 Huanbiao Mo
Huanbiao Mo Rayna Jeter2
Rayna Jeter2 Sophie T. Yount
Sophie T. Yount Hoda Yeganehjoo
Hoda Yeganehjoo- 1Department of Nutrition, Byrdine F. Lewis College of Nursing and Health Professions, Georgia State University, Atlanta, GA, United States
- 2Department of Clinical Nutrition, University of Texas Southwestern Medical Center, Dallas, TX, United States
- 3Department of Chemistry, Georgia State University, Atlanta, GA, United States
- 4Department of Pathology, Texas Tech University Health Sciences Center, Lubbock, TX, United States
The mevalonate pathway provides sterols for membrane structure and nonsterol intermediates for the post-translational modification and membrane anchorage of growth-related proteins, including the Ras, Rac, and Rho GTPase family. Mevalonate-derived products are also essential for the Hedgehog pathway, steroid hormone signaling, and the nuclear localization of Yes-associated protein and transcriptional co-activator with PDZ-binding motif, all of which playing roles in tumorigenesis and cancer stem cell function. The phosphatidylinositol-4,5-bisphosphate 3-kinase-AKT-mammalian target of rapamycin complex 1 pathway, p53 with gain-of-function mutation, and oncoprotein MYC upregulate the mevalonate pathway, whereas adenosine monophosphate-activated protein kinase and tumor suppressor protein RB are the downregulators. The rate-limiting enzyme, 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), is under a multivalent regulation. Sterol regulatory element binding protein 2 mediates the sterol-controlled transcriptional downregulation of HMGCR. UbiA prenyltransferase domain-containing protein-1 regulates the ubiquitination and proteasome-mediated degradation of HMGCR, which is accelerated by 24, 25-dihydrolanosterol and the diterpene geranylgeraniol. Statins, competitive inhibitors of HMGCR, deplete cells of mevalonate-derived intermediates and consequently inhibit cell proliferation and induce apoptosis. Clinical application of statins is marred by dose-limiting toxicities and mixed outcomes on cancer risk, survival and mortality, partially resulting from the statin-mediated compensatory upregulation of HMGCR and indiscriminate inhibition of HMGCR in normal and tumor cells. Tumor HMGCR is resistant to the sterol-mediated transcriptional control; consequently, HMGCR is upregulated in cancers derived from adrenal gland, blood and lymph, brain, breast, colon, connective tissue, embryo, esophagus, liver, lung, ovary, pancreas, prostate, skin, and stomach. Nevertheless, tumor HMGCR remains sensitive to isoprenoid-mediated degradation. Isoprenoids including monoterpenes (carvacrol, L-carvone, geraniol, perillyl alcohol), sesquiterpenes (cacalol, farnesol, β-ionone), diterpene (geranylgeranyl acetone), “mixed” isoprenoids (tocotrienols), and their derivatives suppress the growth of tumor cells with little impact on non-malignant cells. In cancer cells derived from breast, colon, liver, mesothelium, prostate, pancreas, and skin, statins and isoprenoids, including tocotrienols, geraniol, limonene, β-ionone and perillyl alcohol, synergistically suppress cell proliferation and associated signaling pathways. A blend of dietary lovastatin and δ-tocotrienol, each at no-effect doses, suppress the growth of implanted murine B16 melanomas in C57BL6 mice. Isoprenoids have potential as adjuvant agents to reduce the toxicities of statins in cancer prevention or therapy.
Introduction
Statins are competitive inhibitors of 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase (HMGCR), the rate-limiting enzyme in the mevalonate pathway (Goldstein and Brown, 1990). Widely prescribed as hypocholesterolemic agents, statins have been shown to inhibit cell proliferation and induce apoptosis in preclinical studies (Clendening and Penn, 2012). Clinical data suggest inverse association of statin use and risk of some, but not all, cancers. Dose-limiting toxicities of statins attributable to statin-induced compensatory upregulation of HMGCR and indiscriminate inhibition of HMGCR in normal and tumor cells pose constraints on the application of statins in cancer and call for novel approaches in reducing their side effects.
In this review we first summarize the role of the mevalonate pathway in cell proliferation and cancer by highlighting the intertwined relations between the mevalonate pathway and several key signaling molecules in growth regulation (Mullen et al., 2016). The multivalent regulation of HMGCR, including sterol-mediated transcriptional downregulation and nonsterol-induced enhancement of degradation, in normal cells contrasts with the sterol-resistant, dysregulated tumor HMGCR that remains uniquely sensitive to isoprenoid-mediated downregulation (Mo and Elson, 2004). The mechanisms underlying the isoprenoid-mediated tumor suppression is elucidated, followed by studies showing the synergistic effect of statins and isoprenoids and suggesting the potential of isoprenoids in adjuvant therapy to reduce the toxicities of statins.
Role of the Mevalonate Pathway in Cell Proliferation and Cancer
The mevalonate pathway provides the bulk end product, cholesterol, and nonsterol isoprenoids such as heme-A, ubiquinone (or coenzyme Q10), dolichol, isopentenyl adenine, farnesyl pyrophosphate (FPP), and geranylgeranyl pyrophosphate (GGPP) (Goldstein and Brown, 1990). Cholesterol is important for membrane structure, whereas the mevalonate-derived nonsterol compounds play vital roles in iron-containing cofactors of hemoproteins (e.g., hemoglobin, myoglobin, catalase, endothelial nitric oxide synthase, and cytochrome), mitochondrial electron transport and cellular respiration, N-glycosylation of proteins, transfer RNA (Buhaescu and Izzedine, 2007), and post-translational prenylation and membrane anchorage of growth-related proteins, including the Ras, Rac, and Rho GTPase family (Hentschel et al., 2016; Wang and Casey, 2016). The prenylated proteins collectively support cell proliferation and cancer growth (Mullen et al., 2016) (Figure 1).
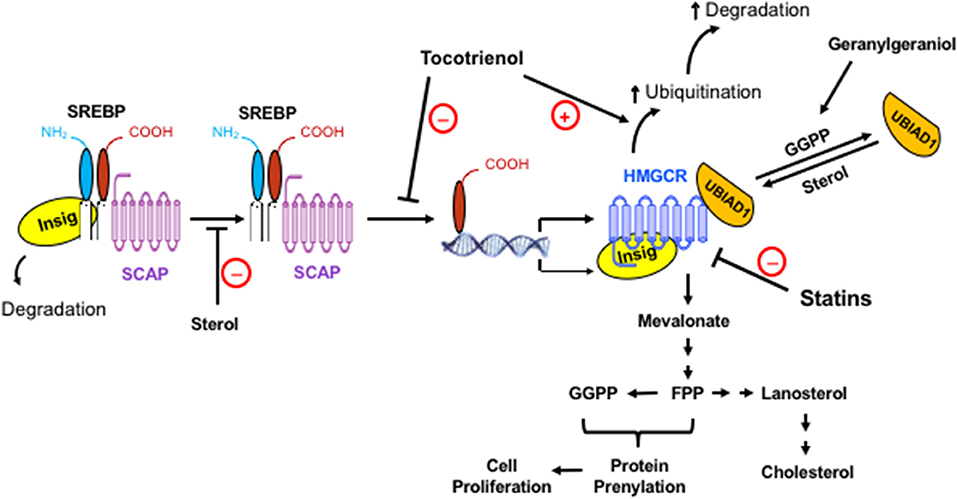
Figure 1. The regulation of the mevalonate pathway and the role of mevalonate-derived metabolites in cell proliferation. When cellular level of sterol is low, Insig dissociates from the SREBP-SCAP complex, allowing the latter to move to the Golgi apparatus. Following proteolytic cleavage of SREBP, its basic helix-loop-helix fragment enters the nucleus, binds to the SRE domain of target genes, and initiates the transcription and synthesis of a number of enzymes in the mevalonate pathway, including its rate-limiting enzyme, HMGCR. The activated mevalonate pathway produces sterols and nonsterols, including FPP and GGPP for protein prenylation and cell proliferation. Sterols block the transport of the SREBP-SCAP complex to the Golgi apparatus and induce the binding of HMGCR to Insig, leading to the ubiquitination and degradation of HMGCR. Exogenous geranylgeraniol is phosphorylated and converted to GGPP, which induces the dissociation of UBIAD1 from HMGCR and facilitates the degradation of HMGCR. The statins competitively inhibit HMGCR. Consequently, the lack of sterols and nonsterols results in a compensatory upregulation of HMGCR. Tocotrienols and potentially other isoprenoids block the processing and nuclear localization of SREBP and enhance the ubiquitination and degradation of HMGCR. HMGCR is upregulated in tumors but remains sensitive to isoprenoid-mediated downregulation. Synergistic impact of isoprenoids and statins on tumor HMGCR may offer a novel approach in enhancing the efficacy of statins with reduced toxicity.
The mevalonate pathway is also intertwined with several signaling pathways with regulatory roles in cancer. The most frequently altered signaling pathway in cancer, the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)-AKT pathway, regulates cell survival and proliferation (Mullen et al., 2016). Through the downstream mammalian target of rapamycin (mTOR) complex 1 signaling (Duvel et al., 2010), PI3K-AKT pathway activates the mevalonate pathway (Ricoult et al., 2016) and concomitantly stimulates glucose uptake and glycolysis that provide acetyl-CoA and NADPH to support the mevalonate pathway. Gain-of-function mutations of p53 (Freed-Pastor et al., 2012), another frequently mutated gene in cancer, and the oncoprotein MYC (Wu et al., 2016), upregulate the transcription of mevalonate pathway genes. In contrast, adenosine monophosphate-activated protein kinase (AMPK), an energy sensor and central regulator of metabolism, downregulates the mevalonate pathway via phosphorylation (Beg et al., 1973, 1978) and transcriptional control (Li et al., 2011) of HMGCR. The tumor suppressor protein RB also downregulates the mevalonate pathway (Shamma et al., 2009). Conversely, the mevalonate pathway provides sterols and nonsterol products for Hedgehog pathway (Eaton, 2008), steroid hormone signaling (Nguyen et al., 2015), and the nuclear localization of Yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) (Sorrentino et al., 2014; Koo and Guan, 2018), all of which have important roles in tumorigenesis and the cancer stem cell function (Mancini et al., 2018).
Regulation of the Mevalonate Pathway in Normal Cells
In non-malignant cells, HMGCR is highly regulated by a multivalent feedback mechanism mediated by sterols and nonsterol isoprenoid end products of mevalonate metabolism. This tight regulatory feedback system, which operates at transcriptional and post-transcriptional levels, ensures proper signaling for sufficient sterol and isoprenoid syntheses and optimal cell growth (Goldstein and Brown, 1990). Transcription regulation, exerted by sterols and mediated by sterol regulatory element (SRE), is the dominant feedback mechanism. The SRE is a promoter sequence in the 5′ flanking region of more than 30 genes involved in lipid biosynthesis and uptake, including those of HMG CoA synthase, HMGCR, and low density lipoprotein (LDL) receptor (Goldstein and Brown, 1990; Brown and Goldstein, 1997; Horton et al., 2002; Goldstein et al., 2006). By providing a binding site for the membrane-bound transcriptional factors called SRE binding proteins (SREBPs), the SRE domain allows for SREBP-mediated transcriptional control of all these SRE-containing genes. Another ER membrane-embedded protein, known as SREBP cleavage activating protein (SCAP), is coupled with SREBP and plays a critical role in the intracellular escort and proteolytic processing of SREBP (Osborne et al., 1985; Gil et al., 1988; Goldstein et al., 2006). SCAP and HMGCR share an intramembrane sequence named sterol-sensing domain that detects the concentration of membrane and intracellular sterols and mediates the interactions of SCAP and HMGCR with insulin–induced gene (Insig) proteins during transcriptional and post-transcriptional regulations (Goldstein et al., 2006; Johnson and DeBose-Boyd, 2018).
When sterol levels diminish in cells, Insig dissociates from the SCAP-SREBP-Insig complex and undergoes ubiquitin-mediated degradation (Gong et al., 2006). The liberated SCAP-SREBP complex is then taken into vesicles coated by COPII proteins (Sec23, Sec24, and Sar1-GTP) and transported to the Golgi apparatus (Brown and Goldstein, 1999; Goldstein et al., 2006; Espenshade and Hughes, 2007). The SREBP is cleaved on the cytosolic surface of Golgi membrane in a two-step proteolytic reaction by site-1 serine protease and site-2 Zn2+ metalloproteinase, producing the transcriptionally active, basic helix-loop-helix fragment of SREBP. This active motif of SREBP enters nucleus, binds to the SRE domain, and initiates transcription of target genes that produce proteins taking part in cholesterol uptake and biosynthesis, including LDL receptor, HMG CoA synthase, HMGCR, Insig-1, and FPP synthase (Goldstein and Brown, 1990; Brown and Goldstein, 1999; Horton et al., 2002).
Once intracellular sterol levels are restored by increased biosynthesis or uptake due to aforementioned SREBP-upregulated events, a convergent feedback inhibition terminates the SREBP-directed transcriptional operation. Two of the SREBP-induced molecules, newly synthesized Insig-1 and newly biosynthesized or collected exogenous cholesterol, bind SCAP simultaneously and induce its conformational changes. As a result, the SCAP-SREBP complex is stabilized and retained in the ER membrane and prevented from undergoing any further transcriptional processing (Yang et al., 2002; Goldstein et al., 2006; Gong et al., 2006). This coordinated process ensures that cells attain sufficient amounts of both cholesterol and nonsterol isoprenoids for their growth and metabolic requirements before the SREBP transcriptional operation is fully halted. Furthermore, this feedback mechanism precludes overaccumulation of potentially toxic sterols.
As one of the genes in the mevalonate pathway under the transcriptional control of SREBP, HMGCR has a secondary line of regulation on its synthesis and degradation. While an earlier study postulated that an unknown mevalonate-derived isoprenoid regulates HMGCR translation via a 5′-untranslated region of HMGCR mRNA (Nakanishi et al., 1988), the mechanisms underlying the degradation of HMGCR have been more clearly elucidated. This post-transcriptional fine-tuning of HMGCR regulation is mediated by two mevalonate-derived compounds, a reduced tetracyclic triterpenoid, 24, 25-dihydrolanosterol, and a nonsterol diterpene, geranylgeraniol (Sever et al., 2003). 24, 25-Dihydrolanosterol and a less effective cholesterol induce binding of Insig to the sterol sensing domain of HMGCR, a process that requires a tetrapeptide sequence YIYF located in the second transmembrane segment of HMGCR (Roitelman and Simoni, 1992; McGee et al., 1996; Ravid et al., 2000; DeBose-Boyd, 2008). This binding is followed by the ubiquitination and proteasome-mediated degradation of HMGCR (Sever et al., 2004; Song et al., 2005; Lange et al., 2008; Nguyen et al., 2009). Addition of HMGCR inhibitors to cultured cells containing adequate amounts of sterols enhanced HMGCR protein expression and reduced its degradation, which was reverted with supplemental nonsterol metabolites (Goldstein and Brown, 1990), suggesting a nonsterol metabolite also regulates the expression and degradation of HMGCR. Using SV589 human fibroblasts that lack the monocarboxylate transporter and consequently the ability for mevalonate uptake, studies showed that geranylgeraniol is more potent than the 15-carbon isoprenoid farnesol in accelerating the sterol-induced and Insig-dependent ubiquitination and degradation of HMGCR (Sever et al., 2003).
More recent studies have shown that geranylgeraniol might be phosphorylated and converted to GGPP, which serves as a potent regulator of HMGCR stability and degradation (Garza et al., 2009; Schumacher et al., 2016, 2018). Buildup of GGPP in ER membranes triggers dissociation of UbiA prenyltransferase domain-containing protein-1 (UBIAD1) from HMGCR, permitting ER-to-Golgi transport and maximal degradation of HMGCR (Schumacher et al., 2018). Additionally, pulse-chase experiments confirmed the synergistic effects of mevalonate or geranylgeraniol, but not farnesol, with sterols in enhancing complete HMGCR degradation (Sever et al., 2003).
