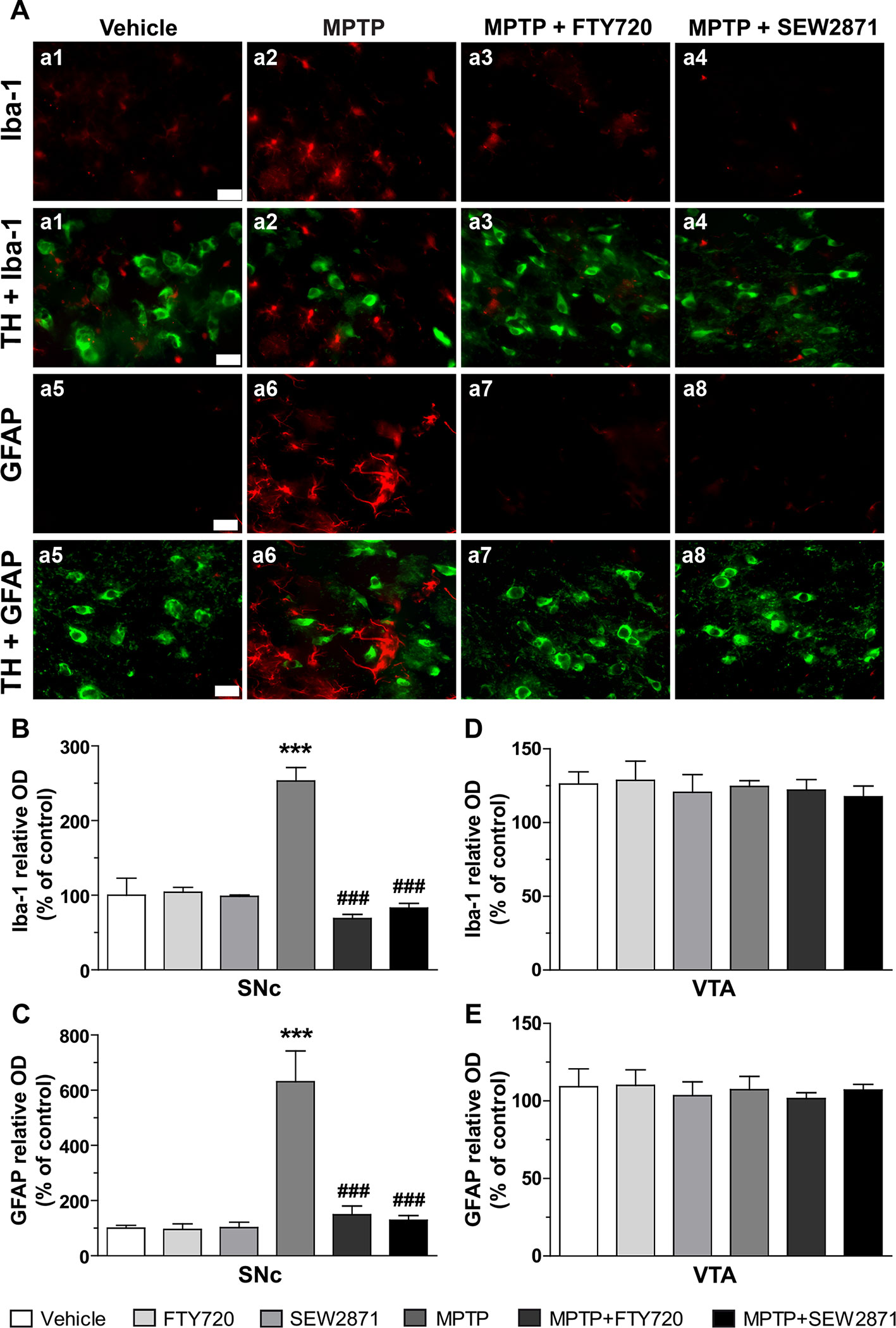
Figure 4 Signs of astrogliosis and microgliosis induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) are prevented by S1P1R modulators. (A) Representative examples of Iba-1 immunoreactive microglia (panel a1 to a4; red color), GFAP-positive astrocytes (panel a5 to a8; red color) and TH-positive neurons (panel a1 to a8; green color) using epifluorescence microscope (40X objective) in the mouse subtantia nigra pars reticulate (SNr; −3.16 mm from the Bregma). Bar equals 50 µm. Fluorescence intensity of Iba-1 and GFAP were measured in the SNc (B, D) as well as in the ventral tegmental area (VTA) (C, E). The data represent the mean of Iba-1 and GFAP relative optical density (expressed as a percentage of control values) ± S.E.M., n = 3–5 mice/group. ***p < 0.001 vs. vehicle + saline; ###p < 0.001 vs. vehicle + MPTP.
S1PRs Modulators Protects Against MPTP-Induced Nigrostriatal Cellular Loss
The extent of MPTP-induced midbrain dopaminergic denervation was estimated by immunofluorescence techniques using two independent antibodies (Figure 5A), one raised against the neuronal marker NeuN and the second raised against TH. This rate-limiting enzyme for dopamine synthesis is widely used as a marker of dopaminergic depletion (Chagniel et al., 2012). In the SNc of MPTP-treated mice, our stereological method revealed a reduction in TH immunopositive cells that was totally prevented by the SEW2871 and FTY720 treatments [Figure 5B; F(5,23) = 5.31, P < 0.01]. In contrast, no effect of either MPTP, SEW2871, or FTY720 administration was observed on TH immunopositive cells of the VTA [Figure 5C; F(5,23) = 0.13, P > 0.05]. These data were replicated using the NeuN marker in the SNc [Figure 5D; F(5,23) = 3.57, P < 0.05] and the VTA [Figure 5E; F(5,23) = 0.15, P > 0.05].
