 Claudia Balducci
Claudia Balducci Gianluigi Forloni
Gianluigi Forloni
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol. , 03 July 2019
Sec. Experimental Pharmacology and Drug Discovery
Volume 10 - 2019 | https://doi.org/10.3389/fphar.2019.00738
This article is part of the Research Topic Alzheimer’s Disease: Original Mechanisms and Translational Impact View all 19 articles
Alzheimer’s disease (AD) is the most widespread form of dementia, affecting about 45 million people worldwide. Although the β-amyloid peptide (Aβ) remains the most acknowledged culprit of AD, the multiple failures of Aβ-centric therapies call for alternative therapeutic approaches. Conceivably, the complexity of the AD neuropathological scenario cannot be solved with single-target therapies, so multiple-target approaches are needed. Core targets of AD to date are soluble oligomeric Aβ species and neuroinflammation, in an intimate detrimental dialogue. Aβ oligomers, the most neurotoxic species, appear to induce synaptic and cognitive dysfunction through the activation of glial cells. Anti-inflammatory drugs can prevent the action of Aβ oligomers. Neuroinflammation is a chronic event whose perpetuation leads to the continuous release of pro-inflammatory cytokines, promoting neuronal cell death and gross brain atrophy. Among the possible multi-target therapeutic alternatives, this review highlights the antibiotic tetracyclines, which besides antimicrobial activity also have pleiotropic action against amyloidosis, neuroinflammation, and oxidative stress. A particular focus will be on doxycycline (Doxy), a second-generation tetracycline that crosses the blood–brain barrier more easily and has a safer clinical profile. Doxy emerged as a promising preventive strategy in prion diseases and gave compelling pre-clinical results in mouse models of AD against Aβ oligomers and neuroinflammation. This strongly supports its therapeutic potential and calls for deciphering its exact mechanisms of action so as to maximize its effects in the clinic.
Alzheimer’s disease (AD) is a subtle and so far incurable neurodegenerative disease that makes patients completely unable to run their daily life activities, remember their past and relatives (Selkoe, 2011). It affects about 45 million people worldwide with an enormous socio-economic burden, likely to increase further because of longer life expectancy, and aging as a major risk factor (Garre-Olmo, 2018).
The brains of AD patients present two main lesions: extracellular senile plaques and intracellular neurofibrillary tangles. Senile plaques, rich in aggregates of the β-amyloid peptide (Aβ), act both as a reservoir of the most neurotoxic Aβ soluble species, namely Aβ oligomers (AβOs), and a determinant of neuritic dystrophy and neuronal network interruption (Mucke and Selkoe, 2012). Neurofibrillary tangles are rich in hyperphosphorylated tau protein, which dissociates from microtubules, causing their destabilization. Mitochondria are also compromised (Desler et al., 2018). Neuroinflammation is a chronic neurotoxic event (Heneka et al., 2015), and the vascular system is damaged due to the accumulation of Aβ on the vessel wall (Daulatzai, 2017). On the functional level, synaptic activity is severely impaired and responsible for the onset of cognitive deficits (Mucke and Selkoe, 2012). Progressive neuronal loss culminates in gross brain atrophy (Pini et al., 2016).
The drama of AD lies in the fact that when cognitive deficits arise bringing patients to clinical attention, their brain is already severely compromised and the pathology has progressed for about 10–15 years. This makes the identification of an efficacious therapy a very hard challenge and suggests that the complexity of AD means we must abandon single-target therapies and move on to multi-level approaches. All the single-target “Aβ-centric” clinical trials so far have failed to produce significant benefits (Panza et al., 2019).
The present review will focus on two vital therapeutic AD targets, AβOs and neuroinflammation, that have recently attracted much attention among scientists fighting AD. We highlight the possibility of counteracting AβOs’ detrimental activities and neuroinflammation with doxycycline (Doxy), a second generation-tetracycline antibiotic, which has anti-AβOs, anti-inflammatory activities (Balducci et al., 2018), a good blood–brain barrier (BBB) penetration, and a safe pharmacological profile.
The amyloid cascade hypothesis was first put forward by Hardy and Higgins in 1992, stating that: “Deposition of the Aβ peptide, the main component of senile plaques, is the primary event in the pathogenesis of AD” (Hardy and Higgins, 1992). About 10 years later this theory underwent a significant revision, with Aβ plaques overtaken by smaller and soluble AβOs (Hardy and Selkoe, 2002).
AβOs are the first species originating from the amyloidogenic process, by which the Aβ peptide, with its remarked hydrophobicity and overload in AD brains, aggregates, leading to the formation of different-sized polymers including soluble oligomers, protofibrils, and insoluble fibrils.
AβOs are the small, soluble aggregates and the most potent toxic conformers of AD (Haass and Selkoe, 2007), as well as the best correlate of disease severity compared to plaques (Kuo et al., 1996; Lue et al., 1999; McLean et al., 1999). The number of Aβ plaques detectable many years before the onset of clinical symptoms (Perrin et al., 2009) does not correlate with the severity of the cognitive loss in patients (Lue et al., 1999; Naslund et al., 2000) and Aβ deposits are also found in cognitively healthy subjects.
