 Mitsuhiro Yamada
Mitsuhiro Yamada Masakazu Ichinose
Masakazu Ichinose
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol., 03 December 2018
Sec. Respiratory Pharmacology
Volume 9 - 2018 | https://doi.org/10.3389/fphar.2018.01426
This article is part of the Research TopicNew Perspectives in Assessment, Phenotyping and Pharmacological Treatment of Chronic Obstructive Pulmonary Disease (COPD)View all 6 articles
In COPD, the activity of the cholinergic system is increased, which is one of the reasons for the airflow limitation caused by the contraction of airway smooth muscles. Therefore, blocking the contractive actions with anticholinergics is a useful therapeutic intervention to reduce the airflow limitation. In addition to the effects of bronchoconstriction and mucus secretion, accumulating evidence from animal models of COPD suggest acetylcholine has a role in inflammation. Experiments using muscarinic M3-receptor deficient mice or M3 selective antagonists revealed that M3-receptors on parenchymal cells, but not on hematopoietic cells, are involved in the pro-inflammatory effect of acetylcholine. Recently, combinations of long-acting β2 adrenergic agonists (LABAs) and long-acting muscarinic antagonists (LAMAs) have become available for COPD treatment. These dual long-acting bronchodilators may have synergistic anti-inflammatory effects because stimulation of β2 adrenergic receptors induces inhibitory effects in inflammatory cells via a different signaling pathway from that by antagonizing M3-receptor, though these anti-inflammatory effects have not been clearly demonstrated in COPD patients. In contrast to the pro-inflammatory effects by ACh via muscarinic receptors, it has been demonstrated that the cholinergic anti-inflammatory pathway, which involves the parasympathetic nervous systems, regulates excessive inflammatory responses to protect organs during tissue injury and infection. Stimulation of acetylcholine via the α7 nicotinic acetylcholine receptor (α7nAChR) exerts inhibitory effects on leukocytes including macrophages and type 2 innate lymphoid cells. Although it remains unclear whether the inhibitory effects of acetylcholine via α7nAChR in inflammatory cells can regulate inflammation in COPD, neuroimmune interactions including the cholinergic anti-inflammatory pathway might serve as potential therapeutic targets.
Acetylcholine (ACh), a neurotransmitter released from nerve terminals of postganglionic parasympathetic neurons, promote the contraction of airway smooth muscles as well as mucus secretion via muscarinic type 3 (M3) receptor in the airways (Roffel et al., 1988; Mak and Barnes, 1990). It has been also revealed that non-neuronal cells including airway epithelial cells can synthesize and release ACh, which may induce biological functions in an autocrine or paracrine manner (Proskocil et al., 2004; Kummer and Krasteva-Christ, 2014). ACh is significantly involved in the pathogenesis of COPD because the activity of the cholinergic system is increased in this disease augmenting the airflow limitation and hypersecretion (Gross and Skorodin, 1984). In addition to these roles in the pathogenesis of COPD, accumulating evidence has revealed that ACh can regulate inflammatory responses. In this review, we summarize recent literature on the roles of cholinergic pathways in inflammation, especially focusing on: (1) pro-inflammatory effects of ACh through mAChRs; (2) possible benefits of the combinations of long-acting β2 adrenergic agonists (LABAs) and long-acting muscarinic antagonists (LAMAs) in terms of regulating inflammation; (3) anti-inflammatory effects of ACh via the α7 nicotinic acetylcholine receptor (α7nAChR); (4) recent advances in clarifying neuroimmune interactions that regulating innate lymphoid cells (ILCs), which could lead to potential pharmacotherapeutic targets for COPD.
In addition to the role of ACh in neurotransmission, the activation of muscles and mucus secretion, accumulating evidence from animal studies using muscarinic antagonists or subtype-specific deficient mice has suggested that ACh can augment airway inflammation through muscarinic ACh receptors. Tiotropium, one of the LAMAs with higher selectivity for M3 receptors, inhibited pulmonary neutrophilic inflammation and the release of leukotriene B4 (LTB4), interleukin-6 (IL-6), keratinocyte-derived chemokine(KC; IL-8 in humans), monocyte chemotactic protein-1 (MCP-1), macrophage inflammatory protein-1alpha and -2 (MIP-1α and MIP-2), and tumor necrosis factor alpha (TNF-α) in a mouse COPD model induced by cigarette smoke (Wollin and Pieper, 2010). Tiotropium also attenuated neutrophil infiltration and the elevation of IL-6 and TNF-α in cigarette smoke and virus-exposed mice (Bucher et al., 2016). Glycopyrronium, another M3-selective LAMA, attenuated the accumulation of neutrophils and macrophages and the elevation of inflammatory cytokines and chemokines induced by both acute and subchronic cigarette smoke exposure (Shen et al., 2014; Hsiao et al., 2018). Aclidinium bromide, another M3-selective LAMA, also attenuated neutrophilic infiltration in the alveolar septa in a cigarette smoke–exposed guinea pig model of COPD (Dominguez-Fandos et al., 2014). Although these LAMAs are kinetically selective for M3 receptors due to their dissociation time from M3 being longer than that from M2 or M1, the binding affinity of these agents for M1, M2, and M3 are similar and have limited selectivity (Casarosa et al., 2009), meaning that it is still unclear which muscarinic receptor subtype was critical for the pro-inflammatory effects in a cigarette smoke-exposed animal model of COPD. To answer this question, gene-targeting in mice for each subtype of muscarinic receptor was employed. M3 receptor-deficient mice showed a decrease in both neutrophil accumulation in the lungs and the release of KC, a neutrophil chemotactic factor, in an acute cigarette smoke-exposed mouse model (Kistemaker et al., 2013). In contrast to M3 receptor-deficient mice, M2 receptor-deficient mice showed an increase in both neutrophil infiltration in the airspaces and the release of KC after exposure to cigarette smoke (Kistemaker et al., 2013). Because the M2 receptor is located on the pre-junctions of pre- and post-ganglionic pulmonary parasympathetic nerves and acts as a feedback inhibitory receptor limiting acetylcholine release (Fryer and Maclagan, 1984; Faulkner et al., 1986; Minette and Barnes, 1988), the levels of ACh are increased in the airway of M2 receptor-deficient mice causing an enhancement of neutrophilic inflammation via the M3 receptor. Mice deficient in the M1 receptor, which is expressed on parasympathetic ganglia and mediate neurotransmission, also shows a remarkable enhancement of neutrophil inflammation after cigarette smoke-exposure (Kistemaker et al., 2013). This seems to be paradoxical because a M1 receptor deficiency would cause a reduction of acetylcholine in the airway, resulting the reduction of inflammation. Because M1 receptors are also expressed on airway epithelial cells and regulate electrolyte and water secretion (Ishihara et al., 1992), a hypothesis explaining this paradox is that the clearance of smoke particles is impaired in M1 deficient mice. In vitro cell culture experiments also supported the pro-inflammatory actions of ACh thorough muscarinic receptors on airway parenchymal cells and inflammatory cells (Table 1). Because M3 receptors are expressed in various types of cells including parenchymal cells and hematopoietic cells in airways (Table 1), the question has arisen whether the pro-inflammatory effects of ACh via M3 receptors are mediated mainly by parenchymal cells or inflammatory cells. To answer this question, bone marrow chimeric mice were established. This investigation revealed that the accumulation of neutrophils in the airways was inhibited in M3 receptor-deficient mice with wild-type bone marrow, whereas wild-type mice with M3 receptor-deficient bone marrow showed a comparable increase in neutrophil accumulation, suggesting that M3 receptors of the airway parenchymal cells are primarily involved in the pro-inflammatory effects of ACh during cigarette smoke-induced inflammation (Kistemaker et al., 2015b).