The E2 conjugating enzyme Ubc7, E3 ubiquitin ligase glycoprotein 78 (gp78), and ATPase VCP/p97 facilitate delivery of ubiquitinated HMGCR from the ER membrane to the proteasomes for proteolysis and degradation (Espenshade and Hughes, 2007). A recent study (Hwang et al., 2016) confirmed that liver employs the same mechanism regulating the accelerated degradation of HMGCR and cholesterol homeostasis initially found in cultured cells. This operation safeguards HMGCR synthesis and stability until the cellular requirements of sterols and isoprenoids are met (Goldstein and Brown, 1990; Peffley and Gayen, 2003; Sever et al., 2003; Song et al., 2005; Goldstein et al., 2006; Lange et al., 2008; Nguyen et al., 2009). It was recently reported that the monoterpene linalool prevents the binding of SREBP-2 to HMGCR promoter and enhances the ubiquitin-mediated degradation of HMGCR (Cho et al., 2011). A second monoterpene geraniol suppresses the protein level and specific activity of HMGCR in mouse liver (Galle et al., 2015) and A549 cells (Galle et al., 2014). These findings may add another layer of complexity to the regulation of HMGCR. It remains unknown whether GGPP mediates the effect of these monoterpenes on HMGCR.
Upregulation of HMGCR in Tumors
Tumorigenesis has been associated with alterations and reprograming of energy, carbohydrate and lipid metabolisms with intertwined relations (Hanahan and Weinberg, 2011; Pavlova and Thompson, 2016). Activated oncogenes including ras and myc, for example, have been associated with alterations in glucose metabolism termed “aerobic glycolysis,” one of the hallmarks of cancer cells that is also known as the “Warburg effect” (Warburg, 1956; Hanahan and Weinberg, 2011). Hypoxia and the Ras oncoprotein that requires mevalonate-derived FPP for its post-translational modification can independently stimulate hypoxia-inducible factor 1α (HIF1α) and HIF2α transcriptional factors, which in turn upregulate glucose transporters and enzymes of the glycolytic pathway (Hanahan and Weinberg, 2011).
One of the metabolic adaptations to satisfy accelerated tumor growth is the upregulation of the mevalonate pathway, which has been implicated in the origin, progression, and phenotype of many human malignancies (Mo and Elson, 2004; Mullen et al., 2016). Tumor cells have augmented demands for nonsterol isoprenoids required for the prenylation of proteins supporting their excessive growth and proliferation. In response, tumors alter the monitory systems of mevalonate pathway that assure a constant and sufficient intracellular pool of sterols and nonsterol isoprenoids for non-malignant cell growth. One of the dysregulations stems from the overexpression and hyperactivity of tumor HMGCR as a result of its resistance to the sterol-dependent transcriptional regulation. Since Siperstein and Fagan (Siperstein and Fagan, 1964) reported the dysregulation of cholesterol synthesis in tumors and tumor-bearing animals, the uncoupling of HMGCR activity from sterol-mediated feedback regulation has been found in diverse tumors, embryonic and differentiating tissues and carcinogen-treated and regenerating liver (Elson et al., 1999).
Table 1 lists the studies showing differential expression of HMGCR in normal and tumor cells. In various human tissues including adrenal gland (Lehoux et al., 1984), blood and lymph (Yachnin and Mannickarottu, 1984; Harwood et al., 1991; Vitols et al., 1994; Hentosh et al., 2001; Kuzu et al., 2016), brain (Maltese, 1983; Laezza et al., 2015), breast (El-Sohemy and Archer, 2000; Celis et al., 2006; Ginestier et al., 2012), connective tissue (Kuzu et al., 2016), colon (Cerda et al., 1995; Hentosh et al., 2001; Notarnicola et al., 2004; Caruso and Notarnicola, 2005; Tyagi, 2005), embryo (Engstrom and Schofield, 1987), esophagus (Shi et al., 2013), liver (Kawata et al., 1990; Tam et al., 1991; Sohda et al., 2013), lung (Bennis et al., 1993), ovary (Zheng et al., 2018), prostate (Chen and Hughes-Fulford, 2001; Ettinger et al., 2004; Krycer et al., 2009; Murtola et al., 2012), skin (Kuzu et al., 2016), and stomach (Caruso et al., 2002), HMGCR mRNA and proteins levels are several-fold higher in tumors than their counterparts in normal tissues. Factors controlling the expression of HMGCR, including SREBP-1a, SREBP-2 and SCAP, were found to be overexpressed in prostate cancer (Krycer et al., 2009), contributing to transcriptional upregulation of HMGCR. Other enzymes in the mevalonate pathways, including farnesyltransferase, FPP synthase (Abate et al., 2017), and GGPP synthase (Wang et al., 2018), are also upregulated, suggesting a well-concerted regulation in raising the available metabolites derived from mevalonate. The augmented biosynthesis of cholesterol observed in tumors is consistent with the resistance of tumor HMGCR and SREBP-2 (Chen and Hughes-Fulford, 2001) to the cholesterol-mediated feedback inhibition that was initially observed in mouse hepatoma BW-7756 (Siperstein and Fagan, 1964). Several tissues in rodents, including bone marrow, breast (Rao et al., 1988; El-Sohemy and Archer, 2000), liver (Kandutsch and Hancock, 1971; Siperstein et al., 1971; Mitchell et al., 1978; Feingold et al., 1983; Gregg et al., 1986; Erickson et al., 1988; Azrolan and Coleman, 1989; Coni et al., 1992; Olsson et al., 1995; Clendening et al., 2010), lymph (Philippot et al., 1977), and pancreas (Rao et al., 1983), showed higher HMGCR expression and activity once they become malignant.
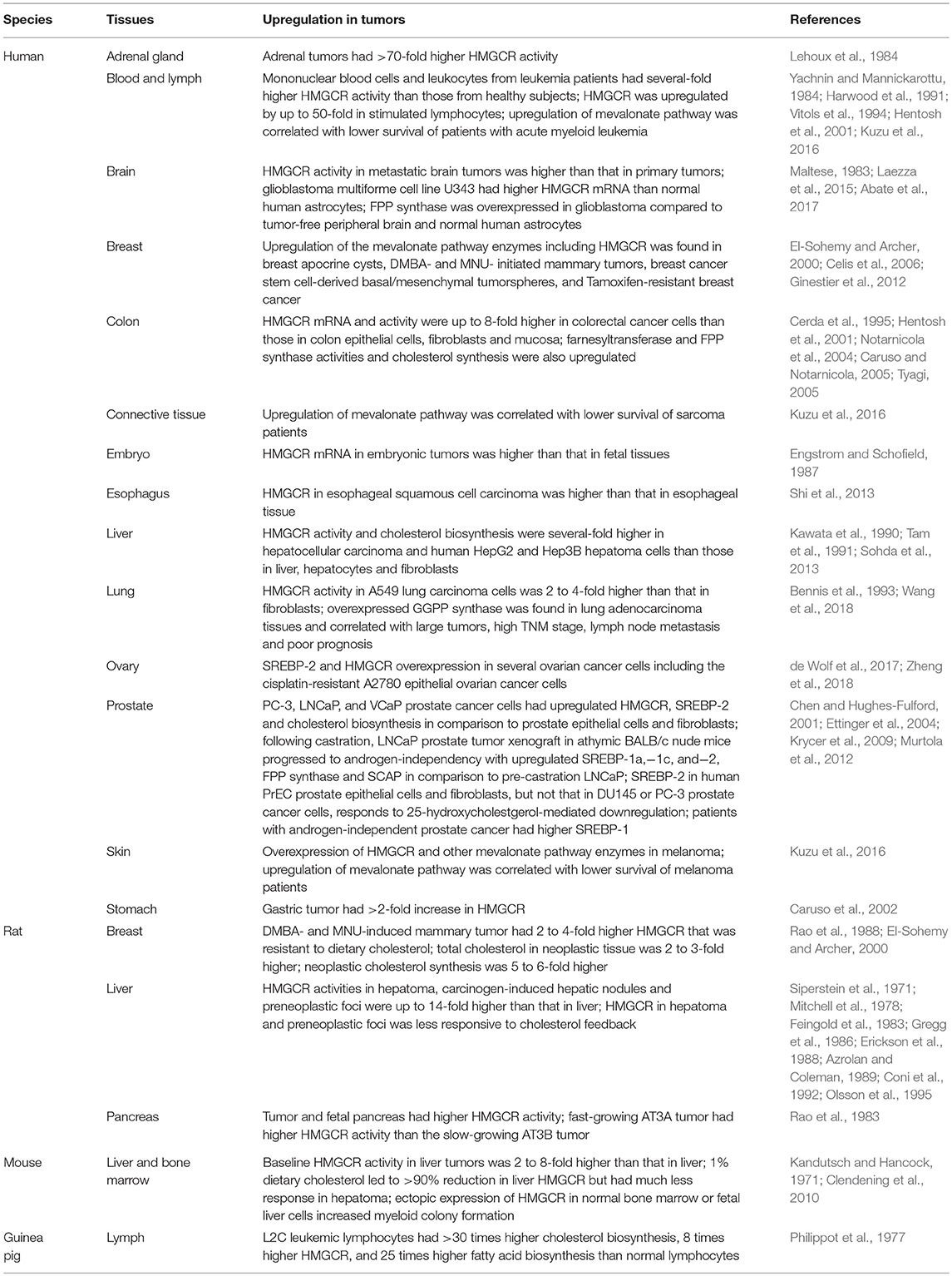
Table 1. Overexpression of HMGCR and upregulation of mevalonate pathway activities in tumors.
Consistent with these observations is the finding that exogenous mevalonate or artificially overexpressed HMGCR promotes growth. Supplemental mevalonate promoted growth of metastatic human breast cancer MDA-MB-435 cells in xenograft-bearing mice (Duncan et al., 2004a). Supplemental mevalonate increased the proliferation of cancer cells by upregulating cyclin-dependent kinase 2 (CDK2) activity and accelerating entry of cells into the S phase of cell division cycle (Duncan et al., 2004a). Ectopic expression of full-length or splice variant of HMGCR promotes transformation (Clendening et al., 2010). Finally, overexpression of mevalonate pathway genes is correlated with poor prognosis of recurrence-free and overall survival in breast cancer patients (Kimbung et al., 2016).
A recent study found that mutant p53-mediated upregulation of the mevalonate pathway is both necessary and sufficient for architectural phenotypes in breast cancer (Freed-Pastor et al., 2012), offering a new perspective for the role of the mevalonate pathway in cancer. The correlation between p53 mutation and highly expressed mevalonate pathway genes likely involves SREBP-2 and to a lesser extent, SREBP-1, which may assist the binding of p53 to the promoter of HMGCR gene. The mutant p53 and the mevalonate pathway also form a feed-forward loop in promoting cell proliferation. Mevalonate kinase upregulates DNAJ heat shock protein family (Hsp40) member A1 (DNAJA1), a type-I Hsp40, which inhibits the ubiquitination and degradation of mutant p53 mediated by the C-terminus of Hsc70-interacting protein (CHIP) E3 ubiquitin ligase (Parrales et al., 2016, 2018). Further implicating the role of the mevalonate pathway in tumorigenesis is the finding that mevalonate pathway inhibitors have cytostatic and cytotoxic effects in von Hippel-Lindau (VHL)-deficient clear cell renal cell carcinoma (CC-RCC) through an HIF-dependent mechanism (Thompson et al., 2018). Overall, these findings demonstrate the cancer promoting effects of excessive mevalonate and HMGCR activity.
Effects of Statins on Cancer Growth With Dose-limiting Toxicities
Statins, competitive inhibitors of HMGCR and cholesterol-lowering drugs, possess anti-cancer properties due to their ability to suppress the mevalonate pathway (Clendening and Penn, 2012). Studies have suggested chemopreventive potential of statins against several types of cancer (Hindler et al., 2006) including those of blood, brain (Girgert et al., 1999; Koyuturk et al., 2004), bone (Kany et al., 2018), head and neck (Dimitroulakos et al., 2001; Knox et al., 2005), liver (Paragh et al., 2005), ovary (Abdullah et al., 2017; Jones et al., 2017), skin (Shellman et al., 2005), and thyroid (Chen et al., 2017). Statins deplete cells of mevalonate-derived isoprene metabolites (FPP and GGPP) that are essential for the prenylation and activation of oncoproteins, including Ras and Rho. Consequently, statins have been shown in preclinical in vitro and in vivo studies to modulate signaling molecules including H-, K-, and N-Ras, Raf-1, nuclear factor kappa B (NFκB), mitogen-activated protein kinases (MAPKs), PI3K/AKT, extracellular signal-regulated kinase (ERK), mTOR, signal transducer and activator of transcription 3 (STAT3), Janus kinase 2 (JAK2) and caspases, suppress cell proliferation and cell cycle progress, and induce tumor cell apoptosis (Hindler et al., 2006; Pisanti et al., 2014; Chen et al., 2015; Ahmadi et al., 2017; Beckwitt et al., 2018; Kong et al., 2018). Furthermore, statins inhibit tumor cell invasion, migration, and metastasis by attenuating the geranylgeranylation and activation of Rho oncoproteins (Al-Haidari et al., 2014; Kato et al., 2018). Conversely, mevalonate and GGPP abolished statin-induced effects on p-AKT, p-ERK, cell cycle arrest, and apoptosis in several tumors including human HL-60 leukemia cells (Chen et al., 2015), ovarian cancer cells (de Wolf et al., 2017), MiaPaCa-2 pancreatic cancer cells (Gbelcova et al., 2017), Caki-1 and KTC-26 renal carcinoma cells (Woschek et al., 2016), and malignant anaplastic thyroid cancer (Chen et al., 2017). By blocking the synthesis of mevalonate-derived metabolites that hinder the ubiquitination and degradation of mutant p53 protein, statins also suppress the growth of mutant p53-expressing cancer cells (Freed-Pastor et al., 2012; Freed-Pastor and Prives, 2016; Parrales et al., 2016). A recent study suggest that the anticancer effect of statins is associated with the epithelial-to-mesenchymal transition phenotype (Yu et al., 2018).
Clinical efficacy of statins in cancer reduction may be tissue specific. Statin use was found to be associated with lower risks of primary liver cancer (McGlynn et al., 2015), hepatocellular carcinoma (Kim et al., 2018), HPV-negative squamous cell carcinoma (SCC) of the larynx, hypopharynx, and nasopharynx (Lebo et al., 2018), and subtypes of non-Hodgkin lymphomas including diffuse large B-cell lymphomas and plasma cell lymphomas (Ye et al., 2018), reduced aggressiveness (Allott et al., 2016) and mortality (Yu et al., 2014) of prostate cancer, and lower cancer specific and all-cause mortalities in esophageal cancer (Nguyen et al., 2018). However, statins do not affect survival after colorectal cancer (Hoffmeister et al., 2015) and small-cell lung cancer (Seckl et al., 2017), the risk of pancreatic cancer (Hamada et al., 2018), or the progression of prostate cancer in certain minority-enriched subpopulations (Allott et al., 2018). The type and hydrophilicity of statins, length of statin use, and ethnicity, lifestyle, and preexisting health condition of subjects may have contributed to the diverse outcome in statin and cancer studies—with some but not all studies showing the anticancer effect of statins (Gong et al., 2017).
Reported dose-limiting toxicities of statins may further deter the use of statins—at least as single therapies—in cancer treatment. Observations in clinical practices note approximately 20% adverse reaction rates to statins (Bruckert et al., 2005; Maningat and Breslow, 2011; Zhang et al., 2013, 2017). Possible adverse effects include diabetes mellitus, hemorrhagic stroke, cognition decline, tendon rupture, interstitial lung disease, and muscle problems (Thompson et al., 2016). The Effect of Statins on Skeletal Muscle Function and Performance (STOMP) study suggested 5–10% myalgia incidence in statin users (Parker et al., 2013). Concerned about the potential adverse effects of statins on cognition, glycemic control, and incident diabetes as well as their wide range of interactions with other medications, the US Food and Drug Administration issued and implemented new safety labeling for statins in 2012.
