 Lei Zhang
Lei Zhang Yanan Wang1†
Yanan Wang1† Suowen Xu
Suowen Xu Xiaojun Feng
Xiaojun Feng Sheng Liu
Sheng Liu- 1The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, China
- 2Aab Cardiovascular Research Institute, University of Rochester, Rochester, NY, United States
Cardiovascular disease is the main cause of death worldwide, but its pathogenesis is not yet clear. Hydrogen sulfide (H2S) is considered to be the third most important endogenous gasotransmitter in the organism after carbon monoxide and nitric oxide. It can be synthesized in mammalian tissues and can freely cross the cell membrane and exert many biological effects in various systems including cardiovascular system. More and more recent studies have supported the protective effects of endogenous H2S and exogenous H2S-releasing compounds (such as NaHS, Na2S, and GYY4137) in cardiovascular diseases, such as cardiac hypertrophy, heart failure, ischemia/reperfusion injury, and atherosclerosis. Here, we provided an up-to-date overview of the mechanistic actions of H2S as well as the therapeutic potential of various classes of H2S donors in treating cardiovascular diseases.
Introduction
Hydrogen sulfide (H2S) is a colorless, smelly water soluble gas (Wang, 2010). It was first described in the 17th century (Wang, 2012). Later in 1989, H2S was first discovered in rat brain (Altaany et al., 2013). H2S is considered to be the third most important endogenous gas molecule in the organism after carbon monoxide (CO) and nitric oxide (NO) (Wang, 2002; Hartle and Pluth, 2016; Szabo, 2016). It can be synthesized in mammalian tissues and can freely cross the cell membrane and exert many biological effects in various systems (Wang, 2012). Studies have found that H2S has played a role in neurophysiology, cardiovascular disease, endocrine regulation, and other physiological and pathological processes (Szabo, 2012; Li et al., 2015a).
Hydrogen sulfide is mainly produced by cystathionine gamma-lyase (CSE) and cystathionine beta-synthase (CBS) from L-cysteine and homocysteine (Chiku et al., 2009). It can also be produced in the presence of alpha-ketoglutarate by PRP-independent 3-mercapto-pyruvate sulfate transferase (3-MST) or cysteine aminotransferase (CAT) (Kabil and Banerjee, 2010; Li L. et al., 2011). Free H2S can be oxidized by sulfhydryl reductase (SQR) in mitochondria, and it can be methylated by sulfhydryl-S-methyltransferase in the cytoplasm (Bouillaud and Blachier, 2011; Levitt et al., 2011). In addition, free H2S is excreted through biological fluids after it is combined with methemoglobin and molecules with metal or disulfide bonds (Yang et al., 2004). In the human cardiovascular system, CSE is the major H2S production enzyme (Hosoki et al., 1997; Zhao et al., 2001), but the main H2S-producing enzyme in rat coronary arteries is 3-MST (Gadalla and Snyder, 2010; Li L. et al., 2011).
H2S levels may be enhanced in vivo with conventional inorganic sulfide salts, organic H2S donors, or phosphodiesterase inhibitors (Bełtowski, 2015). Common H2S donors include: sodium hydrosulfide, P-(4-methoxyphenyl)-p-4-morpholinodithiophosphoric acid (GYY4137) (Bankhele et al., 2018); 4-carboxyphenyl-isothiocyanate acid esters (4CPI) (Testai et al., 2016); SG-1002 (Kondo et al., 2013); cysteine analogs; S-propylcysteine; S-allylcysteine; N-acetylcysteine, and other drug chimeras such as L-DOPA, NOSH-sulindac (AVT-18A), NOSH-aspirin, ACS67 (mixed compound of latanoprost and H2S releasing moiety) (Kashfi and Olson, 2013; Salvi et al., 2016a). Among them, S-propargyl-cysteine (SPRC), which can slowly release H2S, also called ZYZ-802, is an analog of S-allylcysteine (SAC), and SAC is the most abundant component in aged garlic extract (Wen and Zhu, 2015). N-acetylcysteine (NAC) is commonly used as an antioxidant and cell protectant (Cerda and Pluth, 2018). L-cysteine is a substrate for the endogenous production of H2S (Salvi et al., 2016b), mitochondria-targeted anethole dithiolethione (AP39), and (AP123) (Gerő et al., 2016). Please see Table 1 for more information on common H2S donors as well as CBS and CSE inhibitors (Lertratanangkoon et al., 1999; Lima et al., 2006; Szabó, 2007; Li et al., 2008; Kulkarni et al., 2009; Sun et al., 2009; Tyagi et al., 2009; Liu et al., 2010, 2011; Wang et al., 2010, 2016; Kodela et al., 2012; Predmore et al., 2012; Guo et al., 2013; Kondo et al., 2013; Lisjak et al., 2013; Luo et al., 2013; Martelli et al., 2013, 2014; Barr et al., 2015; Fonseca et al., 2015; Iciek et al., 2015, 2016; Liang D. et al., 2015; Polhemus et al., 2015; Tsai et al., 2015; Vannini et al., 2015; Chao et al., 2016; Qian et al., 2016; Salvi et al., 2016a; Tain et al., 2016; Testai et al., 2016; Wu et al., 2016; Ali et al., 2018; Cao et al., 2018; Du et al., 2018; Ezeriņa et al., 2018; Liang et al., 2018; Lin S. et al., 2018; Ning et al., 2018; Powell et al., 2018; Qiu et al., 2018; Shimizu et al., 2018; Sone et al., 2018; Zhao et al., 2018; Zhou et al., 2018).
