
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pharmacol. , 16 April 2019
Sec. Cancer Molecular Targets and Therapeutics
Volume 10 - 2019 | https://doi.org/10.3389/fphar.2019.00400
This article is part of the Research Topic Tumor Microenvironment and Resistance to Current Therapies View all 12 articles
 Julia Sachs1Katja Döhl2Anja Weber3Michele Bonus4Ferdinand Ehlers1Edmond Fleischer5Anette Klinger5Holger Gohlke4,6Jörg Pietruszka3,7Lutz Schmitt2Nicole Teusch1*
Julia Sachs1Katja Döhl2Anja Weber3Michele Bonus4Ferdinand Ehlers1Edmond Fleischer5Anette Klinger5Holger Gohlke4,6Jörg Pietruszka3,7Lutz Schmitt2Nicole Teusch1*Multidrug resistance (MDR) in tumors and pathogens remains a major problem in the efficacious treatment of patients by reduction of therapy options and subsequent treatment failure. Various mechanisms are described to be involved in the development of MDR with overexpression of ATP-binding cassette (ABC) transporters reflecting the most extensively studied. These membrane transporters translocate a wide variety of substrates utilizing energy from ATP hydrolysis leading to decreased intracellular drug accumulation and impaired drug efficacy. One treatment strategy might be inhibition of transporter-mediated efflux by small molecules. Isocoumarins and 3,4-dihydroisocoumarins are a large group of natural products derived from various sources with great structural and functional variety, but have so far not been in the focus as potential MDR reversing agents. Thus, three natural products and nine novel 3,4-dihydroisocoumarins were designed and analyzed regarding cytotoxicity induction and inhibition of human ABC transporters P-glycoprotein (P-gp), multidrug resistance-associated protein 1 (MRP1) and breast cancer resistance protein (BCRP) in a variety of human cancer cell lines as well as the yeast ABC transporter Pdr5 in Saccharomyces cerevisiae. Dual inhibitors of P-gp and BCRP and inhibitors of Pdr5 were identified, and distinct structure-activity relationships for transporter inhibition were revealed. The strongest inhibitor of P-gp and BCRP, which inhibited the transporters up to 80 to 90% compared to the respective positive controls, demonstrated the ability to reverse chemotherapy resistance in resistant cancer cell lines up to 5.6-fold. In the case of Pdr5, inhibitors were identified that prevented substrate transport and/or ATPase activity with IC50 values in the low micromolar range. However, cell toxicity was not observed. Molecular docking of the test compounds to P-gp revealed that differences in inhibition capacity were based on different binding affinities to the transporter. Thus, these small molecules provide novel lead structures for further optimization.
Cancer remains the second most common cause of death worldwide with 8.7 million deaths in 2015 (Fitzmaurice et al., 2017). Despite constant progress in antitumor drug development, multidrug resistance (MDR) poses a major problem in effective patient treatment. MDR is estimated to cause treatment failure in about 90% of patients with recurrent tumors (Longley and Johnston, 2005). Additionally, it becomes a more and more severe problem in the treatment of pathogens. In general, MDR encompasses intrinsic or acquired resistance of cancer cells or pathogens to a spectrum of drugs, finally leading to reduction of treatment options and to therapy failure. Several mechanisms mediating MDR have been described, including mutations in drug targets, alterations in drug metabolism, decreased uptake or increased efflux of the drug (Kaye and Kaye, 2000; Gillet and Gottesman, 2010).
Probably the most prominent and widespread mechanism in eukaryotic cells is covered by increased drug transport from the cytoplasm through overexpression of ATP-binding cassette (ABC) transporters (El-Awady et al., 2016). The ABC transporter superfamily, representing one of the oldest and largest protein families, is expressed from archaea to human. The common mechanism for all ABC transporters encompasses the active membrane translocation of a broad spectrum of substrates using energy from ATP hydrolysis (Szöllõsi et al., 2017).
Regarding MDR development accompanying antitumor therapy, three ABC transporters were identified as the main contributors in humans. P-glycoprotein (P-gp, ABCB1), multidrug resistance-associated protein 1 (MRP1, ABCC1) and breast cancer resistance protein (BCRP, ABCG2) are frequently overexpressed in chemotherapy-resistant tumors, where they facilitate resistance to wide varieties of drugs commonly used in clinical practice (Bugde et al., 2017). Furthermore, recent studies have shown a pronounced influence of ABC transporters in drug resistance mechanisms controlled by cancer stem cells, mainly responsible for recurrence of the disease (Begicevic and Falasca, 2017).
The ABC transporter Pdr5 from Saccharomyces cerevisiae is located in the plasma membrane (PM) and acts in combination with other exporter pumps as a first line of defense against structurally unrelated xenobiotic compounds forming the pleitropic drug resistance (PDR) network (Ernst et al., 2005). Pdr5 is the most abundant exporter pump in yeast and highly homologous to multidrug mediating transporters of clinical relevant fungi such as Candida albicans. Each year billions of people become infected with pathogenic fungi and 1.5 million people are killed because of that (Brown et al., 2012). One of the largest problems is the evolving resistance shift of several fungi against azoles, which prevent one of the most efficient treatments. Therefore, the development of new antifungal drugs is of great importance.
Since the discovery of ABC transporter and characterization of their significant contribution to MDR, pharmacological strategies to overcome transporter-mediated MDR have been in the focus of various drug discovery approaches (Szakács et al., 2006; Tillotson and Theriault, 2013). One focus is the development of small molecule inhibitors interfering with transporter activity in combination with chemotherapeutic drugs, thereby increasing intracellular drug accumulation and efficacy by reversing resistance (Zhang and Ma, 2010; Spengler et al., 2017; Tran-Nguyen et al., 2017). ABC transporters involved in MDR often have overlapping substrate spectra. For example, several tyrosine kinase inhibitors including dasatinib and imatinib were demonstrated to become translocated to the extracellular space by P-gp and BCRP (Dohse et al., 2010). This leads to reduced efficacy of those drugs and subsequent therapy failure.
In most tumors, MDR is not only mediated by overexpression of one ABC transporter, but by expression of several transporters. Furthermore, P-gp and BCRP are co-expressed at the blood–brain barrier and prevent effective treatment of brain tumors (Agarwal et al., 2011). Hence, the design of dual transporter inhibitors appears to be more efficacious in mentioned cases. To date, several dual inhibitors of P-gp and BCRP have been identified including tariquidar and derivatives, aurones and chalcones (Sim et al., 2011; Gu et al., 2014; Li et al., 2015). Tariquidar entered clinical trials up to phase II to circumvent P-gp mediated MDR, but did not reveal sufficient clinical activity (Pusztai et al., 2005).
Natural compounds and their derivatives play an important role in drug discovery and development (Newman and Cragg, 2016; Guo, 2017). In this context, various ABC transporter inhibitors from natural sources were identified and characterized in the last decades (Karthikeyan and Hoti, 2015). Early natural product P-gp inhibitors such as cyclosporine A or its synthetic derivative PSC833 were tested in clinical trials up to phase III, but failed due to toxicity or other unintended side effects (Warner et al., 1995; Baer et al., 2002). Among them were several dual inhibitors of P-gp and BCRP, for example some aurones, chalcones or flavonoids (Boumendjel et al., 2009; Sim et al., 2011; Yuan et al., 2012; Iriti et al., 2017).
Isocoumarins, isomers of coumarin, represent a large class of secondary metabolites, which can be found in bacteria, fungi, lichens, marine sponges and to a lesser extent in higher plants (Saeed, 2016). To date, several 100 different isocoumarins and dihydroisocoumarins from nature have been identified and derivatives have been synthesized. Owing to their great structural diversity, diverse biological and pharmacological activities of isocoumarins including cytotoxicity and antimetastatic effects against various cancer types including breast, colon, melanoma (Cichewicz et al., 2004; Abid et al., 2012; Guimarães et al., 2016; Zhang et al., 2017), inhibition of inflammation (Liang et al., 2011; Ramanan et al., 2016) or different enzymes including aromatase and kallikrein peptidases as potential cancer targets (Endringer et al., 2008; Teixeira et al., 2011) as well as antibacterial, antifungal and antimalarial activities (Hoepfner et al., 2012; Lai et al., 2016; Simic et al., 2016) could be identified. Several natural or synthetic coumarins were identified as inhibitors of either P-gp, BCRP or both, yet activities were mostly moderate, and compound concentrations between 10 and 100 μM had to be applied for transporter inhibition (Barthomeuf et al., 2005; Raad et al., 2006; Combes et al., 2011; Bisi et al., 2017; Sjöstedt et al., 2017). In contrast, isocoumarins have to our knowledge not been characterized as potential MDR-reversing agents to date.
In this study, the three natural compounds 6-methoxymellein (3), angelicoin B (4) and ellagic acid as well as nine novel 3,4-dihydroisocoumarins (Figure 1) were analyzed regarding their cytotoxicity in cancer cells and inhibition of the endogenously expressed human ABC transporters P-gp, BCRP, and MRP1 and of the yeast transporter Pdr5. For further insights into the mechanism of action, Pdr5 ATPase and substrate transport assays were performed. These results were complemented with molecular docking studies that indicate that differences in the inhibitory power of the investigated 3,4-dihydroisocoumarins with respect to P-gp-mediated transport result from differences in the compounds’ binding affinities to P-gp.
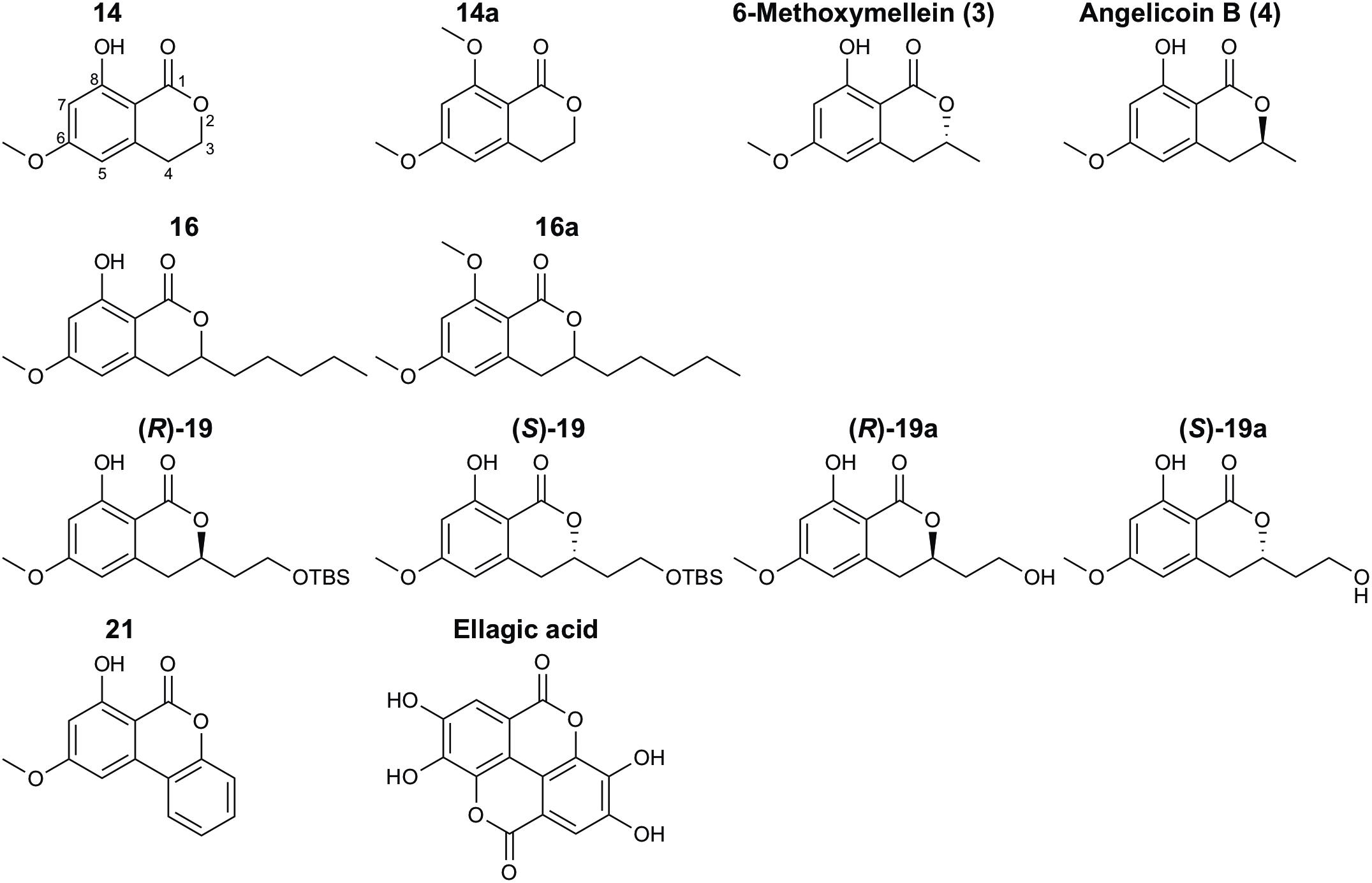
Figure 1. Structures of natural and synthetic 3,4-dihydroisocoumarins.
A549 and HCT-15 cells were purchased from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany) and H69AR cells from the American Type Culture Collection (Manassas, VA, United States). Dr. Erasmus Schneider (Wadsworth Center, New York State Department of Health, Albany, NY, United States) kindly provided the MCF-7/MX cells. Cell culture media and supplements were purchased from Thermo Fisher Scientific (Waltham, MA, United States). Yeast extract, peptone, and D-glucose were purchased from Carl Roth (Karlsruhe, Germany). Hoechst 33342, rhodamine 6G (R6G) and adenosine 5′-triphosphate (ATP) were purchased from Sigma-Aldrich (St. Louis, MO, United States) and dissolved in water. Calcein-AM, PSC833, Ko143, MK-571, doxorubicin, mitoxantrone, and ketoconazole (Sigma-Aldrich, St. Louis, MO, United States) were dissolved in dimethyl sulfoxide (DMSO; Carl Roth, Karlsruhe, Germany or Acros Organics, Geel, Belgium). Hepes buffer was purchased from Lonza (Basel, Switzerland). CellTiter-Glo Luminescent Cell Viability Assay was purchased from Promega (Madison, WI, United States). Microtiter plates were purchased from Greiner Bio-One (Kremsmünster, Austria) or Falcon (Corning, NY, United States).
