 Zhiwei Shao
Zhiwei Shao Sheng Tu2
Sheng Tu2 Anwen Shao
Anwen Shao- 1Department of Hepatobiliary and Pancreatic Surgery, Department of Surgery, Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 2Department of Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
- 3Department of Neurosurgery, Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China
Intracerebral hemorrhage (ICH) is a subtype of hemorrhagic stroke with high mortality and morbidity. The resulting hematoma within brain parenchyma induces a series of adverse events causing primary and secondary brain injury. The mechanism of injury after ICH is very complicated and has not yet been illuminated. This review discusses some key pathophysiology mechanisms in ICH such as oxidative stress (OS), inflammation, iron toxicity, and thrombin formation. The corresponding therapeutic targets and therapeutic strategies are also reviewed.
Introduction
Hemorrhagic stroke, including intracerebral hemorrhage (ICH) and subarachnoid hemorrhage (SAH), is associated with high mortality and morbidity (Keep et al., 2012). Despite significant progress in clinical treatment, the incidence of hemorrhagic stroke has not declined (van Asch et al., 2010). At present, although hematoma can be removed surgically to relieve the mechanical compression of peripheral brain tissue, there are still no efficient treatments for secondary brain injuries (SBIs) such as oxidative stress (OS), inflammatory response, neuronal apoptosis, and thrombin formation caused by hemorrhage (Hu et al., 2016; Zheng et al., 2016; Zhang et al., 2017; Zeng et al., 2018). These mechanisms are related to each other, but they have not been fully illustrated. Obviously, a multi-targeted neuroprotective compound would be a promising strategy to alleviate brain injury after ICH. In this review, we aim to summarize the essential mechanisms and pathological progress after ICH and to comment on potential therapeutic targets.
Primary Injury
The initial pathological damage of cerebral hemorrhage to brain is the mechanical compression caused by hematoma. The hematoma mass can increase intracranial pressure, compressing brain and thereby potentially affecting blood flow, and subsequently leading to brain hernia (Keep et al., 2012). Subsequently, brain hernia and brain edema cause secondary injury, which may be associated with poor outcome and mortality in ICH patients (Yang et al., 2016). Unfortunately, the common treatment of brain edema (steroids, mannitol, glycerol, and hyperventilation) cannot effectively reduce intracranial pressure or prevent secondary brain injury (Cordonnier et al., 2018).
Secondary Brain Injury
Inflammation
Substantial evidence indicates that inflammatory mechanisms are associated with ICH-induced brain injury and microglia/macrophages activation, and polarization is thought to play vital pathophysiological roles (Wan et al., 2016; Koh et al., 2018). Under physiological conditions, microglia/macrophages monitor the surrounding microenvironment and maintain the stability of neurons, blood–brain barrier (BBB) and matrix. When cerebral hemorrhage occurs, it is activated rapidly. Excessive microglia/macrophages will release a large number of inflammatory factors and induce inflammatory waterfall reaction, which will eventually lead to pathological changes such as BBB injury, edema, cell death, and so on (Bhatia et al., 2016). In the early stage of ICH, activated microglia have been proven to produce pro-inflammatory factors and are associated with neurologic functional damage, and inhibiting microglial activation can decrease brain injury and edema (Wu et al., 2008; Scott et al., 2018). After microglia/macrophages activation, there are two types of cells, including classically activated microglia/macrophages (M1 phenotype) and alternative activated microglia/macrophages (M2 phenotype) (Xiong et al., 2016). In the early phase of ICH, microglia/macrophages can be activated by various components in the blood and readily turn into the M1 phenotype. M1 express a large number of toll like receptor 4 (TLR4) and heme oxygenase 1 (HO-1) to clear the hematoma, but they also produce proinflammatory mediators [interleukin (IL)-1β, IL-6, IL-12, IL-23, and tumor necrosis factor alpha (TNF-α)], iron content, and oxidative metabolites, which aggravate brain injury (Varnum and Ikezu, 2012; Ponomarev et al., 2013; Scott et al., 2018). M2 can improve brain recovery by secreting IL-10, CD36, and transforming growth factor-β (TGF-β) to clear cell debris and reduce inflammation, and are associated with tissue remodeling (Pan et al., 2015; Xia et al., 2015; Zheng et al., 2016; Zhang et al., 2018). Therefore, it indicates that promoting M2 phenotype and inhibiting M1 phenotype are beneficial to brain recovery after ICH. Research revealed that pinocembrin can reduce the number of M1-like microglia without affecting M2-like microglia in the surrounding area (Lan et al., 2017). A study showed that advanced glycation end products (AGEs) could cause non-specific neuroinflammation by