 Hua Li
Hua Li Fang Fang
Fang Fang Manting Xu
Manting Xu Zhimei Liu
Zhimei Liu- Department of Neurology, Beijing Children’s Hospital, Capital Medical University, National Center for Children's Health, Beijing, China
Epileptic encephalopathy, caused by mutations in the dynamin-1 (DNM1; NM_004408) gene, is a newly identified neurologic disorder in children. Thus far, the full clinical and electroencephalographic features of children with DNM1 mutation-related epileptic encephalopathy have not been established. The aim of this study is to characterize the phenotypic, genetic, and electroencephalographic features of children with DNM1 mutation-related epileptic encephalopathy. Here, we investigated a patient with a novel pathogenic DNM1 variant, who received treatment in Beijing Children's Hospital and had detailed clinical, EEG, and genetic information. Conversely, we performed an extensive literature search in PubMed, EMBASE, Cochrane Central Register of Controlled Trials, Chinese BioMedical Literature Database, China National Knowledge Infrastructure, and Wanfang Database using the term “DNM1” and were able to find 32 cases reported in nine articles (in English) from January 2013 to December 2018. The clinical features of 33 cases with pathogenic DNM1 variants were analyzed and the results showed that patients carrying pathogenic variants in the GTPase or middle domains present with epileptic encephalopathy and severe neurodevelopmental symptoms. Patients carrying pathogenic variants in both domains exhibited comparable phenotypes.
Introduction
Epileptic encephalopathy, caused by dynamin-1 (DNM1) mutations, is a newly characterized neurologic disorder in children (Kolnikova et al., 2018). The DNM1 gene codes for the DNM1 protein is involved in the synaptic vesicle cycle that facilitates the exocytosis of neurotransmitters during receptor-mediated endocytosis, which is necessary for signaling pathway function and central nervous system development. Pathogenic DNM1 variants affect brain development and function and cause epileptic encephalopathy associated with severe neurodevelopmental complications (Allen et al., 2013; Appenzellar, 2014; Allen et al., 2016; Deng et al., 2016; Nakashima et al., 2016). Previously reported pathogenic variants of DNM1 have been associated with early onset of epileptic encephalopathy (including West and Lennox-Gastaut syndromes) and are present in up to 2% of patients with infantile spasms or Lennox-Gastaut syndrome (Appenzellar, 2014). For many years, considerable attention has been paid to the genetic studies, patients carrying pathogenic variants in the GTPase or middle domains of DNM1 exhibit epileptic encephalopathy and severe neurodevelopmental complications. With no clear and effective treatment, antiepileptic medications, as a limited manner, are often insufficient for seizure control in patients with earlier onset and higher frequency of seizures. Thus far, the specific clinical and electroencephalographic features of children with DNM1 mutation-related epileptic encephalopathy have not been clearly established. Here, we characterized the phenotypic, genetic, and electroencephalographic features of children with DNM1 mutation-related epileptic encephalopathy.
Materials and Methods
Patients
We reported a patient with a novel pathogenic DNM1 variant, who received treatment in Beijing Children's Hospital and had detailed clinical, EEG and genetic information. In addition, we performed an extensive literature search in PubMed, EMBASE, Cochrane Central Register of Controlled Trials, Chinese BioMedical Literature Database, China National Knowledge Infrastructure and Wanfang Database using the term “DNM1,” and were able to find 32 cases reported in nine articles with complete clinical data (in English) from January 2013 to December 2018 (Table 1). We then analyzed the clinical features of 33 cases with pathogenic DNM1 variants, including gender, age at seizure onset, seizure types, development, number of antiepileptic drugs (AEDs) administrated, EEG, and mutations. Seizures and epilepsy syndromes were classified in accordance with the guidelines of the International League Against Epilepsy (Belousova et al., 2017). The analysis of the pathogenic effects of these variations on DNM1 gene was conducted mainly by using three prediction algorithms: SIFT, PolyPhen 2 Hvar, Mutation taster (Table 2).
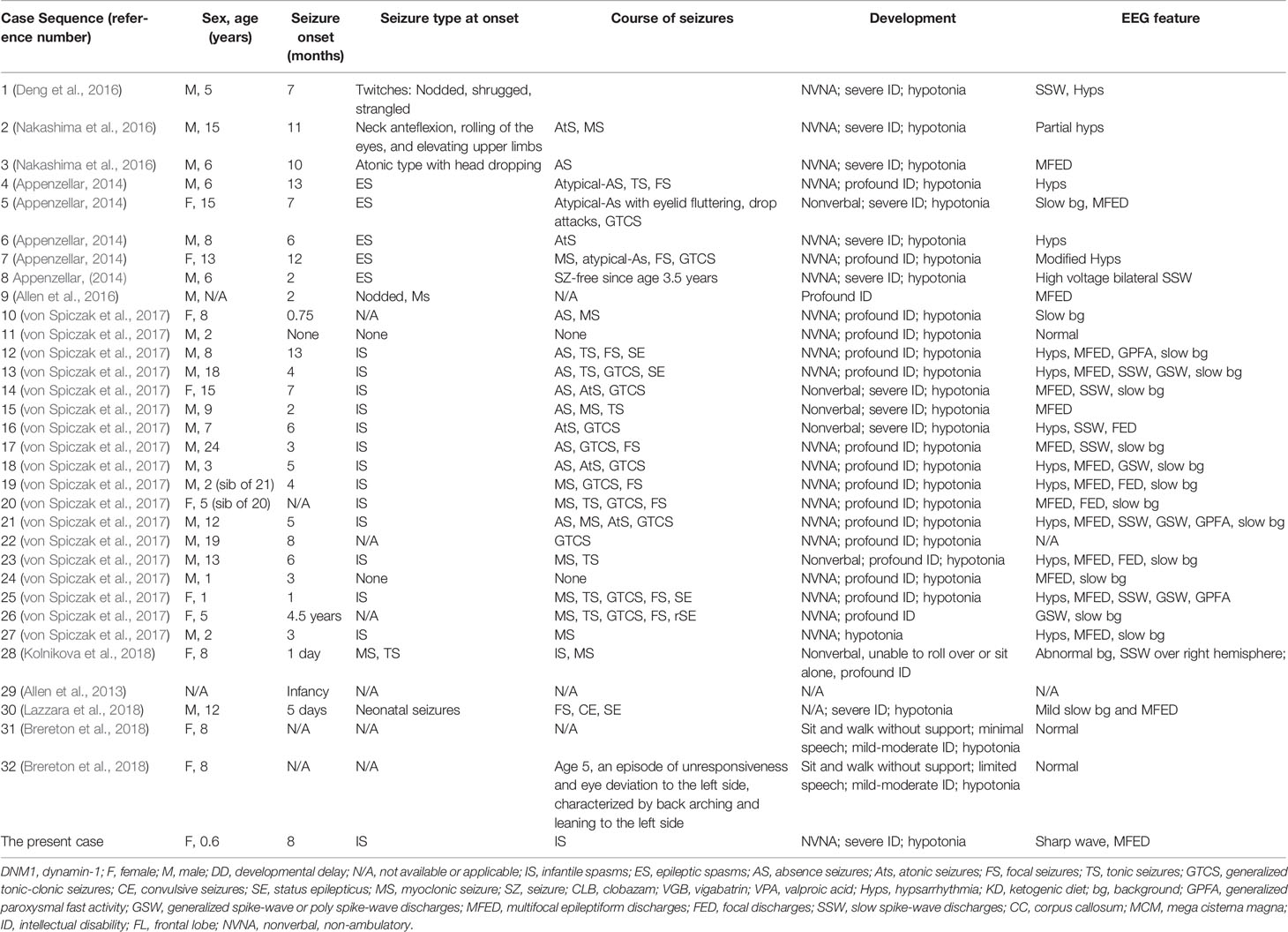
Table 1 Clinical characteristics of patients with pathogenic DNM1 variants.