Many experimental data have supported the important role of AβOs in synaptic dysfunction. From transgenic mouse models of AD, it emerged that the onset of synaptic and cognitive dysfunction preceded plaque deposition (Holcomb et al., 1998; Hsia et al., 1999; Mucke et al., 2000; Balducci et al., 2010b). Ultrastructural examination of AD mouse brains revealed the presence of AβOs in the synaptic compartment before plaque deposition (Balducci et al., 2010b). In vitro and in vivo data also indicated that the application of well-characterized synthetic AβO-enriched solutions, as well as oligomeric species extracted from patient brains or derived from AD mutated cell lines, abolished the formation of new dendritic spines, inhibiting synaptic plasticity and remodeling, thus impairing learning and memory when delivered in the brain of naive mice or rats (Cleary et al., 2005; Lesne et al., 2006; Poling et al., 2008; Shankar et al., 2008; Balducci et al., 2010a; Freir et al., 2011).
Also in humans, biochemical and morphological analyses indicate that AD represents, at least at the more initial stages of the pathology, an attack at the synapses. Indeed, the degree of cognitive decline has been correlated with a decrease of the pre-synaptic marker synaptophysin in the hippocampal area and associated cortices. Notably, a 25% reduction in the expression of synaptophysin was described in the cortex of MCI or very mild AD subjects compared to aged-matched healthy controls (Selkoe, 2002).
In a virtual scenario, AβOs must be visualized as undisturbed dynamic entities, either newly formed or traveling in and out of plaques, perturbing the CNS at many functional levels (Benilova et al., 2012; Forloni et al., 2016).
For many years, synapses have been considered the main AβO target. Initial work supported this, by describing the ability of AβOs to interfere with post-synaptic receptors such as N-methyl D-aspartate receptors (NMDARs) (Balducci et al., 2010b; Yamin, 2009), affecting calcium current, and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs), with the ultimate outcome of inhibition of synaptic plasticity and the induction of memory impairment through the prevention/abolition of new dendritic spine formation where new memories are stored (Chidambaram et al., 2019). Action in the pre-synaptic compartment was also described, through an interaction with the nicotinic acetylcholine receptors α7-nAcChR (Dineley et al., 2001; Puzzo et al., 2008). The cellular prion protein (PrPC) has been suggested to mediate AβO effects, although this remains controversial since we and others have confirmed direct binding between PrPC and AβOs, but not a functional contribution (Forloni and Balducci, 2011). Activation of the apoptotic machinery was also described as an AβO-mediated mechanism for synaptic loss (Jo et al., 2011).
However, compelling new theories have emerged in more recent years, bringing to light an intimate mutual interaction between AβOs and glial cells, responsible for synaptic perturbation and loss.
Neuroinflammation has re-emerged as a driving force of neurodegeneration (McManus and Heneka, 2017). Microglia are important in brain tissue homeostasis, secreting neurotrophic factors, and patrolling the microenvironment through the release of cytokines and chemokines that influence astrocytes and neurons, particularly after infection or cell injury. This triggers inflammatory events normally calling a transient immune response followed by tissue repair. Under pathological conditions, resolution (Serhan et al., 2007; Serhan, 2010) can fail, promoting chronic neuroinflammation and neurodegeneration.
Microglia are found in a chronic activated state in AD around senile plaques, and through the continuous release of pro-inflammatory cytokines they drive neuropathology from the very early disease stages (Heneka et al., 2015). Neuroinflammation in the AD brain is chronic presumably because it is never resolved, as indicated by studies showing low levels of specialized pro-resolving mediators (Wang et al., 2015).
A relation between the detrimental action of AβOs and microglial cells is illustrated by the fact that microglia are also crucial in the control of synapse modelling and activity, and consequently of cognitive functions. Resting-state microglia survey neuronal activity by establishing intimate contacts with neurons. They are highly dynamic and plastic cells which continuously extend and retract their processes and contact synapses in an activity- and experience-dependent manner (Morris et al., 2013). However, under pathological conditions, when microglial cell activation is chronic as in AD, synaptic surveillance is lost and cognitive functions are perturbed.
Substantial data have corroborated the existence of a “dangerous liaison” between AβOs and microglial cells, by which they mutually sustain their detrimental effects on synapses. A series of studies comparing the effect of AβOs with that of Aβ fibrils demonstrated that AβOs foster microglial activation to a greater extent and apparently in a conformation-dependent manner (Heurtaux et al., 2010), with the lightest oligomeric species more likely to induce neuroinflammation. Michelucci et al. (2009) reported that AβOs are stronger inducers of the M1 microglial pro-inflammatory phenotype than fibrils. Then, He et al. (2012) described a more pronounced pro-inflammatory action of AβOs after chronic delivery inside the hippocampus of C57BL/6 mice, closely correlated with more severe cognitive deficit, altered neuronal organization, and ultrastructural changes. They also showed that AβOs increased the expression of toll-like receptor-4 (TLR4) and TNFα.