activating the RAGE/Rho/ROCK pathway and inhibition of RAGE/ROCK not only avoids polarization of pro-inflammatory macrophages (M1) but also promoted shifting of M1 phenotype to M2 (Chen et al., 2017). Furthermore, there are several studies that show activated microglia express high levels of TLR4, which is closely related to neuroinflammation, infiltration of leukocytes, production of cytokines, and chemokines after ICH (Sansing et al., 2011; Lin et al., 2012; Gang et al., 2018). In experimental ICH, blockade of TLR4 reduced neuronal loss and edema formation and improved neurological function (Lin et al., 2012). What’s more, the TLR4 inhibitor ethyl (6R)-6-[N-(2-chloro-4-fluorophenyl) sulfamoyl]-cyclohex-1-ene-1-carboxylate (TAK-242) can promote hematoma resolution and attenuate neurological deficit (Fang et al., 2014). TLR4 is stimulated by exogenous or endogenous ligands through two main signaling pathways, myeloid differentiation factor 88 (MyD88)-dependent pathway and MyD88-independent pathway. One study has showed that MyD88 is the main signaling pathways of TLR4-induced inflammatory reaction (Gang et al., 2018). The intracellular domain of TLR4 binds to the carboxyl terminal of downstream MyD88 or Toll/IR-1 domain containing adaptor protein inducing interferon-beta (TRIF), activates downstream NF-κB induced kinase (NIK), and finally activates NF-κB, which promotes microglial secretion of TNF-α, IL-1β, and IL-6 (Pearson et al., 1999; Vezzani et al., 1999; Kong and Le, 2011; Gang et al., 2018). Lin et al. (2012) have confirmed that the secretion of inflammatory factors and microglia/macrophages infiltration is reduced after ICH in MyD88 gene knockout mice. TNF-α produced by microglia/macrophages plays a central role in neuronal damage after brain injury (Lambertsen et al., 2005; Rodriguez-Yanez and Castillo, 2008). The ICH-induced brain edema is significantly decreased in TNF-α knockout mice compared to wild-type mice, and another set of studies show that treatment with TNF-α antibody after ICH can inhibit microglia/macrophage activation and lead to less brain edema and better recovery of neurological function (Hua et al., 2006; Behrouz, 2016). Another study showed that although TNF-α inhibitors can reduce the degree of brain edema, inflammation, and neurologic impairment, they do not alter hematoma volume (Lei et al., 2013). IL-1β produced by microglia/macrophages is also considered a key mediator of neuronal injury; some studies show that neuroprotection is associated with downregulation of IL-1β (Wu et al., 2010; Bimpis et al., 2015). To sum up, inflammation mediated by TLR4 signaling pathway lead to brain injury after ICH, which provides us with a potential therapeutic target of ICH. We may use TLR4 antagonists and some negative regulators of TLR4 signal pathways or inhibit inflammatory factors to intervene with TLR4 downstream signal transduction, so as to treat ICH and improve the prognosis. However, it is noteworthy that microglia/macrophage-mediated phagocytosis is advantaged for tissue repair and functional recovery (Wang and Tsirka, 2005; Zhao et al., 2007). CD36 is a class II scavenger receptor of microglia/macrophages, which is closely related to phagocytosis (Flores et al., 2016). A study showed that CD36 expression was increased in perihematomal tissues in mice after ICH, while the absorption of hematoma was decreased in CD36(−/−) mice (Fang et al., 2014). Treating animals with peroxisome proliferator-activated receptor gamma (PPARγ) agonists (e.g., rosiglitazone, pioglitazone, or 15d-PGJ2) increased CD36 expression levels and enhanced hematoma resolution after ICH (Flores et al., 2016). Therefore, in addition to blocking microglia-mediated inflammatory response, promoting phagocytosis of microglia is also a therapeutic direction. However, the microglia that ingest more than two red blood cells (RBCs) lead to the release of harmful heme and iron into the extracellular matrix (Kondo et al., 1988). Prostaglandin (PG) E2-mediated inflammatory response is involved in ICH-induced secondary brain injury (Mohan et al., 2012). PGE2 has four different G-protein-coupled receptor subtypes known as EP1–EP4. Compared with the toxicity of the EP1/EP3 receptor, the EP2 receptor has neuroprotection after ICH (Mohan et al., 2012; Wu et al., 2017). Previous study found that misoprostol, an ep2/ep4 receptor agonist, can protect the brain from ICH damage (Wu et al., 2015). In addition, the synthesis of PGE2 is catalyzed by cyclooxygenase and PGE2 synthase. Celecoxib is a selective inhibitor of COX-2 that can reduce ICH-induced brain damage (Shao et al., 2019). However, excessive suppression of inflammation after ICH not only increases the risk of infection but also hinders tissue repair and hematoma clearance (Tapia-Perez et al., 2016). Therefore, regulating the balance between pro-inflammatory and anti-inflammatory responses is the pivotal point (Figure 1).
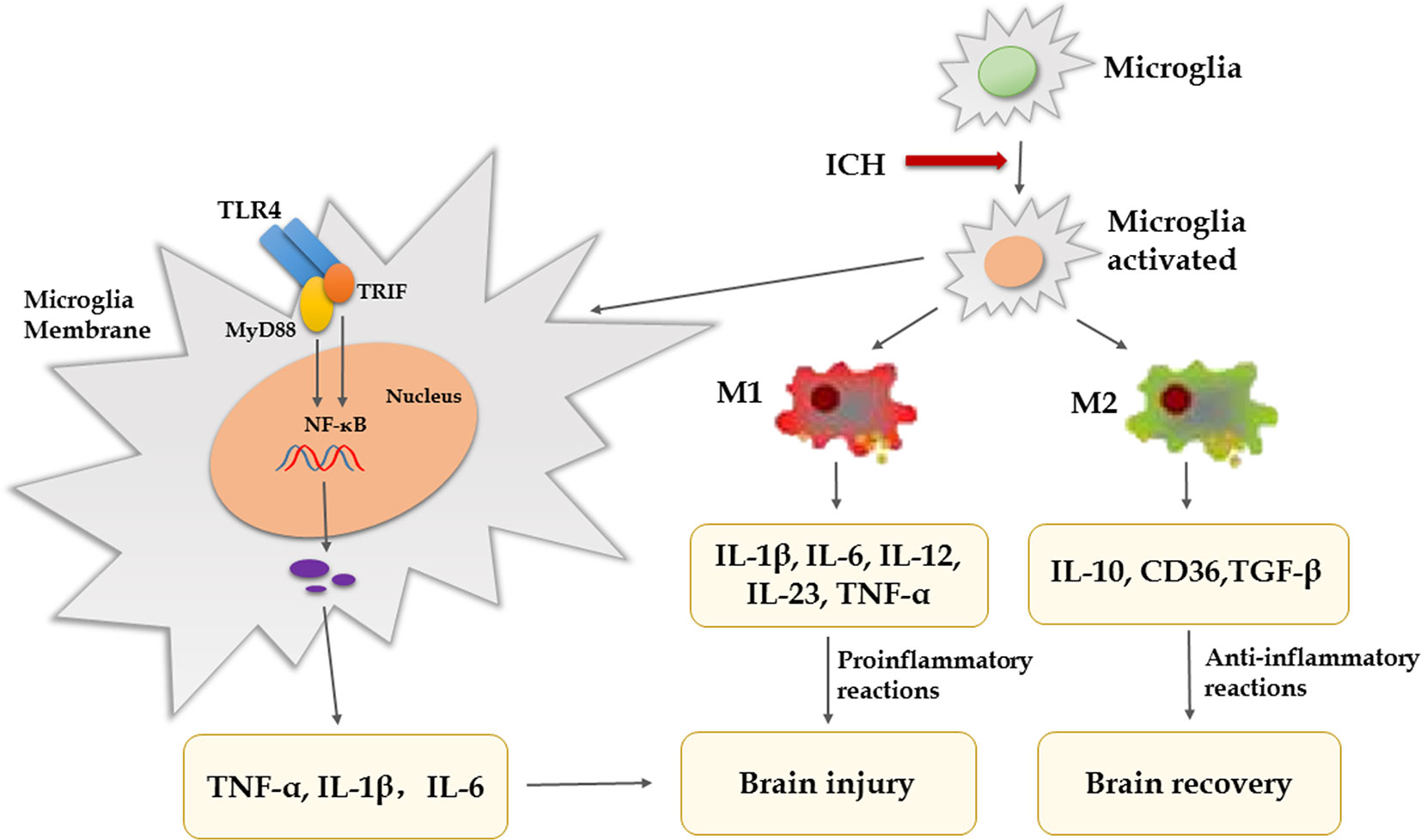
Figure 1 Pro- and anti-inflammatory cytokines in secondary brain injury after ICH.
