 Swayam Prakash Srivastava
Swayam Prakash Srivastava Ahmad F. Hedayat
Ahmad F. Hedayat Keizo Kanasaki
Keizo Kanasaki Julie E. Goodwin
Julie E. Goodwin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol., 16 August 2019
Sec. Renal Pharmacology
Volume 10 - 2019 | https://doi.org/10.3389/fphar.2019.00904
This article is part of the Research TopicSmall Interfering RNAs, Nuclear Oligonucleotides and Oligonucleotide-Based TherapiesView all 6 articles
microRNAs (miRNAs) are small, non-coding nucleotides that regulate diverse biological processes. Altered microRNA biosynthesis or regulation contributes to pathological processes including kidney fibrosis. Kidney fibrosis is characterized by deposition of excess extracellular matrix (ECM), which is caused by infiltration of immune cells, inflammatory cells, altered chemokines, and cytokines as well as activation and accumulation of fibroblasts in the kidney. These activated fibroblasts can arise from epithelial cells via epithelial-to-mesenchymal transition (EMT), from bone marrow-derived M2 phenotype macrophages via macrophage-to-mesenchymal transition (MMT), from endothelial cells via endothelial-to-mesenchymal transition (EndMT), from resident fibroblasts, and from bone marrow-derived monocytes and play a crucial role in fibrotic events. Disrupted microRNA biosynthesis and aberrant regulation contribute to the activation of mesenchymal programs in the kidney. miR-29 regulates the interaction between dipeptidyl peptidase-4 (DPP-4) and integrin β1 and the associated active transforming growth factor β (TGFβ) and pro-EndMT signaling; however, miR-let-7 targets transforming growth factor β receptors (TGFβRs) to inhibit TGFβ signaling. N-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP) is an endogenous anti-fibrotic peptide, which is associated with fibroblast growth factor receptor 1 (FGFR1) phosphorylation and subsequently responsible for the production of miR-let-7. miR-29 and miR-let-7 family clusters participate in crosstalk mechanisms, which are crucial for endothelial cell homeostasis. The physiological level of AcSDKP is vital for the activation of anti-fibrotic mechanisms including restoration of anti-fibrotic microRNA crosstalk and suppression of profibrotic signaling by mitigating DPP-4-associated mesenchymal activation in the epithelial cells, endothelial cells, and M2 phenotype macrophages. The present review highlights recent advancements in the understanding of both the role of microRNAs in the development of kidney disease and their potential as novel therapeutic targets for fibrotic disease states.
Kidney fibrosis is the final outcome of progressive diabetic kidney disease that can lead to end stage renal disease (ESRD) (Parving, 2001; Remuzzi et al., 2002; Alicic et al., 2017; Isaka, 2018; Luyckx et al., 2018; Umanath and Lewis, 2018; Allison, 2019; Cooper and Warren, 2019; Djudjaj and Boor, 2019). It results in the massive destruction of cellular structures and kidney function. Kidney fibrosis is caused by prolonged injury and deregulation of normal wound healing processes in association with excess deposition of extracellular matrix (ECM) (Lee and Kalluri, 2010; Nogueira et al., 2017). In such fibrotic processes, kidney fibroblasts play vital roles, but the origin of fibroblasts still remains unclear and a matter of ongoing debate (Kanasaki et al., 2013a; El Agha et al., 2017; Di Carlo and Peduto, 2018). These debates were based on each report that stick to the idea that one single cell type can explain majority of fibrogenesis events in kidney; renal fibrogenesis is the consequence of the interaction between all the cell types in kidney, either kidney cells or invaded inflammatory cells (Liu, 2011; Medici and Kalluri, 2012; Lebleu et al., 2013; Mack and Yanagita, 2015; Nogueira et al., 2017). Also, complete conversion into mesenchymal cell types is not essential; intermediate phenotypes of mesenchymal programs were sufficient to induce alteration in fibrogenic programs (Kanasaki et al., 2013a; Lebleu et al., 2013; Kim et al., 2017; Xing and Tian, 2019). Activation of resident fibroblasts is the first step in renal fibrogenesis (Grgic et al., 2012; Sato and Yanagita, 2017). Figure 1 depicts the sources of fibroblasts that have been proposed, including from pericytes, fibrocytes, bone marrow-derived monocytes, and fibroblast originating from bone marrow-derived M2 type macrophages via macrophage-to-mesenchymal transition (MMT), fibroblasts originating from epithelial-to-mesenchymal transition (EMT), and endothelial-to-mesenchymal transition (EndMT) (Barnes and Gorin, 2011; Lebleu et al., 2013; Yan et al., 2016; Sato and Yanagita, 2017; Di Carlo and Peduto, 2018; Xiong et al., 2018; Glover et al., 2019). The available treatments for kidney fibrosis are unsatisfactory to address this problem, and approved therapies are not cell-specific in nature (Lee et al., 2015; Quiroga et al., 2015; Breyer and Susztak, 2016). Current treatment strategies may slow the rate of disease progression but cannot prevent progression to ESRD (Brenner et al., 2001; Lee et al., 2015; Quiroga et al., 2015; Johnson et al., 2016; Luyckx et al., 2018); hence, current therapies are ineffective.
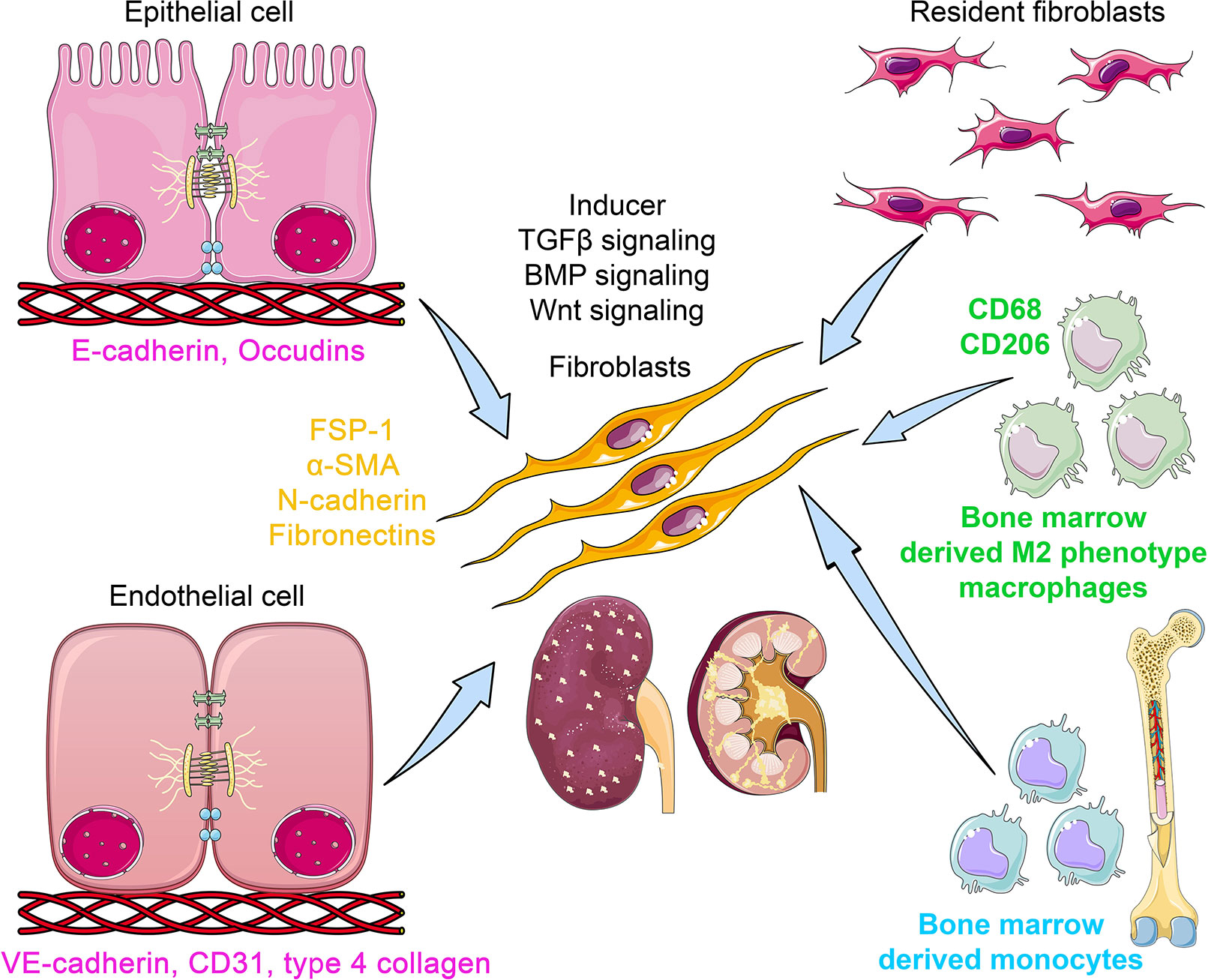
Figure 1 The origins of kidney myofibroblasts. Myofibroblasts are originated from resident fibroblasts, bone marrow-derived monocytes, bone marrow-derived M2 phenotype macrophages via macrophage-to-mesenchymal transition (MMT) process, from epithelial cell via epithelial-to-mesenchymal transition (EMT) program, and from endothelial cells via endothelial to mesenchymal transition (EndMT) program. During the process of EMT, epithelial cells loose the epithelial markers (E-cadherin and occludins) and gain the mesenchymal markers (FSP-1, α-SMA, N-cadherin, and fibronectin). In the EndMT process, endothelial cells lose the endothelial markers (CD31 and VE cadherin) and gain the mesenchymal markers. However, during the process of MMT, the myo-fibroblasts co-express M2 phenotype macrophage markers (CD206) with mesenchymal markers. TGFβ, BMP, and Wnt signaling play a crucial role in the activation of mesenchymal transition processes. The myofibroblasts are polar, pointed, and elongated in shape and ability to migrate and invade the neighbor cells.
EMT involves a series of events through which epithelial cells lose their epithelial characteristics and acquire properties of typical mesenchymal cells (Hills and Squires, 2011; Grande et al., 2015; Lovisa et al., 2015; Marquez-Exposito et al., 2018). Figure 1 displays the unique phenotypes of epithelial, endothelial, and mesenchymal cells. Epithelial cells are normally associated tightly with their neighbor cells, which inhibits their potential to dissociate from the epithelial layer. In contrast, mesenchymal cells do not form a layer of cells or intercellular adhesion complexes (Srivastava et al., 2013). Mesenchymal cells are elongated in shape and exhibit end-to-end polarity and focal adhesions, allowing for increased migratory capacity (Srivastava et al., 2013). In adults, the main function of fibroblasts, which are prototypical mesenchymal cells that exist in many tissues, is to maintain structural integrity by secreting extracellular matrix (ECM). Fibroblast-specific protein 1 (FSP-1; also known as S100A4), alpha-smooth muscle actin (αSMA), fibronectin, and collagen I have proved to be reliable markers to characterize the mesenchymal products generated by EMT that occurs during the development of fibrosis in various organs (Kalluri and Weinberg, 2009; Srivastava et al., 2013; Alidadiani et al., 2018). Inflammatory injury to the mouse kidney can result in the recruitment of a diverse array of cells that can trigger EMT through their release of growth factors, such as transforming growth factor-beta (TGFβ), platelet-derived growth factor (PDGF), epidermal growth factor (EGF), and fibroblast growth factor-2 (FGF-2) (Kalluri and Weinberg, 2009; Alidadiani et al., 2018; Liu et al., 2019b).
Vascular endothelial cells can also originate fibroblasts by undergoing a phenotypic transition, referred to as EndMT (Srivastava et al., 2013; Curci et al., 2014; Li et al., 2017; Lovisa and Kalluri, 2018; Glover et al., 2019). Figure 1 displays the process and contribution of EndMT to fibrogenesis. EndMT is the process that is characterized by the loss of endothelial markers, including cluster of differentiation 31 (CD31) and vascular endothelial cadherin (VE-cadherin), and acquisition of the expression of mesenchymal proteins including αSMA (Zeisberg et al., 2007; Srivastava et al., 2013; Curci et al., 2014; Lovisa and Kalluri, 2018; Glover et al., 2019). EndMT contributes to cardiac fibrogenesis (Zeisberg et al., 2007; Kovacic et al., 2019), pulmonary fibrosis (Good et al., 2015; Cho et al., 2018), idiopathic hypertension (Kitao et al., 2009; Ranchoux et al., 2015), and fibrosis in the cornea (Nakano et al., 2008; Medici, 2016; Lee et al., 2018). Many signaling pathways that govern EMT also regulate EndMT in the embryonic heart, during the development of cardiac fibrosis (Pardali et al., 2017; Man et al., 2019) and pulmonary fibrosis (Guan and Zhou, 2017; Pardali et al., 2017), and in liver fibrogenesis (Dufton et al., 2017; Pardali et al., 2017). Compared to EMT, comparatively little is known about EndMT. The contribution of EndMT to renal fibrosis has been reviewed in recent years (Srivastava et al., 2013; Curci et al., 2014; Medici, 2016; Glover et al., 2019). In the adult organism, pathological conditions such as injury, inflammation, or aging can induce EndMT and influence organ fibrosis (Cho et al., 2018). Zeisberg et al. (2008) performed a seminal experiment that confirmed the contribution of EndMT in renal fibrosis in three mouse models: unilateral ureteral obstruction (UUO), a surgical model used to study progressive tubulointerstitial fibrosis, streptozotocin (STZ)-induced diabetic mice, and α3 chain of collagen type 4 (COL4A3) knockout mice (a mouse model for Alport syndrome). The authors reported that a considerable number of myofibroblasts co-expressed CD31 with αSMA and FSP-1 in all three models (Zeisberg et al., 2008). The authors analyzed the kidneys of diabetic CD-1 mice 6 months after a single injection of STZ and showed that kidneys had progressive glomerular sclerosis and tubulointerstitial fibrosis. The co-immunofluorescence analysis in the kidneys of diabetic CD-1 mice displayed approximately 40% of all FSP-1 positive cells, and 50% of αSMA positive stromal cells were CD31-positive (Zeisberg et al., 2008). Similarly, in the kidneys of COL4A3 knockout mice, 45% of all αSMA-positive fibroblasts and 60% of all FSP-1-positive fibroblasts were CD31-positive, suggesting that these fibroblasts are of endothelial origin and that EndMT might contribute critically to the development and progression of renal fibrosis (Zeisberg et al., 2008). Li et al. (2009) confirmed that EndMT contributes to the activation of myofibroblasts in early diabetic renal fibrosis. In the landmark experiment using endothelial cell-lineage tracing with Tie2-Cre and LoxP-enhanced green fluorescent protein (EGFP) transgenic mice, the authors confirmed a large population of interstitial αSMA-positive cells of endothelial origin in the fibrotic kidneys of STZ-induced diabetic mice (Li et al., 2009). These endothelial cells demonstrated a set of biomarkers including VE-cadherin, CD31, tyrosine kinase with immunoglobulin-like EGF-like domains 1 (TIE1), TEK receptor kinase (TIE2), von Willebrand factor (vWF), and cytokeratins (Srivastava et al., 2013). During the process of EndMT, biochemical changes leads to the decreased expression of endothelial markers and the gain of mesenchymal markers such as FSP-1, αSMA, smooth muscle 22-alpha (SM22α), N-cadherin, fibronectin, vimentin, type I and III collagen, nestin, cluster of differentiation 73 (CD73), matrix metalloproteinase -2 (MMP-2), and matrix metalloproteinase-9 (MMP-9) (Medici and Kalluri, 2012; Srivastava et al., 2013; Srivastava et al., 2016).