TLR4 belong to a well-known family of pattern recognition receptors initiating the innate immune response and are critically involved in AD (Drouin-Ouellet and Cicchetti, 2012). Using an AβO-induced acute mouse model, we also demonstrated that a single intracerebroventricular injection (ICV) of AβOs in C57BL/6 naive mice induced a transient memory impairment in the novel object recognition test (Balducci et al., 2010a), associated with transient activation of glial cells and an increase in the expression of pro-inflammatory cytokines in the hippocampus within a 2–24 h time window (Balducci et al., 2017), a crucial interval in the elaboration, and consolidation of long-term memory (Sutton and Schuman, 2006). Pre-treatment with anti-inflammatory drugs abolished the AβO-mediated memory impairment. While seeking further insight on the molecular mechanisms linking AβOs and microglial activation toward memory impairment, we also confirmed that TLR4 are vital, since neither memory impairment nor glial activation was observed in TLR4 null mice receiving AβOs ICV (Balducci et al., 2017).
Beside synaptic dysfunction and cognitive deficits, microglia activation also comes on stage to explain synapse loss. In a very elegant paper, Hong et al. (2016) demonstrated that microglia mediates abnormal synapse engulfment through C1q and C3 complement factors. C1q is the initiating protein of the classical complement pathway and, together with C3, localizes on synapses to mediate synaptic pruning through microglial phagocytosis (Presumey et al., 2017). In pathological conditions, such as AD, their expression is increased and localized on post-synaptic proteins, exacerbating synapse loss. The fact that this phenomenon was detectable at very early pre-plaque ages in AD-mutated mice suggested that most likely soluble Aβ species were involved. This was confirmed by specifically injecting AβOs ICV in wild-type mice and demonstrating an increase in C1q synaptic deposition, as well as the C1q-mediated C3 opsonization marking synapses for their elimination (Hong et al., 2016).
One of the most difficult challenges in AD is identifying an efficacious therapy to delay the onset, halt its progression, and prevent or reverse cognitive dysfunction. To date most attempts have focused on the Aβ peptide, with scarce or no beneficial effects (Panza et al., 2019). There might be several reasons to explain these multiple failures: wrong treatment timing, inappropriate treatment regimen, and poor or inadequate selection of patients. Although they are all valid possibilities, one of the main problems limiting therapeutic success may lie in the multi-factorial nature of AD, probably requiring multi-target therapies.
We therefore propose the antibiotic tetracyclines as a promising multi-target therapeutic approach, with a special focus on Doxy, a second-generation tetracycline with a safer pharmacological profile and a better passage across the BBB.
Interest in the tetracyclines in AD raised around the early 2000s when it was found, using cell-free approaches, that tetracyclines could inhibit the aggregation of both the synthetic PrP residues 106–126 and 82–146 of human PrP and the Aβ peptide. Facilitation of PrP and Aβ disaggregation as well as the sensitivity of their aggregates to proteases were also described (Tagliavini et al., 2000; Forloni et al., 2001). These anti-amyloidogenic effects were later confirmed for a series of other misfolding proteins responsible for neurodegenerative disorders, including Huntington’s and Parkinson’s disease (reviewed in Stoilova et al., 2013).
Neuroprotective activities of tetracyclines were first demonstrated against PrP in vitro, and in vivo again using the synthetic PrP residues 82–146 and 106–126 and through infection with the pathological form of the PrP, namely PrP scrapie (PrPsc). In vitro, tetracycline prevented the PrP 106–126-mediated neurotoxicity and astroglial proliferation (Tagliavini et al., 2000); in vivo pretreatment with either tetracycline or Doxy in experimental scrapie reduced infectivity, delayed the onset of pathology, and increased survival when intracerebrally injected in Syrian hamsters (De Luigi et al., 2008). Incubation of 263K PrPsc-infected brain homogenate with 1 mM tetracycline or doxycycline resulted in more than 80% reduction in the PK-resistant core of PrPsc. It was also reported that these compounds can interact with partially purified PrPsc from patients with the new variant of Creutzfeldt–Jakob disease (CJD) (Forloni et al., 2002).
At the clinical level, Doxy gave positive results in the initial observational studies in CJD patients, which, however, were not confirmed in a double blind against placebo trial in subjects with a diagnosis of definitive or probable sporadic CJD or genetic forms of the disease (Haik et al., 2014). Later it was reported that an asymptomatic CJD patient given Doxy for 4 years survived longer (Assar et al., 2015; Pocchiari and Ladogana, 2015), and Varges and coworkers (2017) showed a longer survival in early-stage CJD patients, suggesting a preventive action of the drug.