Oxidative Stress
OS has been increasingly recognized as a contributing factor in secondary brain injury (SBI) following ICH. OS is a condition in which there is an overproduction of free radicals, mainly reactive oxygen species (ROS), and it is involved in various important stages of pathophysiological response during ICH (Aronowski and Zhao, 2011). During physiological conditions, the organism relies on the free radical scavenging system to maintain a relative balance between the production and scavenging of free radicals. However, once the oxygen free radicals are overproduced or the clearance is weakened, it will lead to cell damage or death (Duan et al., 2016). Moreover, the central nervous system consumes more oxygen, while its endogenous antioxidant defense capacity is lower than other organs, which makes it more susceptible to OS (Liu et al., 2019). Firstly, the inflammation after ICH can produce large numbers of free radicals, which can lead to brain injury (Keep et al., 2012; Duan et al., 2016; Zheng et al., 2016). During the inflammatory response following ICH, activation of neutrophils leads to outbreak of the respiratory chain, releasing a lot of ROS and nitric oxide, and the superoxide dismutase (SOD) is consumed in large quantities to eliminate free radicals, which ultimately results in excessive lipid peroxidation (Yu et al., 2014). Excessive oxidation of lipids alters the physical properties of cellular membranes and can cause covalent modification of proteins and nucleic acids, leading to brain injury (Gaschler and Stockwell, 2017). Secondly, blood cell decomposition products such as iron ions and heme can directly lead to brain damage by producing a lot of free radicals (Wagner et al., 2003; Li et al., 2017). As a scavenger for oxygen free radicals and antioxidant, edaravone can relieve oxidative damage of neurons by scavenging free radicals and inhibiting lipid peroxidation in mice (Lu et al., 2012; Wu et al., 2014). In a published clinical trial, edaravone significantly improved the NIHSS score in patients with ICH after removal of hematoma with minimally invasive surgery (Zhao and Liu, 2014). Though its neuroprotective function has been confirmed in some preclinical and clinical studies, the effect of edaravone in ICH remains unclear because of a lack of multicenter, randomized, double-blind clinical trials (Yang et al., 2015). Some studies have shown that the nuclear transcription factor Keap1/Nrf2 pathway is the core regulatory hub of antioxidant defense system and plays an important role in the repair of SBI such as cerebral hemorrhage and ischemia (Dang et al., 2012). ROS can activate the Keap1/Nrf2 pathway to counteract oxidative damage after ICH as an adaptive response. When the Nrf2 gene was knocked out, the ICH model of mice showed increased hemorrhage volume, leukocyte infiltration, ROS production, and DNA damage. However, when Nrf2 was overexpressed, the abovementioned manifestations were significantly alleviated (Wang et al., 2007; Zhao et al., 2007). Gene silencing of Nrf2 aggravated brain edema and neuronal degeneration. Isoliquiritigenin (ILG) is a flavonoid with a chalcone structure, which can relieve early brain injury and neurological deficits by activating the Nrf2-mediated antioxidant system (Zeng et al., 2017). Moreover, it has been reported recently that nicotinamide mononucleotide treatment significantly reduced brain edema, brain cell death, OS, neuroinflammation, intercellular adhesion molecule-1 expression, microglia activation, and neutrophil infiltration in brain hemorrhagic area by promoting the activation of Nrf2 signaling pathway and inhibiting OS (Wei et al., 2017). PPARγ agonists also have been reported to function as an anti-oxidant by activating the Nrf2 pathway and increasing catalase and SOD (Zhao et al., 2015). In conclusion, antioxidant therapy is a worthy and promising ICH treatment direction (Figure 2).
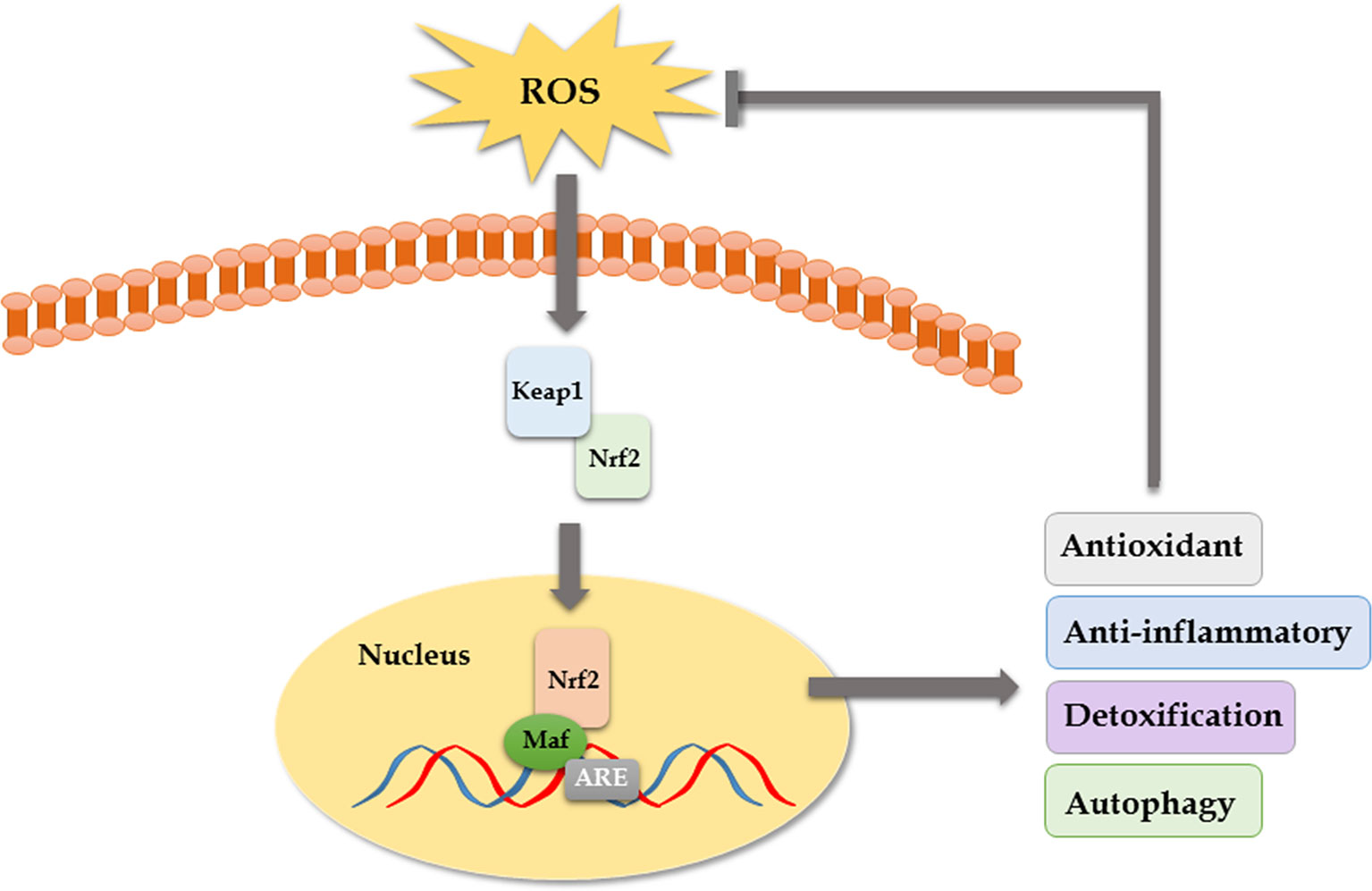
Figure 2 Mechanisms of erythrocyte lysates and thrombin in secondary brain injury after ICH.