3,4-Dihydroisocoumarins were synthesized in three steps starting from α,β-unsaturated δ-lactones (Fischer and Pietruszka, 2007; Böse et al., 2014) and freshly prepared Brassard’s diene. The reaction was catalyzed by AlMe3 as Lewis acid and Tf2CH2 as Brønsted acid leading to the major vinylogous (E)-configured Michael-product and the minor cyclic product. The isolated (E)-configured Michael-product was cyclized with the strong base lithium bis(trimethylsilyl)amide (LHMDS) at -78°C to room temperature for 16 h. Both fractions of the isochromenones from the first and second step were oxidized with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in toluene at room temperature for 4 h to the desired end product. Overall yields were between 30 and 84%.
Deprotection of isocoumarins (R)-19 and (S)-19 by boron trifluoride diethyl ether in dichloromethane at 0°C gave (R)-19a and (S)-19a in good to moderate yields between 36 and 47%. The free hydroxy-groups of the pentyl-derivative 16 and the unsubstituted isocoumarin 14 were protected by dimethylsulfate. This procedure gave 66% of the corresponding methyl-protected pentyl-isocoumarin 16a and 51% of the methyl-protected isocoumarin 14a.
Ellagic acid was provided by MicroCombiChem GmbH (Wiesbaden, Germany). All test compounds were dissolved in DMSO.
The human lung adenocarcinoma cell line A549 was cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin. P-gp-expressing human colon adenocarcinoma cells HCT-15 were cultured in RPMI 1640 medium supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin. BCRP-expressing MCF-7/MX human breast adenocarcinoma cells were cultured in DMEM medium supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin. MRP1-expressing H69AR human small cell lung cancer cells were cultured in RPMI 1640 medium (ATCC modification) supplemented with 20% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin. All cancer cell lines employed have been systematically characterized regarding their respective ABC transporter expression profile (Sachs et al., 2019).
Cells were maintained in a humidified atmosphere at 37°C and 5% CO2 and subcultured when confluency reached 80 to 90%.
The following S. cerevisiae yeast strain was used: YRE1001 (MATa ura3-52 trp1-1 leu2-3,112 his3-11,15 ade2-1 pdr1-3 pdr5pdr5promΔ::TRP1). S. cerevisiae was cultured in YPD medium (10 g/L yeast extract, 20 g/L peptone, 2% glucose) at 30°C and 200 rpm. The yeast strain has been described previously (Ernst et al., 2008; Gupta et al., 2014). Based on these studies it is evident that Pdr5 localizes to the PM and is present in approximately 10% of the overall membrane protein content of the PM.
Cytotoxic activity of test compounds was analyzed after 48 h using the CellTiter-Glo Luminescent Cell Viability Assay published previously (Weber et al., 2017) in 384-well plates with the following cell densities: A549, MCF-7/MX: 2 × 103 cells/well; HCT-15: 3 × 103 cells/well; H69AR: 5 × 103 cells/well. Pipetting was conducted with the CyBi-Well 96-channel simultaneous pipettor (Analytik Jena AG, Jena, Germany). For generation of dose-response curves and calculation of IC50 values, the four-parameter logistic model was applied.
The ability of identified inhibitors of P-gp and BCRP to sensitize HCT-15 or MCF-7/MX cells to chemotherapy was analyzed by co-treatment with test compounds and doxorubicin or mitoxantrone, respectively. Cells were incubated with a serial dilution of the chemotherapeutic drug alone or in combination with fixed concentrations of the test compounds or positive controls (2.5 μM PSC833 and 1 μM Ko143, respectively). Fold change of drug IC50 was calculated with the formula IC50 (drug alone)/IC50 (drug with modulator).
The liquid drug assay was carried out in sterile 96-well plates. 50 μl of yeast cell culture at an OD600 of 0.15 were mixed with 193.75 μl YPD and 6.25 μl of the test compound or DMSO as a negative control. To distinguish between cytotoxic compounds and those which inhibit Pdr5 WT specifically, the Pdr5 substrate ketoconazole was added only to cultures expressing wild-type Pdr5 in a final concentration of 1 μg/ml. At this concentration, inhibition of Pdr5 would result in ketoconazole-mediated cell death. After 48 h at 30°C the growth was measured with an ELISA plate reader (BioRad, Hercules, CA, United States) at 595 nm.
Saccharomyces cerevisiae cells expressing Pdr5 WT or the E1036Q (EQ) mutant were cultured to an OD600 of 1.5 in YPD-media at 25°C. The nitrogen source was replenished by addition of a 10th volume of 5x YP (50 g/l yeast extract, 100 g/l peptone). The cells were grown to an OD of 3.5 and harvested (5000 ×g at 4°C for 15 min). The isolation of Pdr5 containing PMs was performed as described (Kolaczkowski et al., 1996; Ernst et al., 2008).
The transport of calcein-AM by P-gp and its modulation by test compounds was analyzed in the P-gp-expressing HCT-15 cell line. 5 × 104 cells were seeded in black 96-well plates and incubated at 37°C and 5% CO2 for 24 h. The culture medium was removed and replaced by Hank’s balanced salt solution supplemented with 10 mM HEPES. Test compounds, 2.5 μM PSC833 (positive control) or 0.5% DMSO (negative control) were added in triplicates and incubated for 30 min at 37°C and 5% CO2. The substrate calcein-AM was added (final concentration 0.5 μM) and the fluorescence (excitation 485 nm, emission 520 nm) was measured over 3 h with the Infinite m1000 pro microplate reader (Tecan Group AG, Männedorf, Switzerland) at 37°C. The transport activity of P-gp was analyzed by determining the slope of the linear part of the fluorescence-time curve (0–30 min) using linear regression. Normalization of the data to PSC833 (100% inhibition) and DMSO (0% inhibition) was performed.
Breast cancer resistance protein transport activity was determined in the BCRP-expressing MCF-7/MX cell line. Hoechst 33342 was applied as transporter substrate and 1 μM Ko143 as positive control or 0.5% DMSO as negative control and the assay was performed according to the P-gp assay. Fluorescence was measured at an excitation wavelength of 355 nm and emission wavelength of 460 nm. The transport activity of BCRP was analyzed by determining the plateau of the fluorescence-time curve by using non-linear regression (one-phase exponential fit). Normalization of the data to Ko143 (100% inhibition) and DMSO (0% inhibition) was performed.
Transport activity of MRP1 was determined in MPR1-expressing H69AR cells using the substrate calcein-AM and 20 μM MK-571 as the positive control or 0.5% DMSO as negative control. Experimental procedure was according to the P-gp assay, except that 7.5 × 104 cells were seeded per well. The transport activity of MRP1 was analyzed by determining the slope of the linear part of the fluorescence-time curve (0–20 min) using linear regression. Normalization of the data to MK-571 (100% inhibition) and DMSO (0% inhibition) was performed.
Active transport of R6G was measured according to the protocol developed by Kolaczkowski et al. (1996), using a Tecan Infinite 200 pro reader (Tecan Group AG, Männedorf, Switzerland). 6 μg of the isolated PM containing Pdr5 were resuspended in 200 μl of the transport buffer (50 mM HEPES, pH 7.0, 5 mM MgCl2, 10 mM NaN3, and 150 nM R6G) and incubated in a black 96-well plate at 30°C. 5 μl of the test compound were added at the indicated concentrations. Active transport was initiated by the addition of 10 μl 200 mM ATP and fluorescence intensity was recorded in 15 s-intervals for 20 min (excitation wavelength at 524 nm, emission wavelength at 558 nm, number of flashes 30, integration time 2000 μs). IC50 determination was performed with a serial dilution of the test compounds.
Oligomycin (OM)-sensitive ATPase activity was measured from 0.8 μg PM containing Pdr5 incubated with 2 mM ATP, 5 mM MgCl2 in 270 mM TRIS-glycine buffer (pH 9.5) and 2.5 μl of the test compounds or DMSO as a control in a total volume of 100 μl. To reduce the background activities, 0.2 mM ammonium molybdate, 10 mM NaN3 and 50 mM KNO3, respectively, were added (Dufour et al., 1988; Goffeau and Dufour, 1988). In a second assay, OM (20 μg/ml) was added to an otherwise identical setup. After incubation at 30°C for 20 min, the reaction was stopped by adding 25 μl of the reaction to 175 μl 40 mM H2SO4. The amount of released inorganic phosphate was determined by a colometric assay in 96-well plates (Goffeau and Dufour, 1988; Decottignies et al., 1994; Wada et al., 2002). The difference of both assays ( ± OM) corresponds to the specific ATPase activity of Pdr5 (Ernst et al., 2008).
The cryo-EM structure of P-gp (PDB ID: 6FN1, chains A and B) (Alam et al., 2018) was prepared for molecular docking using the Protein Preparation Wizard (Sastry et al., 2013) as implemented in the Maestro GUI1 of the Schrödinger Suite version 2018-21. Protonation states for Asp, Glu, His and Lys, tautomers for His and chi flips for Asp, Glu, and His were calculated at pH 7.4 with the PROPKA (Olsson et al., 2011; Søndergaard et al., 2011) implementation in Maestro. Subsequently, a restrained energy minimization was performed on all hydrogen atoms. To be able to use the structure for later MM-GBSA calculations with implicit membrane, the coordinates of the prepared structure were superimposed onto the coordinates of its respective OPM (Lomize et al., 2012) entry. The membrane region was then defined in the Maestro GUI, using the membrane dimensions of 31.4 Å calculated by the PPM server (Lomize et al., 2012).
Molecular docking was performed using the Glide XP docking protocol (Friesner et al., 2004, 2006) of the Schrödinger Suite version 2018-2. To allow more poses to pass through the initial Glide screens, the initial number of poses per ligand was increased from 5,000 to 50,000 and the scoring window was widened from 100.0 to 500.0. In addition, the number of minimized poses per ligand was increased from 400 to 1,000 and the ‘expanded sampling’ option was enabled. Post-docking minimization was performed on 100 poses, and the resulting best 10 poses per ligand were kept for further evaluation.
Four re-docking experiments were performed to assess the suitability of the selected docking approach: two re-dockings were performed in the presence of the respective other ligand entity (re-docking 1), and two re-dockings were performed in which both ligand entities were docked sequentially (re-docking 2). For re-docking 1, the size of the region in which the geometric center of the ligand can move during docking (‘inner box’) was set to 10 Å, and the size of the region in which ligand atoms may be placed during docking (‘outer box’) was set such that it expands the size of the inner box by 17 Å. For re-docking 2, the center and size of the inner box was set to match the center and size of the smallest possible cuboid that encloses both ligands in the cryo-EM complex. The size of the outer box was set such that it expands the size of the inner box by 15 Å. The same box as for re-docking 2 was also used for the docking of the 3,4-dihydroisocoumarins. Since this docking procedure produced favorably scored poses in two different binding sites, the dockings were repeated for each of these binding sites individually, using docking boxes centered on the geometric center of the poses generated in the corresponding binding pocket. For these dockings, the box sizes were set as for re-docking 1.
In order to obtain a more reliable estimate of the relative affinities of the 3,4-dihydroisocoumarins, for each of the two binding sites, the best scored pose of each docked ligand was post-processed using the MM-GBSA implementation in Prime (see footnote number 1) with the VSGB 2.1 solvation model (Jianing et al., 2011). During this procedure, protein flexibility was taken into account for all residues within 8 Å of the ligand pose. Contributions due to changes in the configurational entropy of the ligand or the receptor upon complex formation were neglected, in order to avoid introducing uncertainty in the computations (Gohlke and Case, 2004; Weis et al., 2006; Hou et al., 2011). For compounds 16 and 16a, the final effective binding energy was expressed as the average over the effective binding energies for both enantiomers. All effective binding energies were then converted to relative effective binding energies.
All experiments were performed in at least three independent replicates. Data were analyzed with GraphPad Prism v. 6.07 (GraphPad Software, Inc., La Jolla, CA, United States) and results are presented as mean ± SEM. Different groups were compared statistically using one-way ANOVA followed by Dunnett’s multiple comparisons test. Differences were considered significant when p < 0.05.
Cytotoxicity of 12 3,4-dihydroisocoumarins was analyzed in the sensitive lung cancer cell line A549 as well as in three resistant cell lines HCT-15 (colon carcinoma), MCF-7/MX (breast carcinoma) and H69AR (lung carcinoma) either expressing the ABC transporter P-gp, BCRP, or MRP1, respectively. Cells were incubated for 48 h with test compounds and cell viability was assessed.
IC50 values of the tested 3,4-dihydroisocoumarins are summarized in Table 1. Most compounds did not display significant cytotoxic activities in the cancer cell lines and IC50 values were above the highest concentrations applied in the assay. However, one exception were the enantiomers (R)-19 and (S)-19. Both compounds displayed comparable cytotoxicity in sensitive A549 cells and the BCRP-overexpressing MCF-7/MX cells. In A549 cells, IC50 value of compound (R)-19 was 47.5 ± 3.2 μM (mean ± SEM) and IC50 value of (S)-19 was 42.0 ± 3.9 μM. In MCF-7/MX cells, both compounds were slightly more toxic with IC50 values of 28.1 ± 0.7 μM and 33.5 ± 7.5 μM, respectively. In contrast, in P-gp expressing HCT-15 cells, only (R)-19 demonstrated a cytotoxic effect, whereas (S)-19 was not toxic. Furthermore, the unprotected derivatives of (R)-19 and (S)-19, (R)-19a and (S)-19a, were analyzed. In both cases, removal of the protecting TBS group led to complete or near complete loss of cytotoxic activity in all cell lines.
