 Sara Grassi
Sara Grassi Laura Mauri†
Laura Mauri† Livia Cabitta
Livia Cabitta Sandro Sonnino
Sandro Sonnino Alessandro Prinetti
Alessandro Prinetti Paola Giussani
Paola Giussani
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Pharmacol. , 23 July 2019
Sec. Experimental Pharmacology and Drug Discovery
Volume 10 - 2019 | https://doi.org/10.3389/fphar.2019.00807
This article is part of the Research Topic New Paradigms in Neuroscience and Related Targets for Drug Discovery, Volume I View all 21 articles
The central nervous system is characterized by a high content of sphingolipids and by a high diversity in terms of different structures. Stage- and cell-specific sphingolipid metabolism and expression are crucial for brain development and maintenance toward adult age. On the other hand, deep dysregulation of sphingolipid metabolism, leading to altered sphingolipid pattern, is associated with the majority of neurological and neurodegenerative diseases, even those totally lacking a common etiological background. Thus, sphingolipid metabolism has always been regarded as a promising pharmacological target for the treatment of brain disorders. However, any therapeutic hypothesis applied to complex amphipathic sphingolipids, components of cellular membranes, has so far failed probably because of the high regional complexity and specificity of the different biological roles of these structures. Simpler sphingosine-based lipids, including ceramide and sphingosine 1-phosphate, are important regulators of brain homeostasis, and, thanks to the relative simplicity of their metabolic network, they seem a feasible druggable target for the treatment of brain diseases. The enzymes involved in the control of the levels of bioactive sphingoids, as well as the receptors engaged by these molecules, have increasingly allured pharmacologists and clinicians, and eventually fingolimod, a functional antagonist of sphingosine 1-phosphate receptors with immunomodulatory properties, was approved for the therapy of relapsing–remitting multiple sclerosis. Considering the importance of neuroinflammation in many other brain diseases, we would expect an extension of the use of such analogs for the treatment of other ailments in the future. Nevertheless, many aspects other than neuroinflammation are regulated by bioactive sphingoids in healthy brain and dysregulated in brain disease. In this review, we are addressing the multifaceted possibility to address the metabolism and biology of bioactive sphingosine 1-phosphate as novel targets for the development of therapeutic paradigms and the discovery of new drugs.
Sphingolipids are a wide group of eukaryotic cellular lipids, whose common structural feature is the presence of a 2-amino-1,3-dihydroxy-octadec-4-ene, trivially known as “sphingosine” (Roisen et al., 1981) (2S,3R,4E). In addition to the C18 molecular species, which is usually the most abundant in complex sphingolipids in mammals, structures with shorter and longer alkyl chains have been identified as minor components in different biological samples, and complex glycosphingolipids containing sphingosine with 20 carbon atoms are relatively abundant in mammalian brain and in cultured neurons (Sonnino and Chigorno, 2000). Usually, the term “sphingolipids” is referred to as the complex amphipathic lipids of this family, whose hydrophobic moiety is represented by ceramide. In ceramide, the sphingosine is linked via an amide bond with a fatty acid, whose structure can greatly vary for its acyl chain length and the presence of double bonds. Ceramide is the precursor for the biosynthesis of amphipathic sphingolipids, characterized by the presence of a polar head group of different nature and chemical complexity linked to the hydroxyl group at the position 1 of ceramide. Phosphocholine is the hydrophilic head group of sphingomyelin (SM) (the only membrane phosphosphingolipid present in mammals). On the other hand, a phosphate group linked to the hydroxyl group is present in sphingosine 1-phosphate and ceramide-1-phosphate. In glycosphingolipids, the polar head group is represented by a single sugar (in cerebrosides) or by an oligosaccharide chain that can be relatively complex, as in polysialogangliosides. In addition to glycosphingolipids containing neutral sugars, two families of glycosphingolipids in eukaryotes are characterized by the presence of acidic residues in the oligosaccharide chain: sulfatides, containing one or more sulfate groups, bond via O-glycosidic linkage on a glucose or galactose residue; and gangliosides, hallmarked by the presence of one or more sialic acids, a family of sugars characterized by the presence of a carboxyl group (Schauer, 1982) (Figure 1).
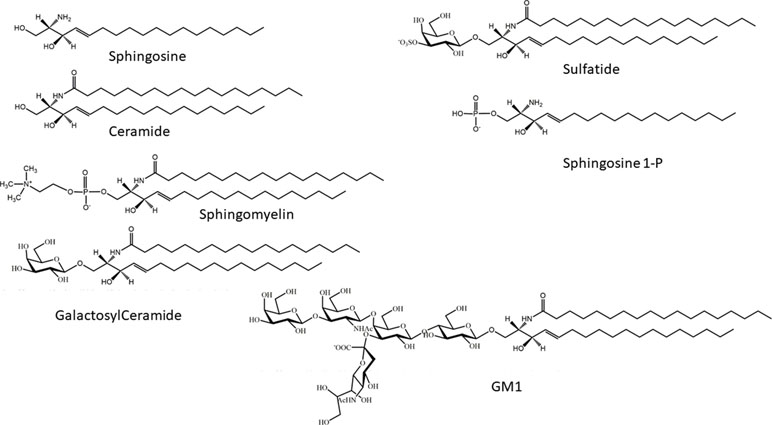
Figure 1 Chemical structures of sphingosine, ceramide, sphingosine 1-phosphate, galactosylceramide, sphingomyelin, and monosialoganglioside GM1.
Sphingolipids are in general minor cell membrane components. Most glycosphingolipids do not form bilayers spontaneously in aqueous environment; however, they can accommodate in the glycerophospholipid bilayer, thanks to their hydrophobic moiety, ceramide. Sphingolipids are ubiquitous components of mammalian cell membranes; however, their distribution is highly organ- and tissue-specific. Indeed, in some cases, imaging mass spectrometry is also revealing a cell-specific distribution. For example, neuron subpopulations located in very close proximity can bear dramatic differences in their sphingolipid repertoire (Hirano-Sakamaki et al., 2015; Sugiyama and Setou, 2018). The nervous system is the mammalian tissue with the highest concentration in sphingolipids. Within the nervous system, SM is highly expressed in oligodendrocytes and in neurons. Galactosylceramide and its sulfated derivative, 3-O-sulfogalactosyceramide (sulfatide) are highly expressed in myelin, whereas polysialogangliosides of the ganglio series are abundant in neurons (we showed that ganglio series gangliosides represent about 5% of total amphipathic lipids in cultured cerebellar granule cells; Prinetti et al., 2000). Even if minor components in average cell membranes, sphingolipids are highly concentrated in the plasma membrane and asymmetrically distributed in the exoplasmic leaflet. Moreover, sphingolipids are not homogeneously distributed at the cell surface, but they tend to cluster, forming sphingolipid-rich membrane areas or “domains” (Sonnino et al., 2007; Sonnino et al., 2014); thus, their local concentration within a specific membrane microenvironment can be very high. Plasma membrane sphingolipids, and in particular glycosphingolipids (Sonnino and Prinetti, 2010), do exert important biological functions, affecting the properties of plasma membrane-associated proteins (e.g., growth factor receptors, adhesion molecules) via direct interactions or via the modulation of the protein membrane microenvironment (Loberto et al., 2005; Rivaroli et al., 2007). Glycosphingolipid-mediated interactions can also involve ligands occurring on adjacent cells (trans interactions). For example, the trans interaction between sulfatide and galactosylceramide clustered in distinct membrane microdomains at the juxtaposed membranes of myelin wrap plays a relevant role in the proper maintenance of mature myelin function, as well as in myelin formation (Boggs et al., 2010). On the axonal side, the polysialogangliosides GD1a and GT1b organized in clusters do engage trans interactions with the glycoprotein MAG on the innermost myelin sheath (Schnaar and Lopez, 2009), contributing to long-term axon–myelin stability.
Ceramide, synthesized in the endoplasmic reticulum (ER), is the common precursor for the biosynthesis of SM and glycosphingolipids (Figure 2). In turn, degradation of SM and glycosphingolipids at the lysosomal level or in another subcellular localization can yield ceramide. Lysosomal ceramide is the key intermediate of complete catabolism of complex sphingolipids. Sphingosine, generated by ceramide hydrolysis, can be phosphorylated from two known sphingosine kinases (SK1 and SK2) to produce sphingosine 1-phosphate (S1P) (Figure 1), which is irreversibly cleaved by an S1P lyase, yielding phosphoethanolamine and a fatty aldehyde (Figure 2). For decades, ceramide, sphingosine, and S1P have been regarded solely as intermediates in complex sphingolipid metabolism. However, from the end of the 1980s, a number of papers appeared, describing the effects of these molecules on a plethora of cellular targets, implying their involvement in several biological processes. For these relatively simple sphingosine-based molecules, the term “bioactive sphingoids” has been coined.
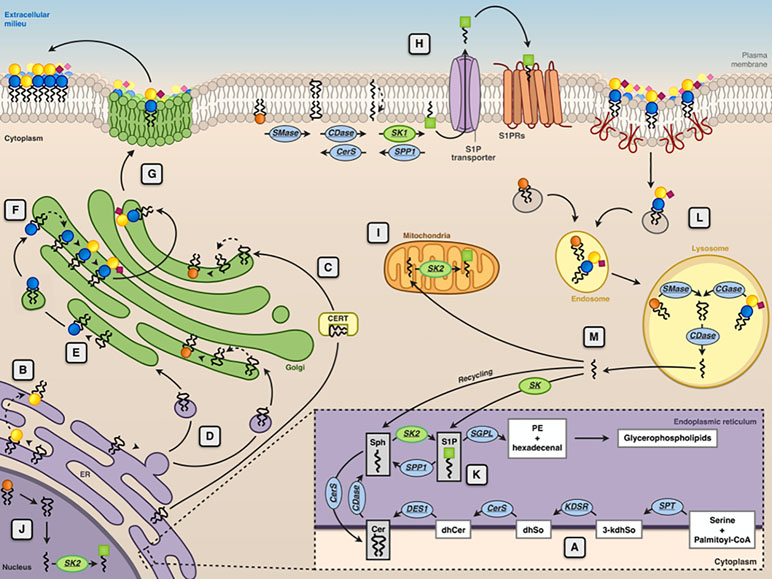
Figure 2 Subcellular compartmentalization of sphingolipid metabolism. The endoplasmic reticulum (ER) is the subcellular site where the de novo ceramide (Cer) synthesis occurs (A). Here, the synthesis of the sphingoid bases (sphingosine) starts with the condensation of palmitoyl-CoA and serine, catalyzed by serine palmitoyltransferase (SPT). The product of this reaction is 3-ketodihydrosphingosine (3-kdhSo), which is later reduced to dihydrosphingosine (dhSo) via the action of 3-ketodihydrosphingosine reductase (KDSR). Then, dhSo is acylated generating dihydroceramide (dhCer). In humans, this reaction occurs through the activities of six different ceramide synthases (CerS). Dihydroceramide is then unsaturated to ceramide by the enzyme dihydroceramide desaturase 1 (DES1). At ER level, Cer is either transformed into GalCer ( ) by CGalT (B) or delivered by ceramide transport protein (CERT) (C) or through vesicular transport (D) to the Golgi apparatus for the synthesis of sphingomyelin (SM
) by CGalT (B) or delivered by ceramide transport protein (CERT) (C) or through vesicular transport (D) to the Golgi apparatus for the synthesis of sphingomyelin (SM  ) by SMS1 and glucosylceramide (GlcCer
) by SMS1 and glucosylceramide (GlcCer  ) by GCS (E). At the Golgi, GlcCer is transformed into lactosylceramide (LacCer
) by GCS (E). At the Golgi, GlcCer is transformed into lactosylceramide (LacCer 
 ) and complex GSLs (e.g., GM3
) and complex GSLs (e.g., GM3 

 ) (F), which are then delivered to the plasma membrane via Golgi vesicular transport (G). At the plasma membrane level, SM can be converted into ceramide by sphingomyelinase (SMase), ceramide can then be transformed into sphingosine (Sph) by ceramidase (CDase), and sphingosine is converted into sphingosine 1-phosphate (S1P
) (F), which are then delivered to the plasma membrane via Golgi vesicular transport (G). At the plasma membrane level, SM can be converted into ceramide by sphingomyelinase (SMase), ceramide can then be transformed into sphingosine (Sph) by ceramidase (CDase), and sphingosine is converted into sphingosine 1-phosphate (S1P  ) by sphingosine kinases (SK1) (H). S1P is then transported across the membrane by specific transporters. Phosphorylation of sphingosine yielding to S1P because of the action of sphingosine kinase 2 (SK2) can occur at the mitochondria (I), nucleus (J), and ER (K). In the ER, S1P can be either irreversibly cleaved by S1P lyase or dephosphorylated back to sphingosine by a specific phosphatase (SPP1). Membrane sphingolipids are internalized via caveolae-dependent endocytosis and, in the lysosome, they are degraded by acidic forms of SMase and by different glycosidases (GCase) yielding ceramide that can be further hydrolyzed by the acid ceramidases (CDase) (L). The sphingosine formed in this reaction can escape the lysosome and can be metabolized to glycerophospholipids after phosphorylation by SK1 and cleavage by S1P lyase (SGPL) or it can be recycled for sphingolipid synthesis in the salvage pathway (M).
) by sphingosine kinases (SK1) (H). S1P is then transported across the membrane by specific transporters. Phosphorylation of sphingosine yielding to S1P because of the action of sphingosine kinase 2 (SK2) can occur at the mitochondria (I), nucleus (J), and ER (K). In the ER, S1P can be either irreversibly cleaved by S1P lyase or dephosphorylated back to sphingosine by a specific phosphatase (SPP1). Membrane sphingolipids are internalized via caveolae-dependent endocytosis and, in the lysosome, they are degraded by acidic forms of SMase and by different glycosidases (GCase) yielding ceramide that can be further hydrolyzed by the acid ceramidases (CDase) (L). The sphingosine formed in this reaction can escape the lysosome and can be metabolized to glycerophospholipids after phosphorylation by SK1 and cleavage by S1P lyase (SGPL) or it can be recycled for sphingolipid synthesis in the salvage pathway (M).
Stimulus-mediated SM hydrolysis by sphingomyelinases (SMases) (Hannun, 1994) was described as the main source of bioactive ceramide. The production of bioactive ceramide is not necessarily a lysosomal event because SMases are present at the plasma membrane or can be recruited to it from intracellular sites as a consequence of different stimuli (Levade and Jaffrezou, 1999; Goni and Alonso, 2002). Ceramide, acting on specific cellular targets (including several signaling protein kinases and phosphoprotein phosphatases) or determining the reorganization of plasma membrane signaling platforms (Zhang et al., 2009), regulates several cellular events, most notably programmed cell death with important consequences in cancer, including inflammation, bacterial infection, and signaling pathways related to Alzheimer’s disease (AD) and other neurodegenerative and neurological disorders (Van Echten-Deckert and Walter, 2012; Czubowicz et al., 2019). On the other hand, ceramide can be phosphorylated by a specific ceramide kinase yielding ceramide 1-phosphate, another bioactive molecule acting on its own cellular targets (Presa et al., 2016). Ceramide hydrolysis by various ceramidases negatively regulates ceramide levels; however, it fuels the production of S1P by providing sphingosine as a substrate for sphingosine kinases. As already mentioned, S1P derived from the catabolism of ceramide and more complex sphingolipids is an important biologically active mediator involved in diverse signal transduction pathways that regulate many different cell functions, in some cases, with effects opposed to those of ceramide. Considering the high numbers of enzymes, transporters, cellular targets, and receptors involved in the regulation of the cellular levels of bioactive sphingoids, and in their cellular actions, they do represent an intriguing druggable target for many pathologies.
As mentioned above, ceramide-derived sphingosine can either be recycled for the resynthesis of sphingolipids or be phosphorylated at C1 with the generation of S1P by two isoforms of sphingosine kinase, sphingosine kinase 1 (SK1), and sphingosine kinase 2 (SK2). S1P can be metabolized through irreversible cleavage by the S1P lyase enzyme (Serra and Saba, 2010) (SGPL1) to a fatty aldehyde (hexadecenal if starting from the most common C18 sphingosine) and phosphoethanolamine; alternatively, S1P can also be dephosphorylated back to sphingosine through a reaction that is catalyzed by lipid phosphatase or S1P-specific phosphatases (Le Stunff et al., 2002; Le Stunff et al., 2007; Pyne et al., 2009; Giussani et al., 2014).
S1P is released from cells in the extracellular milieu through specific transporters [spinster homolog 2 (Spns2) or ABC transporters], then from the extracellular milieu, S1P binds to a family of plasma membrane G protein-coupled receptors, the S1P receptors (S1P1–S1P5) (Giussani et al., 2014; Pyne et al., 2016), triggering different biological responses (Figure 3). On the other hand, S1P generated intracellularly by the action of SK2 can engage to various targets, including HDAC-1/2 (Hait et al., 2009), human telomerase reverse transcriptase (hTERT) (Panneer Selvam et al., 2015), and prohibitin 2 (Strub et al., 2011). Thus, the activity of SK2 seems relevant for diverse and crucial biological events, including epigenetic regulation, aging, and mitochondrial respiratory complex assembly (Pyne et al., 2017) (Figure 3).