In AD, Doxy has been tested in two clinical trials in mild to moderate patients, yielding both positive and negative results (Loeb et al., 2004; Molloy et al., 2013). In the first study there was less decline in cognitive abilities and functional behavior, whereas no benefits were obtained in the second one. The two trials were comparable in terms of patients’ stage of disease at enrolment. Doxy was given orally at the dose of 200 mg/day together with rifampin 300 mg/day in the first study. The same doses were used in the second trial, with the sole difference that Doxy was given at the dose of 100 mg twice a day, rather than one. The main difference lies in the fact that in the former, patients were treated for 3 months, whereas in the latter one, treatment continued for 12 months. As stated by the authors, one possible explanation for the failure in the later study is that Doxy might have some negative properties that become evident when treatment is longer than 3 months. Unfortunately, the therapeutic effects were investigated only at the behavioral and functional levels, with no assessment of Aβ and tau levels in plasma and/or CSF, or of the inflammatory state.
Despite these controversies at the clinical level, preclinical studies indicated the therapeutic potential of Doxy. Initial investigation was done in two simple in vivo models. Diomede et al. (2010) tested Doxy in Caenorhabditis elegans (C. elegans), a simplified invertebrate model of AD where intracellular Aβ deposits caused C. elegans paralysis. Doxy protected against this damage by directly interacting with the Aβ aggregates and reducing the load of AβOs. Subsequently, Costa and colleagues (2011) demonstrated that Doxy treatment of Aβ42-expressing Drosophilae melanogaster did not improve their lifespan but slowed the progression of their locomotor deficits and partially rescued the toxicity of Aβ in the developing eye.
We recently found that Doxy had beneficial effects in acute and chronic mouse models of AD (Balducci et al., 2018). Chronic treatment with 10 mg/kg Doxy for 20 days or 2 months, injected intraperitoneally in APP/PS1dE9 transgenic mice, significantly restored memory independently of plaque reduction, but lowered the expression of the 18-mer oligomeric species. Interestingly, an acute treatment also led to memory recovery. On the basis of this evidence, and the lack of changes in plaque number, we assumed that Doxy was restoring memory by interfering with the oligomeric species. This was confirmed in the AβO-induced acute mouse model described above (Balducci et al., 2010a; Balducci and Forloni, 2014), which demonstrated that C57BL/6 naive mice treated ICV specifically with AβOs and pre-treated with Doxy were no longer impaired in their recognition memory. Moreover, because of the close relation between microglial activation and AβO detrimental cognitive effects (He et al., 2012; Balducci et al., 2017), we further show that the memory protection was associated with abolition of AβO-mediated microglial activation. The anti-inflammatory effect of Doxy together with memory recovery was also proved in the APP/PS1dE9 mice chronically treated with Doxy, and in LPS-treated mice, which present an AβO-independent inflammatory context (Balducci et al., 2018). The anti-inflammatory effect of Doxy has been demonstrated in a series of other pathological contexts (reviewed in Stoilova et al., 2013). Figure 1 depicts all Doxy effects described above in our AD mouse models.
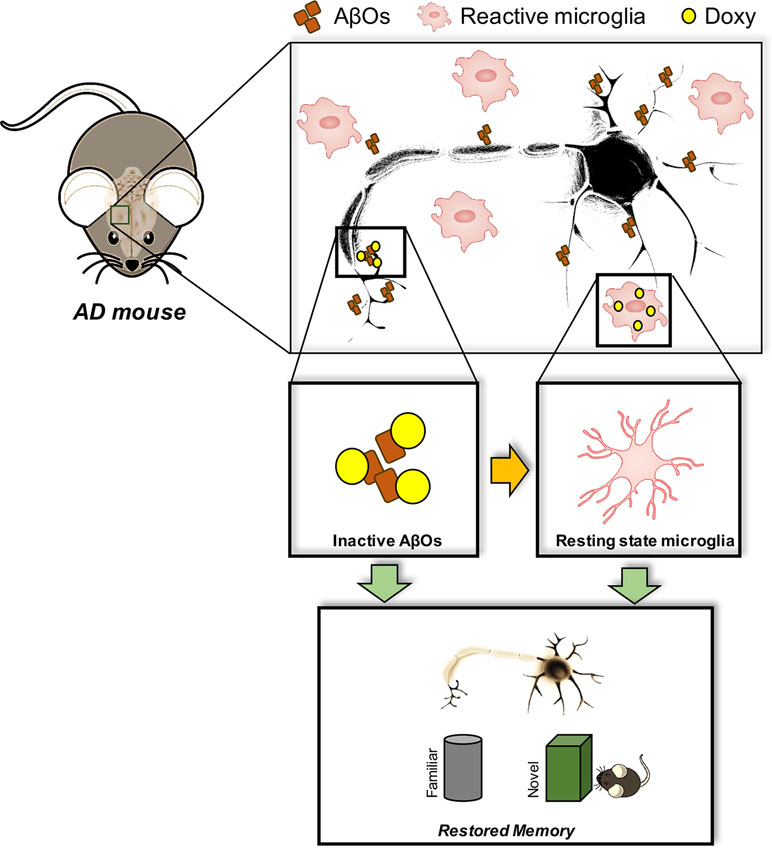
Figure 1 Doxy-mediated effects in AD mouse models. AβOs are the most powerful toxic species in AD brain, which are responsible for the memory impairment. Such detrimental effect is associated with microglial cell activation, a chronic event in AD responsible for both cognitive dysfunction, synaptic loss, and neurodegeneration. Doxy apparently interferes with either the action of AβOs by directly neutralizing their effects at both neuronal and glial level, and/or exerting a direct anti-inflammatory effect. All these actions culminate in a positive outcome at the cognitive level by restoring memory to normal.