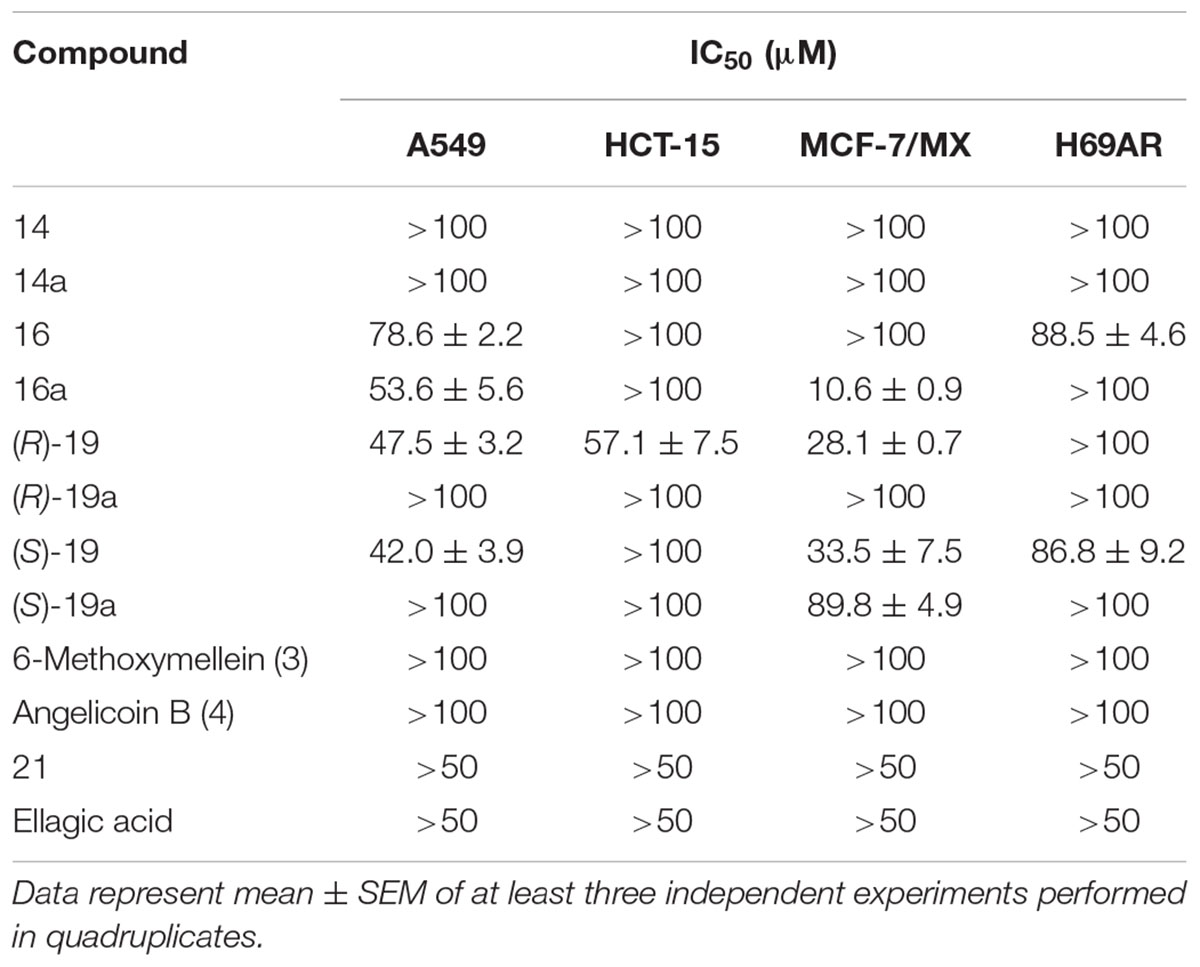
Table 1. Cytotoxicity of 3,4-dihydroisocoumarins on human tumor cell lines.
Compound 16 was not cytotoxic in P-gp expressing HCT-15 cells and BCRP-expressing MCF-7/MX cells. In MRP1-expressing H69AR cells and sensitive A549 cells, only minor cytotoxic activity was observed. IC50 values were 88.5 ± 4.6 μM and 78.6 ± 2.2 μM, respectively. Remarkably, the derivative 16a, which differs from 16 by the methoxy group instead of the hydroxy group at position 8, displayed slightly improved activity in A549 cells with an about 1.5-fold lower IC50 value and a clearly improved activity in BCRP-expressing MCF-7/MX cells. In those cells, the IC50 value was 10.6 ± 0.9 μM. In P-gp or MRP1-expressing cell lines, no cytotoxicity could be observed.
The known natural products 6-methoxymellein (3), angelicoin B (4) and ellagic acid did not show any cytotoxic effects against the tested cancer cell lines.
The 12 3,4-dihydroisocoumarins were tested for their ability to inhibit the transport activity of the human ABC transporters P-gp, BCRP, and MRP1 in cell lines overexpressing the respective transporter. Therefore, intracellular fluorescence of the transporter substrates calcein-AM and Hoechst 33341 were measured over time after cells had been treated with different concentrations of the test compounds. Inhibition rates of the compounds were normalized to the positive controls 2.5 μM PSC833, 1 μM Ko143, and 20 μM MK-571, respectively. As a result, three compounds were identified as dual inhibitors of transport activity of P-gp and BCRP (Figures 2A,B).
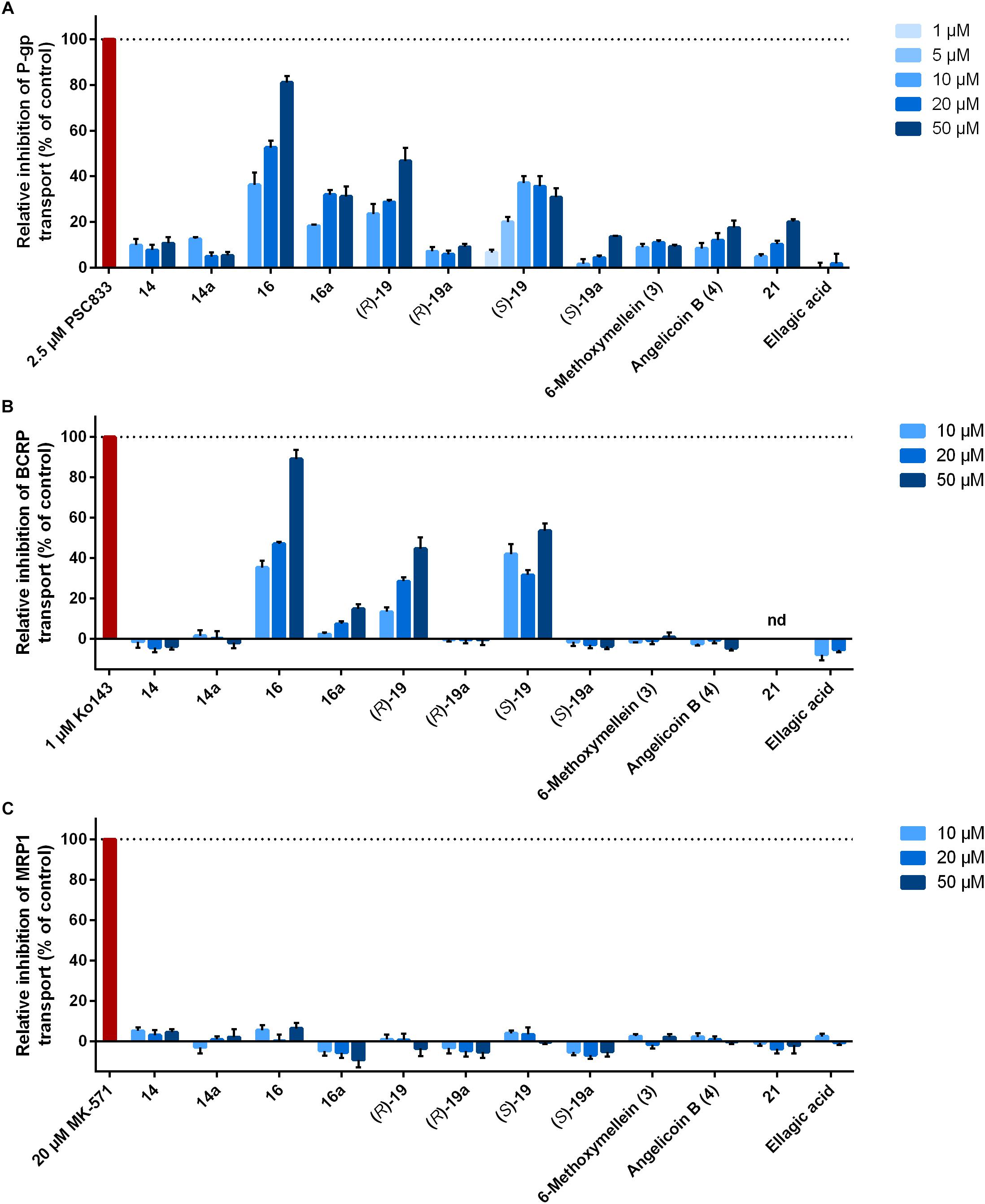
Figure 2. Influence of 3,4-dihydroisocoumarins on ABC transporter activity. Cancer cell lines overexpressing (A) P-gp (HCT-15), (B) BCRP (MCF-7/MX), or (C) MRP1 (H69AR) were incubated with test compounds and intracellular fluorescence of the substrates calcein-AM (P-gp, MRP1) or Hoechst 33342 (BCRP) was measured over 3 h. Data were normalized to DMSO (0% inhibition) and the positive controls PSC833, Ko143, and MK-571, respectively (100% inhibition). Bars represent mean ± SEM of at least three independent experiments. nd, not determined because of intrinsic fluorescence.
Pentyl-derivative 16 was the strongest dose-dependent inhibitor of P-gp and BCRP among the tested compounds. Relative inhibition of P-gp was 36.3 ± 5.3% at a concentration of 10 μM, 52.6 ± 3.1% at 20 μM and 81.2 ± 2.7% at 50 μM (mean ± SEM). Regarding inhibition of BCRP, the rates were comparable to P-gp with relative values between 35.3 ± 3.4% at 10 μM and 89.1 ± 4.6% at 50 μM. Interestingly, derivative 16a, which differs from 16 only by the methoxy group instead of the hydroxy group at position 8, showed reduced inhibition rates against both transporters with a more pronounced effect for BCRP. Relative inhibition was between 18.4 ± 0.5 and 31.9 ± 2.2% for P-gp and 2.3 ± 0.9 and 14.9 ± 2.2% for BCRP.
Two additional compounds were identified as dual P-gp and BCRP inhibitors, (R)-19 and (S)-19, but with lower activities compared to derivative 16. Derivative (R)-19 inhibited P-gp with relative rates between 23.6 ± 4.2 and 46.8 ± 5.7%. Regarding (S)-19, highest P-gp inhibition was achieved at 10 μM (37.1 ± 2.9%). Inhibition of BCRP was comparable. As both compounds contain protective groups at the hydroxy group at the ethyl chain at position 3, inhibition rates were compared to the deprotected derivatives (R)-19a and (S)-19a. In both cases, the deprotected compounds showed clearly reduced inhibitory ability of P-gp with values between about 5 and 15% and completely abolished inhibitory ability of BCRP.
Two compounds without substitution at position 3 were analyzed, 14 and 14a. They only differ in the substituent at position 8, which is either a hydroxy group 14 or methoxy group 14a. Both compounds did not inhibit BCRP and displayed only minor inhibitory activity against P-gp up to about 10% relative inhibition.
Furthermore, the two natural products and enantiomers 6-methoxymellein (3) and angelicoin B (4) were studied. They share the same structure with compound 14, but are substituted with a methyl group at position 3. Relative rates of P-gp inhibition of 6-methoxymellein (3) and angelicoin B (4) did not differ significantly from those of derivative 14. In case of BCRP, no inhibitory activity could be observed, as was the case for derivative 14.
Compound 21, a tricyclic derivative, weakly inhibited P-gp with a relative inhibition rate of 20.0 ± 1.2% at the highest concentration used. Its ability to inhibit BCRP could not be determined due to inherent fluorescence. The natural product ellagic acid did not display any inhibitory activity against P-gp or BCRP. It has to be noted that this compound could not be employed at a concentration of 50 μM due to its low solubility.
Regarding inhibition of MRP1 transport function in H69AR cells, none of the test compounds demonstrated any activity (Figure 2C).
Cancer cells overexpressing MDR related ABC transporters are characterized by reduced susceptibility to chemotherapeutic drugs, which are substrates of the respective transporters. These are for example doxorubicin (P-gp) and mitoxantrone (BCRP). To determine if the identified P-gp and BCRP inhibitor derivative 16 is able to re-sensitize resistant cancer cells to chemotherapy, P-gp expressing HCT-15 cells and BCRP-expressing MCF-7/MX cells were co-treated with doxorubicin or mitoxantrone and different concentrations of compound 16. IC50 values were determined after 48 h and compared to single treatment with the chemotherapeutic drugs. PSC833 (2.5 μM) and Ko143 (1 μM) served as respective positive controls for P-gp and BCRP, respectively.
In both cases, compound 16 was able to significantly sensitize the resistant cancer cells to chemotherapy. In HCT-15 cells, doxorubicin alone had an IC50 value of 8.5 ± 0.7 μM (mean ± SEM; Figure 3A and Table 2). Addition of 20 μM of compound 16 did not lead to a change in IC50, whereas the combination with 50 μM of the compound lead to a 3.7-fold decrease in IC50 to 2.3 ± 0.1 μM. In contrast, compound 14, which displayed minor P-gp inhibiting activity, did not sensitize the cells to doxorubicin. The positive control PSC833 reduced the IC50 of doxorubicin around 28-fold to a value of 0.35 ± 0.08 μM.
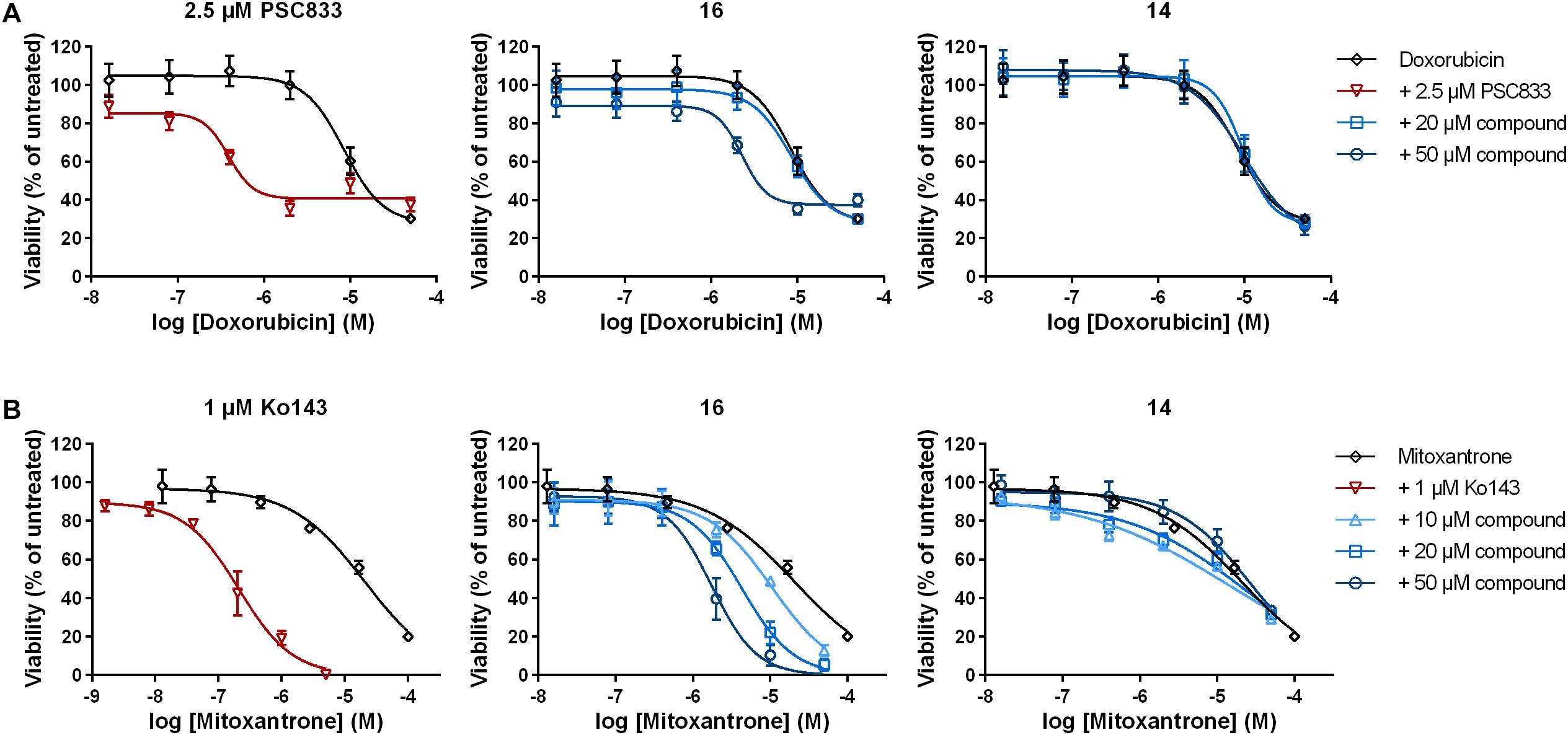
Figure 3. Sensitization of resistant cancer cells to chemotherapy by selected 3,4-dihydroisocoumarins. (A) P-gp expressing HCT-15 and (B) BCRP-expressing MCF-7/MX were treated with either doxorubicin or mitoxantrone alone, in combination with positive controls PSC833 or Ko143 or in combination with varying concentrations of test compounds 16 and 14. Cell viability was determined after 48 h. Data represent mean ± SEM normalized to untreated cells of at least three independent experiments performed in quadruplicates.