Interstitial fibrosis is the key characteristics in chronic renal allograft injury (Boor and Floege, 2015; Bontha et al., 2017). In the chronic renal allograft injury, diverse ranges of immune and nonimmune responses cause the macrophages to undergo macrophage-to-mesenchymal transition (MMT) process (Wang et al., 2019; Zhou et al., 2019). Higher rate of MMT contributes in the development of interstitial fibrosis (Wang et al., 2017). Wang et al. performed the seminal experiments on MMT and identified that the kidneys in the patients and in the experimental chronic renal allograft injury displayed co-expression of macrophage marker (CD68) with myofibroblast marker (α-SMA) (Wang et al., 2017). Approximately 50% cells of total myo-fibroblasts cells in the kidneys were CD68+/α-SMA+ and were associated with interstitial fibrosis after the chronic renal allograft injury (Wang et al., 2017). Moreover, MMT processes were observed mainly in the bone marrow-derived M2-phenotype macrophages (Wang et al., 2017). However, M1-phenotype macrophages are responsible for pro-inflammatory cytokine production and contribute in the graft loss in the kidneys (Ma et al., 2013; Kwan et al., 2014; Salehi and Reed, 2015). These data are in accord with previous observation that showed that bone marrow-derived monocytes and macrophages can contribute in the collagen formation by inducing the MMT processes in the kidneys of mouse model of ureteric obstruction and in the progressive chronic kidney disease (CKD) subjects (Yang et al., 2013; Liu et al., 2018). The MMT processes were dependent on TGFβ-smad3 signaling (Wang et al., 2017). However, M2-to-M1 phenotype conversion can induce cytokines that lead to the higher MMT process and can be TGFβ independent (Wang et al., 2017).
MicroRNAs (miRNAs) are well known for their regulatory role in diseases like diabetes, cancer, and fibrosis (Ruiz and Chakrabarti, 2013; Srivastava et al., 2013; Shah et al., 2016; Miao et al., 2018; Nadeem et al., 2018; Tan et al., 2018). They are small (around 22 nt) evolutionarily conserved, non-coding RNAs that regulate the expression of protein coding genes at the post-transcriptional level by binding to regions complementary to the 3’untranslated regions (UTR) of target mRNA. miRNAs suppress protein expression by either inhibiting mRNA translation or facilitating mRNA degradation (Kaur et al., 2011; Gebert and Macrae, 2019). Differential expression in tissues and tissue-specific selectivity enable them to play an important role in understanding the pathophysiology as well as the potential therapy of kidney diseases (Lv et al., 2018; Nascimento and Domingueti, 2019; Zhao et al., 2019). Modulation of kidney-specific miRNAs may enable renal-specific expression of target proteins that are vital for kidney function (Metzinger-Le Meuth et al., 2019).
Differential miRNA expression data suggest a role of altered miRNA in the pathogenesis of kidney disease (Bhatt et al., 2011; Lorenzen et al., 2011; Zhong et al., 2011; Chau et al., 2012; Chung and Lan, 2015; Van Der Hauwaert et al., 2015; Schauerte et al., 2017; Zhang et al., 2017; Hajarnis et al., 2018; Thomas et al., 2018; Xi et al., 2018; Yang et al., 2018; Zheng et al., 2018; Fujii et al., 2019; Liu et al., 2019a; Liu et al., 2019c; Zhao et al., 2019). The term fibromiR has been suggested for those miRNAs that regulate fibro-proliferative diseases (Pottier et al., 2014). So far, researches in this area have included TGFβ-associated regulation of miRNA expression in diabetic nephropathy (Kato et al., 2007; Kato et al., 2010; Kato et al., 2011; Kolling et al., 2017; Zanchi et al., 2017; Assmann et al., 2018; Zhang et al., 2018; Nascimento and Domingueti, 2019; Regmi et al., 2019), p53 induction of miR-34a in ischemic acute kidney injury (Bhatt et al., 2010), and miR-15a regulation of the cell division cycle regulator Cdc25A (Lee et al., 2008). Natarajan and colleagues reported that TGFβ-induced up-regulation of miR-192, miR-216a, and miR-217 in a diabetic mouse model and in glomerular mesangial cells (Kato et al., 2007; Kato et al., 2010) via targeting smad interacting protein 1 (SIP1), protein-tyrosine phosphatase (PTEN), and y-box binding protein 1 (Ybx1) played critical roles in collagen expression (Kato et al., 2007; Kato et al., 2010). However, clinical studies of diabetic nephropathy display remarkably lower miR-192 expression; further studies are required to explain this discrepancy (Krupa et al., 2010). In other studies, miR-335 and miR-43a encourage renal cell senescence by suppressing mitochondrial antioxidative enzymes (Bai et al., 2011). miR-192 has been shown to mediate lysine deficient protein kinase 1 (WNK1)-regulated sodium and potassium balance (Elvira-Matelot et al., 2010) and TGFβ-induced fibrosis (Chung et al., 2010). Moreover, angiotensin-converting-enzyme inhibitor (lisinopril) treatment caused an anti-fibrotic effect in the kidneys of Munich Wistar Fromter rats (a mouse model of progressive nephropathy) by inhibiting miR-324-3p-dependent suppression of prolyl endopeptidase (POP), a serine peptidase involved in the synthesis of the endogenous antifibrotic peptide AcSDKP, which is critical in the homeostasis of ECM secretion (Macconi et al., 2012). A recent study demonstrated that a feedback loop between miR-21 and programmed cell death protein 4 (PDCD4) and activated protein (AP-1) drives progression in a mouse model of renal fibrosis (Sun et al., 2018). A significant number of reviews have addressed the role of miRNAs in renal fibrosis (Li et al., 2010; Amrouche et al., 2011; Lorenzen et al., 2011; Chandrasekaran et al., 2012; Srivastava et al., 2013; Chung and Lan, 2015; Kato and Natarajan, 2015; Van Der Hauwaert et al., 2015; Zhang et al., 2017; Assmann et al., 2018; Lv et al., 2018; Fujii et al., 2019; Nascimento and Domingueti, 2019; Regmi et al., 2019; Zhao et al., 2019). miRNA actions can be pro-fibrotic or anti-fibrotic depending on the kidney cell type. Figure 2 depicts the altered level of miRNAs in EMT and EndMT processes, which regulates fibroblast synthesis and fibroblast accumulation in kidney.
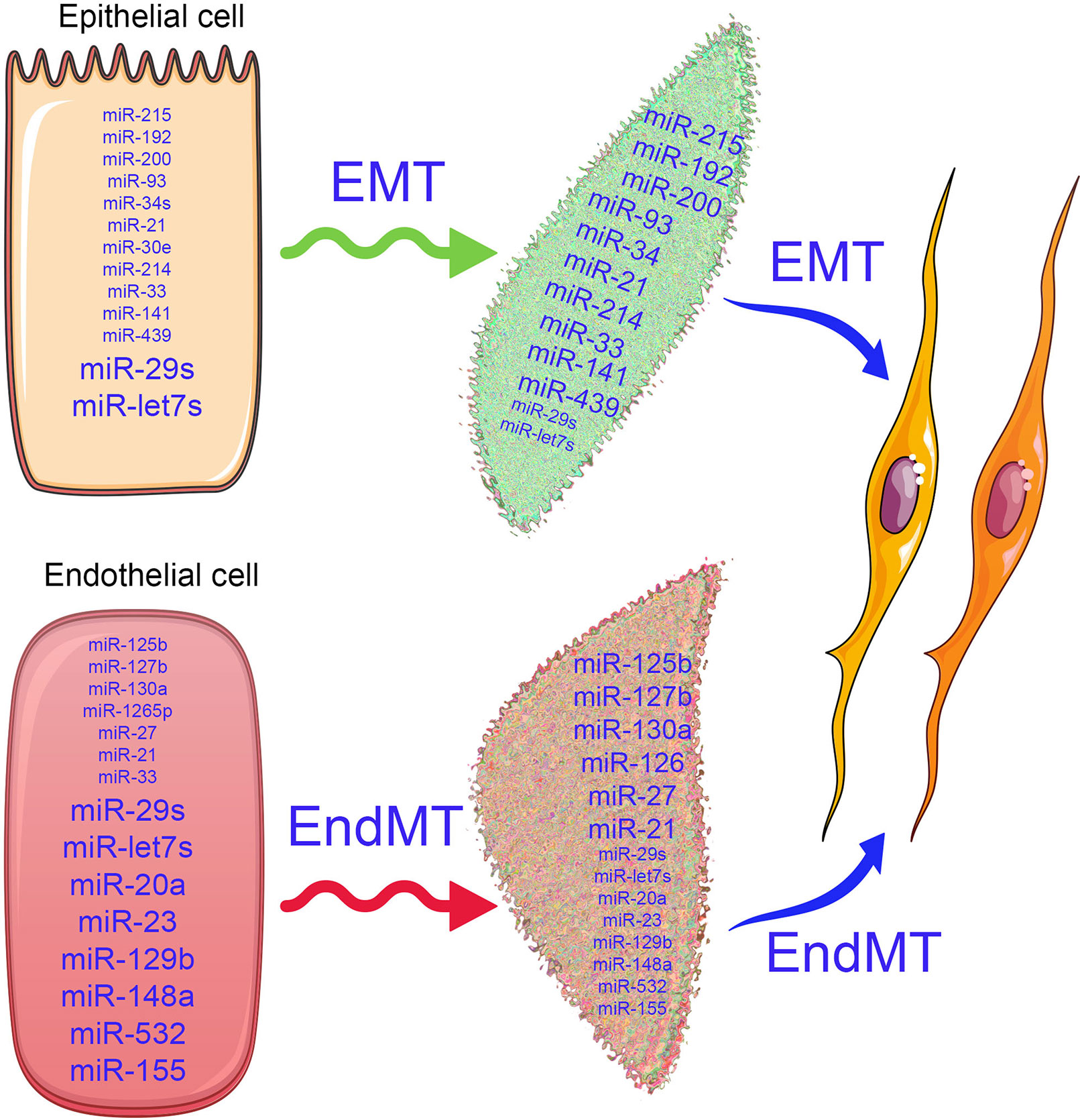
Figure 2 Alterations in microRNA expression influence the EMT and EndMT programs. In the epithelial cells, the antifibrotic miRNAs (miR-29 and let-7s) and profibrotic microRNAs (miR-15, miR-192, miR-200, miR-293, miR-34, miR-33, miR-214, miR-439, and miR-141) are expressed physiologically. Similarly, in the endothelial cells, antifibrotic microRNAs (miR-29, let-7, miR-20, miR-23, miR-129b, miR-148a, miR-532, and miR-155) and profibrotoic microRNAs (miR-125b, miR-127b, miR-130a, miR-27, miR-33, miR-21, and miR-1265) are expressed. The differential expressions of microRNAs in the epithelial cells and endothelial cells regulate the biological pathways and signaling events and maintain the homeostasis. As depicted by changes in font size, when healthy cells undergoes the mesenchymal transition process, the expression of anti-fibrotic microRNAs decreases, while pro-fibrotic microRNA expression increases and disrupts the cellular homeostasis.
The miR-29 family clusters emerge as a major anti-fibrotic player in kidney fibrosis associated with Smad-dependent and Smad-independent pathways (Chung and Lan, 2015). The expression level of members of miR-29 family is significantly suppressed in both renal fibrosis (Lan, 2012; Meng et al., 2013; Srivastava et al., 2014) and diabetic (Srivastava et al., 2016) and hypertensive nephropathy (Wei et al., 2013). miR-29 is downstream of Smad3 and can suppress the upstream TGFβ–Smad3 signaling by miR-29b-mediated negative feedback (He et al., 2013). miR-29b binds to the coding region of TGFβ1 mRNA at exon 3, which blocks the translation of TGFβ1, resulting in the suppression of Smad3-dependent fibrosis (Zhang et al., 2014). miR-29 binds to the promoter region of smad3 and exerts anti-fibrotic properties. In vitro, overexpression of miR-29 inhibited, but knockdown of miR-29 enhanced, TGFβ1-induced expression of collagens I and III in cultured proximal tubular epithelial cells (TECs) (Qin et al., 2011; Wang et al., 2012a; Qi and Yang, 2018). However, ultrasound-mediated gene delivery of miR-29 blocked progressive renal fibrosis in obstructive nephropathy (UUO) (Qin et al., 2011; Qi and Yang, 2018). Data from various studies have shown that members of the miR-29 family target different isoforms of collagen and have an anti-fibrotic role (Wang et al., 2012a; Qi and Yang, 2018). TGFβ1 inhibits the beneficial role of miR-29 family by down-regulating the expression in TECs (Du et al., 2010; Wang et al., 2012a), mesangial cells (Wang et al., 2012a), and podocytes (Wang et al., 2012a). miR-29b suppression contributes to progressive renal injury in several mouse models of chronic kidney disease (CKD) (Qin et al., 2011; Wang et al., 2012a; Ramdas et al., 2013); however, overexpression of miR-29b provides a therapeutic benefit in UUO and db/db mice (Qin et al., 2011; Chen et al., 2014a). In db/db mice, miR-29a has been shown to be elevated in the liver and regulate gluconeogenesis (Pandey et al., 2011). Of note, treatment of rats with losartan caused a remarkable increase in the level of miR-29b expression, which was linked with lower expression of collagen, fibronectin, and laminin, and provided protection from kidney fibrosis (Wang et al., 2012a). miR-29 family clusters also inhibit elevated dipeptidyl dipeptidase-4 (DPP-4) protein levels by targeting the 3’UTR of its mRNA (Kanasaki et al., 2014; Shi et al., 2015). TGFβ2-mediated induction of DPP-4 and down-regulation of miR-29 are associated with EndMT (Kanasaki et al., 2014; Shi et al., 2015). miR-29 and TGFβ signaling exhibit a negative feedback loop and regulate each other, as induction of TGFβ signaling suppresses downstream miR-29 (Kanasaki et al., 2014) and miR-29 suppresses upstream TGFβ signaling (Zhang et al., 2014), This relationship is quite interesting and supports an anti-fibrotic role of miR-29 in kidney fibrosis. The schematic diagram displays the renal protective action of miR-29 in EndMT and associated renal fibrosis (Figure 3).