The dose-limiting toxicities of statins may be at least partially attributed to statin-mediated compensatory upregulation of HMGCR. By mimicking and competing with HMG CoA, the substrate for HMGCR, statins render HMGCR unavailable for catalyzing the formation of mevalonate and downstream sterols and nonsterols including FPP and GGPP. Statins also change the conformation of HMGCR and prevent it from attaining a functional structure, attaining high efficacy and specificity in inhibiting HMGCR (Sirtori, 2014). By removing the sterol and nonsterol products that downregulate HMGCR, persistent exposure to statins induces compensatory overexpression of the HMGCR in vitro and in vivo (Roglans et al., 2002). Elevated HMGCR expression by approximately 6- to 10-folds has been observed in liver microsomes and fibroblasts (Bensch et al., 1978; Sirtori, 2014). In our hands lovastatin also induced upregulation of HMGCR in human DU145 prostate carcinoma cells (Yeganehjoo et al., 2017). Exposure to rosuvastatin, lovastatin, and atorvastatin caused 6-, 11-, and 15-fold greater expression of hepatic HMGCR protein, respectively, and enhanced SREBP-2 processing of some target genes in mice (Schonewille et al., 2016). Since majority of cholesterol synthesis occurs in the liver, the significant hepatic uptake of statins and the limited bioavailability of many statins in extra-hepatic tissues can cause compensatory induction of mevalonate pathway in these tissues. This can, in turn, stimulate overproduction of isoprenoid metabolites (FPP and GGPP) and downstream prenylated proteins (Duncan et al., 2005; Brown, 2007; Solomon and Freeman, 2008). A compensatory increase in HMGCR expression in extrahepatic tissues following cholesterol-lowering statin therapy may provide a mechanistic basis for the elevated risk of statin-related tumorigenesis observed in some studies (Sacks et al., 1996; Coogan et al., 2002; Shepherd et al., 2002; Beck et al., 2003).
In summary, preclinical and mechanistic studies support the anti-cancer property of statins. The indiscriminate inhibition of HMGCR in tumor and non-tumor cells and the resulted overexpression of HMGCR may have contributed to dose-limiting toxicities of statins and mixed outcomes in clinical investigations. Efforts have been devoted to identifying agents capable of synergizing with statins in cancer growth inhibition.
Isoprenoids and Their Mechanisms of Action
Isoprenoids, also known as terpenoids, are a class of naturally occurring phytochemicals found in fruits, vegetables, and unrefined cereal grains. The carbon skeleton of these organic compounds is assembled from one or multiple copies of the five-carbon isoprene unit (X), giving rise to hemiterpenoids (1X, e.g., prenol and isovaleric acid), cyclic monoterpenes (2X, e.g., d-limonene, perillyl alcohol, perillaldehyde, carvacrol, carvone, and thymol), acyclic monoterpenes (2X, e.g., geraniol), sesquiterpenes (3X, e.g., cacalol, farnesol), diterpenes (4X, e.g., geranylgeraniol), triterpenes (6X, e.g., lupeol), tetraterpenes (8X, e.g., lycopene), and polyterpenes. Other isoprenoids such as the tocotrienols, members of the vitamin E family with a farnesyl side chain, are “mixed” isoprenoids with only part of the molecule being derived from the isoprene unit (Bach, 1995; Kumari et al., 2013). Besides the classical mevalonate pathway, an alternative non-mevalonate 2-C-methyl-D-erythritol 4-phosphate/1-deoxy-D-xylulose 5-phosphate (MEP/DOXP) pathway also produces isoprenoid precursors in bacteria, green algae, and some plants (Lichtenthaler, 1999; Hunter, 2007).
Diverse isoprenoids of plant mevalonate metabolism trigger post-transcriptional events to regulate the HMGCR expression and activity. These isoprenoid-mediated modulations include blockade of HMGCR mRNA translation and induction of HMGCR degradation in proteasomes.
Despite the demolished sterol-dependent transcriptional regulation in cancer cells, the isoprenoid-induced post-transcriptional tuning of HMGCR still exists and, in fact, becomes more compelling in tumor cells (Mo and Elson, 1999, 2004). Several isoprenoids such as δ-, γ-, and α-tocotrienol (Parker et al., 1993), farnesol (Meigs et al., 1996), β-ionone (Jones et al., 2013), geraniol (Houten et al., 2003; Peffley and Gayen, 2003), and their derivatives (Bradfute and Simoni, 1994; Pearce et al., 1994) suppress HMGCR activity. The 20-carbon isoprenoid geranylgeraniol effectively hinders HMGCR activity in human lung (Miquel et al., 1996) and prostate (Fernandes et al., 2013) tumor cells. At a concentration (30 μM) insufficient to affect SREBP-2 processing, geranylgeraniol synergized with sterols in shutting down processing of this HMGCR transcriptional factor in human SV589 fibroblasts (Sever et al., 2003). Tocotrienols inhibit mevalonate pathway through an Insig-dependent ubiquitination and degradation of HMGCR (Song and DeBose-Boyd, 2006). This regulation of HMGCR by tocotrienols has endowed these molecules with regulatory effects on cholesterol production and, thus, their hypocholesterolemic properties in mammalian cells (Parker et al., 1993). δ- and γ-tocotrienols, the strongest suppressors of HMGCR (Pearce et al., 1994; Song and DeBose-Boyd, 2006), were initially found to mimic nonsterol isoprenoids in accelerating HMGCR degradation at the post-transcriptional level (Parker et al., 1993). Subsequent studies revealed the direct recognition of these two structures by sterol-sensing systems in the ER membrane and their sterol-like activities in enhancing the Insig-dependent ubiquitination and degradation of HMGCR (Song and DeBose-Boyd, 2006). The same study delineated efficient blocking of SREBP-2 processing by the δ- but not γ- isoform of tocotrienol (Song and DeBose-Boyd, 2006). Additionally, combination of mevalonate or a mevalonate-derived nonsterol product (e.g., geranylgeraniol) with either δ- or γ-tocotrienol maximally eliminated HMGCR expression in these experiments (Song and DeBose-Boyd, 2006). A later study found tocotrienols suppress SREBP-2 activity by degrading mature SREBP-2 in the nucleus via a proteasome-independent mechanism in a human LNCaP-364 androgen-independent prostate cancer cell line (Krycer et al., 2012). Consequently, tocotrienols downregulate the HMGCR mRNA level.
By depleting tumor cells of mevalonate-derived intermediates, such as FPP and GGPP, isoprenoids can interrupt the prenylation and modification of critical signaling proteins including Ras, lamin B, and other growth related proteins and, thus, suppress tumor growth and proliferation (Mo and Elson, 1999, 2004; Yeganehjoo et al., 2017). Numerous studies have been conducted to unveil the significant outcomes of isoprenoid-based manipulations of the mevalonate pathway and HMGCR in cancer cells. γ-Tocotrienol at 0–30 μM downregulated HMGCR, membrane H-, K-, and N-Ras, and Raf-1, p-AKT, and p-ERK in HL-60 cells (Chen et al., 2015), adding to a long list of in vitro and in vivo studies showing isoprenoid-mediated suppression of the growth of cancer cells including those of blood (Shoff et al., 1991; Melnykovych et al., 1992; Mo and Elson, 1999; Lee et al., 2015), breast (Iqbal et al., 2004; Pierpaoli et al., 2010, 2013; Gomide et al., 2013; Ding et al., 2017), cervix (Yazlovitskaya and Melnykovych, 1995; Xu et al., 2017; Potocnjak et al., 2018), colon (Mo and Elson, 1999; Gomide et al., 2013, 2016), liver (Wada et al., 2005; Crespo et al., 2013; Scolastici et al., 2014; Rodenak-Kladniew et al., 2018), lung (Mo and Elson, 1999; Wada et al., 2005; Gomide et al., 2013; Galle et al., 2014), mouth (Liang et al., 2013; Madankumar et al., 2013), pancreas (Hussein and Mo, 2009; Fernandes et al., 2013), prostate (Mo and Elson, 2004; Sundin et al., 2012; Fernandes et al., 2013; Jones et al., 2013; Yeganehjoo et al., 2017), skin (Mo et al., 2000; McAnally et al., 2007; Chang et al., 2009; Chaudhary et al., 2013), and stomach (Dong et al., 2013; Liu et al., 2013). Concomitantly, isoprenoids induce cell cycle arrest and apoptosis in the tumor cells (Mo and Elson, 1999, 2004; Dong et al., 2013; Jones et al., 2013; Yeganehjoo et al., 2017; Rodenak-Kladniew et al., 2018; Sailo et al., 2018).
More importantly, differing from the statin-mediated indiscriminate inhibition of HMGCR in normal and tumor cells, the differential responses of HMGCR to isoprenoid-mediated downregulation render tumor cells more sensitive to isoprenoid-induced growth suppression (Mo et al., 2013). Non-tumor cells including aortic epithelial cells (Adany et al., 1994), fibroblast (Yazlovitskaya and Melnykovych, 1995; Ura et al., 1998; Mo and Elson, 1999; Smalley and Eisen, 2002; Yano et al., 2005), primary hemopoietic cells (Rioja et al., 2000), hepatocytes (Pearce et al., 1992; Ruch and Sigler, 1994; Sakai et al., 2004; Har and Keong, 2005; Yin et al., 2012), mammary epithelial cells (McIntyre et al., 2000; Duncan et al., 2004b; Shun et al., 2004; Yap et al., 2010; Liu et al., 2011; Patel and Thakkar, 2014), myeloid cells (Sahin et al., 1999), pancreatic ductal epithelial cells (Stayrook et al., 1997), prostate cells (Adany et al., 1994; Srivastava and Gupta, 2006), and umbilical vein endothelial cells (HUVEC) (Yoshikawa et al., 2010), are much less susceptible to the isoprenoid-mediated growth inhibition, suggesting that isoprenoids exert tumor-targeted growth inhibitory effects (Elson et al., 1999; Mo and Elson, 2004). Isoprenoid-rich extracts such as tocotrienol-rich fraction (TRF) of palm oil selectively and significantly inhibit growth and induce apoptosis of human prostate cancer LNCaP, DU145, and PC-3 cells with no significant impact on the viability of normal human prostate epithelial cells (Srivastava and Gupta, 2006). Similarly, tocotrienol stimulate growth inhibition, caspase activity, DNA fragmentation, and apoptosis in rat hepatoma dRLh-84 cells without affecting normal RLN-10 hepatocytes (Sakai et al., 2004). Tocotrienols also induce apoptosis of estrogen-non-responsive MDA-MB-435 and estrogen-responsive MCF-7 human breast cancer cells with no or lower levels of impact in normal human mammary epithelial cells and immortalized but non-tumorigenic human MCF-10A cells (McIntyre et al., 2000; Shun et al., 2004; Yap et al., 2010). As summarized in Table 2, isoprenoids such as monoterpenes (carvacrol, L-carvone, geraniol, perillyl alcohol), sesquiterpenes (cacalol, farnesol, β-ionone), diterpene (geranylgeranyl acetone), and “mixed” isoprenoids (tocotrienols) and their derivatives exert tumor-targeted growth inhibition with no-to-minimal impacts on the viability and morphology of normal healthy cells.
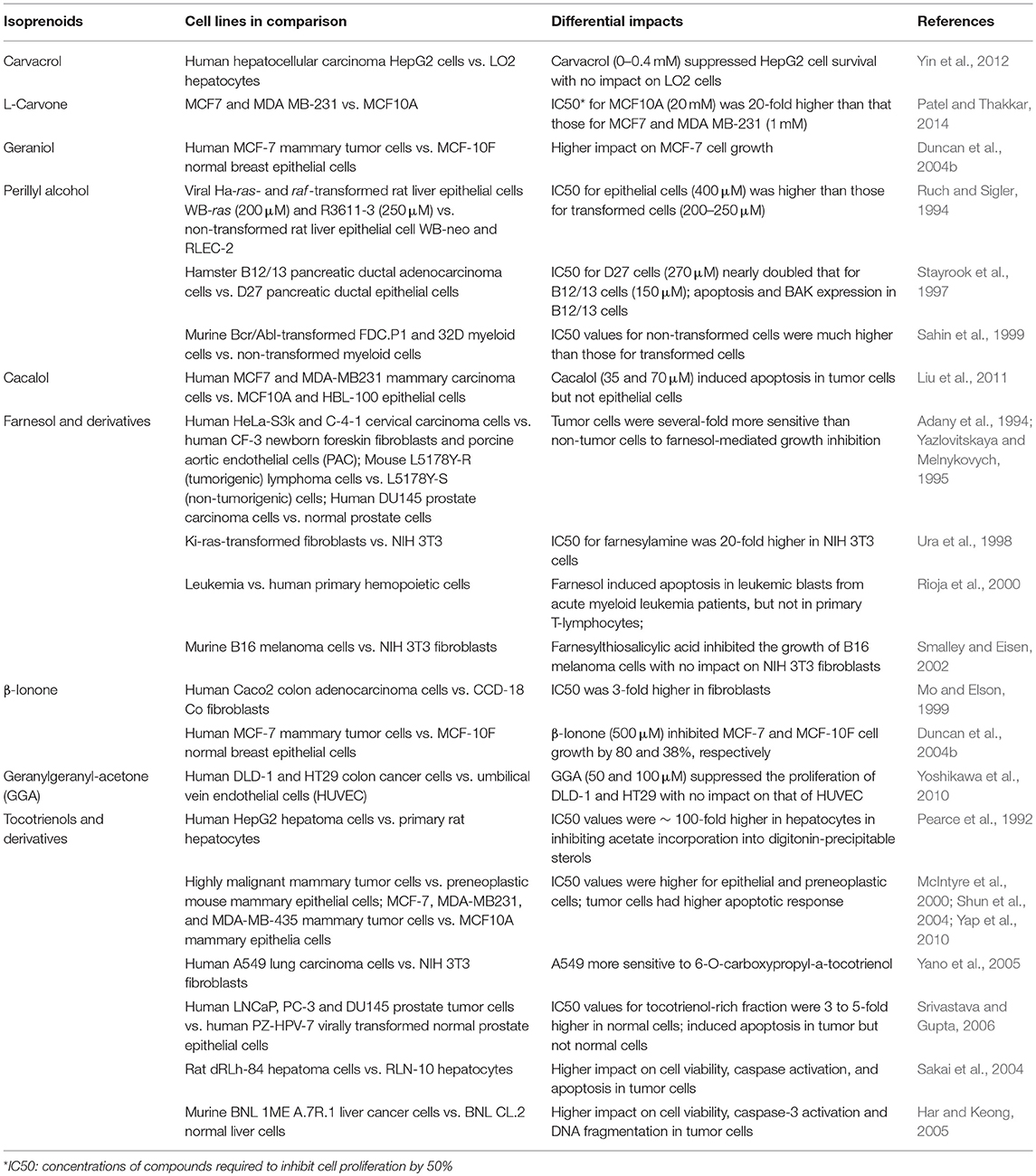
Table 2. Differential sensitivities of tumor and non-tumor cells to isoprenoids-mediated HMGCR downregulation, growth suppression and apoptosis.
The isoprenoid-mediated degradation of HMGCR complements the sterol-mediated transcriptional regulation in normal cells. In tumor cells with HMGCR that is resistant to sterol feedback, post-transcriptional downregulation of HMGCR by isoprenoids leads to suppression of cell proliferation and induction of apoptosis; concomitantly isoprenoids have minimal impact on normal cells. This unique tumor-targeting property of isoprenoids affords them potential in adjuvant therapy with statins.
Synergistic Effect of Statins and Isoprenoids on Tumor Growth may Reduce Statin Toxicity
Since long-term use of a single compound requires a relatively high dose, which tends to be associated with adverse effects, synergistic effects of therapeutic compounds can provide the same or enhanced efficacy with lower doses of each agent and plausibly fewer adverse effects to lesser degrees. Investigations with blends of isoprenoids have shown favorable outcomes. Studies using tumor cells of skin (Mo and Elson, 1999; McAnally et al., 2007), prostate (Mo and Elson, 2004), and pancreas (Hussein and Mo, 2009) have delineated the synergistic or cumulative effects of HMGCR suppressors on mevalonate signaling and tumor cell growth. Geranylgeraniol and d-δ-tocotrienol synergistically suppressed growth of the murine b16 melanoma cells (Katuru et al., 2011). Dietary γ-tocotrienol (2 mmol/kg) and β-ionone (2 mmol/kg) administered individually or together also improved survival in mice bearing implanted melanomas (He et al., 1997).