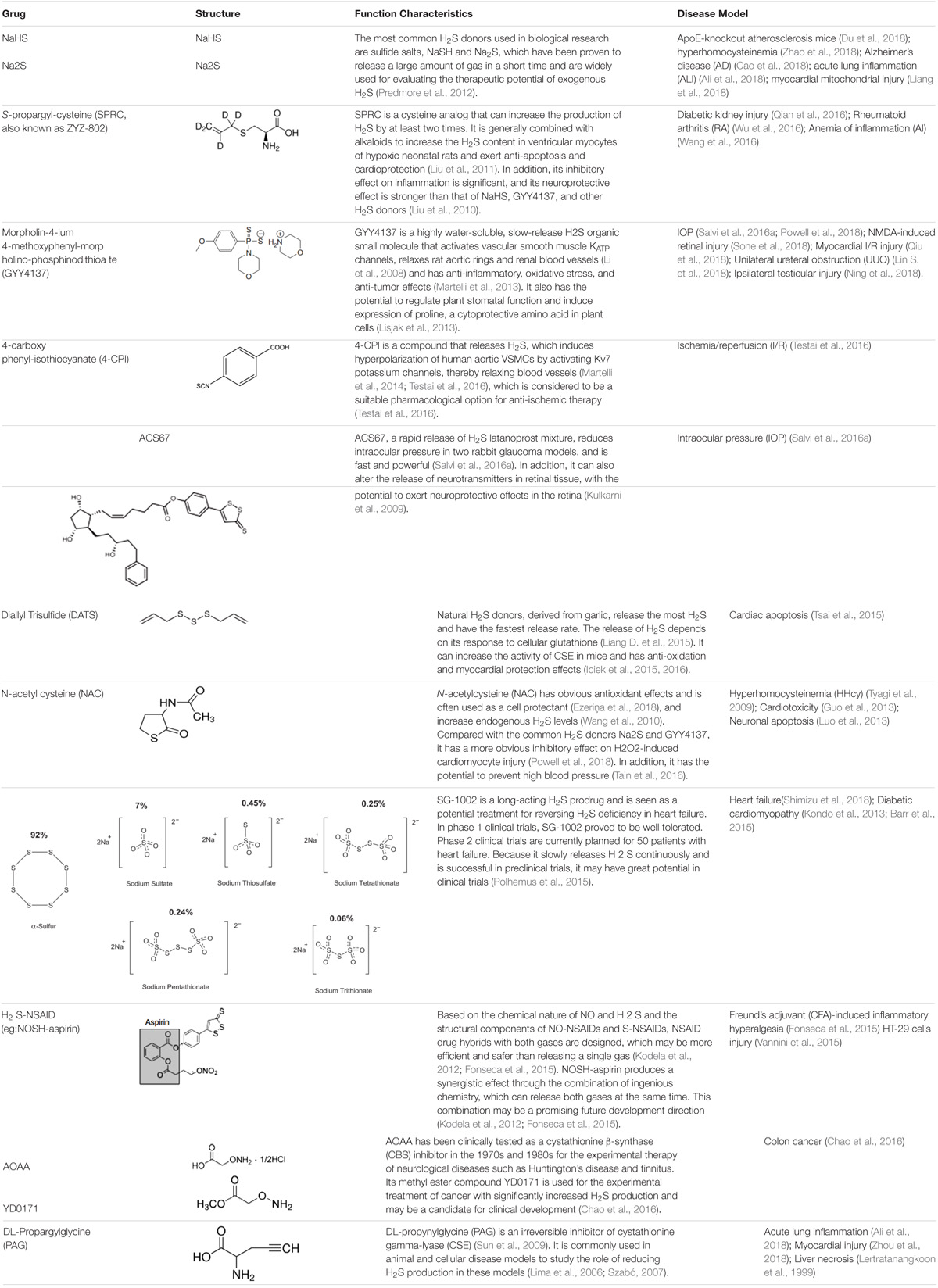
TABLE 1. Structure and characteristics of common H2S donors and inhibitor of CSE or CBS.
To date, there has been a lot of research on the therapeutic effects of H2S in atherosclerosis, cardiac remodeling, and myocardial ischemia-reperfusion injury (Yang et al., 2008; Wang et al., 2009; Calvert et al., 2010; Xu et al., 2014). Its related mechanisms involve anti-oxidation, inhibition of cell apoptosis, pro-angiogenesis, anti-inflammatory, ion channel regulation, and so on (Wang, 2012; Xu et al., 2014).