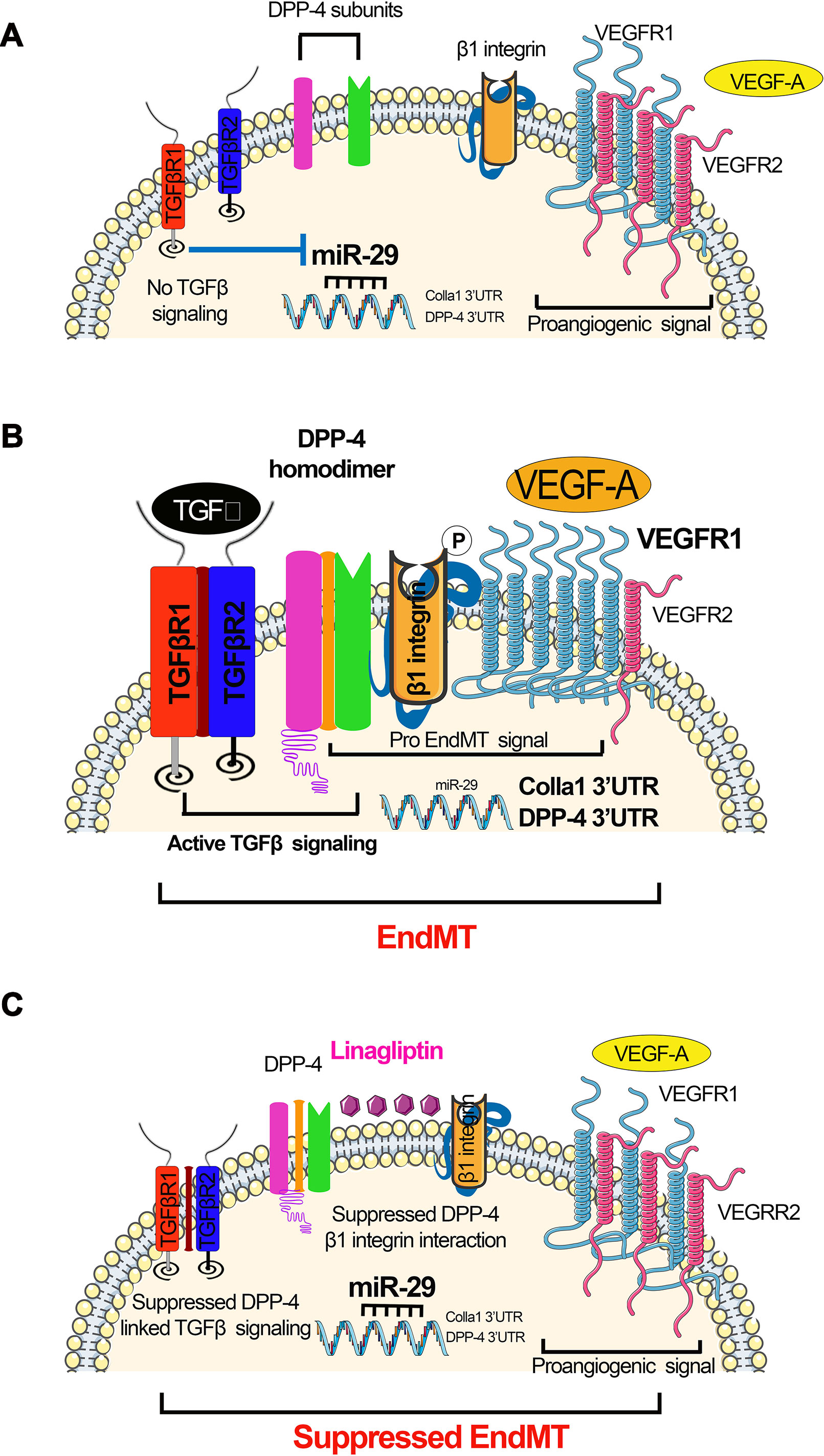
Figure 3 miR-29s regulate DPP-4–integrin β1-associated active TGFβ signaling and switch between VEGFR1 and VEGFR2 in the endothelial cells. (A) Absent TGFβ signaling; in the absence of TGFβ, miR-29 families were expressed at normal level, which targets the 3’UTR of DPP-4 mRNA and 3’UTR of Colla 1 mRNA. Suppressed level of DPP-4–β-integrin further leads to suppression in the TGFβ signaling. (B) Active TGFβ signaling resulting in EndMT. Active TGFβ signaling causes suppression in the miR-29, which results to the higher DPP-4 mRNA and Colla1 transcription. Higher level of DPP-4–β-integrin and VEGF influences the active TGFβ signaling. (C) Suppressed TGFβ signaling mediated by DPP-4 inhibitor (linagliptin) restores miR-29 expression level and reduces the level of VEGF. The elevated level of miR-29 causes the suppression of the DPP-4–β-integrin level. The concomitant effect of reduced level of VEGF suppressed level DPP-4–β-integrin and finally leads to inhibition of active TGFβ signaling and suppression in the EndMT processes.
A new pro-fibrotic molecular mechanism exists, which is associated with the interaction between DPP-4 and integrin β1 and is a therapeutic target for kidney fibrosis during diabetes (Shi et al., 2015). In endothelial cells, miR-29 negatively regulates the DPP-4 and integrin β1 interaction (Shi et al., 2015). This interaction is a key regulator of the switch between vascular endothelial growth factor 1 (VEGFR1) and vascular endothelial growth factor 2 (VEGFR2) (Shi et al., 2015). VEGFR1 is a positive effector of monocyte and macrophage migration and has been reported as a negative regulator of the VEGFR2 signaling capacity of VEGF-A (Olsson et al., 2006). Integrin β1 is involved in several biological processes, including cell migration, cell adhesion, formation of basement membrane, and control of cell cycle (Mulrooney et al., 2001; Tanjore et al., 2008; Kanasaki et al., 2013b). Decreased expression of DPP-4 or integrin β1 inhibits TGFβ2-stimulated heterodimer formation of transforming growth factor β receptors (TGFβRs), thereby abolishing active TGFβ signaling (Figure 3A). Increased expression of TGFβ causes suppression of miR-29 (Qin et al., 2011) and increases the interaction between DPP-4 and integrin β1-induced VEGFR1 expression level with concomitant reduction of VEGFR2 expression levels, leading to active TGFβ and pro-EndMT signaling (Figure 3B). The DPP-4 inhibitor linagliptin is associated with EndMT inhibition by suppressing the interaction between DPP-4 and integrin β1 and elevating the miR-29 level (Kanasaki et al., 2014). TGFβ2 increases VEGFR1 levels, and TGFβ2-induced up-regulation of VEGFR1 can be suppressed by linagliptin (Figure 3C).
miR-let-7 family clusters demonstrate an anti-fibrotic role in lung fibrosis (Pandit et al., 2010; Rajasekaran et al., 2015), cardiac fibrosis (Wang et al., 2015), and renal fibrosis (Brennan et al., 2013; Srivastava et al., 2014; Srivastava et al., 2016). It was shown that TGFβ1 reinforces its signaling by mitigating miR-let-7b production, which targets the 3’UTR of TGFβR1 mRNA in rat TECs (Wang et al., 2014). Down-regulated miR-let-7b expression was found in mouse models of diabetic (Nagai et al., 2014) and non-diabetic renal fibrosis (Brennan et al., 2013). Similarly, miR-let-7c targets TGFβR1, collagen type 1 alpha 1 (COL1A1), collagen type 1 alpha 2 (COL1A2), and thrombospondin in human TECs (Brennan et al., 2013). Lipoxins, which are endogenously produced lipid mediators, decrease renal fibrosis in a UUO model in the rats by elevating miR-let-7c expression (Brennan et al., 2013), promote the resolution of inflammation, and inhibit fibrosis in cultured human proximal tubular epithelial (HK-2) cells (Brennan et al., 2013). Lipoxin A4 (LXA4) has been shown to decrease TGFβ1-induced expression of mesenchymal markers, i.e., fibronectin, N-cadherin, thrombospondin, and the notch ligand jagged-1 in HK-2 cells through a mechanism by inducing of miR-let-7c (Brennan et al., 2013). In the UUO model of renal fibrosis, the expression level of miR-let-7c was up-regulated by treatment with LXA4 analog. LXA4 treatment caused up-regulation of miR-let-7c and inhibited TGFβR1 and its associated signaling. Therefore, LXA4-associated up-regulation of miR-let-7c expression suppresses TGFβ1-induced fibrosis, which is a key pathway that is dysregulated in human renal fibrosis. We discussed the role of lipid mediators in diabetic nephropathy in our previous published review (Srivastava et al., 2014). Protein kinase C (PKC) activation and ceramides are associated with the suppression of antifibrotic microRNAs, and cumulative effects lead to the induction of fibrogenic processes in the kidney; several anti-dyslipidemic drugs have a differential effect on renal outcome (Srivastava et al., 2014). AcSDKP inhibits EndMT-driven renal fibrosis by ameliorating the miR-let-7 family clusters (Nagai et al., 2014; Nitta et al., 2016; Li et al., 2017) and the miR-let-7s-FGFR1 axis inhibits TGFβ signaling in fibrotic kidneys (Nagai et al., 2014). AcSDKP inhibits TGFβ–smad3 signaling and EndMT via activation of the fibroblast growth factor receptor 1 (FGFR1)–mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4) pathway (Li et al., 2017). AcSDKP-associated induction of MAP4K4 signaling inhibits integrin β1 phosphorylation, leading to anti-EndMT signals (Li et al., 2017). AcSDKP exerts anti-EndMT and antifibrotic effects in several mouse models of organ fibrosis (Nagai et al., 2014; Nitta et al., 2016; Srivastava et al., 2016). However, the precise molecular mechanisms by which AcSDKP suppresses TGFβ–smad3 signaling and EndMT are not fully investigated. FGFR1 is a key inhibitor of TGFβ-induced EndMT (Chen et al., 2014b). FGFR1 is critical in the AcSDKP-induced suppression of TGFβ-associated EndMT by elevating the level of miR-let-7 family clusters (Nagai et al., 2014). AcSDKP-associated activation of MAP4K4 suppresses DPP-4–integrin β1 signaling in endothelial cells (Vitorino et al., 2015) and DPP-4–integrin β1 influences TGFβ signaling and EndMT (Shi et al., 2015). MAP4K4 is a crucial downstream protein responsible for the anti-EndMT effect of AcSDKP (Li et al., 2017). Figure 4 depicts the contribution of interactions among AcSDKP, FGFR1, miR-let-7 family clusters, and MAP4K4 in endothelial cell homeostasis. The interaction between AcSDKP and FGFR1 mitigates the TGFβ–smad3 signaling associated EndMT by activating the MAP4K4 signaling pathway and by inducing miR-let-7 production. AcSDKP restores both diabetes-suppressed FGFR1 and MAP4K4 phosphorylation levels. The AcSDKP–FGFR1–MAP4K4 signaling axis offers significant information towards the understanding of endothelial cell homeostasis and provides a future target for the study of EndMT-associated organ fibrosis.
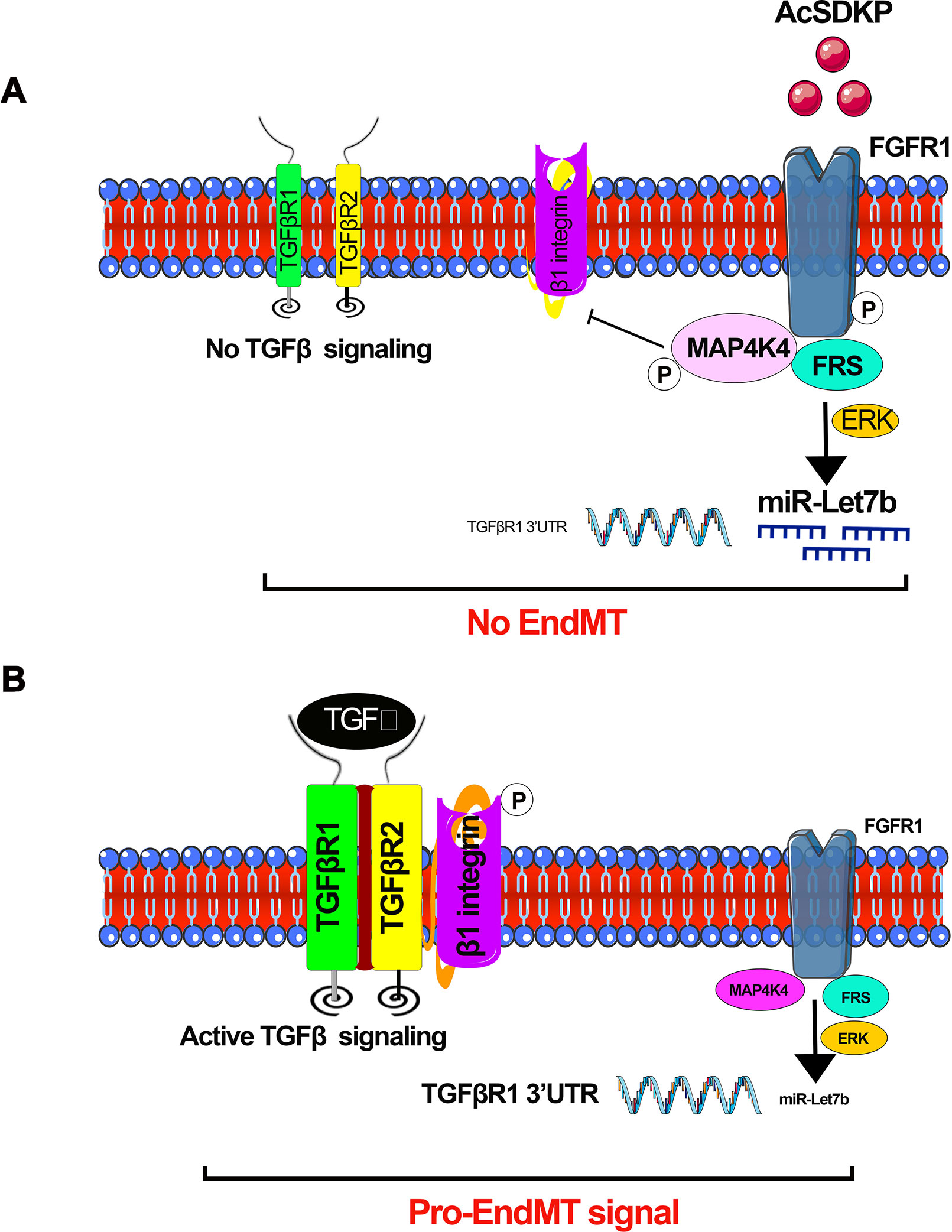
Figure 4 AcSDKP-mediated regulation of active TGFβ signaling and EndMT processes. (A) Phosphorylation of FGFR1 is the key mechanism for AcSDKP action. Phosphorylation of FGFR1 leads to activation of MAP4K4 signaling and miR-let-7 production. miR-let-7b targets the 3’UTR of TGFβR1 mRNA. AcSDKP-associated production of miR-let-7 family clusters and activation of MAP4K4 negatively regulates the DPP-4 integrin β1-associated active TGFβ signaling in the endothelial cells. (B) In the absence of AcSDKP, reduced FGFR1 phosphorylation leads to down-regulation of miR-let-7b gene expression and suppressed MAP4K4 signaling, finally resulting in active TGFβ signaling and activation of pro-EndMT signals.