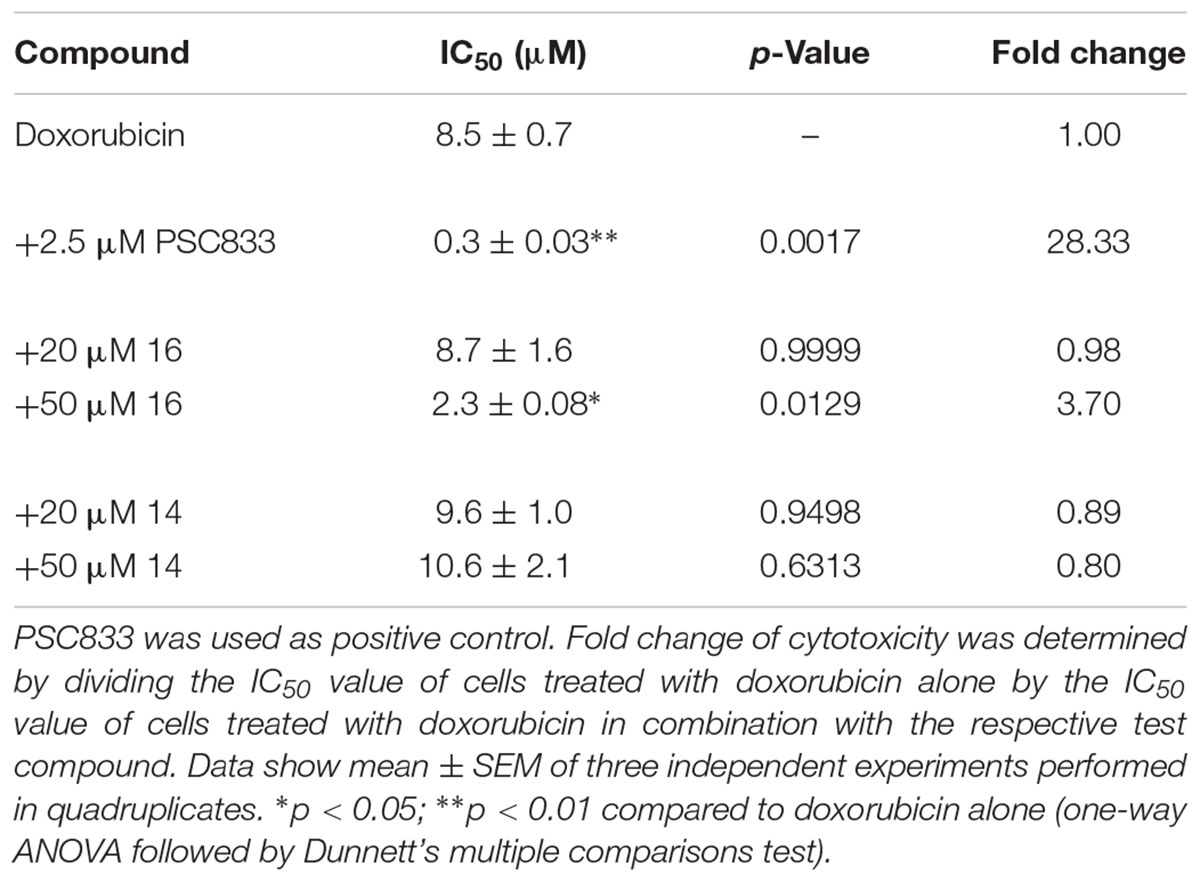
Table 2. Sensitization of HCT-15 cells to doxorubicin treatment by selected 3,4-dihydroisocoumarins.
In BCRP-expressing MCF-7/MX cells, IC50 value of the drug mitoxantrone alone was 19.6 ± 2.4 μM (Figure 3B and Table 3). Addition of compound 16 revealed a dose-dependent decrease in IC50 up to 5.6-fold at 50 μM to 3.5 ± 0.9 μM. As observed above, compound 14, which did not inhibit BCRP transport function, did not have a significant influence on the sensitivity of the cells to mitoxantrone. The positive control Ko143 was able to increase the cytotoxicity of mitoxantrone around 65-fold to an IC50 value of 0.33 ± 0.07 μM.
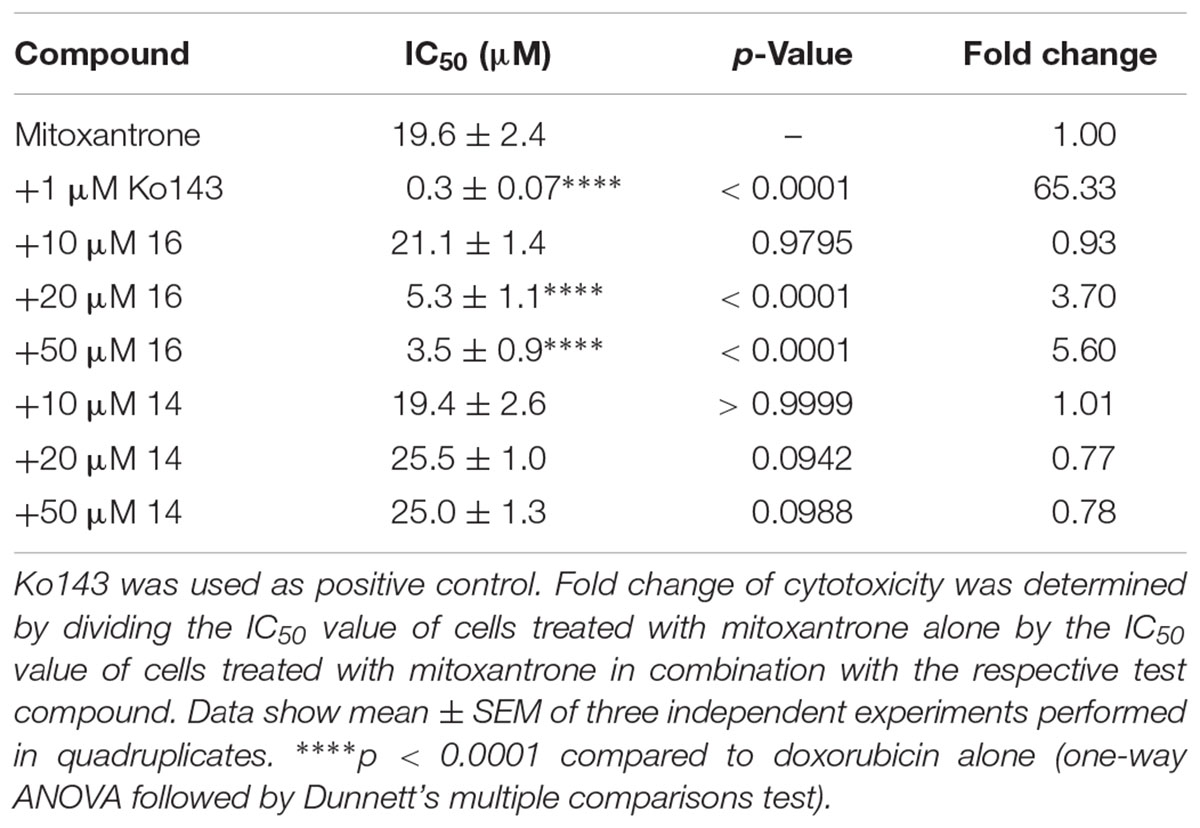
Table 3. Sensitization of MCF-7/MX cells to mitoxantrone treatment by selected 3,4-dihydroisocoumarins.
The 12 3,4-dihydroisocoumarins were tested on two different S. cerevisiae strains to determine their cytotoxicity on yeast. Therefore, a Pdr5 WT and a Pdr5 EQ mutant overexpressing strain were incubated with serial dilutions of the test compounds and OD600 was measured after 48 h of cell growth at 30°C. None of the tested 3,4-dihydroisocoumarins showed an effect on the growth of the cells (Figure 4), except compound 16a, which inhibited the growth of the mutant strain slightly at the highest tested concentration (Figure 4D). However, it had no effect on the wild-type strain (Figure 4C).
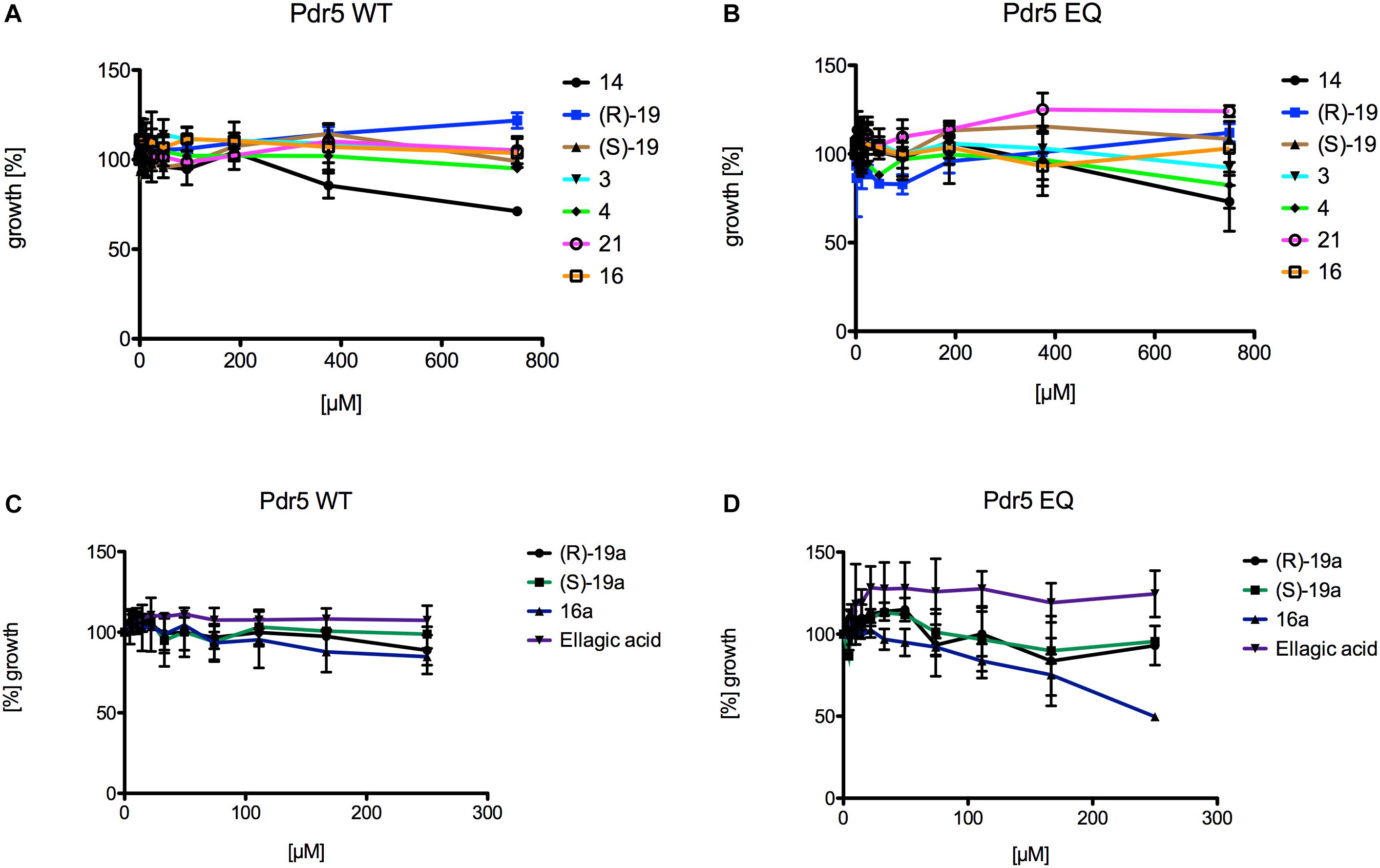
Figure 4. Liquid drug assays of the 12 3,4-dihydroisocoumarins and S. cerevisiae strains expressing Pdr5 WT or the mutant Pdr5 EQ. (A) The influence of 14, (R)-19, (S)-19, 3, 4, 21, and 16 on growth of strains expressing Pdr5 WT. (B) The influence of 14, (R)-19, (S)-19, 3, 4, 21, and 16 on growth of strains expressing Pdr5 EQ. (C) The influence of (R)-19a, (S)-19a, 16a and ellagic acid on growth of strains expressing Pdr5 WT. (D) The influence of (R)-19a, (S)-19a, 16a and ellagic acid on growth of strains expressing Pdr5 EQ. Cells were incubated at different concentrations of the compounds for 48 h at 30 °C and OD600 was determined.