Cytotoxicity of Erythrocyte Lysates
Increasing evidence suggests that hemoglobin and iron release from the hematoma is a major contributor to brain injury induced by ICH (Zhang et al., 2017). After cerebral hemorrhage, a lot of RBCs that contain large numbers of hemoglobin are released into the brain’s parenchyma, and RBCs will be broken down 24 h later, resulting in hemoglobin disintegrating into heme and iron (Wagner et al., 2003). Experiments have shown that infusion of dissolved RBCs into rat striatum results in increased brain water content and neurological injury (Wu et al., 2002). Infusion of hemoglobin and hemin can promote the inflammatory response of brain injury (Huang et al., 2002; Wang et al., 2014). The mechanism of brain injury produced by erythrocyte lysates is multifaceted, and researchers have found that there are four main aspects: inflammation, oxidation, nitric oxide scavenging, and edema. Firstly, overaccumulation of iron can do harm to the brain, and HO-1, the initial enzyme and rate-limiting enzyme of heme metabolism, will be expressed increasingly after ICH, which can exacerbate brain injury by promoting microglial activation and iron deposition (Lin et al., 2012; Zhang et al., 2017). However, recent studies also showed that hemin-induced HO-1 expression in perivascular cells before ICH can attenuate BBB disruption after ICH (Lu et al., 2014). Although the role of HO-1 in ICH has been extensively studied, the effect of HO-1 in ICH remains controversial. Secondly, another mechanism by which iron might cause tissue injury is the generation of free radicals. It is suggested that divalent iron ions can react with lipid to produce ROS and lipid ROS, leading to neurological damage and oxidative brain injury (Katsu et al., 2010; Li et al., 2017). Therefore, the iron chelator deferoxamine mesylate (DFX) is a promising candidate for ICH patients (Yeatts et al., 2013). DFX can cross the BBB and chelate iron ions and generate a stable complex with ferric iron; it also reduces the production of free radicals (Yeatts et al., 2013; Zeng et al., 2018). According to the mechanisms mentioned above, DFX decreases OS and neuronal death and improves functional outcome after ICH (Zhang et al., 2017). However, others claim that, although DFX can diminish total parenchymal iron levels, it does not alleviate damage or improve neurological function (Auriat et al., 2012). Thirdly, nitric oxide is depleted rapidly by hemoglobin, which produces microthrombosis in cerebral vessels in SAH and leads to brain damage (Bulters et al., 2018). Nitric oxide donors have turned out to be beneficial for ICH and SAH (Oldfield et al., 2013). Finally, there are evidences that hemoglobin and its decomposition products are main causes of edema (Wang et al., 2018). Previous study has shown that autologous blood injection will result in BBB disruption and brain edema in the rat model (Yuan et al., 2019). It has recently been demonstrated that estrogen reduces ferrous iron toxicity in vivo and in vitro, indicating that estrogen may be a potential therapeutic drug in ICH (Gu et al., 2010). Some animal experiments showed that administration of ferrostatin-1 can reduce iron deposition induced by hemoglobin and prevent neuronal death in organotypic hippocampal slice cultures (OHSCs) by inhibiting lipid ROS and COX-2 expression (Wu et al., 2011; Li et al., 2017; Zille et al., 2017). Haptoglobin–hemoglobin–CD163 acts as a main pathway in hemoglobin scavenging after ICH. A study has shown that PPAR-agonist significantly reduces hematoma volume, brain edema, and hemoglobin by promoting this signal pathway after ICH onset (Wang et al., 2018). All in all, reducing iron accumulation and erythrocyte lysate toxicity needs further study; however, continues to be an important direction in the treatment of ICH patients (Figure 3).
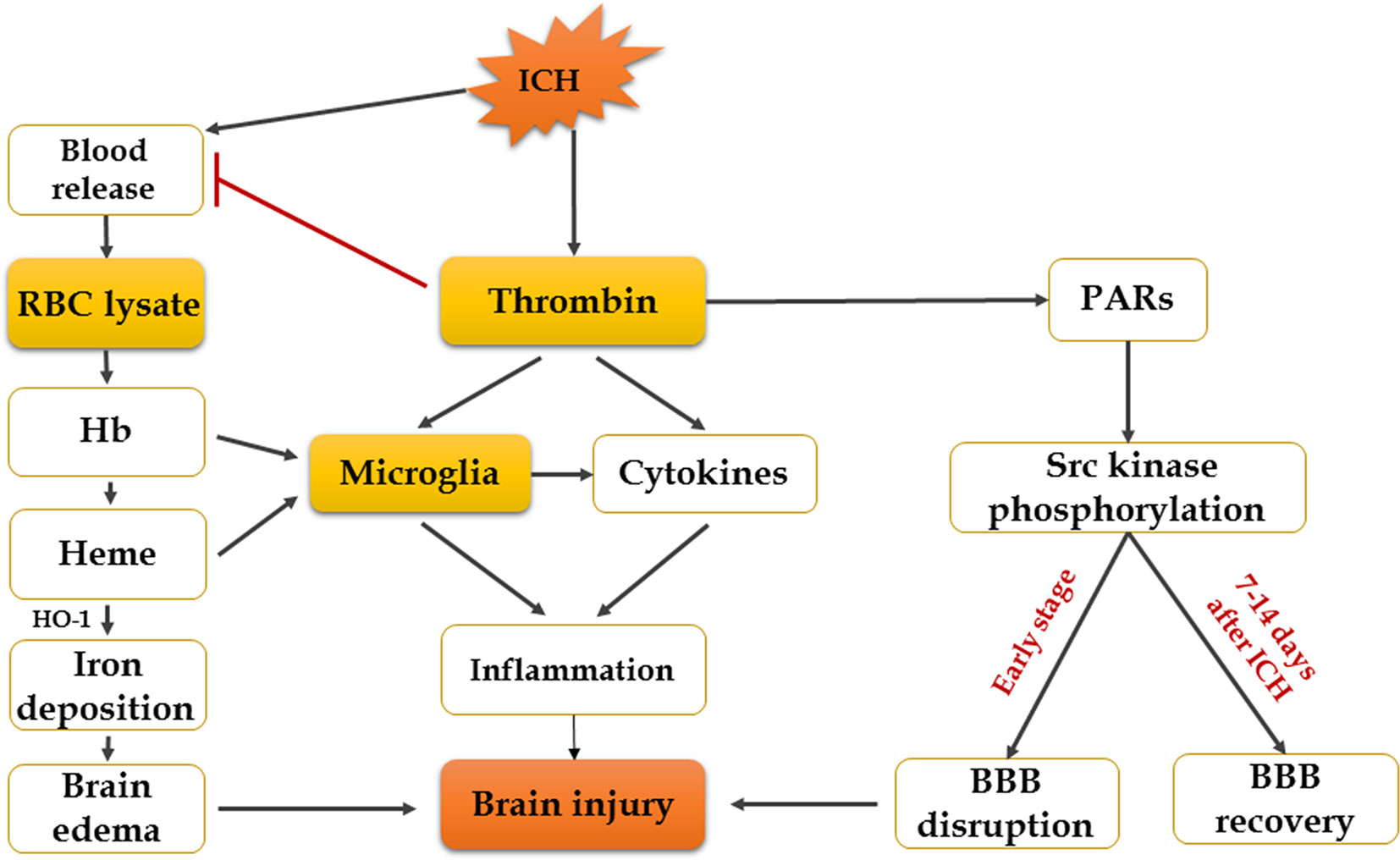
Figure 3 The Keap1–Nrf2–ARE pathway. Keap1 is an OS sensor and negatively regulates Nrf2. Once exposed to ROS, the activated Nrf2 translocates to the nucleus, binds to antioxidant response element (ARE), heterodimerizes with one of the small Maf (musculoaponeurotic fibrosarcoma oncogene homolog) proteins, and enhances the upregulation of cytoprotective, antioxidant, anti-inflammatory, and detoxification genes that mediate cell survival.