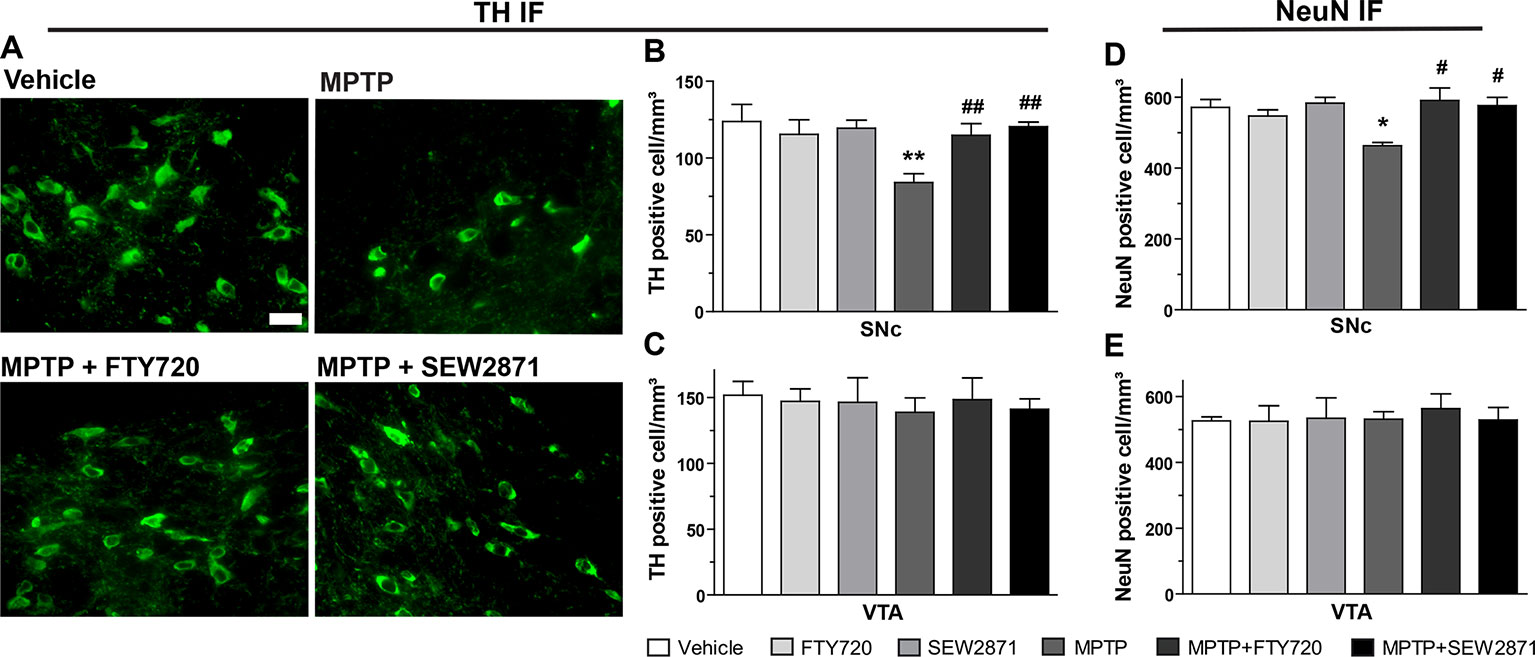
Figure 5 S1PR modulators protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced subtantia nigra pars compacta (SNc) cellular loss. Representative examples of tyrosine hydroxylase (TH)-immunostaining (green) using the 40X objective of the epifluorescence microscope in coronal sections (−3.16 mm from the Bregma) of mouse subtantia nigra pars reticulate (SNr). Bar equals 50 µm (A). Stereological counts of TH- and neuronal nuclear antigen (NeuN)-positive neurons in the SNc (B, D) and ventral tegmental area (VTA) (C, E). The data represent the mean of TH- and NeuN-positive cell numbers/mm3 (expressed as a percentage of control values) ± S.E.M., n = 3-5 mice/group. *p < 0.05, **p < 0.01 vs. vehicle + saline; #p < 0.05, ##p < 0.01 vs. vehicle + MPTP.
In addition to the VTA and SNc, we also investigated the detrimental effects of MPTP on striatal dopamine terminals by using Western blot analysis. Two well-known markers of dopaminergic neuron terminals were employed and robust decreases in TH [Figure 6A; F(5,23) = 5.92, P < 0.001] and DAT [Figure 6B; F(5,23) = 3.98, P < 0.01] levels were observed after MPTP injections. SEW2871 and FTY720 administrations were both able to prevent this effect of MPTP (Figures 6A, B).
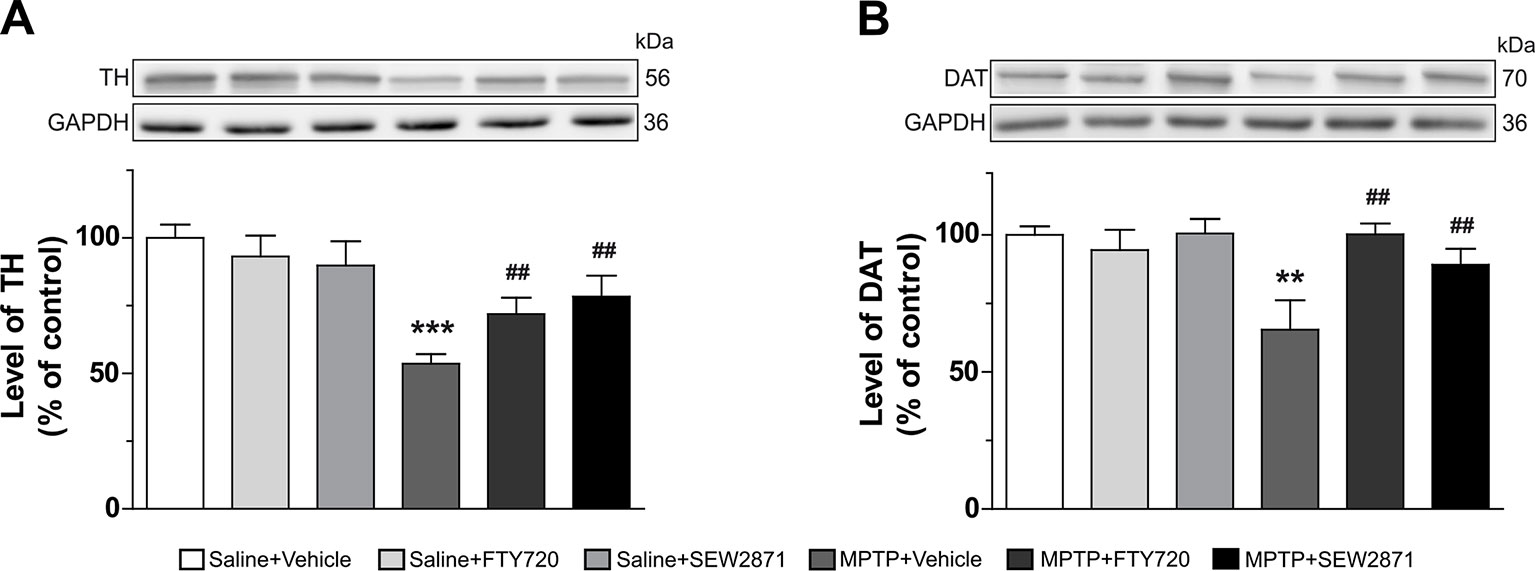
Figure 6 Dopamine terminals were spared in the striatum following S1P1Rs modulators treatments. Levels of tyrosine hydroxylase (TH) (A) and dopamine transporter (DAT) (B) were determined in the mouse striatum by Western blot experiments. The data, expressed relative to GAPDH, represent the mean of relative optical density in triplicate experiments of TH and DAT (expressed as a percentage of control values) ± S.E.M., n = 3–5 mice/group. **p < 0.01, ***p < 0.001 vs. vehicle + saline; ##p < 0.01 vs. vehicle + MPTP.