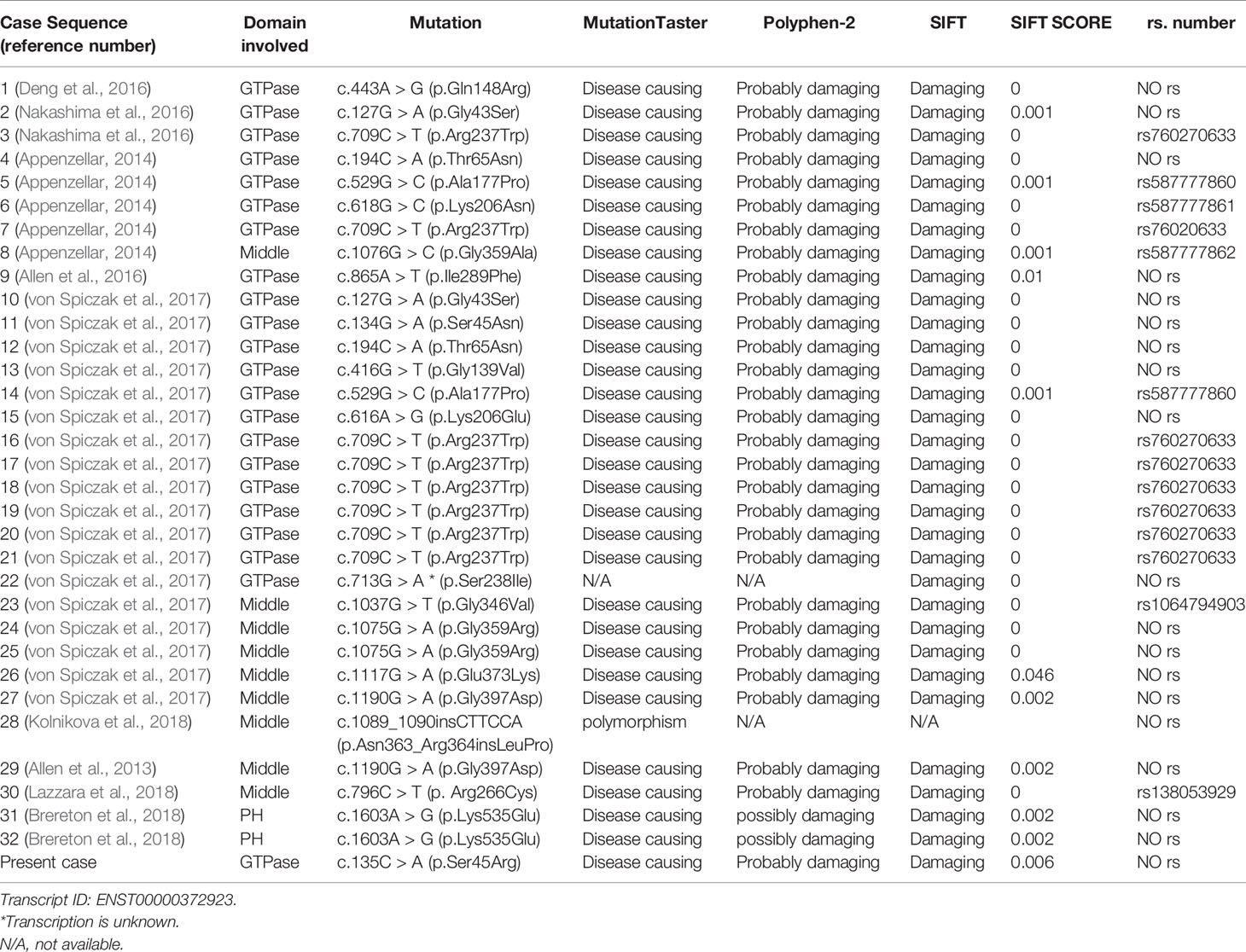
Table 2 Details of the DNM1 mutations variants of 33 cases.
Statistical Analysis
The continuous variables were described by mean or median with range, and the categorical variables by number or percentage. Chi-square tests were used for comparison of phenotypic difference with mutations in different gene domains. P-value below 0.05 was considered significant. Statistical analysis was performed using SPSS 22.0.
Ethics Statement
The present study was approved by the Ethics Committee of Beijing Children's Hospital and informed consents were collected from the participant's parents.
Results
Case Report
A 3-year-old girl presented with severe psychomotor developmental delay; nonverbal and non-ambulatory. She had been delivered at full term after 40 weeks of gestation (birth weight 3,150 g, Apgar score 10), and was the first child of the family. The patient has normal personal and family history. She exhibited limb shaking at 2 months after birth and EEG analysis revealed no epileptiform discharges. Over the following 4 months, she exhibited shaking limbs intermittently upon waking. At 6 months of age, the patient exhibited “binocular vision and tongue vomiting” which gradually increased over time, although the time of onset was unclear. Video-EEG monitoring was performed multiple times, based on the suspicion of non-epileptic seizures. Levetiracetam was administrated during observation. Infantile spasms manifested at 8 months after birth. EEG analysis showed prime spike waves, a small number of multiple spike waves, spike-slow waves, and synchronous or non-synchronous discharge (atypical hypsarrhythmia) onto the bilateral rear head during seizures. Subsequent treatment (beginning at 8 months of age) constituted administration of levetiracetam and topiramate. Changes in EEG are shown in Figures 1–4. The bilateral ventricles were slightly widened on magnetic resonance images beginning at 2 months of age.