H2S and Cardiovascular Diseases
Atherosclerosis
Atherosclerosis is based on lipid metabolism disorder and is the main cause of various cardiovascular and cerebrovascular diseases, including coronary heart disease, cerebral infarction, and peripheral blood vessels. The development of atherosclerosis involves a variety of mechanisms, including endothelial cell damage and dysfunction, inflammatory cell recruitment, foam cell formation, smooth muscle cell proliferation and migration, calcification, fibrous cap rupture, and thrombosis (Bentzon et al., 2014; Nowak et al., 2017). Atherosclerotic lesions start from the intima, usually with accumulation of lipids and complex carbohydrates, hemorrhage and thrombosis, and then fibrous tissue hyperplasia and calcinosis, and gradually gradual metamorphosis and calcification of the arterial layer, leading to thickening of the arterial wall hardened, narrowed blood vessel lumen (Bentzon et al., 2014; Escárcega et al., 2018; Raggi et al., 2018).
Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia due to defects in insulin secretion and/or impaired biological effects. Hyperglycemia, which persists in diabetes, can cause chronic damage, endothelial dysfunction, and atherosclerosis (Bai et al., 2018; Wu T. et al., 2018).
Deregulation of H2S and H2S Producing Enzymes in Human and Mouse Atherosclerosis
Plasma H2S levels were obviously lower in patients with acute coronary syndrome (ACS), compared with patients with non-coronary artery disease (CAD), or stable angina pectoris (SAP). The levels of plasma monocyte chemokine receptor CCL2 and CX3CL1 were significantly increased (Gao et al., 2015b). In addition, studies in 113 patients with chronic hemodialysis showed an increase in cardiovascular risk factors (such as atherosclerosis) and mortality, which may be related to low levels of plasma H2S, activation of PKCβII, and upregulation of VCAM-1/ICAM-1 (Feng et al., 2015). The plasma H2S and aortic H2S levels were decreased in ApoE(-/-) mice. CSE expression was reduced in oxidized LDL (Ox-LDL)-stimulated human aortic endothelial cells (HAEC) and in the aorta of high fat diet-induced ApoE(-/-) mice (Leucker et al., 2017). The infiltration of red blood cells into atherosclerotic plaques is associated with atherosclerosis. Inside the lesion, hemoglobin (Hb) is oxidized to ferrous and sulfhydryl Hb to exhibit pro-oxidative and pro-inflammatory activity. The expression of CSE is mainly up-regulated in macrophages, foam cells, and myofibroblasts from human atherosclerotic lesions of patients with carotid specimens. A similar pattern was observed in aortic lesions of ApoE(-/-) mice on a high-fat diet. H2S can obviously reduce the oxidation of Hb and inhibit the progression of atherosclerosis (Potor et al., 2018). In mouse and human atherosclerosis, CSE expression is upregulated, but circulating and plaque levels of H2S are reduced, a phenomenon that can be attributed to inhibition of CSE enzyme activity (Bibli et al., 2018).
In adipose tissue macrophages (ATM) isolated from diet-induced obese mice, the intracellular concentration of H2S was lower than that of H2S in ATM from lean mice. The intracellular H2S concentration in the mouse macrophage cell line RAW264.7 was decreased during the inflammatory reaction induced by lipopolysaccharide (LPS). Production of pro-inflammatory cytokines in RAW264.7 cells and ATM from obese mice can be inhibited by exogenous H2S (Velmurugan et al., 2015). Homocysteine (Hcy) is a precursor of H2S, which forms H2S by a transsulfide pathway catalyzed by CBS and CSE (Li et al., 2015b). Hcy inhibits CSE expression by increasing DNA methylation in the CSE promoter region, whereas DNA methyltransferase (DNMT) knockout reverses the reduction of CSE transcription in Hcy-induced macrophages (Li et al., 2015b; Du et al., 2016). MicroRNAs (miRNAs) are small, siRNA-like molecules encoded by the genome of higher eukaryotes and are approximately 22 nucleotides in length. The miRNA induces silencing complex (RISC) to degrade mRNA or hinder translation by base pairing with the target gene mRNA (Bartel, 2009). CSE/H2S obviously increased ATP-binding cassette transporter A1 (ABCA1) expression and regulated cholesterol efflux in human THP-1 macrophage-derived foam cells, and MiR-216a significantly reduces CSE expression by directly targeting its 3′ untranslated region, thereby increasing cholesterol levels in THP-1 macrophage-derived foam cells (Gong et al., 2016). Similarly, MiR-186 can also directly inhibit CSE expression by targeting its 3′ untranslated region to promote THP-1 macrophage pro-inflammatory cytokine secretion and lipid accumulation (Yao et al., 2016).