Motor Abilities Were Preserved With FTY720 and SEW2871 Administrations
To assess the effect of treatments on motor abilities, we performed two behavioral tests, namely the pole and beam tests. In particular, the pole test estimated bradykinesia and motor coordination whereas the beam test analyzed skilled walking and overall coordination. It is noteworthy that mice were pre-trained in order to remove the learning variables associated to these tests. We observed that while SEW2871 or FTY720 treatments alone were without effects on motor behaviors, they prevented the MPTP-induced motor deficits in all tasks. We observed impaired performances of MPTP treated mice at the pole test [Figure 7A; F(5,23) = 3,47, P < 0.05] as well as an increase in the time taken to go across the beam [Figure 7B; F(5,23) = 2,81, P < 0.05] and in the number of stepping errors at the beam test [Figure 7C; F(5,23) = 13,48, P < 0.001]. The time required to perform the pole test and the beam test, as well as the average foot-faults were returning to control values when MPTP mice received either SEW2871 or FTY720 administrations. Note that statistical significance was not reached for the time required to perform the beam test in both groups.
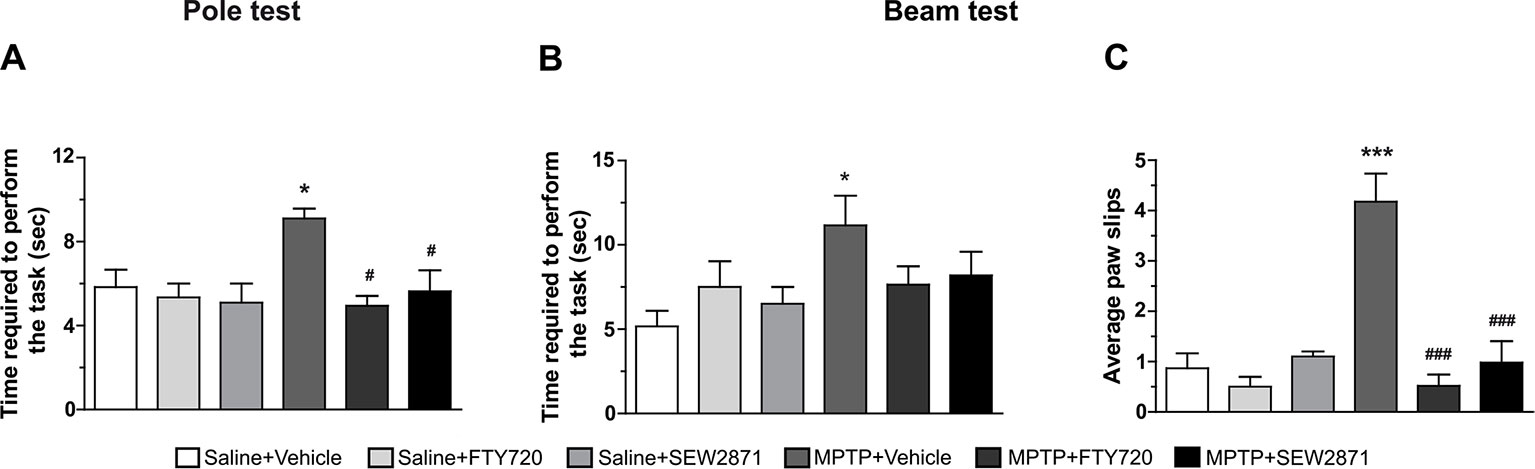
Figure 7 Motor behavioral outcomes. Motor abilities on the pole and beam tests were evaluated. Data represent the mean time require to perform the pole (A) and beam (B) tests and the mean number of foot-faults occurring during the beam test (C) ± S.E.M., n = 3–5 mice/group. *p < 0.05, ***p < 0.001 vs. vehicle + saline; #p < 0.05, ###p < 0.001 vs. vehicle + MPTP.
Discussion
The present study demonstrates that oral treatments with FTY720 and SEW2871 provide protection against loss of dopaminergic neurons and motor deficits in a mouse model of PD. In addition to PD, FTY720 was known to exert several beneficial pharmacological effects on central nervous system (CNS) cells contributing to the treatment of various neuropathological conditions, such as stroke (Brunkhorst et al., 2014), Huntington’s (Di Pardo et al., 2014), and Alzheimer’s diseases (Martin and Sospedra, 2014). However, this study is the first to demonstrate such CNS positive outcomes for the S1P1R-selective agonist SEW2871. In addition, we provide evidence that FTY720 and SEW2871 treatments have the capacity to prevent activation of astrocytes and glial cells, as well as prevent the decrease of BDNF levels, in regions of the brain involved in the control of motor functions. Our finding propose that drugs acting via the S1P1R have the capacity to provide neuroprotection to the detrimental effects of MPTP treatments.