TABLE 1. Pro-inflammatory effects of ACh on parenchymal and leukocytes via muscarinic receptors.
Although there is accumulating evidence for a significant role of ACh as a pro-inflammatory factor in COPD as well as clinical evidence showing that LAMAs decrease exacerbations of COPD, stronger evidence for its pro-inflammatory role in COPD patients is still limited. Treatment with tiotropium in COPD patients decreased the annual rate of exacerbations. However, the amount of IL-6 and myeloperoxidase in sputum did not decrease. Moreover, the concentration of IL-8 and MMP-9 increased in the sputum of the group treated with tiotropium (Powrie et al., 2007; Perng et al., 2009). Tiotropium treatment also did not significantly decrease IL-6 or C-reactive protein in the serum (Powrie et al., 2007). This is possibly due to the fact that tiotropium decreased the amount of mucus secretion, causing a higher concentration of the mediators in sputum. It has been reported that targeted lung denervation of parasympathetic nerves in patients with COPD induces a reduction of neutrophil accumulation and inflammatory mediators including IL-8 (Kistemaker et al., 2015a). This is the first direct evidence showing that ACh acts a pro-inflammatory factor in actual COPD patients. Further investigations including histological evaluations by bronchial biopsy will be needed to elucidate whether antimuscarinic drugs actually effect inflammation in COPD patients.
Several clinical studies have reported that the combination of LABA and LAMA reduces exacerbations more than the use of LABA or LAMA alone (Wedzicha et al., 2013; Calverley et al., 2018). LABA/LAMA combinations decrease hyperinflation (Beeh et al., 2014; Vincken et al., 2014) and symptom severity (Mahler et al., 2014; Buhl et al., 2015; Oba et al., 2016), and improve sputum clearance (Powrie et al., 2007; Meyer et al., 2011; Tagaya et al., 2016), all of which may contribute to decreasing exacerbations. Although the effects of the LABA/LAMA combination therapy against inflammation have not been demonstrated in patients with COPD, nor has LAMA or LABA alone, several mechanisms are possibly involved in the synergistic actions of the LABA/LAMA combination against inflammation (Figure 1). One of the possible mechanisms is that LABA and LAMA synergistically inhibit the release of non-neurogenic acetylcholine from bronchial epithelial cells. A recent report showed that, although single administration of glycopyrronium or indacaterol did not modify the release of acetylcholine from primary human bronchial epithelial cells, the combination of glycopyrronium and indacaterol significantly reduced the release of acetylcholine (Cazzola et al., 2016). This effect may originate through the modulation of organic cation transporter 1, a transporter for the release of acetylcholine from airway epithelial cells (Salomon et al., 2015). This mechanism may contribute to the anti-inflammatory effects by reducing the levels of acetylcholines, which have pro-inflammatory effects via M3 receptors. Another possible mechanism is that LABA and LAMA synergistically inhibit the release of pro-inflammatory factors from parenchymal cells or inflammatory cells. Tiotropium or olodaterol attenuates the production of reactive oxygen species (ROS), NOX-4 protein expression, and IL-8 release from 16HBE cells induced by cigarette smoke extract (CSE), and the combination of LABA and LAMA provides stronger effects compared to LAMA or LABA alone (Albano et al., 2018). Tiotropium or olodaterol reduces neutrophil adhesion and the expression of macrophage-1 antigen (MAC-1) adhesion protein on neutrophils stimulated by sputum supernatants from patients with COPD. The combination of these agents augments these effects. Olodaterol attenuates neutrophil accumulation and pro-inflammatory mediators in the airway of cigarette smoke or LPS-induced murine and guinea pig lung inflammation models, and also attenuates the release of TNF-α from blood leukocytes and reduces CD11b adhesion molecule expression on granulocytes in vitro (Wex et al., 2015). Recent studies showed that β2-receptor agonists attenuated the release of TNF-α and MCP-1 induced by LPS from a mouse macrophage cell line by mediating the cAMP-dependent inhibition of ERK and p38MAPK pathways (Keranen et al., 2016, 2017). Because a M3 mAChR antagonist attenuated the production of TNF-α induced by LPS from alveolar macrophages via mediating the NF-κB signaling pathway, combinations of these drugs may possibly inhibit the release of pro-inflammatory factors synergistically also in terms of intracellular signaling (Xu et al., 2012). To confirm these suggested mechanisms in the synergistic effects by the LABA/LAMA combination in COPD patients, further clinical investigations will be needed.
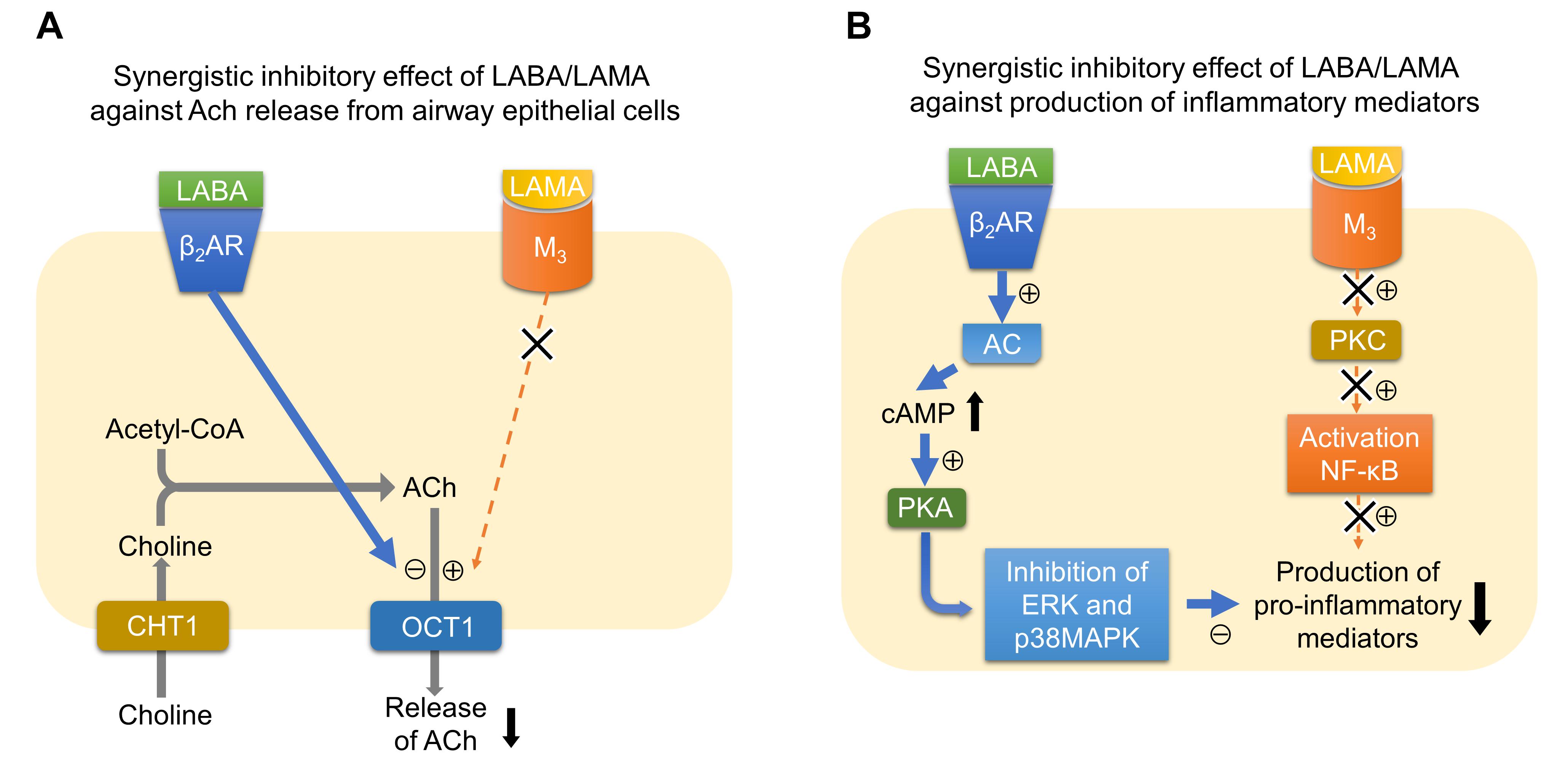
FIGURE 1. Possible mechanisms of synergistic actions of the LABA/LAMA combination against inflammation. (A) Synergistic inhibitory effect of LABA/LAMA against ACh release from airway epithelial cells. Signaling via β2AR inhibits the function of OCT1, a transporter for the release of acetylcholine from airway epithelial cells, whereas signaling via M3 possibly upregulates the function of OCT1. Therefore, the LABA/LAMA combination synergistically inhibits the release of ACh. (B) Synergistic inhibitory effect of LABA/LAMA against the production of inflammatory mediators. Signaling via β2AR activates AC-cAMP-PKA signaling pathway. PKA inhibits ERK and p38MAPK pathways. In contrast, signaling via M3 activates PKC, which results in the activation of NF-κB. The combination of LABA and LAMA therefore possibly inhibits the production of pro-inflammatory mediators synergistically via inhibiting both MAPK and NF-κB pathways. ACh, acetylcholine; AC, adenylate cyclase; β2AR, beta-2 adrenergic receptor; cAMP, cyclic adenosine monophosphate; CHT1, high-affinity choline transporter 1; ERK, extracellular signal-regulated kinase; LABA, long-acting β2 agonist; LAMA, long-acting muscarinic antagonist; MAPK, mitogen-activated protein kinase; M3, acetylcholine muscarinic M3 receptor; NF-κB, nuclear factor-kappa B; OCT1, organic cationic transporter 1; PKA, protein kinase A; PKC, protein kinase C.