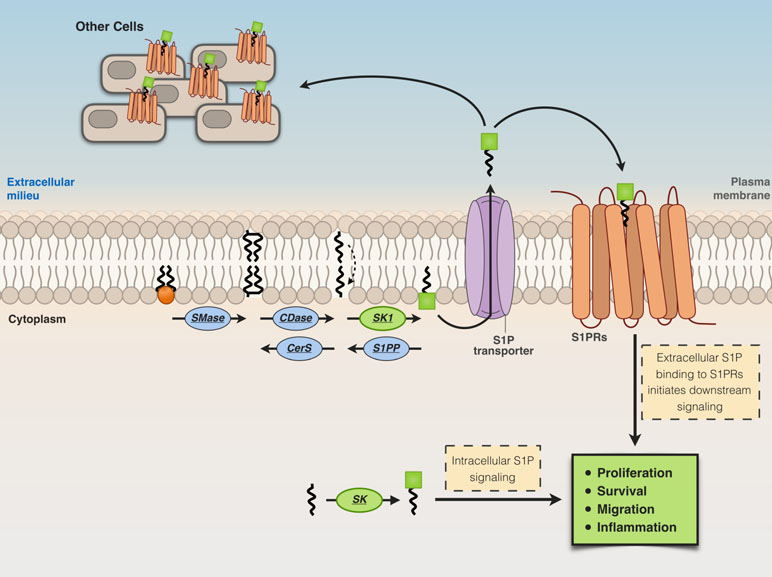
Figure 3 Extracellular and intracellular actions of S1P. Cells release S1P in the extracellular milieu through specific transporters [spinster homolog 2 (Spns2) or ABC transporters]. S1P in the extracellular milieu then binds to S1P receptors (S1P1–S1P5) located at the plasma membrane, thus inducing biological responses.
It is now known that S1P metabolism is tightly regulated. In particular, new pieces of evidence indicating specific roles for SK1 and SK2 in different diseases, including cancer, cardiovascular system and central nervous system (CNS) affections, inflammation, and diabetes, have been recently published (Pyne et al., 2016; Cattaneo et al., 2018). In particular, the role of S1P/SK2 pathway in regulating the survival of the dopaminergic neurons has emerged, suggesting that dysregulation of this pathway might be relevant to the clinical development of Parkinson’s disease (PD); in addition, a role for SK1 and SK2 has also been suggested in the onset of AD (Pyne et al., 2016). Moreover, Hagen et al., (2009) demonstrated that there is an additional aspect of S1P generated by SK2 that may exert a neurotoxic effect, inducing apoptosis in lyase-deficient neurons.
Moreover, the involvement of S1P/S1P lyase in cystic fibrosis pathology is supported by the evidence that in a murine cystic fibrosis model, treatment with a specific SGPL1 inhibitor is able to alter S1P metabolism and results in a reduction of the lung inflammatory response to Pseudomonas aeruginosa (Veltman et al., 2016).
S1P is a positive regulator of cellular survival, proliferation, and motility in glial cells (Bassi et al., 2006). In particular, it has been shown that S1P is able to mediate calcium signaling in cultured cerebellar astrocytes, whereas it failed to trigger any calcium-mediated response in differentiated cerebellar neurons. This effect could be important for neuron–glia communication in the cerebellum (Giussani et al., 2007).
On this basis, it is crucial to continue research to develop isoform-selective SK inhibitors (Table 1).
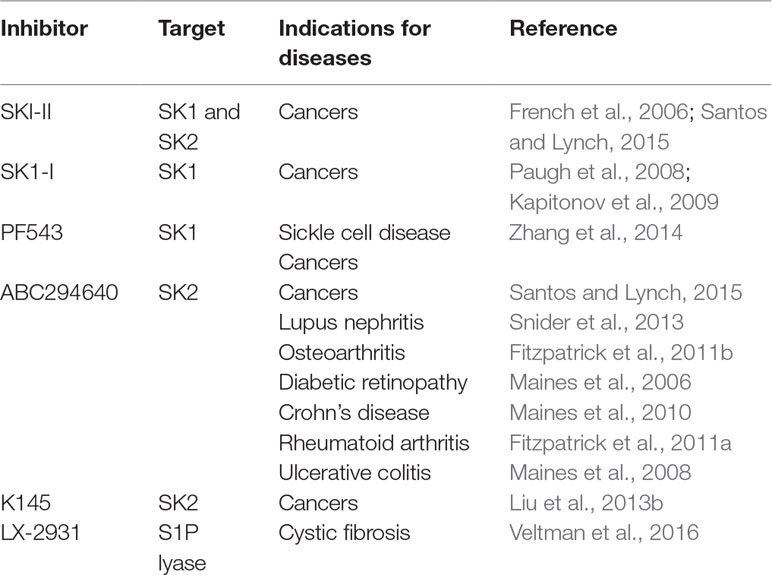
Table 1 Research and clinical use of enzymes of S1P metabolism.
Literature has recently shown a cytotoxic effect of S1P (somewhat in contrast with the mainstream ceramide/S1P rheostat theory) in neurons (Hagen et al., 2011) and in beta-cells of the pancreas (Hahn et al., 2017). In particular, it has been demonstrated that S1P induced i) apoptosis in hippocampal neurons (Moore et al., 1999) as well as ii) accumulation of S1P-induced apoptosis in neurons lacking S1P lyase (Hagen et al., 2009). Moreover, Hagen et al. (2011) have shown that cerebellar neurons with abundant S1P lyase expression are the first to degenerate in S1P lyase-deficient mice. These findings give light to an important role of S1P lyase, which is ubiquitously expressed, in the regulation of physiological and pathophysiological aspects, as suggested also in the review by Choi and Saba (2019).
Most acute or chronic neurodegenerative diseases are associated with changes in the total levels and/or in the composition of sphingolipids in different areas and cell populations in the nervous system. However, several questions remain without any answer on these sphingolipid alterations. Some sphingolipids are bioactive sphingolipids that are involved in the regulation of cell fate. Among them, S1P mediates crucial signaling pathways relevant to neurodegeneration (Hagen et al., 2011; Assi et al., 2013; Blaho and Hla, 2014; Proia and Hla, 2015; Pyne et al., 2018; Wang and Bieberich, 2018). In the brain, S1P regulates several fundamental processes, such as proliferation, survival, differentiation, and migration of all the different cell populations, including neurons, astrocytes, microglia, and oligodendrocytes, as well as infiltration of peripheral immune cells in the CNS during neuroinflammation (Kleger et al., 2007; Riccitelli et al., 2013; Smith et al., 2013; Marfia et al., 2014; Chiricozzi et al., 2018; Grassi et al., 2019). S1P can act both as an extracellular and as an intracellular mediator (Hait et al., 2009; Alvarez et al., 2010; Blaho and Hla, 2014; Proia and Hla, 2015; Park et al., 2016). It regulates different signal transduction pathways in a cell type- and context-specific manner. When acting as an extracellular mediator, S1P effects are dependent on the type and expression levels of the different S1P receptor(s). Different S1P receptors are coupled to different G-proteins, thus regulating specific signaling pathways, including those mediated by MAPK, PI3K/Akt, and phospholipase C (Spiegel et al., 1998; Huang et al., 2011; Spiegel and Milstien, 2011; Kunkel et al., 2013; Blaho and Hla, 2014; Proia and Hla, 2015). On the other hand, different S1P intracellular targets have been recently discovered in several cell types, including neurons. It has been demonstrated in different cell types that S1P regulates the function of some proteins in different cellular compartments, such as the ER and nuclei; for example, HDACs, TRAF2, Hsp90, and HRP94 (Hait et al., 2009; Alvarez et al., 2010; Blaho and Hla, 2014; Proia and Hla, 2015; Park et al., 2016). In particular, three different nuclear transcription factors seem to be regulated by S1P-dependent pathways: 1) FOXO3a, which in PC12 cells is inhibited by the PI3K/Akt pathway (Safarian et al., 2015); 2) AP-1, regulated by the Jnk/p38/ERK pathway (Hsu et al., 2015; Jazvinscak Jembrek et al., 2015) and, in turn, controlling the mutual coregulation between sphingolipid-related genes (O’Neill et al., 2011; Huang et al., 2014; Wegner et al., 2014); 3) NF-κB, which is regulated via the tumor necrosis factor (TNF) receptor-associated factor TRAF2. In fact, TRAF2 can directly interact with SK1 and, on the other hand, it can be regulated by S1P as a receptor cofactor (Xia et al., 2002; Alvarez et al., 2010). In addition, histone deacetylases (HDAC1 and HDAC2), which can negatively regulate NF-κB via its deacetylation (Dai et al., 2005), are inhibited through S1P binding (Hait et al., 2009). The final effect of NF-κB influence on cell fate may vary, depending on the particular signaling context and by the underlying immune activation, among many other factors. Finally, S1P, by acting on its receptors and affecting the downstream PI3K/Akt, can inhibit GSK-3β (the crucial tau kinase) (Wang et al., 2015) and the proapoptotic protein Bad (Czubowicz et al., 2019).
It is now known that S1P has physiological functions in the CNS; several papers demonstrate that S1P plays an essential role in brain development (Mizugishi et al., 2005; Blaho and Hla, 2014; Proia and Hla, 2015). Mizugishi et al. (2005) demonstrated that SK1 is highly expressed in mouse brain during normal development. Moreover, S1P depletion in SK1/SK2-double knockout mice (Mizugishi et al., 2005) caused severe defects in neural cell survival and in neurogenesis, leading to increased neural cell apoptosis and impaired neural tube closure, ultimately leading to an embryonic lethal phenotype. Remarkably, S1P1-null mice show a very similar neural phenotype, suggesting a pivotal role of S1P signaling in neurogenesis and brain development (Mizugishi et al., 2005; Blaho and Hla, 2014; Proia and Hla, 2015).
Abundant literature suggests important roles of S1P also for neuronal survival and for the maintenance of neuronal functions in adult brains. Different papers demonstrate that in hippocampal neurons, S1P is important for the stability of the presynaptic structure, for synaptic strength, and for the availability of synaptic vesicles (Brailoiu et al., 2002; Camoletto et al., 2009; Darios et al., 2009; Kanno et al., 2010; Riganti et al., 2016). On the other hand, under certain circumstances, S1P might become harmful. In fact, Mitroi et al. (2016) demonstrated that a certain S1P threshold concentration causes an increase in basal calcium in neurons that impairs presynaptic architecture most probably via a UPS-mediated mechanism.
Moruno Manchon et al. (2015, 2016) demonstrated that S1P promotes prosurvival neuronal autophagy; in particular, they have shown that SK1 enhances autophagic activity, whereas SGPL1 reduces this activity. Autophagy plays a crucial role for neuron survival; in fact, this process allows neurons to get rid of damaged and aggregated proteins and organelles during different conditions, such as aging and diseases, in particular, to counteract cell death induced by ceramide or other pathogens (Moruno Manchon et al., 2015; Moruno Manchon et al., 2016; Moruno-Manchon et al., 2018).
However, the role of S1P metabolism in brain autophagy is very complex. In fact, in contrast to these results, Mitroi et al. (2017) demonstrated that the deficiency of SGPL exerted a block on the autophagic flux. The authors demonstrated that depletion of SGPL, increasing S1P levels while decreasing ethanolamine phosphate and consequently phosphatidylethanolamine, blocks autophagy at the initial stages (Mitroi et al., 2017). These findings suggest that in neurons, the increase of S1P and the simultaneous decrease of phosphatidylethanolamine caused by the modulation of SGLP both affect autophagy even if in an antagonistic way (Mitroi et al., 2017).
Altogether, the role of S1P as a bioactive lipid in the nervous system appears to be dual (Van Echten-Deckert and Alam, 2018). On one hand, as described above, it is essential for proper brain development; on the other hand, its detrimental effects and cytotoxic effects on some particular neuronal populations have been documented (Van Echten-Deckert et al., 2014).
The regulation of S1P concentration in the different cerebral areas suggests a specific function of S1P in different brain regions.
Recently, thanks to the approval by the Food and Drug Administration (FDA) of the sphingosine analog and S1P1 functional antagonist fingolimod {FTY720, Gilenya, 2-amino-2[2-(4-octylphenyl)ethyl]-1,3-propanediol}, the functions of S1P in neuroinflammation processes regarding neurodegeneration attracted the attention of the scientific community. Microglia activation is emerging as one of the crucial factors contributing to the onset of neuroinflammation associated with different neurodegenerative diseases.
Nayak et al. (2010) demonstrated that in activated microglia, SK1 expression is upregulated. In turn, SK1 affects the production of proinflammatory cytokines and nitric oxide in lipopolysaccharide (LPS)-treated microglia (Nayak et al., 2010). In other cell types, treatment with LPS was able to activate the SK1/S1P signaling pathway by inducing the translocation of SK1 to the plasma membrane, leading to increased production of S1P (Hammad et al., 2008; Fernandez-Pisonero et al., 2012). The administration of exogenous S1P to activated microglia increased the inflammatory response, inducing the production of proinflammatory cytokines and neurotoxins (Assi et al., 2013; Lv et al., 2016). Altogether, data in the literature suggest that the S1P/SK1 pathway, acting as an autocrine or paracrine factor, is involved in the inflammatory response of activated microglia, regulating the release of proinflammatory factors by microglia.
On the other hand, it has been shown that under proinflammatory conditions, SK1 and S1P3 in astrocytes are functionally upregulated (Fischer et al., 2011). Moreover, treatment of astrocytes with IL-1 induces the expression of SK1 and, in turn, exposure to exogenous S1P induces astrogliosis (Sorensen et al., 2003; Paugh et al., 2009). In a mouse model of multiple sclerosis (MS), it has been shown that particular clusters of astrocytes are activated, and astrocyte activation progressively expand along white matter tracts. Moreover, the loss of astrocytic S1P1 limited the extent of astrocyte activation (Choi et al., 2011; Dusaban et al., 2017). The role of S1P1 in astrocytes seems to be essential not only for astrocyte’s own functions but also for the interplay between astrocytes and other cell types. Indeed, modulation of S1P1 by fingolimod in astrocytes effectively suppressed different neurodegeneration-inducing pathways mediated by astrocytes, but also by microglia, and by CNS-infiltrating activated leukocytes (Karunakaran and Van Echten-Deckert, 2017; Rothhammer et al., 2017; Wang and Bieberich, 2018).
Considering the role of S1P in the development and physiological homeostasis of the nervous system and its emerging importance in neuroinflammation, it is not surprising that dysregulation of S1P metabolism and S1P-mediated signaling is emerging as a common trait and an important causative factor in various neurodegenerative diseases, including MS, AD, PD, and Huntington disease (HD). The knowledge about the role of S1P in these affections is briefly outlined in the following sections of this review.
MS is a chronic inflammatory disease of the CNS, in which the inflammatory process is associated with a destruction of myelin, leading to the appearance of large focal lesions of demyelination. Axonal damage and loss as consequences of the inflammatory demyelination also occur, even if at variable extents. Active remyelination processes can at least in part repair myelin lesions, whereas axonal loss is permanent and irreversible.
MS is primarily considered an autoimmune neurodegenerative disease, that is, a disease caused by an adaptive immune response to self-antigens. In MS, activated myelin-reactive T cells [in particular, T helper 1 cells (Th1)] are recruited from the periphery to the CNS, leading to the activation of microglia and to the recruitment of circulating macrophages. Consistently with this, fingolimod has proven to be an effective disease-modifying drug for the treatment of relapsing–remitting MS (RR-MS).
Fingolimod, a structural analog of sphingosine, is converted in vivo to fingolimod-P, a structural analog of S1P, which acts as a nonselective agonist for S1P1, S1P3, S1P4, and S1P5 receptors (lacking activity on S1P2). It acts as a functional antagonist of S1P receptors, causing the irreversible internalization and degradation of bound S1P receptors (thus preventing their recycling back to the cell surface) (Figure 4).
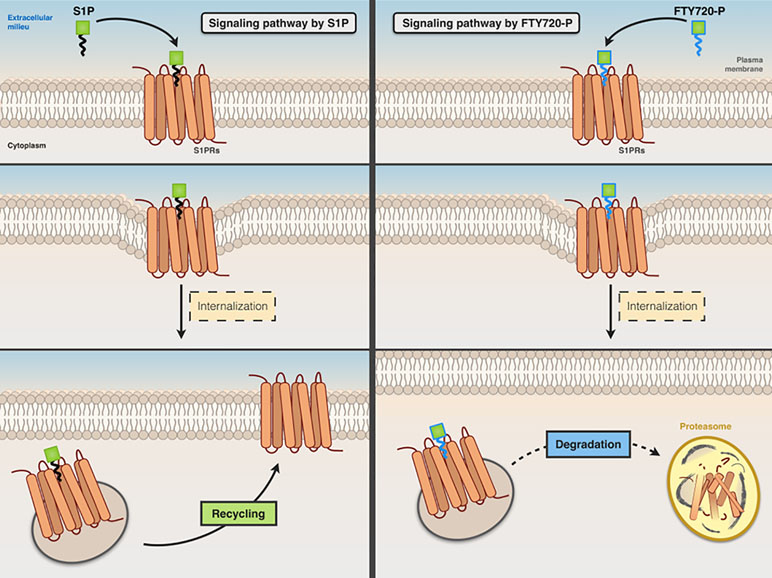
Figure 4 Comparative signaling pathways of S1P and FTY720-P. Both S1P (left panel) and FTY720-P (right panel) stimulate the internalization of S1P receptor. The receptor that binds S1P is recycled back to the cell surface, whereas FTY720-P causes irreversible internalization and degradation of bound S1P receptor.
Fingolimod is effective on MS by preventing the recruitment of T cells, expressing S1P1 and S1P3 receptors, with their consequent redistribution to secondary lymphoid organs, and preventing invasion of auto-aggressive T cells to the CNS. However, the heterogeneity of human MS and the comparison between the human disease and the different animal models suggest that additional factors other than Th1-mediated autoimmunity are relevant to lead from primary demyelination to a chronic inflammatory lesion. It is worth to note that primates, differently from mice, do not develop demyelination because of pure T cell-mediated inflammation. In fact, fingolimod effectively decreased astrocyte and microglia activation in a non-T cell animal model of demyelination, the cuprizone mouse model (Kim et al., 2011).