Neurotoxicity of Thrombin
Thrombin is an essential component in the clotting cascade, and it is produced in the brain immediately after ICH induction (Zheng et al., 2016). However, thrombin can also participate in ICH-induced injury. The deleterious or protective effect of thrombin depends on its concentration (Zhu et al., 2019). It may provide neuroprotective effects against OS and ischemic injury at very low concentrations, while direct infusion into the brain of large doses of thrombin causes inflammatory cells to infiltrate the brain, proliferation of mesenchymal cells, formation of scar tissue and brain edema, and seizures (Xi et al., 2003). In the cleavage of fibrinogen to fibrin, the effects of thrombin can be mediated by non-receptors, while in the activation of p44/42 mitogen activated protein kinase, it is receptor-mediated, which contains three protease-activated receptors (PARs): PAR-1, PAR-3, and PAR-4. Protease-activated receptors, especially PAR-1, are closely related to brain damage after ICH (Guan et al., 2004; Cheng et al., 2014). Thrombin can also activate Src kinase, which might contribute to the disruption of BBB and formation of edema through PARs (Liu et al., 2010). From the above, it proves that inhibition of thrombin, PAR-1 activation, or Src kinase is a potential effective way to relieve secondary injury after ICH. In fact, a thrombin inhibitor, argatroban, has been proven to exhibit a significant neuroprotective effect and suppress the pathological progression following ICH by preventing thrombin cytotoxicity in vitro (Ohnishi et al., 2007). In addition, thrombin-induced injury to the BBB after ICH can be blocked by acute administration of hirudin (a direct peptide mimetic thrombin inhibitor) (Liu et al., 2010). However, thrombin inhibitors may not be an assured form of treatment, because these might affect clotting and hemostatic functions (Zheng et al., 2016). It is becoming clear that the balance between the hemostatic and pro-hemorrhagic actions of thrombin is likely dependent on multiple factors such as site (intra- or extravascular) and mode of action (activation of which type of receptor) and so on (Cheng et al., 2014). Therefore, it is critical to block the neurotoxic effects of thrombin without inhibiting its hemostasis effect. A study suggested that acute administration of the Src inhibitor PP2 can block the thrombin pathway and reduce brain edema following ICH without affecting coagulation (Liu et al., 2010; Liu and Sharp, 2011). Another set of studies showed that systemic administration of PP2 or intraventricular injection of siRNA-Fyn, a Src family kinase family member, prevented hippocampal neuronal loss and spatial memory deficits following intraventricular hemorrhage (Liu et al., 2017). Thus, the targeted use of PAR-1 or Src kinase inhibitors may possibly represent a future therapeutic method to relieve the toxic consequences of thrombin in ICH (Liu and Sharp, 2011; Gao et al., 2014; Liu et al., 2017). However, Src kinase proto-oncogene members can also promote BBB repair and brain edema resolution in the recovery stage (7–14 days) after ICH (Liu and Sharp, 2011). More studies are required to address which specific Src family members may mediate ICH-induced brain injuries.
Conclusion
The pathophysiology mechanism of injury after ICH is very complicated, which contains OS, inflammation, nerve cell toxicity, thrombin formation, and so on. Preclinical and clinical research evidence in ICH has further revealed the progress of pathophysiology in cerebral hemorrhage, and these works have resulted in much new information about injury mechanisms and potential therapeutic targets. However, truly effective clinical treatments are very limited, mainly because the problem of transforming preclinical research into clinical application has not yet been solved. Therefore, a multi-target neuroprotective therapy will make clinically effective treatment strategies possible, but also requires further study.
Author Contributions
ZS and ST drafted the manuscript. AS reviewed and modified the manuscript. All authors agreed on the final version.
Funding
This work was funded by the China Postdoctoral Science Foundation (2017M612010) and the National Natural Science Foundation of China (81701144).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aronowski, J., Zhao, X. (2011). Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke 42, 1781–1786. doi: 10.1161/STROKEAHA.110.596718
Auriat, A. M., Silasi, G., Wei, Z., Paquette, R., Paterson, P., Nichol, H., et al. (2012). Ferric iron chelation lowers brain iron levels after intracerebral hemorrhage in rats but does not improve outcome. Exp. Neurol. 234, 136–143. doi: 10.1016/j.expneurol.2011.12.030
Behrouz, R. (2016). Re-exploring tumor necrosis factor alpha as a target for therapy in intracerebral hemorrhage. Transl. Stroke Res. 7, 93–96. doi: 10.1007/s12975-016-0446-x
Bhatia, H. S., Baron, J., Hagl, S., Eckert, G. P., Fiebich, B. L. (2016). Rice bran derivatives alleviate microglia activation: possible involvement of MAPK pathway. J. Neuroinflammation 13, 148. doi: 10.1186/s12974-016-0615-6
Bimpis, A., Papalois, A., Voumvourakis, K., Olah, O., Tiszlavicz, L., Liapi, C. (2015). Neuronal tumour necrosis factor-alpha and interleukin-1beta expression in a porcine model of intracerebral haemorrhage: modulation by U-74389G. Brain Res. 1615, 98–105. doi: 10.1016/j.brainres.2015.04.034
Bulters, D., Gaastra, B., Zolnourian, A., Alexander, S., Ren, D., Blackburn, S. L., et al. (2018). Haemoglobin scavenging in intracranial bleeding: biology and clinical implications. Nat. Rev. Neurol. 14, 416–432. doi: 10.1038/s41582-018-0020-0
Chen, J., Sun, Z., Jin, M., Tu, Y., Wang, S., Yang, X., et al. (2017). Inhibition of AGEs/RAGE/Rho/ROCK pathway suppresses non-specific neuroinflammation by regulating BV2 microglial M1/M2 polarization through the NF-kappaB pathway. J. Neuroimmunol. 305, 108–114. doi: 10.1016/j.jneuroim.2017.02.010