At this point it is important to emphasize that the MPTP treatment (5 days, 30 mg/kg, once daily) used in this mouse study is a well validated model and that the results obtained in our experiments are in line with previous observations (Jackson-Lewis et al., 2012; Wang et al., 2015). The loss of nigrostriatal dopaminergic neurons is parallel by a robust increase of glial and astrocyte reactivity in the SNc and striatum, in addition to impaired motor behavior. It is interesting that no alteration at the striatal levels of phosphorylated ERK and Akt, two markers of molecular adaptations of striatal spiny projection neurons, are observed after MPTP treatments. This finding was somewhat expected as variation in the activity of these striatal kinases is associated with high dopaminergic depletion; observed for instance in unilateral 6-hydroxydopamine (OHDA) treated mice (Zhao et al., 2017) or more severe MPTP mouse model (Motyl et al., 2018). In our study, the degree of dopaminergic depletion is of 50%, as evaluated by TH levels in the striatum of MPTP mice, which is reminiscent of a mild dopaminergic-depletion. Limitations of MPTP administrations as a model of PD has been well documented previously (Bezard et al., 2013). Part of the problem with the MPTP toxin models is their acute nature, which is completely different from the insidious progression of PD observed in patients. Compensatory changes may arise in patients over the course of the disease that would not have an opportunity to occur in the acute animal models. In addition, PD occurs most frequently in elderly patients, usually around the age of 60 or older. Unfortunately, because of the inconvenience and cost of housing the animals for an extended period our study did not use older animals. In addition, a closer look at the differences in behavior, physiology, and gene expression between rodents and humans partially reveals why the animal studies do not translate well to clinical studies. With that said, while it may not be realistic to obtain a single animal model that completely reproduces every feature of a human disease as complex as PD, it would still be interesting to verify whether FTY720 and SEW2871 provide also protection during the formation of α-synuclein aggregates that resemble Lewy bodies or Lewy neurites. For instance, chronic administration of neurotoxins induces progressive PD rodent models that include α-synuclein aggregates in addition to motor deficits and rapid nigrostriatal dopaminergic cell death (Creed and Golberg, 2018). Furthermore, genetic-based approaches including transgenic models, viral vector-mediated models based on genes linked to monogenic PD as well as introduction of α-synuclein preformed fibrils all induce toxic protein aggregates in rodents. It would have been interesting to investigate the effects of FTY720 and SEW2871 in these particular models or in a combination of models to study the interplay between genetics and environment in an attempt to untangle the heterogeneity and mechanisms underlying PD.
One striking original finding in our study is the demonstration that SEW2871 and FTY720 prevent the occurrence of neuroinflammatory signs associated with MPTP treatments in mice. In particular, we establish that the SEW2871 and FTY720 treatments have the capacity to prevent the robust increase in GFAP and TNF-α expression levels in the striatum as well as the increased expression of GFAP and Iba-1 of the SNc observed in the MPTP treated mice. While the mechanism of action is still unclear, some multiple sclerosis studies performed in vitro and in animal models have demonstrated that FTY720 reduced microglia mediated inflammation and also diminished astrocyte activation in association to the neuronal protection (Choi et al., 2011; Miguez et al., 2015; Rothhammer et al., 2017; O'Sullivan et al., 2018). Whether the ongoing astrocytosis and microgliosis in the brain of our MPTP treated mice is responsible for the dopamine neuronal loss is unknown, this question is still under extensive debate in PD field (Kaur et al., 2017). However, in the VTA, a region where no effect of the MPTP treatment on dopaminergic neurons is noticed, no signs of astrocytosis or microgliosis are observed. This is at least one evidence showing that neuroinflammatory signs are selectively detected in brain regions where neuronal death is observed (Mosharov et al., 2009). Our data support a new role for S1P1R agonists in mediating anti-inflammatory effects in the MPTP mouse model of PD.
Treatments with FTY720 and SEW2871 did not alter the levels of phosphorylated ERK or Akt in the striatum of MPTP treated mice. In model cell lines transfected with the S1P1R subtype, the natural ligand S1P, FTY720, and SEW2871 appear to activate ERK and Akt pathways (Jo et al., 2005). In mouse models of PD, two recent studies have respectively reported that FTY720 increases levels of p-ERK or p-Akt in the striatum of 6-OHDA or MPTP treated mice (Zhao et al., 2017; Motyl et al., 2018). Again, the mild dopaminergic depletion in our MPTP mice may be responsible for the divergence with previous findings. Another interesting possibility is the fact that FTY720 and SEW2871 may have different effects on S1P1 expression on neuronal versus glial cells. However, we believe that this particular question would necessitate further research as only few studies have addressed this topic and the effect of SEW2871 is not assessed in vivo or in the context of PD. It is however clear from our data that activation of ERK and Akt pathways would play a minor role in the beneficial effects of FTY720 and SEW2871 on dopamine neuronal survival and neuroinflammation. On the other hand, both treatments have the capacity to prevent the reduction of BDNF levels observed in the striatum of MPTP mice. The link between FTY720, increased BDNF levels and neuroprotection has been well described in cell cultured and animal models of Rett syndrome (Deogracias et al., 2012), HD (Miguez et al., 2015), AD (Doi et al., 2013; Fukumoto et al., 2014) and PD (Giasson et al., 2002). For instance, in cultured neurons, FTY720 increases BDNF levels and counteracts N-methyl-d-aspartate (NMDA)-induced neuronal death in a BDNF-dependent manner (Di Menna et al., 2013; Cipriani et al., 2015). In PD animal models, BDNF treatment has been shown to reduce the loss of dopaminergic neurons (Tsukahara et al., 1995; Levivier et al., 1995). Our findings are suggesting that the decrease of BDNF levels may be one contributing factor responsible for the observed dopaminergic neuronal loss, and that rescuing this deficit may underlies the protective effect of FTY720 and SEW2871 treatments.