A significant contribution to the progression of the disease is likely given by circulating demyelinating antibodies against myelin surface components, most notably the anti-myelin oligodendrocyte glycoprotein antibodies, occurring in a significant proportion of MS patients (Linington et al., 1988; Ramanathan et al., 2016; Spadaro et al., 2018). In addition, cell populations resident in the lesion niche, most notably astrocytes and microglia, might critically affect both oligodendrocyte injury and axonal degeneration (Mayo et al., 2012). Indeed, changes in the expression of 13 different growth factors, known to regulate the development and maintenance of oligodendrocytes, were highlighted during demyelination and remyelination in the murine cuprizone model of toxic demyelination (Gudi et al., 2011). In particular, in lesion areas, IGF-1 and CNTF were elevated in astrocytes, whereas GDNF, IGF-1, and FGF were detected at high levels in microglia.
In MS, oligodendrocyte loss and myelin damage can be observed in early lesions, even in the absence of infiltrates of immune cells from the periphery. On the other hand, early activation and proliferation of microglia and astrocytes are consistently present in MS lesions. Several pieces of evidence suggest that S1P signaling in these cell populations might be relevant to the progression of the disease, opening up new perspectives for drugs acting on the S1P axis in the therapy of MS (reviewed in Groves et al., 2013; Farez and Correale, 2016).
Altered sphingolipid metabolism and altered sphingolipid-dependent signaling in reactive astrocytes might contribute to oligodendrocyte damage in MS (Kim et al., 2012). Ceramide accumulated in reactive astrocytes in active lesions of MS and in a non-T cell animal model of demyelination (the cuprizone mouse model). Ceramide accumulation was consequent to the upregulation of serine palmitoyltransferase in reactive astrocytes. In culture, ceramide acted synergistically with TNF, inducing apoptosis of oligodendrocytes (an astrocyte-dependent event). Concomitantly, sphingosine was accumulated whereas S1P levels were decreased. These alterations in sphingolipid metabolism were restored upon active remyelination.
It has been shown that sphingosine kinase 1 and S1P3 are upregulated in reactive astrocytes present at the lesion site or in cultured rat astrocytes treated with LPS. S1P induced secretion of CXCL1 in astrocytes, and secretion was increased in astrocytes pretreated with LPS. Thus, ceramide/S1P pathway in astrocytes is relevant for astrocyte activation, and in MS, it could be detrimental, enhancing astrogliosis, or beneficial, through increased remyelination sustained by CXCL1 (Fischer et al., 2011).
As mentioned above, the main therapeutic effect of fingolimod in MS seems to be related to its ability to prevent migration of auto-aggressive lymphocytes to the CNS. However, because S1P receptors are widely expressed in the CNS, fingolimod easily crosses the blood–brain barrier (BBB) and the effect on MS is at least in part independent of the effect on the migration of immune cells from the periphery (Meno-Tetang et al., 2006; Foster et al., 2007; Chun and Hartung, 2010).
In fact, emerging evidence indicates that the efficacy of fingolimod in MS is at least in part caused by its direct effects on the CNS. S1P signaling effects relevant for MS likely involve different neural cell types (astrocytes, oligodendrocytes, neurons, microglia, and dendritic cells); however, very strong evidence indicates a primary role of astrocytes in the direct effects of fingolimod on the CNS. Astrocytes express S1P receptors, mainly S1P1 and S1P3. S1P1 and S1P3 are upregulated in reactive astrocytes present in demyelinating and chronic MS lesions. S1P modifies astrocyte morphology and increases the expression of GFAP, marker of astrogliosis. Fingolimod stimulates migration of cultured astrocytes, whereas it acts in vivo as a functional antagonist of astrocyte S1P1.
In experimental autoimmune encephalomyelitis (EAE), an experimental paradigm of Th1-mediated demyelinating disease, fingolimod is highly effective; however, its effects are abolished in animals selectively lacking the expression of S1P1 in astrocytes (even if the receptor expression and function in immune cells are preserved) (Choi et al., 2011). As an endpoint, fingolimod appears to be able to promote remyelination by acting on oligodendrocytes, microglia, or astrocytes. In addition, fingolimod was effective in increasing remyelination in a rat CNS reaggregate spheroid cell culture model (a CNS environment devoid of immune system effects) upon lysoPC-induced transient demyelination. Increased remyelination was associated with partial inhibition of microglia activation (Jackson et al., 2011). In summary, downmodulation of S1P1 receptors in astrocytes upon fingolimod treatment causes reduced astrocyte activation and improves the communication of astrocytes with other CNS cells, resulting in reduced demyelination or improved remyelination.
S1P signaling as a pharmacological target in MS is relevant not only in astrocytes but also in microglia. As mentioned above, microglia can contribute to the release of factors positively affecting remyelination. On the other hand, fingolimod in cultured microglia was able to suppress via binding of S1P1 the production of relevant proinflammatory cytokines, such as TNFα, IL-1β, and IL-6 (Choi et al., 2011).
A further layer of complexity in the possibility to affect the S1P/S1P receptor axis in MS is revealed by studies suggesting that fingolimod might be effective in modifying some disease parameters by acting as an agonist, rather than a functional antagonist, of given S1P receptors. In addition to motor deficits, over 50% of MS patients suffer from neuropathic pain, a debilitating symptom that dramatically affects the patients’ quality of life. In EAE mice, painlike behavior appears early on and well before the motor symptoms develop. In this model, fingolimod was able to reduce hyperalgesia by acting as an S1P1 agonist at the central level, inhibiting spinal nociceptive processing (Doolen et al., 2018). This would suggest that novel site- or receptor-specific drugs could be effective in ameliorating the central neuropathic pain.
Finally, despite the prevailing view of T cell-mediated inflammation as the main culprit in the pathogenesis of MS lesions, some data suggest that the inflammatory response and more in general the contribution of the immune system might have a protective role or a role in the repair of myelin lesions. For example, it has been shown that inflammatory cells within MS lesions might release neurotrophic factors (Kerschensteiner et al., 1999) or factors able to stimulate the physiological, per se insufficient, mechanism or remyelination (Diemel et al., 1998). On the other hand, some naturally occurring autoantibodies, a subset of monoclonal antibodies that is a part of the normal immunoglobulin repertoire, were able to stimulate remyelination in CNS demyelinating diseases (Rodriguez et al., 2009). rHIgM22, the recombinant form of a naturally occurring antibody isolated from a patient with Waldenström macroglobulinemia, was able to bind selectively to myelin in vitro and to the surface of cultured oligodendrocytes. It was shown to enter the CNS and to accumulate at the lesion sites in MS (Pirko et al., 2004) and to promote remyelination in Theiler’s murine encephalomyelitis virus (Warrington et al., 2000)-, lysolecithin (Bieber et al., 2002)-, and cuprizone-induced (Mullin et al., 2017) demyelination models of MS. A 16-site Phase 1 clinical trial in MS patients completed in 2015 (NCT0183867) showed no dose-limiting toxicities, no serious treatment-emergent adverse events, and the presence of the antibody in the CFS in all patients (Eisen et al., 2017). Although some studies suggest that the rHIgM22 remyelination-promoting effect is exerted directly on myelin-producing cells (Watzlawik et al., 2010; Watzlawik et al., 2013), others suggest that it acts on other cell types present in the lesion niche, including astrocytes (Paz Soldan et al., 2003) and microglia (Zorina et al., 2018). We recently showed that rHIgM22 stimulated the proliferation of astrocytes in mixed glial cells because of the increased production and release of S1P, suggesting that S1P signaling might be significant in the interplay of different cell types involved in the complex series of events eventually leading to myelin repair (Grassi et al., 2019).
AD is hallmarked by the accumulation of intraneuronal aggregates of hyperphosphorylated tau protein and deposition of extracellular oligomer amyloid beta peptide (predominantly Aβ42 and Aβ40). Evidence strongly suggests a critical role of Aβ, which derives from the proteolytic processing of the amyloid precursor protein (APP), in the initiation of AD (Kozlov et al., 2017).
The possible role of S1P in AD is currently quite controversial. There is evidence suggesting a direct role of S1P in the initiation and progression of AD. S1P was found to directly interact and stimulate the proteolytic activity of the β-secretase BACE1, the rate-limiting enzyme for Aβ production. Moreover, overexpression of SGPL1 and inhibition or knockdown of SKs reduced BACE1 activity, in vitro and in vivo, with consequent decreased Aβ secretion, suggesting a correlation between levels of S1P and those of Aβ (Hagen et al., 2011; Takasugi et al., 2011). A study of SGPL1-deficient mice also revealed that S1P elevated levels to exert a neurotoxic effect mediated by a calcium/calpain/CDK5 mechanism (Hagen et al., 2011). Moreover, SGPL1 deficiency was also associated with hyperphosphorylation of tau, accumulation of APP, and impaired lysosomal activity (Hagen et al., 2011; Karaca et al., 2014). AD brains also showed an increased SK2 activity (Takasugi et al., 2011), and ERK and Fyn kinase, both known modulators of this enzyme, have been implicated in Aβ-mediated neurotoxicity (Olivera et al., 2006; Hait et al., 2007; Crews and Masliah, 2010), which could suggest that SK2 upregulation might be mediated by aberrant phosphorylation by Fyn and/or ERK. On the other hand, consistently with the prosurvival and antiapoptotic effects of S1P, the S1P/sphingosine ratio is decreased in postmortem AD brains and hippocampus, and the decrease is associated with the reduction of SK1 activity. Increased levels of SGPL1 and S1P phosphatase have also been reported in these brains (Katsel et al., 2007; He et al., 2010; Ceccom et al., 2014; Couttas et al., 2014), indicating a protective role of S1P in AD. SKs have also been shown to exert a protective influence in Aβ toxicity. Their overexpression reduced the toxic effects of Aβ (Gomez-Brouchet et al., 2007) whereas SK1 silencing in APP/PS1 mice led to an increased secretion of Aβ and, consequently, an increased Aβ-induced cell death (Zhang et al., 2013). Suppression of SK1 by miRNA 125b, which is abundant in AD, correlated with an increased Aβ production in vitro (Jin et al., 2018).
The exact mechanism through which S1P could exert its protective role is still unclear; however, a few studies (Gomez-Munoz et al., 2003; Malaplate-Armand et al., 2006; He et al., 2010) suggest that S1P might act by inhibiting the activation of acid sphingomyelinase, whose activity has been found to be increased in AD brains (Gomez-Munoz et al., 2003).
SK2 also has a controversial role in AD. Some authors, as previously mentioned, showed an increase in SK2 activity in the frontal cortex of AD brains (Takasugi et al., 2011), whereas others reported a decreased activity in temporal cortex and hippocampus (Couttas et al., 2014). These results, however, may simply reflect the complexity of SK regulation and function, and subcellular localization may play a role in SK2 expression in AD. Interestingly it has been observed that in AD brains, the equilibrium between the nuclear and the cytoplasmic SK2 was altered. It has been reported that SK2 in these brains preferentially localized in the nucleus, whereas the cytoplasmic expression of the enzyme inversely correlated to Aβ deposits (Dominguez et al., 2018).
Fingolimod, the immunomodulatory analogue of S1P, has been tested in AD models. Fingolimod was able to reduce the S1P Aβ-induced neuronal damage in rat hippocampus and improve the consequent cognitive impairment (Asle-Rousta et al., 2013a, Asle-Rousta et al., 2013b). In a mouse model of AD, treatment with fingolimod lad to a reduction of soluble and insoluble Aβ and to decreased Aβ plaque density (Aytan et al., 2016). Furthermore, both fingolimod and KRP203, another S1P analogue, decrease Aβ generation in neuronal cells (Takasugi et al., 2013). The effect of fingolimod was also tested in transgenic mice overexpressing APP and presenilin 1 (APP/PS1 mice). In this model, Aβ accumulation and loss of neuronal function are coupled with augmented BBB permeability and with the activation of astrocytes and microglia (Kelly et al., 2013; Mcmanus et al., 2014; Minogue et al., 2014). A reduction in both accumulation of Aβ and astroglial activation was observed after treatment with fingolimod. Moreover, the treatment also increased the phagocytic activity of astrocytes, suggesting that the decrease in Aβ accumulation in the treated mice could be a consequence of this enhanced phagocytic function (Mcmanus et al., 2017).
Fingolimod’s exact mechanism in AD however remains to be elucidated; however, the protective effect of fingolimod in neurons was associated with an altered expression of MAPKs and some inflammatory markers (Hemmati et al., 2013).
PD is one of the most common neurodegenerative disorders, second only to AD, and is clinically defined by the degeneration or death of dopaminergic neurons in the substantia nigra accompanied by the formation of aggregates of alpha-synuclein and ubiquitin, called Lewy bodies (Beitz, 2014).
In neuronal cells (SH-SY5Y) treated with 1-methyl-4-phenylpyridinium (MPP+), an active metabolite of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), there is a reduction in SK1 gene expression and protein levels, with a concomitant increase of SGPL1 that leads to enhanced reactive oxygen species (ROS) generation (Pyszko and Strosznajder, 2014a; Pyszko and Strosznajder, 2014b). Moreover, pharmacological inhibition of SK1 was shown to suppress prosurvival PI3K/Akt phosphorylation, activation and upregulation of proapoptotic proteins, cytochrome c release from mitochondria, and caspase-dependent apoptosis in these cells (Pyszko and Strosznajder, 2014a; Pyszko and Strosznajder, 2014b). In the same experimental model, inhibition of SK1 and SK2 also led to an increased secretion of alpha-synuclein, a protein able to negatively regulate S1P1 signalling (Pyszko and Strosznajder, 2014a; Pyszko and Strosznajder, 2014b; Badawy et al., 2018). Interestingly, treatment with pramipexole, a D2/D3 receptor agonist commonly used in PD therapy, partially reversed SK1 inhibition in the MPP+ model (Motyl et al., 2018).
In MPP+-treated neuronal cells, S1P administration showed neuroprotective effects. The treatment with exogenous S1P in fact significantly increased cell viability mainly through activation of S1P1 receptor-mediated signalling, reduced the mRNA level of proapoptotic proteins such as Bax and Hrk, and decreased cytochrome c levels in a mitochondrial fraction, leading to caspase-dependent poly(ADP-ribose) polymerase-1 proteolysis (Pyszko and Strosznajder, 2014a; Pyszko and Strosznajder, 2014b). Interestingly, S1P 1 was recently proposed as a candidate gene for a newly identified PD susceptibility locus (Hill-Burns et al., 2014), supporting the potential role for S1P1 signalling in regulating midbrain dopaminergic neuron survival and/or function. Phospholipid phosphatase 3, an integral membrane glycoprotein involved in the modulation of S1P metabolism and signalling in the brain (Lopez-Juarez et al., 2011), has also been implicated in PD, and its inactivation in CNS progenitor cells caused a severe downregulation of S1P1 in the adult ventral midbrain and in the cerebellum (Lopez-Juarez et al., 2011; Gomez-Lopez et al., 2016).
A marked decrease of SK2 levels has been reported in the substantia nigra of the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated C57BL/6 mice, a PD mouse model (Sivasubramanian et al., 2015). Moreover, SK2 inhibition in MN6D dopaminergic neurons decreased the expression of genes for the regulation of mitochondrial function and led to marked ATP depletion and reduction of superoxide dismutase 2 levels. An increase in the ROS levels was also observed (Sivasubramanian et al., 2015).
Administration of fingolimod protected against neurodegeneration and behavioral effects in mouse PD models induced by MPTP, 6-hydroxydopamine, or rotenone through S1P1 signalling and probably Akt (Ren et al., 2017; Zhao et al., 2017; Motyl et al., 2018); however, the S1P analogue was not effective in a PD model induced by subacute administration of MPTP (Komnig et al., 2018). Cellular studies applying newly modified versions of fingolimod referred to as C2 moiety (FTY720 C2 or FTY720-Mitoxy) have shown that these compounds increase BDNF levels, activate protein phosphatase 2A, whose activity is impaired in PD, and protect MN9D cells against TNFα-induced cell death (Vargas-Medrano et al., 2014). Long-term oral administration of fingolimod reduced alpha-synuclein aggregation and increased BDNF levels in transgenic mice overexpressing mutant human alpha-synuclein (Vidal-Martinez et al., 2016). There are also several reports indicating a fingolimod neuroprotective effect in PD models induced by toxins. For example, in C57BL/6 mice subjected to acute MPTP intoxication, there was a reduced expression and activity of SK1. This study also showed that fingolimod exerts neuroprotective effects comparable to pramipexole, a dopamine D2/D3 receptor agonist (Motyl et al., 2018).
HD is an inherited neurodegenerative brain disease characterized by the progressive degeneration of the striatum and cortex, with consequent motor, cognitive, and behavioral symptoms. It is caused by a dominant mutation, which leads to the expansion of a CAG trinucleotide repeat in the gene encoding for the huntingtin protein. The consequence of the mutation is the expression of a protein with a polyglutamine stretch in the N-terminal region much longer than in the wild type (Jimenez-Sanchez et al., 2017). The exact biological function of huntingtin is poorly understood, but it is highly expressed in brain where it seems to be relevant for neuronal development and metabolism.