H2S Donors Prevents Atherosclerosis in vivo
Endogenous H2S production is increased in aortic tissues of ApoE knockout and CSE gene overexpression mice (Tg/KO), atherosclerotic plaque size is reduced, and plasma lipid profile is reduced. Thus, activation of the CSE gene attenuates atherosclerotic symptoms in ApoE(-/-) mice (Cheung et al., 2014). GYY4137 reduced aortic atherosclerotic plaque in ApoE(-/-) mice and improved aortic endothelium-dependent relaxation. The specific mechanism is that GYY4137 reduces the expression of aortic ICAM-1, TNF-α, IL6, and LOX-1, and increases eNOS phosphorylation and PI3K expression (Liu et al., 2013). NaHS reduces atherosclerotic plaque in atherosclerotic rats and reduces ET-1 production in rat aortic endothelium (Liu et al., 2013). H2S can exert its cytoprotective effect through cysteine S-thiol to scavenge free radicals and inhibit oxidative stress, thereby inhibiting atherosclerosis (Cheung and Lau, 2018). In a mouse model of atherosclerosis induced by partial ligation of the left common carotid artery (LCA), NaHS administration significantly reduced the severity of atherosclerosis. This may be due to H2S up-regulating the expression of angiotensin converting enzyme 2 (ACE2) in the carotid artery, thereby converting angiotensin II to angiotensin 1-7 (Lin et al., 2017). H2S can also inhibit atherosclerosis in ApoE(-/-) mice as well as the proliferation and migration of vascular smooth muscle cells (VSMCs) by upregulating plasma NO (Lin et al., 2016). High-fat diet induced a significant decrease in plasma H2S levels and atrial natriuretic peptide (ANP) levels in rats with atherosclerosis, and elevated adrenomedullin (ADM) levels. Treatment with NaHS for 8 weeks reversed these changes in atherosclerotic rats (Li et al., 2015c). NaHS can also up-regulate the expression of ABCA1 by promoting the nuclear translocation of PPARα, which significantly reduces serum triglyceride (TG), cholesterol (TC), low-density lipids protein (LDL) levels, and atherosclerotic plaque size in high-fat diet-fed ApoE(-/-) mice (Li et al., 2016). SHR rats exhibit vascular remodeling and collagen accumulation. H2S regulates vascular collagen and inhibits VSMCs proliferation and collagen production (Zhao et al., 2008).
S-sulfuration is a signaling pathway for H2S, which is thought to be an antiatherogenic molecule that can prevent atherosclerosis. H2S induces S-sulfuration of glutathione peroxidase 1 and further reduces lipid peroxidation and increases antioxidant defense in the aorta by promoting glutathione synthesis (Cheung and Lau, 2018). NaHS or GYY4137 reduces SIRT1 degradation by direct s-sulfhydration, thereby reducing atherosclerotic plaque area, macrophage infiltration, aortic inflammation, and plasma lipid levels in ApoE(-/-) mice (Du et al., 2018). H2S can reduce aortic atherosclerotic plaque formation in streptozotocin (STZ)-treated LDLr(-/-) mice, but not LDLr and Nrf2 double knockout mice. This inhibitory effect of H2S may be related to Nrf2 activation through Keap1 s-sulfhydration (Xie et al., 2016). In oscillatory shear stress (OSS)-induced atherosclerosis, CSE expression is down-regulated, and NaHS activates eNOS and decreases the expression of intercellular ICAM-1, thereby inhibiting OSS-promoting atherosclerosis (Go et al., 2012). Estrogen can increase the production of H2S in the liver and vascular tissue by increasing the activity of CSE, thereby inhibiting atherosclerosis in female mice (Li et al., 2017a). Similarly, the estrogen 17β-estradiol (E2) activates CSE/H2S through PKG in endothelial cells, thereby dilating blood vessels and attenuating atherosclerosis in mice (Zhou et al., 2013).
VSMC plays a significant role in diseases such as atherosclerosis and restenosis after invasive intervention. S-Diclofenac is a novel molecule containing H2S (H2S is linked to diclofenac via an ester bond), inhibits smooth muscle cell proliferation, and may play a role in restenosis in vascular injury sites (Baskar et al., 2008). Similarly, atherosclerotic lesions were induced in rabbits, and we treated rabbits in a similar manner to balloon angioplasty (BA). NaHS treatment significantly reduced VSMCs proliferation in the neointimal, while DL-propargyl glycine (an inhibitor of H2S synthase) significantly induced VSMCs proliferation. Thus, H2S attenuates neointimal hyperplasia and inhibits restenosis after BA (Kuiper et al., 2001). In ApoE(-/-) mice, H2S inhibits proliferation and migration of VSMCs and inhibits the development of atherosclerosis by increasing plasma NO levels and increasing levels of S-nitrosylated proteins in VSMCs (Lin et al., 2016). Farnesyl pyrophosphate synthase (FPPS) plays an important role in the mevalonate pathway, and the FPPS inhibitor alendronate can alleviate diabetes-induced atherosclerosis and inhibit high glucose-induced proliferation of VSMCs in vitro. Specific mechanisms may be through reducing H2S metabolism and inhibiting small GTPase (Rac1, RhoA and Ras) activities (Chen et al., 2017).