Cheng, Y., Xi, G., Jin, H., Keep, R. F., Feng, J., Hua, Y. (2014). Thrombin-induced cerebral hemorrhage: role of protease-activated receptor-1. Transl. Stroke Res. 5, 472–475. doi: 10.1007/s12975-013-0288-8
Cordonnier, C., Demchuk, A., Ziai, W., Anderson, C. S. (2018). Intracerebral haemorrhage: current approaches to acute management. Lancet 392, 1257–1268. doi: 10.1016/S0140-6736(18)31878-6
Dang, J., Brandenburg, L. O., Rosen, C., Fragoulis, A., Kipp, M., Pufe, T., et al. (2012). Nrf2 expression by neurons, astroglia, and microglia in the cerebral cortical penumbra of ischemic rats. J. Mol. Neurosci. 46, 578–584. doi: 10.1007/s12031-011-9645-9
Duan, X., Wen, Z., Shen, H., Shen, M., Chen, G. (2016). Intracerebral hemorrhage, oxidative stress, and antioxidant therapy. Oxid. Med. Cell Longev. 2016, 1203285. doi: 10.1155/2016/1203285
Fang, H., Chen, J., Lin, S., Wang, P., Wang, Y., Xiong, X., et al. (2014). CD36-mediated hematoma absorption following intracerebral hemorrhage: negative regulation by TLR4 signaling. J. Immunol. 192, 5984–5992. doi: 10.4049/jimmunol.1400054
Flores, J. J., Klebe, D., Rolland, W. B., Lekic, T., Krafft, P. R., Zhang, J. H. (2016). PPARgamma-induced upregulation of CD36 enhances hematoma resolution and attenuates long-term neurological deficits after germinal matrix hemorrhage in neonatal rats. Neurobiol. Dis. 87, 124–133. doi: 10.1016/j.nbd.2015.12.015
Gang, X., Han, Q., Zhao, X., Liu, Q., Wang, Y. (2018). Dynamic changes in toll-like receptor 4 in human perihematoma tissue after intracerebral hemorrhage. World Neurosurg. 118, e593–e600. doi: 10.1016/j.wneu.2018.06.247
Gao, F., Liu, F., Chen, Z., Hua, Y., Keep, R. F., Xi, G. (2014). Hydrocephalus after intraventricular hemorrhage: the role of thrombin. J. Cereb. Blood Flow Metab. 34, 489–494. doi: 10.1038/jcbfm.2013.225
Gaschler, M. M., Stockwell, B. R. (2017). Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 482, 419–425. doi: 10.1016/j.bbrc.2016.10.086
Gu, Y., Xi, G., Liu, W., Keep, R. F., Hua, Y. (2010). Estrogen reduces iron-mediated brain edema and neuronal death. Acta Neurochir. Suppl. 106, 159–162. doi: 10.1007/978-3-211-98811-4_29
Guan, J., Sun, S., Cao, X., Chen, Z. (2004). Experimental study on the PAR-1 expression around hemotoma following intracerebral hemorrhage in rats. J. Huazhong Univ. Sci. Technol. Med. Sci. 24, 266–268. doi: 10.1007/BF02832008
Hu, X., Tao, C., Gan, Q., Zheng, J., Li, H., You, C. (2016). Oxidative stress in intracerebral hemorrhage: sources, mechanisms, and therapeutic targets. Oxid. Med. Cell Longev. 2016, 3215391. doi: 10.1155/2016/3215391
Hua, Y., Wu, J., Keep, R. F., Nakamura, T., Hoff, J. T., Xi, G. (2006). Tumor necrosis factor-alpha increases in the brain after intracerebral hemorrhage and thrombin stimulation. Neurosurgery 58, 542–550. doi: 10.1227/01.NEU.0000197333.55473.AD
Huang, F. P., Xi, G., Keep, R. F., Hua, Y., Nemoianu, A., Hoff, J. T. (2002). Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J. Neurosurg. 96, 287–293. doi: 10.3171/jns.2002.96.2.0287
Katsu, M., Niizuma, K., Yoshioka, H., Okami, N., Sakata, H., Chan, P. H. (2010). Hemoglobin-induced oxidative stress contributes to matrix metalloproteinase activation and blood–brain barrier dysfunction in vivo. J. Cereb. Blood Flow Metab. 30, 1939–1950. doi: 10.1038/jcbfm.2010.45
Keep, R. F., Hua, Y., Xi, G. (2012). Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 11, 720–731. doi: 10.1016/S1474-4422(12)70104-7
Koh, Y. C., Yang, G., Lai, C. S., Weerawatanakorn, M., Pan, M. H. (2018). Chemopreventive effects of phytochemicals and medicines on M1/M2 polarized macrophage role in inflammation-related diseases. Int. J. Mol. Sci. 19. doi: 10.3390/ijms19082208
Kondo, H., Saito, K., Grasso, J. P., Aisen, P. (1988). Iron metabolism in the erythrophagocytosing Kupffer cell. Hepatology 8, 32–38. doi: 10.1002/hep.1840080108
Kong, Y., Le, Y. (2011). Toll-like receptors in inflammation of the central nervous system. Int. Immunopharmacol. 11, 1407–1414. doi: 10.1016/j.intimp.2011.04.025
Lambertsen, K. L., Meldgaard, M., Ladeby, R., Finsen, B. (2005). A quantitative study of microglial–macrophage synthesis of tumor necrosis factor during acute and late focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 25, 119–135. doi: 10.1038/sj.jcbfm.9600014
Lan, X., Han, X., Li, Q., Li, Q., Gao, Y., Cheng, T., et al. (2017). Pinocembrin protects hemorrhagic brain primarily by inhibiting toll-like receptor 4 and reducing M1 phenotype microglia. Brain Behav. Immun. 61, 326–339. doi: 10.1016/j.bbi.2016.12.012
Lei, B., Dawson, H. N., Roulhac-Wilson, B., Wang, H., Laskowitz, D. T., James, M. L. (2013). Tumor necrosis factor alpha antagonism improves neurological recovery in murine intracerebral hemorrhage. J. Neuroinflammation 10, 103. doi: 10.1186/1742-2094-10-103
Li, Q., Han, X., Lan, X., Gao, Y., Wan, J., Durham, F., et al. (2017). Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2, e90777. doi: 10.1172/jci.insight.90777
Li, Q., Wan, J., Lan, X., Han, X., Wang, Z., Wang, J. (2017). Neuroprotection of brain-permeable iron chelator VK-28 against intracerebral hemorrhage in mice. J. Cereb. Blood Flow Metab. 37, 3110–3123. doi: 10.1177/0271678X17709186
Lin, S., Yin, Q., Zhong, Q., Lv, F. L., Zhou, Y., Li, J. Q., et al. (2012). Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J. Neuroinflammation 9, 46. doi: 10.1186/1742-2094-9-46
Liu, D. Z., Sharp, F. R. (2011). The dual role of SRC kinases in intracerebral hemorrhage. Acta Neurochir. Suppl. 111, 77–81. doi: 10.1007/978-3-7091-0693-8_13
Liu, D. Z., Ander, B. P., Xu, H., Shen, Y., Kaur, P., Deng, W., et al. (2010). Blood–brain barrier breakdown and repair by Src after thrombin-induced injury. Ann. Neurol. 67, 526–533. doi: 10.1002/ana.21924
Liu, D. Z., Waldau, B., Ander, B. P., Zhan, X., Stamova, B., Jickling, G. C., et al. (2017). Inhibition of Src family kinases improves cognitive function after intraventricular hemorrhage or intraventricular thrombin. J. Cereb. Blood Flow Metab. 37, 2359–2367. doi: 10.1177/0271678X16666291