In contrast to the pro-inflammatory effects by ACh through muscarinic ACh receptors, it has been elucidated that there is a cholinergic anti-inflammatory pathway which includes parasympathetic nerves that regulate the inflammatory responses to maintain the body during tissue injury and infection. An investigation with gene-deficient animal models revealed that α7nAChR on splenic macrophages was critical for attenuating the inflammatory response by ACh in an LPS-induced animal septic model (Wang et al., 2003). Further investigations revealed that the efferent vagus nerve stimulates sympathetic neurons of the splenic nerve, and then NE produced by the splenic nerve stimulates splenic T cells expressing β2-adrenergic receptors causing the production of ACh, which can bind to α7nAChR on splenic macrophages (Rosas-Ballina et al., 2008; Rosas-Ballina et al., 2011; Vida et al., 2011).
In airways, ACh is derived from parasympathetic postganglionic neurons or non-neuronal cells including epithelial cells and can possibly bind to α7nAChR on macrophages and other inflammatory cells attenuating inflammation. An experiment using a mouse model of acid-induced acute lung injury or sepsis-induced acute lung injury showed that α7nAChR is expressed on alveolar macrophages and neutrophils, and activation of α7nAChR by nicotine or PNU-282987, a specific agonist, reduced the inflammatory responses and attenuated the severity of lung injury, whereas the lung injury became worse in α7nAChR-deficient mice (Su et al., 2007, 2010). In a rodent lung injury model of ventilator-induced lung injury, intestinal ischemia-reperfusion, severe acute pancreatitis and cardiopulmonary bypass-induced lung ischemia-reperfusion, the activation of α7nAChR provided protective effects against lung injury (Dos Santos et al., 2011; He et al., 2016; Ma et al., 2016; Ge et al., 2017). The exact mechanism by which the activation of α7nAChR induced anti-inflammatory effects is still not clear. The activation of a7 nAChR by a specific agonist in monocytes epigenetically increases histone deacetylation, which decreases in the NF-κB p65 activity of transcription factors including NF-κB, and the transcription of pro-inflammatory cytokine genes (Yang et al., 2017). In E. coli and LPS-induced lung injury mouse models, vagotomy augmented the lung recruitment of α7nAChR+ CD11b+ monocytes and neutrophils. The activation of α7nAChR by an agonist suppressed the recruitment of α7nAChR+ CD11b+ monocytes and neutrophils. The phosphorylation of serine 473 of AKT1 via α7nAChR signaling is involved in the attenuation of the lung recruitment of these inflammatory cells (Zhao et al., 2017). It has been also reported that activation of α7nAChR attenuates the polarization of alveolar macrophages to the pro-inflammatory M1 phenotype, whereas M2 macrophages were increased in an LPS-induced lung injury model (Pinheiro et al., 2017; Wang et al., 2018).
In contrast to acute lung injury models showing the protective effect of α7nAChR against inflammation, there is a report suggesting that signaling via α7nAChR may worsen fibrosis in the lungs. In a mouse bleomycin-induced lung fibrosis model, the severity of lung fibrosis, including histological scoring and the expression of fibrogenic genes, was attenuated in α7nAChR deficient mice. The activation of α7nAChR by GTS-21 (an α7nAChR agonist) increased the TGF-β-induced phosphorylation of Smad2/3 and transcription of fibrogenic genes in lung fibroblasts (Sun et al., 2017). Another report showed that nicotine stimulated collagen type I mRNA and protein expression in primary lung fibroblasts. This effect of nicotine was not observed in α7nAChR-deficient primary lung fibroblasts. In vivo administration of nicotine increased collagen type I expression in the wild type, but not in α7nAChR-deficient mice (Vicary et al., 2017). These reports suggest that α7nAChR signaling in lung fibroblasts is involved in the pathogenesis of fibrotic lung diseases and airway remodeling.
It has been reported that airway epithelial cells also express α7nAChR (Su et al., 2007; Gahring et al., 2017). Reporter gene analyses for α7nAChR expression in genetically modified mice revealed that α7nAChRs are expressed in club cells and alveolar type II cells as well as alveolar macrophages (Gahring et al., 2017). α7nAChRs are likely involved in the regulation of proliferation, ion transport and responses to inflammatory stimuli (Nastrucci and Russo, 2012; Maouche et al., 2013; Gahring et al., 2017). Moreover, α7nAChRs are expressed in non-small cell lung cancer cells and participate in tumor growth and epidermal-mesenchymal transition (Chernyavsky et al., 2015; Schaal and Chellappan, 2016; Bordas et al., 2017; Zhang et al., 2017; Mucchietto et al., 2018). Therefore, these tumor-promoting actions as well as pro-fibrotic actions via α7nAChRs could be serious barriers to employing α7nAChR agonists to attenuate inflammation in chronic inflammatory diseases such as COPD and asthma, which require continuous drug administration.