H2S Donors Prevents Atherosclerosis in vitro
Improving endothelial dysfunction
Endothelial dysfunction is a vital event event in the early stages of atherosclerosis (Peng et al., 2017). Hyperglycemia is a key factor in the development of diabetic complications, such as atherosclerosis (Lin J. et al., 2018). Receptor interacting protein 3 (RIP3) mediates necrotic apoptosis and is involved in the development of atherosclerosis. NaHS significantly attenuated high-glucose (HG)-induced apoptosis of HUVECs by inhibiting the expression of RIP3 (Lin J. et al., 2018). NaHS also reduces atherosclerotic plaque in rats by protecting vascular endothelial cells and reducing the production of aortic endothelium ET-1 (Li et al., 2015d). In HUVEC, H2S inhibits H2O2-mediated mitochondrial dysfunction by maintaining levels of intracellular antioxidant enzymes (Go and Jones, 2005). In HUVEC, NaHS promotes expression of eNOS protein and NO production by increasing the expression of miR-455-3 (Li et al., 2017b). Similarly, in HUVEC, H2S inhibits TNF-α stimulated ICAM-1 expression by inhibiting NF-κB pathway (Wang et al., 2009). Two new mitochondrial targeting H2S donors AP39 and AP123 (30–300 nM) inhibit HG-induced damage by reducing hyperpolarization of endothelial cell mitochondrial membranes and inhibiting mitochondrial oxidant production. These mitochondria-targeted donors have an >1000-fold increase in potency in inhibiting HG-induced endothelial damage. This suggests that these compounds are useful for combating diabetic vascular complications (Gerő et al., 2016). Under the condition of glucose oxidase-induced oxidative stress, endothelial cells have enhanced oxidative stress, inhibited cell bioenergetic function, and decreased cell viability. AP39 pretreatment significantly attenuated the above reaction (Szczesny et al., 2014).
Tet methylcytosine dioxygenase 2 (TET2) is a DNA demethylase. In human umbilical vein endothelial cells (HUVECs), oxLDL treatment down-regulates TET2 and CSE/H2S, whereas TET2 overexpression up-regulates CSE/H2S by DNA demethylation of the CSE gene promoter, thereby inhibiting oxLDL stimulated NF-κB activation and ICAM-1 expression (Peng et al., 2017). Zofenoprilat is an active metabolite of zofenopril, which inhibits interleukin-1β (IL-1β)-induced inflammatory responses in HUVECs via the CSE/H2S pathway (e.g., NF-κB/COX-2 activation) (Monti et al., 2016). Its inflammation inhibition is also verified in vascular smooth muscle cells and fibroblasts (Monti et al., 2016). In addition, the HDAC6 inhibitor tubacin and HDAC6-specific siRNA inhibited OxLDL-induced decrease in endothelial cell CSE expression and improved endothelial function (Leucker et al., 2017).
Improving VSMCs dysfunction
CSE knockout (CSE-KO) mice’s mesenteric artery VSMCs, compared with CSE-WT cells, CSE-KO cells showed redox imbalance and abnormal mitochondrial activity, and were also more sensitive to hypoxia-induced cell death. It indicates that the endogenous CSE/H2S pathway significantly regulate the normal function of VSMCs (Bryan et al., 2011). Insulin-like growth factor-1 (IGF-1) exerts a variety of physiological and pathophysiological effects on the vascular system, including stimulation of VSMCs proliferation and migration. For VSMCs isolated from mesenteric arteries of wild type and CSE knockout mice, IGF-1 increases the proliferation of VSMCs, and the effect is more pronounced in CSE knockout-VSMCs. In addition, H2S significantly down-regulates IGF-1R expression, stimulates IGF-1R S-sulfation, and impairs IGF-1 binding to IGF-1R, thereby inhibiting IGF-1-induced VSMCs proliferation (Shuang et al., 2018). In VSMCs, H2S significantly reversed the decrease in iNOS expression and NO production induced by oxidized low-density lipoprotein and increased the protein S-nitrosylation level of VSMCs (Lin et al., 2016).
T-type channels (Cav3.1, 3.2, and 3.3) obviously affect the proliferation of VSMCs and H2S selectively inhibits T-channel Cav3.2. H2S induces concentration-dependent proliferation inhibition in the human coronary artery smooth muscle cells and smooth muscle cell line A7r5, however, this mechanism of suppressing cell proliferation is independent of selective inhibition of T-type channels by H2S (Elies et al., 2015). H2S reduces myogenic tension and causes relaxation of the phenylephrine (PE) mesenteric artery. H2S relaxes blood vessels by activating the large-conductance Ca(2+)-activated potassium channel (BKCa) and Cyp2C, a novel vasodilation pathway of emerging signaling molecules (Jackson-Weaver et al., 2013). H2S can also dilate blood vessels by increasing the KATP channel opening in VSMCs (Tang et al., 2005). In addition, maintaining cardiovascular homeostasis requires normal arterial baroreflex, and its sensitivity decreases during vascular calcification (VC). H2S can promote baroreflex sensitivity in hypertensive rats. H2S can directly promote the damage of baroreceptors in VSMCs calcification VC rats and improve VC (Li et al., 2017c). Vitamin D3 plus nicotine (VDN) induces VC and phenotypic conversion of VSMC in rats. H2S may alleviate VC and VSMC phenotypic changes by reducing endoplasmic reticulum stress (ERS) (Yang et al., 2016). In addition, NaHS enhances the expression of NADPH dehydrogenase 1 (NQO1) by enhancing nuclear factor (erythrocyte-derived 2)-like 2 (NRF2) activity, which in vitro attenuates calcineurin-induced calcification of VSMCs by cyclic troponin particles (CPP) (Aghagolzadeh et al., 2017).