Isoprenoids have also shown synergistic effect with statins (Table 3). Figure 2 lists the structures of representative isoprenoids that potentiate the anti-cancer efficacy of statins in vitro and in vivo. All three two-way blends of tocotrienols, lovastatin, and β-ionone synergistically suppressed the proliferation of murine B16 melanoma and human A549 lung carcinoma, A2058 melanoma, MIA PaCa-2 pancreatic carcinoma, and DU145 prostate carcinoma cells (He et al., 1997; Mo and Elson, 1999; McAnally et al., 2007; Fernandes et al., 2010; Katuru et al., 2011). In human MCF-7 mammary tumor cells resistant to doxorubicin and tamoxifen, simvastatin and γ-tocotrienol synergistically eliminated cancer stem-like cells and reduced mammosphere formation and pStat-3 signaling; mevalonate supplementation attenuated the effect of the blend (Gopalan et al., 2013), suggesting that mevalonate depletion mediates the effect. Treatment of HepG2 hepatoma cells with a combination of 5 μM simvastatin and 50 μM geraniol, doses that cannot inhibit proliferation individually, significantly trapped tumor proliferation and inhibited cholesterol biosynthesis (Polo et al., 2011). Co-administration of pravastatin, a HMGCR inhibitor, and d-limonene, an inhibitor of protein prenylation, inhibited the growth of human HepG2 hepatoma cells by blocking post-translational modulation of p21ras rather than suppression of cholesterol and dolichol biosynthesis (Kawata et al., 1994). An additive effect on cell growth and protein prenylation was shown with d-limonene and lovastatin in murine CT26 colon tumor cells (Broitman et al., 1996). Farnesyl-O-acetylhydroquinone, a farnesyl derivative, also synergized with lovastatin to suppress the proliferation and cell cycle progression of B16 melanoma cells (McAnally et al., 2003). Blends of statins and tocotrienols synergistically impacted an array of signaling molecules regulating cell cycle, cell proliferation, and apoptosis, including cyclin D1, CDK2, p27, MAPK, ERK, AKT, c-Jun N-terminal kinase (JNK), Rab, Rap1A, RhoA, caspase-3, and Ki-67, in human MCF-7 and MDA-MB-231 mammary tumor cells, + SA highly malignant mammary epithelial cells (Wali and Sylvester, 2007; Wali et al., 2009a,b; Ali et al., 2010; Alayoubi et al., 2012), MSTO, H2452, H2052, and H28 malignant mesothelioma cells (Tuerdi et al., 2013), and HT29 and HCT116 colon cancer cells (Yang et al., 2010). Blends of dietary lovastatin and d-δ-tocotrienol, but not individual agents, reduced the growth of implanted B16 melanoma in C57BL6 mice (McAnally et al., 2007).
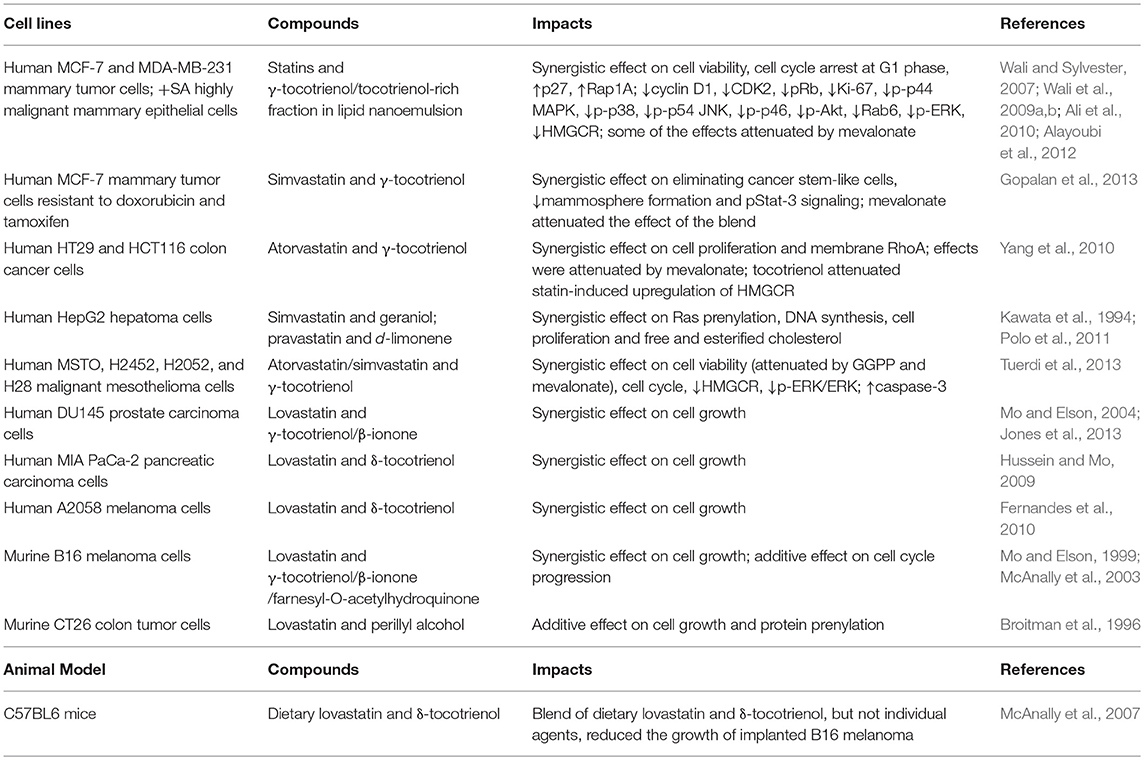
Table 3. In vitro and in vivo studies showing that isoprenoids potentiate the tumor-suppressive effect of statins.
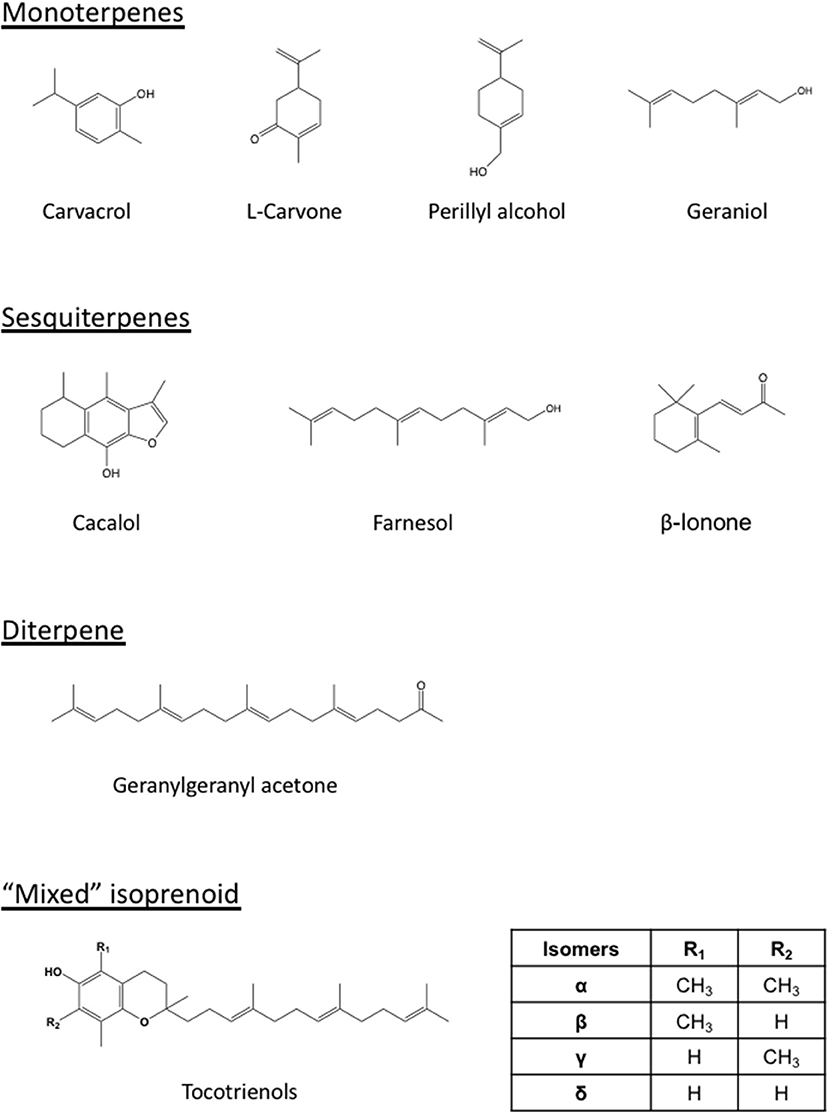
Figure 2. Structures of representative isoprenoids that impact tumors or potentiate the statin-mediated tumor suppression. Differing from the monoterpenes (carvacrol, carvone, perillyl alcohol, geraniol), sesquiterpenes (cacalol, farnesol, β-ionone), and diterpene (geranylgeranyl acetone), the tocotrienols are “mixed” isoprenoids with only part of their structure derived from the mevalonate pathway. The number and location of methyl group (R1 and R2) on the chromanol ring vary among the members (α, β, γ, and δ) of tocotrienols.
The effect of tocotrienols on cell viability correlates with that on SREBP-2 activity, consistent with the finding that downregulation of SREBP2 and subsequent lipid biosynthesis effectively suppresses the growth of colon cancer proliferation and tumor spheroid formation (Wen et al., 2018). SREBP-2 and HMGCR were overexpressed in cisplatin-resistant A2780 epithelial ovarian cancer cells (Zheng et al., 2018). SREBP-2 promotes the expression of Staphylococcal nuclease and tudor domain containing 1 gene that is associated with the progression and malignancy of colon, breast, prostate, lung, glioma, skin, and liver cancers (Armengol et al., 2017), providing potential new applications for tocotrienols in cisplatin-resistant cancers. Mevalonate rescues the effect of statin, but not that of tocotrienol, on prostate cancer cell viability, consistent with tocotrienol-mediated increase in the degradation of nuclear, mature SREBP-2, which maintains cholesterol homeostasis through transcriptional regulation of enzymes in cholesterol biosynthesis and uptake beyond HMGCR (Krycer et al., 2012). The tocotrienol-enhanced degradation of SREBP-2 suggests that tocotrienol may impact, in addition to HMGCR, multiple enzymes of the mevalonate pathway, offering another aspect of synergy with statins, which specifically inhibit HMGCR. Such suitable and optimal combinations of statins and tocotrienol could lead to favorable synergistic effects with lower effective doses of statins with minimized or no side effects. The pair's combined lipid-lowering effects have been shown in chickens (Qureshi and Peterson, 2001) and hypercholesterolemic humans (Qureshi et al., 2001).
Conclusion and Future Directions
The widely prescribed statins, originally designed for hypercholesterolemia, hold promises for cancer therapy as the multiple roles of the mevalonate pathway in growth support and regulation unfold. The repurposing of this class of well-established drugs for cancer stems from their ability to deplete mevalonate-derived sterol and nonsterol products essential for the prenylation of proteins involved in growth regulation and signaling pathways for cell proliferation. Nevertheless, the indiscriminate inhibition of HMGCR in normal and tumor cells and compensatory upregulation of HMGCR induced by statins may have contributed to their dose-limiting toxicities and conflicting clinical outcomes. The upregulated and sterol-resistant tumor HMGCR remains responsive to the isoprenoid-mediated downregulation, offering isoprenoids tumor-targeted growth suppression with little toxicity to normal cells. Isoprenoids also attenuate statin-induced compensatory upregulation of HMGCR via augmented, proteasome-mediated degradation of HMGCR, providing a new approach in enhancing the efficacy of statins with reduced adverse effects associated with lower statin doses. Most studies on the combination of isoprenoids and statins, however, are limited to in vitro models. A major gap hence remains in the lack of clinical data on the bioavailability, pharmacokinetics, pharmacodynamics, efficacy and mechanisms of action of the isoprenoids. The activities of the vast majority of the estimated 22,000 isoprenoids (Bach, 1995) remain unexplored. New formulation including those employing nanoparticles may improve the bioavailability of isoprenoids. Isoprenoid derivatives (Jung et al., 1998; Mo et al., 2000) with improved potencies and lower toxicities may provide more opportunities for optimal formulations. The combinations of isoprenoids and statins with complementary mechanisms and synergistic actions hold promise for cancer prevention and therapy and warrant further clinical studies.
Author Contributions
HM, C-LS, and HY conceptualized the review. HM, RJ, AB, SY, and HY performed literature search. HM and SY constructed the figures. All authors participated in the writing and final editing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AMPK, adenosine monophosphate-activated protein kinase; CC-RCC, clear cell renal cell carcinoma; CDK2, cyclin dependent kinase 2; CHIP, Hsc70-interacting protein; DMBA, 7, 12-Dimethylbenz[a]anthracene; DNAJA1, DNAJ heat shock protein family (Hsp40) member A1; ERK, extracellular signal-regulated kinase; FPP, farnesol pyrophosphate; GGA, geranylgeranyl acetone; GGPP, geranylgeranyl pyrophosphate; Gp78, glycoprotein 78; HIF1α, hypoxia inducible factor 1α; HMG CoA, 3-hydroxy-3-methylglutaryl coenzyme A; HMGCR, 3-hydroxy-3-methylglutaryl coenzyme A reductase; HUVEC, umbilical vein endothelial cell; Insig, insulin-induced gene; JAK2, Janus Kinase 2; JNK, c-Jun N-terminal kinase; LDL, low density lipoprotein; MAPK, mitogen-activated protein kinase; MEP/DOXP, 2-C-methyl-D-erythritol 4-phosphate/1-deoxy-D-xylulose 5-phosphate; MNU, N-methyl-N-nitrosourea; mTOR, mammalian target of rapamycin; NFκB, nuclear factor kappa B; PI3K, phosphatidylinositol-4, 5-bisphosphate 3-kinase; SCAP, sterol regulatory element binding proteins cleavage protein; SRE, sterol regulatory element; SREBPs, sterol regulatory element binding proteins; STAT3, Signal transducer and activator of transcription 3; STOMP, Effect of Statins on Skeletal Muscle Function and Performance; TAZ, PDZ binding motif; TNM, Tumor, Node, Metastasis; TRF, tocotrienol-rich fraction; Ubc7, Ubiquitin-conjugating enzyme E2 7; UBIAD1, UbiA prenyltransferase domain-containing-protein-1; Ubiquinone, coenzyme Q10; VCP, valosin containing protein; VHL, Von Hippel-Lindau; YAP, yes-associated protein.