Recent studies indicate that the expression of S1P-metabolizing enzymes is altered in several HD models, including animal models, human brain tissues, and cultured cells (Di Pardo et al., 2017; Pirhaji et al., 2017). Upregulation of SGPL1 and reduced expression of SK1 are detectable in human postmortem brains and in striatal tissues of two of the most commonly used HD animal models (R6/2 and YAC128 mice), even at an early stage of the disease (Di Pardo et al., 2017; Pirhaji et al., 2017). Moreover, evidence indicates that miRNA 125b expression, which is downregulated in R6/2 mice, negatively regulates gene expression of SGPL1 (Ghose et al., 2011; Yang et al., 2016). Furthermore, it has been demonstrated that cytoplasmic SK1 is involved in the regulation of huntingtin degradation (Moruno Manchon et al., 2015). R6/2 mice also exhibit increased SK2 and reduced S1P levels (Di Pardo et al., 2017; Pirhaji et al., 2017). This reduction of S1P levels could be a potential therapeutic target. In fact, fingolimod was able to improve neuronal activity and motor function, reduce brain atrophy, and increase R6/2 animal survival (Di Pardo et al., 2014). Moreover, both fingolimod and the pharmacological activator K6PC-6 significantly reduced apoptosis in an HD cellular model and increased the activation of Akt and Erk signalling pathways (Di Pardo et al., 2017), whose regulation is known to be defective in HD (Bowles and Jones, 2014). In R6/2 mice, fingolimod also enhanced phosphorylation of huntingtin at the serine-13/16 residues. This might contribute to the protective effect of fingolimod because this posttranslational modification was associated with reduced toxicity of mutated huntingtin (Atwal et al., 2011; Di Pardo et al., 2014). Moreover, reduced synthesis of GM1 gangliosides was found in HD patients and animal models. Lower GM1 levels were associated with higher sensitivity to neuronal death, and artificial reduction of GM1 synthesis increases the apoptotic rate in normal striatal neurons. It has been shown that GM1 administration to HD mice was able to improve motor symptoms (Di Pardo et al., 2012). Remarkably, the molecular mechanism of this effect seems to be mediated by the GM1-induced phosphorylation of huntingtin. On the other hand, fingolimod was able to restore normal levels of GM1 in HD mice (Maglione et al., 2010; Di Pardo et al., 2014). This is an intriguing finding, suggesting that fingolimod’s effects on sphingolipid metabolism might be much more complex than argued on the basis of its efficacy as a functional antagonist of S1P receptors.
Fingolimod also improved synaptic plasticity and memory in the R6/1 mouse model of HD by regulating BDNF signalling and astroglial reactivity. Fingolimod administration prevented overactivation of NF-κB signalling in R6/1 hippocampus, leading to a decrease in TNFα and induced nitric oxide synthase (iNOS) levels. This reduction correlates with the normalization of p75NTR expression in the hippocampus, consequently preventing p75NTR/TrkB imbalance, a critical mechanism for memory and synaptic function in HD (Brito et al., 2014; Miguez et al., 2015). Moreover, fingolimod increased cAMP levels and promoted phosphorylation of CREB and RhoA in the hippocampus of R6/1 mice, supporting its role in the enhancement of synaptic plasticity (Miguez et al., 2015). Interestingly, these results also provide a possible explanation for fingolimod-induced cognitive benefits in AD because both TNFα and p75NTR are overexpressed in AD patients, and p75NTR downregulation is able to prevent cognitive and neurite dysfunction in an AD mouse model (Hu et al., 2002; Alvarez et al., 2007; Asle-Rousta et al., 2013a; Knowles et al., 2013; Fukumoto et al., 2014).
Modulation of SK2 and SGPL1 may also exert protective effects in HD. Administration of two SK2 selective inhibitors (K145 and EMD56773) markedly reduced apoptosis (Di Pardo et al., 2017). On the other hand, inhibition of SGPL1 (using 2-acetyl-5-tetrahydroxybutyl imidazole or 4-deoxypryridoxine) also reduced cell death. This effect was associated with the inhibition of HDAC activity, which in turn resulted in increased levels of histone H3 acetylation. Because histone H3 deacetylation was previously reported to be reduced in several HD models, this could explain the protective effect of SGPL1 inhibition in this disease (Buckley et al., 2010; Di Pardo et al., 2017; Pirhaji et al., 2017).
SK2 has also been implicated in HDAC regulation. In fact, it has been reported that increasing levels of S1P in the nucleus by overexpressing SK2 lead to histone deacetylases 1 and 2 inhibition. S1P directly binds to these deacetylases while SK2 forms a complex with them, which inhibits their activity (Hait et al., 2009; Hait et al., 2014). Whereas SK2 might exert a neuroprotective role through HDAC inhibition, it has been observed that its overexpression is neurotoxic for cultured striatal and cortical neurons in a dose-dependent manner. In these neuronal models and in the BACHD mouse model of HD, SK2 is hyperphosphorylated and promotes the formation of double-strand breaks. Interestingly, a small molecule inhibitor of SK2, ABC294640, was able to mitigate DNA damage and neurotoxicity and protect against degeneration on two mouse models of HD (Moruno-Manchon et al., 2017).
Stimulation of S1P5 in the R6/2 models using the selective agonist A-971432 was also considered as a potential therapeutic approach for HD. Chronic administration of the agonist slowed down the progression of the disease and prolonged the life span of R6/2 mice. These effects were associated to the activation of the prosurvival pathways (AKT, BDNF, ERK) and with a reduction of mutant huntingtin aggregation. Moreover, A-971432 also protected BBB homeostasis in these mice, and when administered early in the disease, it completely protected them from the classic progressive motor deficit and preserved BBB integrity (Di Pardo et al., 2018).
S1P5 is known to be involved in the regulation of BBB tight junctions, and the effect of its agonist A-971432 was associated with an increased expression of the tight junction proteins occludin and claudin-5 (Feldman et al., 2005; Piontek et al., 2008; Van Doorn et al., 2012).
The immunomodulatory drug fingolimod (FTY720) is the first drug approved for oral treatment of relapsing-remitting MS (RR-MS) (Kappos et al., 2006; Brinkmann et al., 2010) because of its impressive efficacy and good tolerability. Fingolimod is phosphorylated to form fingolimod-P, very similar to S1P.
Pharmacologically, fingolimod acts as a nonselective agonist of S1P receptors, with the exception of S1P2, and as a selective functional antagonist of the S1P1 subtype, the main S1P receptor expressed in different tissues, including brain and immune system cells (Blaho and Hla, 2014), inducing receptor downregulation (Pyne et al., 2016). Fingolimod-P initially binds to and activates S1P1. After being engaged by fingolimod-P, S1P1 is subsequently internalized and degraded (Huwiler and Zangemeister-Wittke, 2018).
Although effective in ameliorating the symptoms and delaying the worsening in RR-MS, fingolimod has very limited effect on the progressive forms of MS (Farez and Correale, 2016). For this reason, various selective S1P receptor modulators are currently under investigation in preclinical studies and in some cases in clinical trials to test their clinical use in primary or secondary progressive MS (Mao-Draayer et al., 2017).
Seeking for valuable alternatives to fingolimod, new-generation S1P receptor modulators have been developed, characterized by a higher selectivity and improved pharmacokinetic performance and tolerability (Pyne et al., 2016). A number of S1P receptor drugs in clinical trials of MS are reported in Table 2 (Pyne et al., 2016; O’sullivan and Dev, 2017). Fingolimod is used in MS; however, ongoing preclinical studies suggest the use of fingolimod for different brain diseases, including AD, HD, and PD. In Table 3, fingolimod current clinical trials related to other brain diseases other than MS are summarized (O’sullivan and Dev, 2017).
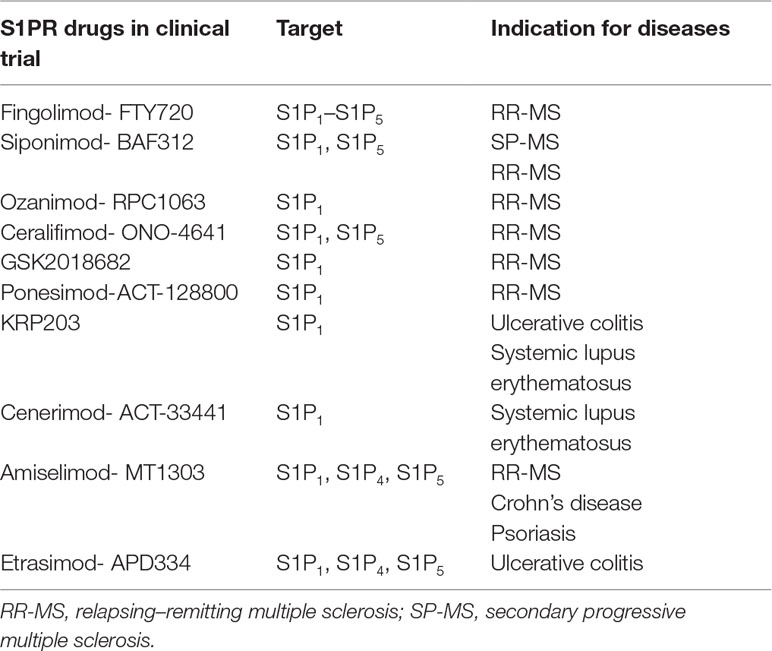
Table 2 Use of S1PR drugs in clinical trial for central nervous system (CNS) disease.

Table 3 Research and clinical use of FTY720/fingolimod.
The role played by S1P in the development of neurodegenerative diseases like MS makes S1P receptors the most interesting pharmacological targets.
As mentioned above, the first molecule used as therapeutic agent for MS is fingolimod, which acts as an S1P1,3,4,5 modulator (Huwiler and Zangemeister-Wittke, 2018). Fingolimod, which in 2010 has been approved by the FDA for the treatment of RR-MS, is a prodrug whose chemical structure is very similar to that of sphingosine. Its phosphorylation turns it into (S)-FTY720-monophosphate, an S1P analog (Sushil and Batra, 2012). This phosphate-derivative undergoes the action of the same phosphatases (SPP1 and SPP2) that dephosphorylate S1P. To overcome this problem and in the wake of fingolimod’s success, structure-activity relationship (SAR) studies performed on FTY720-phosphate have been used to design several fingolimod-phosphate derivatives and nonhydrolyzable phosphonate (reviewed in Marciniak et al., 2018, and Stepanovska and Huwiler, 2019).
The key features of the agonists obtained after these modifications can be summarized as follows: polar head, aromatic region, and lipophilic tail (Liu et al., 2019). Indeed, not only molecules strictly similar to S1P are able to interact with S1P receptors.
Introduction of a carboxylic acid head group that does not require activation for bioavailability led to the development of a compound designated as BAF312, commercially known as siponimod (Figure 5), which entered phase III clinical studies on secondary progressive MS (SPMS) patients. This molecule displays potent agonism on S1P1 and S1P5 while completely sparing S1P3, and the highest selectivity toward S1P1 and S1P5 is caused by the increased rigidity in the lipophilic alkyl chain of fingolimod (Pan et al., 2013).
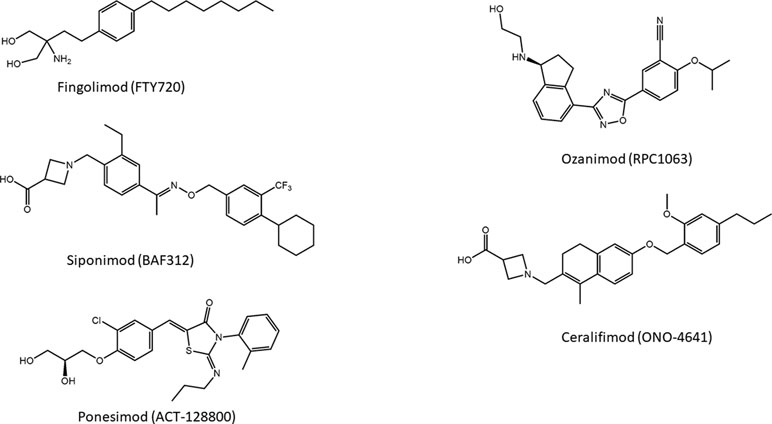
Figure 5 Novel modulators of S1P receptors.
Another S1PR modulator and MS drug candidate is ONO-4641, commercially named ceralifimod (Figure 5). This molecule has a chemical structure similar to that of siponimod and also acts specifically on S1P1 and S1P5. A phase II clinical trial was concluded in 2012; however, at the moment, it is not clear what future developments the molecule will have (Subei and Cohen, 2015).
About the series of oxadiazole-based S1P agonists with high selectivity for S1P1 and S1P5 receptor, the derivative disubstituted on terminal benzene ring and with a terminal hydroxyl group RPC1063, named ozanimod (Figure 5) (Scott et al., 2016), has entered phase III clinical trials. Ozanimod, one of the most promising molecules for MS, has minor adverse effects and shows superior receptor specificity if compared to fingolimod, siponimod, and ponesimod (Westad et al., 2017).
Ponesimod (ACT-128800) (Figure 5) is another class of S1P1 receptor agonists based on the 2-imino-thiazolidin-4-one scaffold, with a diol substituent that does not require phosphorylation for the acidity. This compound has been selected for clinical development (Bolli et al., 2010), and its selectivity for S1P1 is approximately 650-fold higher compared with the natural ligand S1P (D’ambrosio et al., 2016).
Several other molecules have been developed and entered in phase I clinical trials, but other studies are needed to design more potent and selective immunomodulatory drugs without causing major adverse effects.
Deregulation of sphingolipid metabolism is a common feature of a number of brain diseases, even in the absence of specific known defects of sphingolipid metabolic enzymes and/or sphingolipid intracellular traffic (Piccinini et al., 2010). Thus, researchers in this field have frequently explored the concept of targeting sphingolipid metabolism to address brain diseases. However, the translation of basic research to therapeutic opportunities in the case of complex glycosphingolipids has been so far unsuccessful. The only notable exception is represented by the potential use of a glucosylceramide synthase inhibitor, GZ667161, for the treatment of PD patients associated with mutations of the gene encoding for the glucocerebrosidase (Sardi et al., 2017). Dysregulation of S1P signaling and of the metabolic machinery involved in the control of S1P levels has been largely demonstrated in neuroinflammatory and neurodegenerative diseases. The translational research in the field of S1P has been definitely more successful; in particular, addressing the family of S1P receptors as therapeutic target has led to the introduction of fingolimod for the therapy of RR-MS. Fingolimod is recognized as a very efficient drug in RR-MS, and pharmacological research in this sense has continued, leading to analogues with improved pharmacokinetic features and with a potential usefulness in the treatment of other forms of MS, where the neuroinflammation still is a major player. The complexity of fingolimod action in the brain suggests that this or similar drugs might be useful to treat CNS illnesses other than MS. Since discovery of fingolimod, new-generation S1P receptor drugs are also being developed to target more specific S1P receptors. Overall, the family of S1PRs thus appears worthy of continued study and may provide significant therapeutic opportunities. Not only S1P receptors but also many of the enzymes involved in S1P metabolism bear the potential of promising therapeutic targets. From this point of view, in addition to the sphingosine kinases, the enzymes involved in the control of S1P levels along the catabolic pathways have been recently emerged as crucial in mediating the pathological role of S1P in brain disease. As an example, it is worth to remind that mutations in the S1P lyase are associated with neural toxicity (Choi and Saba, 2019). Thus, the array of possible druggable targets is considerably widening, and we would expect exciting developments in this field in the near future.
AP and PG contributed to the conception and design of the paper. AP wrote the first draft of the paper. AP, SG, LM, SP, and PG wrote sections of the paper. LC designed the graphics. All authors contributed to critical analysis of the literature, contributed to manuscript revision, and read and approved the submitted version. SG and LM equally contributed to the paper, and their names are listed in alphabetical order.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
3-kdhSo, 3-ketodihydrosphingosine; AD, Alzheimer’s disease; APP, amyloid precursor protein; BBB, blood brain barrier; CerS, ceramide synthases; CDase, ceramidase; CNS, central nervous system; DES1, dihydroceramide desaturase 1; dhCer, dihydroceramide; dhSo, dihydrosphingosine; EAE, experimental autoimmune encephalomyelitis; GCase, glycosidases; HD, Huntington disease; HDAC(s), histone deacetylase(s); KDSR, 3-ketodihydrosphingosine reductase; LPS, lipopolysaccharide; MS, multiple sclerosis; NF-κB, nuclear factor κB; PD, Parkinson’s disease; PI3K, phosphoinositide 3-kinase; PS1, presenilin-1; ROS, reactive oxygen species; S1P, sphingosine 1-phosphate; S1P1-5, S1P receptor 1 to 5; SGPL1, sphingosine 1-phosphate lyase; SK1, sphingosine kinase 1; SK2, sphingosine kinase 2; SM, sphingomyelin; SMase, sphingomyelinase; SPP1, S1P phosphatase; SPT, serine palmitoyltransferase; RR-MS, relapsing–remitting multiple sclerosis; SP-MS, secondary progressive multiple sclerosis.
Agudo-Lopez, A., Miguel, B. G., Fernandez, I., Martinez, A. M. (2010). Involvement of mitochondria on neuroprotective effect of sphingosine-1-phosphate in cell death in an in vitro model of brain ischemia. Neurosci. Lett. 470, 130–133. doi: 10.1016/j.neulet.2009.12.070
Akahoshi, N., Ishizaki, Y., Yasuda, H., Murashima, Y. L., Shinba, T., Goto, K., et al. (2011). Frequent spontaneous seizures followed by spatial working memory/anxiety deficits in mice lacking sphingosine 1-phosphate receptor 2. Epilepsy. Behav. 22, 659–665. doi: 10.1016/j.yebeh.2011.09.002
Alvarez, A., Cacabelos, R., Sanpedro, C., Garcia-Fantini, M., Aleixandre, M. (2007). Serum TNF-alpha levels are increased and correlate negatively with free IGF-I in Alzheimer disease. Neurobiol. Aging 28, 533–536. doi: 10.1016/j.neurobiolaging.2006.02.012
Alvarez, S. E., Harikumar, K. B., Hait, N. C., Allegood, J., Strub, G. M., Kim, E. Y., et al. (2010). Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 465, 1084–1088. doi: 10.1038/nature09128
Asle-Rousta, M., Kolahdooz, Z., Dargahi, L., Ahmadiani, A., Nasoohi, S. (2014). Prominence of central sphingosine-1-phosphate receptor-1 in attenuating abeta-induced injury by fingolimod. J. Mol. Neurosci. 54, 698–703. doi: 10.1007/s12031-014-0423-3
Asle-Rousta, M., Kolahdooz, Z., Oryan, S., Ahmadiani, A., Dargahi, L. (2013a). FTY720 (fingolimod) attenuates beta-amyloid peptide (Abeta42)-induced impairment of spatial learning and memory in rats. J. Mol. Neurosci. 50, 524–532. doi: 10.1007/s12031-013-9979-6
Asle-Rousta, M., Oryan, S., Ahmadiani, A., Rahnema, M. (2013b). Activation of sphingosine 1-phosphate receptor-1 by SEW2871 improves cognitive function in Alzheimer’s disease model rats. EXCLI. J. 12, 449–461.