Previous reports show that TGFβ down-regulates anti-fibrotic miRNAs such as miR-29 family clusters (Wang et al., 2012a). TGFβ1-regulated crosstalk of miRNAs was de-regulated early in type 1 diabetes subjects who had accelerated rates of progression to ESRD (Pezzolesi et al., 2015). In addition, clusters of the miR-29 family and the miR-let-7 family displayed crosstalk regulation. AcSDKP is a key peptide for the homeostasis of this crosstalk in HMVECs (Srivastava et al., 2016) (Figure 5). Interestingly, miR-29 family clusters have shown negative, bidirectional regulation with TGFβRs. miRNAs could be regulating gene expression of each other directly or indirectly. Such a novel crosstalk phenomenon could be associated with maintenance of an anti-fibrotic milieu in the kidney, and disruption of such a mechanism could accelerate renal fibrosis. Pharmacological interventions that prevent the disruption of this crosstalk may be beneficial in renal fibrosis. The DPP-4 inhibitor (linagliptin) has been shown to suppress EndMT-driven TGFβ signaling in STZ-induced renal fibrosis in diabetic CD-1 mice by inducing miR-29 family clusters (Kanasaki et al., 2014). miR-29 directly targets DPP-4; therefore, DPP-4 inhibition is proposed for the therapy of diabetic nephropathy (Kanasaki et al., 2014). The development of renal fibrosis in mice is largely dependent upon strain types (Srivastava et al., 2018). The CD-1 mouse is well-known as a fibrotic mouse strain, while 129sv and C57Bl6 mouse strains are less fibrotic (Srivastava et al., 2016; Srivastava et al., 2018). The suppression of miR-29 and miR-let-7 family clusters and the induction of TGFβ–smad3 signaling were observed in the fibrotic kidneys of diabetic CD-1 mice; however, such alterations were not observed in the less fibrotic kidneys of diabetic 129sv mice, suggesting that miR-29 and miR-let-7 family clusters play key roles in regulation of TGFβ signaling (Srivastava et al., 2016).
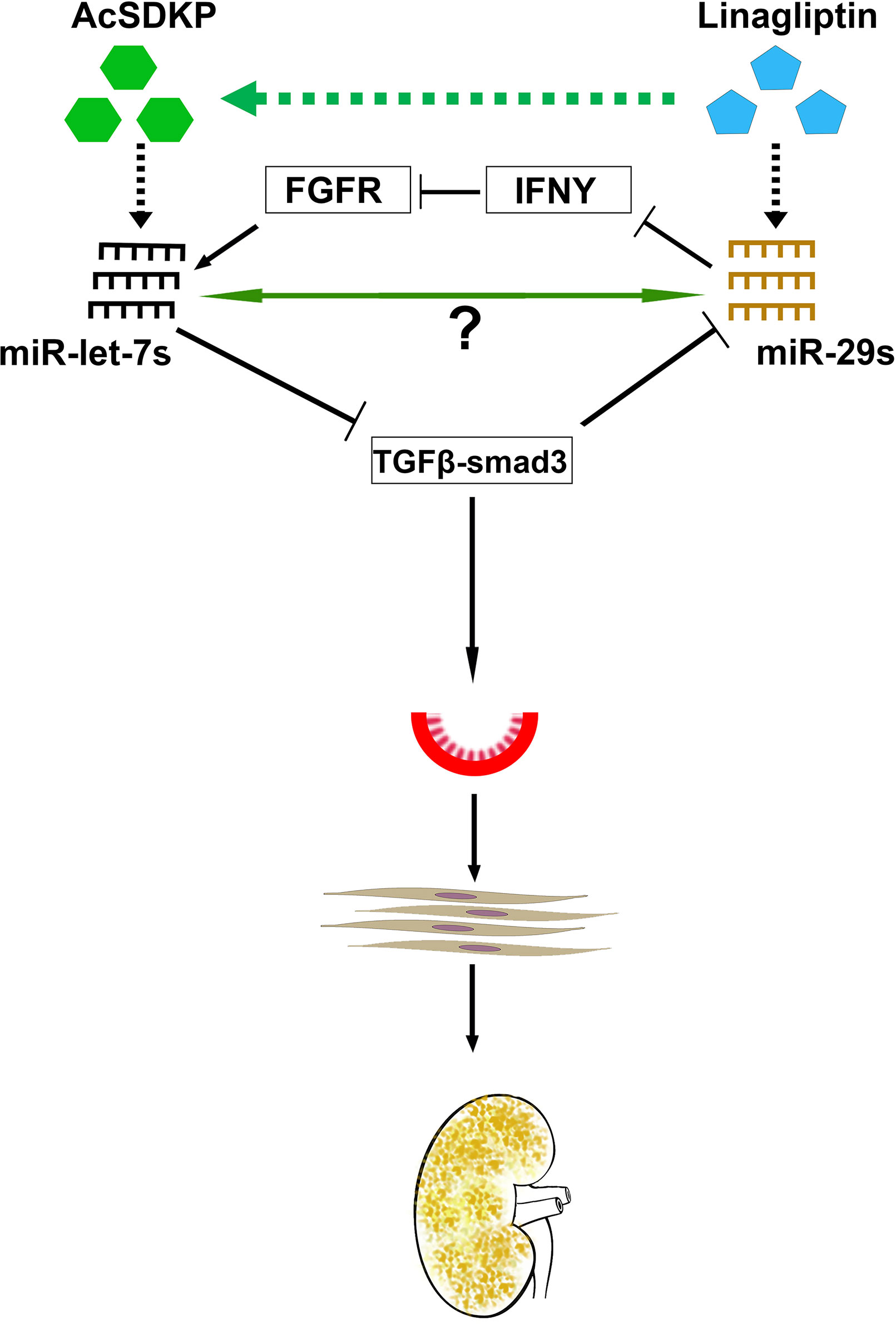
Figure 5 Antifibrotic microRNA crosstalk between miR-29s and miR-let7s is crucial for endothelial cell homeostasis. MiR-29 and miR-let-7 families show crosstalk regulation by inducing FGFR1 phosphorylation and targeting TGFβR1. AcSDKP potentiates crosstalk regulation in the endothelial cells, which is required for endothelial cell homeostasis. DPP-4 inhibition and/or AcSDKP elevate the crosstalk regulation in the endothelial cells. AcSDKP induces the production of miR-let-7 families; miR-let-7b targets TGFβR1 and TGFβ signaling. Suppressed levels of TGFβ signaling results in up-regulation of miR-29 gene expression, which in turn causes the FGFR1 phosphorylation. FGFR1 phosphorylation is critical for miR-let-7 production. In the presence of higher DPP-4 activity level or absence of AcSDKP, miR-let-7 families are down-regulated, which in turn causes activation of TGFβ signaling. Higher levels of TGFβ signaling results in suppression of miR-29 family expression and finally influences EndMT and fibrogenesis.
MiR-let-7 has been shown to inhibit TGFβR1 (Chen et al., 2012), and TGFβ–smad3 signaling has been demonstrated as an inhibitory pathway of miR-29 (Qin et al., 2011; Blahna and Hata, 2012; Wang et al., 2012a; Kanasaki et al., 2014); therefore, it was expected that miR-let-7 could induce the expression level of miR-29. An alternative mechanism of miR-29-associated miR-let-7 expression was explained by the interferon-gamma (IFNγ)–FGFR1 axis. miR-29 targets IFN-γ mRNA (Ma et al., 2011). However, IFN-γ has been shown to inhibit FGFR1. FGFR1 exhibits vital roles in the production of miR-let-7 family clusters (Chen et al., 2012). Suppressed miR-29 causes elevation of IFN -γ; subsequently, higher levels of synthesized IFN-γ discourage FGFR1 and FGFR1-associated expression of miR-let-7 family clusters. This suppression of miR-let-7 causes induction of TGFβR1 protein expression. Triggering TGF-β/smad3 signaling, in turn, inhibits the expression of miR-29 family clusters (Kanasaki et al., 2014). This series of events limits control over the crosstalk regulation between miR-29 and miR-let-7 during fibrotic events in kidneys of diabetic mice. AcSDKP contributes to kidney homeostasis, at least in part, by maintaining the anti-fibrotic crosstalk regulation between miR-29 and miR-let-7.
TGFβ1-linked renal fibrosis has been shown to associate with miR-192 and miR-200; however, TGFβ1 has shown inconsistent effects on miR-192 expression in various in vivo and in vitro models (Meng et al., 2015). TGFβ1 has been shown to have diverse regulation of miR-192 in cultured mesangial cells and cultured TECs (Kato et al., 2007; Chung et al., 2010; Wang et al., 2010; Putta et al., 2012). Likewise, higher expression levels of miR-192 were found in fibrotic kidneys of mice after UUO (Chung et al., 2010). Conversely, lower expression of miR-192 was found in the fibrotic kidneys from a rat 5/6 nephrectomy model (Chung et al., 2010; Sun et al., 2011); however, the expression level of miR-192 was both up-regulated and down-regulated in experimental mouse models of diabetic nephropathy (Kato et al., 2007; Wang et al., 2010; Putta et al., 2012). These conflicting results were due to variations in the animal models, differences in the disease stage analyzed, and/or the diverse in vitro experimental conditions used. In the early stages, biphasic induction of miR-192 by TGFβ1 in mouse mesangial cells involves the smad dependent pathway, which is followed by an induction of the concomitant mechanism that causes expression miRNA by loosening the compacted chromatin structure of the miR-192 gene via Ets1 and histone H3 acetylation (Kato et al., 2013). An identical finding was shown in the glomeruli of db/db mice. In contrast, normal levels of miR-192 expression are found in mouse mesangial cells and human TECSs. In this case, both hepatocyte nuclear factor (HNF) and p53 constitutive binding regions are present in its promoter. TGFβ1 suppresses miR-192 transcription by reducing the binding between HNF and the miR-192 gene (Jenkins et al., 2012). HNF expression is restricted to the tubular compartment; however, it is expressed neither in mesangial cells nor in podocytes (Igarashi et al., 2005), thus substantiating the cell-specific regulation of miR-192.
TGFβ1 treatment of mouse mesangial cells and glomeruli from diabetic mice leads to up-regulation of miR-192 and miR-200b/c expression; TGFβ1 treatment causes induction of Col1a2 and Col4a1 by suppressing the E-box repressors Zeb1 and Zeb2 (Kato et al., 2007; Kato et al., 2011; Putta et al., 2012). Clinical studies in Southwestern American Indians with type 2 diabetes have suggested that expression of miR-192 inversely correlates with Zeb1 and Zeb2 expression levels (Deshpande et al., 2013). In contrast, TGFβ1-induced down-regulation of the miR-200 family (Tang et al., 2013), miR-192 (Krupa et al., 2010; Wang et al., 2010), and miR-215 (Krupa et al., 2010) causes diminished levels of E-cadherin (as miRNA targets E-cadherin transcriptional repressors Zeb1 and Zeb2) in TECs, UUO models of fibrosis (Jenkins et al., 2012), and diabetic models of fibrosis (Wang et al., 2010).
miRNA-regulated circuits in mouse mesangial cells and in the glomeruli of diabetic mice cause amplification of TGFβ1 signaling by forming an auto-regulatory loop involving TGFβ1, miR-192, and miR-200 family members (Kato et al., 2011). TGFβ1 induces crosstalk between p53 and miR-192. Since miR-192 targets Zeb2, this crosstalk has been explained as an auto-regulatory loop in mesangial cells and glomeruli from the kidneys of diabetic mice (Deshpande et al., 2013).
To date, around 20 clinical trials have been launched using miRNA and siRNA-based therapeutics against several diseases (Chakraborty et al., 2017). SPC3649 (miravirsen, Santaris Pharama Denmark), which is an antagomir of miR-122, is the only miRNA-based therapeutic available for the treatment of hepatitis C virus infection (Janssen et al., 2013; Gebert et al., 2014). In recent years, therapeutic microRNAs are some significant biopharmaceuticals that are (or will be) in the commercial space as future medicine for the treatment of kidney diseases (Brandenburger et al., 2018). A recent advance in miRNA-based therapeutics (RG-012) is now in the pipeline to initiate a phase 2 clinical trial. RG-012 (anti-miR-21) is being developed by Regulas Therapeutics for the treatment of Alport nephropathy and its complication (Chau et al., 2012; Gomez et al., 2015).