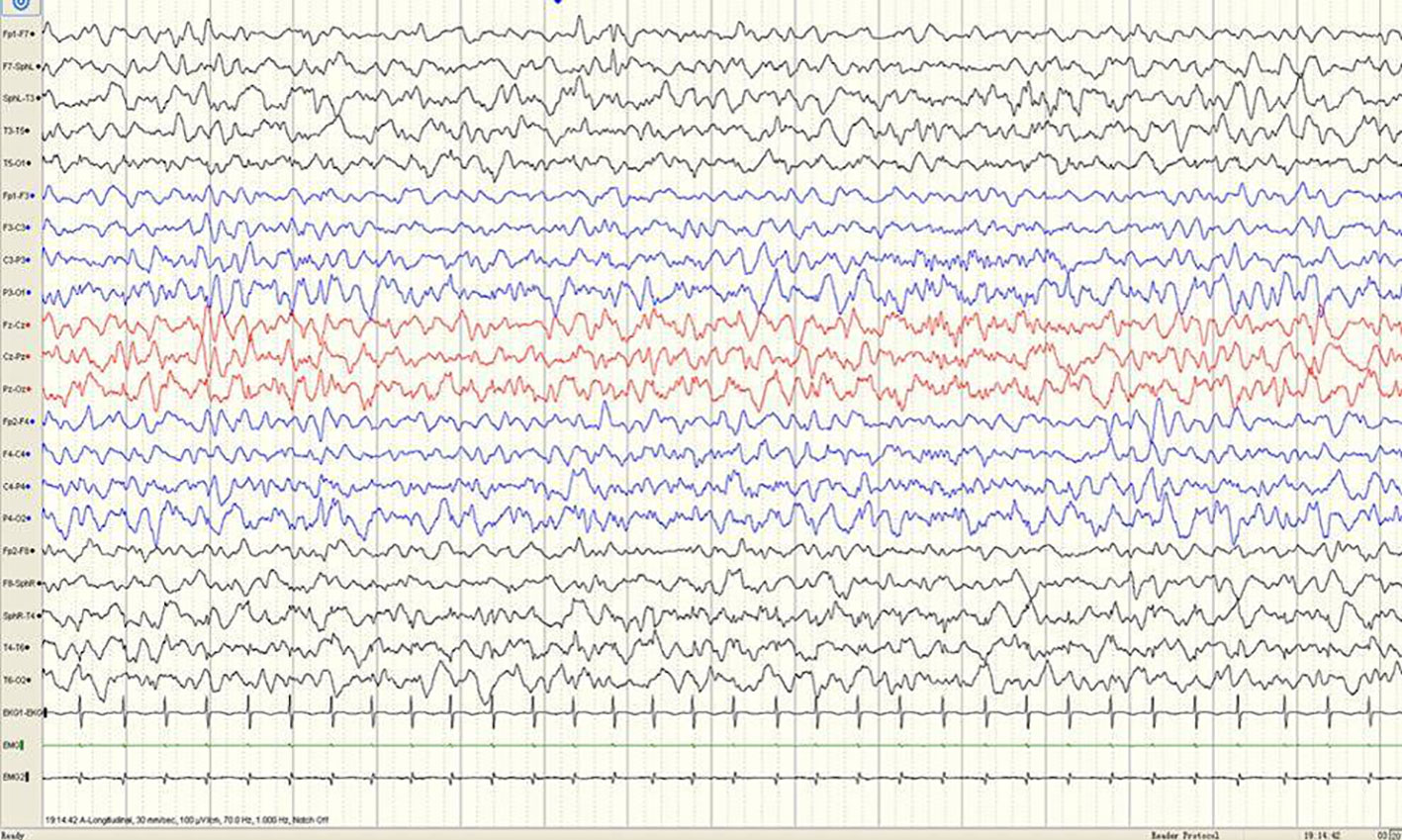
Figure 1 A sample EEG recording of 4-month-old child in this study. The interictal EEG recording shows a sharp slow wave discharge in the left anterior temporal region.
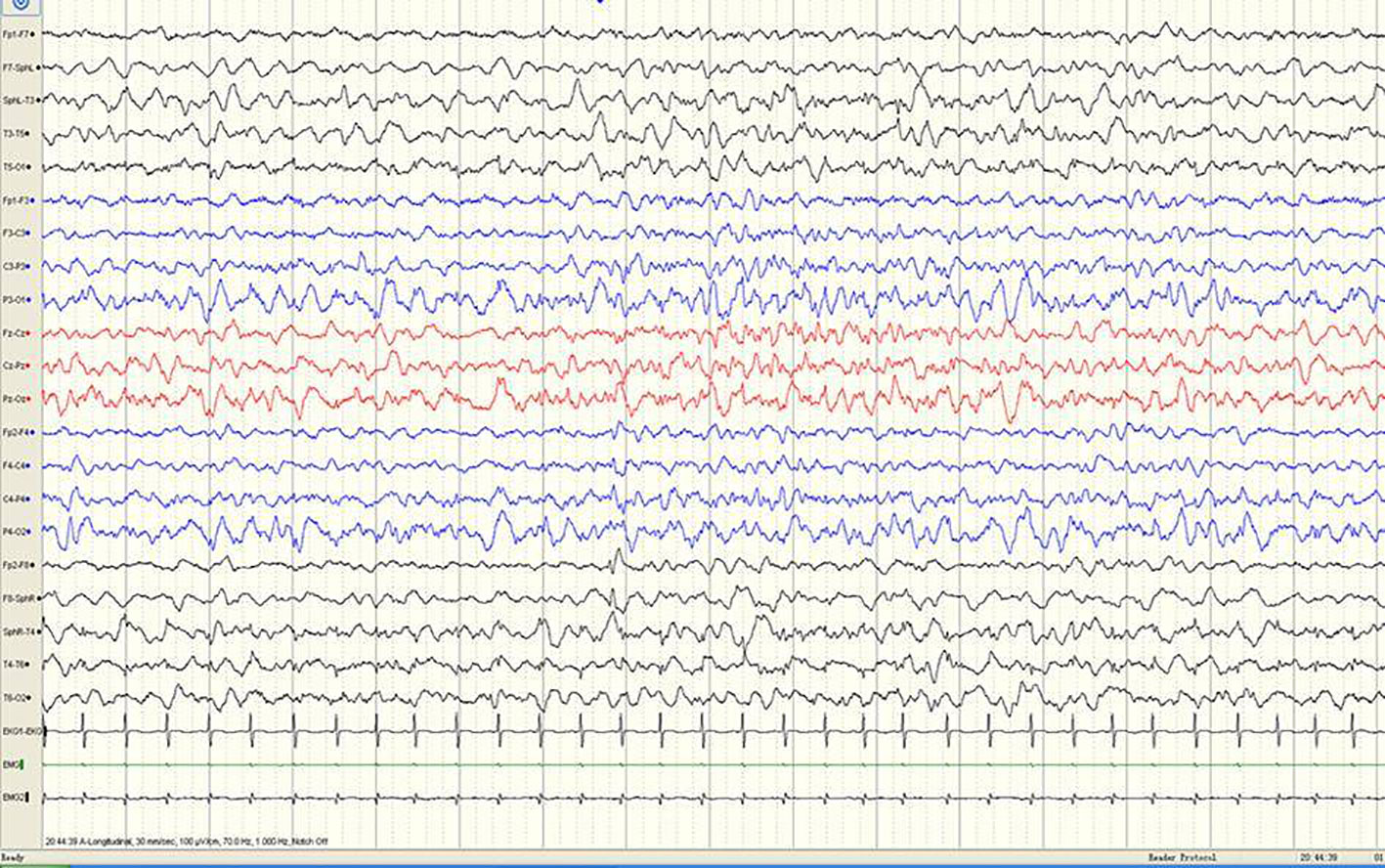
Figure 2 A sample EEG recording of 4-month-old child in this study. The interictal EEG recording shows a sharp slow wave discharge in the right anterior temporal region.