Although we did not find direct AβO-Doxy binding, we assume that—as described for AβOs and tetracycline—an atypical supramolecular interaction might occur, which will result in the formation of colloid structures sequestering and abolishing AβO toxicity in vitro (Airoldi et al., 2011). Accordingly, Costa et al. (2011), using transmission electron microscopy, dynamic light scattering, and thioflavin T binding, demonstrated that Doxy leads to the formation of smaller, non-amyloid and non-toxic Aβ aggregates. Figure 2 summarizes the expected Doxy-mediated changes in the brain of AD patients.
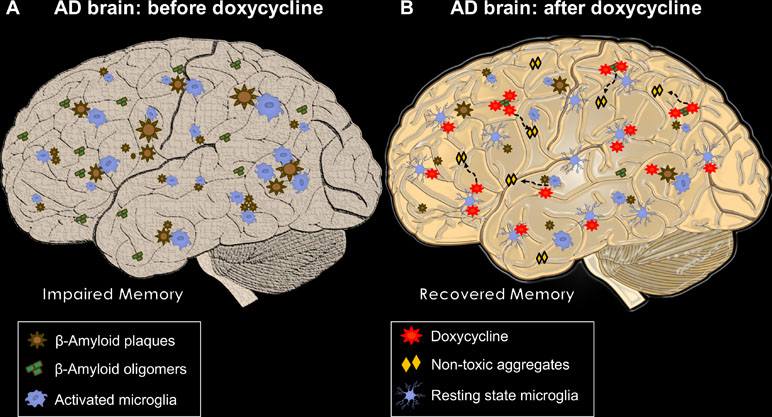
Figure 2 The multiple beneficial actions of Doxy in AD brains. In (A),an AD brain before treatment with Doxy. Different-sized Aβ plaques are widely deposited with activated microglial cells surrounding them. AβOs are freely circulating entities closer to or far from plaques, which, in concert with neuroinflammation, lead to memory impairment. (B) A Doxy-treated AD brain, where the beneficial effects of the drug are summarized. Plaque load can be reduced by long treatment. AβOs interact with Doxy, probably producing non-amyloidogenic and non-toxic structures; microglial cells move closer to a resting state. Both reduction in AβO load and microglial activation may be responsible for the Doxy-mediated memory recovery.
Beside their anti-amyloidogenic and anti-inflammatory effects, tetracyclines also have anti-oxidative and anti-apoptotic activities (Stoilova et al., 2013; Santa-Cecília et al., 2019). Oxidative stress and apoptosis are typical features of AD, whose resolution in the intricate pathological scenario will help to better restore brain physiology.
This evidence and the favorable pharmacological features of Doxy in a translational prospect suggest that this drug holds a considerable therapeutic potential for AD and other neurodegenerative diseases. A recently published comprehensive review describes well the protective effect of Doxy also in Parkinson’s disease and multiple sclerosis (Santa-Cecília et al., 2019).
Despite the beneficial effects of Doxy, clinical trials tell us that not all treatment protocols are effective, or stages of disease adequate for patient enrollment. Patients with too advanced disease are apparently unlikely to respond (Assar et al., 2015; Pocchiari and Ladogana, 2015). This does not necessarily imply that the drug is ineffective, just that it must be used more appropriately. Because of this, Doxy deserves one more chance in AD therapy, more likely with application at a prodromal stage, and a “precision medicine” approach. The latter is strongly recommended, since it will enable us to define the clinical profile (i.e., inflammatory profile) of responders compared to non-responders.