Several issues have been noticed during the design of miRNA-based therapeutics related to the absorption, distribution, metabolism, and excretion (ADME) of new chemical molecules (Caldwell, 2000; Ruiz-Garcia et al., 2008). miRNA-based therapies are often less efficient candidates in terms of absorption (Khatsenko et al., 2000); a more efficient delivery system and more research is needed. Importantly, the delivery of miRNA-based therapy to the target tissues is challenging because of poor pharmacological properties including off targeting, low serum stability, and poor innate immune response (Miller, 2013; Hong and Nam, 2014). Recent advances in the available delivery systems of miRNA-based therapy, such as PEGylated liposome vesicles, are 50–100 nm, which prevents the medicine from being filtered by the kidneys (Love et al., 2010; Broderick and Zamore, 2011; Hong and Nam, 2014). Liposomal encapsulation technology can improve the half-life of therapeutic miRNAs in blood; this is an area of active research and development at pharmaceutical companies.
Some miRNAs display down-regulated status in kidney disease, suggesting protective roles. Anti-fibrotic mechanisms of miRNAs could be dependent on signaling molecules in TGFβ pathways or independent from TGFβ pathways, i.e., targeting signaling molecules of ECM-secreting pathways. miRNA-based therapeutics are superior to those of conventional drug approaches because they are able to target complex pathogenic gene networks. Further benefits include sustained outcomes, expansion of drug-ridden targets to virtually any miRNA, rapid drug development, and limited potential for drug interactions (Pottier et al., 2014; Morishita et al., 2015). Using efficient delivery methods such as liposome-based delivery or nanoparticle-based delivery systems can minimize both the dose required and the toxicity level, both of which could be beneficial for the treatment of kidney diseases.
microRNAs can be used as biomarkers and therapeutic targets for kidney diseases (Fujii et al., 2019; Nascimento and Domingueti, 2019). The challenges to translate their therapeutic potential to clinical applications are a subject of ongoing research. miRNA-based therapies offer a significant promise for the treatment of kidney diseases. miR-let-7c-5p and miR-29a-3p were significantly linked with protection against rapid progression of renal fibrosis, whereas miR-let-7b-5p and miR-21-5p were linked with higher risk of ESRD. Controlling HgbA1c and other covariates, miR-let-7c-5p and miR-29a-3p were associated with significant (>50%) decline in increased progression, whereas miR-let-7b-5p and miR-21-5p were linked with more than a 2.5-fold higher rapid risk of ESRD (Pezzolesi et al., 2015). Some microRNAs need further investigation to establish their potential. miR-200b pre-cursor has been shown to be anti-fibrotic and its mimic can ameliorate renal interstitial fibrosis in UUO kidneys (Oba et al., 2010). Similarly, urinary expression of levels of miR-29b and miR-29c is linked to proteinuria and kidney function in immunoglobulin A (IgA) nephropathy, while urinary levels of miR-93 are coordinated with glomerular scarring (Wang et al., 2012b). MicroRNAs regulating the M2-to-M1 phenotype macrophages regulate MMT processes in kidneys. MiR-9, miR-125b, miR-127, and miR-155 induce the M1 polarization, whereas miR-124, miR-233, miR-34a, miR-132, miR-146a, and miR-125a induce M2 polarization (Essandoh et al., 2016). MicroRNAs regulating the TGFβ signaling (miR-let-7 family and miR-29 family) or smad3 dependent suppression in the antifibrotic microRNAs can be crucial in the regulation of MMT processes. Moreover, this new area needs further investigation.
Altered metabolic states can alter the expression level of pro-fibrotic and anti-fibrotic microRNAs. Figure 6 depicts a hypothetical representation showing possible crosstalk among pro-fibrotic miRNAs (miR-33 and miR-21) and anti-fibrotic microRNAs though which mesenchymal activation is regulated. Hyperglycemia and hyperlipidemia up-regulate pro-fibrotic miRNAs, which could be a result of up-regulated TGFβ/smad3 signaling (Zhong et al., 2011; Chau et al., 2012; Kumarswamy et al., 2012; Nishiga et al., 2017). TGFβ signaling has been shown to up-regulate miR-21 (Liu et al., 2016; Kolling et al., 2017; Schauerte et al., 2017; Chau et al., 2012; Wang et al., 2012c; Kim, 2018; Sun et al., 2018) and down-regulate miR-29 (Qin et al., 2011). Identification of novel miRNA crosstalk mechanisms in the kidney is quite relevant to the understating of renal health and disease. Restoring anti-fibrotic miRNA crosstalk mechanisms provides renal protection. Physiologically relevant anti-fibrotic crosstalk may potentially be useful in combating diabetic kidney disease.
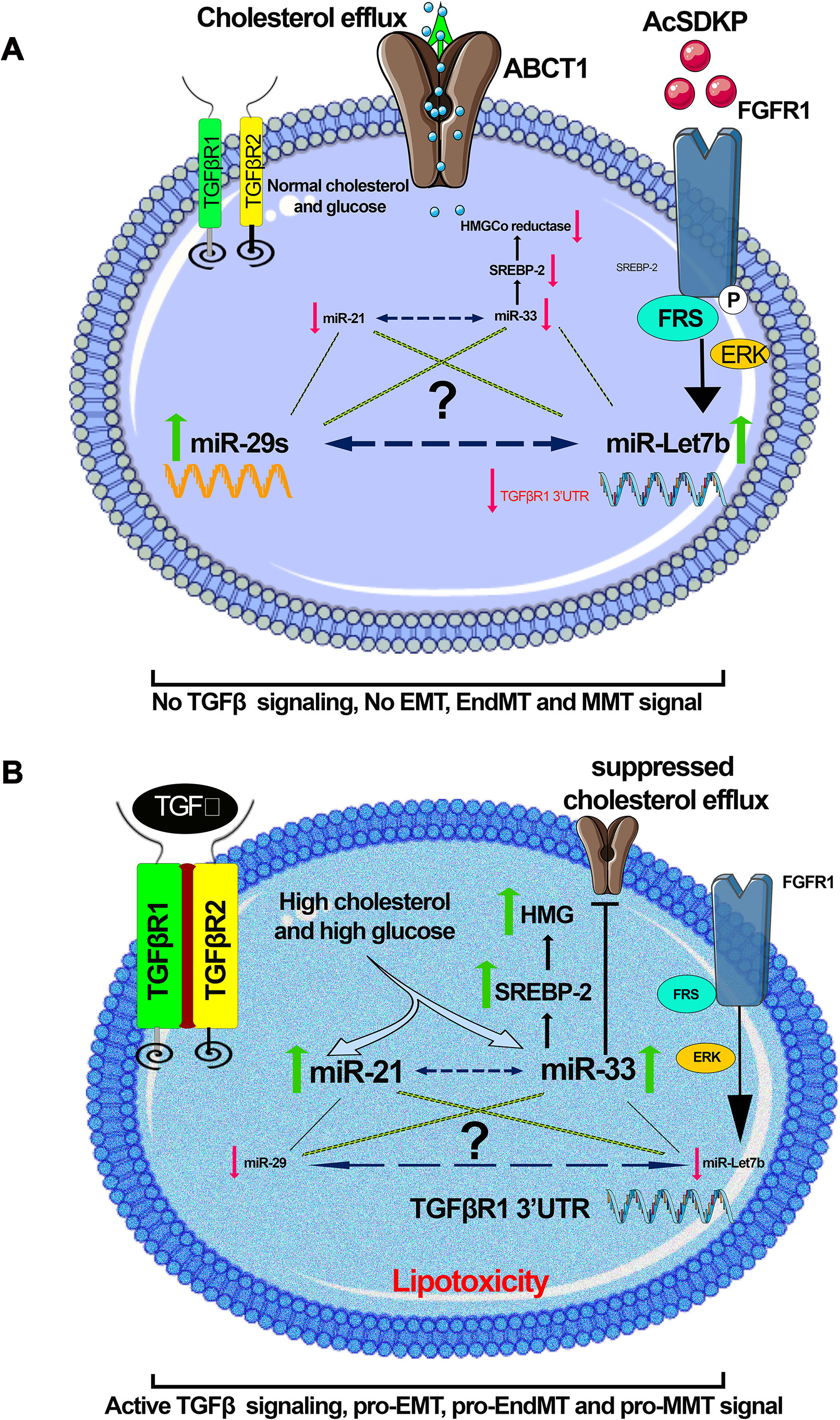
Figure 6 A hypothetical schematic diagram demonstration. (A) Anti-fibrotic; in the presence of AcSDKP, normal glucose, and lipid level, miR-29 and let-7 are found at normal expression level, whereas the expression level of miR-21 and miR-33 is down-regulated. (B) Profibrotic; in the absence of AcSDKP, the presence of hyperglycemia and hyperlipidemia suppresses the expression level of miR-29 and miR-let-7 but induces the expression level of profibrotic microRNA (miR-21 and miR-33) and influences mesenchymal activation in epithelial cells, endothelial cells, and M2 phenotype macrophages. Hyperglycemia is linked with up-regulation in the expression level of miR-21, whereas hyperlipidemia is found to be associated with miR-33. There would be a possibility that a kind of crosstalk mechanism exists among anti-fibrotic and pro-fibrotic microRNAs, which influences the EMT, EndMT, and MMT processes, and the endogenous peptide AcSDKP regulates such profibrotic mechanisms. Figures were created using the Servier Medical Art illustration resources.
SS wrote the manuscript, made the figures, and provided intellectual output. AH helped in editing. KK and JG provided intellectual output in the manuscript.
JG is supported by the National Heart, Lung and Blood Institute Grant R01-HL-131952.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Alicic, R. Z., Rooney, M. T., Tuttle, K. R. (2017). Diabetic kidney disease: challenges, progress, and possibilities. Clin. J. Am. Soc. Nephrol. 12, 2032–2045. doi: 10.2215/CJN.11491116
Alidadiani, N., Ghaderi, S., Dilaver, N., Bakhshamin, S., Bayat, M. (2018). Epithelial mesenchymal transition transcription factor (TF): the structure, function and microRNA feedback loop. Gene 674, 115–120. doi: 10.1016/j.gene.2018.06.049
Allison, S. J. (2019). Ubiquitylation of PTEN drives fibrosis in diabetic kidney disease. Nat. Rev. Nephrol. 15, 254. doi: 10.1038/s41581-019-0130-y
Amrouche, L., Bonifay, R., Anglicheau, D. (2011). MicroRNAs in pathophysiology of renal disease: an increasing interest. Med. Sci. (Paris) 27, 398–404. doi: 10.1051/medsci/2011274398
Assmann, T. S., Recamonde-Mendoza, M., De Souza, B. M., Bauer, A. C., Crispim, D. (2018). MicroRNAs and diabetic kidney disease: systematic review and bioinformatic analysis. Mol. Cell. Endocrinol. 477, 90–102. doi: 10.1016/j.mce.2018.06.005
Bai, X. Y., Ma, Y., Ding, R., Fu, B., Shi, S., Chen, X. M. (2011). miR-335 and miR-34a Promote renal senescence by suppressing mitochondrial antioxidative enzymes. J. Am. Soc. Nephrol. 22, 1252–1261. doi: 10.1681/ASN.2010040367
Barnes, J. L., Gorin, Y. (2011). Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int. 79, 944–956. doi: 10.1038/ki.2010.516
Bhatt, K., Mi, Q. S., Dong, Z. (2011). microRNAs in kidneys: biogenesis, regulation, and pathophysiological roles. Am. J. Physiol. Ren. Physiol. 300, F602–F610. doi: 10.1152/ajprenal.00727.2010
Bhatt, K., Zhou, L., Mi, Q. S., Huang, S., She, J. X., Dong, Z. (2010). MicroRNA-34a is induced via p53 during cisplatin nephrotoxicity and contributes to cell survival. Mol. Med. 16, 409–416. doi: 10.2119/molmed.2010.00002
Blahna, M. T., Hata, A. (2012). Smad-mediated regulation of microRNA biosynthesis. FEBS Lett 586, 1906–1912. doi: 10.1016/j.febslet.2012.01.041
Bontha, S. V., Maluf, D. G., Archer, K. J., Dumur, C. I., Dozmorov, M. G., King, A. L., et al. (2017). Effects of DNA methylation on progression to interstitial fibrosis and tubular atrophy in renal allograft biopsies: a multi-omics approach. Am. J. Transplant. 17, 3060–3075. doi: 10.1111/ajt.14372
Boor, P., Floege, J. (2015). Renal allograft fibrosis: biology and therapeutic targets. Am. J. Transplant. 15, 863–886. doi: 10.1111/ajt.13180
Brandenburger, T., Salgado Somoza, A., Devaux, Y., Lorenzen, J. M. (2018). Noncoding RNAs in acute kidney injury. Kidney Int. 94, 870–881. doi: 10.1016/j.kint.2018.06.033
Brennan, E. P., Nolan, K. A., Borgeson, E., Gough, O. S., Mcevoy, C. M., Docherty, N. G., et al. (2013). Lipoxins attenuate renal fibrosis by inducing let-7c and suppressing TGFbetaR1. J. Am. Soc. Nephrol. 24, 627–637. doi: 10.1681/ASN.2012060550
Brenner, B. M., Cooper, M. E., De Zeeuw, D., Keane, W. F., Mitch, W. E., Parving, H. H., et al. (2001). Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 345, 861–869. doi: 10.1056/NEJMoa011161
Breyer, M. D., Susztak, K. (2016). The next generation of therapeutics for chronic kidney disease. Nat. Rev. Drug Discov. 15, 568–588. doi: 10.1038/nrd.2016.67
Broderick, J. A., Zamore, P. D. (2011). MicroRNA therapeutics. Gene Ther. 18, 1104–1110. doi: 10.1038/gt.2011.50
Caldwell, G. W. (2000). Compound optimization in early- and late-phase drug discovery: acceptable pharmacokinetic properties utilizing combined physicochemical, in vitro and in vivo screens. Curr. Opin. Drug Discov. Devel. 3, 30–41.