References
Abate, M., Laezza, C., Pisanti, S., Torelli, G., Seneca, V., Catapano, G., et al. (2017). Deregulated expression and activity of Farnesyl Diphosphate Synthase (FDPS) in Glioblastoma. Sci. Rep. 7:14123. doi: 10.1038/s41598-017-14495-6
Abdullah, M. I., Abed, M. N., and Richardson, A. (2017). Inhibition of the mevalonate pathway augments the activity of pitavastatin against ovarian cancer cells. Sci. Rep. 7:8090. doi: 10.1038/s41598-017-08649-9
Adany, I., Yazlovitskaya, E. M., Haug, J. S., Voziyan, P. A., and Melnykovych, G. (1994). Differences in sensitivity to farnesol toxicity between neoplastically- and non-neoplastically-derived cells in culture. Cancer Lett. 79, 175–179. doi: 10.1016/0304-3835(94)90257-7
Ahmadi, Y., Ghorbanihaghjo, A., and Argani, H. (2017). The balance between induction and inhibition of mevalonate pathway regulates cancer suppression by statins: a review of molecular mechanisms. Chem. Biol. Interact. 273, 273–285. doi: 10.1016/j.cbi.2017.06.026
Alayoubi, A. Y., Anderson, J. F., Satyanarayanajois, S. D., Sylvester, P. W., and Nazzal, S. (2012). Concurrent delivery of tocotrienols and simvastatin by lipid nanoemulsions potentiates their antitumor activity against human mammary adenocarcenoma cells. Eur. J. Pharm. Sci. 48, 385–392. doi: 10.1016/j.ejps.2012.12.011
Al-Haidari, A. A., Syk, I., and Thorlacius, H. (2014). HMG-CoA reductase regulates CCL17-induced colon cancer cell migration via geranylgeranylation and RhoA activation. Biochem. Biophys. Res. Commun. 446, 68–72. doi: 10.1016/j.bbrc.2014.02.078
Ali, H., Shirode, A. B., Sylvester, P. W., and Nazzal, S. (2010). Preparation, characterization, and anticancer effects of simvastatin-tocotrienol lipid nanoparticles. Int. J. Pharm. 389, 223–231. doi: 10.1016/j.ijpharm.2010.01.018
Allott, E. H., Farnan, L., Steck, S. E., Arab, L., Su, L. J., Mishel, M., et al. (2016). Statin use and prostate cancer aggressiveness: results from the population-based North Carolina-Louisiana Prostate Cancer Project. Cancer Epidemiol. Biomarkers Prev. 25, 670–677. doi: 10.1158/1055-9965.EPI-15-0631
Allott, E. H., Farnan, L., Steck, S. E., Song, L. X., Arab, L., Su, L. J., et al. (2018). Statin use, high cholesterol and prostate cancer progression; results from HCaP-NC. Prostate 78, 857–864. doi: 10.1002/pros.23644
Armengol, S., Arretxe, E., Enzunza, L., Llorente, I., Mendibil, U., Navarro-Imaz, H., et al. (2017). SREBP-2-driven transcriptional activation of human SND1 oncogene. Oncotarget 8, 108181–108194. doi: 10.18632/oncotarget.22569
Azrolan, N. I., and Coleman, P. S. (1989). A discoordinate increase in the cellular amount of 3-hydroxy-3-methylglutaryl-CoA reductase results in the loss of rate-limiting control over cholesterogenesis in a tumour cell-free system. Biochem. J. 258, 421–425. doi: 10.1042/bj2580421
Bach, T. J. (1995). Some new aspects of isoprenoid biosynthesis in plants–a review. Lipids 30, 191–202. doi: 10.1007/BF02537822
Beck, P., Wysowski, D. K., Downey, W., and Butler-Jones, D. (2003). Statin use and the risk of breast cancer. J. Clin. Epidemiol. 56, 280–285. doi: 10.1016/S0895-4356(02)00614-5
Beckwitt, C. H., Shiraha, K., and Wells, A. (2018). Lipophilic statins limit cancer cell growth and survival, via involvement of Akt signaling. PLoS ONE 13:e0197422. doi: 10.1371/journal.pone.0197422
Beg, Z. H., Allmann, D. W., and Gibson, D. M. (1973). Modulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity with cAMP and wth protein fractions of rat liver cytosol. Biochem. Biophys. Res. Commun. 54, 1362–1369. doi: 10.1016/0006-291X(73)91137-6
Beg, Z. H., Stonik, J. A., and Brewer, H. B. Jr. (1978). 3-Hydroxy-3-methylglutaryl coenzyme A reductase: regulation of enzymatic activity by phosphorylation and dephosphorylation. Proc. Natl. Acad. Sci. U.S.A. 75, 3678–3682. doi: 10.1073/pnas.75.8.3678
Bennis, F., Favre, G., Le Gaillard, F., and Soula, G. (1993). Importance of mevalonate-derived products in the control of HMG-CoA reductase activity and growth of human lung adenocarcinoma cell line A549. Int. J. Cancer 55, 640–645. doi: 10.1002/ijc.2910550421
Bensch, W. R., Ingebritsen, T. S., and Diller, E. R. (1978). Lack of correlation between the rate of cholesterol biosynthesis and the activity of 3-hydroxy-3-methylgutaryl coenzyme A reductase in rats and in fibroblasts treated with ML-236B. Biochem. Biophys. Res. Commun. 82, 247–254. doi: 10.1016/0006-291X(78)90602-2
Bradfute, D. L., and Simoni, R. D. (1994). Non-sterol compounds that regulate cholesterogenesis. Analogues of farnesyl pyrophosphate reduce 3-hydroxy-3-methylglutaryl-coenzyme A reductase levels. J. Biol. Chem. 269, 6645–6650.
Broitman, S. A., Wilkinson, J. T., Cerda, S., and Branch, S. K. (1996). Effects of monoterpenes and mevinolin on murine colon tumor CT-26 in vitro and its hepatic metastases in vivo. Adv. Exp. Med. Biol. 401, 111–130. doi: 10.1007/978-1-4613-0399-2_9
Brown, A. J. (2007). Cholesterol, statins and cancer. Clin. Exp. Pharmacol. Physiol. 34, 135–141. doi: 10.1111/j.1440-1681.2007.04565.x
Brown, M. S., and Goldstein, J. L. (1997). The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340. doi: 10.1016/S0092-8674(00)80213-5
Brown, M. S., and Goldstein, J. L. (1999). A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. U.S.A. 96, 11041–11048. doi: 10.1073/pnas.96.20.11041
Bruckert, E., Hayem, G., Dejager, S., Yau, C., and Begaud, B. (2005). Mild to moderate muscular symptoms with high-dosage statin therapy in hyperlipidemic patients–the PRIMO study. Cardiovasc. Drugs Ther. 19, 403–414. doi: 10.1007/s10557-005-5686-z
Buhaescu, I., and Izzedine, H. (2007). Mevalonate pathway: a review of clinical and therapeutical implications. Clin. Biochem. 40, 575–584. doi: 10.1016/j.clinbiochem.2007.03.016
Caruso, M. G., and Notarnicola, M. (2005). Biochemical changes of mevalonate pathway in human colorectal cancer. Anticancer Res. 25, 3393–3397.
Caruso, M. G., Notarnicola, M., Cavallini, A., and Di Leo, A. (2002). 3-Hydroxy-3-methylglutaryl coenzyme A reductase activity and low-density lipoprotein receptor expression in diffuse-type and intestinal-type human gastric cancer. J. Gastroenterol. 37, 504–508. doi: 10.1007/s005350200078
Celis, J. E., Gromov, P., Moreira, J. M., Cabezon, T., Friis, E., Vejborg, I. M., et al. (2006). Apocrine cysts of the breast: biomarkers, origin, enlargement, and relation with cancer phenotype. Mol. Cell. Proteomics 5, 462–483. doi: 10.1074/mcp.M500348-MCP200
Cerda, S. R., Wilkinson, J. T., and Broitman, S. A. (1995). Regulation of cholesterol synthesis in four colonic adenocarcinoma cell lines. Lipids 30, 1083–1092. doi: 10.1007/BF02536608
Chang, P. N., Yap, W. N., Lee, D. T., Ling, M. T., Wong, Y. C., and Yap, Y. L. (2009). Evidence of γ-tocotrienol as an apoptosis-inducing, invasion-suppressing, and chemotherapy drug-sensitizing agent in human melanoma cells. Nutr. Cancer 61, 357–366. doi: 10.1080/01635580802567166
Chaudhary, S. C., Siddiqui, M. S., Athar, M., and Alam, M. S. (2013). Geraniol inhibits murine skin tumorigenesis by modulating COX-2 expression, Ras-ERK1/2 signaling pathway and apoptosis. J. Appl. Toxicol. 33, 828–837. doi: 10.1002/jat.2739
Chen, C. C., Liu, T. Y., Huang, S. P., Ho, C. T., and Huang, T. C. (2015). Differentiation and apoptosis induction by lovastatin and γ-tocotrienol in HL-60 cells via Ras/ERK/NF-kappaB and Ras/Akt/NF-kappaB signaling dependent down-regulation of glyoxalase 1 and HMG-CoA reductase. Cell. Signal. 27, 2182–2190. doi: 10.1016/j.cellsig.2015.07.014
Chen, M. C., Tsai, Y. C., Tseng, J. H., Liou, J. J., Horng, S., Wen, H. C., et al. (2017). Simvastatin inhibits cell proliferation and migration in human anaplastic thyroid cancer. Int. J. Mol. Sci. 18:2690. doi: 10.3390/ijms18122690
Chen, Y., and Hughes-Fulford, M. (2001). Human prostate cancer cells lack feedback regulation of low-density lipoprotein receptor and its regulator, SREBP2. Int. J. Cancer 91, 41–45. doi: 10.1002/1097-0215(20010101)91:1<41::AID-IJC1009>3.0.CO;2-2
Cho, S. Y., Jun, H. J., Lee, J. H., Jia, Y., Kim, K. H., and Lee, S. J. (2011). Linalool reduces the expression of 3-hydroxy-3-methylglutaryl CoA reductase via sterol regulatory element binding protein-2- and ubiquitin-dependent mechanisms. FEBS Lett. 585, 3289–3296. doi: 10.1016/j.febslet.2011.09.012
Clendening, J. W., Pandyra, A., Boutros, P. C., El Ghamrasni, S., Khosravi, F., Trentin, G. A., et al. (2010). Dysregulation of the mevalonate pathway promotes transformation. Proc. Natl. Acad. Sci. U.S.A. 107, 15051–15056. doi: 10.1073/pnas.0910258107
Clendening, J. W., and Penn, L. Z. (2012). Targeting tumor cell metabolism with statins. Oncogene 31, 4967–4978. doi: 10.1038/onc.2012.6
Coni, P., Pang, J., Pichiri-Coni, G., Hsu, S., Rao, P. M., Rajalakshmi, S., et al. (1992). Hypomethylation of β-hydroxy-β-methyl-glutaryl coenzyme A reductase gene and its expression during hepatocarcinogenesis in the rat. Carcinogenesis 13, 497–499. doi: 10.1093/carcin/13.3.497
Coogan, P. F., Rosenberg, L., Palmer, J. R., Strom, B. L., Zauber, A. G., and Shapiro, S. (2002). Statin use and the risk of breast and prostate cancer. Epidemiology 13, 262–267. doi: 10.1097/00001648-200205000-00005
Crespo, R., Montero Villegas, S., Abba, M. C., de Bravo, M. G., and Polo, M. P. (2013). Transcriptional and posttranscriptional inhibition of HMGCR and PC biosynthesis by geraniol in 2 Hep-G2 cell proliferation linked pathways. Biochem. Cell Biol. 91, 131–139. doi: 10.1139/bcb-2012-0076
de Wolf, E., Abdullah, M. I., Jones, S. M., Menezes, K., Moss, D. M., Drijfhout, F. P., et al. (2017). Dietary geranylgeraniol can limit the activity of pitavastatin as a potential treatment for drug-resistant ovarian cancer. Sci. Rep. 7:5410. doi: 10.1038/s41598-017-05595-4
DeBose-Boyd, R. A. (2008). Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res. 18, 609–621. doi: 10.1038/cr.2008.61
Dimitroulakos, J., Ye, L. Y., Benzaquen, M., Moore, M. J., Kamel-Reid, S., Freedman, M. H., Yeger, H., and Penn, L. Z. (2001). Differential sensitivity of various pediatric cancers and squamous cell carcinomas to lovastatin-induced apoptosis: therapeutic implications. Clin. Cancer Res. 7, 158–167.
Ding, Y., Peng, Y., Deng, L., Fan, J., and Huang, B. (2017). Gamma-tocotrienol reverses multidrug resistance of breast cancer cells with a mechanism distinct from that of atorvastatin. J. Steroid Biochem. Mol. Biol. 167, 67–77. doi: 10.1016/j.jsbmb.2016.11.009
Dong, H. W., Zhang, S., Sun, W. G., Liu, Q., Ibla, J. C., Soriano, S. G., et al. (2013). beta-Ionone arrests cell cycle of gastric carcinoma cancer cells by a MAPK pathway. Arch. Toxicol. 87, 1797–1808. doi: 10.1007/s00204-013-1041-5
Duncan, R. E., El-Sohemy, A., and Archer, M. C. (2004a). Mevalonate promotes the growth of tumors derived from human cancer cells in vivo and stimulates proliferation in vitro with enhanced cyclin-dependent kinase-2 activity. J. Biol. Chem. 279, 33079–33084. doi: 10.1074/jbc.M400732200
Duncan, R. E., El-Sohemy, A., and Archer, M. C. (2005). Statins and cancer development. Cancer Epidemiol. Biomarkers Prev. 14, 1897–1898. doi: 10.1158/1055-9965.EPI-05-0027
Duncan, R. E., Lau, D., El-Sohemy, A., and Archer, M. C. (2004b). Geraniol and β-ionone inhibit proliferation, cell cycle progression, and cyclin-dependent kinase 2 activity in MCF-7 breast cancer cells independent of effects on HMG-CoA reductase activity. Biochem. Pharmacol. 68, 1739–1747. doi: 10.1016/j.bcp.2004.06.022
Duvel, K., Yecies, J. L., Menon, S., Raman, P., Lipovsky, A. I., Souza, A. L., et al. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171–183. doi: 10.1016/j.molcel.2010.06.022
Eaton, S. (2008). Multiple roles for lipids in the Hedgehog signalling pathway. Nat. Rev. Mol. Cell Biol. 9, 437–445. doi: 10.1038/nrm2414
El-Sohemy, A., and Archer, M. C. (2000). Inhibition of N-methyl-N-nitrosourea- and 7,12-dimethylbenz[a] anthracene-induced rat mammary tumorigenesis by dietary cholesterol is independent of Ha-Ras mutations. Carcinogenesis 21, 827–831. doi: 10.1093/carcin/21.4.827
Elson, C. E., Peffley, D. M., Hentosh, P., and Mo, H. (1999). Isoprenoid-mediated inhibition of mevalonate synthesis: potential application to cancer. Proc. Soc. Exp. Biol. Med. 221, 294–311. doi: 10.3181/00379727-221-44413
Engstrom, W., and Schofield, P. N. (1987). Expression of the 3-hydroxy-3-methylglutaryl coenzyme A-reductase and LDL-receptor genes in human embryonic tumours and in normal foetal tissues. Anticancer Res. 7, 337–342.
Erickson, S. K., Cooper, A. D., Barnard, G. F., Havel, C. M., Watson, J. A., Feingold, K. R., et al. (1988). Regulation of cholesterol metabolism in a slow-growing hepatoma in vivo. Biochim. Biophys. Acta 960, 131–138. doi: 10.1016/0005-2760(88)90058-6
Espenshade, P. J., and Hughes, A. L. (2007). Regulation of sterol synthesis in eukaryotes. Ann. Rev. Genet. 41, 401–427. doi: 10.1146/annurev.genet.41.110306.130315
Ettinger, S. L., Sobel, R., Whitmore, T. G., Akbari, M., Bradley, D. R., Gleave, M. E., et al. (2004). Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res. 64, 2212–2221. doi: 10.1158/0008-5472.CAN-2148-2
Feingold, K. R., Wiley, M. H., Moser, A. H., and Siperstein, M. D. (1983). Altered activation state of hydroxymethylglutaryl-coenzyme A reductase in liver tumors. Arch. Biochem. Biophys. 226, 231–241. doi: 10.1016/0003-9861(83)90289-8
Fernandes, N. V., Guntipalli, P. K., and Mo, H. (2010). d-δ-Tocotrienol-mediated cell cycle arrest and apoptosis in human melanoma cells. Anticancer Res. 30, 4937–4944.