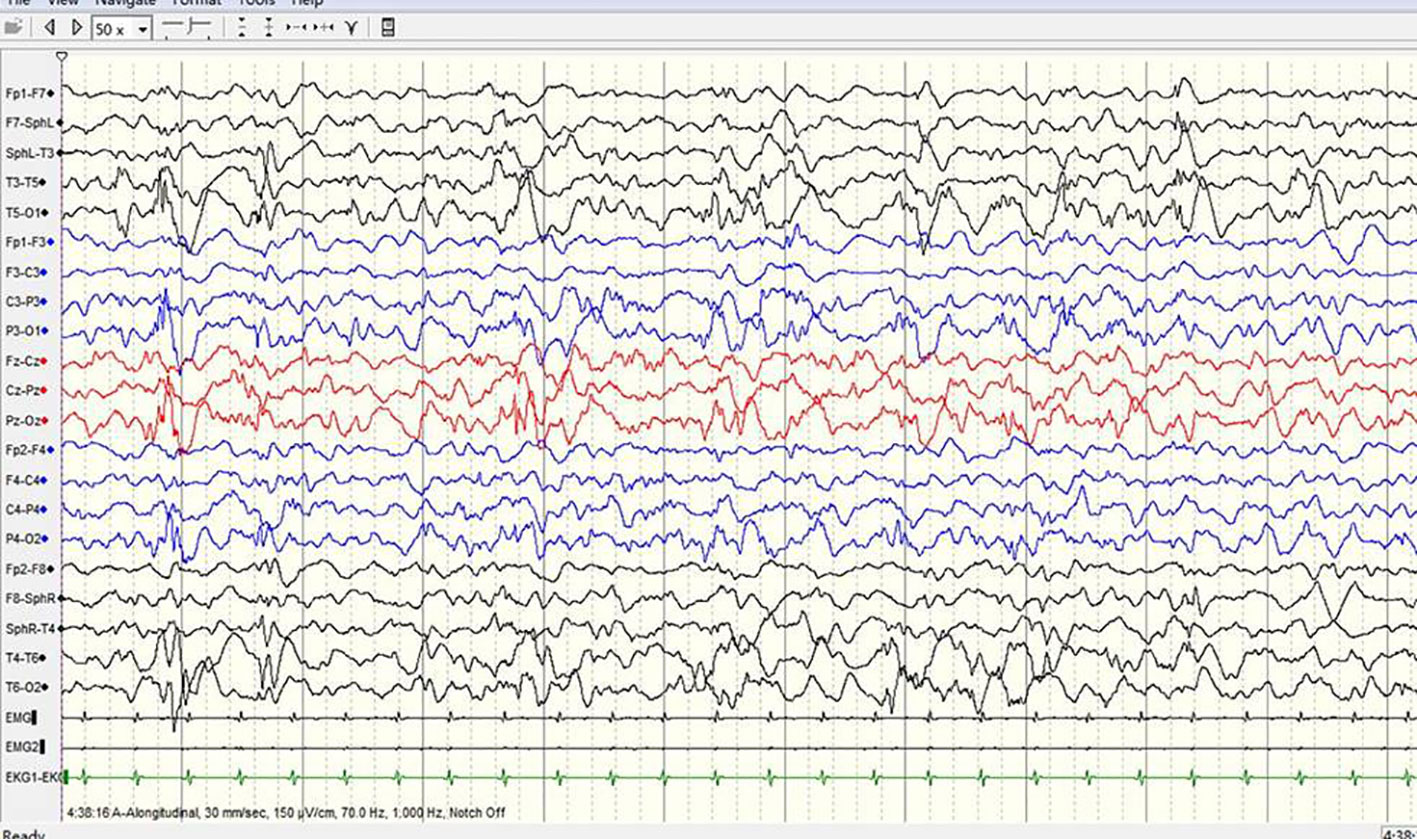
Figure 3 A sample EEG recording of 8-month-old child in this study. The interictal EEG recording shows prime spike waves, a small number of multiple spike waves, spike-slow waves, and multiple spike-slow wave synchronous or non-synchronous discharge (atypical hypsarrhythmia) onto the bilateral rear head.
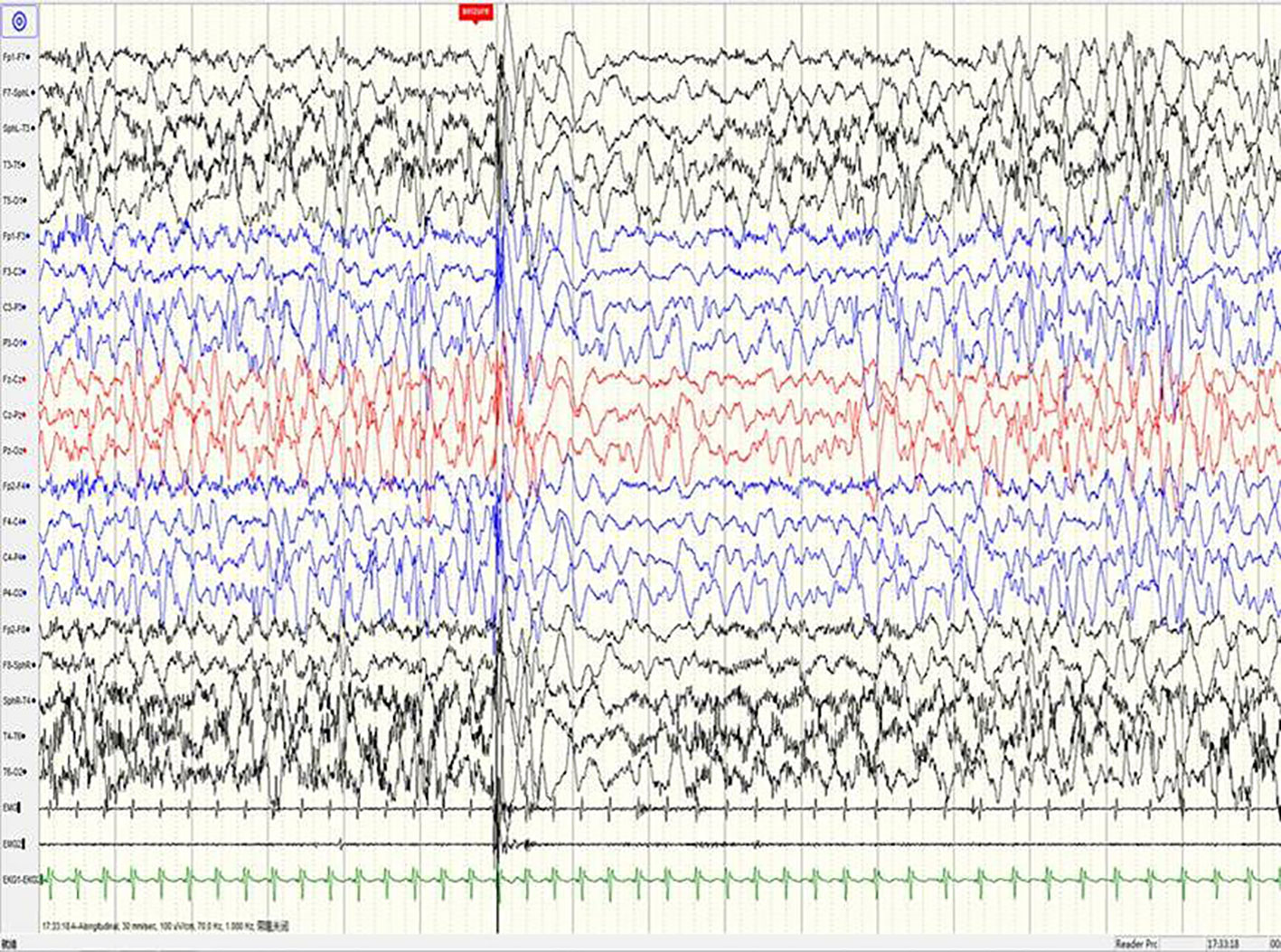
Figure 4 A sample EEG recording of 8-month-old child in this study. The seizures are characterized by left oblique movement of both eyes and limb flexion uplift; the same period of the EEG signals show bilateral lead extensive spike-slow wave and myoelectric outbreak in the bilateral deltoid muscle.
After obtaining informed consent from the parents, whole exome sequencing (WES) of the patient and parental samples were analyzed by a trio-based analysis, which identified a variant, c.135C > A, in the DNM1 gene (NM_004408), with the amino acid changes of p.Ser45Arg (Figure 5), which was confirmed by Sanger sequencing. To the best of our knowledge, this was an unreported de novo mutation. This missense mutation is absent in gnomAD, ExAC, 1000 Genomes, and ESP 6500 databases; moreover, it is predicted to be a disease variant by Polyphen-2 (score of 0.988), MutationTaster (disease causing), SIFT (score of 0.006), and M-CAP (score of 0.979). According to sequence alignment, the Ser45 residue is highly conserved across species, indicating evolutionary importance (Figure 6).