Liu, L., Anderson, G. A., Fernandez, T. G., Dore, S. (2019). Efficacy and mechanism of Panax ginseng in experimental stroke. Front. Neurosci. 13, 294. doi: 10.3389/fnins.2019.00294
Lu, F., Nakamura, T., Toyoshima, T., Liu, Y., Hirooka, K., Kawai, N., et al. (2012). Edaravone, a free radical scavenger, attenuates behavioral deficits following transient forebrain ischemia by inhibiting oxidative damage in gerbils. Neurosci. Lett. 506, 28–32. doi: 10.1016/j.neulet.2011.10.041
Lu, X., Chen-Roetling, J., Regan, R. F. (2014). Systemic hemin therapy attenuates blood–brain barrier disruption after intracerebral hemorrhage. Neurobiol. Dis. 70, 245–251. doi: 10.1016/j.nbd.2014.06.005
Mohan, S., Ahmad, A. S., Glushakov, A. V., Chambers, C., Dore, S. (2012). Putative role of prostaglandin receptor in intracerebral hemorrhage. Front. Neurol. 3, 145. doi: 10.3389/fneur.2012.00145
Ohnishi, M., Katsuki, H., Fujimoto, S., Takagi, M., Kume, T., Akaike, A. (2007). Involvement of thrombin and mitogen-activated protein kinase pathways in hemorrhagic brain injury. Exp. Neurol. 206, 43–52. doi: 10.1016/j.expneurol.2007.03.030
Oldfield, E. H., Loomba, J. J., Monteith, S. J., Crowley, R. W., Medel, R., Gress, D. R., et al. (2013). Safety and pharmacokinetics of sodium nitrite in patients with subarachnoid hemorrhage: a phase IIa study. J. Neurosurg. 119, 634–641. doi: 10.3171/2013.3.JNS13266
Pan, J., Jin, J. L., Ge, H. M., Yin, K. L., Chen, X., Han, L. J., et al. (2015). Malibatol a regulates microglia M1/M2 polarization in experimental stroke in a PPARgamma-dependent manner. J. Neuroinflammation 12, 51. doi: 10.1186/s12974-015-0270-3
Pearson, V. L., Rothwell, N. J., Toulmond, S. (1999). Excitotoxic brain damage in the rat induces interleukin-1beta protein in microglia and astrocytes: correlation with the progression of cell death. Glia 25, 311–323. doi: 10.1002/(SICI)1098-1136(19990215)25:4<311::AID-GLIA1>3.0.CO;2-E
Ponomarev, E. D., Veremeyko, T., Weiner, H. L. (2013). MicroRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS. Glia 61, 91–103. doi: 10.1002/glia.22363
Rodriguez-Yanez, M., Castillo, J. (2008). Role of inflammatory markers in brain ischemia. Curr. Opin. Neurol. 21, 353–357. doi: 10.1097/WCO.0b013e3282ffafbf
Sansing, L. H., Harris, T. H., Welsh, F. A., Kasner, S. E., Hunter, C. A., Kariko, K. (2011). Toll-like receptor 4 contributes to poor outcome after intracerebral hemorrhage. Ann. Neurol. 70, 646–656. doi: 10.1002/ana.22528
Scott, G., Zetterberg, H., Jolly, A., Cole, J. H., De Simoni, S., Jenkins, P. O., et al. (2018). Minocycline reduces chronic microglial activation after brain trauma but increases neurodegeneration. Brain 141, 459–471. doi: 10.1093/brain/awx339
Shao, A., Zhu, Z., Li, L., Zhang, S., Zhang, J. (2019). Emerging therapeutic targets associated with the immune system in patients with intracerebral haemorrhage (ICH): from mechanisms to translation. EBioMedicine 45, 615–623. doi: 10.1016/j.ebiom.2019.06.012
Tapia-Perez, J. H., Karagianis, D., Zilke, R., Koufuglou, V., Bondar, I., Schneider, T. (2016). Assessment of systemic cellular inflammatory response after spontaneous intracerebral hemorrhage. Clin. Neurol. Neurosurg. 150, 72–79. doi: 10.1016/j.clineuro.2016.07.010
van Asch, C. J., Luitse, M. J., Rinkel, G. J., van der Tweel, I., Algra, A., Klijn, C. J. (2010). Incidence, case fatality, and functional outcome of intracerebral haemorrhage over time, according to age, sex, and ethnic origin: a systematic review and meta-analysis. Lancet Neurol. 9, 167–176. doi: 10.1016/S1474-4422(09)70340-0
Varnum, M. M., Ikezu, T. (2012). The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch. Immunol. Ther. Exp. (Warsz). 60, 251–266. doi: 10.1007/s00005-012-0181-2
Vezzani, A., Conti, M., De Luigi, A., Ravizza, T., Moneta, D., Marchesi, F., et al. (1999). Interleukin-1beta immunoreactivity and microglia are enhanced in the rat hippocampus by focal kainate application: functional evidence for enhancement of electrographic seizures. J. Neurosci. 19, 5054–5065. doi: 10.1523/JNEUROSCI.19-12-05054.1999
Wagner, K. R., Sharp, F. R., Ardizzone, T. D., Lu, A., Clark, J. F. (2003). Heme and iron metabolism: role in cerebral hemorrhage. J. Cereb. Blood Flow Metab. 23, 629–652. doi: 10.1097/01.WCB.0000073905.87928.6D
Wan, S., Cheng, Y., Jin, H., Guo, D., Hua, Y., Keep, R. F., et al. (2016). Microglia activation and polarization after intracerebral hemorrhage in mice: the role of protease-activated receptor-1. Transl. Stroke Res. 7, 478–487. doi: 10.1007/s12975-016-0472-8
Wang, J., Tsirka, S. E. (2005). Tuftsin fragment 1–3 is beneficial when delivered after the induction of intracerebral hemorrhage. Stroke 36, 613–618. doi: 10.1161/01.STR.0000155729.12931.8f
Wang, J., Fields, J., Zhao, C., Langer, J., Thimmulappa, R. K., Kensler, T. W., et al. (2007). Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic. Biol. Med. 43, 408–414. doi: 10.1016/j.freeradbiomed.2007.04.020
Wang, Y. C., Zhou, Y., Fang, H., Lin, S., Wang, P. F., Xiong, R. P., et al. (2014). Toll-like receptor 2/4 heterodimer mediates inflammatory injury in intracerebral hemorrhage. Ann. Neurol. 75, 876–889. doi: 10.1002/ana.24159
Wang, G., Li, T., Duan, S. N., Dong, L., Sun, X. G., Xue, F. (2018). PPAR-gamma promotes hematoma clearance through haptoglobin-hemoglobin-CD163 in a rat model of intracerebral hemorrhage. Behav. Neurol. 2018, 7646104. doi: 10.1155/2018/7646104
Wang, G., Wang, L., Sun, X. G., Tang, J. (2018). Haematoma scavenging in intracerebral haemorrhage: from mechanisms to the clinic. J. Cell Mol. Med. 22, 768–777. doi: 10.1111/jcmm.13441
Wei, C. C., Kong, Y. Y., Li, G. Q., Guan, Y. F., Wang, P., Miao, C. Y. (2017). Nicotinamide mononucleotide attenuates brain injury after intracerebral hemorrhage by activating Nrf2/HO-1 signaling pathway. Sci. Rep. 7, 717. doi: 10.1038/s41598-017-00851-z
Wu, B., Ma, Q., Khatibi, N., Chen, W., Sozen, T., Cheng, O., et al. (2010). Ac-YVAD-CMK decreases blood–brain barrier degradation by inhibiting caspase-1 activation of interleukin-1beta in intracerebral hemorrhage mouse model. Transl. Stroke Res. 1, 57–64. doi: 10.1007/s12975-009-0002-z