Improving macrophage dysfunction
Cystathionine gamma-lyase expression and H2S production were reduced in oxidized ox-LDL-stimulated macrophages. Overexpression of CSE decreased ox-LDL-induced TNFα production by inhibiting the JNK/NF-κB signaling pathway (Wang et al., 2013). The NADPH oxidase member NADPH oxidase 4 (Nox4) is closely related to the production of reactive oxygen species (ROS). CSE knockdown expands inflammation by up-regulating Nox4-ROS signaling in sepsis mice and macrophages, and CSE overexpression reduces macrophage-enhanced inflammatory mediator production (Wang et al., 2018). GYY4137 inhibited the increase in TNF-α and IL-1β in HHcy mouse plasma and Hcy-stimulated RAW264.7 cells, whereas the CSE inhibitor PAG aggregated it (Li et al., 2015b). RAW264.7 or mouse peritoneal macrophages were stimulated with NaHS or saline and then induced by interferon-gamma (IFN-γ) or LPS. NaHS obviously attenuated the expression of aortic CX3CR1 and CX3CL1, as well as aortic plaque by modulating proliferator-activated receptor-gamma (PPAR-γ) and NF-κB activities (Zhang H. et al., 2012). The novel slow release H2S release compound FW1256 reduces the levels of TNFα, IL6, PGE, and NO in LPS-stimulated RAW264.7 macrophages (Huang et al., 2016).
Hypoxia-inducible factor 1-alpha (HIF-1α) plays an important regulatory role in inflammation. In THP-1 macrophages, H2S activates the HIF-1α/nuclear factor E2-related factor 2 (Nrf2) signaling pathway through the p38-MAPK pathway to reduce inflammation (Lohninger et al., 2015). H2S inhibits ox-LDL-stimulated macrophage inflammation response by inhibiting NF-κB recruitment to the monocyte chemoattractant protein 1 (MCP-1) promoter. Its molecular mechanism may be the s-sulfhydration of p65 (Du et al., 2014). In macrophages, S-sulfation of c-Jun by H2S enhances its transcription of p62 and SIRT3, which leads to a reduction in mtROS production and NLRP3 inflammasome activation (Lin et al., 2018). In addition, the absence of glutathione (GSH) and the upregulation of IL-1β are associated with the progression of vascular inflammation and atherosclerosis. H2S up-regulates GSH and suppresses IL-1β in U937 monocytes (Jain et al., 2014). H2S also inhibits histone acetylation in macrophages induced by LPS and inhibits transcription of various pro-inflammatory cytokines (Rios et al., 2015). The Jumonji domain protein 3 (JMJD3) is a histone demethylase with a target of histone 3 Lys27. JMJD3 knockdown attenuates LPS-induced inflammatory responses. Over-expression of CSE can reduce the inflammatory mediators produced by macrophages and thereby attenuate LPS-induced inflammatory responses by regulating JMJD3 expression in sepsis mice (Liu et al., 2018). Foam cell formation is a hallmark of atherosclerosis (Xu et al., 2011). H2S supplementation also inhibits foam cell formation in macrophages stimulated with pro-atherogenic factors, such as HG and oxLDL (Zhou et al., 2014; Liu et al., 2013; Xie et al., 2016).
Inhibiting platelet activation and thrombus formation
The relationship between H2S and platelet aggregation is not yet clear, and its mechanism of action needs further study. GYY4137 reduces thrombus stability by reducing platelet-leukocyte aggregation, thereby promoting endogenous thrombolysis (Grambow et al., 2017). NaHS may inhibit ADP or thrombin-induced platelet aggregation at least in part by inhibiting gap junctional cell-cell communication, and H2S released by H2S-releasing aspirin derivative (ACS14) may promote other antiplatelet effects in vitro compared to aspirin (Gao et al., 2015a). NaHS can inhibit collagen-induced platelet aggregation, and the mechanism is related to change of platelet [Ca]2+ (Zhong et al., 2014). Antithrombotic effects of GYY4137 in mice may modulate thrombosis by interfering with adhesion molecule-induced aggregation and platelet activation (Grambow et al., 2014). NaHS inhibits platelet aggregation in rabbits, which is dependent on cAMP (Nishikawa et al., 2013). H2S and dithionite inhibit platelet aggregation stimulated by ADF and collagen (Zaichko and Pentiuk, 2009). NaHS also significantly reversed endothelial cell (EC) damage and platelet aggregation induced by hyperhomocysteinemia (Wang et al., 2017).