Despite that our study raised the interesting possibility that prevention of neuroinflammation and striatal BDNF levels recovery, rather than striatal Akt and ERK signaling modulation, are responsible for the neuroprotective effects of FTY720 and SEW2871 in the MPTP mouse model, the exact target mechanisms remain to be determined. For instance, we cannot exclude that brain inflammation could rely on peripheral mechanisms. Yang et al. (2018) demonstrate that lymphocyte infiltration contributes to brain neurodegeneration in the MPTP mouse model. On that line and, given the ability of FTY720 and SEW2871 to produce lymphopenia (Sanna et al., 2004; Morris et al., 2005), we cannot exclude that the neuroprotection we observed from these drugs in the MPTP-treated mice is dependent on reduced neuroinflammatory processes subsequent to peripheral mechanisms. Administration of S1P modulators after MPTP exposure would have help separating out an effect of drugs on peripheral immune cell recruitment and direct glial modulation. Of course, the precise contributions of central and peripheral mechanisms to the benefit of FTY720 and SEW2871 in PD pathology warrants further investigations. On the other hand, our data suggest that the anti-inflammatory effects of FTY720 and SEW2871 may be responsible for rescuing BDNF levels in the striatum of MPTP-treated animals. This scenario is consistent with the recent literature indicating that production of TNFα and subsequent activation of TNFα receptor 1 can be responsible for down-regulating BDNF expression in the hippocampus. Precisely, Liu et al. (2017) demonstrated that hippocampal BDNF expression is markedly reduced in mice after peripheral nerve injury, an effect that was totally abolished following genetic deletion of TNFα receptor 1. In addition, it has been documented that BDNF is synthesized not only in neurons, but also in astrocytes (Saha et al., 2006). It is therefore tempting to propose the interesting possibility that astrocytes are also mediating these effects given that S1PR expression are enriched in these cells in addition to their capacity to produce BDNF. It would be of interest to find out to what extent the beneficial effects on BDNF expression exerted by FTY720 and SEW2871 are dependent on S1PR and TNFα receptor activation in the striatum of MPTP-treated mice.
Our results show that similar beneficial effects observed with either SEW2871 or FTY720 treatments in the MPTP mouse model of PD suggest a crucial role for the subtype 1 of S1PRs. The specificity of SEW2871 to bind to S1P1R is undeniably well recognized (Bolick et al., 2005; Blaho and Hla, 2014). On the other hand, FTY720 is less selective to that particular S1PR. FTY720 need to be phosphorylated in vivo by sphingosine kinase-2 to form the active moiety FTY720-phosphate known to bind subtypes −1, −3, −4, and −5 (Brinkmann et al., 2002; Soliven et al., 2011). At the exception of S1PR4, all S1PRs subtypes are expressed in the central nervous system and S1P1R and three levels are substantially high in the brain (MacLennan et al., 1994; Zhang et al., 1999). Interestingly, S1P1R is located on astrocytes, oligodendrocytes, microglial and neuronal cells throughout the brain, including region of the brain associated with the control of motor function (Chae et al., 2004; Aktas et al., 2010). Notably, S1P1R responses in astrocytes has been well studied in human and rodent studies (Healy et al., 2013; Wu et al., 2013). For instance, our finding confirmed S1P1R protein expression in the striatum structure. Whether S1P1R is exclusively responsible for the neuroprotection we report using SEW2871 or FTY720 treatments is undetermined, but some evidence are supporting this contention in the literature. For instance, the role of S1P1R in the beneficial action of FTY720 is reported in animal models of stroke and PD (Dev et al., 2008; Hasegawa et al., 2010; Imeri et al., 2014; Brunkhorst et al., 2014; Sun et al., 2016; Zhao et al., 2017; Motyl et al., 2018). These studies all demonstrate that the effect of FTY720 is largely mediated by the S1P1R.
Consistent with other investigations, we observed that FTY720 could exert beneficial effects in PD mouse model (Zhao et al., 2017; Motyl et al., 2018). This drug has been shown to exert several pharmacological effects on CNS cells which may contribute to the treatment of various neuropathological conditions, such as stroke (Brunkhorst et al., 2014) Huntington’s (Di Pardo et al., 2014) and AD (Martin and Sospedra, 2014). In that line, it was recently found to reduce the density of pathological plaques and decreased the number of pro-inflammatory cells in animal models of AD (Fukumoto et al., 2014; Aytan et al., 2016). Unfortunately, both in vitro and in vivo experiments have demonstrated that FTY720, accumulating above a certain threshold in the brain, becomes less effective (Aytan et al., 2016) and even neurotoxic (van Echten-Deckert et al., 2014). In fact, studies have documented the possibility that FTY720 can alter normal brain physiology via a mechanism involving hyperphosphorylation of Tau proteins (Attiori Essis et al., 2015).
In conclusion, we observed that pharmacological targeting of S1P1R with the specific agonist SEW2871 conferred strong resistance to MPTP-induced dopaminergic depletion, inflammation processes and motor dysfunctions. From a clinical perspective, it seems thus plausible that selective activation of S1P1R by SEW2871 (or other chemical analogs) might be more effective and probably safer, knowing that this drug is not prone to induce Tau hyperphosphorylation (St-Cyr Giguere et al., 2017). In addition, many adverse events, including hypertension, macular edema, pulmonary toxicity, and hepatotoxicity, have been associated with FTY720, because of its off-target interactions with other S1PRs subtypes, particularly with S1PR3. The activation of S1PR3 by FTY720 was at least partially responsible for its side effects on the cardiovascular system and organ fibrosis, which may cause prominent safety issues (Cohen and Chun, 2011). Several drugs acting more selectively on S1PRs subtypes have been developed in recent years (Guerrero et al., 2016) and future experimentation are required to test for there efficiencies and safeties in the context of PD.
Data Availability Statement
All datasets generated for this study are included in the article.
Ethics Statement
This study was carried out in accordance with the recommendations of the UQTR Institutional Animal Care and Use Committee and performed in accordance with the Canadian Council on Animal Care.