It has been revealed that ILCs, which originate from common lymphoid cells but do not have antigen receptors, exist in various tissues of mammals including human, and can produce various cytokines in response to various stimuli including alarmins. ILCs are classified into type 1, type 2, and type 3 ILCs according to their functions and the cytokines produced (Vivier et al., 2018). Recently, neurotransmitters including acetylcholine have been shown to likely participate in the regulation of proliferation and activation of ILCs (Figure 2). Type 2 innate lymphoid cells (ILC2s) produce significant amounts of Th2 cytokines and participate in airway allergic inflammation. A recent report described that α7nAChRs are expressed on ILC2s. The activation of α7nAChRs on ILC2s suppresses IL-5 and IL-13 production in ILC2s and ameliorates ILC2-induced airway hyperreactivity. α7nAChR agonist attenuates the expression of GATA3 and NF-κB in ILC2s as well as the proliferation of ILC2s. The activation of α7nAChR by an agonist also prevents human ILC2 mediated airway hyperreactivity as well as type 2 cytokine production and eosinophilic inflammation. These observations may suggest a protective role of α7nAChR signaling in ILC2s against ILC2-mediated allergic inflammation and the possibility that α7nAChR could be a potential therapeutic target for the treatment of ILC2-mediated asthma.
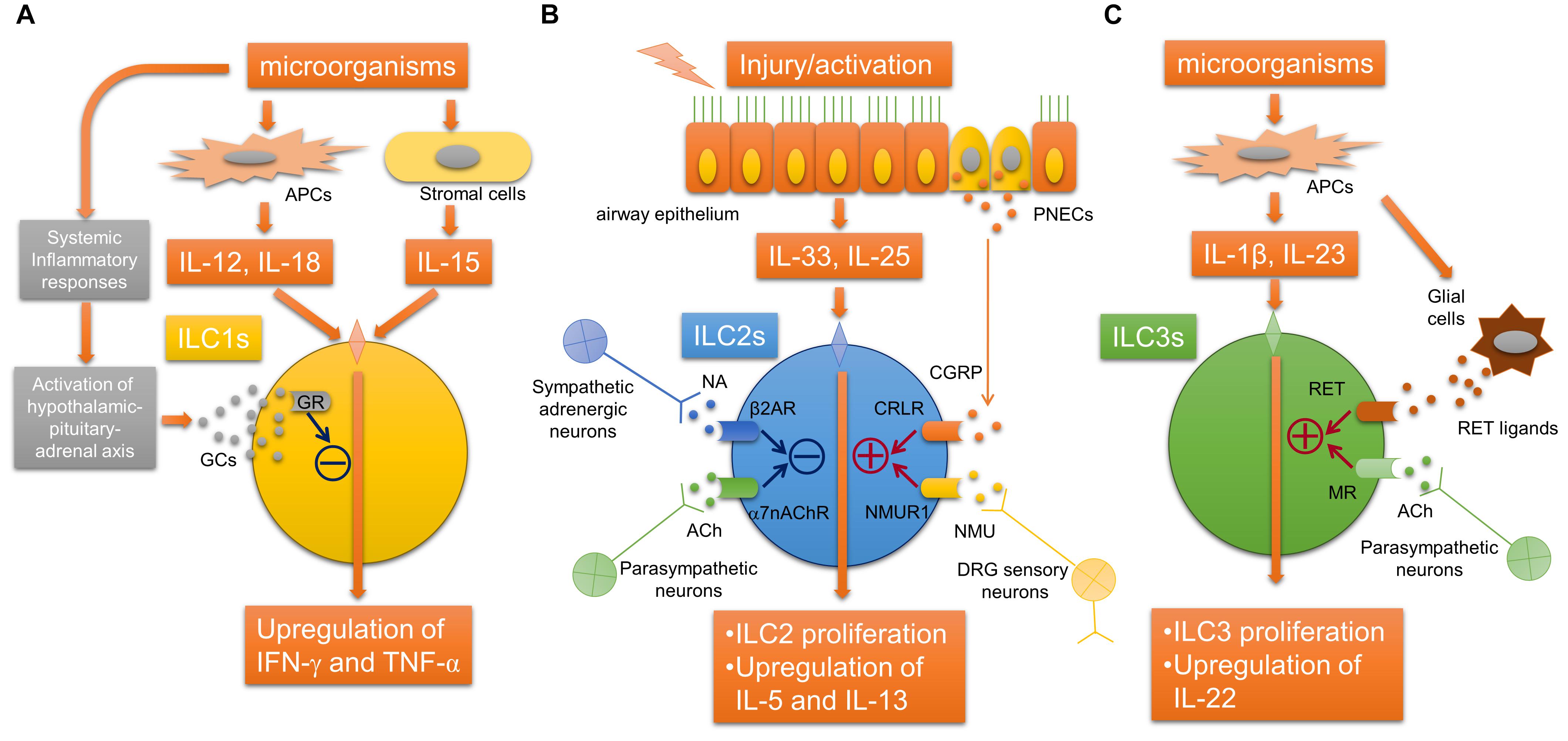
FIGURE 2. Neuroimmune interaction regulating innate lymphoid cells. (A) Glucocorticoids derived from hypothalamic-pituitary-adrenal axis stimulated by inflammatory mediators during systemic inflammation bind to GRs in ILC1s, which inhibits the release of IFN-γ. (B) The activation of ILC2s by epithelial alarmin cytokines is inhibited by ACh derived from parasympathetic neurons and possibly non-neuronal cells via α7nAChR, or NA derived from sympathetic neurons via β2AR. In contrast, neuromedin U derived from DRG sensory neurons or CGRP derived from PNECs augments the activation of ILC2s via NMUR1 or CRLR, respectively. (C) RET ligand derived from glial cells or acetylcholine derived from parasympathetic neurons arguments the proliferation and activation ILC3s via RET or muscarinic receptors, respectively. α7nAChR, α7 nicotinic acetylcholine receptor; ACh, acetylcholine; APC, antigen-presenting cell; β2AR, beta-2 adrenergic receptor; CGRP, calcitonin gene-related peptide; CRLR, calcitonin receptor-like receptor; DRG, dorsal root ganglia; GCs, glucocorticoids; IFN-γ, interferon-gamma; ILCs, innate lymphoid cells; MR, acetylcholine muscarinic receptor; NA, norepinephrine; NMU, neuromedin U; NMUR1, neuromedin-U receptor 1; PNEC, pulmonary neuroendocrine cell; TNF-α, tumor necrosis factor-alpha.
Other recent studies have shown that the neuropeptide neuromedin U (NMU) released from neural cells activates ILC2s and amplifies ILC2-driven allergic inflammation (Cardoso et al., 2017; Klose et al., 2017; Wallrapp et al., 2017). In an airway allergic mouse model induced by intranasal administration of IL-25 and IL-33, single-cell RNA sequencing analyses were performed to profile mouse lung-resident ILCs and revealed that NMUR1, a receptor for NMU, was specifically expressed on ILC2s (Wallrapp et al., 2017). Immunofluorescence microscopic analysis suggested that ILC2s were likely located close to nerve fibers. Sensory neurons in the dorsal root ganglia, but not parasympathetic neurons in the nodose/jugular ganglion, expressed NMU. IL-13 induced the upregulation of NMU in cultured dorsal root ganglia neurons. NMU can activate ILC2s and induce type 2 cytokine productions in vitro. Intranasal co-administration of NMU with IL-25 strongly amplified airway allergic inflammation. Moreover, NMUR1-deficient mice showed an attenuation of both the ILC2 frequency and effector function during airway inflammation from house dust mites. These findings indicated that NMU derived from sensory neurons innervated into airways can activate ILC2s, and this newly identified neuroimmune interaction is involved in the modulation of airway allergic inflammation (Wallrapp et al., 2017).