In addition, NaHS inhibits platelet aggregation stimulated by collagen, ADP, arachidonic acid, epinephrine, thromboxane mimetics, U46619, and thrombin. However, this study considered that this effect of H2S does not depend on NO production, cAMP/cGMP production, or opening of potassium channels (Zagli et al., 2007). The above studies support H2S inhibition of platelet aggregation; however, there is also the opposite conclusion: NaHS significantly increases platelet aggregation collected in healthy volunteers induced by peptide-6 amide (a thrombin receptor activator) (d’Emmanuele et al., 2013).
Cardiac Hypertrophy and Heart Failure
Cardiac remodeling, including progressive pathological changes in heart and vessel size, shape, structure, and function, is characterized by progressive cardiac hypertrophy, ventricular dilatation, cardiac fibrosis, apoptosis, vascular dysfunction, and ultimately heart failure (HF) (Pfeffer and Braunwald, 1990; Rizzello et al., 2009). Prevention or reversal of cardiac remodeling is a key strategy for the treatment of HF (Konstam et al., 2011). The mechanisms of cardiac remodeling are complex, including the renin-angiotensin-aldosterone system (RAAS), autophagy, apoptosis, inflammation, matrix metalloproteinases, miRNAs, transcriptional, and post-transcriptional modifications (Swynghedauw, 1999; Maytin and Colucci, 2002; Fan et al., 2017). To illustrate the role of H2S in cardiac remodeling, we describe cardiac remodeling according to its inducing factors.
Effect of H2S on Cardiac Remodeling Induced by Renin–Angiotensin Receptor Agonists
Hyperstimulation of the β-adrenergic receptor (β-AR), which produces a hypertrophic effect in cardiomyocytes, can rapidly reduce endogenous H2S levels. Glucose-6-phosphate dehydrogenase (G6PD) is the rate-limiting enzyme that produce NADPH, and H2S may inhibit cardiac hypertrophy induced by adrenergic overstimulation by enhancing G6PD activity (Chhabra et al., 2018). Exogenous H2S also inhibits cardiac apoptosis and inhibit isoprenaline (ISO)-induced cardiac remodeling by maintaining mitochondrial membrane potential and reducing ROS production in mitochondria (Lu et al., 2013). In addition, exogenous administration of H2S inhibits ISO-induced left ventricular hypertrophy (LVH) by up-regulating CSE mRNA/H2S, while reducing systolic blood pressure and pulse wave velocity (Ahmad et al., 2018). ZYZ-802, a novel synthetic HS-NO hybrid molecule that decomposes H2S and NO, attenuates ISO-induced heart failure by increasing vascular endothelial growth factor (VEGF) levels and cyclic guanosine 5′-monophosphate (cGMP) levels (Wu D. et al., 2018). H2S also improve ISO-induced heart failure by inhibiting mast cell infiltration and renin degranulation to inhibit local renin levels (Liu et al., 2014).
In addition, sodium thiosulfate (STS), a clinically applicable H2S donor, and NaHS reduce Ang II-induced hypertension, cardiac hypertrophy, tissue fibrosis, and oxidative stress in rats (Snijder et al., 2015). In neonatal rat cardiomyocytes, H2S prevent Ang-II-induced cardiac hypertrophy by activating the Nrf2 pathway and reducing oxidative stress (Shao et al., 2017). H2S even improves glucose utilization in cardiomyocytes, including increased glucose uptake and glycolysis and citric acid cycle efficiency, inhibiting phenylephrine-induced cardiomyocyte hypertrophy (Liang M. et al., 2015).
Effect of H2S on Pressure Overload-Induced Cardiac Remodeling
Krüppel-like factor 5 (KLF5) exerts multiple functions in the cardiovascular system (McConnell and Yang, 2010). KLF5 knockout mice may reduce Ang II-induced inflammatory vascular responses and cardiac hypertrophy (Shindo et al., 2002). GYY4137 regulates KLF5 transcriptional activity through specific protein 1 S-sulfhydration to inhibit cardiac remodeling in spontaneously hypertensive rats (Meng et al., 2016). GYY4137 also inhibits cardiac fibrosis in hypertensive rats (SHR) by inhibiting TGF-β1/Smad2 signaling pathways, and inhibiting Alpha-smooth muscle actin (α-SMA) expression in cardiac fibroblasts (Meng et al., 2015b). Animal and human studies have shown that in LVH, changes in the expression of connexin 43 (Cx43) and disorganization of gap junctions are the basis for the occurrence and development of arrhythmia (Danik et al., 2004; Teunissen et al., 2004). H2S obviously inhibit cardiac hypertrophy and fibrosis caused by coarctation of the abdominal aorta by reducing the activity of cardiac Ang-II and up-regulating the expression of Cx43 (Huang et al., 2012). H2S induces angiogenesis and promotes blood vessel growth in the context of hindlimb ischemia and reduces left ventricular remodeling and dysfunction induced by lateral aortic coarctation in mice by promoting the growth of new blood vessels (Polhemus et al., 2013). SG-1002 inhibits myocardial remodeling induced by transverse aortic constriction in mice by up-regulating the expression of endothelial nitric oxide synthase (eNOS) and the production of NO (Kondo et al., 2013).