Chakraborty, C., Sharma, A. R., Sharma, G., Doss, C. G. P., Lee, S. S. (2017). Therapeutic miRNA and siRNA: moving from bench to clinic as next generation medicine. Mol. Ther. Nucleic Acids 8, 132–143. doi: 10.1016/j.omtn.2017.06.005
Chandrasekaran, K., Karolina, D. S., Sepramaniam, S., Armugam, A., Wintour, E. M., Bertram, J. F., et al. (2012). Role of microRNAs in kidney homeostasis and disease. Kidney Int. 81, 617–627. doi: 10.1038/ki.2011.448
Chau, B. N., Xin, C., Hartner, J., Ren, S., Castano, A. P., Linn, G., et al. (2012). MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci. Transl. Med. 4, 121ra118. doi: 10.1126/scitranslmed.3003205
Chen, H. Y., Zhong, X., Huang, X. R., Meng, X. M., You, Y., Chung, A. C., et al. (2014a). MicroRNA-29b inhibits diabetic nephropathy in db/db mice. Mol. Ther. 22, 842–853. doi: 10.1038/mt.2013.235
Chen, P. Y., Qin, L., Barnes, C., Charisse, K., Yi, T., Zhang, X., et al. (2012). FGF regulates TGF-beta signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep. 2, 1684–1696. doi: 10.1016/j.celrep.2012.10.021
Chen, P. Y., Qin, L., Tellides, G., Simons, M. (2014b). Fibroblast growth factor receptor 1 is a key inhibitor of TGFbeta signaling in the endothelium. Sci. Signal 7, ra90. doi: 10.1126/scisignal.2005504
Cho, J. G., Lee, A., Chang, W., Lee, M. S., Kim, J. (2018). Endothelial to mesenchymal transition represents a key link in the interaction between inflammation and endothelial dysfunction. Front. Immunol. 9, 294. doi: 10.3389/fimmu.2018.00294
Chung, A. C., Lan, H. Y. (2015). MicroRNAs in renal fibrosis. Front. Physiol. 6, 50. doi: 10.3389/fphys.2015.00050
Chung, A. C., Huang, X. R., Meng, X., Lan, H. Y. (2010). miR-192 mediates TGF-beta/Smad3-driven renal fibrosis. J. Am. Soc. Nephrol. 21, 1317–1325. doi: 10.1681/ASN.2010020134
Cooper, M., Warren, A. M. (2019). A promising outlook for diabetic kidney disease. Nat. Rev. Nephrol. 15, 68–70. doi: 10.1038/s41581-018-0092-5
Curci, C., Castellano, G., Stasi, A., Divella, C., Loverre, A., Gigante, M., et al. (2014). Endothelial-to-mesenchymal transition and renal fibrosis in ischaemia/reperfusion injury are mediated by complement anaphylatoxins and Akt pathway. Nephrol. Dial Transplant. 29, 799–808. doi: 10.1093/ndt/gft516
Deshpande, S. D., Putta, S., Wang, M., Lai, J. Y., Bitzer, M., Nelson, R. G., et al. (2013). Transforming growth factor-beta-induced cross talk between p53 and a microRNA in the pathogenesis of diabetic nephropathy. Diabetes 62, 3151–3162. doi: 10.2337/db13-0305
Di Carlo, S. E., Peduto, L. (2018). The perivascular origin of pathological fibroblasts. J. Clin. Invest. 128, 54–63. doi: 10.1172/JCI93558
Djudjaj, S., Boor, P. (2019). Cellular and molecular mechanisms of kidney fibrosis. Mol. Aspects Med. 65, 16–36. doi: 10.1016/j.mam.2018.06.002
Du, B., Ma, L. M., Huang, M. B., Zhou, H., Huang, H. L., Shao, P., et al. (2010). High glucose down-regulates miR-29a to increase collagen IV production in HK-2 cells. FEBS Lett. 584, 811–816. doi: 10.1016/j.febslet.2009.12.053
Dufton, N. P., Peghaire, C. R., Osuna-Almagro, L., Raimondi, C., Kalna, V., Chuahan, A., et al. (2017). Dynamic regulation of canonical TGFbeta signalling by endothelial transcription factor ERG protects from liver fibrogenesis. Nat. Commun. 8, 895. doi: 10.1038/s41467-017-01169-0
El Agha, E., Kramann, R., Schneider, R. K., Li, X., Seeger, W., Humphreys, B. D., et al. (2017). Mesenchymal stem cells in fibrotic disease. Cell Stem Cell 21, 166–177. doi: 10.1016/j.stem.2017.07.011
Elvira-Matelot, E., Zhou, X. O., Farman, N., Beaurain, G., Henrion-Caude, A., Hadchouel, J., et al. (2010). Regulation of WNK1 expression by miR-192 and aldosterone. J. Am. Soc. Nephrol. 21, 1724–1731. doi: 10.1681/ASN.2009111186
Essandoh, K., Li, Y., Huo, J., Fan, G. C. (2016). MiRNA-mediated macrophage polarization and its potential role in the regulation of inflammatory response. Shock 46, 122–131. doi: 10.1097/SHK.0000000000000604
Fujii, R., Yamada, H., Munetsuna, E., Yamazaki, M., Ohashi, K., Ishikawa, H., et al. (2019). Associations of circulating microRNAs (miR-17, miR-21, and miR-150) and chronic kidney disease in a Japanese population. J. Epidemiol. doi: 10.2188/jea.JE20180233
Gebert, L. F., Rebhan, M. A., Crivelli, S. E., Denzler, R., Stoffel, M., Hall, J. (2014). Miravirsen (SPC3649) can inhibit the biogenesis of miR-122. Nucleic Acids Res. 42, 609–621. doi: 10.1093/nar/gkt852
Gebert, L. F. R., Macrae, I. J. (2019). Regulation of microRNA function in animals. Nat. Rev. Mol. Cell. Biol. 20, 21–37. doi: 10.1038/s41580-018-0045-7
Glover, E. K., Jordan, N., Sheerin, N. S., Ali, S. (2019). Regulation of endothelial-to-mesenchymal transition by microRNAs in chronic allograft dysfunction. Transplantation 103, e64–e73. doi: 10.1097/TP.0000000000002589
Gomez, I. G., Mackenna, D. A., Johnson, B. G., Kaimal, V., Roach, A. M., Ren, S., et al. (2015). Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J. Clin. Invest. 125, 141–156. doi: 10.1172/JCI75852
Good, R. B., Gilbane, A. J., Trinder, S. L., Denton, C. P., Coghlan, G., Abraham, D. J., et al. (2015). Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary arterial hypertension. Am. J. Pathol. 185, 1850–1858. doi: 10.1016/j.ajpath.2015.03.019
Grande, M. T., Sanchez-Laorden, B., Lopez-Blau, C., De Frutos, C. A., Boutet, A., Arevalo, M., et al. (2015). Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 21, 989–997. doi: 10.1038/nm.3901
Grgic, I., Duffield, J. S., Humphreys, B. D. (2012). The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr. Nephrol. 27, 183–193. doi: 10.1007/s00467-011-1772-6
Guan, S., Zhou, J. (2017). CXCR7 attenuates the TGF-beta-induced endothelial-to-mesenchymal transition and pulmonary fibrosis. Mol. Biosyst. 13, 2116–2124. doi: 10.1039/C7MB00247E
Hajarnis, S., Yheskel, M., Williams, D., Brefort, T., Glaudemans, B., Debaix, H., et al. (2018). Suppression of microRNA activity in kidney collecting ducts induces partial loss of epithelial phenotype and renal fibrosis. J. Am. Soc. Nephrol. 29, 518–531. doi: 10.1681/ASN.2017030334
He, Y., Huang, C., Lin, X., Li, J. (2013). MicroRNA-29 family, a crucial therapeutic target for fibrosis diseases. Biochimie 95, 1355–1359. doi: 10.1016/j.biochi.2013.03.010
Hills, C. E., Squires, P. E. (2011). The role of TGF-beta and epithelial-to mesenchymal transition in diabetic nephropathy. Cytokine Growth Factor Rev. 22, 131–139. doi: 10.1016/j.cytogfr.2011.06.002
Hong, C. A., Nam, Y. S. (2014). Functional nanostructures for effective delivery of small interfering RNA therapeutics. Theranostics 4, 1211–1232. doi: 10.7150/thno.8491
Igarashi, P., Shao, X., Mcnally, B. T., Hiesberger, T. (2005). Roles of HNF-1beta in kidney development and congenital cystic diseases. Kidney Int. 68, 1944–1947. doi: 10.1111/j.1523-1755.2005.00625.x
Isaka, Y. (2018). Targeting TGF-beta signaling in kidney fibrosis. Int. J. Mol. Sci. 19, 1–13. doi: 10.3390/ijms19092532
Janssen, H. L., Reesink, H. W., Lawitz, E. J., Zeuzem, S., Rodriguez-Torres, M., Patel, K., et al. (2013). Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 368, 1685–1694. doi: 10.1056/NEJMoa1209026
Jenkins, R. H., Martin, J., Phillips, A. O., Bowen, T., Fraser, D. J. (2012). Transforming growth factor beta1 represses proximal tubular cell microRNA-192 expression through decreased hepatocyte nuclear factor DNA binding. Biochem. J. 443, 407–416. doi: 10.1042/BJ20111861
Johnson, D. S., Kapoian, T., Taylor, R., Meyer, K. B. (2016). Going upstream: coordination to improve CKD care. Semin. Dial 29, 125–134. doi: 10.1111/sdi.12461
Kalluri, R., Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428. doi: 10.1172/JCI39104
Kanasaki, K., Shi, S., Kanasaki, M., He, J., Nagai, T., Nakamura, Y., et al. (2014). Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes 63, 2120–2131. doi: 10.2337/db13-1029
Kanasaki, K., Taduri, G., Koya, D. (2013a). Diabetic nephropathy: the role of inflammation in fibroblast activation and kidney fibrosis. Front. Endocrinol. (Lausanne) 4, 7. doi: 10.3389/fendo.2013.00007
Kanasaki, K., Yu, W., Von Bodungen, M., Larigakis, J. D., Kanasaki, M., Ayala De La Pena, F., et al. (2013b). Loss of beta1-integrin from urothelium results in overactive bladder and incontinence in mice: a mechanosensory rather than structural phenotype. FASEB J. 27, 1950–1961. doi: 10.1096/fj.12-223404
Kato, M., Arce, L., Wang, M., Putta, S., Lanting, L., Natarajan, R. (2011). A microRNA circuit mediates transforming growth factor-beta1 autoregulation in renal glomerular mesangial cells. Kidney Int. 80(4), 358–368. doi: 10.1038/ki.2011.43
Kato, M., Dang, V., Wang, M., Park, J. T., Deshpande, S., Kadam, S., et al. (2013). TGF-beta induces acetylation of chromatin and of Ets-1 to alleviate repression of miR-192 in diabetic nephropathy. Sci. Signal 6, ra43. doi: 10.1126/scisignal.2003389
Kato, M., Natarajan, R. (2015). MicroRNAs in diabetic nephropathy: functions, biomarkers, and therapeutic targets. Ann. N. Y. Acad. Sci. 1353(1), 72–88. doi: 10.1111/nyas.12758
Kato, M., Wang, L., Putta, S., Wang, M., Yuan, H., Sun, G., et al. (2010). Post-transcriptional up-regulation of Tsc-22 by Ybx1, a target of miR-216a, mediates TGF-{beta}-induced collagen expression in kidney cells. J. Biol. Chem. 285, 34004–34015. doi: 10.1074/jbc.M110.165027
Kato, M., Zhang, J., Wang, M., Lanting, L., Yuan, H., Rossi, J. J., et al. (2007). MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc. Natl. Acad. Sci. U. S. A. 104, 3432–3437. doi: 10.1073/pnas.0611192104
Kaur, K., Pandey, A. K., Srivastava, S., Srivastava, A. K., Datta, M. (2011). Comprehensive miRNome and in silico analyses identify the Wnt signaling pathway to be altered in the diabetic liver. Mol. Biosyst. 7, 3234–3244. doi: 10.1039/c1mb05041a
Khatsenko, O., Morgan, R., Truong, L., York-Defalco, C., Sasmor, H., Conklin, B., et al. (2000). Absorption of antisense oligonucleotides in rat intestine: effect of chemistry and length. Antisense Nucleic Acid Drug Dev. 10, 35–44. doi: 10.1089/oli.1.2000.10.35
Kim, D. H., Xing, T., Yang, Z., Dudek, R., Lu, Q., Chen, Y. H. (2017). Epithelial mesenchymal transition in embryonic development, tissue repair and cancer: a comprehensive overview. J. Clin. Med. 7, 1–25. doi: 10.3390/jcm7010001
Kim, J. (2018). MicroRNAs as critical regulators of the endothelial to mesenchymal transition in vascular biology. BMB Rep. 51, 65–72. doi: 10.5483/BMBRep.2018.51.2.011
Kitao, A., Sato, Y., Sawada-Kitamura, S., Harada, K., Sasaki, M., Morikawa, H., et al. (2009). Endothelial to mesenchymal transition via transforming growth factor-beta1/Smad activation is associated with portal venous stenosis in idiopathic portal hypertension. Am. J. Pathol. 175, 616–626. doi: 10.2353/ajpath.2009.081061
Kolling, M., Kaucsar, T., Schauerte, C., Hubner, A., Dettling, A., Park, J. K., et al. (2017). Therapeutic miR-21 silencing ameliorates diabetic kidney disease in mice. Mol. Ther. 25, 165–180. doi: 10.1016/j.ymthe.2016.08.001
Kovacic, J. C., Dimmeler, S., Harvey, R. P., Finkel, T., Aikawa, E., Krenning, G., et al. (2019). Endothelial to mesenchymal transition in cardiovascular disease: JACC state-of-the-art review. J. Am. Coll. Cardiol. 73, 190–209. doi: 10.1016/j.jacc.2018.09.089
Krupa, A., Jenkins, R., Luo, D. D., Lewis, A., Phillips, A., Fraser, D. (2010). Loss of MicroRNA-192 promotes fibrogenesis in diabetic nephropathy. J. Am. Soc. Nephrol. 21, 438–447. doi: 10.1681/ASN.2009050530
Kumarswamy, R., Volkmann, I., Jazbutyte, V., Dangwal, S., Park, D. H., Thum, T. (2012). Transforming growth factor-beta-induced endothelial-to-mesenchymal transition is partly mediated by microRNA-21. Arterioscler. Thromb. Vasc. Biol. 32, 361–369. doi: 10.1161/ATVBAHA.111.234286