Fernandes, N. V., Yeganehjoo, H., Katuru, R., DeBose-Boyd, R., Morris, L. L., Michon, R., et al. (2013). Geranylgeraniol suppresses the viability of human DU145 prostate carcinoma cells and the level of HMG CoA reductase. Exp. Biol. Med. 238, 1265–1274. doi: 10.1177/1535370213492693
Freed-Pastor, W., and Prives, C. (2016). Targeting mutant p53 through the mevalonate pathway. Nat. Cell Biol. 18, 1122–1124. doi: 10.1038/ncb3435
Freed-Pastor, W. A., Mizuno, H., Zhao, X., Langerød, A., Moon, S.-H., Rodriguez-Barrueco, R., et al. (2012). Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148, 244–258. doi: 10.1016/j.cell.2011.12.017
Galle, M., Crespo, R., Kladniew, B. R., Villegas, S. M., Polo, M., and de Bravo, M. G. (2014). Suppression by geraniol of the growth of A549 human lung adenocarcinoma cells and inhibition of the mevalonate pathway in culture and in vivo: potential use in cancer chemotherapy. Nutr. Cancer 66, 888–895. doi: 10.1080/01635581.2014.916320
Galle, M., Kladniew, B. R., Castro, M. A., Villegas, S. M., Lacunza, E., Polo, M., et al. (2015). Modulation by geraniol of gene expression involved in lipid metabolism leading to a reduction of serum-cholesterol and triglyceride levels. Phytomedicine 22, 696–704. doi: 10.1016/j.phymed.2015.04.005
Garza, R. M., Tran, P. N., and Hampton, R. Y. (2009). Geranylgeranyl pyrophosphate is a potent regulator of HRD-dependent 3-hydroxy-3-methylglutaryl-CoA reductase degradation in yeast. J. Biol. Chem. 284, 35368–353680. doi: 10.1074/jbc.M109.023994
Gbelcova, H., Rimpelova, S., Knejzlik, Z., Sachova, J., Kolar, M., Strnad, H., et al. (2017). Isoprenoids responsible for protein prenylation modulate the biological effects of statins on pancreatic cancer cells. Lipids Health Dis. 16:250. doi: 10.1186/s12944-017-0641-0
Gil, G., Smith, J. R., Goldstein, J. L., Slaughter, C. A., Orth, K., Brown, M. S., et al. (1988). Multiple genes encode nuclear factor 1-like proteins that bind to the promoter for 3-hydroxy-3-methylglutaryl-coenzyme A reductase. Proc. Natl. Acad. Sci. U.S.A. 85, 8963–8967. doi: 10.1073/pnas.85.23.8963
Ginestier, C., Monville, F., Wicinski, J., Cabaud, O., Cervera, N., Josselin, E., et al. (2012). Mevalonate metabolism regulates basal breast cancer stem cells and is a potential therapeutic target. Stem Cells 30, 1327–1337. doi: 10.1002/stem.1122
Girgert, R., Vogt, Y., Becke, D., Bruchelt, G., and Schweizer, P. (1999). Growth inhibition of neuroblastoma cells by lovastatin and L-ascorbic acid is based on different mechanisms. Cancer Lett. 137, 167–172. doi: 10.1016/S0304-3835(98)00355-3
Goldstein, J. L., and Brown, M. S. (1990). Regulation of the mevalonate pathway. Nature 343, 425–430. doi: 10.1038/343425a0
Goldstein, J. L., DeBose-Boyd, R. A., and Brown, M. S. (2006). Protein sensors for membrane sterols. Cell 124, 35–46. doi: 10.1016/j.cell.2005.12.022
Gomide, M., Lemos, F., Reis, D., José, G., Lopes, M., Machado, M. A., et al. (2016). Identification of dysregulated microRNA expression and their potential role in the antiproliferative effect of the essential oils from four different Lippia species against the CT26.WT colon tumor cell line. Revista Brasileira Farmacognosia 26, 627–633. doi: 10.1016/j.bjp.2016.04.003
Gomide, M.d. S., Lemos, F. d. O., Lopes, M. T. P., Alves, T. M. d. A., Viccini, L. F., and Coelho, C. M. (2013). The effect of the essential oils from five different Lippia species on the viability of tumor cell lines. Revista Brasileira Farmacognosia 23, 895–902. doi: 10.1590/S0102-695X2013000600006
Gong, J., Sachdev, E., Robbins, L. A., Lin, E., Hendifar, A. E., and Mita, M. M. (2017). Statins and pancreatic cancer. Oncol. Lett. 13, 1035–1040. doi: 10.3892/ol.2017.5572
Gong, Y., Lee, J. N., Lee, P. C., Goldstein, J. L., Brown, M. S., and Ye, J. (2006). Sterol-regulated ubiquitination and degradation of Insig-1 creates a convergent mechanism for feedback control of cholesterol synthesis and uptake. Cell Metab. 3, 15–24. doi: 10.1016/j.cmet.2005.11.014
Gopalan, A., Yu, W., Sanders, B. G., and Kline, K. (2013). Eliminating drug resistant breast cancer stem-like cells with combination of simvastatin and gamma-tocotrienol. Cancer Lett. 328, 285–296. doi: 10.1016/j.canlet.2012.10.003
Gregg, R. G., Davidson, M., and Wilce, P. A. (1986). Cholesterol synthesis and HMG CoA reductase activity during hepatocarcinogenesis in rats. Int. J. Biochem. 18, 389–393. doi: 10.1016/0020-711X(86)90046-7
Hamada, T., Khalaf, N., Yuan, C., Babic, A., Morales-Oyarvide, V., Qian, Z. R., et al. (2018). Statin use and pancreatic cancer risk in two prospective cohort studies. J. Gastroenterol. 53, 959–966. doi: 10.1007/s00535-018-1430-x
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi: 10.1016/j.cell.2011.02.013
Har, C. H., and Keong, C. K. (2005). Effects of tocotrienols on cell viability and apoptosis in normal murine liver cells (BNL CL.2) and liver cancer cells (BNL 1ME A.7R.1), in vitro. Asia Pac. J. Clin. Nutr. 14, 374–380.
Harwood, H. J. Jr., Alvarez, I. M., Noyes, W. D., and Stacpoole, P. W. (1991). In vivo regulation of human leukocyte 3-hydroxy-3-methylglutaryl coenzyme A reductase: increased enzyme protein concentration and catalytic efficiency in human leukemia and lymphoma. J. Lipid Res. 32, 1237–1252.
He, L., Mo, H., Hadisusilo, S., Qureshi, A. A., and Elson, C. E. (1997). Isoprenoids suppress the growth of murine B16 melanomas in vitro and in vivo. J. Nutr. 127, 668–674. doi: 10.1093/jn/127.5.668
Hentosh, P., Yuh, S. H., Elson, C. E., and Peffley, D. M. (2001). Sterol-independent regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase in tumor cells. Mol. Carcinog. 32, 154–166. doi: 10.1002/mc.1074
Hentschel, A., Zahedi, R. P., and Ahrends, R. (2016). Protein lipid modifications–More than just a greasy ballast. Proteomics 16, 759–782. doi: 10.1002/pmic.201500353
Hindler, K., Cleeland, C. S., Rivera, E., and Collard, C. D. (2006). The role of statins in cancer therapy. Oncologist 11, 306–315. doi: 10.1634/theoncologist.11-3-306
Hoffmeister, M., Jansen, L., Rudolph, A., Toth, C., Kloor, M., Roth, W., et al. (2015). Statin use and survival after colorectal cancer: the importance of comprehensive confounder adjustment. J. Natl. Cancer Inst. 107:djv045. doi: 10.1093/jnci/djv045
Horton, J. D., Goldstein, J. L., and Brown, M. S. (2002). SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131. doi: 10.1172/JCI0215593
Houten, S. M., Schneiders, M. S., Wanders, R. J., and Waterham, H. R. (2003). Regulation of isoprenoid/cholesterol biosynthesis in cells from mevalonate kinase-deficient patients. J. Biol. Chem. 278, 5736–5743. doi: 10.1074/jbc.M206564200
Hunter, W. N. (2007). The non-mevalonate pathway of isoprenoid precursor biosynthesis. J. Biol. Chem. 282, 21573–21577. doi: 10.1074/jbc.R700005200
Hussein, D., and Mo, H. (2009). d-δ-Tocotrienol-mediated suppression of the proliferation of human PANC-1, MIA PaCa2 and BxPC-3 pancreatic carcinoma cells. Pancreas 38, e124–e136. doi: 10.1097/MPA.0b013e3181a20f9c
Hwang, S., Hartman, I. Z., Calhoun, L. N., Garland, K., Young, G. A., Mitsche, M. A., et al. (2016). Contribution of accelerated degradation to feedback regulation of 3-Hydroxy-3-methylglutaryl coenzyme A reductase and cholesterol metabolism in the liver. J. Biol. Chem. 291, 13479–13494. doi: 10.1074/jbc.M116.728469
Iqbal, J., Minhajuddin, M., and Beg, Z. H. (2004). Suppression of diethylnitrosamine and 2-acetylaminofluorene-induced hepatocarcinogenesis in rats by tocotrienol-rich fraction isolated from rice bran oil. Eur. J. Cancer Prev. 13, 515–520. doi: 10.1097/00008469-200412000-00009
Johnson, B. M., and DeBose-Boyd, R. A. (2018). Underlying mechanisms for sterol-induced ubiquitination and ER-associated degradation of HMG CoA reductase. Semin. Cell Dev. Biol. 81, 121–128. doi: 10.1016/j.semcdb.2017.10.019
Jones, H. M., Fang, Z. W., Sun, W. C., Clark, L. H., Stine, J. E., Tran, A. Q., et al. (2017). Atorvastatin exhibits anti-tumorigenic and anti-metastatic effects in ovarian cancer in vitro. Am. J. Cancer Res. 7, 2478–2490.
Jones, S., Fernandes, N. V., Yeganehjoo, H., Katuru, R., Qu, H., Yu, Z. L., et al. (2013). β-Ionone induces cell cycle arrest and apoptosis in human prostate tumor cells. Nutr. Cancer 65, 600–610. doi: 10.1080/01635581.2013.776091
Jung, M., Mo, H., and Elson, C. E. (1998). Synthesis and biological activity of β-ionone-derived alcohols for cancer chemoprevention. Anticancer Res. 18, 189–192.
Kandutsch, A. A., and Hancock, R. L. (1971). Regulation of the rate of sterol synthesis and the level of β-hydroxy-β-methylglutaryl coenzyme A reductase activity in mouse liver and hepatomas. Cancer Res. 31, 1396–1401.
Kany, S., Woschek, M., Kneip, N., Sturm, R., Kalbitz, M., Hanschen, M., et al. (2018). Simvastatin exerts anticancer effects in osteosarcoma cell lines via geranylgeranylation and c-Jun activation. Int. J. Oncol. 52, 1285–1294. doi: 10.3892/ijo.2018.4288
Kato, S., Liberona, M. F., Cerda-Infante, J., Sanchez, M., Henriquez, J., Bizama, C., et al. (2018). Simvastatin interferes with cancer 'stem-cell' plasticity reducing metastasis in ovarian cancer. Endocr. Relat. Cancer 25, 821–836. doi: 10.1530/ERC-18-0132
Katuru, R., Fernandes, N. V., Elfakhani, M., Dutta, D., Mills, N., Hynds, D. L., et al. (2011). Mevalonate depletion mediates the suppressive impact of geranylgeraniol on murine B16 melanoma cells. Exp. Biol. Med. 236, 604–613. doi: 10.1258/ebm.2011.010379
Kawata, S., Nagase, T., Yamasaki, E., Ishiguro, H., and Matsuzawa, Y. (1994). Modulation of the mevalonate pathway and cell growth by pravastatin and d-limonene in a human hepatoma cell line (Hep G2). Br. J. Cancer 69, 1015–1020. doi: 10.1038/bjc.1994.199
Kawata, S., Takaishi, K., Nagase, T., Ito, N., Matsuda, Y., Tamura, S., et al. (1990). Increase in the active form of 3-hydroxy-3-methylglutaryl coenzyme A reductase in human hepatocellular carcinoma: possible mechanism for alteration of cholesterol biosynthesis. Cancer Res. 50, 3270–3273.
Kim, G., Jang, S. Y., Nam, C. M., and Kang, E. S. (2018). Statin use and the risk of hepatocellular carcinoma in patients at high risk: a nationwide nested case-control study. J. Hepatol. 68, 476–484. doi: 10.1016/j.jhep.2017.10.018
Kimbung, S., Lettiero, B., Feldt, M., Bosch, A., and Borgquist, S. (2016). High expression of cholesterol biosynthesis genes is associated with resistance to statin treatment and inferior survival in breast cancer. Oncotarget 7, 59640–59651. doi: 10.18632/oncotarget.10746
Knox, J. J., Siu, L. L., Chen, E., Dimitroulakos, J., Kamel-Reid, S., Moore, M. J., et al. (2005). A Phase I trial of prolonged administration of lovastatin in patients with recurrent or metastatic squamous cell carcinoma of the head and neck or of the cervix. Eur. J. Cancer 41, 523–530. doi: 10.1016/j.ejca.2004.12.013
Kong, Y. F., Cheng, L. J., Mao, F. Y., Zhang, Z. Z., Zhang, Y. Q., Farah, E., et al. (2018). Inhibition of cholesterol biosynthesis overcomes enzalutamide resistance in castration-resistant prostate cancer (CRPC). J. Biol. Chem. 293, 14328–14341. doi: 10.1074/jbc.RA118.004442
Koo, J. H., and Guan, K. L. (2018). Interplay between YAP/TAZ and metabolism. Cell Metab. 28, 196–206. doi: 10.1016/j.cmet.2018.07.010
Koyuturk, M., Ersoz, M., and Altiok, N. (2004). Simvastatin induces proliferation inhibition and apoptosis in C6 glioma cells via c-jun N-terminal kinase. Neurosci. Lett. 370, 212–217. doi: 10.1016/j.neulet.2004.08.020
Krycer, J. R., Kristiana, I., and Brown, A. J. (2009). Cholesterol homeostasis in two commonly used human prostate cancer cell-lines, LNCaP and PC-3. PLoS ONE 4:e8496. doi: 10.1371/journal.pone.0008496
Krycer, J. R., Phan, L., and Brown, A. J. (2012). A key regulator of cholesterol homeostasis, SREBP-2, can be targeted in prostate cancer cells with natural products. Biochem. J. 446, 191–201. doi: 10.1042/BJ20120545
Kumari, S., Priya, P., Misra, G., and Yadav, G. (2013). Structural and biochemical perspectives in plant isoprenoid biosynthesis. Phytochem. Rev. 12, 255–291. doi: 10.1007/s11101-013-9284-6
Kuzu, O. F., Noory, M. A., and Robertson, G. P. (2016). The role of cholesterol in cancer. Cancer Res. 76, 2063–2070. doi: 10.1158/0008-5472.CAN-15-2613
Laezza, C., D'Alessandro, A., Di Croce, L., Picardi, P., Ciaglia, E., Pisanti, S., et al. (2015). p53 regulates the mevalonate pathway in human glioblastoma multiforme. Cell Death Dis. 6:e1909. doi: 10.1038/cddis.2015.279
Lange, Y., Ory, D. S., Ye, J., Lanier, M. H., Hsu, F. F., and Steck, T. L. (2008). Effectors of rapid homeostatic responses of endoplasmic reticulum cholesterol and 3-hydroxy-3-methylglutaryl-CoA reductase. J. Biol. Chem. 283, 1445–1455. doi: 10.1074/jbc.M706967200
Lebo, N. L., Griffiths, R., Hall, S., Dimitroulakos, J., and Johnson-Obaseki, S. (2018). Effect of statin use on oncologic outcomes in head and neck squamous cell carcinoma. Head Neck J. Sci. Spec. 40, 1697–1706. doi: 10.1002/hed.25152
Lee, J. H., Kim, C., S.-Kim, H., Sethi, G., and Ahn, K. S. (2015). Farnesol inhibits tumor growth and enhances the anticancer effects of bortezomib in multiple myeloma xenograft mouse model through the modulation of STAT3 signaling pathway. Cancer Lett. 360, 280–293. doi: 10.1016/j.canlet.2015.02.024
Lehoux, J. G., Kandalaft, N., Belisle, S., Bellabarba, D., Benard, B., and Lefebvre, A. (1984). Increased 3-hydroxy-3-methyl-glutaryl coenzyme A reductase activity in a virilizing adrenal carcinoma. J. Steroid Biochem. 21, 329–342. doi: 10.1016/0022-4731(84)90308-X
Li, Y., Xu, S., Mihaylova, M. M., Zheng, B., Hou, X., Jiang, B., et al. (2011). AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 13, 376–388. doi: 10.1016/j.cmet.2011.03.009
Liang, W. Z., Chou, C. T., Lu, T., Chi, C. C., Tseng, L. L., Pan, C. C., et al. (2013). The mechanism of carvacrol-evoked [Ca2+]i rises and non-Ca2+-triggered cell death in OC2 human oral cancer cells. Toxicology 303, 152–161. doi: 10.1016/j.tox.2012.10.026
Lichtenthaler, H. K. (1999). The 1-deoxy-d-xylulose-5-phosphate pathway of isoprenoid biosynthesis in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 50, 47–65. doi: 10.1146/annurev.arplant.50.1.47
Liu, Q., Dong, H. W., Sun, W. G., Liu, M., Ibla, J. C., Liu, L. X., et al. (2013). Apoptosis initiation of beta-ionone in SGC-7901 gastric carcinoma cancer cells via a PI3K-AKT pathway. Arch. Toxicol. 87, 481–490. doi: 10.1007/s00204-012-0962-8
Liu, W., Furuta, E., Shindo, K., Watabe, M., Xing, F., Pandey, P. R., et al. (2011). Cacalol, a natural sesquiterpene, induces apoptosis in breast cancer cells by modulating Akt-SREBP-FAS signaling pathway. Breast Cancer Res. Treat. 128, 57–68. doi: 10.1007/s10549-010-1076-8
Madankumar, A., Jayakumar, S., Gokuladhas, K., Rajan, B., Raghunandhakumar, S., Asokkumar, S., et al. (2013). Geraniol modulates tongue and hepatic phase I and phase II conjugation activities and may contribute directly to the chemopreventive activity against experimental oral carcinogenesis. Eur. J. Pharmacol. 705, 148–155. doi: 10.1016/j.ejphar.2013.02.048
Maltese, W. A. (1983). 3-hydroxy-3-methylglutaryl coenzyme A reductase in human brain tumors. Neurology 33, 1294–1299. doi: 10.1212/WNL.33.10.1294
Mancini, R., Noto, A., Pisanu, M. E., De Vitis, C., Maugeri-Saccà, M., and Ciliberto, G. (2018). Metabolic features of cancer stem cells: the emerging role of lipid metabolism. Oncogene 37, 2367–2378. doi: 10.1038/s41388-018-0141-3
Maningat, P., and Breslow, J. L. (2011). Needed: pragmatic clinical trials for statin-intolerant patients. N. Engl. J. Med. 365, 2250–2251. doi: 10.1056/NEJMp1112023
McAnally, J. A., Gupta, J., Sodhani, S., Bravo, L., and Mo, H. (2007). Tocotrienols potentiate lovastatin-mediated growth suppression in vitro and in vivo. Exp. Biol. Med. 232, 523–531.