CB wrote the manuscript and created figures. GF revised the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Airoldi, C., Colombo, L., Manzoni, C., Sironi, E., Natalello, A., Doglia, S. M., et al. (2011). Tetracycline prevents Aβ oligomer toxicity through an atypical supramolecular interaction. Org. Biomol. Chem. 9, 463–472. doi: 10.1039/C0OB00303D
Assar, H., Topakian, R., Weis, S., Rahimi, J., Trenkler, J., Hoftberger, R., et al. (2015). A case of variably protease-sensitive prionopathy treated with doxycyclin. J. Neurol. Neurosurg. Psychiatry 86, 816–818. doi: 10.1136/jnnp-2014-309871
Balducci, C., Forloni, G. (2014). In vivo application of beta amyloid oligomers: a simple tool to evaluate mechanisms of action and new therapeutic approaches. Curr Pharm. Des 20, 2491–505. doi: 10.2174/13816128113199990497
Balducci, C., Beeg, M., Stravalaci, M., Bastone, A., Sclip, A., Biasini, E., et al. (2010a). Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. U. S. A. 107, 2295–2300. doi: 10.1073/pnas.0911829107
Balducci, C., Tonini, R., Zianni, E., Nazzaro, C., Fiordaliso, F., Salio, M., et al. (2010b). Cognitive deficits associated with alteration of synaptic metaplasticity precede plaque deposition in AbetaPP23 transgenic mice. J. Alzheimers Dis. 21, 1367–1381. doi: 10.3233/JAD-2010-100675
Balducci, C., Frasca, A., Zotti, M., La Vitola, P., Mhillaj, E., Grigoli, E., et al. (2017). Toll-like receptor 4-dependent glial cell activation mediates the impairment in memory establishment induced by β-amyloid oligomers in an acute mouse model of Alzheimer’s disease. Brain. Behav. Immun. 60, 188–197. doi: 10.1016/j.bbi.2016.10.012
Balducci, C., Santamaria, G., La Vitola, P., Brandi, E., Grandi, F., Viscomi, A. R., et al. (2018). Doxycycline counteracts neuroinflammation restoring memory in Alzheimer’s disease mouse models. Neurobiol. Aging 70, 128–139. doi: 10.1016/j.neurobiolaging.2018.06.002
Benilova, I., Karran, E., De Strooper, B. (2012). The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357. doi: 10.1038/nn.3028
Chidambaram, S. B., Rathipriya, A. G., Bolla, S. R., Bhat, A., Ray, B., Mahalakshmi, A. M., et al. (2019). Dendritic spines: revisiting the physiological role. Prog. Neuropsychopharmacol. Biol. Psychiatry 92, 161–193. doi: 10.1016/j.pnpbp.2019.01.005
Cleary, J. P., Walsh, D. M., Hofmeister, J. J., Shankar, G. M., Kuskowski, M. A., Selkoe, D. J., et al. (2005). Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci. 8, 79–84. doi: 10.1038/nn1372
Costa, R., Speretta, E., Crowther, D. C., Cardoso, I. (2011). Testing the therapeutic potential of doxycycline in a Drosophila melanogaster model of Alzheimer disease. J. Biol. Chem. 286, 41647–41655. doi: 10.1074/jbc.M111.274548
Daulatzai, M. A. (2017). Cerebral hypoperfusion and glucose hypometabolism: key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer’s disease. J. Neurosci. Res. 95, 943–972. doi: 10.1002/jnr.23777
De Luigi, A., Colombo, L., Diomede, L., Capobianco, R., Mangieri, M., Miccolo, C., et al. (2008). The efficacy of tetracyclines in peripheral and intracerebral prion infection. PLoS ONE 3, e1888. doi: 10.1371/journal.pone.0001888
Desler, C., Lillenes, M. S., Tønjum, T., Rasmussen, L. J. (2018). The role of mitochondrial dysfunction in the progression of alzheimer’s disease. Curr. Med. Chem. 25, 5578–5587. doi: 10.2174/0929867324666170616110111
Dineley, K. T., Westerman, M., Bui, D., Bell, K., Ashe, K. H., Sweatt, J. D. (2001). Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 21, 4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001
Diomede, L., Cassata, G., Fiordaliso, F., Salio, M., Ami, D., Natalello, A., et al. (2010). Tetracycline and its analogues protect Caenorhabditis elegans from beta amyloid-induced toxicity by targeting oligomers. Neurobiol. Dis. 40, 424–431. doi: 10.1016/j.nbd.2010.07.002
Drouin-Ouellet, J., Cicchetti, F. (2012). Inflammation and neurodegeneration: the story “retolled.” Trends Pharmacol. Sci. 33, 542–551. doi: 10.1016/j.tips.2012.07.002
Forloni, G., Artuso, V., La Vitola, P., Balducci, C. (2016). Oligomeropathies and pathogenesis of Alzheimer and Parkinson’s diseases. Mov. Disord. 31, 771–781. doi: 10.1002/mds.26624
Forloni, G., Balducci, C. (2011). β-amyloid oligomers and prion protein: fatal attraction? Prion 5, 10–5. doi: 10.4161/pri.5.1.14367
Forloni, G., Colombo, L., Girola, L., Tagliavini, F., Salmona, M. (2001). Anti-amyloidogenic activity of tetracyclines: studies in vitro. FEBS Lett. 487, 404–407. doi: 10.1016/S0014-5793(00)02380-2