In a first initial screen, the effect of the highest concentrations (10 mM) of the 3,4-dihydroisocoumarins on R6G transport and ATPase activity of Pdr5 in PM preparations was analyzed (Figure 5). In the case of the transport assay, Pdr5 containing PM preparations were incubated simultaneously with R6G and the test compound. If Pdr5 actively transports R6G, the concentration of the substrate increases in one of the membrane leaflets, which leads to a self-quenching effect. This can be measured as a decrease of fluorescence intensity in real time. Compound 14, 6-methoxymellein (3) and angelicoin B (4) did not interfere with Pdr5-mediated R6G transport (Figure 5A) as the observed changes in fluorescence were comparable to R6G transport of the wild-type protein in the absence of the compound. In contrast, compounds 21, (R)-19a and (S)-19a inhibited transport activity to 49, 43, and 37%, respectively. Only compound (R)-19, (S)-19, 16 and 16a were able to inhibit the transport of R6G completely.
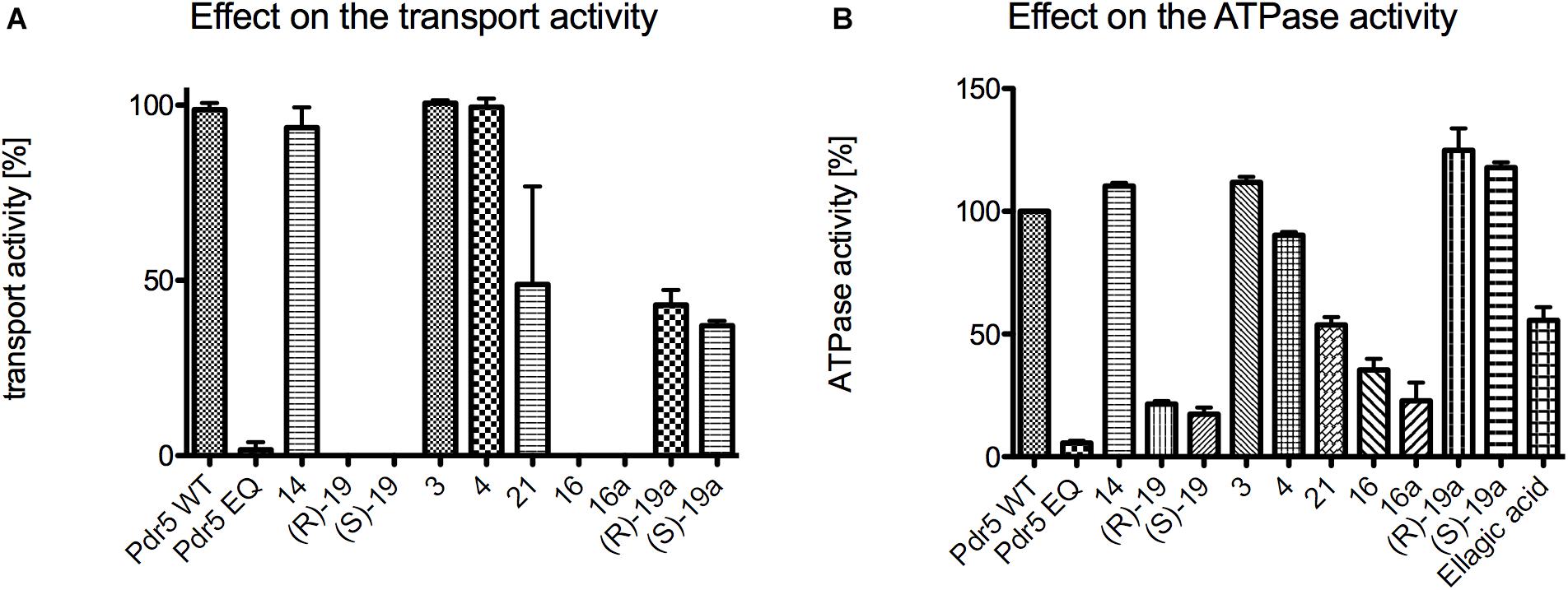
Figure 5. Initial screening of the 12 test compounds to determine their effect on (A) the transport activity of Pdr5 WT and (B) their effect on the ATPase activity. Within the transport assay compound 14, 3 and 4 had no effect on both activities. Compound 21, (R)-19a and (S)-19a were able to inhibit the transport activity to around 50%. Only compound (R)-19, (S)-19, 16 and 16a were able to inhibit the transport activity of Pdr5 WT completely. In contrast, compounds 14, 3, 4, (R)-19a and (S)-19a had nearly no effect on the ATPase activity. Compound 21 and ellagic acid were able to inhibit the ATPase activity of Pdr5 WT to 50%. Only compound (R)-19, (S)-19, 16 and 16a were able to inhibit ATPase activity below 40% in comparison to Pdr5 WT in the absence of any compound.
Figure 5B summarizes the effect of the 3,4-dihydroisocoumarins on ATPase activity of Pdr5 WT. Again, compound 14, 6-methoxymellein (3) and angelicoin B (4) had no inhibitory effect. In contrast to the capability of (R)-19a and (S)-19a to inhibit transport of R6G, no inhibition of the ATPase activity was observed. Ellagic acid and compound 21 inhibited the ATPase activity to 56 and 54%, respectively. Compounds (R)-19, (S)-19, 16, and 16a showed the highest inhibition potential on the Pdr5 WT ATPase activity.
Finally, IC50 values were determined for those inhibitors that displayed the highest potential in the transport and the ATPase assay ((R)-19, (S)-19, 16, and 16a) (Figures 6, 7). Figure 6 summarizes the IC50 measurements of the transport assays. The lowest IC50 was determined for compound (S)-19 with 5.3 ± 0.6 μM. Compound 16 and 16a showed IC50 values of 15.1 ± 2.1 and 14.6 ± 3.9 μM, respectively. The highest IC50 value was detected for compound (R)-19 with 26.0 ± 3.5 μM. Figure 7 summarizes the IC50 measurements with respect to inhibition of ATPase activity. Due to the solubility limit of these compounds, no reliable determination of IC50 values could be performed.
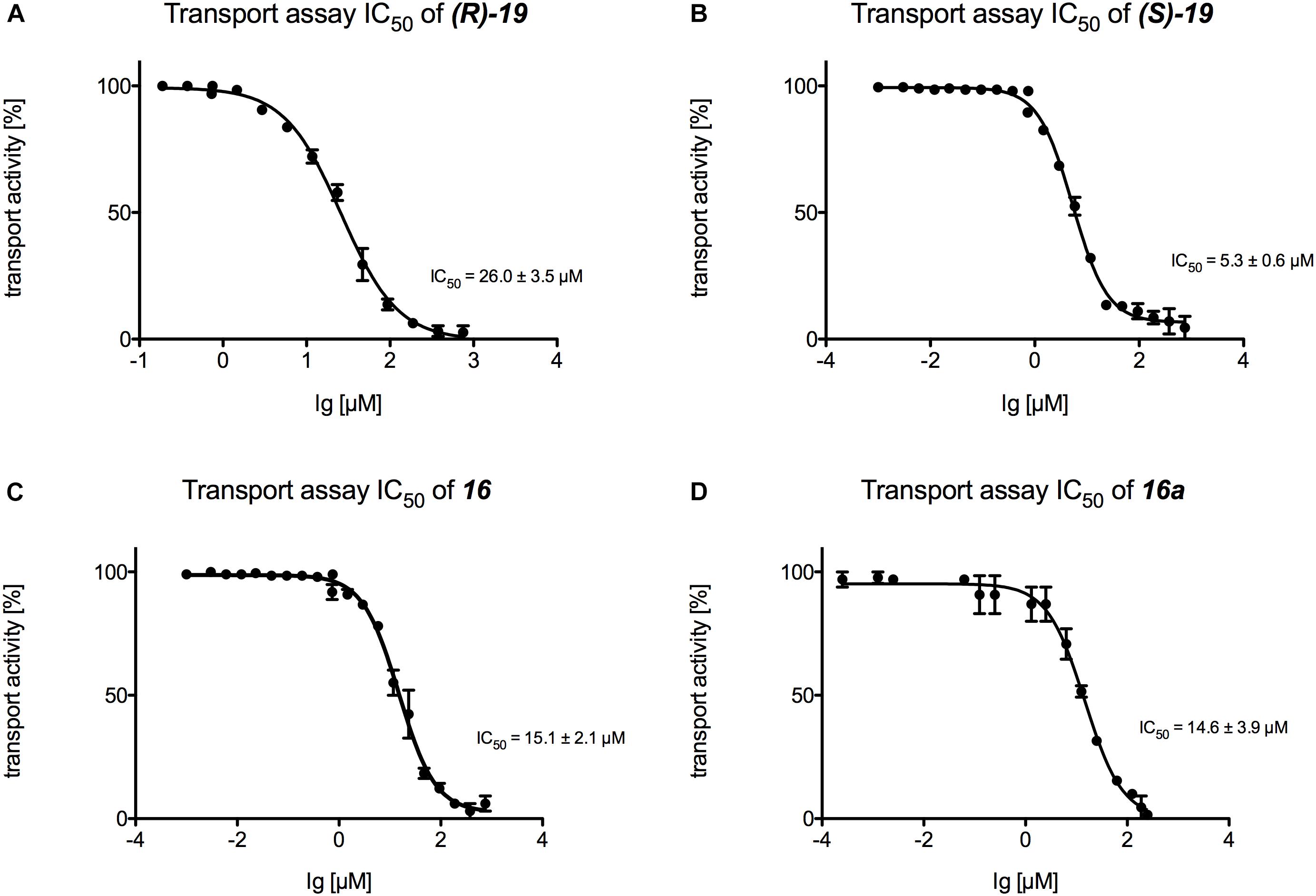
Figure 6. Determination of the transport IC50 values for (A) compound (R)-19, (B) compound (S)-19, (C) compound 16 and (D) compound 16a. For each compound a serial dilution was used to determine the IC50 curve. Membranes were incubated with the compound and active transport was detected with the Tecan Infinite M200.
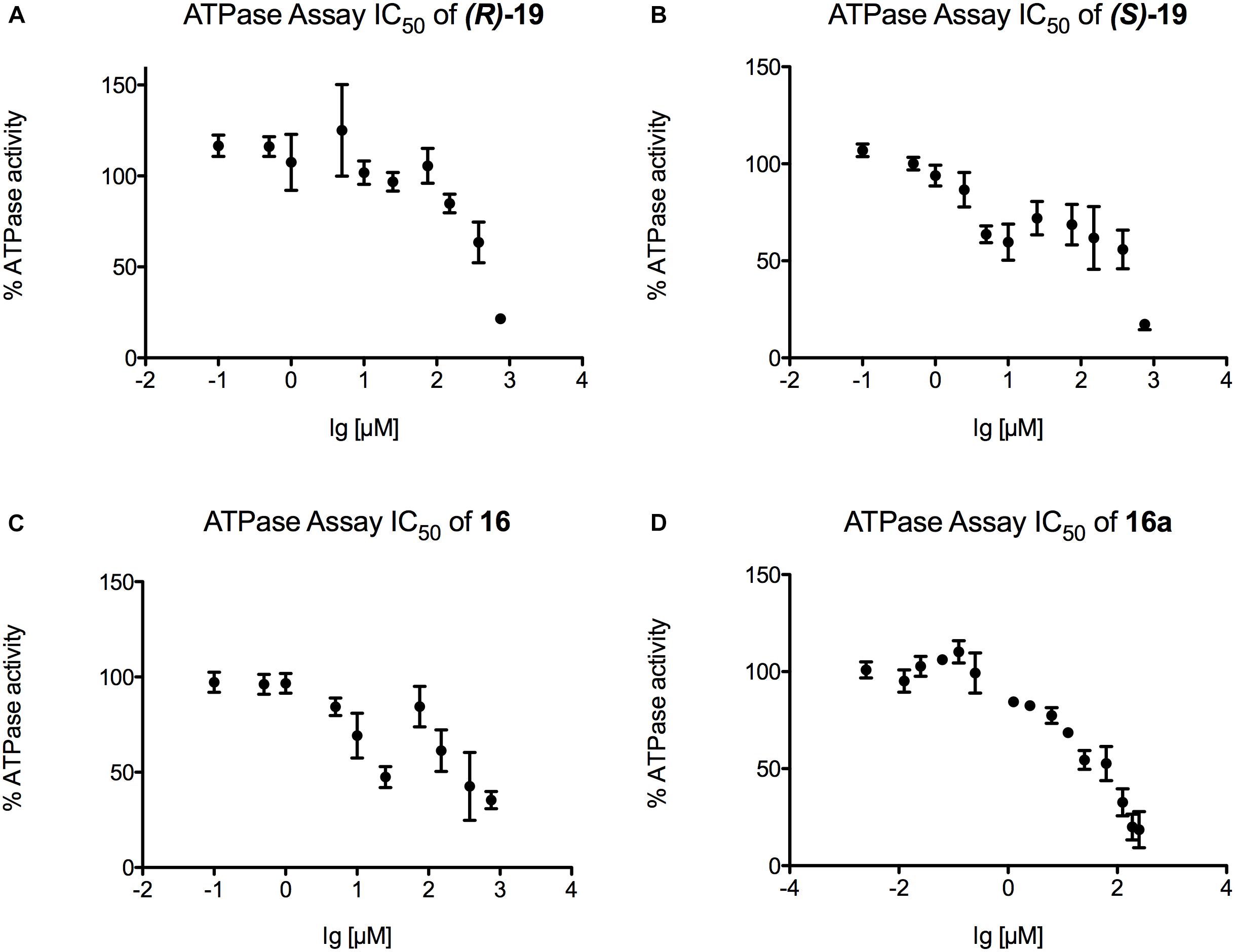
Figure 7. Determination of the ATPase IC50 values for (A) compound (R)-19, (B) compound (S)-19, (C) compound 16 and (D) compound 16a. For each compound a serial dilution was used to display the IC50 curve. Pdr5 plasma membranes (PMs) were incubated with the compound for 20 min and release of inorganic phosphate was detected with an Elisa PlateReader at 595 nm.