Another study using RNA sequencing analyses of ILC2s revealed that mouse and human ILC2s express β2-adrenergic receptor (β2AR) (Moriyama et al., 2018). β2AR-deficient mice showed amplified ILC2 activation and airway allergic inflammation induced by intranasal stimulation with IL-33 or Alternaria extract, whereas the administration of β2 agonist attenuated the ILC2-responses and allergic inflammation. RNA-sequence analyses further showed that β2AR signaling pathway inhibits the cell proliferation and effector function of ILC2s. This study suggests that sympathetic adrenergic neurons that innervate the airways negatively regulate ILC2s via β2AR.
Pulmonary neuroendocrine cells (PNECs) are a unique but poorly understood cell population in the lungs. Although it has been reported that PNECs are increased in pulmonary diseases, including small-cell lung cancer, the precise role of PNECs in pulmonary diseases remains unknown. A recent report showed that PNECs have a significant role in the mucosal type 2 response in models of allergic asthma (Sui et al., 2018). PNECs act through calcitonin gene-related peptide (CGRP) to stimulate ILC2s and induce downstream immune responses, and also act through GABA to induce goblet-cell hyperplasia. These new findings demonstrated a novel neuroimmune system in the airway that augments the type 2 allergic response. It is still unknown how PNECs are activated following allergen challenge. PNECs are innervated by sensory afferents mostly from nodose ganglion neurons and vagal efferents, and this innervation of PNEC is required for the increased production of GABA (Barrios et al., 2017), suggesting that stimulating efferent neurons that innervate PNECs may be involved in the activation of PNECs.
Compared with ILC2s, neuroimmune interactions that regulate the other ILCs, ILC3 and ILC1, in airways remain unclear, although it has been reported that their possible interactions may regulate other ILCs during systemic inflammation or inflammation of other organs. In gut, ILC3s express RET, a neuroregulatory receptor, and are localized close to glial cells expressing RET-ligands. Glial cells sense microenvironmental changes and release RET-ligands, causing ILC3 activation and the release of IL-22 (Ibiza et al., 2016). In a mouse E. coli peritonitis model, acetylcholine, probably via muscarinic receptors, upregulates ILC3 numbers, causing peritoneal macrophage production of a protective mediator, protectin conjugates in tissue regeneration 1 (PCTR1), and accelerate the resolution of inflammation induced by bacterial infection (Dalli et al., 2017). In a study using an LPS-induced septic model, group 1 ILCs expressed glucocorticoid receptor and stimulation with glucocorticoids prevented IFN-γ production by ILC1s, which cause the development of IL-10-dependent tolerance to endotoxin (Quatrini et al., 2017).
These on-going investigations of neuroimmune crosstalk, including the cholinergic pathways in airways, may lead to new potential therapeutic targets not only for acute inflammation but also for chronic inflammatory respiratory diseases including COPD. However, we should also note that neurotransmitters participating in neuroimmune interactions also have other fundamental functions including neurotransmission, which means blocking these neurotransmitters could possibly cause critical adverse effects. Accordingly, we also have to take into account the delivery system for candidate drugs that regulate neuroimmune interactions.
Accumulating evidence from in vitro and in vivo animal model investigations suggests that acetylcholine derived from parasympathetic neurons and non-neuronal cells can modulate airway inflammation. The pro-inflammatory actions of acetylcholine, mainly via M3 receptors, and possible synergistic anti-inflammatory effects of β2AR signaling may provide evidence for the benefit of treatment with LAMA or the combination of LABA/LAMA, although it remains unclear whether these treatments really have anti-inflammatory effects in COPD patients. Therefore, further clinical investigation will be needed to clarify the anti-inflammatory effects. Anti-inflammatory effects by acetylcholine via α7nAChR, which may modulate lung inflammation, have been revealed especially in systemic inflammation. However, we should be careful about other biological effects of α7nAChR signaling, including pro-fibrotic actions and tumor-promoting actions in terms of therapeutic utilization for chronic respiratory diseases including COPD. Recent on-going studies has revealed critical interactions between ILCs and neurotransmitters including acetylcholine. These newly found neuroimmune interactions may reveal therapeutic targets for airway inflammatory diseases. Further investigations will reveal novel effects of neurotransmitters including acetylcholine on immune systems and inflammation and may provide new therapeutic clues for clarifying airway inflammatory diseases such as COPD.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We very much appreciate Mr. Brent K. Bell for critical reading of the manuscript.
Albano, G. D., Bonanno, A., Moscato, M., Anzalone, G., Di Sano, C., Riccobono, L., et al. (2018). Crosstalk between mAChRM3 and beta2AR, via acetylcholine PI3/PKC/PBEP1/Raf-1 MEK1/2/ERK1/2 pathway activation, in human bronchial epithelial cells after long-term cigarette smoke exposure. Life Sci. 192, 99–109. doi: 10.1016/j.lfs.2017.11.034
Anzalone, G., Gagliardo, R., Bucchieri, F., Albano, G. D., Siena, L., Montalbano, A. M., et al. (2016). IL-17A induces chromatin remodeling promoting IL-8 release in bronchial epithelial cells: effect of Tiotropium. Life Sci. 152, 107–116. doi: 10.1016/j.lfs.2016.03.031
Barrios, J., Patel, K. R., Aven, L., Achey, R., Minns, M. S., Lee, Y., et al. (2017). Early life allergen-induced mucus overproduction requires augmented neural stimulation of pulmonary neuroendocrine cell secretion. FASEB J. 31, 4117–4128. doi: 10.1096/fj.201700115R
Beeh, K. M., Korn, S., Beier, J., Jadayel, D., Henley, M., D’andrea, P., et al. (2014). Effect of QVA149 on lung volumes and exercise tolerance in COPD patients: the BRIGHT study. Respir. Med. 108, 584–592. doi: 10.1016/j.rmed.2014.01.006
Bordas, A., Cedillo, J. L., Arnalich, F., Esteban-Rodriguez, I., Guerra-Pastrian, L., De Castro, J., et al. (2017). Expression patterns for nicotinic acetylcholine receptor subunit genes in smoking-related lung cancers. Oncotarget 8, 67878–67890. doi: 10.18632/oncotarget.18948
Bucher, H., Duechs, M. J., Tilp, C., Jung, B., and Erb, K. J. (2016). Tiotropium attenuates virus-induced pulmonary inflammation in cigarette smoke-exposed mice. J. Pharmacol. Exp. Ther. 357, 606–618. doi: 10.1124/jpet.116.232009
Buhl, R., Maltais, F., Abrahams, R., Bjermer, L., Derom, E., Ferguson, G., et al. (2015). Tiotropium and olodaterol fixed-dose combination versus mono-components in COPD (GOLD 2-4). Eur. Respir. J. 45, 969–979. doi: 10.1183/09031936.00136014
Buhling, F., Lieder, N., Kuhlmann, U. C., Waldburg, N., and Welte, T. (2007). Tiotropium suppresses acetylcholine-induced release of chemotactic mediators in vitro. Respir. Med. 101, 2386–2394. doi: 10.1016/j.rmed.2007.06.009
Calverley, P. M. A., Anzueto, A. R., Carter, K., Gronke, L., Hallmann, C., Jenkins, C., et al. (2018). Tiotropium and olodaterol in the prevention of chronic obstructive pulmonary disease exacerbations (DYNAGITO): a double-blind, randomised, parallel-group, active-controlled trial. Lancet Respir. Med. 6, 337–344. doi: 10.1016/S2213-2600(18)30102-4
Cardoso, V., Chesne, J., Ribeiro, H., Garcia-Cassani, B., Carvalho, T., Bouchery, T., et al. (2017). Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 549, 277–281. doi: 10.1038/nature23469
Casarosa, P., Bouyssou, T., Germeyer, S., Schnapp, A., Gantner, F., and Pieper, M. (2009). Preclinical evaluation of long-acting muscarinic antagonists: comparison of tiotropium and investigational drugs. J. Pharmacol. Exp. Ther. 330, 660–668. doi: 10.1124/jpet.109.152470
Cazzola, M., Calzetta, L., Puxeddu, E., Ora, J., Facciolo, F., Rogliani, P., et al. (2016). Pharmacological characterisation of the interaction between glycopyrronium bromide and indacaterol fumarate in human isolated bronchi, small airways and bronchial epithelial cells. Respir. Res. 17:70. doi: 10.1186/s12931-016-0386-8
Chernyavsky, A. I., Shchepotin, I. B., Galitovkiy, V., and Grando, S. A. (2015). Mechanisms of tumor-promoting activities of nicotine in lung cancer: synergistic effects of cell membrane and mitochondrial nicotinic acetylcholine receptors. BMC Cancer 15:152. doi: 10.1186/s12885-015-1158-4
Costa, L., Roth, M., Miglino, N., Keglowich, L., Zhong, J., Lardinois, D., et al. (2014). Tiotropium sustains the anti-inflammatory action of olodaterol via the cyclic AMP pathway. Pulm. Pharmacol. Ther. 27, 29–37. doi: 10.1016/j.pupt.2013.11.001
Dalli, J., Colas, R. A., Arnardottir, H., and Serhan, C. N. (2017). Vagal regulation of group 3 innate lymphoid cells and the immunoresolvent PCTR1 controls infection resolution. Immunity 46, 92–105. doi: 10.1016/j.immuni.2016.12.009
Dominguez-Fandos, D., Ferrer, E., Puig-Pey, R., Carreno, C., Prats, N., Aparici, M., et al. (2014). Effects of aclidinium bromide in a cigarette smoke-exposed Guinea pig model of chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 50, 337–346.