Assi, E., Cazzato, D., De Palma, C., Perrotta, C., Clementi, E., Cervia, D. (2013). Sphingolipids and brain resident macrophages in neuroinflammation: an emerging aspect of nervous system pathology. Clin. Dev. Immunol. 2013, 309302. doi: 10.1155/2013/309302
Atwal, R. S., Desmond, C. R., Caron, N., Maiuri, T., Xia, J., Sipione, S., et al. (2011). Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat. Chem. Biol. 7, 453–460. doi: 10.1038/nchembio.582
Aytan, N., Choi, J. K., Carreras, I., Brinkmann, V., Kowall, N. W., Jenkins, B. G., et al. (2016). Fingolimod modulates multiple neuroinflammatory markers in a mouse model of Alzheimer’s disease. Sci. Rep. 6, 24939. doi: 10.1038/srep24939
Badawy, S. M. M., Okada, T., Kajimoto, T., Hirase, M., Matovelo, S. A., Nakamura, S., et al. (2018). Extracellular alpha-synuclein drives sphingosine 1-phosphate receptor subtype 1 out of lipid rafts, leading to impaired inhibitory G-protein signaling. J. Biol. Chem. 293, 8208–8216. doi: 10.1074/jbc.RA118.001986
Bassi, R., Anelli, V., Giussani, P., Tettamanti, G., Viani, P., Riboni, L. (2006). Sphingosine-1-phosphate is released by cerebellar astrocytes in response to bFGF and induces astrocyte proliferation through Gi-protein-coupled receptors. Glia 53, 621–630. doi: 10.1002/glia.20324
Beitz, J. M. (2014). Parkinson’s disease: a review. Front. Biosci. (Schol. Ed.) 6, 65–74. doi: 10.2741/S415
Bieber, A. J., Warrington, A., Asakura, K., Ciric, B., Kaveri, S. V., Pease, L. R., et al. (2002). Human antibodies accelerate the rate of remyelination following lysolecithin-induced demyelination in mice. Glia 37, 241–249. doi: 10.1002/glia.10033
Blaho, V. A., Hla, T. (2014). An update on the biology of sphingosine 1-phosphate receptors. J. Lipid. Res. 55, 1596–1608. doi: 10.1194/jlr.R046300
Boggs, J. M., Gao, W., Zhao, J., Park, H. J., Liu, Y., Basu, A. (2010). Participation of galactosylceramide and sulfatide in glycosynapses between oligodendrocyte or myelin membranes. FEBS. Lett. 584, 1771–1778. doi: 10.1016/j.febslet.2009.11.074
Bolli, M. H., Abele, S., Binkert, C., Bravo, R., Buchmann, S., Bur, D., et al. (2010). 2-imino-thiazolidin-4-one derivatives as potent, orally active S1P1 receptor agonists. J. Med. Chem. 53, 4198–4211. doi: 10.1021/jm100181s
Bowles, K. R., Jones, L. (2014). Kinase signalling in Huntington’s disease. J. Huntingtons. Dis. 3, 89–123. doi: 10.3233/JHD-140106
Brailoiu, E., Cooper, R. L., Dun, N. J. (2002). Sphingosine 1-phosphate enhances spontaneous transmitter release at the frog neuromuscular junction. Br. J. Pharmacol. 136, 1093–1097. doi: 10.1038/sj.bjp.0704839
Brinkmann, V., Billich, A., Baumruker, T., Heining, P., Schmouder, R., Francis, G., et al. (2010). Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 9, 883–897. doi: 10.1038/nrd3248
Brito, V., Giralt, A., Enriquez-Barreto, L., Puigdellivol, M., Suelves, N., Zamora-Moratalla, A., et al. (2014). Neurotrophin receptor p75(NTR) mediates Huntington’s disease-associated synaptic and memory dysfunction. J. Clin. Invest. 124, 4411–4428. doi: 10.1172/JCI74809
Buckley, N. J., Johnson, R., Zuccato, C., Bithell, A., Cattaneo, E. (2010). The role of REST in transcriptional and epigenetic dysregulation in Huntington’s disease. Neurobiol. Dis. 39, 28–39. doi: 10.1016/j.nbd.2010.02.003
Camoletto, P. G., Vara, H., Morando, L., Connell, E., Marletto, F. P., Giustetto, M., et al. (2009). Synaptic vesicle docking: sphingosine regulates syntaxin1 interaction with Munc18. PLoS One 4, e5310. doi: 10.1371/journal.pone.0005310
Cattaneo, M. G., Vanetti, C., Samarani, M., Aureli, M., Bassi, R., Sonnino, S., et al. (2018). Cross-talk between sphingosine-1-phosphate and EGFR signaling pathways enhances human glioblastoma cell invasiveness. FEBS. Lett. 592, 949–961. doi: 10.1002/1873-3468.13000
Ceccom, J., Loukh, N., Lauwers-Cances, V., Touriol, C., Nicaise, Y., Gentil, C., et al. (2014). Reduced sphingosine kinase-1 and enhanced sphingosine 1-phosphate lyase expression demonstrate deregulated sphingosine 1-phosphate signaling in Alzheimer’s disease. Acta. Neuropathol. Commun. 2, 12. doi: 10.1186/2051-5960-2-12
Chen, Y. J., Hill, S., Huang, H., Taraboletti, A., Cho, K., Gallo, R., et al. (2014). Inflammation triggers production of dimethylsphingosine from oligodendrocytes. Neuroscience 279, 113–121. doi: 10.1016/j.neuroscience.2014.08.011
Chiricozzi, E., Loberto, N., Schiumarini, D., Samarani, M., Mancini, G., Tamanini, A., et al. (2018). Sphingolipids role in the regulation of inflammatory response: From leukocyte biology to bacterial infection. J. Leukoc. Biol. 103, 445–456. doi: 10.1002/JLB.3MR0717-269R
Choi, J. W., Gardell, S. E., Herr, D. R., Rivera, R., Lee, C. W., Noguchi, K., et al. (2011). FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. U. S. A. 108, 751–756. doi: 10.1073/pnas.1014154108
Choi, Y. J., Saba, J. D. (2019). Sphingosine phosphate lyase insufficiency syndrome (SPLIS): a novel inborn error of sphingolipid metabolism. Adv. Biol. Regul. 71, 128–140. doi: 10.1016/j.jbior.2018.09.004
Chun, J., Hartung, H. P. (2010). Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin. Neuropharmacol. 33, 91–101. doi: 10.1097/WNF.0b013e3181cbf825
Contreras, M. A., Ries, W. L., Shanmugarajan, S., Arboleda, G., Singh, I., Singh, A. K. (2010). Factors that affect postnatal bone growth retardation in the twitcher murine model of Krabbe disease. Biochim. Biophys. Acta. 1802, 601–608. doi: 10.1016/j.bbadis.2010.04.006
Coste, O., Pierre, S., Marian, C., Brenneis, C., Angioni, C., Schmidt, H., et al. (2008). Antinociceptive activity of the S1P-receptor agonist FTY720. J. Cell. Mol. Med. 12, 995–1004. doi: 10.1111/j.1582-4934.2008.00160.x
Couttas, T. A., Kain, N., Daniels, B., Lim, X. Y., Shepherd, C., Kril, J., et al. (2014). Loss of the neuroprotective factor Sphingosine 1-phosphate early in Alzheimer’s disease pathogenesis. Acta. Neuropathol. Commun. 2, 9. doi: 10.1186/2051-5960-2-9
Crews, L., Masliah, E. (2010). Molecular mechanisms of neurodegeneration in Alzheimer’s disease. Hum. Mol. Genet. 19, R12–20. doi: 10.1093/hmg/ddq160
Czech, B., Pfeilschifter, W., Mazaheri-Omrani, N., Strobel, M. A., Kahles, T., Neumann-Haefelin, T., et al. (2009). The immunomodulatory sphingosine 1-phosphate analog FTY720 reduces lesion size and improves neurological outcome in a mouse model of cerebral ischemia. Biochem. Biophys. Res. Commun. 389, 251–256. doi: 10.1016/j.bbrc.2009.08.142
Czubowicz, K., Cieslik, M., Pyszko, J., Strosznajder, J. B., Strosznajder, R. P. (2015). Sphingosine-1-phosphate and its effect on glucose deprivation/glucose reload stress: from gene expression to neuronal survival. Mol. Neurobiol. 51, 1300–1308. doi: 10.1007/s12035-014-8807-5
Czubowicz, K., Jesko, H., Wencel, P., Lukiw, W. J., Strosznajder, R. P. (2019). The role of ceramide and sphingosine-1-phosphate in Alzheimer’s disease and other neurodegenerative disorders. Mol. Neurobiol. [Epub ahead of print]. doi: 10.1007/s12035-018-1448-3
D’ambrosio, D., Freedman, M. S., Prinz, J. (2016). Ponesimod, a selective S1P1 receptor modulator: a potential treatment for multiple sclerosis and other immune-mediated diseases. Ther. Adv. Chronic. Dis. 7, 18–33. doi: 10.1177/2040622315617354
Dai, Y., Rahmani, M., Dent, P., Grant, S. (2005). Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappaB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol. Cell. Biol. 25, 5429–5444. doi: 10.1128/MCB.25.13.5429-5444.2005
Darios, F., Wasser, C., Shakirzyanova, A., Giniatullin, A., Goodman, K., Munoz-Bravo, J. L., et al. (2009). Sphingosine facilitates SNARE complex assembly and activates synaptic vesicle exocytosis. Neuron 62, 683–694. doi: 10.1016/j.neuron.2009.04.024
Deogracias, R., Yazdani, M., Dekkers, M. P., Guy, J., Ionescu, M. C., Vogt, K. E., et al. (2012). Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc Natl. Acad. Sci. U. S. A. 109, 14230–14235. doi: 10.1073/pnas.1206093109
Di Pardo, A., Amico, E., Basit, A., Armirotti, A., Joshi, P., Neely, M. D., et al. (2017). Defective sphingosine-1-phosphate metabolism is a druggable target in Huntington’s disease. Sci. Rep. 7, 5280. doi: 10.1038/s41598-017-05709-y
Di Pardo, A., Amico, E., Favellato, M., Castrataro, R., Fucile, S., Squitieri, F., et al. (2014). FTY720 (fingolimod) is a neuroprotective and disease-modifying agent in cellular and mouse models of Huntington disease. Hum. Mol. Genet. 23, 2251–2265. doi: 10.1093/hmg/ddt615
Di Pardo, A., Castaldo, S., Amico, E., Pepe, G., Marracino, F., Capocci, L., et al. (2018). Stimulation of S1PR5 with A-971432, a selective agonist, preserves blood-brain barrier integrity and exerts therapeutic effect in an animal model of Huntington’s disease. Hum. Mol. Genet. 27, 2490–2501. doi: 10.1093/hmg/ddy153
Di Pardo, A., Maglione, V., Alpaugh, M., Horkey, M., Atwal, R. S., Sassone, J., et al. (2012). Ganglioside GM1 induces phosphorylation of mutant huntingtin and restores normal motor behavior in Huntington disease mice. Proc. Natl. Acad. Sci U. S. A. 109, 3528–3533. doi: 10.1073/pnas.1114502109
Diemel, L. T., Copelman, C. A., Cuzner, M. L. (1998). Macrophages in CNS remyelination: friend or foe? Neurochem. Res. 23, 341–347. doi: 10.1023/A:1022405516630
Dominguez, G., Maddelein, M. L., Pucelle, M., Nicaise, Y., Maurage, C. A., Duyckaerts, C., et al. (2018). Neuronal sphingosine kinase 2 subcellular localization is altered in Alzheimer’s disease brain. Acta. Neuropathol. Commun. 6, 25. doi: 10.1186/s40478-018-0527-z
Doolen, S., Iannitti, T., Donahue, R. R., Shaw, B. C., Grachen, C. M., Taylor, B. K. (2018). Fingolimod reduces neuropathic pain behaviors in a mouse model of multiple sclerosis by a sphingosine-1 phosphate receptor 1-dependent inhibition of central sensitization in the dorsal horn. Pain 159, 224–238. doi: 10.1097/j.pain.0000000000001106
Doyle, T., Finley, A., Chen, Z., Salvemini, D. (2011). Role for peroxynitrite in sphingosine-1-phosphate-induced hyperalgesia in rats. Pain 152, 643–648. doi: 10.1016/j.pain.2010.12.011
Dusaban, S. S., Chun, J., Rosen, H., Purcell, N. H., Brown, J. H. (2017). Sphingosine 1-phosphate receptor 3 and RhoA signaling mediate inflammatory gene expression in astrocytes. J. Neuroinflammation. 14, 111. doi: 10.1186/s12974-017-0882-x
Eisen, A., Greenberg, B. M., Bowen, J. D., Arnold, D. L., Caggiano, A. O. (2017). A double-blind, placebo-controlled, single ascending-dose study of remyelinating antibody rHIgM22 in people with multiple sclerosis. Mult. Scler. J. Exp. Transl. Clin. 3, 2055217317743097. doi: 10.1177/2055217317743097
Estrada-Bernal, A., Palanichamy, K., Ray Chaudhury, A., Van Brocklyn, J. R. (2012). Induction of brain tumor stem cell apoptosis by FTY720: a potential therapeutic agent for glioblastoma. Neuro. Oncol. 14, 405–415. doi: 10.1093/neuonc/nos005
Farez, M. F., Correale, J. (2016). Sphingosine 1-phosphate signaling in astrocytes: Implications for progressive multiple sclerosis. J. Neurol. Sci. 361, 60–65. doi: 10.1016/j.jns.2015.12.022
Feldman, G. J., Mullin, J. M., Ryan, M. P. (2005). Occludin: structure, function and regulation. Adv. Drug Deliv. Rev. 57, 883–917. doi: 10.1016/j.addr.2005.01.009
Fernandez-Pisonero, I., Duenas, A. I., Barreiro, O., Montero, O., Sanchez-Madrid, F., Garcia-Rodriguez, C. (2012). Lipopolysaccharide and sphingosine-1-phosphate cooperate to induce inflammatory molecules and leukocyte adhesion in endothelial cells. J. Immunol. 189, 5402–5410. doi: 10.4049/jimmunol.1201309
Fischer, I., Alliod, C., Martinier, N., Newcombe, J., Brana, C., Pouly, S. (2011). Sphingosine kinase 1 and sphingosine 1-phosphate receptor 3 are functionally upregulated on astrocytes under pro-inflammatory conditions. PLoS One 6, e23905. doi: 10.1371/journal.pone.0023905
Fitzpatrick, L. R., Green, C., Frauenhoffer, E. E., French, K. J., Zhuang, Y., Maines, L. W., et al. (2011a). Attenuation of arthritis in rodents by a novel orally-available inhibitor of sphingosine kinase. Inflammopharmacology 19, 75–87. doi: 10.1007/s10787-010-0060-6
Fitzpatrick, L. R., Green, C., Maines, L. W., Smith, C. D. (2011b). Experimental osteoarthritis in rats is attenuated by ABC294640, a selective inhibitor of sphingosine kinase-2. Pharmacology 87, 135–143. doi: 10.1159/000323911
Foster, C. A., Howard, L. M., Schweitzer, A., Persohn, E., Hiestand, P. C., Balatoni, B., et al. (2007). Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: consequences for mode of action in multiple sclerosis. J. Pharmacol. Exp. Ther. 323, 469–475. doi: 10.1124/jpet.107.127183
French, K. J., Upson, J. J., Keller, S. N., Zhuang, Y., Yun, J. K., Smith, C. D. (2006). Antitumor activity of sphingosine kinase inhibitors. J. Pharmacol. Exp. Ther. 318, 596–603. doi: 10.1124/jpet.106.101345
Fukumoto, K., Mizoguchi, H., Takeuchi, H., Horiuchi, H., Kawanokuchi, J., Jin, S., et al. (2014). Fingolimod increases brain-derived neurotrophic factor levels and ameliorates amyloid beta-induced memory impairment. Behav. Brain. Res. 268, 88–93. doi: 10.1016/j.bbr.2014.03.046
Gao, F., Liu, Y., Li, X., Wang, Y., Wei, D., Jiang, W. (2012). Fingolimod (FTY720) inhibits neuroinflammation and attenuates spontaneous convulsions in lithium-pilocarpine induced status epilepticus in rat model. Pharmacol. Biochem. Behav. 103, 187–196. doi: 10.1016/j.pbb.2012.08.025
Ghose, J., Sinha, M., Das, E., Jana, N. R., Bhattacharyya, N. P. (2011). Regulation of miR-146a by RelA/NFκB and p53 in STHdh(Q111)/Hdh(Q111) cells, a cell model of Huntington’s disease. PLoS One 6, e23837. doi: 10.1371/journal.pone.0023837
Giussani, P., Ferraretto, A., Gravaghi, C., Bassi, R., Tettamanti, G., Riboni, L., et al. (2007). Sphingosine-1-phosphate and calcium signaling in cerebellar astrocytes and differentiated granule cells. Neurochem. Res. 32, 27–37. doi: 10.1007/s11064-006-9219-3
Giussani, P., Tringali, C., Riboni, L., Viani, P., Venerando, B. (2014). Sphingolipids: key regulators of apoptosis and pivotal players in cancer drug resistance. Int. J. Mol. Sci. 15, 4356–4392. doi: 10.3390/ijms15034356
Gomez-Brouchet, A., Pchejetski, D., Brizuela, L., Garcia, V., Altie, M. F., Maddelein, M. L., et al. (2007). Critical role for sphingosine kinase-1 in regulating survival of neuroblastoma cells exposed to amyloid-beta peptide. Mol. Pharmacol. 72, 341–349. doi: 10.1124/mol.106.033738
Gomez-Lopez, S., Martinez-Silva, A. V., Montiel, T., Osorio-Gomez, D., Bermudez-Rattoni, F., Massieu, L., et al. (2016). Neural ablation of the PARK10 candidate Plpp3 leads to dopaminergic transmission deficits without neurodegeneration. Sci. Rep. 6, 24028. doi: 10.1038/srep24028
Gomez-Munoz, A., Kong, J., Salh, B., Steinbrecher, U. P. (2003). Sphingosine-1-phosphate inhibits acid sphingomyelinase and blocks apoptosis in macrophages. FEBS Lett. 539, 56–60. doi: 10.1016/S0014-5793(03)00197-2
Goni, F. M., Alonso, A. (2002). Sphingomyelinases: enzymology and membrane activity. FEBS Lett. 531, 38–46. doi: 10.1016/S0014-5793(02)03482-8
Grassi, S., Giussani, P., Prioni, S., Button, D., Cao, J., Hakimi, I., et al. (2019). Human remyelination promoting antibody stimulates astrocytes proliferation through modulation of the sphingolipid rheostat in primary rat mixed glial cultures. Neurochem. Res. 44, 1460–1474. doi: 10.1007/s11064-018-2701-x
Groves, A., Kihara, Y., Chun, J. (2013). Fingolimod: direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J. .Neurol. Sci. 328, 9–18. doi: 10.1016/j.jns.2013.02.011
Gudi, V., Skuljec, J., Yildiz, O., Frichert, K., Skripuletz, T., Moharregh-Khiabani, D., et al. (2011). Spatial and temporal profiles of growth factor expression during CNS demyelination reveal the dynamics of repair priming. PLoS One 6, e22623. doi: 10.1371/journal.pone.0022623
Guy, J., Hendrich, B., Holmes, M., Martin, J. E., Bird, A. (2001). A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 27, 322–326. doi: 10.1038/85899
Hagen, N., Hans, M., Hartmann, D., Swandulla, D., Van Echten-Deckert, G. (2011). Sphingosine-1-phosphate links glycosphingolipid metabolism to neurodegeneration via a calpain-mediated mechanism. Cell. Death. Differ. 18, 1356–1365. doi: 10.1038/cdd.2011.7
Hagen, N., Van Veldhoven, P. P., Proia, R. L., Park, H., Merrill, A. H., Jr., Van Echten-Deckert, G. (2009). Subcellular origin of sphingosine 1-phosphate is essential for its toxic effect in lyase-deficient neurons. J. Biol. Chem. 284, 11346–11353. doi: 10.1074/jbc.M807336200
Hahn, C., Tyka, K., Saba, J. D., Lenzen, S., Gurgul-Convey, E. (2017). Overexpression of sphingosine-1-phosphate lyase protects insulin-secreting cells against cytokine toxicity. J. Biol. Chem. 292, 20292–20304. doi: 10.1074/jbc.M117.814491
Hait, N. C., Allegood, J., Maceyka, M., Strub, G. M., Harikumar, K. B., Singh, S. K., et al. (2009). Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325, 1254–1257. doi: 10.1126/science.1176709
Hait, N. C., Bellamy, A., Milstien, S., Kordula, T., Spiegel, S. (2007). Sphingosine kinase type 2 activation by ERK-mediated phosphorylation. J. Biol. Chem. 282, 12058–12065. doi: 10.1074/jbc.M609559200
Hait, N. C., Wise, L. E., Allegood, J. C., O’brien, M., Avni, D., Reeves, T. M., et al. (2014). Active, phosphorylated fingolimod inhibits histone deacetylases and facilitates fear extinction memory. Nat. Neurosci. 17, 971–980. doi: 10.1038/nn.3728
Hammad, S. M., Crellin, H. G., Wu, B. X., Melton, J., Anelli, V., Obeid, L. M. (2008). Dual and distinct roles for sphingosine kinase 1 and sphingosine 1 phosphate in the response to inflammatory stimuli in RAW macrophages. Prostaglandins Other Lipid Mediat. 85, 107–114. doi: 10.1016/j.prostaglandins.2007.11.002
Hannun, Y. A. (1994). The sphingomyelin cycle and the second messenger function of ceramide. J. Biol. Chem. 269, 3125–3128.