The selected docking protocol was able to reproduce the binding modes of Zosuquidar (ZQU) in P-gp, regardless of whether the second ligand entity was retained in the structure (RMSDZQU1304: 0.93 Å, RMSDZQU1305: 1.14 Å, Figure 8A) or not (RMSDZQU1305: 1.53 Å, RMSDZQU1304: 3.21 Å, Figure 8B), suggesting that it is also suitable for predicting the binding modes of other molecules, such as the 3,4-dihydroisocoumarins. When two molecules of ZQU were sequentially docked to the P-gp structure, all poses obtained during the first run corresponded to the binding mode of ZQU1305 in the cryo-EM structure (Figure 8B), indicating that the subpocket occupied by this ligand entity has a higher affinity toward ZQU than the subpocket occupied by the ligand entity ZQU1304. In line with this, the pose obtained for re-docking of ZQU1305 with ZQU1304 present was scored markedly better than the pose obtained for re-docking of ZQU1304 with ZQU1305 present (Glide Docking Scores: -10.40 and -8.60 kcal mol-1, respectively). Similarly, initial docking of the 3,4-dihydroisocoumarins revealed two spatially distinct binding sites (BS1 and BS2) overlapping with those of ZQU1304 and ZQU1305, respectively, to which all ligands were docked separately during the final docking experiment (Figure 8C). Since, on average, the calculated effective binding energies were 7.44 ± 3.45 kcal mol-1 (21.64 ± 6.01 kcal mol-1 for the active molecules 16, (R)-19 and (S)-19) more favorable in BS2 (Table 4), only the poses in BS2 were used for the final evaluation.
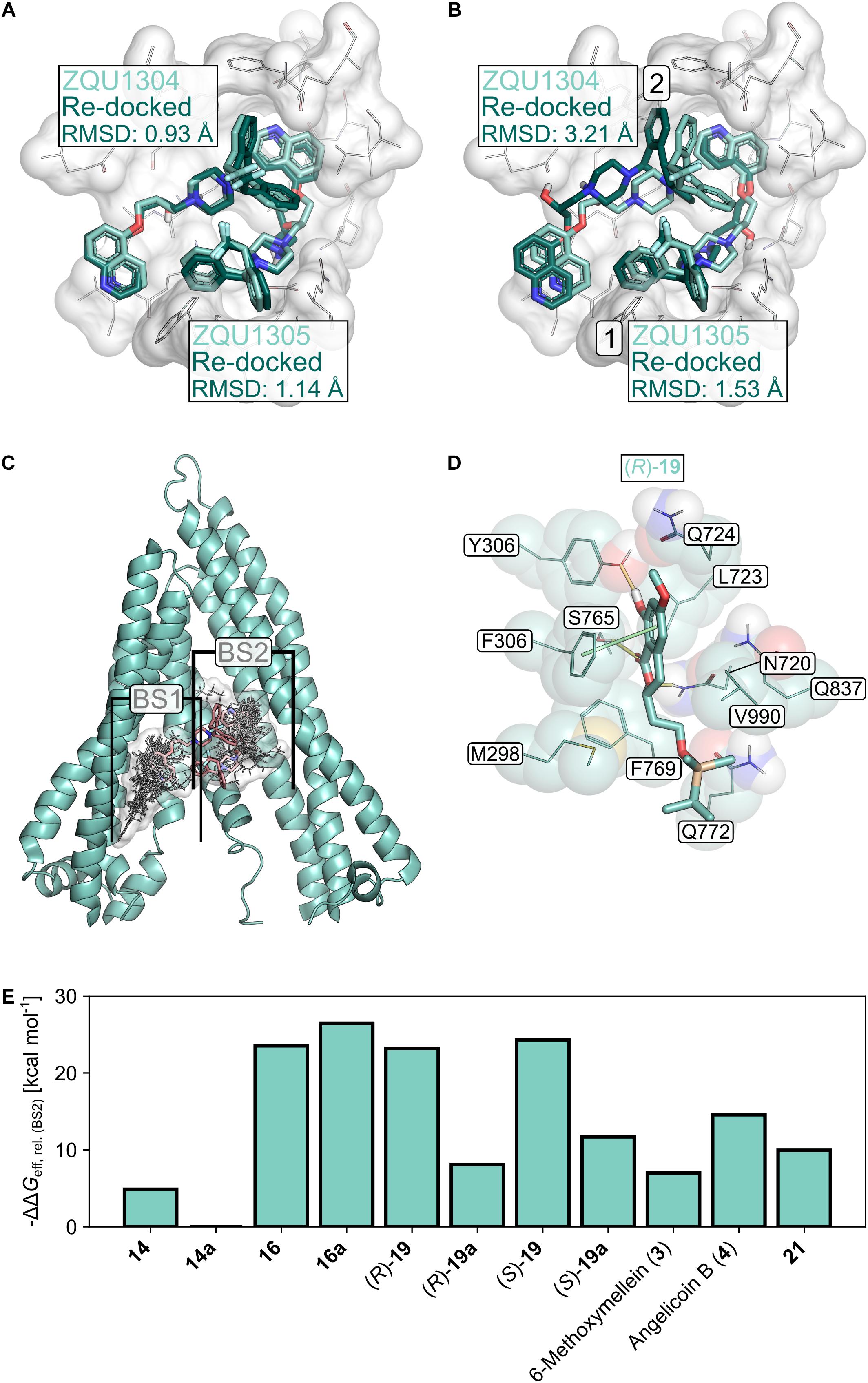
Figure 8. Molecular docking studies. (A) Re-docking of Zosuquidar (ZQU, darker turquoise, sticks representation) into the cryo-EM structure (PDB ID: 6FN1; Alam et al., 2018) of P-gp (white, sticks and cartoon representation, only part of the protein is shown for clarity) while the coordinates of the respective second ZQU molecule (lighter turquoise) were retained from the cryo-EM structure. (B) Sequential re-docking of ZQU into the cryo-EM structure of P-gp. The result of the first and second runs are indicated with a 1 and 2, respectively. Colors and representations are as in (A). (C) Binding sites (denoted as BS1 and BS2) for 3,4-dihydroisocoumarins found during docking. The molecular structures of the 3,4-dihydroisocoumarins are displayed as lines, and their joint molecular surface is rendered in white. Zosuquidar is shown as red sticks. The relevant region in the transmembrane domain of P-gp is shown in cartoon representation (D) Binding mode of (R)-19 in BS2 of P-gp. Hydrogen bonds are displayed as yellow lines, π-stacking interactions as green lines. (E) Relative effective binding energies for 3,4-dihydroisocoumarins in P-gp, calculated by the MM-GBSA approach. To allow for a better comparison with the P-gp transport assay, the energies are expressed as -ΔΔGeff.
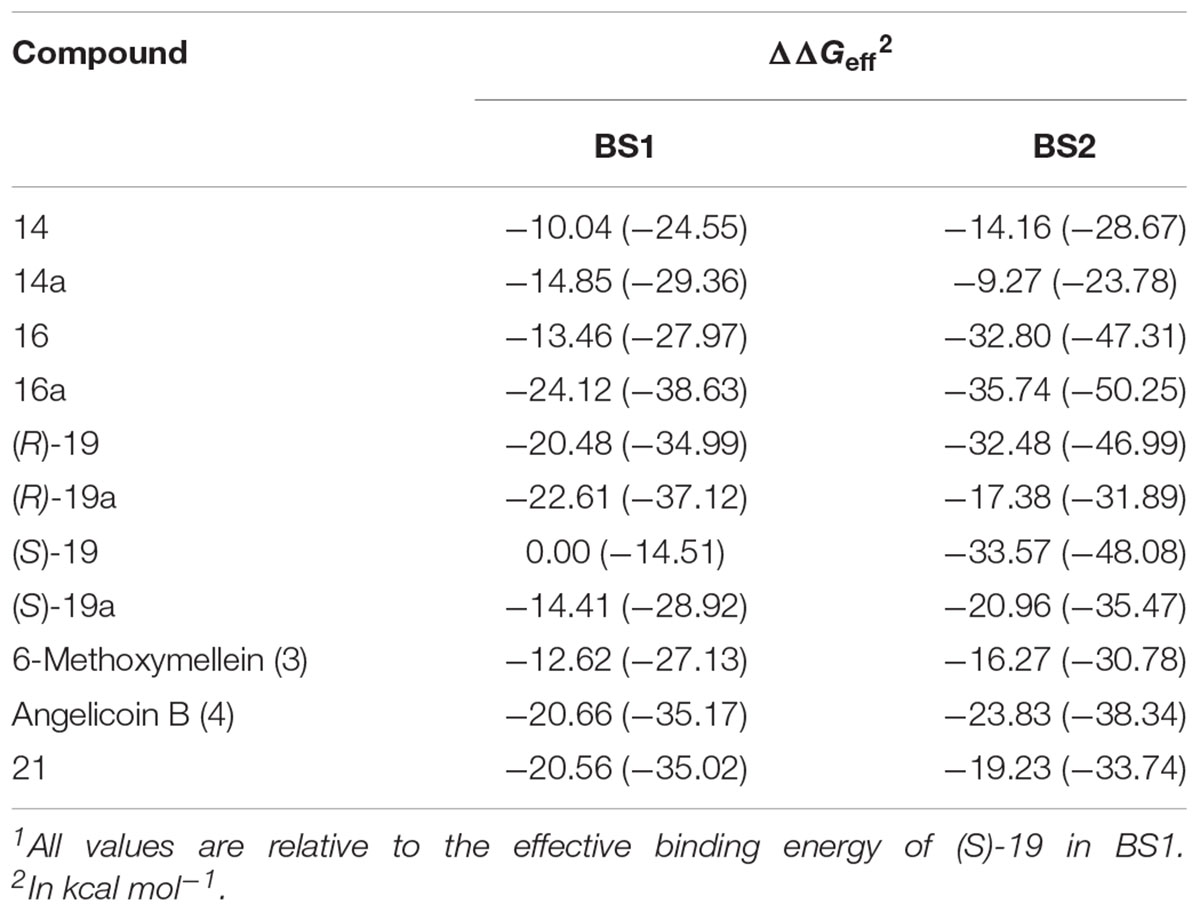
Table 4. Relative effective energies of 3,4-dihydroisocoumarins binding to P-gp.1
The calculated effective binding energies of the 3,4-dihydroisocoumarins agree very well with their ability to inhibit P-gp-mediated transport: Compounds 16, 16a, (R)-19 and (S)-19, which all inhibited substrate transport by > 20% at concentrations of 20 μM (Figure 2A), also showed significantly more favorable effective binding energies than the inactive compounds (ΔGeff = -48.16 ± 0.73 kcal mol-1 versus ΔGeff = -31.81 ± 1.79 kcal mol-1, p < 10-4) (Figure 8C and Table 4). Note, though, that configurational entropy differences were neglected here, such that the difference in the binding free energies of the two classes may be smaller due to enthalpy-entropy compensation effects. These results validate the obtained binding modes for the 3,4-dihydroisocoumarins in P-gp and corroborate the experimental findings on the compounds’ ability to inhibit P-gp-mediated substrate transport.
In this study, 3,4-dihydroisocoumarins, which have so far not been in the focus as potential MDR reversing agents, were evaluated as potential inhibitors of human and yeast ABC transporters. Notably, among the tested derivatives, three novel 3,4-dihydroisocoumarins were identified as dual inhibitors of two human transporters, P-gp and BCRP. Furthermore, the most potent inhibitor, derivative 16, demonstrated inhibition of both transporters between 80 and 90% compared to the respective positive controls at the highest test concentration (50 μM). As proof of concept, P-gp expressing HCT-15 cells and BCRP-expressing MCF-7/MX cells (Sachs et al., 2019) were treated with compound 16 in combination with the chemotherapeutic drugs doxorubicin and mitoxantrone, broadly described as transporter substrates for P-gp and BCRP. Subsequently, co-treatment with 50 μM of derivative 16 sensitized chemotherapy-resistant cancer cells HCT-15 colon carcinoma cells to doxorubicin by decreasing the IC50 value of doxorubicin-induced cytotoxicity by 3.7-fold and sensitized MCF-7/MX breast carcinoma cells to mitoxantrone by decreasing the IC50 value of mitoxantrone-induced cytotoxicity by 5.6-fold. Moreover, enantiomers (R)-19 and (S)-19 inhibited transport function of P-gp and BCRP, but with lower potency compared to derivative 16. Noteworthy, distinct structure-activity relationships for transporter inhibition could be identified, and they run in parallel for P-gp and BCRP: our data show that the hydroxy group at position 6 is mandatory for transporter inhibition and substitution by a methoxy group clearly reduces activity. Furthermore, a hydrophobic carbon chain at position 3 is indispensable for inhibition and this substituent seems to need a certain chain length. Compounds without alkyl substituent (derivative 14) or with a methyl group [6-methoxymellein (3) and angelicoin B (4)] as well as compounds with a hydrophilic chain (derivatives (R)-19a and (S)-19a) demonstrated significantly decreased inhibition of both transporters. Those effects were more pronounced in BCRP inhibition than in P-gp inhibition.
Molecular docking can provide an explanation at the atomistic level for the observed structure-activity relationship. Here, we focused on docking of 3,4-dihydroisocoumarins to P-gp because for this system it was possible to validate the suitability of our docking protocol. The predicted binding modes of the active molecules 16, (R)-19 and (S)-19 close to the binding sites of ZQU are largely identical (Figures 8D,E, 9A,B) and reveal that the hydroxy group at position 8 stabilizes the binding mode via hydrogen bonding to the hydroxy group in Y306. Introduction of a bulky methyl group, as realized in 16a, not only abolishes this interaction, but also leads to a steric clash with Y306, forcing the molecule to invert its orientation (Figure 9B). Furthermore, the hydrophobic OTBS moiety in (R)-19 and (S)-19 protrudes into a predominantly hydrophobic subpocket, formed by residues M298, N720, F769, Q772, Q837, and V990. Removal of this group, as realized in (R)-19a and (S)-19a, leads to an unsatisfied hydrogen bond donor in this subpocket, which disfavors binding. Finally, compounds 3, 4, 14, 14a, and 21 adopt binding modes that either resemble the inverted orientation of 16a, or are dissimilar to those of the active molecules 16, (R)-19 and (S)-19 (Figure 9C). Taken together, these results indicate that differences in the inhibitory power of the investigated 3,4-dihydroisocoumarins with respect to P-gp-mediated transport result from differences in the compounds’ binding affinities to P-gp rather than their ability to modulate P-gp’s transport cycle or to affect P-gp function via a different mechanism. They further support the recent finding (Alam et al., 2018) that the central cavity in P-gp can accommodate multiple molecules in different subpockets and, thereby, substantiate the concept of a binding site with high plasticity (Wise, 2012; McCormick et al., 2015).