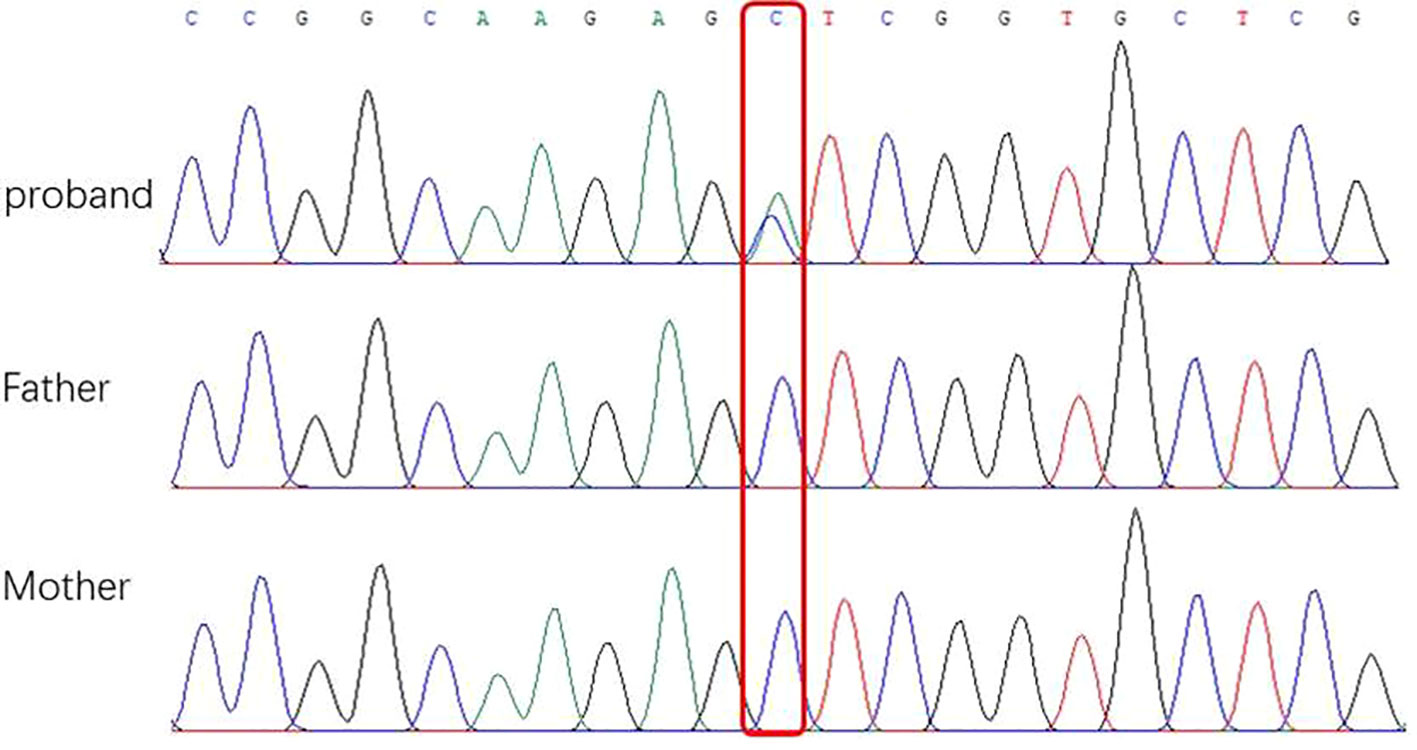
Figure 5 Sequencing of DNM1 gene mutations of children and their parents in this study. (A) Base 135 of the coding sequence of the DNA of a patient in this group shows a missense mutation c.135C > A (p.GluS45R) (arrow). (B) and (C) are the corresponding gene sites in the father and mother, respectively; these sites (arrow) do not show the mutation.
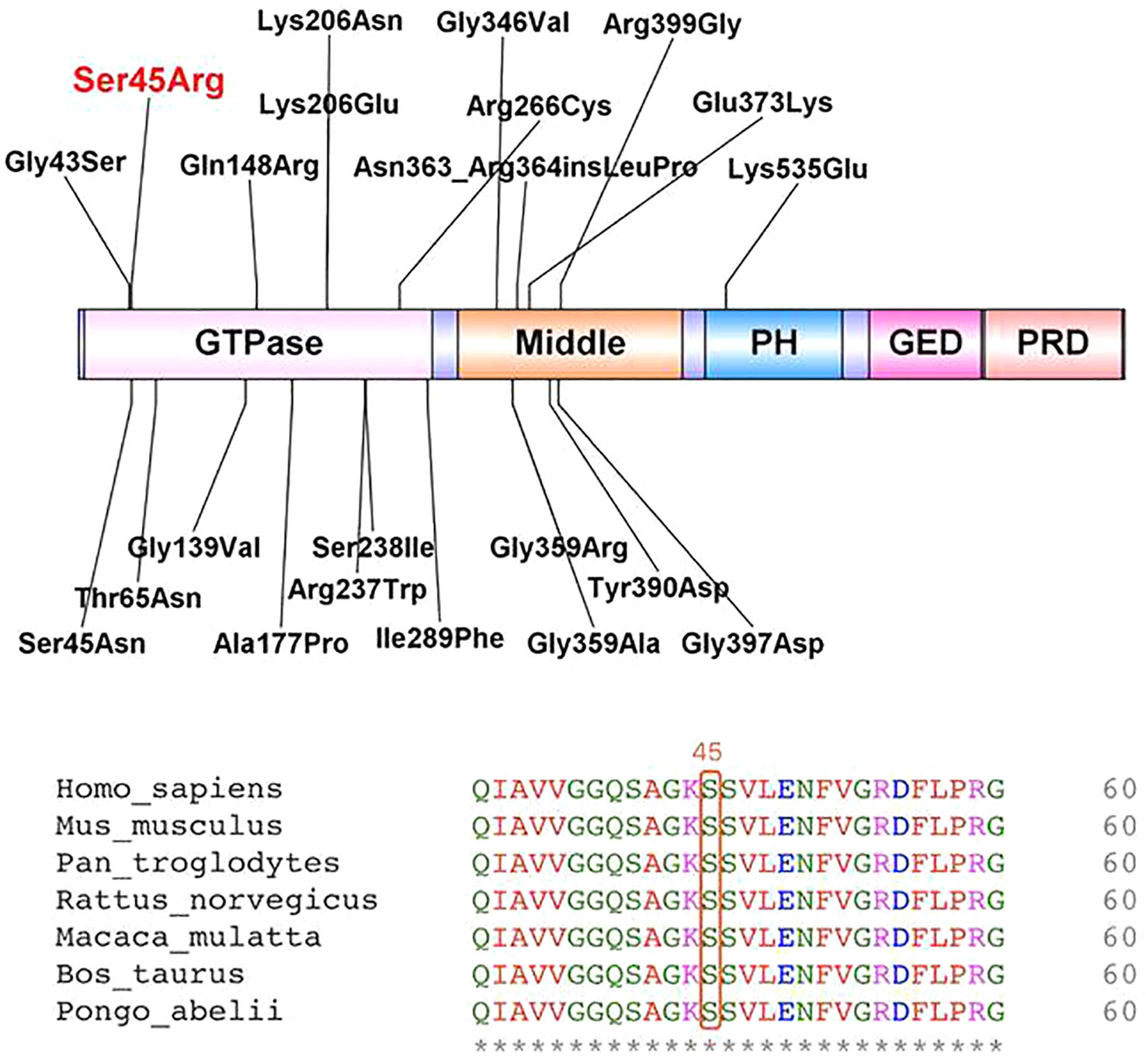
Figure 6 Locations of identified variants in dynamin-1 protein structure (top) and amino acid sequence of the present variant (bottom).