He, X., Huang, Y., Li, B., Gong, C. X., Schuchman, E. H. (2010). Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol. Aging 31, 398–408. doi: 10.1016/j.neurobiolaging.2008.05.010
Hemmati, F., Dargahi, L., Nasoohi, S., Omidbakhsh, R., Mohamed, Z., Chik, Z., et al. (2013). Neurorestorative effect of FTY720 in a rat model of Alzheimer’s disease: comparison with memantine. Behav. Brain Res. 252, 415–421. doi: 10.1016/j.bbr.2013.06.016
Hill-Burns, E. M., Wissemann, W. T., Hamza, T. H., Factor, S. A., Zabetian, C. P., Payami, H. (2014). Identification of a novel Parkinson’s disease locus via stratified genome-wide association study. BMC Genomics 15, 118. doi: 10.1186/1471-2164-15-118
Hirano-Sakamaki, W., Sugiyama, E., Hayasaka, T., Ravid, R., Setou, M., Taki, T. (2015). Alzheimer’s disease is associated with disordered localization of ganglioside GM1 molecular species in the human dentate gyrus. FEBS Lett. 589, 3611–3616. doi: 10.1016/j.febslet.2015.09.033
Hodgson, D. M., Taylor, A. N., Zhang, Z., Rosenberg, A. (1999). Lysosphingomyelin prevents behavioral aberrations and hippocampal neuron loss induced by the metabotropic glutamate receptor agonist quisqualate. Prog. Neuropsychopharmacol. Biol. Psychiatry. 23, 877–892. doi: 10.1016/S0278-5846(99)00047-0
Hsu, C. K., Lee, I. T., Lin, C. C., Hsiao, L. D., Yang, C. M. (2015). Sphingosine-1-phosphate mediates COX-2 expression and PGE2/IL-6 secretion via c-Src-dependent AP-1 activation. J. Cell Physiol. 230, 702–715. doi: 10.1002/jcp.24795
Hu, X. Y., Zhang, H. Y., Qin, S., Xu, H., Swaab, D. F., Zhou, J. N. (2002). Increased p75(NTR) expression in hippocampal neurons containing hyperphosphorylated tau in Alzheimer patients. Exp. Neurol. 178, 104–111. doi: 10.1006/exnr.2002.8018
Huang, K., Huang, J., Chen, C., Hao, J., Wang, S., Huang, J., et al. (2014). AP-1 regulates sphingosine kinase 1 expression in a positive feedback manner in glomerular mesangial cells exposed to high glucose. Cell Signal. 26, 629–638. doi: 10.1016/j.cellsig.2013.12.002
Huang, Y. L., Huang, W. P., Lee, H. (2011). Roles of sphingosine 1-phosphate on tumorigenesis. World J. Biol. Chem. 2, 25–34. doi: 10.4331/wjbc.v2.i2.25
Huwiler, A., Zangemeister-Wittke, U. (2018). The sphingosine 1-phosphate receptor modulator fingolimod as a therapeutic agent: recent findings and new perspectives. Pharmacol. Ther. 185, 34–49. doi: 10.1016/j.pharmthera.2017.11.001
Ichijo, M., Ishibashi, S., Li, F., Yui, D., Miki, K., Mizusawa, H., et al. (2015). Sphingosine-1-phosphate receptor-1 selective agonist enhances collateral growth and protects against subsequent stroke. PLoS One 10, e0138029. doi: 10.1371/journal.pone.0138029
Jackson, S. J., Giovannoni, G., Baker, D. (2011). Fingolimod modulates microglial activation to augment markers of remyelination. J. Neuroinflammation. 8, 76. doi: 10.1186/1742-2094-8-76
Jang, S., Kim, D., Lee, Y., Moon, S., Oh, S. (2011). Modulation of sphingosine 1-phosphate and tyrosine hydroxylase in the stress-induced anxiety. Neurochem. Res. 36, 258–267. doi: 10.1007/s11064-010-0313-1
Jazvinscak Jembrek, M., Hof, P. R., Simic, G. (2015). Ceramides in Alzheimer’s disease: key mediators of neuronal apoptosis induced by oxidative stress and abeta accumulation. Oxid. Med. Cell. Longev. 2015, 346783. doi: 10.1155/2015/346783
Jimenez-Sanchez, M., Licitra, F., Underwood, B. R., Rubinsztein, D. C. (2017). Huntington’s disease: mechanisms of pathogenesis and therapeutic strategies. Cold. Spring. Harb. Perspect. Med. 7, a024240. doi: 10.1101/cshperspect.a024240
Jin, Y., Tu, Q., Liu, M. (2018). MicroRNA125b regulates Alzheimer’s disease through SphK1 regulation. Mol. Med. Rep. 18, 2373–2380. doi: 10.3892/mmr.2018.9156
Kanno, T., Nishizaki, T., Proia, R. L., Kajimoto, T., Jahangeer, S., Okada, T., et al. (2010). Regulation of synaptic strength by sphingosine 1-phosphate in the hippocampus. Neuroscience 171, 973–980. doi: 10.1016/j.neuroscience.2010.10.021
Kapitonov, D., Allegood, J. C., Mitchell, C., Hait, N. C., Almenara, J. A., Adams, J. K., et al. (2009). Targeting sphingosine kinase 1 inhibits Akt signaling, induces apoptosis, and suppresses growth of human glioblastoma cells and xenografts. Cancer Res. 69, 6915–6923. doi: 10.1158/0008-5472.CAN-09-0664
Kappos, L., Antel, J., Comi, G., Montalban, X., O’connor, P., Polman, C. H., et al. (2006). Oral fingolimod (FTY720) for relapsing multiple sclerosis. N. Engl. J. Med. 355, 1124–1140. doi: 10.1056/NEJMoa052643
Karaca, I., Tamboli, I. Y., Glebov, K., Richter, J., Fell, L. H., Grimm, M. O., et al. (2014). Deficiency of sphingosine-1-phosphate lyase impairs lysosomal metabolism of the amyloid precursor protein. J. Biol. Chem. 289, 16761–16772. doi: 10.1074/jbc.M113.535500
Karunakaran, I., Van Echten-Deckert, G. (2017). Sphingosine 1-phosphate - A double edged sword in the brain. Biochim. Biophys. Acta. Biomembr. 1859, 1573–1582. doi: 10.1016/j.bbamem.2017.03.008
Katsel, P., Li, C., Haroutunian, V. (2007). Gene expression alterations in the sphingolipid metabolism pathways during progression of dementia and Alzheimer’s disease: a shift toward ceramide accumulation at the earliest recognizable stages of Alzheimer’s disease? Neurochem. Res. 32, 845–856. doi: 10.1007/s11064-007-9297-x
Kelly, R. J., Minogue, A. M., Lyons, A., Jones, R. S., Browne, T. C., Costello, D. A., et al. (2013). Glial activation in AbetaPP/PS1 Mice is associated with Infiltration of IFNgamma-producing cells. J. Alzheimers. Dis. 37, 63–75. doi: 10.3233/JAD-130539
Kerschensteiner, M., Gallmeier, E., Behrens, L., Leal, V. V., Misgeld, T., Klinkert, W. E., et al. (1999). Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J. Exp. Med. 189, 865–870. doi: 10.1084/jem.189.5.865
Kim, H. J., Miron, V. E., Dukala, D., Proia, R. L., Ludwin, S. K., Traka, M., et al. (2011). Neurobiological effects of sphingosine 1-phosphate receptor modulation in the cuprizone model. FASEB J. 25, 1509–1518. doi: 10.1096/fj.10-173203
Kim, S., Steelman, A. J., Zhang, Y., Kinney, H. C., Li, J. (2012). Aberrant upregulation of astroglial ceramide potentiates oligodendrocyte injury. Brain Pathol. 22, 41–57. doi: 10.1111/j.1750-3639.2011.00501.x
Kleger, A., Busch, T., Liebau, S., Prelle, K., Paschke, S., Beil, M., et al. (2007). The bioactive lipid sphingosylphosphorylcholine induces differentiation of mouse embryonic stem cells and human promyelocytic leukaemia cells. Cell Signal. 19, 367–377. doi: 10.1016/j.cellsig.2006.07.015
Knowles, J. K., Simmons, D. A., Nguyen, T. V., Vander Griend, L., Xie, Y., Zhang, H., et al. (2013). Small molecule p75NTR ligand prevents cognitive deficits and neurite degeneration in an Alzheimer’s mouse model. Neurobiol. Aging 34, 2052–2063. doi: 10.1016/j.neurobiolaging.2013.02.015
Komnig, D., Dagli, T. C., Habib, P., Zeyen, T., Schulz, J. B., Falkenburger, B. H. (2018). Fingolimod (FTY720) is not protective in the subacute MPTP mouse model of Parkinson’s disease and does not lead to a sustainable increase of brain-derived neurotrophic factor. J. Neurochem. 147, 678–691. doi: 10.1111/jnc.14575
Kozlov, S., Afonin, A., Evsyukov, I., Bondarenko, A. (2017). Alzheimer’s disease: as it was in the beginning. Rev. Neurosci. 28, 825–843. doi: 10.1515/revneuro-2017-0006
Kunkel, G. T., Maceyka, M., Milstien, S., Spiegel, S. (2013). Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug Discov. 12, 688–702. doi: 10.1038/nrd4099
Le Stunff, H., Giussani, P., Maceyka, M., Lepine, S., Milstien, S., Spiegel, S. (2007). Recycling of sphingosine is regulated by the concerted actions of sphingosine-1-phosphate phosphohydrolase 1 and sphingosine kinase 2. J. Biol. Chem. 282, 34372–34380. doi: 10.1074/jbc.M703329200
Le Stunff, H., Peterson, C., Thornton, R., Milstien, S., Mandala, S. M., Spiegel, S. (2002). Characterization of murine sphingosine-1-phosphate phosphohydrolase. J. Biol. Chem. 277, 8920–8927. doi: 10.1074/jbc.M109968200
Levade, T., Jaffrezou, J. P. (1999). Signalling sphingomyelinases: which, where, how and why? Biochim. Biophys. Acta 1438, 1–17. doi: 10.1016/S1388-1981(99)00038-4
Liesz, A., Sun, L., Zhou, W., Schwarting, S., Mracsko, E., Zorn, M., et al. (2011). FTY720 reduces post-ischemic brain lymphocyte influx but does not improve outcome in permanent murine cerebral ischemia. PLoS One 6, e21312. doi: 10.1371/journal.pone.0021312
Linington, C., Bradl, M., Lassmann, H., Brunner, C., Vass, K. (1988). Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am. J. Pathol. 130, 443–454.