Dos Santos, C. C., Shan, Y., Akram, A., Slutsky, A. S., and Haitsma, J. J. (2011). Neuroimmune regulation of ventilator-induced lung injury. Am. J. Respir. Crit. Care Med. 183, 471–482. doi: 10.1164/rccm.201002-0314OC
Faulkner, D., Fryer, A. D., and Maclagan, J. (1986). Postganglionic muscarinic inhibitory receptors in pulmonary parasympathetic nerves in the guinea-pig. Br. J. Pharmacol. 88, 181–187. doi: 10.1111/j.1476-5381.1986.tb09485.x
Fryer, A. D., and Maclagan, J. (1984). Muscarinic inhibitory receptors in pulmonary parasympathetic nerves in the guinea-pig. Br. J. Pharmacol. 83, 973–978. doi: 10.1111/j.1476-5381.1984.tb16539.x
Fujii, Y. X., Tashiro, A., Arimoto, K., Fujigaya, H., Moriwaki, Y., Misawa, H., et al. (2007). Diminished antigen-specific IgG1 and interleukin-6 production and acetylcholinesterase expression in combined M1 and M5 muscarinic acetylcholine receptor knockout mice. J. Neuroimmunol. 188, 80–85. doi: 10.1016/j.jneuroim.2007.05.017
Gahring, L. C., Myers, E. J., Dunn, D. M., Weiss, R. B., and Rogers, S. W. (2017). Nicotinic alpha 7 receptor expression and modulation of the lung epithelial response to lipopolysaccharide. PLoS One 12:e0175367. doi: 10.1371/journal.pone.0175367
Ge, J., Tian, J., Yang, H., Hou, L., Wang, Z., He, Z., et al. (2017). Alpha7 nicotine acetylcholine receptor agonist PNU-282987 attenuates acute lung injury in a cardiopulmonary bypass model in rats. Shock 47, 474–479. doi: 10.1097/SHK.0000000000000744
Gosens, R., Rieks, D., Meurs, H., Ninaber, D. K., Rabe, K. F., Nanninga, J., et al. (2009). Muscarinic M3 receptor stimulation increases cigarette smoke-induced IL-8 secretion by human airway smooth muscle cells. Eur. Respir. J. 34, 1436–1443. doi: 10.1183/09031936.00045209
Gross, N. J., and Skorodin, M. S. (1984). Role of the parasympathetic system in airway-obstruction due to emphysema. N. Engl. J. Med. 311, 421–425. doi: 10.1056/NEJM198408163110701
He, Y., Ye, Z. Q., Li, X., Zhu, G. S., Liu, Y., Yao, W. F., et al. (2016). Alpha7 nicotinic acetylcholine receptor activation attenuated intestine-derived acute lung injury. J. Surg. Res. 201, 258–265. doi: 10.1016/j.jss.2015.10.046
Hsiao, Y. H., Tseng, C. M., Su, K. C., Chen, W. C., Wu, M. T., Wu, Y. C., et al. (2018). Glycopyrronium bromide inhibits lung inflammation and small airway remodeling induced by subchronic cigarette smoke exposure in mice. Respir. Physiol. Neurobiol. 249, 16–22. doi: 10.1016/j.resp.2017.12.005
Ibiza, S., Garcia-Cassani, B., Ribeiro, H., Carvalho, T., Almeida, L., Marques, R., et al. (2016). Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature 535, 440–443. doi: 10.1038/nature18644
Ishihara, H., Shimura, S., Satoh, M., Masuda, T., Nonaka, H., Kase, H., et al. (1992). Muscarinic receptor subtypes in feline tracheal submucosal gland secretion. Am. J. Physiol. 262, L223–L228. doi: 10.1152/ajplung.1992.262.2.L223
Keranen, T., Hommo, T., Hamalainen, M., Moilanen, E., and Korhonen, R. (2016). Anti-inflammatory effects of beta2-receptor agonists salbutamol and terbutaline are mediated by MKP-1. PLoS One 11:e0148144. doi: 10.1371/journal.pone.0148144
Keranen, T., Hommo, T., Moilanen, E., and Korhonen, R. (2017). beta2-receptor agonists salbutamol and terbutaline attenuated cytokine production by suppressing ERK pathway through cAMP in macrophages. Cytokine 94, 1–7. doi: 10.1016/j.cyto.2016.07.016
Kistemaker, L. E., Bos, I. S., Hylkema, M. N., Nawijn, M. C., Hiemstra, P. S., Wess, J., et al. (2013). Muscarinic receptor subtype-specific effects on cigarette smoke-induced inflammation in mice. Eur. Respir. J. 42, 1677–1688. doi: 10.1183/09031936.00112412
Kistemaker, L. E., Slebos, D. J., Meurs, H., Kerstjens, H. A., and Gosens, R. (2015a). Anti-inflammatory effects of targeted lung denervation in patients with COPD. Eur. Respir. J. 46, 1489–1492. doi: 10.1183/13993003.00413-2015
Kistemaker, L. E., Van Os, R. P., Dethmers-Ausema, A., Bos, I. S., Hylkema, M. N., Van Den Berge, M., et al. (2015b). Muscarinic M3 receptors on structural cells regulate cigarette smoke-induced neutrophilic airway inflammation in mice. Am. J. Physiol. Lung. Cell Mol. Physiol. 308, L96–L103. doi: 10.1152/ajplung.00259.2014
Klose, C. S. N., Mahlakoiv, T., Moeller, J. B., Rankin, L. C., Flamar, A. L., Kabata, H., et al. (2017). The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 549, 282–286. doi: 10.1038/nature23676
Koarai, A., Traves, S. L., Fenwick, P. S., Brown, S. M., Chana, K. K., Russell, R. E., et al. (2012). Expression of muscarinic receptors by human macrophages. Eur. Respir. J. 39, 698–704. doi: 10.1183/09031936.00136710
Kummer, W., and Krasteva-Christ, G. (2014). Non-neuronal cholinergic airway epithelium biology. Curr. Opin. Pharmacol. 16, 43–49. doi: 10.1016/j.coph.2014.03.001
Ma, P., Yu, K., Yu, J., Wang, W., Ding, Y., Chen, C., et al. (2016). Effects of nicotine and vagus nerve in severe acute pancreatitis-associated lung injury in rats. Pancreas 45, 552–560. doi: 10.1097/MPA.0000000000000575
Mahler, D. A., Decramer, M., D’urzo, A., Worth, H., White, T., Alagappan, V. K., et al. (2014). Dual bronchodilation with QVA149 reduces patient-reported dyspnoea in COPD: the BLAZE study. Eur. Respir. J. 43, 1599–1609. doi: 10.1183/09031936.00124013
Mak, J. C., and Barnes, P. J. (1990). Autoradiographic visualization of muscarinic receptor subtypes in human and guinea pig lung. Am. Rev. Respir. Dis. 141, 1559–1568. doi: 10.1164/ajrccm/141.6.1559
Maouche, K., Medjber, K., Zahm, J. M., Delavoie, F., Terryn, C., Coraux, C., et al. (2013). Contribution of alpha7 nicotinic receptor to airway epithelium dysfunction under nicotine exposure. Proc. Natl. Acad. Sci. U.S.A. 110, 4099–4104. doi: 10.1073/pnas.1216939110
Meyer, T., Reitmeir, P., Brand, P., Herpich, C., Sommerer, K., Schulze, A., et al. (2011). Effects of formoterol and tiotropium bromide on mucus clearance in patients with COPD. Respir. Med. 105, 900–906. doi: 10.1016/j.rmed.2011.02.007