McAnally, J. A., Jung, M., and Mo, H. (2003). Farnesyl-O-acetylhydroquinone and geranyl-O-acetylhydroquinone suppress the proliferation of murine B16 melanoma cells, human prostate and colon adenocarcinoma cells, human lung carcinoma cells, and human leukemia cells. Cancer Lett. 202, 181–192. doi: 10.1016/j.canlet.2003.08.008
McGee, T. P., Cheng, H. H., Kumagai, H., Omura, S., and Simoni, R. D. (1996). Degradation of 3-hydroxy-3-methylglutaryl-CoA reductase in endoplasmic reticulum membranes is accelerated as a result of increased susceptibility to proteolysis. J. Biol. Chem. 271, 25630–25638. doi: 10.1074/jbc.271.41.25630
McGlynn, K. A., Hagberg, K., Chen, J., Graubard, B. I., London, W. T., Jick, S., et al. (2015). Statin use and risk of primary liver cancer in the Clinical Practice Research Datalink. J. Natl. Cancer Inst. 107:djv009. doi: 10.1093/jnci/djv009
McIntyre, B. S., Briski, K. P., Gapor, A., and Sylvester, P. W. (2000). Antiproliferative and apoptotic effects of tocopherols and tocotrienols on preneoplastic and neoplastic mouse mammary epithelial cells. Proc. Soc. Exp. Biol. Med. 224, 292–301. doi: 10.1046/j.1525-1373.2000.22434.x
Meigs, T. E., Roseman, D. S., and Simoni, R. D. (1996). Regulation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase degradation by the nonsterol mevalonate metabolite farnesol in vivo. J. Biol. Chem. 271, 7916–7922. doi: 10.1074/jbc.271.14.7916
Melnykovych, G., Haug, J. S., and Goldner, C. M. (1992). Growth inhibition of leukemia cell line CEM-C1 by farnesol: effects of phosphatidylcholine and diacylglycerol. Biochem. Biophys. Res. Commun. 186, 543–548. doi: 10.1016/S0006-291X(05)80842-3
Miquel, K., Pradines, A., and Favre, G. (1996). Farnesol and geranylgeraniol induce actin cytoskeleton disorganization and apoptosis in A549 lung adenocarcinoma cells. Biochem. Biophys. Res. Commun. 225, 869–876. doi: 10.1006/bbrc.1996.1265
Mitchell, A. D., Pugh, T. D., and Goldfarb, S. (1978). Partial feedback control of β-hydroxy-β-methylglutaryl coenzyme A reductase activity in primary hepatocellular carcinomas. Cancer Res. 38, 4474–4477.
Mo, H., Elfakhani, M., Shah, A., and Yeganehjoo, H. (2013). “Mevalonate-suppressive tocotrienols for cancer chemoprevention and adjuvant therapy,” in Tocotrienols: vitamin E beyond tocopherols, eds R. R. Watson, V. R. Preedy, and B. Tan (Boca Raton, FL: CRC Press), 135–149.
Mo, H., and Elson, C. E. (1999). Apoptosis and cell-cycle arrest in human and murine tumor cells are initiated by isoprenoids. J. Nutr. 129, 804–813. doi: 10.1093/jn/129.4.804
Mo, H., and Elson, C. E. (2004). Studies of the isoprenoid-mediated inhibition of mevalonate synthesis applied to cancer chemotherapy and chemoprevention. Exp. Biol. Med. 229, 567–585. doi: 10.1177/153537020422900701
Mo, H., Tatman, D., Jung, M., and Elson, C. E. (2000). Farnesyl anthranilate suppresses the growth, in vitro and in vivo, of murine B16 melanomas. Cancer Lett. 157, 145–153. doi: 10.1016/S0304-3835(00)00490-0
Mullen, P. J., Yu, R., Longo, J., Archer, M. C., and Penn, L. Z. (2016). The interplay between cell signalling and the mevalonate pathway in cancer. Nat. Rev. Cancer 16, 718–731. doi: 10.1038/nrc.2016.76
Murtola, T. J., Syvala, H., Pennanen, P., Blauer, M., Solakivi, T., Ylikomi, T., et al. (2012). The importance of LDL and cholesterol metabolism for prostate epithelial cell growth. PLoS ONE 7:e39445. doi: 10.1371/journal.pone.0039445
Nakanishi, M., Goldstein, J. L., and Brown, M. S. (1988). Multivalent control of 3-hydroxy-3-methylglutaryl coenzyme A reductase. Mevalonate-derived product inhibits translation of mRNA and accelerates degradation of enzyme. J. Biol. Chem. 263, 8929–8937.
Nguyen, A. D., Lee, S. H., and DeBose-Boyd, R. A. (2009). Insig-mediated, sterol-accelerated degradation of the membrane domain of hamster 3-hydroxy-3-methylglutaryl-coenzyme A reductase in insect cells. J. Biol. Chem. 284, 26778–26788. doi: 10.1074/jbc.M109.032342
Nguyen, T., Khan, A., Liu, Y., El-Serag, H. B., and Thrift, A. P. (2018). The association between statin use after diagnosis and mortality risk in patients with esophageal cancer: a retrospective cohort study of United States veterans. Am. J. Gastroenterol. 113, 1310–1318. doi: 10.1038/s41395-018-0169-6
Nguyen, V. T., Barozzi, I., Faronato, M., Lombardo, Y., Steel, J. H., Patel, N., et al. (2015). Differential epigenetic reprogramming in response to specific endocrine therapies promotes cholesterol biosynthesis and cellular invasion. Nat. Commun. 6:10044. doi: 10.1038/ncomms10044
Notarnicola, M., Messa, C., Pricci, M., Guerra, V., Altomare, D. F., Montemurro, S., et al. (2004). Up-regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in left-sided human colon cancer. Anticancer Res. 24, 3837–3842.
Olsson, J. M., Schedin, S., Teclebrhan, H., Eriksson, L. C., and Dallner, G. (1995). Enzymes of the mevalonate pathway in rat liver nodules induced by 2-acetylaminofluorene treatment. Carcinogenesis 16, 599–605. doi: 10.1093/carcin/16.3.599
Osborne, T. F., Goldstein, J. L., and Brown, M. S. (1985). 5' end of HMG CoA reductase gene contains sequences responsible for cholesterol-mediated inhibition of transcription. Cell 42, 203–212. doi: 10.1016/S0092-8674(85)80116-1
Paragh, G., Foris, G., Paragh, G. Jr., Seres, I., Karanyi, Z., Fulop, P., et al. (2005). Different anticancer effects of fluvastatin on primary hepatocellular tumors and metastases in rats. Cancer Lett. 222, 17–22. doi: 10.1016/j.canlet.2004.09.028
Parker, B. A., Capizzi, J. A., Grimaldi, A. S., Clarkson, P. M., Cole, S. M., Keadle, J., et al. (2013). Effect of statins on skeletal muscle function. Circulation 127, 96–103. doi: 10.1161/CIRCULATIONAHA.112.136101
Parker, R. A., Pearce, B. C., Clark, R. W., Gordon, D. A., and Wright, J. J. (1993). Tocotrienols regulate cholesterol production in mammalian cells by post-transcriptional suppression of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J. Biol. Chem. 268, 11230–11238.
Parrales, A., Ranjan, A., Iyer, S. V., Padhye, S., Weir, S. J., Roy, A., et al. (2016). DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 18, 1233–1243. doi: 10.1038/ncb3427
Parrales, A., Thoenen, E., and Iwakuma, T. (2018). The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. 25, 460–470. doi: 10.1038/s41418-017-0026-y
Patel, P. B., and Thakkar, V. R. (2014). L-carvone induces p53, caspase 3 mediated apoptosis and inhibits the migration of breast cancer cell lines. Nutr. Cancer 66, 453–462. doi: 10.1080/01635581.2014.884230
Pavlova, N. N., and Thompson, C. B. (2016). The emerging hallmarks of cancer metabolism. Cell Metab. 23, 27–47. doi: 10.1016/j.cmet.2015.12.006
Pearce, B. C., Parker, R. A., Deason, M. E., Dischino, D. D., Gillespie, E., Qureshi, A. A., et al. (1994). Inhibitors of cholesterol biosynthesis. 2. Hypocholesterolemic and antioxidant activities of benzopyran and tetrahydronaphthalene analogues of the tocotrienols. J. Med. Chem. 37, 526–541. doi: 10.1021/jm00030a012
Pearce, B. C., Parker, R. A., Deason, M. E., Qureshi, A. A., and Wright, J. J. (1992). Hypocholesterolemic activity of synthetic and natural tocotrienols. J. Med. Chem. 35, 3595–3606. doi: 10.1021/jm00098a002
Peffley, D. M., and Gayen, A. K. (2003). Plant-derived monoterpenes suppress hamster kidney cell 3-hydroxy-3-methylglutaryl coenzyme a reductase synthesis at the post-transcriptional level. J. Nutr. 133, 38–44. doi: 10.1093/jn/133.1.38
Philippot, J. R., Cooper, A. G., and Wallach, D. F. (1977). Regulation of cholesterol biosynthesis by normal and leukemic (L2C) guinea pig lymphocytes. Proc. Natl. Acad. Sci. U.S.A. 74, 956–960. doi: 10.1073/pnas.74.3.956
Pierpaoli, E., Viola, V., Barucca, A., Orlando, F., Galli, F., and Provinciali, M. (2013). Effect of annatto-tocotrienols supplementation on the development of mammary tumors in HER-2/neu transgenic mice. Carcinogenesis 34, 1352–1360. doi: 10.1093/carcin/bgt064
Pierpaoli, E., Viola, V., Pilolli, F., Piroddi, M., Galli, F., and Provinciali, M. (2010). γ- and δ-tocotrienols exert a more potent anticancer effect than α-tocopheryl succinate on breast cancer cell lines irrespective of HER-2/neu expression. Life Sci. 86, 668–675. doi: 10.1016/j.lfs.2010.02.018
Pisanti, S., Picardi, P., Ciaglia, E., D'Alessandro, A., and Bifulco, M. (2014). Novel prospects of statins as therapeutic agents in cancer. Pharmacol. Res. 88, 84–98. doi: 10.1016/j.phrs.2014.06.013
Polo, M. P., Crespo, R., and de Bravo, M. G. (2011). Geraniol and simvastatin show a synergistic effect on a human hepatocarcinoma cell line. Cell Biochem. Funct. 29, 452–458. doi: 10.1002/cbf.1772
Potocnjak, I., Gobin, I., and Domitrovic, R. (2018). Carvacrol induces cytotoxicity in human cervical cancer cells but causes cisplatin resistance: involvement of MEK-ERK activation. Phytother. Res. 32, 1090–1097. doi: 10.1002/ptr.6048
Qureshi, A. A., and Peterson, D. M. (2001). The combined effects of novel tocotrienols and lovastatin on lipid metabolism in chickens. Atherosclerosis 156, 39–47. doi: 10.1016/S0021-9150(00)00612-2
Qureshi, A. A., Sami, S. A., Salser, W. A., and Khan, F. A. (2001). Synergistic effect of tocotrienol-rich fraction (TRF(25)) of rice bran and lovastatin on lipid parameters in hypercholesterolemic humans. J. Nutr. Biochem. 12, 318–329. doi: 10.1016/S0955-2863(01)00144-9
Rao, K. N., Kottapally, S., and Shinozuka, H. (1983). Lipid composition and 3-hydroxy-3-methylglutaryl-CoA reductase activity of acinar cell carcinoma of rat pancreas. Biochim. Biophys. Acta 759, 74–80. doi: 10.1016/0304-4165(83)90191-5
Rao, K. N., Melhem, M. F., Gabriel, H. F., Eskander, E. D., Kazanecki, M. E., and Amenta, J. S. (1988). Lipid composition and de novo cholesterogenesis in normal and neoplastic rat mammary tissues. J. Natl. Cancer Inst. 80, 1248–1253. doi: 10.1093/jnci/80.15.1248
Ravid, T., Doolman, R., Avner, R., Harats, D., and Roitelman, J. (2000). The ubiquitin-proteasome pathway mediates the regulated degradation of mammalian 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J. Biol. Chem. 275, 35840–35847. doi: 10.1074/jbc.M004793200
Ricoult, S. J., Yecies, J. L., Ben-Sahra, I., and Manning, B. D. (2016). Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 35, 1250–1260. doi: 10.1038/onc.2015.179
Rioja, A., Pizzey, A. R., Marson, C. M., and Thomas, N. S. (2000). Preferential induction of apoptosis of leukaemic cells by farnesol. FEBS Lett. 467, 291–295. doi: 10.1016/S0014-5793(00)01168-6
Rodenak-Kladniew, B., Castro, A., Starkel, P., De Saeger, C., de Bravo, M. G., and Crespo, R. (2018). Linalool induces cell cycle arrest and apoptosis in HepG2 cells through oxidative stress generation and modulation of Ras/MAPK and Akt/mTOR pathways. Life Sci. 199, 48–59. doi: 10.1016/j.lfs.2018.03.006
Roglans, N., Verd, J. C., Peris, C., Alegret, M., Vazquez, M., Adzet, T., et al. (2002). High doses of atorvastatin and simvastatin induce key enzymes involved in VLDL production. Lipids 37, 445–454. doi: 10.1007/s11745-002-0916-0
Roitelman, J., and Simoni, R. D. (1992). Distinct sterol and nonsterol signals for the regulated degradation of 3-hydroxy-3-methylglutaryl-CoA reductase. J. Biol. Chem. 267, 25264–25273.
Ruch, R. J., and Sigler, K. (1994). Growth inhibition of rat liver epithelial tumor cells by monoterpenes does not involve Ras plasma membrane association. Carcinogenesis 15, 787–789. doi: 10.1093/carcin/15.4.787
Sacks, F. M., Pfeffer, M. A., Moye, L. A., Rouleau, J. L., Rutherford, J. D., Cole, T. G., et al. (1996). The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and recurrent events trial investigators. N. Engl. J. Med. 335, 1001–1009. doi: 10.1056/NEJM199610033351401
Sahin, M. B., Perman, S. M., Jenkins, G., and Clark, S. S. (1999). Perillyl alcohol selectively induces G0/G1 arrest and apoptosis in Bcr/Abl-transformed myeloid cell lines. Leukemia 13, 1581–1591. doi: 10.1038/sj.leu.2401536
Sailo, B. L., Banik, K., Padmavathi, G., Javadi, M., Bordoloi, D., and Kunnumakkara, A. B. (2018). Tocotrienols: the promising analogues of vitamin E for cancer therapeutics. Pharmacol. Res. 130, 259–272. doi: 10.1016/j.phrs.2018.02.017
Sakai, M., Okabe, M., Yamasaki, M., Tachibana, H., and Yamada, K. (2004). Induction of apoptosis by tocotrienol in rat hepatoma dRLh-84 cells. Anticancer Res. 24, 1683–1688.