Liu, J., Zhang, C., Tao, W., Liu, M. (2013a). Systematic review and meta-analysis of the efficacy of sphingosine-1-phosphate (S1P) receptor agonist FTY720 (fingolimod) in animal models of stroke. Int. J. Neurosci. 123, 163–169. doi: 10.3109/00207454.2012.749255
Liu, K., Guo, T. L., Hait, N. C., Allegood, J., Parikh, H. I., Xu, W., et al. (2013b). Biological characterization of 3-(2-amino-ethyl)-5-[3-(4-butoxyl-phenyl)-propylidene]-thiazolidine-2,4-dione (K145) as a selective sphingosine kinase-2 inhibitor and anticancer agent. PLoS One 8, e56471. doi: 10.1371/journal.pone.0056471
Liu, T., Jin, J., Chen, Y., Xi, Q., Hu, J., Jia, W., et al. (2019). Identification and Structure-Activity Relationship (SAR) of potent and selective oxadiazole-based agonists of sphingosine-1-phosphate receptor (S1P1). Bioorg. Chem. 82, 41–57. doi: 10.1016/j.bioorg.2018.09.008
Loberto, N., Prioni, S., Bettiga, A., Chigorno, V., Prinetti, A., Sonnino, S. (2005). The membrane environment of endogenous cellular prion protein in primary rat cerebellar neurons. J. Neurochem. 95, 771–783. doi: 10.1111/j.1471-4159.2005.03397.x
Lopez-Juarez, A., Morales-Lazaro, S., Sanchez-Sanchez, R., Sunkara, M., Lomeli, H., Velasco, I., et al. (2011). Expression of LPP3 in Bergmann glia is required for proper cerebellar sphingosine-1-phosphate metabolism/signaling and development. Glia 59, 577–589. doi: 10.1002/glia.21126
Lv, M., Zhang, D., Dai, D., Zhang, W., Zhang, L. (2016). Sphingosine kinase 1/sphingosine-1-phosphate regulates the expression of interleukin-17A in activated microglia in cerebral ischemia/reperfusion. Inflamm. Res. 65, 551–562. doi: 10.1007/s00011-016-0939-9
Maclennan, A. J., Carney, P. R., Zhu, W. J., Chaves, A. H., Garcia, J., Grimes, J. R., et al. (2001). An essential role for the H218/AGR16/Edg-5/LP(B2) sphingosine 1-phosphate receptor in neuronal excitability. Eur. J. Neurosci. 14, 203–209. doi: 10.1046/j.0953-816x.2001.01634.x
Maglione, V., Marchi, P., Di Pardo, A., Lingrell, S., Horkey, M., Tidmarsh, E., et al. (2010). Impaired ganglioside metabolism in Huntington’s disease and neuroprotective role of GM1. J. Neurosci. 30, 4072–4080. doi: 10.1523/JNEUROSCI.6348-09.2010
Maines, L. W., Fitzpatrick, L. R., Green, C. L., Zhuang, Y., Smith, C. D. (2010). Efficacy of a novel sphingosine kinase inhibitor in experimental Crohn’s disease. Inflammopharmacology 18, 73–85. doi: 10.1007/s10787-010-0032-x
Maines, L. W., Fitzpatrick, L. R., French, K. J., Zhuang, Y., Xia, Z., Keller, S. N., et al. (2008). Suppression of ulcerative colitis in mice by orally available inhibitors of sphingosine kinase. Dig. Dis. Sci. 53, 997–1012. doi: 10.1007/s10620-007-0133-6
Maines, L. W., French, K. J., Wolpert, E. B., Antonetti, D. A., Smith, C. D. (2006). Pharmacologic manipulation of sphingosine kinase in retinal endothelial cells: implications for angiogenic ocular diseases. Invest. Ophthalmol. Vis. Sci. 47, 5022–5031. doi: 10.1167/iovs.05-1236
Mair, N., Benetti, C., Andratsch, M., Leitner, M. G., Constantin, C. E., Camprubi-Robles, M., et al. (2011). Genetic evidence for involvement of neuronally expressed S1P(1) receptor in nociceptor sensitization and inflammatory pain. PLoS One 6, e17268. doi: 10.1371/journal.pone.0017268
Malaplate-Armand, C., Florent-Bechard, S., Youssef, I., Koziel, V., Sponne, I., Kriem, B., et al. (2006). Soluble oligomers of amyloid-beta peptide induce neuronal apoptosis by activating a cPLA2-dependent sphingomyelinase-ceramide pathway. Neurobiol. Dis. 23, 178–189. doi: 10.1016/j.nbd.2006.02.010
Mao-Draayer, Y., Sarazin, J., Fox, D., Schiopu, E. (2017). The sphingosine-1-phosphate receptor: A novel therapeutic target for multiple sclerosis and other autoimmune diseases. Clin. Immunol. 175, 10–15. doi: 10.1016/j.clim.2016.11.008
Marciniak, A., Camp, S. M., Garcia, J. G. N., Polt, R. (2018). An update on sphingosine-1-phosphate receptor 1 modulators. Bioorg. Med. Chem. Lett. 28, 3585–3591. doi: 10.1016/j.bmcl.2018.10.042
Marfia, G., Campanella, R., Navone, S. E., Di Vito, C., Riccitelli, E., Hadi, L. A., et al. (2014). Autocrine/paracrine sphingosine-1-phosphate fuels proliferative and stemness qualities of glioblastoma stem cells. Glia 62, 1968–1981. doi: 10.1002/glia.22718
Mayo, L., Quintana, F. J., Weiner, H. L. (2012). The innate immune system in demyelinating disease. Immunol. Rev. 248, 170–187. doi: 10.1111/j.1600-065X.2012.01135.x
Mcmanus, R. M., Finucane, O. M., Wilk, M. M., Mills, K. H. G., Lynch, M. A. (2017). FTY720 Attenuates Infection-Induced Enhancement of Abeta Accumulation in APP/PS1 Mice by Modulating Astrocytic Activation. J. Neuroimmune. Pharmacol. 12, 670–681. doi: 10.1007/s11481-017-9753-6
Mcmanus, R. M., Higgins, S. C., Mills, K. H., Lynch, M. A. (2014). Respiratory infection promotes T cell infiltration and amyloid-beta deposition in APP/PS1 mice. Neurobiol. Aging 35, 109–121. doi: 10.1016/j.neurobiolaging.2013.07.025
Mencl, S., Hennig, N., Hopp, S., Schuhmann, M. K., Albert-Weissenberger, C., Siren, A. L., et al. (2014). FTY720 does not protect from traumatic brain injury in mice despite reducing posttraumatic inflammation. J. Neuroimmunol. 274, 125–131. doi: 10.1016/j.jneuroim.2014.07.010
Meno-Tetang, G. M., Li, H., Mis, S., Pyszczynski, N., Heining, P., Lowe, P., et al. (2006). Physiologically based pharmacokinetic modeling of FTY720 (2-amino-2[2-(-4-octylphenyl)ethyl]propane-1,3-diol hydrochloride) in rats after oral and intravenous doses. Drug. Metab. Dispos. 34, 1480–1487. doi: 10.1124/dmd.105.009001
Miguez, A., Garcia-Diaz Barriga, G., Brito, V., Straccia, M., Giralt, A., Gines, S., et al. (2015). Fingolimod (FTY720) enhances hippocampal synaptic plasticity and memory in Huntington’s disease by preventing p75NTR up-regulation and astrocyte-mediated inflammation. Hum. Mol. Genet. 24, 4958–4970. doi: 10.1093/hmg/ddv218
Mikati, M. A., Abi-Habib, R. J., El Sabban, M. E., Dbaibo, G. S., Kurdi, R. M., Kobeissi, M., et al. (2003). Hippocampal programmed cell death after status epilepticus: evidence for NMDA-receptor and ceramide-mediated mechanisms. Epilepsia 44, 282–291. doi: 10.1046/j.1528-1157.2003.22502.x
Minogue, A. M., Jones, R. S., Kelly, R. J., Mcdonald, C. L., Connor, T. J., Lynch, M. A. (2014). Age-associated dysregulation of microglial activation is coupled with enhanced blood-brain barrier permeability and pathology in APP/PS1 mice. Neurobiol. Aging 35, 1442–1452. doi: 10.1016/j.neurobiolaging.2013.12.026
Mitroi, D. N., Deutschmann, A. U., Raucamp, M., Karunakaran, I., Glebov, K., Hans, M., et al. (2016). Sphingosine 1-phosphate lyase ablation disrupts presynaptic architecture and function via an ubiquitin- proteasome mediated mechanism. Sci. Rep. 6, 37064. doi: 10.1038/srep37064
Mitroi, D. N., Karunakaran, I., Graler, M., Saba, J. D., Ehninger, D., Ledesma, M. D., et al. (2017). SGPL1 (sphingosine phosphate lyase 1) modulates neuronal autophagy via phosphatidylethanolamine production. Autophagy 13, 885–899. doi: 10.1080/15548627.2017.1291471
Mizugishi, K., Yamashita, T., Olivera, A., Miller, G. F., Spiegel, S., Proia, R. L. (2005). Essential role for sphingosine kinases in neural and vascular development. Mol. Cell. Biol. 25, 11113–11121. doi: 10.1128/MCB.25.24.11113-11121.2005
Moore, A. N., Kampfl, A. W., Zhao, X., Hayes, R. L., Dash, P. K. (1999). Sphingosine-1-phosphate induces apoptosis of cultured hippocampal neurons that requires protein phosphatases and activator protein-1 complexes. Neuroscience 94, 405–415. doi: 10.1016/S0306-4522(99)00288-2
Moruno-Manchon, J. F., Uzor, N. E., Ambati, C. R., Shetty, V., Putluri, N., Jagannath, C., et al. (2018). Sphingosine kinase 1-associated autophagy differs between neurons and astrocytes. Cell. Death. Dis. 9, 521. doi: 10.1038/s41419-018-0599-5
Moruno-Manchon, J. F., Uzor, N. E., Blasco-Conesa, M. P., Mannuru, S., Putluri, N., Furr-Stimming, E. E., et al. (2017). Inhibiting sphingosine kinase 2 mitigates mutant Huntingtin-induced neurodegeneration in neuron models of Huntington disease. Hum Mol Genet. 26, 1305–1317. doi: 10.1093/hmg/ddx046
Moruno Manchon, J. F., Uzor, N. E., Dabaghian, Y., Furr-Stimming, E. E., Finkbeiner, S., Tsvetkov, A. S. (2015). Cytoplasmic sphingosine-1-phosphate pathway modulates neuronal autophagy. Sci Rep. 5, 15213. doi: 10.1038/srep15213
Moruno Manchon, J. F., Uzor, N. E., Finkbeiner, S., Tsvetkov, A. S. (2016). SPHK1/sphingosine kinase 1-mediated autophagy differs between neurons and SH-SY5Y neuroblastoma cells. Autophagy 12, 1418–1424. doi: 10.1080/15548627.2016.1183082
Motyl, J., Przykaza, L., Boguszewski, P. M., Kosson, P., Strosznajder, J. B. (2018). Pramipexole and Fingolimod exert neuroprotection in a mouse model of Parkinson’s disease by activation of sphingosine kinase 1 and Akt kinase. Neuropharmacology 135, 139–150. doi: 10.1016/j.neuropharm.2018.02.023
Muhle, C., Reichel, M., Gulbins, E., Kornhuber, J. (2013). Sphingolipids in psychiatric disorders and pain syndromes. Handb. Exp. Pharmacol. 216, 431–456. doi: 10.1007/978-3-7091-1511-4_22
Mullin, A. P., Cui, C., Wang, Y., Wang, J., Troy, E., Caggiano, A. O., et al. (2017). rHIgM22 enhances remyelination in the brain of the cuprizone mouse model of demyelination. Neurobiol. Dis. 105, 142–155. doi: 10.1016/j.nbd.2017.05.015
Munoz-Saez, E., De Munck Garcia, E., Arahuetes Portero, R. M., Vicente, F., Ortiz-Lopez, F. J., Cantizani, J., et al. (2015). Neuroprotective role of sphingosine-1-phosphate in L-BMAA treated neuroblastoma cells (SH-SY5Y). Neurosci. Lett. 593, 83–89. doi: 10.1016/j.neulet.2015.03.010
Nayak, D., Huo, Y., Kwang, W. X., Pushparaj, P. N., Kumar, S. D., Ling, E. A., et al. (2010). Sphingosine kinase 1 regulates the expression of proinflammatory cytokines and nitric oxide in activated microglia. Neuroscience 166, 132–144. doi: 10.1016/j.neuroscience.2009.12.020
Novgorodov, S. A., Riley, C. L., Yu, J., Borg, K. T., Hannun, Y. A., Proia, R. L., et al. (2014). Essential roles of neutral ceramidase and sphingosine in mitochondrial dysfunction due to traumatic brain injury. J. Biol. Chem. 289, 13142–13154. doi: 10.1074/jbc.M113.530311
O’neill, S. M., Houck, K. L., Yun, J. K., Fox, T. E., Kester, M. (2011). AP-1 binding transcriptionally regulates human neutral ceramidase. Arch. Biochem. Biophys. 511, 31–39. doi: 10.1016/j.abb.2011.04.009
O’sullivan, C., Dev, K. K. (2015). Galactosylsphingosine (psychosine)-induced demyelination is attenuated by sphingosine 1-phosphate signalling. J. Cell Sci. 128, 3878–3887. doi: 10.1242/jcs.169342
O’sullivan, S., Dev, K. K. (2017). Sphingosine-1-phosphate receptor therapies: Advances in clinical trials for CNS-related diseases. Neuropharmacology 113, 597–607. doi: 10.1016/j.neuropharm.2016.11.006
Olivera, A., Urtz, N., Mizugishi, K., Yamashita, Y., Gilfillan, A. M., Furumoto, Y., et al. (2006). IgE-dependent activation of sphingosine kinases 1 and 2 and secretion of sphingosine 1-phosphate requires Fyn kinase and contributes to mast cell responses. J. Biol. Chem. 281, 2515–2525. doi: 10.1074/jbc.M508931200
Pan, S., Gray, N. S., Gao, W., Mi, Y., Fan, Y., Wang, X., et al. (2013). Discovery of BAF312 (Siponimod), a potent and selective S1P receptor modulator. ACS. Med. Chem. Lett. 4, 333–337. doi: 10.1021/ml300396r
Panneer Selvam, S., De Palma, R. M., Oaks, J. J., Oleinik, N., Peterson, Y. K., Stahelin, R. V., et al. (2015). Binding of the sphingolipid S1P to hTERT stabilizes telomerase at the nuclear periphery by allosterically mimicking protein phosphorylation. Sci. Signal. 8, ra58. doi: 10.1126/scisignal.aaa4998
Park, K., Ikushiro, H., Seo, H. S., Shin, K. O., Kim, Y. I., Kim, J. Y., et al. (2016). ER stress stimulates production of the key antimicrobial peptide, cathelicidin, by forming a previously unidentified intracellular S1P signaling complex. Proc. Natl. Acad. Sci. U. S. A. 113, E1334–1342. doi: 10.1073/pnas.1504555113
Paugh, B. S., Bryan, L., Paugh, S. W., Wilczynska, K. M., Alvarez, S. M., Singh, S. K., et al. (2009). Interleukin-1 regulates the expression of sphingosine kinase 1 in glioblastoma cells. J. Biol. Chem. 284, 3408–3417. doi: 10.1074/jbc.M807170200
Paugh, S. W., Paugh, B. S., Rahmani, M., Kapitonov, D., Almenara, J. A., Kordula, T., et al. (2008). A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood 112, 1382–1391. doi: 10.1182/blood-2008-02-138958
Paz Soldan, M. M., Warrington, A. E., Bieber, A. J., Ciric, B., Van Keulen, V., Pease, L. R., et al. (2003). Remyelination-promoting antibodies activate distinct Ca2+ influx pathways in astrocytes and oligodendrocytes: relationship to the mechanism of myelin repair. Mol. Cell. Neurosci. 22, 14–24. doi: 10.1016/S1044-7431(02)00018-0
Piccinini, M., Scandroglio, F., Prioni, S., Buccinna, B., Loberto, N., Aureli, M., et al. (2010). Deregulated sphingolipid metabolism and membrane organization in neurodegenerative disorders. Mol. Neurobiol. 41, 314–340. doi: 10.1007/s12035-009-8096-6
Piontek, J., Winkler, L., Wolburg, H., Muller, S. L., Zuleger, N., Piehl, C., et al. (2008). Formation of tight junction: determinants of homophilic interaction between classic claudins. FASEB J. 22, 146–158. doi: 10.1096/fj.07-8319com
Pirhaji, L., Milani, P., Dalin, S., Wassie, B. T., Dunn, D. E., Fenster, R. J., et al. (2017). Identifying therapeutic targets by combining transcriptional data with ordinal clinical measurements. Nat. Commun. 8, 623. doi: 10.1038/s41467-017-00353-6
Pirko, I., Ciric, B., Gamez, J., Bieber, A. J., Warrington, A. E., Johnson, A. J., et al. (2004). A human antibody that promotes remyelination enters the CNS and decreases lesion load as detected by T2-weighted spinal cord MRI in a virus-induced murine model of MS. FASEB J. 18, 1577–1579. doi: 10.1096/fj.04-2026fje
Potenza, R. L., De Simone, R., Armida, M., Mazziotti, V., Pezzola, A., Popoli, P., et al. (2016). Fingolimod: a disease-modifier drug in a mouse model of amyotrophic lateral sclerosis. Neurotherapeutics 13, 918–927. doi: 10.1007/s13311-016-0462-2
Presa, N., Gomez-Larrauri, A., Rivera, I. G., Ordonez, M., Trueba, M., Gomez-Munoz, A. (2016). Regulation of cell migration and inflammation by ceramide 1-phosphate. Biochim. Biophys. Acta 1861, 402–409. doi: 10.1016/j.bbalip.2016.02.007
Prinetti, A., Chigorno, V., Tettamanti, G., Sonnino, S. (2000). Sphingolipid-enriched membrane domains from rat cerebellar granule cells differentiated in culture. A compositional study. J. Biol. Chem. 275, 11658–11665. doi: 10.1074/jbc.275.16.11658
Proia, R. L., Hla, T. (2015). Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J. Clin. Invest. 125, 1379–1387. doi: 10.1172/JCI76369
Puglielli, L., Ellis, B. C., Saunders, A. J., Kovacs, D. M. (2003). Ceramide stabilizes beta-site amyloid precursor protein-cleaving enzyme 1 and promotes amyloid beta-peptide biogenesis. J. Biol. Chem. 278, 19777–19783. doi: 10.1074/jbc.M300466200
Pyne, N. J., Adams, D. R., Pyne, S. (2017). Sphingosine Kinase 2 in Autoimmune/Inflammatory Disease and the Development of Sphingosine Kinase 2 Inhibitors. Trends. Pharmacol. Sci. 38, 581–591. doi: 10.1016/j.tips.2017.04.003
Pyne, S., Adams, D. R., Pyne, N. J. (2016). Sphingosine 1-phosphate and sphingosine kinases in health and disease: Recent advances. Prog. Lipid. Res. 62, 93–106. doi: 10.1016/j.plipres.2016.03.001
Pyne, S., Adams, D. R., Pyne, N. J. (2018). Sphingosine kinases as druggable targets. Handb. Exp. Pharmacol. [Epub ahead of print]. doi: 10.1007/164_2018_96
Pyne, S., Lee, S. C., Long, J., Pyne, N. J. (2009). Role of sphingosine kinases and lipid phosphate phosphatases in regulating spatial sphingosine 1-phosphate signalling in health and disease. Cell. Signal. 21, 14–21. doi: 10.1016/j.cellsig.2008.08.008