Kwan, T., Wu, H., Chadban, S. J. (2014). Macrophages in renal transplantation: roles and therapeutic implications. Cell. Immunol. 291, 58–64. doi: 10.1016/j.cellimm.2014.05.009
Lan, H. Y. (2012). Transforming growth factor-beta/Smad signalling in diabetic nephropathy. Clin. Exp. Pharmacol. Physiol. 39, 731–738. doi: 10.1111/j.1440-1681.2011.05663.x
Lebleu, V. S., Taduri, G., O’connell, J., Teng, Y., Cooke, V. G., Woda, C., et al. (2013). Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 19, 1047–1053. doi: 10.1038/nm.3218
Lee, J. G., Jung, E., Heur, M. (2018). Fibroblast growth factor 2 induces proliferation and fibrosis via SNAI1-mediated activation of CDK2 and ZEB1 in corneal endothelium. J. Biol. Chem. 293, 3758–3769. doi: 10.1074/jbc.RA117.000295
Lee, S. B., Kalluri, R. (2010). Mechanistic connection between inflammation and fibrosis. Kidney Int. Suppl. 119, S22–S26. doi: 10.1038/ki.2010.418
Lee, S. O., Masyuk, T., Splinter, P., Banales, J. M., Masyuk, A., Stroope, A., et al. (2008). MicroRNA15a modulates expression of the cell-cycle regulator Cdc25A and affects hepatic cystogenesis in a rat model of polycystic kidney disease. J. Clin. Invest. 118, 3714–3724. doi: 10.1172/JCI34922
Lee, S. Y., Kim, S. I., Choi, M. E. (2015). Therapeutic targets for treating fibrotic kidney diseases. Transl. Res. 165, 512–530. doi: 10.1016/j.trsl.2014.07.010
Li, J., Qu, X., Bertram, J. F. (2009). Endothelial-myofibroblast transition contributes to the early development of diabetic renal interstitial fibrosis in streptozotocin-induced diabetic mice. Am. J. Pathol. 175, 1380–1388. doi: 10.2353/ajpath.2009.090096
Li, J., Shi, S., Srivastava, S. P., Kitada, M., Nagai, T., Nitta, K., et al. (2017). FGFR1 is critical for the anti-endothelial mesenchymal transition effect of N-acetyl-seryl-aspartyl-lysyl-proline via induction of the MAP4K4 pathway. Cell. Death. Dis. 8, e2965. doi: 10.1038/cddis.2017.353
Li, J. Y., Yong, T. Y., Michael, M. Z., Gleadle, J. M. (2010). Review: the role of microRNAs in kidney disease. Nephrology (Carlton) 15, 599–608. doi: 10.1111/j.1440-1797.2010.01363.x
Liu, T., Liu, L., Liu, M., Du, R., Dang, Y., Bai, M., et al. (2019a). MicroRNA-493 targets STMN-1 and promotes hypoxia-induced epithelial cell cycle arrest in G2/M and renal fibrosis. FASEB J. 33, 1565–1577. doi: 10.1096/fj.201701355RR
Liu, X., Hong, Q., Wang, Z., Yu, Y., Zou, X., Xu, L. (2016). Transforming growth factor-beta-sphingosine kinase 1/S1P signaling upregulates microRNA-21 to promote fibrosis in renal tubular epithelial cells. Exp. Biol. Med. (Maywood) 241, 265–272. doi: 10.1177/1535370215605586
Liu, X., Sun, N., Mo, N., Lu, S., Song, E., Ren, C., et al. (2019b). Quercetin inhibits kidney fibrosis and the epithelial to mesenchymal transition of the renal tubular system involving suppression of the Sonic Hedgehog signaling pathway. Food Funct. 10, 3782–3797. doi: 10.1039/C9FO00373H
Liu, Y. (2011). Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 7, 684–696. doi: 10.1038/nrneph.2011.149
Liu, Y., Bi, X., Xiong, J., Han, W., Xiao, T., Xu, X., et al. (2019c). MicroRNA-34a promotes renal fibrosis by downregulation of Klotho in tubular epithelial cells. Mol. Ther. 27(5), 1051–1065. doi: 10.1016/j.ymthe.2019.02.009
Liu, Y., Wang, K., Liang, X., Li, Y., Zhang, Y., Zhang, C., et al. (2018). Complement C3 Produced by Macrophages Promotes Renal Fibrosis via IL-17A Secretion. Front. Immunol. 9, 2385. doi: 10.3389/fimmu.2018.02385
Lorenzen, J. M., Haller, H., Thum, T. (2011). MicroRNAs as mediators and therapeutic targets in chronic kidney disease. Nat. Rev. Nephrol. 7, 286–294. doi: 10.1038/nrneph.2011.26
Love, K. T., Mahon, K. P., Levins, C. G., Whitehead, K. A., Querbes, W., Dorkin, J. R., et al. (2010). Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. U. S. A. 107, 1864–1869. doi: 10.1073/pnas.0910603106
Lovisa, S., Kalluri, R. (2018). Fatty acid oxidation regulates the activation of endothelial-to-mesenchymal transition. Trends Mol. Med. 24, 432–434. doi: 10.1016/j.molmed.2018.03.003
Lovisa, S., Lebleu, V. S., Tampe, B., Sugimoto, H., Vadnagara, K., Carstens, J. L., et al. (2015). Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 21, 998–1009. doi: 10.1038/nm.3902
Luyckx, V. A., Tonelli, M., Stanifer, J. W. (2018). The global burden of kidney disease and the sustainable development goals. Bull. World Health Organ 96, 414–422D. doi: 10.2471/BLT.17.206441
Lv, W., Fan, F., Wang, Y., Gonzalez-Fernandez, E., Wang, C., Yang, L., et al. (2018). Therapeutic potential of microRNAs for the treatment of renal fibrosis and CKD. Physiol. Genomics 50, 20–34. doi: 10.1152/physiolgenomics.00039.2017
Ma, F., Xu, S., Liu, X., Zhang, Q., Xu, X., Liu, M., et al. (2011). The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-gamma. Nat. Immunol. 12, 861–869. doi: 10.1038/ni.2073
Ma, F. Y., Woodman, N., Mulley, W. R., Kanellis, J., Nikolic-Paterson, D. J. (2013). Macrophages contribute to cellular but not humoral mechanisms of acute rejection in rat renal allografts. Transplantation 96, 949–957. doi: 10.1097/TP.0b013e3182a4befa
Macconi, D., Tomasoni, S., Romagnani, P., Trionfini, P., Sangalli, F., Mazzinghi, B., et al. (2012). MicroRNA-324-3p promotes renal fibrosis and is a target of ACE inhibition. J. Am. Soc. Nephrol. 23, 1496–1505. doi: 10.1681/ASN.2011121144
Mack, M., Yanagita, M. (2015). Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 87, 297–307. doi: 10.1038/ki.2014.287
Man, S., Sanchez Duffhues, G., Ten Dijke, P., Baker, D. (2019). The therapeutic potential of targeting the endothelial-to-mesenchymal transition. Angiogenesis 22, 3–13. doi: 10.1007/s10456-018-9639-0
Marquez-Exposito, L., Lavoz, C., Rodrigues-Diez, R. R., Rayego-Mateos, S., Orejudo, M., Cantero-Navarro, E., et al. (2018). Gremlin regulates tubular epithelial to mesenchymal transition via VEGFR2: potential role in renal fibrosis. Front. Pharmacol. 9, 1195. doi: 10.3389/fphar.2018.01195
Medici, D. (2016). Endothelial-mesenchymal transition in regenerative medicine. Stem Cells Int. 2016, 6962801. doi: 10.1155/2016/6962801
Medici, D., Kalluri, R. (2012). Endothelial-mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin. Cancer Biol. 22, 379–384. doi: 10.1016/j.semcancer.2012.04.004
Meng, X. M., Chung, A. C., Lan, H. Y. (2013). Role of the TGF-beta/BMP-7/Smad pathways in renal diseases. Clin. Sci. (Lond.) 124, 243–254. doi: 10.1042/CS20120252
Meng, X. M., Tang, P. M., Li, J., Lan, H. Y. (2015). TGF-beta/Smad signaling in renal fibrosis. Front. Physiol. 6, 82. doi: 10.3389/fphys.2015.00082
Metzinger-Le Meuth, V., Fourdinier, O., Charnaux, N., Massy, Z. A., Metzinger, L. (2019). The expanding roles of microRNAs in kidney pathophysiology. Nephrol. Dial Transplant. 34, 7–15. doi: 10.1093/ndt/gfy140
Miao, C., Zhang, G., Xie, Z., Chang, J. (2018). MicroRNAs in the pathogenesis of type 2 diabetes: new research progress and future direction. Can. J. Physiol. Pharmacol. 96, 103–112. doi: 10.1139/cjpp-2017-0452
Miller, A. D. (2013). Delivery of RNAi therapeutics: work in progress. Expert Rev. Med. Devices 10, 781–811. doi: 10.1586/17434440.2013.855471
Morishita, Y., Imai, T., Yoshizawa, H., Watanabe, M., Ishibashi, K., Muto, S., et al. (2015). Delivery of microRNA-146a with polyethylenimine nanoparticles inhibits renal fibrosis in vivo. Int. J. Nanomed. 10, 3475–3488. doi: 10.2147/IJN.S82587
Mulrooney, J. P., Hong, T., Grabel, L. B. (2001). Serine 785 phosphorylation of the beta1 cytoplasmic domain modulates beta1A-integrin-dependent functions. J. Cell. Sci. 114, 2525–2533.
Nadeem, A., Ashraf, M. R., Javed, M., Hussain, T., Tariq, M. S., Babar, M. E. (2018). Review—MicroRNAs: a new paradigm towards mechanistic insight of diseases. Pak. J. Pharm. Sci. 31, 2017–2026.
Nagai, T., Kanasaki, M., Srivastava, S. P., Nakamura, Y., Ishigaki, Y., Kitada, M., et al. (2014). N-acetyl-seryl-aspartyl-lysyl-proline inhibits diabetes-associated kidney fibrosis and endothelial-mesenchymal transition. Biomed. Res. Int. 2014, 696475. doi: 10.1155/2014/696475
Nakano, Y., Oyamada, M., Dai, P., Nakagami, T., Kinoshita, S., Takamatsu, T. (2008). Connexin43 knockdown accelerates wound healing but inhibits mesenchymal transition after corneal endothelial injury in vivo. Invest. Ophthalmol. Vis. Sci. 49, 93–104. doi: 10.1167/iovs.07-0255
Nascimento, L. R. D., Domingueti, C. P. (2019). MicroRNAs: new biomarkers and promising therapeutic targets for diabetic kidney disease. J. Bras. Nefrol. doi: 10.1590/2175-8239-jbn-2018-0165
Nishiga, M., Horie, T., Kuwabara, Y., Nagao, K., Baba, O., Nakao, T., et al. (2017). MicroRNA-33 controls adaptive fibrotic response in the remodeling heart by preserving lipid raft cholesterol. Circ. Res. 120, 835–847. doi: 10.1161/CIRCRESAHA.116.309528
Nitta, K., Shi, S., Nagai, T., Kanasaki, M., Kitada, M., Srivastava, S. P., et al. (2016). Oral administration of N-acetyl-seryl-aspartyl-lysyl-proline ameliorates kidney disease in both type 1 and type 2 diabetic mice via a therapeutic regimen. Biomed. Res. Int. 2016, 9172157. doi: 10.1155/2016/9172157
Nogueira, A., Pires, M. J., Oliveira, P. A. (2017). Pathophysiological mechanisms of renal fibrosis: a review of animal models and therapeutic strategies. In Vivo 31, 1–22. doi: 10.21873/invivo.11019
Oba, S., Kumano, S., Suzuki, E., Nishimatsu, H., Takahashi, M., Takamori, H., et al. (2010). miR-200b precursor can ameliorate renal tubulointerstitial fibrosis. PLoS One 5, e13614. doi: 10.1371/journal.pone.0013614
Olsson, A. K., Dimberg, A., Kreuger, J., Claesson-Welsh, L. (2006). VEGF receptor signalling—in control of vascular function. Nat. Rev. Mol. Cell. Biol. 7, 359–371. doi: 10.1038/nrm1911
Pandey, A. K., Verma, G., Vig, S., Srivastava, S., Srivastava, A. K., Datta, M. (2011). miR-29a levels are elevated in the db/db mice liver and its overexpression leads to attenuation of insulin action on PEPCK gene expression in HepG2 cells. Mol. Cell. Endocrinol. 332, 125–133. doi: 10.1016/j.mce.2010.10.004
Pandit, K. V., Corcoran, D., Yousef, H., Yarlagadda, M., Tzouvelekis, A., Gibson, K. F., et al. (2010). Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 182, 220–229. doi: 10.1164/rccm.200911-1698OC
Pardali, E., Sanchez-Duffhues, G., Gomez-Puerto, M. C., Ten Dijke, P. (2017). TGF-beta-induced endothelial-mesenchymal transition in fibrotic diseases. Int. J. Mol. Sci. 18, 1–22. doi: 10.3390/ijms18102157
Parving, H. H. (2001). Diabetic nephropathy: prevention and treatment. Kidney Int. 60, 2041–2055. doi: 10.1046/j.1523-1755.2001.00020.x
Pezzolesi, M. G., Satake, E., Mcdonnell, K. P., Major, M., Smiles, A. M., Krolewski, A. S. (2015). Circulating TGF-beta1-regulated miRNAs and the risk of rapid progression to ESRD in type 1 diabetes. Diabetes. 64(9), 3285–3293. doi: 10.2337/db15-0116
Pottier, N., Cauffiez, C., Perrais, M., Barbry, P., Mari, B. (2014). FibromiRs: translating molecular discoveries into new anti-fibrotic drugs. Trends Pharmacol. Sci. 35, 119–126. doi: 10.1016/j.tips.2014.01.003
Putta, S., Lanting, L., Sun, G., Lawson, G., Kato, M., Natarajan, R. (2012). Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J. Am. Soc. Nephrol. 23, 458–469. doi: 10.1681/ASN.2011050485
Qi, R., Yang, C. (2018). Renal tubular epithelial cells: the neglected mediator of tubulointerstitial fibrosis after injury. Cell. Death. Dis. 9, 1126. doi: 10.1038/s41419-018-1157-x
Qin, W., Chung, A. C., Huang, X. R., Meng, X. M., Hui, D. S., Yu, C. M., et al. (2011). TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol. 22, 1462–1474. doi: 10.1681/ASN.2010121308
Quiroga, B., Arroyo, D., De Arriba, G. (2015). Present and future in the treatment of diabetic kidney disease. J. Diabetes Res. 2015, 801348. doi: 10.1155/2015/801348