Clinical Characteristics of Patients With Pathogenic DNM1 Variants
Our data showed that patients carrying pathogenic variants in the GTPase or middle domains present with epileptic encephalopathy and severe neurodevelopmental symptoms. For the analysis of DNM1-related encephalopathy, 31 out of the 33 patients were included (9 females, 21 males, the sex of one patient was not available). The age range of the patients was 0.6–24 years, the median age was, at inclusion, 8 years. Pregnancy and delivery were unremarkable in all patients with normal birth parameters. Patient 25 died at 2 years of age, before enrolment in this study. The clinical characteristics of DNM1 mutation-related epileptic encephalopathy patients were analyzed as follows:
Seizures
From the 31 patients with DNM1 mutation-related epileptic encephalopathy, 29 (93.5%) experienced epileptic seizures. Patient 11 did not have seizures, and patient 24 showed only subcortical, nonepileptic myoclonic jerks. Seizures began at a median age of 5 months (range 1 day to 4.5 years). Patient 26 was an outlier with onset at 4.5 years with a febrile infection related epilepsy syndrome phenotype. During the course of the disease, 23 (74.2%) patients had spasm seizures, 12 (38.7%) patients had absence seizures, 9 (29.0%) patients had tonic seizures, 12 (38.7%) patients had myoclonic seizures, 5 (16.1%) patients had atonic seizures, 13 (41.9%) patients had generalized tonic-clonic seizures, and 9 (29.0%) patients had focal seizures (Table 1). Fourteen out of 15 patients (93.3%) presented with infantile spasms initially, whereas 1 patient presented with myoclonic seizures, tonic seizures, generalized tonic-clonic seizures (GTCS), and focal seizures. Information was not available for one patient.
Development
All patients with DNM1 mutation-related epileptic encephalopathy were nonverbal except for two patients, which were not mentioned in literature, with severe to profound intellectual disability. In 24 out of the 31 patients (77.4%), the developmental delay was apparent before seizure onset. Except for six patients who had normal development until the onset of refractory seizures, all patients had considerable developmental delays in the first year of life. Twenty-eight out of the 31 patients (90.3%) were non-ambulatory.
Response to Treatment
Seizure outcome was assessed in 31 patients: 24 out of 31 patients (77.4%) had refractory seizures. Three patients (9.7%) became seizure-free post treatment. Patient 8 became seizure-free after placement on ketogenic diet at the age of 3.5 years while patient 6 had some response to ketogenic diet. Seizures in patients 1, 2, and 5 were controlled with valproic acid, clobazam, or vigabatrin over a period of 5 years between the ages 3 to 8 years.
In addition, patients 31 and 32, carrying pathogenic variants in the pleckstrin homology domain exhibited milder phenotypes without epilepsy. The two girls, 8 years old, were monozygotic triplet sisters who presented for evaluation of developmental delay, autism spectrum disorder, some dysmorphic features, and hypotonia without repeated seizures.
EEG Results
The EEG results of patients 31 and 32 were normal. EEG results were abnormal in 30 out of 31 (96.7%) patients with DNM1 mutation-related epileptic encephalopathy. The EEG patterns of five patients reveal varied epileptiform discharges initiating as hypsarrhythmia, and evolving from slow generalized spike-wave discharges to paroxysmal fast activity. One patient exhibited non-specific background activity. Among the 31 patients, 15 (48.4%) had epileptiform discharge and background slowing. Twenty-eight (90.3%) patients had epileptiform discharge, of which multifocal discharge was the most common. There were 18 (58.1%) patients with multifocal discharge and 14 (45.2%) patients with hypsarrhythmia (Figure 3). Regarding other epileptiform discharges, there were nine (29.0%) patients with slow-spike wave complex, three (9.7%) patients with fast-wave activity, three (9.7%) patients with extensive spike activity, and four (12.9%) patients with focal epileptiform discharge. In addition, there were two (6.5%) patients with multifocal discharge or hypsarrhythmia and slow-spike wave complex.
DNM1 Mutation Results
The current study reviewed data from 33 patients, including 31 sporadic patients and a sibling pair (patients 20 and 21), resulting in a total of 20 independent mutations (Table 2). The most common mutation was c.709C.T (p.Arg237Trp), which was found in 8 out of the 33 independent patients (24.2%). All mutations were confirmed to be de novo, except for the affected sibling pair. Twelve of 20 mutations (60.0%), including the recurrent c.709C.T (p.Arg237Trp) mutation, occurred in the GTPase domain of DNM1.Seven out of the 20 mutations (35.0%) occurred in the middle domain of DNM1. One (5.0%) occurred in the PH domain of DNM1. Twenty novel missense/frame-insertion mutations were predicted as pathogenic using the in silico prediction tools Mutation Taster Server, Polyphen-2, and SIFT.
Comparison of Genetic and Clinical Phenotypes of Children With DNM1 Mutation-Related Encephalopathy
As shown in Table 3, sex (female vs. male, P = 0.5935), age at seizure onset (< 6 months vs. > 12 months vs. 6–12 months, P = 0.4007), seizure type at onset (infantile/epileptic spasms vs. other type, P = 0.5491), seizure outcome (intractable vs. seizure-free, P = 0.1145), and intellectual disability (profound vs. severe, P = 0.5523) showed no significant associations with the GTPase or middle domains.
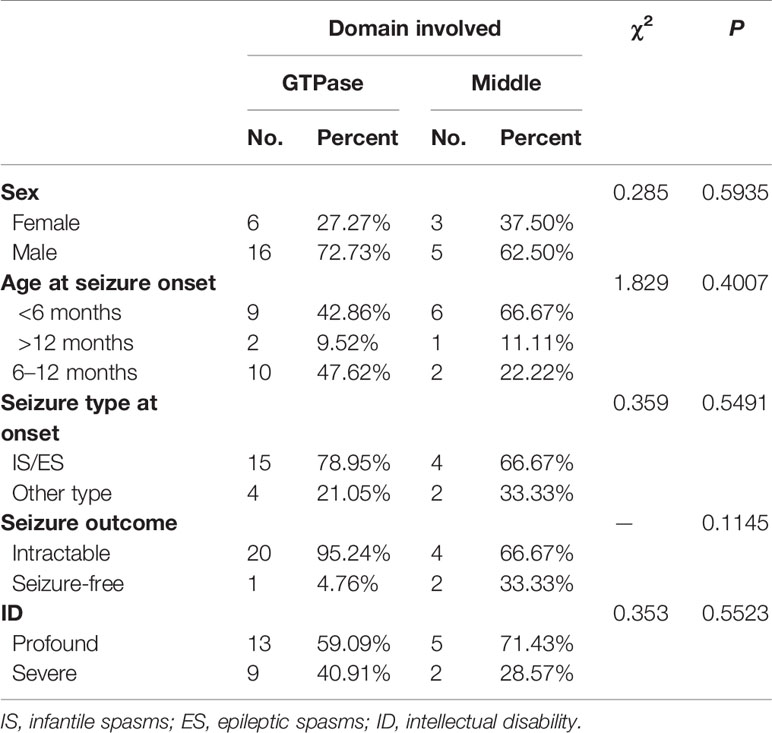
Table 3 Comparison of gene domains and clinical features of 31 cases with mutation-related epileptic encephalopathy.