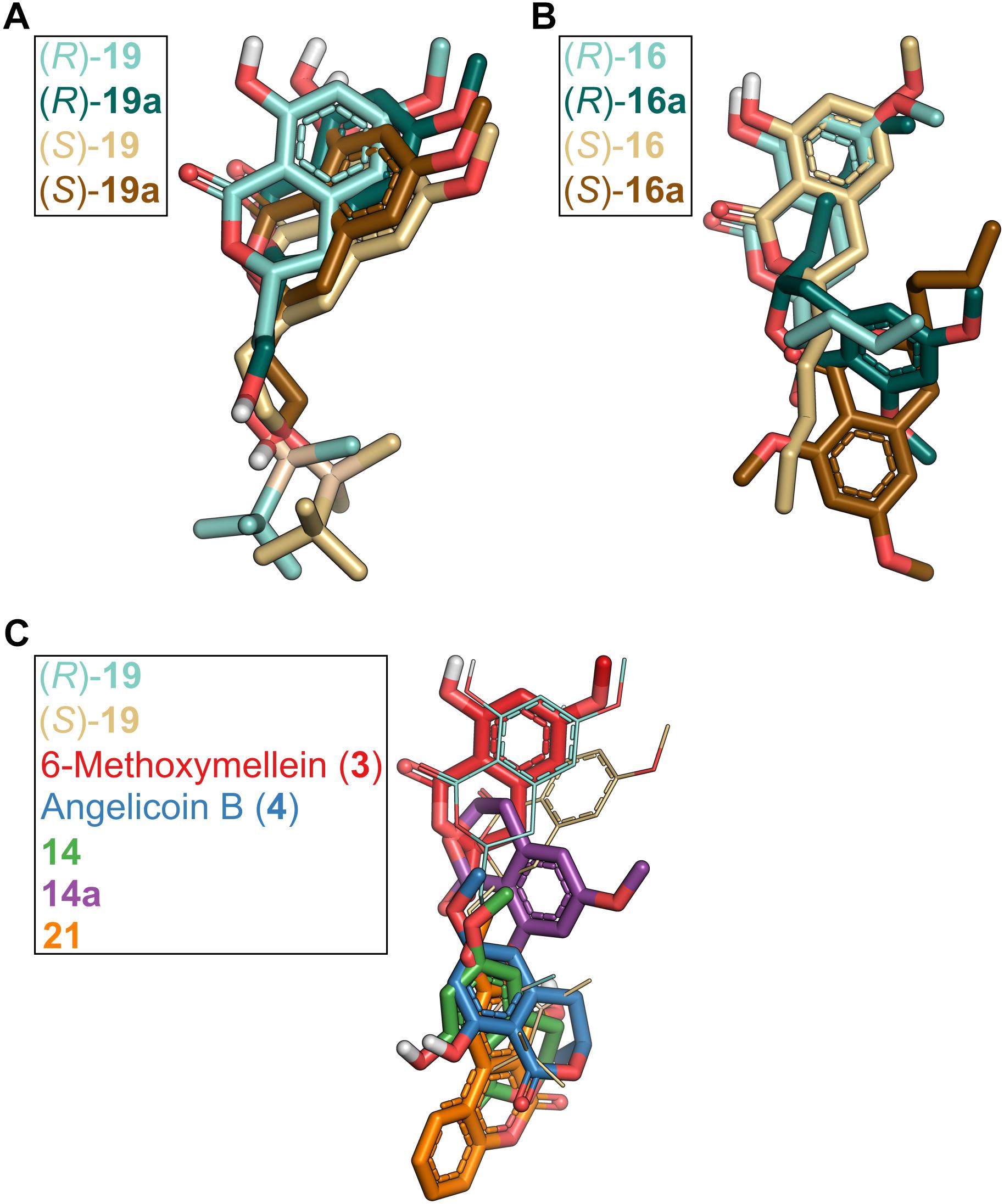
Figure 9. Comparison of binding poses of 3,4-dihydroisocoumarins in P-gp obtained by molecular docking. (A) Binding poses of compounds carrying a TBS group in the side chain [(R)-19, (S)-19] versus compounds in which the TBS group was removed [(R)-19a, (S)-19a]. (B) Binding poses of compounds that carry a hydroxy group at position 8 [(R)-16, (S)-16] versus compounds that carry a methoxy group at position 8 [(R)-16a, (S)-16a]. (C) Binding modes of compounds 3, 4, 14, 14a, and 21 in comparison to (R)-19 and (S)-19.
Regarding these results, the 3,4-dihydroisocoumarin derivatives that we identified as dual P-gp and BCRP inhibitors with moderate activity might serve as a promising starting point for further development of novel inhibitors for overcoming cancer MDR. In future work, it will be important to identify additional substituents at other positions of the molecule to significantly improve the potency of the current chemical lead structure.
In a first initial screening (R)-19, (S)-19, 16 and 16a were identified as potential inhibitors of the yeast ABC transporter Pdr5. With these four compounds IC50 measurements with respect to R6G transport as well as the ATPase activity were performed. In the case of the transport assay the highest IC50 value was determined for (R)-19 with 26.0 ± 3.5 μM. Interestingly the lowest IC50 was detected for its enantiomer (S)-19 with 5.3 ± 0.6 μM. The transport IC50 values for 16 and 16a were nearly similar with 15.1 ± 2.1 and 14.6 ± 3.9 μM, respectively. For the ATPase activity it was not possible to reliably determine any IC50 value because of the limited solubility of the compounds. The highest ATPase activity inhibition of Pdr5 wild-type was detected for (S)-19 with an inhibition down to 17.3% activity in comparison to Pdr5 WT without any compound. For (R)-19 the ATPase activity was inhibited to 21.5% and for 16 and 16a the activity was inhibited to 35.4 and 22.8%, respectively. Our data furthermore suggest that these four coumarins are substrates of Pdr5, which are transported in competition to the fluorophore R6G and become inhibitory at higher concentrations, a phenomena often observed for P-gp (Al-Shawi et al., 2003) and Pdr5 (Ernst et al., 2008). In contrast to P-gp, it is obviously not important if there is a hydroxy or methoxy group at position 6, but it seems important that a longer hydrophobic side chain is present at position 3. The two natural substrates 6-methoxymellein (3) and angelicoin (4) with only a small methyl group at position 3 or compound 14 with no alkyl side chain at all displayed no effect on neither transport nor ATPase activity. Furthermore, in comparison to that the two compounds with a longer alkyl side chain with a hydrophilic group ((R)-19a and (S)-19a) showed only inhibition to around 40%. In addition to transporter inhibition, the cytotoxic activity of the test compounds was analyzed in sensitive A549 lung carcinoma cells as well as resistant HCT-15 colon carcinoma, H69AR lung carcinoma and MCF-7/MX breast carcinoma cells. Most compounds did not exhibit any toxic effects in cancer cells. One exception was derivative 16a, which proved to be selectively cytotoxic in BCRP-expressing MCF-7/MX cells with an IC50 value of 10.6 μM, but not in P-gp or MRP1-expressing cells. In sensitive A549, cytotoxicity was significantly lower with an IC50 value of 53.6 μM. At this point, it would be interesting to further characterize if compound 16a specifically kills multidrug-resistant cells by analyzing its activity in parental, sensitive MCF-7 cells. If this is the case, gaining insights into the mechanism of action and target would be helpful to find out more about collateral sensitivity in BCRP-overexpressing cancer cells and how to exploit this feature in tumor therapy (Pluchino et al., 2012; Rao et al., 2014).
JS, KD, and MB performed experiments, analyzed data, and wrote the manuscript. AW synthesized test compounds. EF and AK provided ellagic acid. NT, LS, HG, JP, EF, and AK contributed to study conception and design and received funding. All authors revised the manuscript.
This work was funded by the German Federal Ministry for Economic Affairs and Energy (‘ZIM Kooperationsprojekt’ KF3279X01AJ3 to EF, AK, JP, LS, and NT) and supported by a scholarship of the Studienstiftung des Deutschen Volkes for AW. The Ph.D. training of JS was financed by the graduate program in Pharmacology and Experimental Therapeutics at the University of Cologne, which is financially and scientifically supported by Bayer. Funding by Deutsche Forschungsgemeinschaft (DFG) (INST 208/704-1 FUGG) to purchase the hybrid computer cluster used in this study is gratefully acknowledged.
EF and AK were employed by company MicroCombiChem GmbH.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Dr. Erasmus Schneider (Wadsworth Center, New York State Department of Health, Albany, NY, United States) for kindly providing the MCF-7/MX cell line and Janina Betz and Vanessa Mundorf for excellent technical assistance. We are grateful for computational support by the “Zentrum für Informations und Medientechnologie” at the Heinrich Heine University and the computing time provided by the John von Neumann Institute for Computing (NIC) to HG on the supercomputer JURECA at Jülich Supercomputing Centre (JSC) (user ID: HKF7).
Abid, O. U. R., Khalid, M., Hussain, M. T., Hanif, M., Qadeer, G., Rama, N. H., et al. (2012). Synthesis and anti-cancer, anti-metastatic evaluation of some new fluorinated isocoumarins and 3,4-dihydroisocoumarins. J. Fluor. Chem. 135, 240–245. doi: 10.1016/j.jfluchem.2011.11.011
Agarwal, S., Hartz, A. M. S., Elmquist, W. F., and Bauer, B. (2011). Breast cancer resistance protein and P-glycoprotein in brain cancer: two gatekeepers team up. Curr. Pharm. Des. 17, 2793–2802. doi: 10.2174/138161211797440186
Alam, A., Küng, R., Kowal, J., McLeod, R. A., Tremp, N., Broude, E. V., et al. (2018). Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc. Natl. Acad. Sci. 115, E1973–E1982. doi: 10.1073/pnas.1717044115
Al-Shawi, M. K., Polar, M. K., Omote, H., and Figler, R. A. (2003). Transition state analysis of the coupling of drug transport to ATP hydrolysis by P-glycoprotein. J. Biol. Chem. 278, 52629–52640. doi: 10.1074/jbc.M308175200
Baer, M. R., George, S. L., Dodge, R. K., O’Loughlin, K. L., Minderman, H., Caligiuri, M. A., et al. (2002). Phase 3 study of the multidrug resistance modulator PSC-833 in previously untreated patients 60 years of age and older with acute myeloid leukemia: Cancer and leukemia group B Study 9720. Blood 100, 1224–1232.
Barthomeuf, C., Grassi, J., Demeule, M., Fournier, C., Boivin, D., and Béliveau, R. (2005). Inhibition of P-glycoprotein transport function and reversion of MDR1 multidrug resistance by cnidiadin. Cancer Chemother. Pharmacol. 56, 173–181. doi: 10.1007/s00280-004-0914-y
Begicevic, R.-R., and Falasca, M. (2017). ABC transporters in cancer stem cells: beyond chemoresistance. Int. J. Mol. Sci. 18:2362. doi: 10.3390/ijms18112362
Bisi, A., Cappadone, C., Rampa, A., Farruggia, G., Sargenti, A., Belluti, F., et al. (2017). Coumarin derivatives as potential antitumor agents: growth inhibition, apoptosis induction and multidrug resistance reverting activity. Eur. J. Med. Chem. 127, 577–585. doi: 10.1016/j.ejmech.2017.01.020
Böse, D., Niesobski, P., Lübcke, M., and Pietruszka, J. (2014). A diastereoselective one-pot, three-step cascade toward α-substituted allylboronic esters. J. Org. Chem. 79, 4699–4703. doi: 10.1021/jo5004168
Boumendjel, A., McLeer-Florin, A., Champelovier, P., Allegro, D., Muhammad, D., Souard, F., et al. (2009). A novel chalcone derivative which acts as a microtubule depolymerising agent and an inhibitor of P-gp and BCRP in in-vitro and in-vivo glioblastoma models. BMC Cancer 9:242. doi: 10.1186/1471-2407-9-242
Brown, G. D., Denning, D. W., Gow, N. A. R., Levitz, S. M., Netea, M. G., and White, T. C. (2012). Hidden killers: human fungal infections. Sci. Transl. Med. 4:165rv13. doi: 10.1126/scitranslmed.3004404
Bugde, P., Biswas, R., Merien, F., Lu, J., Liu, D.-X., Chen, M., et al. (2017). The therapeutic potential of targeting ABC transporters to combat multi-drug resistance. Expert Opin. Ther. Targets 21, 511–530. doi: 10.1080/14728222.2017.1310841
Cichewicz, R. H., Valeriote, F. A., and Crews, P. (2004). Psymberin, a potent sponge-derived cytotoxin from Psammocinia distantly related to the pederin family. Org. Lett. 6, 1951–1954. doi: 10.1021/ol049503q
Combes, S., Barbier, P., Douillard, S., McLeer-Florin, A., Bourgarel-Rey, V., Pierson, J.-T., et al. (2011). Synthesis and biological evaluation of 4-arylcoumarin analogues of combretastatins. Part 2. J. Med. Chem. 54, 3153–3162. doi: 10.1021/jm901826e
Decottignies, A., Kolaczkowski, M., Balzi, E., and Goffeau, A. (1994). Solubilization and characterization of the overexpressed PDR5 multidrug resistance nucleotide triphosphatase of yeast. J. Biol. Chem. 269, 12797–12803.