Author Contributions
ÉP and MC designed the study. ÉP conducted this research. TJ and GL contributed for the behavioral experiments. ÉP, MC, and GM wrote the manuscript. MC and GM contributed to the conceptual frame of the study and edited the manuscript.
Funding
This work was supported by the Natural Sciences and Engineering Research Council of Canada (Grant #2017-06411). EP is the holder of a Fonds de recherche du Québec–Santé (FRQS) studentship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aktas, O., Kury, P., Kieseier, B., Hartung, H. P. (2010). Fingolimod is a potential novel therapy for multiple sclerosis. Nat. Rev. Neurol. 6 (7), 373–382. doi: 10.1038/nrneurol.2010.76
Attiori Essis, S., Laurier-Laurin, M. E., Pepin, E., Cyr, M., Massicotte, G. (2015). GluN2B-containing NMDA receptors are upregulated in plasma membranes by the sphingosine-1-phosphate analog FTY720P. Brain Res. 1624, 349–358. doi: 10.1016/j.brainres.2015.07.055
Aytan, N., Choi, J. K., Carreras, I., Brinkmann, V., Kowall, N. W., Jenkins, B. G., et al. (2016). Fingolimod modulates multiple neuroinflammatory markers in a mouse model of Alzheimer’s disease. Sci. Rep. 6, 24939. doi: 10.1038/srep24939
Bergeron, Y., Bureau, G., Laurier-Laurin, M. E., Asselin, E., Massicotte, G., Cyr, M. (2017). Genetic Deletion of Akt3 induces an endophenotype reminiscent of psychiatric manifestations in mice. Front. Mol. Neurosci. 10, 102. doi: 10.3389/fnmol.2017.00102
Bezard, E., Zhenyu, Y., Deniz, K., Spillantini, M. G. (2013). Animal models of parkinson’s disease: limits and relevance to neuroprotection studies. Mov. Disord. 28 (1), 61–70. doi: 10.1002/mds.25108
Blaho, V. A., Hla, T. (2014). An update on the biology of sphingosine 1-phosphate receptors. J. Lipid Res. 55 (8), 1596–1608. doi: 10.1194/jlr.R046300
Bolick, D. T., Srinivasan, S., Kim, K. W., Hatley, M. E., Clemens, J. J., Whetzel, A., et al. (2005). Sphingosine-1-phosphate prevents tumor necrosis factor-{alpha}-mediated monocyte adhesion to aortic endothelium in mice. Arterioscler. Thromb. Vasc. Biol. 25 (5), 976–981. doi: 10.1161/01.ATV.0000162171.30089.f6
Brinkmann, V., Davis, M. D., Heise, C. E., Albert, R., Cottens, S., Hof, R., et al. (2002). The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J. Biol. Chem. 277 (24), 21453–21457. doi: 10.1074/jbc.C200176200
Brunkhorst, R., Vutukuri, R., Pfeilschifter, W. (2014). Fingolimod for the treatment of neurological diseases-state of play and future perspectives. Front. Cell Neurosci. 8, 283. doi: 10.3389/fncel.2014.00283
Calabrese, F., Rossetti, A. C., Racagni, G., Gass, P., Riva, M. A., Molteni, R. (2014). Brain-derived neurotrophic factor: a bridge between inflammation and neuroplasticity. Front. Cell Neurosci. 8, 430. doi: 10.3389/fncel.2014.00430
Chae, S. S., Proia, R. L., Hla, T. (2004). Constitutive expression of the S1P1 receptor in adult tissues. Prostaglandins Other Lipid Mediat. 73 (1-2), 141–150. doi: 10.1016/j.prostaglandins.2004.01.006
Chagniel, L., Robitaille, C., Lacharite-Mueller, C., Bureau, G., Cyr, M. (2012). Partial dopamine depletion in MPTP-treated mice differentially altered motor skill learning and action control. Behav. Brain Res. 228 (1), 9–15. doi: 10.1016/j.bbr.2011.11.019
Chagniel, L., Bergeron, Y., Bureau, G., Massicotte, G., Cyr, M. (2016). Correction: regulation of tyrosine phosphatase STEP61 by protein kinase a during motor skill learning in mice. PloS One 11 (3), e0150220. doi: 10.1371/journal.pone.0150220
Choi, J. W., Gardell, S. E., Herr, D. R., Rivera, R., Lee, C. W., Noguchi, K., et al. (2011). FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. U. S. A. 108 (2), 751–756. doi: 10.1073/pnas.1014154108
Cipriani, R., Chara, J. C., Rodriguez-Antiguedad, A., Matute, C. (2015). FTY720 attenuates excitotoxicity and neuroinflammation. J. Neuroinflammation 12, 86. doi: 10.1186/s12974-015-0308-6
Cohen, J. A., Chun, J. (2011). Mechanisms of fingolimod's efficacy and adverse effects in multiple sclerosis. Ann. Neurol. 69 (5), 759–777. doi: 10.1002/ana.22426
Creed, R. B., Golberg, M. S. (2018). New developments in genetic rat models of parkinson’s disease. Mov. Disord. 33 (5), 717–729. doi: 10.1002/mds.27296
Deogracias, R., Yazdani, M., Dekkers, M. P., Guy, J., Ionescu, M. C., Vogt, K. E., et al. (2012). Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U. S. A 109 (35), 14230–14235. doi: 10.1073/pnas.1206093109
Dev, K. K., Mullershausen, F., Mattes, H., Kuhn, R. R., Bilbe, G., Hoyer, D., et al. (2008). Brain sphingosine-1-phosphate receptors: implication for FTY720 in the treatment of multiple sclerosis. Pharmacol. Ther. 117 (1), 77–93. doi: 10.1016/j.pharmthera.2007.08.005
Di Menna, L., Molinaro, G., Di Nuzzo, L., Riozzi, B., Zappulla, C., Pozzilli, C., et al. (2013). Fingolimod protects cultured cortical neurons against excitotoxic death. Pharmacol. Res. 67 (1), 1–9. doi: 10.1016/j.phrs.2012.10.004