Forloni, G., Iussich, S., Awan, T., Colombo, L., Angeretti, N., Girola, L., et al. (2002). Tetracyclines affect prion infectivity. Proc. Natl. Acad. Sci. U. S. A. 99, 10849–10854. doi: 10.1073/pnas.162195499
Freir, D. B., Fedriani, R., Scully, D., Smith, I. M., Selkoe, D. J., Walsh, D. M., et al. (2011). Aβ oligomers inhibit synapse remodelling necessary for memory consolidation. Neurobiol. Aging 32, 2211–2218. doi: 10.1016/j.neurobiolaging.2010.01.001
Garre-Olmo, J. (2018). Epidemiology of Alzheimer’s disease and other dementias. Rev. Neurol. 66, 377–386. doi: 10.33588/rn.6611.2017519
Haass, C., Selkoe, D. J. (2007). Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. doi: 10.1038/nrm2101
Haik, S., Marcon, G., Mallet, A., Tettamanti, M., Welaratne, A., Giaccone, G., et al. (2014). Doxycycline in Creutzfeldt-Jakob disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 13, 150–158. doi: 10.1016/S1474-4422(13)70307-7
Hardy, J. A., Higgins, G. A. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Hardy, J., Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
He, Y., Zheng, M. M., Ma, Y., Han, X. J., Ma, X. Q., Qu, C. Q., et al. (2012). Soluble oligomers and fibrillar species of amyloid beta-peptide differentially affect cognitive functions and hippocampal inflammatory response. Biochem. Biophys. Res. Commun. 429, 125–130. doi: 10.1016/j.bbrc.2012.10.129
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Heneka, M. T., Golenbock, D. T., Latz, E. (2015). Innate immunity in Alzheimer’s disease. Nat. Immunol. 16, 229–236. doi: 10.1038/ni.3102
Heurtaux, T., Michelucci, A., Losciuto, S., Gallotti, C., Felten, P., Dorban, G., et al. (2010). Microglial activation depends on beta-amyloid conformation: role of the formylpeptide receptor 2. J. Neurochem. 114, 576–586. doi: 10.1111/j.1471-4159.2010.06783.x
Holcomb, L., Gordon, M. N., McGowan, E., Yu, X., Benkovic, S., Jantzen, P., et al. (1998). Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 4, 97–100. doi: 10.1038/nm0198-097
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hsia, A. Y., Masliah, E., McConlogue, L., Yu, G. Q., Tatsuno, G., Hu, K., et al. (1999). Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. U. S. A. 96, 3228–3233. doi: 10.1073/pnas.96.6.3228
Jo, J., Whitcomb, D. J., Olsen, K. M., Kerrigan, T. L., Lo, S.-C., Bru-Mercier, G., et al. (2011). Aβ(1-42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3β. Nat. Neurosci. 14, 545–547. doi: 10.1038/nn.2785
Kuo, Y. M., Emmerling, M. R., Vigo-Pelfrey, C., Kasunic, T. C., Kirkpatrick, J. B., Murdoch, G. H., et al. (1996). Water-soluble Abeta (N-40, N-42) oligomers in normal and Alzheimer disease brains. J. Biol. Chem. 271, 4077–4081. doi: 10.1074/jbc.271.8.4077
Lesne, S., Koh, M. T., Kotilinek, L., Kayed, R., Glabe, C. G., Yang, A., et al. (2006). A specific amyloid-beta protein assembly in the brain impairs memory. Nature 440, 352–7. doi: 10.1038/nature04533
Loeb, M. B., Molloy, D. W., Smieja, M., Standish, T., Goldsmith, C. H., Mahony, J., et al. (2004). A randomized, controlled trial of doxycycline and rifampin for patients with Alzheimer’s disease. J. Am. Geriatr. Soc. 52, 381–387. doi: 10.1111/j.1532-5415.2004.52109.x
Lue, L. F., Kuo, Y. M., Roher, A. E., Brachova, L., Shen, Y., Sue, L., et al. (1999). Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 155, 853–862. doi: 10.1016/S0002-9440(10)65184-X
McLean, C. A., Cherny, R. A., Fraser, F. W., Fuller, S. J., Smith, M. J., Beyreuther, K., et al. (1999). Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 46, 860–866. doi: 10.1002/1531-8249(199912)46:6<860::AID-ANA8>3.0.CO;2-M
McManus, R. M., Heneka, M. T. (2017). Role of neuroinflammation in neurodegeneration: new insights. Alzheimers Res. Ther. 9, 14. doi: 10.1186/s13195-017-0241-2
Michelucci, A., Heurtaux, T., Grandbarbe, L., Morga, E., Heuschling, P. (2009). Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: effects of oligomeric and fibrillar amyloid-beta. J. Neuroimmunol. 210, 3–12. doi: 10.1016/j.jneuroim.2009.02.003
Molloy, D. W., Standish, T. I., Zhou, Q., Guyatt, G. (2013). A multicenter, blinded, randomized, factorial controlled trial of doxycycline and rifampin for treatment of Alzheimer’s disease: the DARAD trial. Int. J. Geriatr. Psychiatry. 28, 463–470. doi: 10.1002/gps.3846
Morris, G. P., Clark, I. A., Zinn, R., Vissel, B. (2013). Microglia: a new frontier for synaptic plasticity, learning and memory, and neurodegenerative disease research. Neurobiol. Learn. Mem. 105, 40–53. doi: 10.1016/j.nlm.2013.07.002