Dohse, M., Scharenberg, C., Shukla, S., Robey, R. W., Volkmann, T., Deeken, J. F., et al. (2010). Comparison of ATP-binding cassette transporter interactions with the tyrosine kinase inhibitors imatinib, nilotinib, and dasatinib. Drug Metab. Dispos. 38, 1371–1380. doi: 10.1124/dmd.109.031302
Dufour, J.-P., Amory, A., and Goffeau, A. (1988). Plasma membrane ATPase from the yeast Schizosaccharomyces pombe. Methods Enzymol. 157, 513–528. doi: 10.1016/0076-6879(88)57100-8
El-Awady, R., Saleh, E., Hashim, A., Soliman, N., Dallah, A., Elrasheed, A., et al. (2016). The role of eukaryotic and prokaryotic abc transporter family in failure of chemotherapy. Front. Pharmacol. 7:535. doi: 10.3389/fphar.2016.00535
Endringer, D. C., Guimarães, K. G., Kondratyuk, T. P., Pezzuto, J. M., and Braga, F. C. (2008). Selective inhibition of aromatase by a dihydroisocoumarin from Xyris pterygoblephara. J. Nat. Prod. 71, 1082–1084. doi: 10.1021/np800098f
Ernst, R., Klemm, R., Schmitt, L., and Kuchler, K. (2005). Yeast ATP-binding cassette transporters: cellular cleaning pumps. Methods Enzymol. 400, 460–484. doi: 10.1016/S0076-6879(05)00026-1
Ernst, R., Kueppers, P., Klein, C. M., Schwarzmueller, T., Kuchler, K., and Schmitt, L. (2008). A mutation of the H-loop selectively affects rhodamine transport by the yeast multidrug ABC transporter Pdr5. Proc. Natl. Acad. Sci. 105, 5069–5074. doi: 10.1073/pnas.0800191105
Fischer, T., and Pietruszka, J. (2007). Efficient synthesis of either enantiomer of ethyl 5-hydroxyhept-6-enoate. Adv. Synth. Catal. 349, 1533–1536. doi: 10.1002/adsc.200700086
Fitzmaurice, C., Allen, C., Barber, R. M., Barregard, L., Bhutta, Z. A., Brenner, H., et al. (2017). Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol. 3, 524–548. doi: 10.1001/jamaoncol.2016.5688
Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Klicic, J. J., Mainz, D. T., et al. (2004). Glide: a new approach for rapid, accurate docking and scoring. 1. method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749. doi: 10.1021/jm0306430
Friesner, R. A., Murphy, R. B., Repasky, M. P., Frye, L. L., Greenwood, J. R., Halgren, T. A., et al. (2006). Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49, 6177–6196. doi: 10.1021/jm051256o
Gillet, J.-P., and Gottesman, M. M. (2010). “Mechanisms of Multidrug Resistance in Cancer,” in Multi-Drug Resistance in Cancer, ed. J. Zhou (Totowa, NJ: Humana Press), 47–76. doi: 10.1007/978-1-60761-416-6_4
Goffeau, A., and Dufour, J.-P. (1988). Plasma membrane ATPase from the yeast Saccharomyces cerevisiae. Methods Enzymol. 157, 528–533. doi: 10.1016/0076-6879(88)57101-X
Gohlke, H., and Case, D. A. (2004). Converging free energy estimates: MM-PB(GB)SA studies on the protein-protein complex Ras-Raf. J. Comput. Chem. 25, 238–250. doi: 10.1002/jcc.10379
Gu, X., Ren, Z., Peng, H., Peng, S., and Zhang, Y. (2014). Bifendate-chalcone hybrids: A new class of potential dual inhibitors of P-glycoprotein and breast cancer resistance protein. Biochem. Biophys. Res. Commun. 455, 318–322. doi: 10.1016/j.bbrc.2014.11.016
Guimarães, K. G., Freitas, R. P., de Ruiz, A. L. T. G., Fiorito, G. F., Carvalho, J. E., de da Cunha, E. F. F., et al. (2016). Synthesis, antiproliferative activities, and computational evaluation of novel isocoumarin and 3,4-dihydroisocoumarin derivatives. Eur. J. Med. Chem. 111, 103–113. doi: 10.1016/j.ejmech.2016.01.051
Guo, Z. (2017). The modification of natural products for medical use. Acta. Pharm. Sin. B 7, 119–136. doi: 10.1016/j.apsb.2016.06.003
Gupta, R. P., Kueppers, P., Hanekop, N., and Schmitt, L. (2014). Generating symmetry in the asymmetric ABC transporter Pdr5 from Saccharomyces cerevisiae. J. Biol. Chem. 289, 15272–15279. doi: 10.1074/jbc.M114.553065
Hoepfner, D., McNamara, C. W., Lim, C. S., Studer, C., Riedl, R., Aust, T., et al. (2012). Selective and specific inhibition of the Plasmodium falciparum lysyl-tRNA synthetase by the fungal secondary metabolite cladosporin. Cell Host Microbe 11, 654–663. doi: 10.1016/j.chom.2012.04.015
Hou, T., Wang, J., Li, Y., and Wang, W. (2011). Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. the accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 51, 69–82. doi: 10.1021/ci100275a
Iriti, M., Kubina, R., Cochis, A., Sorrentino, R., Varoni, E. M., Kabała-Dzik, A., et al. (2017). Rutin, a quercetin glycoside, restores chemosensitivity in human breast cancer cells. Phytother. Res. 31, 1529–1538. doi: 10.1002/ptr.5878
Jianing, L., Robert, A., Kai, Z., Yixiang, C., Suwen, Z., and Richard, A. F. (2011). The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 79, 2794–2812. doi: 10.1002/prot.23106
Karthikeyan, S., and Hoti, S. (2015). Development of fourth generation ABC inhibitors from natural products: a novel approach to overcome cancer multidrug resistance. Anticancer Agents Med. Chem. 15, 605–615. doi: 10.2174/1871520615666150113103439
Kaye, K. S., and Kaye, D. (2000). Multidrug-resistant pathogens: mechanisms of resistance and epidemiology. Curr. Infect. Dis. Rep. 2, 391–398. doi: 10.1007/s11908-000-0065-1
Kolaczkowski, M., van der Michel, R., Cybularz-Kolaczkowska, A., Soumillion, J.-P., Konings, W. N., and André, G. (1996). Anticancer drugs, ionophoric peptides, and steroids as substrates of the yeast multidrug transporter Pdr5p. J. Biol. Chem. 271, 31543–31548. doi: 10.1074/jbc.271.49.31543
Lai, D., Wang, A., Cao, Y., Zhou, K., Mao, Z., Dong, X., et al. (2016). Bioactive dibenzo-α-pyrone derivatives from the endophytic fungus Rhizopycnis vagum Nitaf22. J. Nat. Prod. 79, 2022–2031. doi: 10.1021/acs.jnatprod.6b00327
Li, X.-Q., Wang, L., Lei, Y., Hu, T., Zhang, F.-L., Cho, C.-H., et al. (2015). Reversal of P-gp and BCRP-mediated MDR by tariquidar derivatives. Eur. J. Med. Chem. 101, 560–572. doi: 10.1016/j.ejmech.2015.06.049
Liang, Q., Wu, Q., Jiang, J., Duan, J. A., Wang, C., Smith, M. D., et al. (2011). Characterization of sparstolonin B, a Chinese herb-derived compound, as a selective Toll-like receptor antagonist with potent anti-inflammatory properties. J. Biol. Chem. 286, 26470–26479. doi: 10.1074/jbc.M111.227934
Lomize, M. A., Pogozheva, I. D., Joo, H., Mosberg, H. I., and Lomize, A. L. (2012). OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376. doi: 10.1093/nar/gkr703
Longley, D. B., and Johnston, P. G. (2005). Molecular mechanisms of drug resistance. J. Pathol. 205, 275–292. doi: 10.1002/path.1706
McCormick, J. W., Vogel, P. D., and Wise, J. G. (2015). Multiple drug transport pathways through human p-glycoprotein. Biochemistry 54, 4374–4390. doi: 10.1021/acs.biochem.5b00018
Newman, D. J., and Cragg, G. M. (2016). Natural poducts as sources of new drugs from 1981 to 2014. J. Nat. Prod. 79, 629–661. doi: 10.1021/acs.jnatprod.5b01055
Olsson, M. H. M., Søndergaard, C. R., Rostkowski, M., and Jensen, J. H. (2011). PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 7, 525–537. doi: 10.1021/ct100578z
Pluchino, K. M., Hall, M. D., Goldsborough, A. S., Callaghan, R., and Gottesman, M. M. (2012). Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist. Updat. 15, 98–105. doi: 10.1016/j.drup.2012.03.002
Pusztai, L., Wagner, P., Ibrahim, N., Rivera, E., Theriault, R., Booser, D., et al. (2005). Phase II study of tariquidar, a selective P-glycoprotein inhibitor, in patients with chemotherapy-resistant, advanced breast carcinoma. Cancer 104, 682–691. doi: 10.1002/cncr.21227
Raad, I., Terreux, R., Richomme, P., Matera, E.-L., Dumontet, C., Raynaud, J., et al. (2006). Structure-activity relationship of natural and synthetic coumarins inhibiting the multidrug transporter P-glycoprotein. Bioorg. Med. Chem. 14, 6979–6987. doi: 10.1016/j.bmc.2006.06.026
Ramanan, M., Sinha, S., Sudarshan, K., Aidhen, I. S., and Doble, M. (2016). Inhibition of the enzymes in the leukotriene and prostaglandin pathways in inflammation by 3-aryl isocoumarins. Eur. J. Med. Chem. 124, 428–434. doi: 10.1016/j.ejmech.2016.08.066
Rao, D. K., Liu, H., Ambudkar, S. V., and Mayer, M. (2014). A combination of curcumin with either gramicidin or ouabain selectively kills cells that express the multidrug resistance-linked ABCG2 transporter. J. Biol. Chem. 289, 31397–31410. doi: 10.1074/jbc.M114.576819
Sachs, J., Kadioglu, O., Weber, A., Mundorf, V., Betz, J., Efferth, T., et al. (2019). Selective inhibition of P-gp transporter by goniothalamin derivatives sensitizes resistant cancer cells to chemotherapy. J. Nat. Med. 73, 226–235. doi: 10.1007/s11418-018-1230-x
Saeed, A. (2016). Isocoumarins, miraculous natural products blessed with diverse pharmacological activities. Eur. J. Med. Chem. 116, 290–317. doi: 10.1016/j.ejmech.2016.03.025
Sastry, G. M., Adzhigirey, M., Day, T., Annabhimoju, R., and Sherman, W. (2013). Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aid. Mol. Des. 27, 221–234. doi: 10.1007/s10822-013-9644-8
Sim, H. M., Loh, K. Y., Yeo, W. K., Lee, C. Y., and Go, M. L. (2011). Aurones as modulators of ABCG2 and ABCB1: synthesis and structure-activity relationships. ChemMedChem 6, 713–724. doi: 10.1002/cmdc.201000520
Simic, M., Paunovic, N., Boric, I., Randjelovic, J., Vojnovic, S., Nikodinovic-Runic, J., et al. (2016). Functionalised isocoumarins as antifungal compounds: synthesis and biological studies. Bioorg. Med. Chem. Lett. 26, 235–239. doi: 10.1016/j.bmcl.2015.08.086
Sjöstedt, N., Holvikari, K., Tammela, P., and Kidron, H. (2017). Inhibition of breast cancer resistance protein and multidrug resistance associated protein 2 by natural compounds and their derivatives. Mol. Pharma. 14, 135–146. doi: 10.1021/acs.molpharmaceut.6b00754
Søndergaard, C. R., Olsson, M. H. M., Rostkowski, M., and Jensen, J. H. (2011). Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pka values. J. Chem. Theory Comput. 7, 2284–2295. doi: 10.1021/ct200133y
Spengler, G., Kincses, A., Gajdács, M., and Amaral, L. (2017). New roads leading to old destinations: efflux pumps as targets to reverse multidrug resistance in bacteria. Molecules 22:468. doi: 10.3390/molecules22030468
Szakács, G., Paterson, J. K., Ludwig, J. A., Booth-Genthe, C., and Gottesman, M. M. (2006). Targeting multidrug resistance in cancer. Nat. Rev. Drug. Discov. 5, 219–234. doi: 10.1038/nrd1984
Szöllõsi, D., Rose-Sperling, D., Hellmich, U. A., and Stockner, T. (2017). Comparison of mechanistic transport cycle models of ABC exporters. Biochim. Biophys. Acta 1860, 818–832. doi: 10.1016/j.bbamem.2017.10.028
Teixeira, T. S. P., Freitas, R. F., Abrahão, O., Devienne, K. F., Souza, L. R., de Blaber, S. I., et al. (2011). Biological evaluation and docking studies of natural isocoumarins as inhibitors for human kallikrein 5 and 7. Bioorg. Med. Chem. Lett. 21, 6112–6115. doi: 10.1016/j.bmcl.2011.08.044
Tillotson, G. S., and Theriault, N. (2013). New and alternative approaches to tackling antibiotic resistance. F1000Prime Rep. 5:51. doi: 10.12703/P5-51
Tran-Nguyen, V.-K., Prasad, R., Falson, P., and Boumendjel, A. (2017). Modulators of the efflux pump Cdr1p of Candida albicans: mechanisms of action and chemical features. Curr. Med. Chem. 24, 3242–3253. doi: 10.2174/0929867324666170523102244
Wada, S. I., Niimi, M., Niimi, K., Holmes, A. R., Monk, B. C., Cannon, R. D., et al. (2002). Candida glabrata ATP-binding cassette transporters Cdr1p and Pdh1p expressed in a Saccharomyces cerevisiae strain deficient in membrane transporters show phosphorylation-dependent pumping properties. J. Biol. Chem. 277, 46809–46821. doi: 10.1074/jbc.M207817200
Warner, E., Tobe, S. W., Andrulis, I. L., Pei, Y., Trachtenberg, J., and Skorecki, K. L. (1995). Phase I-II study of vinblastine and oral cyclosporin A in metastatic renal cell carcinoma. Am. J. Clin. Oncol. 18, 251–256. doi: 10.1097/00000421-199506000-00013
Weber, A., Döhl, K., Sachs, J., Nordschild, A. C. M., Schröder, D., Kulik, A., et al. (2017). Synthesis and cytotoxic activities of goniothalamins and derivatives. Bioorg. Med. Chem. 25, 6115–6125. doi: 10.1016/j.bmc.2017.02.004
Weis, A., Katebzadeh, K., Söderhjelm, P., Nilsson, I., and Ryde, U. (2006). Ligand affinities predicted with the MM/PBSA method: Dependence on the simulation method and the force field. J. Med. Chem. 49, 6596–6606. doi: 10.1021/jm0608210
Wise, J. G. (2012). Catalytic transitions in the human MDR1 P-glycoprotein drug binding sites. Biochemistry 51, 5125–5141. doi: 10.1021/bi300299z
Yuan, J., Wong, I. L. K., Jiang, T., Wang, S. W., Liu, T., Wen, B. J., et al. (2012). Synthesis of methylated quercetin derivatives and their reversal activities on P-gp- and BCRP-mediated multidrug resistance tumour cells. Eur. J. Med. Chem. 54, 413–422. doi: 10.1016/j.ejmech.2012.05.026
Zhang, G.-J., Li, B., Chen, L., Tian, Y., Liu, S.-J., Cui, H.-M., et al. (2017). Isocoumarin derivatives and monoterpene glycoside from the seeds of Orychophragmus violaceus. Fitoterapia 125, 111–116. doi: 10.1016/j.fitote.2017.12.025
Keywords: multidrug resistance, cancer chemotherapy, 3,4-dihydroisocoumarin, P-glycoprotein, breast cancer resistance protein, Pdr5
Citation: Sachs J, Döhl K, Weber A, Bonus M, Ehlers F, Fleischer E, Klinger A, Gohlke H, Pietruszka J, Schmitt L and Teusch N (2019) Novel 3,4-Dihydroisocoumarins Inhibit Human P-gp and BCRP in Multidrug Resistant Tumors and Demonstrate Substrate Inhibition of Yeast Pdr5. Front. Pharmacol. 10:400. doi: 10.3389/fphar.2019.00400
Received: 26 September 2018; Accepted: 01 April 2019;
Published: 16 April 2019.
Edited by:
Ahmed Lasfar, Rutgers University – The State University of New Jersey, United StatesReviewed by:
Chun Hei Antonio Cheung, National Cheng Kung University, TaiwanCopyright © 2019 Sachs, Döhl, Weber, Bonus, Ehlers, Fleischer, Klinger, Gohlke, Pietruszka, Schmitt and Teusch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicole Teusch, nicole.teusch@uni-osnabrueck.de
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.