Di Pardo, A., Amico, E., Favellato, M., Castrataro, R., Fucile, S., Squitieri, F., et al. (2014). FTY720 (fingolimod) is a neuroprotective and disease-modifying agent in cellular and mouse models of Huntington disease. Hum. Mol. Genet. 23 (9), 2251–2265. doi: 10.1093/hmg/ddt615
Doi, Y., Takeuchi, H., Horiuchi, H., Hanyu, T., Kawanokuchi, J., Jin, S., et al. (2013). Fingolimod phosphate attenuates oligomeric amyloid beta-induced neurotoxicity via increased brain-derived neurotrophic factor expression in neurons. PloS One 8 (4), e61988. doi: 10.1371/journal.pone
Dong, J., Wang, H., Wu, G., Zhao, J., Zhang, L., Zuo, L., et al. (2014). Oral treatment with SEW2871, a sphingosine-1-phosphate type 1 receptor agonist, ameliorates experimental colitis in interleukin-10 gene deficient mice. Clin. Exp. Immunol. 177 (1), 94–101. doi: 10.1111/cei.12304
Fukumoto, K., Mizoguchi, H., Takeuchi, H., Horiuchi, H., Kawanokuchi, J., Jin, S., et al. (2014). Fingolimod increases brain-derived neurotrophic factor levels and ameliorates amyloid beta-induced memory impairment. Behav. Brain Res. 268, 88–93. doi: 10.1016/j.bbr.2014.03.046
Giasson, B. I., Duda, J. E., Quinn, S. M., Zhang, B., Trojanowski, J. Q., Lee, V. M. (2002). Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34 (4), 521–533. doi: 10.1016/S0896-6273(02)00682-7
Guerrero, M., Urbano, M., Roberts, E. (2016). Sphingosine 1-phosphate receptor 1 agonists: a patent review, (2013-2015). Expert Opin. Ther. Pat. 26 (4), 455–470. doi: 10.1517/13543776.2016.1157165
Hasegawa, Y., Suzuki, H., Sozen, T., Rolland, W., Zhang, J. H. (2010). Activation of sphingosine 1-phosphate receptor-1 by FTY720 is neuroprotective after ischemic stroke in rats. Stroke 41 (2), 368–374. doi: 10.1161/STROKEAHA.109.568899
Healy, L. M., Sheridan, G. K., Pritchard, A. J., Rutkowska, A., Mullershausen, F., Dev, K. K. (2013). Pathway specific modulation of S1P1 receptor signalling in rat and human astrocytes. Br. J. Pharmacol. 169 (5), 1114–1129. doi: 10.1111/bph.12207
Imeri, F., Fallegger, D., Zivkovic, A., Schwalm, S., Enzmann, G., Blankenbach, K., et al. (2014). Novel oxazolo-oxazole derivatives of FTY720 reduce endothelial cell permeability, immune cell chemotaxis and symptoms of experimental autoimmune encephalomyelitis in mice. Neuropharmacology 85, 314–327. doi: 10.1016/j.neuropharm.2014.05.012
Jackson-Lewis, V., Blesa, J., Przedborski, S. (2012). Animal models of Parkinson’s disease. Parkinsonism Relat. Disord. 18 Suppl 1, S183–S185. doi: 10.1016/S1353-8020(11)70057-8
Jo, E., Sanna, M. G., Gonzalez-Cabrera, P. J., Thangada, S., Tigyi, G., Osborne, D. A., et al. (2005). S1P1-selective in vivo-active agonists from high-throughput screening: off-the-shelf chemical probes of receptor interactions, signaling, and fate. Chem. Biol. 12 (6), 703–715. doi: 10.1016/j.chembiol.2005.04.019
Kaur, K., Gill, J. S., Bansal, P. K., Deshmukh, R. (2017). Neuroinflammation - A major cause for striatal dopaminergic degeneration in Parkinson’s disease. J. Neurol. Sci. 381, 308–314. doi: 10.1016/j.jns.2017.08.3251
Levivier, M., Przedborski, S., Bencsics, C., Kang, U. J. (1995). Intrastriatal implantation of fibroblasts genetically engineered to produce brain-derived neurotrophic factor prevents degeneration of dopaminergic neurons in a rat model of Parkinson’s disease. J. Neurosci. 15 (12), 7810–7820. doi: 10.1523/JNEUROSCI.15-12-07810.1995
Liu, Y., Zhou, L. J., Wang, J., Li, D., Ren, W. J., Peng, J., et al. (2017). TNF-α differentially regulates synaptic plasticity in the hippocampus and spinal cord by microglia-dependent mechanisms after peripheral nerve injury. J. Neurosci. 37 (4), 871–881. doi: 10.1523/JNEUROSCI.2235-16.2016
MacLennan, A. J., Browe, C. S., Gaskin, A. A., Lado, D. C., Shaw, G. (1994). Cloning and characterization of a putative G-protein coupled receptor potentially involved in development. Mol. Cell Neurosci. 5 (3), 201–209. doi: 10.1006/mcne.1994.1024
Martin, R., Sospedra, M. (2014). Sphingosine-1 phosphate and central nervous system. Curr. Top. Microbiol. Immunol. 378, 149–170. doi: 10.1007/978-3-319-05879-5_7
Miguez, A., Garcia-Diaz Barriga, G., Brito, V., Straccia, M., Giralt, A., Gines, S., et al. (2015). Fingolimod (FTY720) enhances hippocampal synaptic plasticity and memory in Huntington’s disease by preventing p75NTR up-regulation and astrocyte-mediated inflammation. Hum. Mol. Genet. 24 (17), 4958–4970. doi: 10.1093/hmg/ddv218
Morris, M. A., Gibb, D. R., Picard, F., Brinkmann, V., Straume, M., Ley K, K. (2005). Transient T cell accumulation in lymph nodes and sustained lymphopenia in mice treated with FTY720. Eur. J. Immunol. 35 (12), 3570–3580. doi: 10.1002/eji.200526218
Mosharov, E. V., Larsen, K. E., Kanter, E., Phillips, K. A., Wilson, K., Schmitz, Y., et al. (2009). Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 62 (2), 218–229. doi: 10.1016/j.neuron.2009.01.033