Rajasekaran, S., Rajaguru, P., Sudhakar Gandhi, P. S. (2015). MicroRNAs as potential targets for progressive pulmonary fibrosis. Front. Pharmacol. 6, 254. doi: 10.3389/fphar.2015.00254
Ramdas, V., Mcbride, M., Denby, L., Baker, A. H. (2013). Canonical transforming growth factor-beta signaling regulates disintegrin metalloprotease expression in experimental renal fibrosis via miR-29. Am. J. Pathol. 183, 1885–1896. doi: 10.1016/j.ajpath.2013.08.027
Ranchoux, B., Antigny, F., Rucker-Martin, C., Hautefort, A., Pechoux, C., Bogaard, H. J., et al. (2015). Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 131, 1006–1018. doi: 10.1161/CIRCULATIONAHA.114.008750
Regmi, A., Liu, G., Zhong, X., Hu, S., Ma, R., Gou, L., et al. (2019). Evaluation of serum microRNAs in patients with diabetic kidney disease: a nested case-controlled study and bioinformatics analysis. Med .Sci. Monit. 25, 1699–1708. doi: 10.12659/MSM.913265
Remuzzi, G., Schieppati, A., Ruggenenti, P. (2002). Clinical practice. Nephropathy in patients with type 2 diabetes. N. Engl. J. Med. 346, 1145–1151. doi: 10.1056/NEJMcp011773
Ruiz, M. A., Chakrabarti, S. (2013). MicroRNAs: the underlying mediators of pathogenetic processes in vascular complications of diabetes. Can. J. Diabetes 37, 339–344. doi: 10.1016/j.jcjd.2013.07.003
Ruiz-Garcia, A., Bermejo, M., Moss, A., Casabo, V. G. (2008). Pharmacokinetics in drug discovery. J. Pharm. Sci. 97, 654–690. doi: 10.1002/jps.21009
Salehi, S., Reed, E. F. (2015). The divergent roles of macrophages in solid organ transplantation. Curr. Opin. Organ Transplant. 20, 446–453. doi: 10.1097/MOT.0000000000000209
Sato, Y., Yanagita, M. (2017). Resident fibroblasts in the kidney: a major driver of fibrosis and inflammation. Inflamm. Regen. 37, 17. doi: 10.1186/s41232-017-0048-3
Schauerte, C., Hubner, A., Rong, S., Wang, S., Shushakova, N., Mengel, M., et al. (2017). Antagonism of profibrotic microRNA-21 improves outcome of murine chronic renal allograft dysfunction. Kidney Int. 92, 646–656. doi: 10.1016/j.kint.2017.02.012
Shah, M. Y., Ferrajoli, A., Sood, A. K., Lopez-Berestein, G., Calin, G. A. (2016). microRNA therapeutics in cancer—an emerging concept. EBioMedicine 12, 34–42. doi: 10.1016/j.ebiom.2016.09.017
Shi, S., Srivastava, S. P., Kanasaki, M., He, J., Kitada, M., Nagai, T., et al. (2015). Interactions of DPP-4 and integrin beta1 influences endothelial-to-mesenchymal transition. Kidney Int. 88, 479–489. doi: 10.1038/ki.2015.103
Srivastava, S. P., Koya, D., Kanasaki, K. (2013). MicroRNAs in kidney fibrosis and diabetic nephropathy: roles on EMT and EndMT. Biomed. Res. Int. 2013, 125469. doi: 10.1155/2013/125469
Srivastava, S. P., Li, J., Kitada, M., Fujita, H., Yamada, Y., Goodwin, J. E., et al. (2018). SIRT3 deficiency leads to induction of abnormal glycolysis in diabetic kidney with fibrosis. Cell. Death. Dis. 9, 997. doi: 10.1038/s41419-018-1057-0
Srivastava, S. P., Shi, S., Kanasaki, M., Nagai, T., Kitada, M., He, J., et al. (2016). Effect of antifibrotic microRNAs crosstalk on the action of N-acetyl-seryl-aspartyl-lysyl-proline in diabetes-related kidney fibrosis. Sci. Rep. 6, 29884. doi: 10.1038/srep29884
Srivastava, S. P., Shi, S., Koya, D., Kanasaki, K. (2014). Lipid mediators in diabetic nephropathy. Fibrogenesis Tissue Repair 7, 12. doi: 10.1186/1755-1536-7-12
Sun, L., Zhang, D., Liu, F., Xiang, X., Ling, G., Xiao, L., et al. (2011). Low-dose paclitaxel ameliorates fibrosis in the remnant kidney model by down-regulating miR-192. J. Pathol. 225, 364–377. doi: 10.1002/path.2961
Sun, Q., Miao, J., Luo, J., Yuan, Q., Cao, H., Su, W., et al. (2018). The feedback loop between miR-21, PDCD4 and AP-1 functions as a driving force for renal fibrogenesis. J. Cell. Sci. 131, 1–13. doi: 10.1242/jcs.202317
Tan, W., Liu, B., Qu, S., Liang, G., Luo, W., Gong, C. (2018). MicroRNAs and cancer: key paradigms in molecular therapy. Oncol. Lett. 15, 2735–2742. doi: 10.3892/ol.2017.7638
Tang, O., Chen, X. M., Shen, S., Hahn, M., Pollock, C. A. (2013). MiRNA-200b represses transforming growth factor-beta1-induced EMT and fibronectin expression in kidney proximal tubular cells. Am. J. Physiol. Ren. Physiol. 304, F1266–F1273. doi: 10.1152/ajprenal.00302.2012
Tanjore, H., Zeisberg, E. M., Gerami-Naini, B., Kalluri, R. (2008). Beta1 integrin expression on endothelial cells is required for angiogenesis but not for vasculogenesis. Dev. Dyn. 237, 75–82. doi: 10.1002/dvdy.21385
Thomas, M. J., Fraser, D. J., Bowen, T. (2018). Biogenesis, stabilization, and transport of microRNAs in kidney health and disease. Noncoding RNA 4, 1–13. doi: 10.3390/ncrna4040030
Umanath, K., Lewis, J. B. (2018). Update on diabetic nephropathy: core curriculum 2018. Am. J. Kidney Dis. 71, 884–895. doi: 10.1053/j.ajkd.2017.10.026
Van Der Hauwaert, C., Savary, G., Hennino, M. F., Pottier, N., Glowacki, F., Cauffiez, C. (2015). MicroRNAs in kidney fibrosis. Nephrol. Ther. 11(6), 474–482.
Vitorino, P., Yeung, S., Crow, A., Bakke, J., Smyczek, T., West, K., et al. (2015). MAP4K4 regulates integrin-FERM binding to control endothelial cell motility. Nature 519, 425–430. doi: 10.1038/nature14323
Wang, B., Herman-Edelstein, M., Koh, P., Burns, W., Jandeleit-Dahm, K., Watson, A., et al. (2010). E-cadherin expression is regulated by miR-192/215 by a mechanism that is independent of the profibrotic effects of transforming growth factor-beta. Diabetes 59, 1794–1802. doi: 10.2337/db09-1736
Wang, B., Jha, J. C., Hagiwara, S., Mcclelland, A. D., Jandeleit-Dahm, K., Thomas, M. C., et al. (2014). Transforming growth factor-beta1-mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let-7b. Kidney Int. 85, 352–361. doi: 10.1038/ki.2013.372
Wang, B., Komers, R., Carew, R., Winbanks, C. E., Xu, B., Herman-Edelstein, M., et al. (2012a). Suppression of microRNA-29 expression by TGF-beta1 promotes collagen expression and renal fibrosis. J. Am. Soc. Nephrol. 23, 252–265. doi: 10.1681/ASN.2011010055
Wang, G., Kwan, B. C., Lai, F. M., Chow, K. M., Li, P. K., Szeto, C. C. (2012b). Urinary miR-21, miR-29, and miR-93: novel biomarkers of fibrosis. Am. J. Nephrol. 36, 412–418. doi: 10.1159/000343452
Wang, T., Zhang, L., Shi, C., Sun, H., Wang, J., Li, R., et al. (2012c). TGF-beta-induced miR-21 negatively regulates the antiproliferative activity but has no effect on EMT of TGF-beta in HaCaT cells. Int. J. Biochem. Cell. Biol. 44, 366–376. doi: 10.1016/j.biocel.2011.11.012
Wang, X., Wang, H. X., Li, Y. L., Zhang, C. C., Zhou, C. Y., Wang, L., et al. (2015). MicroRNA Let-7i negatively regulates cardiac inflammation and fibrosis. Hypertension 66, 776–785. doi: 10.1161/HYPERTENSIONAHA.115.05548
Wang, Y., Pang, L., Zhang, Y., Lin, J., Zhou, H. (2019). Fenofibrate improved interstitial fibrosis of renal allograft through inhibited epithelial-mesenchymal transition induced by oxidative stress. Oxid. Med. Cell. Longev. 2019, 8936856. doi: 10.1155/2019/8936856
Wang, Y. Y., Jiang, H., Pan, J., Huang, X. R., Wang, Y. C., Huang, H. F., et al. (2017). Macrophage-to-myofibroblast transition contributes to interstitial fibrosis in chronic renal allograft injury. J. Am. Soc. Nephrol. 28, 2053–2067. doi: 10.1681/ASN.2016050573
Wei, Q., Mi, Q. S., Dong, Z. (2013). The regulation and function of microRNAs in kidney diseases. IUBMB Life 65, 602–614. doi: 10.1002/iub.1174
Xi, W., Zhao, X., Wu, M., Jia, W., Li, H. (2018). Lack of microRNA-155 ameliorates renal fibrosis by targeting PDE3A/TGF-beta1/Smad signaling in mice with obstructive nephropathy. Cell. Biol. Int. 42, 1523–1532. doi: 10.1002/cbin.11038
Xing, J., Tian, X. J. (2019). Investigating epithelial-to-mesenchymal transition with integrated computational and experimental approaches. Phys. Biol. 16, 031001. doi: 10.1088/1478-3975/ab0032
Xiong, J., Kawagishi, H., Yan, Y., Liu, J., Wells, Q. S., Edmunds, L. R., et al. (2018). A metabolic basis for endothelial-to-mesenchymal transition. Mol. Cell. 69, 689-698, e687. doi: 10.1016/j.molcel.2018.01.010
Yan, J., Zhang, Z., Jia, L., Wang, Y. (2016). Role of bone marrow-derived fibroblasts in renal fibrosis. Front. Physiol. 7, 61. doi: 10.3389/fphys.2016.00061
Yang, J., Lin, S. C., Chen, G., He, L., Hu, Z., Chan, L., et al. (2013). Adiponectin promotes monocyte-to-fibroblast transition in renal fibrosis. J. Am. Soc. Nephrol. 24, 1644–1659. doi: 10.1681/ASN.2013030217
Yang, S., Abdulla, R., Lu, C., Zhang, L. (2018). Inhibition of microRNA-376b protects against renal interstitial fibrosis via inducing macrophage autophagy by upregulating Atg5 in mice with chronic kidney disease. Kidney Blood Press. Res. 43, 1749–1764. doi: 10.1159/000495394
Zanchi, C., Macconi, D., Trionfini, P., Tomasoni, S., Rottoli, D., Locatelli, M., et al. (2017). MicroRNA-184 is a downstream effector of albuminuria driving renal fibrosis in rats with diabetic nephropathy. Diabetologia 60, 1114–1125. doi: 10.1007/s00125-017-4248-9
Zeisberg, E. M., Potenta, S. E., Sugimoto, H., Zeisberg, M., Kalluri, R. (2008). Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 19, 2282–2287. doi: 10.1681/ASN.2008050513
Zeisberg, E. M., Tarnavski, O., Zeisberg, M., Dorfman, A. L., Mcmullen, J. R., Gustafsson, E., et al. (2007). Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13, 952–961. doi: 10.1038/nm1613
Zhang, L., Li, R., He, J., Yang, Q., Wu, Y., Huang, J., et al. (2017). Co-expression analysis among microRNAs, long non-coding RNAs, and messenger RNAs to understand the pathogenesis and progression of diabetic kidney disease at the genetic level. Methods 124, 46–56. doi: 10.1016/j.ymeth.2017.05.023
Zhang, Y., Huang, X. R., Wei, L. H., Chung, A. C., Yu, C. M., Lan, H. Y. (2014). miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-beta/Smad3 signaling. Mol. Ther. 22, 974–985. doi: 10.1038/mt.2014.25
Zhang, Y., Zhao, S., Wu, D., Liu, X., Shi, M., Wang, Y., et al. (2018). MicroRNA-22 promotes renal tubulointerstitial fibrosis by targeting PTEN and suppressing autophagy in diabetic nephropathy. J. Diabetes Res. 2018, 4728645. doi: 10.1155/2018/4728645
Zhao, H., Ma, S. X., Shang, Y. Q., Zhang, H. Q., Su, W. (2019). microRNAs in chronic kidney disease. Clin. Chim. Acta 491, 59–65. doi: 10.1016/j.cca.2019.01.008
Zheng, S. B., Zheng, Y., Jin, L. W., Zhou, Z. H., Li, Z. Y. (2018). Microvesicles containing microRNA-21 secreted by proximal tubular epithelial cells are involved in renal interstitial fibrosis by activating AKT pathway. Eur. Rev. Med. Pharmacol. Sci. 22, 707–714. doi: 10.26355/eurrev_201802_14297
Zhong, X., Chung, A. C., Chen, H. Y., Meng, X. M., Lan, H. Y. (2011). Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J. Am. Soc. Nephrol. 22, 1668–1681. doi: 10.1681/ASN.2010111168
Keywords: microRNAs, diabetic kidney disease, kidney fibrosis, microRNA crosstalk, epithelial-to-mesenchymal transition, endothelial-to-mesenchymal transition, macrophage-to-mesenchymal transition
Citation: Srivastava SP, Hedayat AF, Kanasaki K and Goodwin JE (2019) microRNA Crosstalk Influences Epithelial-to-Mesenchymal, Endothelial-to-Mesenchymal, and Macrophage-to-Mesenchymal Transitions in the Kidney. Front. Pharmacol. 10:904. doi: 10.3389/fphar.2019.00904
Received: 25 January 2019; Accepted: 18 July 2019;
Published: 16 August 2019.
Edited by:
Johan M. Lorenzen, University of Zurich, SwitzerlandReviewed by:
Regalla Kumarswamy, Centre for Cellular and Molecular Biology (CSIR), IndiaCopyright © 2019 Srivastava, Hedayat, Kanasaki and Goodwin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Swayam Prakash Srivastava, c3dheWFtLnNyaXZhc3RhdmFAeWFsZS5lZHU=; Keizo Kanasaki, a2thbmFzYWtAbWVkLnNoaW1hbmUtdS5hYy5qcA==; Julie Goodwin, anVsaWUuZ29vZHdpbkB5YWxlLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.