Discussion
Neurotransmission in the central system relies on synaptic vesicle transport. DNM1 is a protein involved in the synaptic vesicle cycle, which facilitates the exocytosis of neurotransmitters necessary for normal signaling pathways and development in the central nervous system. Dynamin proteins have five domains; the GTPase domain is the largest and best understood, followed by a middle domain, a pleckstrin homology domain, a GTPase effector domain, and a proline-rich domain (McNiven et al., 2000). The pleckstrin homology domain is thought to interact directly with the lipid bilayer. The DNM1 gene is mainly expressed in the central nervous system (Romeu and Arola, 2014), which explains the neurological phenotypes in DNM1-related disorders. Next-generation sequencing has been rapidly implemented into routine clinical practice, where it has improved the diagnostic rate of patients with neuromuscular diseases. The widespread application of next-generation sequencing has greatly facilitated the understanding of the underlying mechanisms of epileptic encephalopathy (Fang et al., 2017; Ni and Shi, 2017). Previous publications have characterized the functional consequences of DNM1 mutations and found that the seizure phenotype is largely due to the deleterious effects of DNM1 mutations in GABAergic interneurons, while behavioral locomotor phenotypes may be due to the effect of the mutation in pyramidal cells (Asinof et al., 2015; Asinof et al., 2016).
Based on the collected mutation pattern and clinical information, we analyzed the relationship between genotypes and phenotypes. Previous research interpreted the molecular mechanisms of DNM1 mutations and inferred the connection between genotypes and phenotypes to certain extent. It has been reported that mutations in different domains lead to distinct clinical phenotypes. Patients carrying pathogenic variants in the GTPase or middle domains present with epileptic encephalopathy and severe neurodevelopmental symptoms. These mutations have been reported in association with early onset epileptic encephalopathy (Appenzellar, 2014), intractable seizures (seizure onset in DNM1 patients ranges from 2–13 months of and usually presents with infantile spasms. The seizure type manifests in various forms as the patient ages, ranging from absence seizures to generalized tonic-clonic seizures.), motor impairments, and severe to profound intellectual disability. In this study, 24 patients (77.4%) had refractory seizures. During the course of the disease, 23 (74.2%) patients initially had spasm seizures; all patients had severe to profound intellectual disability and considerable developmental delay in the first year of life. Other clinical features reported in some affected individuals included hypotonia, developmental regression, movement disorder, autism, cortical visual impairment, behavioral concerns, and microcephaly. Patients carrying pathogenic variants in both domains exhibited comparable phenotypes (Table 3), although the mechanism of protein disruption was distinct from that of patients with variants in a single domain. Most variants in the GTPase domain were predicted to impair hydrolysis of GTP, but not its binding to the synaptic vesicle; this resulted in integrated oligomeric assembly and impaired vesicle scission. However, middle domain variants were predicted to impair the ability of the DNM1 protein to form larger oligomeric assemblies. In the case of the patients, the dominant negative effect of DNM1 results in a generally similar overall phenotype suggesting a similar pathway (von Spiczak et al., 2017). Patients carrying pathogenic variants in the pleckstrin homology domain exhibited milder phenotypes without epilepsy. These patients were 8-year-old identical twin sisters who had no seizures and exhibited mild-to-moderate developmental delay/intellectual disability and autism spectrum disorder (Brereton et al., 2018). The de novo p.Lys535Glu mutation is a likely pathogenic novel variant in exon 15 of DNM1. However, this was reported in a single patient without an epilepsy phenotype. Therefore, reports of additional patients are needed to define the relationship between genotype and phenotype in DNM1 mutation-related epileptic encephalopathy (Jia and Shi, 2017).
EEG is an important tool for assessment of the diagnosis and prognosis of epileptic encephalopathy in patients carrying DNM1 mutations. The EEG patterns are consistent with changes in the electrical activities of the brain in patients with infantile epileptic encephalopathy. The EEG patterns reveal varied epileptiform discharges initiating as hypsarrhythmia and evolving from slow generalized spike-wave discharges to paroxysmal fast activity. In this study of epileptic encephalopathies in 31 patients carrying DNM1 mutations (Table 1), approximately 96.7% of patients' recordings portrayed abnormal EEG; multifocal discharge was most common (58.1%), followed by hypsarrhythmia (45.2%). Other epileptiform discharges were characterized by slow-spike and slow-wave complex, fast-wave activity, extensive spike activity, and focal epileptic discharge. The results of this study were consistent with those of a retrospective study published in 2017. The specific EEG pattern remains the basis for the diagnosis. Series EEG with video and video EEG with electromyogram electrodes is also recommended. The association of characteristic multiple seizure types and intellectual disability represents the classic hallmark of Lennox-Gastaut syndrome. This diagnostic triad may not be completely present at the onset of seizures; therefore, an accurate diagnosis of Lennox-Gastaut syndrome often requires further disease development over time (Markand, 2003; Arzimanoglou et al., 2009; Camfield, 2011; Bourgeois et al., 2014). The patient in the present case underwent a series of video EEG monitoring, which initially showed focal discharge, followed by atypical hypsarrhythmia and infantile spasms; this suggested evolution of the disease and provided clues for diagnosis and treatment.
The long-term outcomes of patients with DNM1 mutation-related epileptic encephalopathy were often disappointing. The choice of AEDs at the onset of seizures was tailored to seizure type, clinical presentation, and EEG pattern. Thus far, there are no international guidelines for the pharmacological treatment of DNM1 mutation-related epileptic encephalopathy because of the limited efficacy of antiepileptic medications. In the present study, three patients had been given sodium valproate and became seizure-free; however, as the disease progressed, they developed drug-refractory epilepsy. Therefore, we analyzed differences in the response of the same gene mutation to drug treatment. First, we speculated that the choice of treatment time or the natural process of disease might influence the response. Then, we investigated whether the type and site distributions of DNM1 gene mutations were associated with clinical phenotype, potentially providing clues for clinical diagnosis and treatment. Eight patients carried the p.Arg237Trp mutation (Tables 1 and 2). Given that DNM1 mutations are present in up to 2% of patients with severe epilepsy (Kolnikova et al., 2018), this mutation is particularly frequent in patients with epileptic encephalopathy. The relatively homogeneous phenotype and predicted dominant-negative mechanism of this mutation make DNM1-associated encephalopathy has the potential of being an effective therapeutic target. Gene therapy might also be an effective means to restore DNM1 function (Kolnikova et al., 2018). We presume that treatment methods and strategies will be further refined with additional studies involving more patients and investigations into the molecular basis of the disease.
In conclusion, to the best of our knowledge, this is the first integrated analysis of the phenotypic, genetic, and electroencephalographic features of children with DNM1 mutation-related encephalopathy. Our study highlighted the role of series EEG and video EEG of children with DNM1 mutation-related encephalopathy; EEG patterns may aid in providing clues for treatment.
There were several limitations to this study, such as the fact that it is a retrospective and summary study. Due to the small number of cases, we have not come to a definite conclusion; the pathogenic variant in this study needs to be confirmed by functional experiments. To determine the association of phenotype and genotype of children with DNM1 mutation-related encephalopathy, further analysis of additional patients is needed.
Ethics Statement
The present study was approved by the Ethics Committee of Beijing Children's Hospital and the patient have gave written informed consent.