Schonewille, M., de Boer, J. F., Mele, L., Wolters, H., Bloks, V. W., Wolters, J. C., et al. (2016). Statins increase hepatic cholesterol synthesis and stimulate fecal cholesterol elimination in mice. J. Lipid Res. 57, 1455–1464. doi: 10.1194/jlr.M067488
Schumacher, M. M., Jun, D. J., Jo, Y., Seemann, J., and DeBose-Boyd, R. A. (2016). Geranylgeranyl-regulated transport of the prenyltransferase UBIAD1 between membranes of the ER and Golgi. J. Lipid Res. 57, 1286–1299. doi: 10.1194/jlr.M068759
Schumacher, M. M., Jun, D. J., Johnson, B. M., and DeBose-Boyd, R. A. (2018). UbiA prenyltransferase domain-containing protein-1 modulates HMG-CoA reductase degradation to coordinate synthesis of sterol and nonsterol isoprenoids. J. Biol. Chem. 293, 312–323. doi: 10.1074/jbc.RA117.000423
Scolastici, C., de Conti, A., Cardozo, M. T., Ong, T. P., Purgatto, E., Horst, M. A., et al. (2014). β-Ionone inhibits persistent preneoplastic lesions during the early promotion phase of rat hepatocarcinogenesis: TGF-alpha, NF-kappaB, and p53 as cellular targets. Nutr. Cancer 66, 234–241. doi: 10.1080/01635581.2014.863364
Seckl, M. J., Ottensmeier, C. H., Cullen, M., Schmid, P., Ngai, Y., Muthukumar, D., et al. (2017). Multicenter, phase III, randomized, double-blind, placebo-controlled trial of pravastatin added to first-line standard chemotherapy in small-cell lung cancer (LUNGSTAR). J. Clin. Oncol. 35, 1506–1514. doi: 10.1200/JCO.2016.69.7391
Sever, N., Lee, P. C., Song, B. L., Rawson, R. B., and Debose-Boyd, R. A. (2004). Isolation of mutant cells lacking Insig-1 through selection with SR-12813, an agent that stimulates degradation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J. Biol. Chem. 279, 43136–43147. doi: 10.1074/jbc.M406406200
Sever, N., Song, B. L., Yabe, D., Goldstein, J. L., Brown, M. S., and DeBose-Boyd, R. A. (2003). Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J. Biol. Chem. 278, 52479–52490. doi: 10.1074/jbc.M310053200
Shamma, A., Takegami, Y., Miki, T., Kitajima, S., Noda, M., Obara, T., et al. (2009). Rb regulates DNA damage response and cellular senescence through E2F-dependent suppression of N-ras isoprenylation. Cancer Cell 15, 255–269. doi: 10.1016/j.ccr.2009.03.001
Shellman, Y. G., Ribble, D., Miller, L., Gendall, J., Vanbuskirk, K., Kelly, D., et al. (2005). Lovastatin-induced apoptosis in human melanoma cell lines. Melanoma Res. 15, 83–89. doi: 10.1097/00008390-200504000-00001
Shepherd, J., Blauw, G. J., Murphy, M. B., Bollen, E. L., Buckley, B. M., Cobbe, S. M., et al. (2002). Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 360, 1623–1630. doi: 10.1016/S0140-6736(02)11600-X
Shi, J., Zhu, J., Zhao, H., Zhong, C., Xu, Z., and Yao, F. (2013). Mevalonate pathway is a therapeutic target in esophageal squamous cell carcinoma. Tumour Biol. 34, 429–435. doi: 10.1007/s13277-012-0567-0
Shoff, S. M., Grummer, M., Yatvin, M. B., and Elson, C. E. (1991). Concentration-dependent increase of murine P388 and B16 population doubling time by the acyclic monoterpene geraniol. Cancer Res. 51, 37–42.
Shun, M. C., Yu, W., Gapor, A., Parsons, R., Atkinson, J., Sanders, B. G., et al. (2004). Pro-apoptotic mechanisms of action of a novel vitamin E analog (alpha-TEA) and a naturally occurring form of vitamin E (delta-tocotrienol) in MDA-MB-435 human breast cancer cells. Nutr. Cancer 48, 95–105. doi: 10.1207/s15327914nc4801_13
Siperstein, M. D., and Fagan, V. M. (1964). Deletion of the cholesterol-negative feedback system in liver tumors. Cancer Res. 24, 1108–1115.
Siperstein, M. D., Gyde, A. M., and Morris, H. P. (1971). Loss of feedback control of hydroxymethylglutaryl coenzyme A reductase in hepatomas. Proc. Natl. Acad. Sci. U.S.A. 68, 315–317. doi: 10.1073/pnas.68.2.315
Sirtori, C. R. (2014). The pharmacology of statins. Pharmacol. Res. 88, 3–11. doi: 10.1016/j.phrs.2014.03.002
Smalley, K. S., and Eisen, T. G. (2002). Farnesyl thiosalicylic acid inhibits the growth of melanoma cells through a combination of cytostatic and pro-apoptotic effects. Int. J. Cancer 98, 514–522. doi: 10.1002/ijc.10213
Sohda, T., Iwata, K., Hirano, G., Sakurai, K., Yokoyama, K., Morihara, D., et al. (2013). 3-Hydroxyl-3-methylglutaryl-coenzyme A reductase is up regulated in hepatocellular carcinoma associated with paraneoplastic hypercholesterolemia. Med. Mol. Morphol. 46, 239–242. doi: 10.1007/s00795-013-0042-z
Solomon, K. R., and Freeman, M. R. (2008). Do the cholesterol-lowering properties of statins affect cancer risk? Trends Endocrinol. Metab. 19, 113–121. doi: 10.1016/j.tem.2007.12.004
Song, B. L., and DeBose-Boyd, R. A. (2006). Insig-dependent ubiquitination and degradation of 3-hydroxy-3-methylglutaryl coenzyme a reductase stimulated by delta- and gamma-tocotrienols. J. Biol. Chem. 281, 25054–25061. doi: 10.1074/jbc.M605575200
Song, B. L., Javitt, N. B., and DeBose-Boyd, R. A. (2005). Insig-mediated degradation of HMG CoA reductase stimulated by lanosterol, an intermediate in the synthesis of cholesterol. Cell Metab. 1, 179–189. doi: 10.1016/j.cmet.2005.01.001
Sorrentino, G., Ruggeri, N., Specchia, V., Cordenonsi, M., Mano, M., Dupont, S., et al. (2014). Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 16, 357–366. doi: 10.1038/ncb2936
Srivastava, J. K., and Gupta, S. (2006). Tocotrienol-rich fraction of palm oil induces cell cycle arrest and apoptosis selectively in human prostate cancer cells. Biochem. Biophys. Res. Commun. 346, 447–453. doi: 10.1016/j.bbrc.2006.05.147
Stayrook, K. R., McKinzie, J. H., Burke, Y. D., Burke, Y. A., and Crowell, P. L. (1997). Induction of the apoptosis-promoting protein Bak by perillyl alcohol in pancreatic ductal adenocarcinoma relative to untransformed ductal epithelial cells. Carcinogenesis 18, 1655–1658. doi: 10.1093/carcin/18.8.1655
Sundin, T., Peffley, D. M., Gauthier, D., and Hentosh, P. (2012). The isoprenoid perillyl alcohol inhibits telomerase activity in prostate cancer cells. Biochimie 94, 2639–2648. doi: 10.1016/j.biochi.2012.07.028
Tam, S. P., Brissette, L., Ramharack, R., and Deeley, R. G. (1991). Differences between the regulation of 3-hydroxy-3-methylglutaryl-coenzyme A reductase and low density lipoprotein receptor in human hepatoma cells and fibroblasts reside primarily at the translational and post-translational levels. J. Biol. Chem. 266, 16764–16773.
Thompson, J. M., Alvarez, A., Singha, M. K., Pavesic, M. W., Nguyen, Q. H., Nelson, L. J., et al. (2018). Targeting the mevalonate pathway suppresses VHL-deficient CC-RCC through an HIF-dependent mechanism. Mol. Cancer Ther. 17, 1781–1792. doi: 10.1158/1535-7163.MCT-17-1076
Thompson, P. D., Panza, G., Zaleski, A., and Taylor, B. (2016). Statin-associated side effects. J. Am. Coll. Cardiol. 67, 2395–2410. doi: 10.1016/j.jacc.2016.02.071
Tuerdi, G., Ichinomiya, S., Sato, H., Siddig, S., Suwa, E., Iwata, H., et al. (2013). Synergistic effect of combined treatment with gamma-tocotrienol and statin on human malignant mesothelioma cells. Cancer Lett. 339, 116–127. doi: 10.1016/j.canlet.2013.07.015
Tyagi, P. (2005). Current data with HMG-CoA reductase inhibitors (statins) for colorectal cancer prevention. Clin. Colorectal Cancer 5, 245–246. doi: 10.1016/S1533-0028(11)70190-6
Ura, H., Obara, T., Shudo, R., Itoh, A., Tanno, S., Fujii, T., et al. (1998). Selective cytotoxicity of farnesylamine to pancreatic carcinoma cells and Ki-ras-transformed fibroblasts. Mol. Carcinog. 21, 93–99. doi: 10.1002/(SICI)1098-2744(199802)21:2<93::AID-MC3>3.0.CO;2-P
Vitols, S., Norgren, S., Juliusson, G., Tatidis, L., and Luthman, H. (1994). Multilevel regulation of low-density lipoprotein receptor and 3-hydroxy-3-methylglutaryl coenzyme A reductase gene expression in normal and leukemic cells. Blood 84, 2689–2698.
Wada, S., Satomi, Y., Murakoshi, M., Noguchi, N., Yoshikawa, T., and Nishino, H. (2005). Tumor suppressive effects of tocotrienol in vivo and in vitro. Cancer Lett. 229, 181–191. doi: 10.1016/j.canlet.2005.06.036
Wali, V. B., Bachawal, S. V., and Sylvester, P. W. (2009a). Combined treatment of γ-tocotrienol with statins induce mammary tumor cell cycle arrest in G1. Exp. Biol. Med. 234, 639–650. doi: 10.3181/0810-RM-300
Wali, V. B., Bachawal, S. V., and Sylvester, P. W. (2009b). Suppression in mevalonate synthesis mediates antitumor effects of combined statin and γ-tocotrienol treatment. Lipids 44, 925–934. doi: 10.1007/s11745-009-3344-0
Wali, V. B., and Sylvester, P. W. (2007). Synergistic antiproliferative effects of γ-tocotrienol and statin treatment on mammary tumor cells. Lipids 42, 1113–1123. doi: 10.1007/s11745-007-3102-0
Wang, M., and Casey, P. J. (2016). Protein prenylation: unique fats make their mark on biology. Nat. Rev. Mol. Cell Biol. 17, 110–122. doi: 10.1038/nrm.2015.11
Wang, X., Xu, W., Zhan, P., Xu, T., Jin, J., Miu, Y., et al. (2018). Overexpression of geranylgeranyl diphosphate synthase contributes to tumour metastasis and correlates with poor prognosis of lung adenocarcinoma. J. Cell. Mol. Med. 22, 2177–2189. doi: 10.1111/jcmm.13493
Warburg, O. (1956). On the origin of cancer cells. Science 123, 309–314. doi: 10.1126/science.123.3191.309
Wen, Y. A., Xiong, X., Zaytseva, Y. Y., Napier, D. L., Vallee, E., Li, A. T., et al. (2018). Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. 9:265. doi: 10.1038/s41419-018-0330-6
Woschek, M., Kneip, N., Jurida, K., Marzi, I., and Relja, B. (2016). Simvastatin reduces cancerogenic potential of renal cancer cells via geranylgeranyl pyrophosphate and mevalonate pathway. Nutr. Cancer 68, 420–427. doi: 10.1080/01635581.2016.1152383
Wu, Y., Chen, K., Liu, X., Huang, L., Zhao, D., Li, L., et al. (2016). Srebp-1 interacts with c-Myc to enhance somatic cell reprogramming. Stem Cells 34, 83–92. doi: 10.1002/stem.2209
Xu, W., Mi, Y., He, P., He, S., and Niu, L. (2017). gamma-Tocotrienol inhibits proliferation and induces apoptosis via the mitochondrial pathway in human cervical cancer hela cells. Molecules 22:1299. doi: 10.3390/molecules22081299
Yachnin, S., and Mannickarottu, V. (1984). Increased 3-hydroxy-3-methylglutaryl coenzyme A reductase activity and cholesterol biosynthesis in freshly isolated hairy cell leukemia cells. Blood 63, 690–693.
Yang, T., Espenshade, P. J., Wright, M. E., Yabe, D., Gong, Y., Aebersold, R., et al. (2002). Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 110, 489–500. doi: 10.1016/S0092-8674(02)00872-3
Yang, Z., Xiao, H., Jin, H., Koo, P. T., Tsang, D. J., and Yang, C. S. (2010). Synergistic actions of atorvastatin with γ-tocotrienol and celecoxib against human colon cancer HT29 and HCT116 cells. Int. J. Cancer 126, 852–863. doi: 10.1002/ijc.24766
Yano, Y., Satoh, H., Fukumoto, K., Kumadaki, I., Ichikawa, T., Yamada, K., et al. (2005). Induction of cytotoxicity in human lung adenocarcinoma cells by 6-O-carboxypropyl-alpha-tocotrienol, a redox-silent derivative of alpha-tocotrienol. Int. J. Cancer 115, 839–846. doi: 10.1002/ijc.20809
Yap, W. N., Zaiden, N., Tan, Y. L., Ngoh, C. P., Zhang, X. W., Wong, Y. C., et al. (2010). Id1, inhibitor of differentiation, is a key protein mediating anti-tumor responses of gamma-tocotrienol in breast cancer cells. Cancer Lett. 291, 187–199. doi: 10.1016/j.canlet.2009.10.012
Yazlovitskaya, E. M., and Melnykovych, G. (1995). Selective farnesol toxicity and translocation of protein kinase C in neoplastic HeLa-S3K and non-neoplastic CF-3 cells. Cancer Lett. 88, 179–183. doi: 10.1016/0304-3835(94)03635-V
Ye, X. B., Zhang, G., Righolt, C., Johnston, J. B., Banerji, V., Gibson, S. B., et al. (2018). Associations between statin use and risk of non-Hodgkin lymphomas by subtype. Int. J. Cancer 143, 971–979. doi: 10.1002/ijc.31373
Yeganehjoo, H., McFarlin, B. K., and Mo, H. (2017). Synergistic impact of d-δ-tocotrienol and geranylgeraniol on the growth and HMG CoA reductase of human DU145 prostate carcinoma cells. Nutr. Cancer 69, 682–691. doi: 10.1080/01635581.2017.1299876
Yin, Q. H., Yan, F. X., Zu, X. Y., Wu, Y. H., Wu, X. P., Liao, M. C., et al. (2012). Anti-proliferative and pro-apoptotic effect of carvacrol on human hepatocellular carcinoma cell line HepG-2. Cytotechnology 64, 43–51. doi: 10.1007/s10616-011-9389-y
Yoshikawa, N., Tsuno, N. H., Okaji, Y., Kawai, K., Shuno, Y., Nagawa, H., et al. (2010). Isoprenoid geranylgeranylacetone inhibits human colon cancer cells through induction of apoptosis and cell cycle arrest. Anticancer. Drugs 21, 850–860. doi: 10.1097/CAD.0b013e32833e53cf
Yu, O., Eberg, M., Benayoun, S., Aprikian, A., Batist, G., Suissa, S., et al. (2014). Use of statins and the risk of death in patients with prostate cancer. J. Clin. Oncol. 32, 5–11. doi: 10.1200/JCO.2013.49.4757
Yu, R., Longo, J., van Leeuwen, J. E., Mullen, P. J., Ba-Alawi, W., Haibe-Kains, B., et al. (2018). Statin-induced cancer cell death can be mechanistically uncoupled from prenylation of RAS family proteins. Cancer Res. 78, 1347–1357. doi: 10.1158/0008-5472.CAN-17-1231
Zhang, H., Plutzky, J., Shubina, M., and Turchin, A. (2017). Continued statin prescriptions after adverse reactions and patient outcomes: a cohort study. Ann. Intern. Med. 167, 221–227. doi: 10.7326/M16-0838
Zhang, H., Plutzky, J., Skentzos, S., Morrison, F., Mar, P., Shubina, M., et al. (2013). Discontinuation of statins in routine care settings: a cohort study. Ann. Intern. Med. 158, 526–534. doi: 10.7326/0003-4819-158-7-201304020-00004
Keywords: isoprenoids, HMG CoA reductase, mevalonate, SREBP, statin, synergy, cancer
Citation: Mo H, Jeter R, Bachmann A, Yount ST, Shen C-L and Yeganehjoo H (2019) The Potential of Isoprenoids in Adjuvant Cancer Therapy to Reduce Adverse Effects of Statins. Front. Pharmacol. 9:1515. doi: 10.3389/fphar.2018.01515
Received: 04 September 2018; Accepted: 11 December 2018;
Published: 04 January 2019.
Edited by:
Zhiling Yu, Hong Kong Baptist University, Hong KongReviewed by:
Ting L. I., Shaanxi Normal University, ChinaYonghua Wang, South China University of Technology, China
Copyright © 2019 Mo, Jeter, Bachmann, Yount, Shen and Yeganehjoo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huanbiao Mo, aG1vQGdzdS5lZHU=