Milara, J., Cervera, A., De Diego, A., Sanz, C., Juan, G., Gavalda, A., et al. (2016). Non-neuronal cholinergic system contributes to corticosteroid resistance in chronic obstructive pulmonary disease patients. Respir. Res. 17:145. doi: 10.1186/s12931-016-0467-8
Minette, P. A., and Barnes, P. J. (1988). Prejunctional inhibitory muscarinic receptors on cholinergic nerves in human and guinea pig airways. J. Appl. Physiol. 64, 2532–2537. doi: 10.1152/jappl.1988.64.6.2532
Moriyama, S., Brestoff, J. R., Flamar, A. L., Moeller, J. B., Klose, C. S. N., Rankin, L. C., et al. (2018). beta2-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science 359, 1056–1061. doi: 10.1126/science.aan4829
Mucchietto, V., Fasoli, F., Pucci, S., Moretti, M., Benfante, R., Maroli, A., et al. (2018). alpha9- and alpha7-containing receptors mediate the pro-proliferative effects of nicotine in the A549 adenocarcinoma cell line. Br. J. Pharmacol. 175, 1957–1972. doi: 10.1111/bph.13954
Nastrucci, C., and Russo, P. (2012). alpha7 nAChR in airway respiratory epithelial cells. Curr. Drug Targets 13, 666–670. doi: 10.2174/138945012800398865
Oba, Y., Sarva, S. T., and Dias, S. (2016). Efficacy and safety of long-acting beta-agonist/long-acting muscarinic antagonist combinations in COPD: a network meta-analysis. Thorax 71, 15–25. doi: 10.1136/thoraxjnl-2014-206732
Perng, D. W., Tao, C. W., Su, K. C., Tsai, C. C., Liu, L. Y., and Lee, Y. C. (2009). Anti-inflammatory effects of salmeterol/fluticasone, tiotropium/fluticasone or tiotropium in COPD. Eur. Respir. J. 33, 778–784. doi: 10.1183/09031936.00115308
Pinheiro, N. M., Santana, F. P., Almeida, R. R., Guerreiro, M., Martins, M. A., Caperuto, L. C., et al. (2017). Acute lung injury is reduced by the alpha7nAChR agonist PNU-282987 through changes in the macrophage profile. FASEB J. 31, 320–332. doi: 10.1096/fj.201600431R
Powrie, D. J., Wilkinson, T. M., Donaldson, G. C., Jones, P., Scrine, K., Viel, K., et al. (2007). Effect of tiotropium on sputum and serum inflammatory markers and exacerbations in COPD. Eur. Respir. J. 30, 472–478. doi: 10.1183/09031936.00023907
Profita, M., Bonanno, A., Montalbano, A. M., Ferraro, M., Siena, L., Bruno, A., et al. (2011). Cigarette smoke extract activates human bronchial epithelial cells affecting non-neuronal cholinergic system signalling in vitro. Life Sci. 89, 36–43. doi: 10.1016/j.lfs.2011.04.025
Profita, M., Bonanno, A., Siena, L., Ferraro, M., Montalbano, A. M., Pompeo, F., et al. (2008). Acetylcholine mediates the release of IL-8 in human bronchial epithelial cells by a NFkB/ERK-dependent mechanism. Eur. J. Pharmacol. 582, 145–153. doi: 10.1016/j.ejphar.2007.12.029
Proskocil, B. J., Sekhon, H. S., Jia, Y. B., Savchenko, V., Blakely, R. D., Lindstrom, J., et al. (2004). Acetylcholine is an autocrine or paracrine hormone synthesized and secreted by airway bronchial epithelial cells. Endocrinology 145, 2498–2506. doi: 10.1210/en.2003-1728
Quatrini, L., Wieduwild, E., Guia, S., Bernat, C., Glaichenhaus, N., Vivier, E., et al. (2017). Host resistance to endotoxic shock requires the neuroendocrine regulation of group 1 innate lymphoid cells. J. Exp. Med. 214, 3531–3541. doi: 10.1084/jem.20171048
Reinheimer, T., Mohlig, T., Zimmermann, S., Hohle, K. D., and Wessler, I. (2000). Muscarinic control of histamine release from airways. Inhibitory M1-receptors in human bronchi but absence in rat trachea. Am. J. Respir. Crit. Care Med. 162, 534–538. doi: 10.1164/ajrccm.162.2.9911094
Roffel, A. F., Elzinga, C. R. S., Vanamsterdam, R. G. M., Dezeeuw, R. A., and Zaagsma, J. (1988). Muscarinic M2 receptors in bovine tracheal smooth-muscle – Discrepancies between binding and function. Eur. J. Pharmacol. 153, 73–82. doi: 10.1016/0014-2999(88)90589-4
Rosas-Ballina, M., Ochani, M., Parrish, W. R., Ochani, K., Harris, Y. T., Huston, J. M., et al. (2008). Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc. Natl. Acad. Sci. U.S.A. 105, 11008–11013. doi: 10.1073/pnas.0803237105
Rosas-Ballina, M., Olofsson, P. S., Ochani, M., Valdes-Ferrer, S. I., Levine, Y. A., Reardon, C., et al. (2011). Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334, 98–101. doi: 10.1126/science.1209985
Salomon, J. J., Hagos, Y., Petzke, S., Kuhne, A., Gausterer, J. C., Hosoya, K., et al. (2015). Beta-2 adrenergic agonists are substrates and inhibitors of human organic cation transporter 1. Mol. Pharm. 12, 2633–2641. doi: 10.1021/mp500854e
Sato, E., Koyama, S., Okubo, Y., Kubo, K., and Sekiguchi, M. (1998). Acetylcholine stimulates alveolar macrophages to release inflammatory cell chemotactic activity. Am. J. Physiol. 274, L970–L979. doi: 10.1152/ajplung.1998.274.6.L970
Schaal, C., and Chellappan, S. (2016). Nicotine-mediated regulation of nicotinic acetylcholine receptors in non-small cell lung adenocarcinoma by E2F1 and STAT1 transcription factors. PLoS One 11:e0156451. doi: 10.1371/journal.pone.0156451
Shen, L. L., Liu, Y. N., Shen, H. J., Wen, C., Jia, Y. L., Dong, X. W., et al. (2014). Inhalation of glycopyrronium inhibits cigarette smoke-induced acute lung inflammation in a murine model of COPD. Int. Immunopharmacol. 18, 358–364. doi: 10.1016/j.intimp.2013.12.021
Su, X., Lee, J. W., Matthay, Z. A., Mednick, G., Uchida, T., Fang, X., et al. (2007). Activation of the alpha7 nAChR reduces acid-induced acute lung injury in mice and rats. Am. J. Respir. Cell Mol. Biol. 37, 186–192. doi: 10.1165/rcmb.2006-0240OC
Su, X., Matthay, M. A., and Malik, A. B. (2010). Requisite role of the cholinergic alpha7 nicotinic acetylcholine receptor pathway in suppressing gram-negative sepsis-induced acute lung inflammatory injury. J. Immunol. 184, 401–410. doi: 10.4049/jimmunol.0901808
Sui, P., Wiesner, D. L., Xu, J., Zhang, Y., Lee, J., Van Dyken, S., et al. (2018). Pulmonary neuroendocrine cells amplify allergic asthma responses. Science 360:eaan8546.