Author Contributions
All authors contributed to the study design, critically reviewed the manuscript, and approved the final version. HL performed literature search and analysis, and wrote the manuscript. MX, ZL, JZ, XiaohW, XiaofW, and TH performed literature search and analysis. FF revised the manuscript.
Funding
This work was supported by National Natural Science Foundation of China(81541115), the Capital Health Research and Development Fund(2018-2-2096) and Beijing Municipal Administration of Hospitals Incubating Program(PX2017065).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Editage (http://editage.com/frontiers/) for editing a draft of this manuscript. We also acknowledge the financial support of the Open Access Publication Fund of Beijing Children's Hospital.
References
Allen, A. S., Berkovic, S. F., Cossette, P., Delanty, N., Dlugos, D., Eichler, E. E., et al. (2013). De novo mutations in epileptic encephalopathies. Nature 501, 217–221. doi: 10.1038/nature12439
Allen, N. M., Conroy, J., Shahwan, A., Lynch, B., Correa, R. G., Pena, S. D., et al. (2016). Unexplained early onset epileptic encephalopathy: exome screening and phenotype expansion. Epilepsia 57, e12–e17. doi: 10.1111/epi.13250
Appenzellar (2014). De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am. J. Hum. Genet. 95, 360–370. doi: 10.1016/j.ajhg.2014.08.013
Arzimanoglou, A., French, J., Blume, W. T., Cross, J. H., Ernst, J. P., Feucht, M., et al. (2009). Lennox-Gastaut syndrome: a consensus approach on diagnosis, assessment, management, and trial methodology. Lancet Neurol. 8, 82–93. doi: 10.1016/S1474-4422(08)70292-8
Asinof, S. K., Sukoff Rizzo, S. J., Buckley, A. R., Beyer, B. J., Letts, V. A., Frankel, W. N., et al. (2015). Independent neuronal origin of seizures and behavioral comorbidities in an animal model of a severe childhood genetic epileptic encephalopathy. PloS Genet. 11, e1005347. doi: 10.1371/journal.pgen.1005347
Asinof, S., Mahaffey, C., Beyer, B., Frankel, W. N., Boumil, R. (2016). Dynamin 1 isoform roles in a mouse model of severe childhood epileptic encephalopathy. Neurobiol. Dis. 95, 1–11. doi: 10.1016/j.nbd.2016.06.014
Belousova, E. D., Zavadenko, N. N., Kholin, A. A., Sharkov, A. A. (2017). New classifications of epilepsies and seizure types created by the International League against Epilepsy (2017). Zh. Nevrol. Psikhiatr. Im. S. S. Korsakova 117, 99–106. doi: 10.17116/jnevro20171177199-106
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The Protein Data Bank. Nucleic Acids Res. 28, 235–242. doi: 10.1093/nar/28.1.235
Bourgeois, B. F., Douglass, L. M., Sankar, R. (2014). Lennox-Gastaut syndrome: a consensus approach to differential diagnosis. Epilepsia 55 Suppl 4, 4–9. doi: 10.1111/epi.12567
Brereton, E., Fassi, E., Araujo, G. C., Dodd, J., Telegrafi, A., Pathak, S. J., et al. (2018). Mutations in the PH Domain of DNM1 are associated with a nonepileptic phenotype characterized by developmental delay and neurobehavioral abnormalities. Mol. Genet. Genomic Med. 6, 294–300. doi: 10.1002/mgg3.362
Camfield, P. R. (2011). Definition and natural history of Lennox-Gastaut syndrome. Epilepsia 52 Suppl 5, 3–9. doi: 10.1111/j.1528-1167.2011.03177.x
Chappie, J. S., Acharya, S., Leonard, M., Schmid, S. L., Dyda, F. (2010). G domain dimerization controls dynamin's assembly-stimulated GTPase activity. Nature 465, 435–440. doi: 10.1038/nature09032
Deng, X. L., Yin, F., Zhang, C. L., Ma, Y. P., He, F., Wu, L. W., et al. (2016). [Dynamin-1-related infantile spasms: a case report and review of literature]. Zhonghua Er. Ke. Za. Zhi. 54, 856–859. doi: 10.3760/cma.j.issn.0578-1310.2016.11.014
Fang, F., Liu, Z., Fang, H., Wu, J., Shen, D., Sun, S., et al. (2017). The clinical and genetic characteristics in children with mitochondrial disease in China. Sci. China Life Sci. 60, 746–757. doi: 10.1007/s11427-017-9080-y
Farwell, K. D., Shahmirzadi, L., El-Khechen, D., Powis, Z., Chao, E. C., Tippin, D. B., et al. (2015). Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet. Med. 17, 578–586. doi: 10.1038/gim.2014.154
Jia, J., Shi, T. (2017). Towards efficiency in rare disease research: what is distinctive and important? Sci. China Life Sci. 60, 686–691. doi: 10.1007/s11427-017-9099-3
Kolnikova, M., Skopkova, M., Ilencikova, D., Foltan, T., Payerova, J., Danis, D., et al. (2018). DNM1 encephalopathy—atypical phenotype with hypomyelination due to a novel de novo variant in the DNM1 gene. Seizure 56, 31–33. doi: 10.1016/j.seizure.2018.01.020
Lazzara, A., Asghar, S., Zacharia, T., Byler, D. (2018). DNM1 Mutation in a child associated with progressive bilateral mesial temporal sclerosis. Clin. Case Rep. 6, 2037–2039. doi: 10.1002/ccr3.1793
Markand, O. N. (2003). Lennox-Gastaut syndrome (childhood epileptic encephalopathy). J. Clin. Neurophysiol. 20, 426–441. doi: 10.1097/00004691-200311000-00005
McNiven, M. A., Cao, H., Pitts, K. R., Yoon, Y. (2000). The dynamin family of mechanoenzymes: pinching in new places. Trends Biochem. Sci. 25, 115–120. doi: 10.1016/S0968-0004(99)01538-8
Nakashima, M., Kouga, T., Lourenco, C. M., Shiina, M., Goto, T., Tsurusaki, Y., et al. (2016). De novo DNM1 mutations in two cases of epileptic encephalopathy. Epilepsia 57, e18–e23. doi: 10.1111/epi.13257
Ni, X., Shi, T. (2017). The challenge and promise of rare disease diagnosis in China. Sci. China Life Sci. 60, 681–685. doi: 10.1007/s11427-017-9100-1
Reubold, T. F., Faelber, K., Plattner, N., Posor, Y., Ketel, K., Curth, U., et al. (2015). Crystal structure of the dynamin tetramer. Nature 525, 404–408. doi: 10.1038/nature14880
Romeu, A., Arola, L. (2014). Classical dynamin DNM1 and DNM3 genes attain maximum expression in the normal human central nervous system. BMC Res. Notes 7, 188. doi: 10.1186/1756-0500-7-188
Keywords: epileptic encephalopathy, dynamin-1, mutation, electroencephalogram, children
Citation: Li H, Fang F, Xu M, Liu Z, Zhou J, Wang X, Wang X and Han T (2019) Clinical Assessments and EEG Analyses of Encephalopathies Associated With Dynamin-1 Mutation. Front. Pharmacol. 10:1454. doi: 10.3389/fphar.2019.01454
Received: 03 January 2019; Accepted: 13 November 2019;
Published: 04 December 2019.
Edited by:
Tieliu Shi, East China Normal University, ChinaReviewed by:
Alberto Spalice, Policlinico Umberto I, ItalyLi Zhang, East China Normal University, China
Copyright © 2019 Li, Fang, Xu, Liu, Zhou, Wang, Wang and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang Fang, MTM5MTAxNTAzODlAMTYzLmNvbQ==