Motyl, J., Przykaza, L., Boguszewski, P. M., Kosson, P., Strosznajder, J. B. (2018). Pramipexole and Fingolimod exert neuroprotection in a mouse model of Parkinson’s disease by activation of sphingosine kinase 1 and Akt kinase. Neuropharmacology 135, 139–150. doi: 10.1016/j.neuropharm.2018.02.023
Nagatsu, T., Sawada, M. (2007). Biochemistry of postmortem brains in Parkinson’s disease: historical overview and future prospects. J. Neural Transm. Suppl (72), 113–120. doi: 10.1007/978-3-211-73574-9_14
O'Sullivan, S. A., O'Sullivan, C., Healy, L. M., Dev, K. K., Sheridan, G. K. (2018). Sphingosine 1-phosphate receptors regulate TLR4-induced CXCL5 release from astrocytes and microglia. J. Neurochem. 144 (6), 736–747. doi: 10.1111/jnc.14313
Rothhammer, V., Kenison, J. E., Tjon, E., Takenaka, M. C., de Lima, K. A., Borucki, D. M., et al. (2017). Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl. Acad. Sci. U. S. A 114 (8), 2012–2017. doi: 10.1073/pnas.1615413114
Saha, R. N., Liu, X., Pahan, K. (2006). Up-regulation of BDNF in astrocytes by TNF-alpha: A case for the neuroprotective role of cytokine. J. Neuroimmune. Pharmacol. 1, 212–222. doi: 10.1007/s11481-006-9020-8
Sanna, M. G., Liao, J., Jo, E., Alfonso, C., Ahn, M. Y., Peterson, M. S., et al. (2004). Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J. Biol. Chem. 279 (14), 13839–13848. doi: 10.1074/jbc.M311743200
Shen, T., You, Y., Joseph, C., Mirzaei, M., Klistorner, A., Graham, S. L., et al. (2018). BDNF polymorphism: a review of its diagnostic and clinical relevance in neurodegenerative disorders. Aging Dis. 9 (3), 523–536. doi: 10.14336/AD.2017.0717
Soliven, B., Miron, V., Chun, J. (2011). The neurobiology of sphingosine 1-phosphate signaling and sphingosine 1-phosphate receptor modulators. Neurology 76 (8 Suppl 3), S9–14. doi: 10.1212/WNL.0b013e31820d9507
St-Cyr Giguere, F., Attiori Essis, S., Chagniel, L., Germain, M., Cyr, M., Massicotte, G. (2017). The sphingosine-1-phosphate receptor 1 agonist SEW2871 reduces Tau-Ser262 phosphorylation in rat hippocampal slices. Brain Res. 1658, 51–59. doi: 10.1016/j.brainres.2017.01.014
Sun, N., Shen, Y., Han, W., Shi, K., Wood, K., Fu, Y., et al. (2016). Selective Sphingosine-1-phosphate receptor 1 modulation attenuates experimental intracerebral hemorrhage. Stroke 47 (7), 1899–1906. doi: 10.1161/STROKEAHA.115.012236
Tsukahara, T., Takeda, M., Shimohama, S., Ohara, O., Hashimoto., N. (1995). Effects of brain-derived neurotrophic factor on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in monkeys. Neurosurgery 37 (4), 733–741. doi: 10.1227/00006123-199510000-00018
van Echten-Deckert, G., Hagen-Euteneuer, N., Karaca, I., Walter, J. (2014). Sphingosine-1-phosphate: boon and bane for the brain. Cell Physiol. Biochem. 34 (1), 148–157. doi: 10.1159/000362991
Wang, Q., Liu, Y., Zhou, J. (2015). Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 4, 19. doi: 10.1186/s40035-015-0042-0
Wu, C., Leong, S. Y., Moore, C. S., Cui, Q. L., Gris, P., Bernier, L. P., et al. (2013). Dual effects of daily FTY720 on human astrocytes in vitro: relevance for neuroinflammation. J. Neuroinflammation 10, 41. doi: 10.1186/1742-2094-10-41
Xiao-Feng, L., Wen-Ting, Z., Yuan-Yuan, X., Chong-Fa, L., Lu, Z., Jin-Jun, R., et al. (2016). Protective role of 6-Hydroxy-1-H-Indazole in an MPTP-induced mouse model of Parkinson’s disease. Eur. J. Pharmacol. 791, 348–354. doi: 10.1016/j.ejphar.2016.08.011
Yang, X., Ren, H., Wood, K., Li, M., Qiu, S., Shi, F. D., et al. (2018). Depletion of microglia augments the dopaminergic neurotoxicity of MPTP. FASEB J. 32 (6), 3336–3345. doi: 10.1096/fj.201700833RR
Yazdi, A., Baharvand, H., Javan, M. (2015). Enhanced remyelination following lysolecithin-induced demyelination in mice under treatment with fingolimod (FTY720). Neuroscience 311, 34–44. doi: 10.1016/j.neuroscience.2015.10.013
Zhang, Q., Peyruchaud, O., French, K. J., Magnusson, M. K., Mosher, D. F. (1999). Sphingosine 1-phosphate stimulates fibronectin matrix assembly through a Rho-dependent signal pathway. Blood 93 (9), 2984–2990. doi: 10.1182/blood.V93.9.2984
Keywords: S1P receptors, FTY720, SEW2871, MPTP, neuroinflammation, neuroprotection
Citation: Pépin É, Jalinier T, Lemieux GL, Massicotte G and Cyr M (2020) Sphingosine-1-Phosphate Receptors Modulators Decrease Signs of Neuroinflammation and Prevent Parkinson’s Disease Symptoms in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model. Front. Pharmacol. 11:77. doi: 10.3389/fphar.2020.00077
Received: 21 March 2019; Accepted: 27 January 2020;
Published: 21 February 2020.
Edited by:
Carlos Alberto Manssour Fraga, Federal University of Rio de Janeiro, BrazilReviewed by:
Zhude Tu, Washington University School of Medicine in St. Louis, United StatesLuke Michael Healy, McGill University, Canada
Copyright © 2020 Pépin, Jalinier, Lemieux, Massicotte and Cyr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michel Cyr, bWljaGVsLmN5ckB1cXRyLmNh