Sun, P., Li, L., Zhao, C., Pan, M., Qian, Z., and Su, X. (2017). Deficiency of alpha7 nicotinic acetylcholine receptor attenuates bleomycin-induced lung fibrosis in mice. Mol. Med. 23, 34–39. doi: 10.2119/molmed.2016.00083
Tagaya, E., Yagi, O., Sato, A., Arimura, K., Takeyama, K., Kondo, M., et al. (2016). Effect of tiotropium on mucus hypersecretion and airway clearance in patients with COPD. Pulm. Pharmacol. Ther. 39, 81–84. doi: 10.1016/j.pupt.2016.06.008
Vicary, G. W., Ritzenthaler, J. D., Panchabhai, T. S., Torres-Gonzalez, E., and Roman, J. (2017). Nicotine stimulates collagen type I expression in lung via alpha7 nicotinic acetylcholine receptors. Respir. Res. 18:115. doi: 10.1186/s12931-017-0596-8
Vida, G., Pena, G., Kanashiro, A., Thompson-Bonilla Mdel, R., Palange, D., Deitch, E. A., et al. (2011). beta2-Adrenoreceptors of regulatory lymphocytes are essential for vagal neuromodulation of the innate immune system. FASEB J. 25, 4476–4485. doi: 10.1096/fj.11-191007
Vincken, W., Aumann, J., Chen, H., Henley, M., Mcbryan, D., and Goyal, P. (2014). Efficacy and safety of coadministration of once-daily indacaterol and glycopyrronium versus indacaterol alone in COPD patients: the GLOW6 study. Int. J. Chron. Obstruct. Pulmon Dis. 9, 215–228. doi: 10.2147/COPD.S51592
Vivier, E., Artis, D., Colonna, M., Diefenbach, A., Di Santo, J. P., Eberl, G., et al. (2018). Innate lymphoid cells: 10 years on. Cell 174, 1054–1066. doi: 10.1016/j.cell.2018.07.017
Wallrapp, A., Riesenfeld, S. J., Burkett, P. R., Abdulnour, R. E., Nyman, J., Dionne, D., et al. (2017). The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 549, 351–356. doi: 10.1038/nature24029
Wang, H., Yu, M., Ochani, M., Amella, C. A., Tanovic, M., Susarla, S., et al. (2003). Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421, 384–388. doi: 10.1038/nature01339
Wang, J., Li, R., Peng, Z., Zhou, W., Hu, B., Rao, X., et al. (2018). GTS-21 reduces inflammation in acute lung injury by regulating M1 polarization and function of alveolar macrophages. Shock doi: 10.1097/SHK.0000000000001144[Epub ahead of print].
Wedzicha, J. A., Decramer, M., Ficker, J. H., Niewoehner, D. E., Sandstrom, T., Taylor, A. F., et al. (2013). Analysis of chronic obstructive pulmonary disease exacerbations with the dual bronchodilator QVA149 compared with glycopyrronium and tiotropium (SPARK): a randomised, double-blind, parallel-group study. Lancet Respir. Med. 1, 199–209. doi: 10.1016/S2213-2600(13)70052-3
Wex, E., Kollak, I., Duechs, M. J., Naline, E., Wollin, L., and Devillier, P. (2015). The long-acting beta2 -adrenoceptor agonist olodaterol attenuates pulmonary inflammation. Br. J. Pharmacol. 172, 3537–3547. doi: 10.1111/bph.13143
Wollin, L., and Pieper, M. P. (2010). Tiotropium bromide exerts anti-inflammatory activity in a cigarette smoke mouse model of COPD. Pulm. Pharmacol. Ther. 23, 345–354. doi: 10.1016/j.pupt.2010.03.008
Xu, Z. P., Yang, K., Xu, G. N., Zhu, L., Hou, L. N., Zhang, W. H., et al. (2012). Role of M3 mAChR in in vivo and in vitro models of LPS-induced inflammatory response. Int. Immunopharmacol. 14, 320–327. doi: 10.1016/j.intimp.2012.07.020
Yang, X., Zhao, C., Chen, X., Jiang, L., and Su, X. (2017). Monocytes primed with GTS-21/alpha7 nAChR (nicotinic acetylcholine receptor) agonist develop anti-inflammatory memory. QJM 110, 437–445.
Zhang, C., Yu, P., Zhu, L., Zhao, Q., Lu, X., and Bo, S. (2017). Blockade of alpha7 nicotinic acetylcholine receptors inhibit nicotine-induced tumor growth and vimentin expression in non-small cell lung cancer through MEK/ERK signaling way. Oncol. Rep. 38, 3309–3318.
Keywords: acetylcholine, cholinergic anti-inflammatory pathway, chronic obstructive pulmonary disease, neuroimmune interactions, α7 nicotinic acetylcholine receptor
Citation: Yamada M and Ichinose M (2018) The Cholinergic Pathways in Inflammation: A Potential Pharmacotherapeutic Target for COPD. Front. Pharmacol. 9:1426. doi: 10.3389/fphar.2018.01426
Received: 20 August 2018; Accepted: 19 November 2018;
Published: 03 December 2018.
Edited by:
Paolo Montuschi, Università Cattolica del Sacro Cuore, ItalyReviewed by:
Isaac Kirubakaran Sundar, University of Rochester, United StatesCopyright © 2018 Yamada and Ichinose. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Masakazu Ichinose, aWNoaW5vc2VAcm0ubWVkLnRvaG9rdS5hYy5qcA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.