Mucke, L., Selkoe, D. J. (2012). Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2, a006338. doi: 10.1101/cshperspect.a006338
Mucke, L., Masliah, E., Yu, G. Q., Mallory, M., Rockenstein, E. M., Tatsuno, G., et al. (2000). High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000
Naslund, J., Haroutunian, V., Mohs, R., Davis, K. L., Davies, P., Greengard, P., et al. (2000). Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA 283, 1571–1577. doi: 10.1001/jama.283.12.1571
Panza, F., Lozupone, M., Logroscino, G., Imbimbo, B. P. (2019). A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 15, 73–88. doi: 10.1038/s41582-018-0116-6
Perrin, R. J., Fagan, A. M., Holtzman, D. M. (2009). Multimodal techniques for diagnosis and prognosis of Alzheimer’s disease. Nature 461, 916–922. doi: 10.1038/nature08538
Pini, L., Pievani, M., Bocchetta, M., Altomare, D., Bosco, P., Cavedo, E., et al. (2016). Brain atrophy in Alzheimer’s Disease and aging. Ageing Res. Rev. 30, 25–48. doi: 10.1016/j.arr.2016.01.002
Pocchiari, M., Ladogana, A. (2015). Rethinking of doxycycline therapy in Creutzfeldt-Jakob disease. J. Neurol. Neurosurg. Psychiatry 86, 705. doi: 10.1136/jnnp-2014-310127
Poling, A., Morgan-Paisley, K., Panos, J. J., Kim, E. M., O’Hare, E., Cleary, J. P., et al. (2008). Oligomers of the amyloid-beta protein disrupt working memory: confirmation with two behavioral procedures. Behav. Brain Res. 193, 230–234. doi: 10.1016/j.bbr.2008.06.001
Presumey, J., Bialas, A. R., Carroll, M. C. (2017). Complement System in Neural Synapse Elimination in Development and Disease. Adv. Immunol. 135, 53–79. doi: 10.1016/bs.ai.2017.06.004
Puzzo, D., Privitera, L., Leznik, E., Fa, M., Staniszewski, A., Palmeri, A., et al. (2008). Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J. Neurosci. 28, 14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008
Santa-Cecília, F. V., Leite, C. A., Del-Bel, E., Raisman-Vozari, R. (2019). The neuroprotective effect of doxycycline on neurodegenerative diseases. Neurotox. Res. 35, 981–986. doi: 10.1007/s12640-019-00015-z
Selkoe, D. J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Selkoe, D. J. (2011). Alzheimer’s disease. Cold Spring Harb. Perspect Biol. 3 (7), a004457. doi: 10.1101/cshperspect.a004457
Serhan, C. N. (2010). Novel lipid mediators and resolution mechanisms in acute inflammation: to resolve or not? Am. J. Pathol. 177, 1576–1591. doi: 10.2353/ajpath.2010.100322
Serhan, C. N., Brain, S. D., Buckley, C. D., Gilroy, D. W., Haslett, C., O’Neill, L. A., et al. (2007). Resolution of inflammation: state of the art, definitions and terms. FASEB J. 21, 325–32. doi: 10.1096/fj.06-7227rev
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Stoilova, T., Colombo, L., Forloni, G., Tagliavini, F., Salmona, M. (2013). A new face for old antibiotics: tetracyclines in treatment of amyloidoses. J. Med. Chem. 56, 5987–6006. doi: 10.1021/jm400161p
Sutton, M. A., Schuman, E. M. (2006). Dendritic protein synthesis, synaptic plasticity, and memory. Cell 127, 49–58. doi: 10.1016/j.cell.2006.09.014
Tagliavini, F., Forloni, G., Colombo, L., Rossi, G., Girola, L., Canciani, B., et al. (2000). Tetracycline affects abnormal properties of synthetic PrP peptides and PrP(Sc) in vitro. J. Mol. Biol. 300, 1309–1322. doi: 10.1006/jmbi.2000.3840
Varges, D., Manthey, H., Heinemann, U., Ponto, C., Schmitz, M., Schulz-Schaeffer, W. J., et al. (2017). Doxycycline in early CJD: a double-blinded randomised phase II and observational study. J. Neurol. Neurosurg. Psychiatry 88, 119–125. doi: 10.1136/jnnp-2016-313541
Wang, X., Zhu, M., Hjorth, E., Cortes-Toro, V., Eyjolfsdottir, H., Graff, C., et al. (2015). Resolution of inflammation is altered in Alzheimer’s disease. Alzheimers Dement. 11, 40–50. doi: 10.1016/j.jalz.2013.12.024
Keywords: Alzheimer’s disease, beta-amyloid oligomers, neuroinflammation, tetracycline, memory
Citation: Balducci C and Forloni G (2019) Doxycycline for Alzheimer’s Disease: Fighting β-Amyloid Oligomers and Neuroinflammation. Front. Pharmacol. 10:738. doi: 10.3389/fphar.2019.00738
Received: 30 April 2019; Accepted: 07 June 2019;
Published: 03 July 2019.
Edited by:
Cesare Mancuso, Catholic University of the Sacred Heart, ItalyCopyright © 2019 Balducci and Forloni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudia Balducci, Y2xhdWRpYS5iYWxkdWNjaUBtYXJpb25lZ3JpLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.