Pyszko, J., Strosznajder, J. B. (2014a). Sphingosine kinase 1 and sphingosine-1-phosphate in oxidative stress evoked by 1-methyl-4-phenylpyridinium (MPP+) in human dopaminergic neuronal cells. Mol. Neurobiol. 50, 38–48. doi: 10.1007/s12035-013-8622-4
Pyszko, J. A., Strosznajder, J. B. (2014b). The key role of sphingosine kinases in the molecular mechanism of neuronal cell survival and death in an experimental model of Parkinson’s disease. Folia. Neuropathol. 52, 260–269. doi: 10.5114/fn.2014.45567
Ramanathan, S., Dale, R. C., Brilot, F. (2016). Anti-MOG antibody: the history, clinical phenotype, and pathogenicity of a serum biomarker for demyelination. Autoimmun. Rev. 15, 307–324. doi: 10.1016/j.autrev.2015.12.004
Ren, M., Han, M., Wei, X., Guo, Y., Shi, H., Zhang, X., et al. (2017). FTY720 attenuates 6-OHDA-associated dopaminergic degeneration in cellular and mouse parkinsonian models. Neurochem. Res. 42, 686–696. doi: 10.1007/s11064-016-2125-4
Riccitelli, E., Giussani, P., Di Vito, C., Condomitti, G., Tringali, C., Caroli, M., et al. (2013). Extracellular sphingosine-1-phosphate: a novel actor in human glioblastoma stem cell survival. PLoS One 8, e68229. doi: 10.1371/journal.pone.0068229
Riganti, L., Antonucci, F., Gabrielli, M., Prada, I., Giussani, P., Viani, P., et al. (2016). Sphingosine-1-Phosphate (S1P) impacts presynaptic functions by regulating synapsin I localization in the Presynaptic Compartment. J. Neurosci. 36, 4624–4634. doi: 10.1523/JNEUROSCI.3588-15.2016
Rivaroli, A., Prioni, S., Loberto, N., Bettiga, A., Chigorno, V., Prinetti, A., et al. (2007). Reorganization of prion protein membrane environment during low potassium-induced apoptosis in primary rat cerebellar neurons. J. Neurochem. 103, 1954–1967. doi: 10.1111/j.1471-4159.2007.04890.x
Rodriguez, M., Warrington, A. E., Pease, L. R. (2009). Invited article: human natural autoantibodies in the treatment of neurologic disease. Neurology 72, 1269–1276. doi: 10.1212/01.wnl.0000345662.05861.e4
Roisen, F. J., Bartfeld, H., Nagele, R., Yorke, G. (1981). Ganglioside stimulation of axonal sprouting in vitro. Science 214, 577–578. doi: 10.1126/science.7291999
Rothhammer, V., Kenison, J. E., Tjon, E., Takenaka, M. C., De Lima, K. A., Borucki, D. M., et al. (2017). Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl. Acad. Sci. U. S. A. 114, 2012–2017. doi: 10.1073/pnas.1615413114
Safarian, F., Khallaghi, B., Ahmadiani, A., Dargahi, L. (2015). Activation of S1P(1) receptor regulates PI3K/Akt/FoxO3a pathway in response to oxidative stress in PC12 cells. J. Mol. Neurosci. 56, 177–187. doi: 10.1007/s12031-014-0478-1
Santos, W. L., Lynch, K. R. (2015). Drugging sphingosine kinases. ACS. Chem. Biol. 10, 225–233. doi: 10.1021/cb5008426
Sardi, S. P., Viel, C., Clarke, J., Treleaven, C. M., Richards, A. M., Park, H., et al. (2017). Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc. Natl. Acad. Sci. U. S. A. 114, 2699–2704. doi: 10.1073/pnas.1616152114
Schauer, R. (1982). Chemistry, metabolism, and biological functions of sialic acids. Adv. Carbohydr. Chem. Biochem. 40, 131–234. doi: 10.1016/S0065-2318(08)60109-2
Schnaar, R. L., Lopez, P. H. (2009). Myelin-associated glycoprotein and its axonal receptors. J. Neurosci. Res. 87, 3267–3276. doi: 10.1002/jnr.21992
Scott, F. L., Clemons, B., Brooks, J., Brahmachary, E., Powell, R., Dedman, H., et al. (2016). Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1) and receptor-5 (S1P5) agonist with autoimmune disease-modifying activity. Br. J. Pharmacol. 173, 1778–1792. doi: 10.1111/bph.13476
Serra, M., Saba, J. D. (2010). Sphingosine 1-phosphate lyase, a key regulator of sphingosine 1-phosphate signaling and function. Adv. Enzyme. Regul. 50, 349–362. doi: 10.1016/j.advenzreg.2009.10.024
Shichita, T., Sugiyama, Y., Ooboshi, H., Sugimori, H., Nakagawa, R., Takada, I., et al. (2009). Pivotal role of cerebral interleukin-17-producing gammadeltaT cells in the delayed phase of ischemic brain injury. Nat. Med. 15, 946–950. doi: 10.1038/nm.1999
Sivasubramanian, M., Kanagaraj, N., Dheen, S. T., Tay, S. S. (2015). Sphingosine kinase 2 and sphingosine-1-phosphate promotes mitochondrial function in dopaminergic neurons of mouse model of Parkinson’s disease and in MPP+ -treated MN9D cells in vitro. Neuroscience 290, 636–648. doi: 10.1016/j.neuroscience.2015.01.032
Smith, G. S., Kumar, A., Saba, J. D. (2013). Sphingosine phosphate lyase regulates murine embryonic stem cell proliferation and pluripotency through an S1P2/STAT3 signaling pathway. Biomolecules 3, 351–368. doi: 10.3390/biom3030351
Snider, A. J., Ruiz, P., Obeid, L. M., Oates, J. C. (2013). Inhibition of sphingosine kinase-2 in a murine model of lupus nephritis. PLoS One 8, e53521. doi: 10.1371/journal.pone.0053521
Sonnino, S., Aureli, M., Grassi, S., Mauri, L., Prioni, S., Prinetti, A. (2014). Lipid rafts in neurodegeneration and neuroprotection. Mol. Neurobiol. 50, 130–148. doi: 10.1007/s12035-013-8614-4
Sonnino, S., Chigorno, V. (2000). Ganglioside molecular species containing C18- and C20-sphingosine in mammalian nervous tissues and neuronal cell cultures. Biochim. Biophys. Acta 1469, 63–77. doi: 10.1016/S0005-2736(00)00210-8
Sonnino, S., Mauri, L., Chigorno, V., Prinetti, A. (2007). Gangliosides as components of lipid membrane domains. Glycobiology 17, 1R–13R. doi: 10.1093/glycob/cwl052
Sonnino, S., Prinetti, A. (2010). Gangliosides as regulators of cell membrane organization and functions. Adv. Exp. Med. Biol. 688, 165–184. doi: 10.1007/978-1-4419-6741-1_12
Sorensen, S. D., Nicole, O., Peavy, R. D., Montoya, L. M., Lee, C. J., Murphy, T. J., et al. (2003). Common signaling pathways link activation of murine PAR-1, LPA, and S1P receptors to proliferation of astrocytes. Mol. Pharmacol. 64, 1199–1209. doi: 10.1124/mol.64.5.1199
Spadaro, M., Winklmeier, S., Beltran, E., Macrini, C., Hoftberger, R., Schuh, E., et al. (2018). Pathogenicity of human antibodies against myelin oligodendrocyte glycoprotein. Ann. Neurol. 84, 315–328. doi: 10.1002/ana.25291
Spiegel, S., Cuvillier, O., Edsall, L., Kohama, T., Menzeleev, R., Olivera, A., et al. (1998). Roles of sphingosine-1-phosphate in cell growth, differentiation, and death. Biochemistry (Mosc) 63, 69–73. doi: 10.1111/j.1749-6632.1998.tb09658.x
Spiegel, S., Milstien, S. (2011). The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 11, 403–415. doi: 10.1038/nri2974
Stepanovska, B., Huwiler, A. (2019). Targeting the S1P receptor signaling pathways as a promising approach for treatment of autoimmune and inflammatory diseases. Pharmacol Res. S1043-6618 (18), 32039-32035. doi: 10.1016/j.phrs.2019.02.009
Strub, G. M., Paillard, M., Liang, J., Gomez, L., Allegood, J. C., Hait, N. C., et al. (2011). Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 25, 600–612. doi: 10.1096/fj.10-167502
Subei, A. M., Cohen, J. A. (2015). Sphingosine 1-phosphate receptor modulators in multiple sclerosis. CNS Drugs 29, 565–575. doi: 10.1007/s40263-015-0261-z
Sugiyama, E., Setou, M. (2018). Visualization of brain gangliosides using MALDI imaging mass spectrometry. Methods. Mol. Biol. 1804, 223–229. doi: 10.1007/978-1-4939-8552-4_10
Sushil, S., Batra, V. S. (2012). Modification of red mud by acid treatment and its application for CO removal. J Hazard Mater. 203–204, 264–273. doi: 10.1016/j.jhazmat.2011.12.007
Takasugi, N., Sasaki, T., Ebinuma, I., Osawa, S., Isshiki, H., Takeo, K., et al. (2013). FTY720/fingolimod, a sphingosine analogue, reduces amyloid-beta production in neurons. PLoS One 8, e64050. doi: 10.1371/journal.pone.0064050
Takasugi, N., Sasaki, T., Suzuki, K., Osawa, S., Isshiki, H., Hori, Y., et al. (2011). BACE1 activity is modulated by cell-associated sphingosine-1-phosphate. J. Neurosci. 31, 6850–6857. doi: 10.1523/JNEUROSCI.6467-10.2011
Van Doorn, R., Lopes Pinheiro, M. A., Kooij, G., Lakeman, K., Van Het Hof, B., Van Der Pol, S. M., et al. (2012). Sphingosine 1-phosphate receptor 5 mediates the immune quiescence of the human brain endothelial barrier. J Neuroinflammation 9, 133. doi: 10.1186/1742-2094-9-133
Van Echten-Deckert, G., Alam, S. (2018). Sphingolipid metabolism - an ambiguous regulator of autophagy in the brain. Biol. Chem. 399, 837–850. doi: 10.1515/hsz-2018-0237
Van Echten-Deckert, G., Hagen-Euteneuer, N., Karaca, I., Walter, J. (2014). Sphingosine-1-phosphate: boon and bane for the brain. Cell. Physiol. Biochem. 34, 148–157. doi: 10.1159/000362991
Van Echten-Deckert, G., Walter, J. (2012). Sphingolipids: critical players in Alzheimer’s disease. Prog. Lipid. Res. 51, 378–393. doi: 10.1016/j.plipres.2012.07.001
Vargas-Medrano, J., Krishnamachari, S., Villanueva, E., Godfrey, W. H., Lou, H., Chinnasamy, R., et al. (2014). Novel FTY720-based compounds stimulate neurotrophin expression and Phosphatase activity in dopaminergic cells. ACS Med. Chem. Lett. 5, 782–786. doi: 10.1021/ml500128g
Veltman, M., Stolarczyk, M., Radzioch, D., Wojewodka, G., De Sanctis, J. B., Dik, W. A., et al. (2016). Correction of lung inflammation in a F508del CFTR murine cystic fibrosis model by the sphingosine-1-phosphate lyase inhibitor LX2931. Am. J. Physiol. Lung Cell Mol. Physiol. 311, L1000–L1014. doi: 10.1152/ajplung.00298.2016
Vidal-Martinez, G., Vargas-Medrano, J., Gil-Tommee, C., Medina, D., Garza, N. T., Yang, B., et al. (2016). FTY720/Fingolimod reduces synucleinopathy and improves gut motility in A53T Mice: contributions of pro-brain-derived neurotrophic factor (PRO-BDNF) and mature bdnf. J. Biol. Chem. 291, 20811–20821. doi: 10.1074/jbc.M116.744029
Wacker, B. K., Freie, A. B., Perfater, J. L., Gidday, J. M. (2012). Junctional protein regulation by sphingosine kinase 2 contributes to blood-brain barrier protection in hypoxic preconditioning-induced cerebral ischemic tolerance. J. Cereb. Blood. Flow. Metab. 32, 1014–1023. doi: 10.1038/jcbfm.2012.3
Wang, G., Bieberich, E. (2018). Sphingolipids in neurodegeneration (with focus on ceramide and S1P). Adv. Biol. Regul. 70, 51–64. doi: 10.1016/j.jbior.2018.09.013
Wang, Y., Yang, R., Gu, J., Yin, X., Jin, N., Xie, S., et al. (2015). Cross talk between PI3K-AKT-GSK-3beta and PP2A pathways determines tau hyperphosphorylation. Neurobiol. Aging 36, 188–200. doi: 10.1016/j.neurobiolaging.2014.07.035
Warrington, A. E., Asakura, K., Bieber, A. J., Ciric, B., Van Keulen, V., Kaveri, S. V., et al. (2000). Human monoclonal antibodies reactive to oligodendrocytes promote remyelination in a model of multiple sclerosis. Proc. Natl. Acad. Sci. U. S. A. 97, 6820–6825. doi: 10.1073/pnas.97.12.6820
Watzlawik, J., Holicky, E., Edberg, D. D., Marks, D. L., Warrington, A. E., Wright, B. R., et al. (2010). Human remyelination promoting antibody inhibits apoptotic signaling and differentiation through Lyn kinase in primary rat oligodendrocytes. Glia 58, 1782–1793. doi: 10.1002/glia.21048
Watzlawik, J. O., Warrington, A. E., Rodriguez, M. (2013). PDGF is required for remyelination-promoting IgM stimulation of oligodendrocyte progenitor cell proliferation. PLoS One 8, e55149. doi: 10.1371/journal.pone.0055149
Wegner, M. S., Wanger, R. A., Oertel, S., Brachtendorf, S., Hartmann, D., Schiffmann, S., et al. (2014). Ceramide synthases CerS4 and CerS5 are upregulated by 17beta-estradiol and GPER1 via AP-1 in human breast cancer cells. Biochem. Pharmacol. 92, 577–589. doi: 10.1016/j.bcp.2014.10.007
Wei, Y., Yemisci, M., Kim, H. H., Yung, L. M., Shin, H. K., Hwang, S. K., et al. (2011). Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann. Neurol. 69, 119–129. doi: 10.1002/ana.22186
Westad, A., Venugopal, A., Snyder, E. (2017). The multiple sclerosis market. Nat. Rev. Drug. Discov. 16, 675–676. doi: 10.1038/nrd.2017.107
Xia, P., Wang, L., Moretti, P. A., Albanese, N., Chai, F., Pitson, S. M., et al. (2002). Sphingosine kinase interacts with TRAF2 and dissects tumor necrosis factor-alpha signaling. J. Biol. Chem. 277, 7996–8003. doi: 10.1074/jbc.M111423200
Yang, W., Wang, A., Zhao, C., Li, Q., Pan, Z., Han, X., et al. (2016). miR-125b Enhances IL-8 production in early-onset severe preeclampsia by targeting sphingosine-1-phosphate lyase 1. PLoS One 11, e0166940. doi: 10.1371/journal.pone.0166940
Yoshida, Y., Nakada, M., Sugimoto, N., Harada, T., Hayashi, Y., Kita, D., et al. (2010). Sphingosine-1-phosphate receptor type 1 regulates glioma cell proliferation and correlates with patient survival. Int. J. Cancer 126, 2341–2352. doi: 10.1002/ijc.24933
Yung, L. M., Wei, Y., Qin, T., Wang, Y., Smith, C. D., Waeber, C. (2012). Sphingosine kinase 2 mediates cerebral preconditioning and protects the mouse brain against ischemic injury. Stroke 43, 199–204. doi: 10.1161/STROKEAHA.111.626911
Zhang, L., Wang, H., Ding, K., Xu, J. (2015). FTY720 induces autophagy-related apoptosis and necroptosis in human glioblastoma cells. Toxicol. Lett. 236, 43–59. doi: 10.1016/j.toxlet.2015.04.015
Zhang, Y., Berka, V., Song, A., Sun, K., Wang, W., Zhang, W., et al. (2014). Elevated sphingosine-1-phosphate promotes sickling and sickle cell disease progression. J. Clin. Invest. 124, 2750–2761. doi: 10.1172/JCI74604
Zhang, Y., Li, X., Becker, K. A., Gulbins, E. (2009). Ceramide-enriched membrane domains–structure and function. Biochim. Biophys. Acta 1788, 178–183. doi: 10.1016/j.bbamem.2008.07.030
Zhang, Y., Yu, Q., Lai, T. B., Yang, Y., Li, G., Sun, S. G. (2013). Effects of small interfering RNA targeting sphingosine kinase-1 gene on the animal model of Alzheimer’s disease. J. Huazhong Univ. Sci. Technol. Med. Sci. 33, 427–432. doi: 10.1007/s11596-013-1136-5
Zhang, Z., Fauser, U., Artelt, M., Burnet, M., Schluesener, H. J. (2007). FTY720 attenuates accumulation of EMAP-II+ and MHC-II+ monocytes in early lesions of rat traumatic brain injury. J. Cell. Mol. Med. 11, 307–314. doi: 10.1111/j.1582-4934.2007.00019.x
Zhang, Z., Fauser, U., Schluesener, H. J. (2008). Early attenuation of lesional interleukin-16 up-regulation by dexamethasone and FTY720 in experimental traumatic brain injury. Neuropathol. Appl. Neurobiol. 34, 330–339. doi: 10.1111/j.1365-2990.2007.00893.x
Zhao, P., Yang, X., Yang, L., Li, M., Wood, K., Liu, Q., et al. (2017). Neuroprotective effects of fingolimod in mouse models of Parkinson’s disease. FASEB J. 31, 172–179. doi: 10.1096/fj.201600751R
Zheng, S., Wei, S., Wang, X., Xu, Y., Xiao, Y., Liu, H., et al. (2015). Sphingosine kinase 1 mediates neuroinflammation following cerebral ischemia. Exp. Neurol. 272, 160–169. doi: 10.1016/j.expneurol.2015.03.012
Zhou, Y., Lekic, T., Fathali, N., Ostrowski, R. P., Martin, R. D., Tang, J., et al. (2010). Isoflurane posttreatment reduces neonatal hypoxic-ischemic brain injury in rats by the sphingosine-1-phosphate/phosphatidylinositol-3-kinase/Akt pathway. Stroke 41, 1521–1527. doi: 10.1161/STROKEAHA.110.583757
Keywords: sphingosine 1-phosphate, fingolimod, FTY720, sphingosine 1-phosphate receptors, sphingosine kinase, sphingosine 1-phosphate phosphatase, sphingosine 1-phosphate lyase
Citation: Grassi S, Mauri L, Prioni S, Cabitta L, Sonnino S, Prinetti A and Giussani P (2019) Sphingosine 1-Phosphate Receptors and Metabolic Enzymes as Druggable Targets for Brain Diseases. Front. Pharmacol. 10:807. doi: 10.3389/fphar.2019.00807
Received: 21 March 2019; Accepted: 21 June 2019;
Published: 23 July 2019.
Edited by:
Salvatore Salomone, University of Catania, ItalyReviewed by:
Gerhild Van Echten-Deckert, University of Bonn, GermanyCopyright © 2019 Grassi, Mauri, Prioni, Cabitta, Sonnino, Prinetti and Giussani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandro Prinetti, YWxlc3NhbmRyby5wcmluZXR0aUB1bmltaS5pdA==
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.