Wu, H., Wu, T., Han, X., Wan, J., Jiang, C., Chen, W., et al. (2017). Cerebroprotection by the neuronal PGE2 receptor EP2 after intracerebral hemorrhage in middle-aged mice. J. Cereb. Blood Flow Metab. 37, 39–51. doi: 10.1177/0271678X15625351
Wu, H., Wu, T., Hua, W., Dong, X., Gao, Y., Zhao, X., et al. (2015). PGE2 receptor agonist misoprostol protects brain against intracerebral hemorrhage in mice. Neurobiol. Aging 36, 1439–1450. doi: 10.1016/j.neurobiolaging.2014.12.029
Wu, J., Hua, Y., Keep, R. F., Schallert, T., Hoff, J. T., Xi, G. (2002). Oxidative brain injury from extravasated erythrocytes after intracerebral hemorrhage. Brain Res. 953, 45–52. doi: 10.1016/S0006-8993(02)03268-7
Wu, J., Yang, S., Xi, G., Song, S., Fu, G., Keep, R. F., et al. (2008). Microglial activation and brain injury after intracerebral hemorrhage. . Suppl. 105, 59–65. doi: 10.1007/978-3-211-09469-3_13
Wu, S., Sena, E., Egan, K., Macleod, M., Mead, G. (2014). Edaravone improves functional and structural outcomes in animal models of focal cerebral ischemia: a systematic review. Int. J. Stroke 9, 101–106. doi: 10.1111/ijs.12163
Wu, T., Wu, H., Wang, J., Wang, J. (2011). Expression and cellular localization of cyclooxygenases and prostaglandin E synthases in the hemorrhagic brain. . Neuroinflammation 8, 22. doi: 10.1186/1742-2094-8-22
Xi, G., Reiser, G., Keep, R. F. (2003). The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: deleterious or protective? J. Neurochem. 84, 3–9. doi: 10.1046/j.1471-4159.2003.01268.x
Xia, C. Y., Zhang, S., Gao, Y., Wang, Z. Z., Chen, N. H. (2015). Selective modulation of microglia polarization to M2 phenotype for stroke treatment. Int. Immunopharmacol. 25, 377–382. doi: 10.1016/j.intimp.2015.02.019
Xiong, X. Y., Liu, L., Yang, Q. W. (2016). Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 142, 23–44. doi: 10.1016/j.pneurobio.2016.05.001
Yang, J., Cui, X., Li, J., Zhang, C., Zhang, J., Liu, M. (2015). Edaravone for acute stroke: meta-analyses of data from randomized controlled trials. Dev. Neurorehabil. 18, 330–335. doi: 10.3109/17518423.2013.830153
Yang, Y., Zhang, Y., Wang, Z., Wang, S., Gao, M., Xu, R., et al. (2016). Attenuation of acute phase injury in rat intracranial hemorrhage by cerebrolysin that inhibits brain edema and inflammatory response. Neurochem. Res. 41, 748–757. doi: 10.1007/s11064-015-1745-4
Yeatts, S. D., Palesch, Y. Y., Moy, C. S., Selim, M. (2013). High dose deferoxamine in intracerebral hemorrhage (HI-DEF) trial: rationale, design, and methods. Neurocrit. Care 19, 257–266. doi: 10.1007/s12028-013-9861-y
Yu, Y. P., Chi, X. L., Liu, L.J. (2014). A hypothesis: hydrogen sulfide might be neuroprotective against subarachnoid hemorrhage induced brain injury. Sci. World J. 2014, 432318. doi: 10.1155/2014/432318
Yuan, S., Yu, Z., Zhang, Z., Zhang, J., Zhang, P., Li, X., et al. (2019). RIP3 participates in early brain injury after experimental subarachnoid hemorrhage in rats by inducing necroptosis. Neurobiol. Dis. 129, 144–158. doi: 10.1016/j.nbd.2019.05.004
Zeng, J., Chen, Y., Ding, R., Feng, L., Fu, Z., Yang, S., et al. (2017). Isoliquiritigenin alleviates early brain injury after experimental intracerebral hemorrhage via suppressing ROS- and/or NF-kappaB-mediated NLRP3 inflammasome activation by promoting Nrf2 antioxidant pathway. J. Neuroinflammation 14, 119. doi: 10.1186/s12974-017-0895-5
Zeng, L., Tan, L., Li, H., Zhang, Q., Li, Y., Guo, J. (2018). Deferoxamine therapy for intracerebral hemorrhage: a systematic review. PLoS One 13, e0193615. doi: 10.1371/journal.pone.0193615
Zhang, B., Wei, Y. Z., Wang, G. Q., Li, D. D., Shi, J. S., Zhang, F. (2018). Targeting MAPK pathways by naringenin modulates microglia M1/M2 polarization in lipopolysaccharide-stimulated cultures. Front. Cell Neurosci. 12, 531. doi: 10.3389/fncel.2018.00531
Zhang, Z., Zhang, Z., Lu, H., Yang, Q., Wu, H., Wang, J. (2017). Microglial polarization and inflammatory mediators after intracerebral hemorrhage. Mol. Neurobiol. 54, 1874–1886. doi: 10.1007/s12035-016-9785-6
Zhao, F., Liu, Z. (2014). Beneficial effects of edaravone on the expression of serum matrix metalloproteinase-9 after cerebral hemorrhage. Neurosci. (Riyadh) 19, 106–110.
Zhao, X., Sun, G., Zhang, J., Strong, R., Dash, P. K., Kan, Y. W., et al. (2007). Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke 38, 3280–3286. doi: 10.1161/STROKEAHA.107.486506
Zhao, X., Sun, G., Zhang, J., Strong, R., Song, W., Gonzales, N., et al. (2007). Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann. Neurol. 61, 352–362. doi: 10.1002/ana.21097
Zhao, X. R., Gonzales, N., Aronowski, J. (2015). Pleiotropic role of PPARgamma in intracerebral hemorrhage: an intricate system involving Nrf2, RXR, and NF-kappaB. CNS Neurosci. Ther. 21, 357–366. doi: 10.1111/cns.12350
Zheng, H., Chen, C., Zhang, J., Hu, Z. (2016). Mechanism and therapy of brain edema after intracerebral hemorrhage. Cerebrovasc. Dis. 42, 155–169. doi: 10.1159/000445170
Zhu, H., Wang, Z., Yu, J., Yang, X., He, F., Liu, Z., et al. (2019). Role and mechanisms of cytokines in the secondary brain injury after intracerebral hemorrhage. Prog. Neurobiol. 178, 101610. doi: 10.1016/j.pneurobio.2019.03.003
Keywords: intracerebral hemorrhage, inflammation, oxidative stress, Nrf2, iron, thrombin
Citation: Shao Z, Tu S and Shao A (2019) Pathophysiological Mechanisms and Potential Therapeutic Targets in Intracerebral Hemorrhage. Front. Pharmacol. 10:1079. doi: 10.3389/fphar.2019.01079
Received: 21 November 2018; Accepted: 26 August 2019;
Published: 19 September 2019.
Edited by:
Anna Rita Bilia, University of Florence, ItalyReviewed by:
Shekher Mohan, Manchester University, United StatesRajeev Kumar Garg, Rush University, United States
Copyright © 2019 Shao, Tu and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anwen Shao, MjExMTgxMTZAemp1LmVkdS5jbg==; YW53ZW5zaGFvQHNpbmEuY29t