CSE knockout mice and heart-specific overexpressed mice were prepared using knock-out or transgenic techniques. After transverse aortic coarctation surgery, cardiac hypertrophy was significantly aggravated in CSE-knockout mice, but cardiac hypertrophy was significantly reduced in overexpressing mice. Mechanism studies have shown that CSE upregulates vascular endothelial growth factor-Akt-eNOS-NO-cGMP pathway, maintain mitochondrial function, weaken oxidative stress, and increase myocardial vascular density (Kondo et al., 2013). In addition, H2S improves heart function in chronic heart failure rats induced by coarctation of abdominal aorta by dilating blood vessels and affecting extracellular collagen metabolism (Especially type I collagen) (Li X.H. et al., 2011). H2S also induces matrix metalloproteinases (MMP)-2 to enhance VEGF synthesis and angiogenesis, while inhibiting TIMP-3 and MMP-9 levels, reducing anti-angiogenesis factors, and reducing intracardiac fibrosis and cardiac remodeling in pressure-overloaded mice (Givvimani et al., 2011). Heme oxygenase-1 (HO-1) is up-regulated by many oxidative stress in the cardiovascular system (Immenschuh and Schröder, 2006). HO-1 also reduce atherosclerotic lesions, ischemic myocardial injury, and modulated blood pressure (Johnson et al., 1997; Ndisang et al., 2002; Otterbein et al., 2003; Yet et al., 2003; Fujimoto et al., 2004; Liu et al., 2006). H2S inhibits volume overload-induced chronic heart failure (CHF) through up-regulation of HO-1 expression (Zhang et al., 2013).
Effects of H2S on Ischemic Injury-Induced Cardiac Remodeling
Hydrogen sulfide has beneficial effects on left ventricular hypertrophy after myocardial infarction (MI) in mice. H2S improves ischemic heart failure in mice by inhibiting oxidation, increasing mitochondrial biogenesis, and reducing apoptosis (Wu et al., 2017). In the heart failure (HF) model after MI induced by left coronary artery ligation in the left anterior descending coronary artery, NaHS inhibits heart cell apoptosis and improve mitochondrial dysfunction in HF hearts (Calvert et al., 2010). SPRC is a new type of endogenous H2S controlled-release preparation that protects rat HF from left coronary occlusion by maintaining levels of antioxidant molecules such as GSH, CAT, and SOD (Huang et al., 2013). NaNO2 significantly improved ischemia-induced left ventricular function in CHF mice by increasing H2S bioavailability, Nrf2 activation, and antioxidant defense (Donnarumma et al., 2016b). In addition, Na2S treatment inhibits ischemic heart failure in mice by inhibiting nuclear export of histone deacetylase 4 and apoptotic signaling kinase-1 signaling in a thioredoxin 1 dependent manner (Nicholson et al., 2013). Na2S also enhances cardiac proteasome activity and function through Nrf2-dependent manner, thereby inhibiting cardiac dysfunction (Shimizu et al., 2016).
The Effect of H2S on Other Types of Cardiac Remodeling
The level of H2S in mice with diabetic cardiomyopathy was reduced. H2S is able to improve the energy metabolism of cardiac tissue in db/db mice by up-regulating the expression and activity of SIRT3 (Sun et al., 2018). H2S also relieves streptozotocin-induced diabetic diabetic cardiomyopathy (DCM) development by reducing inflammation, oxidative stress, and apoptosis (Zhou et al., 2015a).
In the high-fat diet (HFD)-induced mouse cardiomyopathy model, circulating and cardiac H2S levels are also reduced, and SG-1002 treatment restores adiponectin levels and inhibit cardiac endoplasmic reticulum stress (Barr et al., 2015). Elevated homocysteine levels in hyperhomocysteinemia (HHcy) are the inducing factors of pathological cardiac remodeling. HHcy induces cardiac hypertrophy by facilitating MEF2C-HDAC1 complex formation, inactivating MEF2C, and inhibiting miR-133a in cardiomyocytes. H2S inhibits myocardial hypertrophy by activating MEF2C and inducing miR-133a in cardiomyocytes (Kesherwani et al., 2015). There may be negative feedback regulation between CBS and CSE enzymes, and in vivo studies using CBS(±) mice show that CBS deficiency increases cardiac CSE (Nandi and Mishra, 2017). In addition, H2S donor therapy reduces arteriovenous fistula (AVF)-induced cardiac cell fibrosis and apoptosis in mice by reducing oxidative and proteolytic pressures (Mishra et al., 2010). In AVF-induced heart failure model in mice, sodium thiosulfate (STS) partially improves cardiac dysfunction in mice by increasing H2S production (Sen et al., 2008). Passive smoking established rat left ventricular remodeling model. H2S may produce anti-oxidation through PI3K/Akt-dependent activation of Nrf2 signaling, thereby reducing ventricular remodeling (Zhou et al., 2015b). H2S improves endothelin-induced cardiac hypertrophy and fibrosis in rats (Yang et al., 2014). NaHS stimulates ANP secretion and reduces atrial pressure (AP) in the rat atrium through the K-channel and PI3K/Akt signaling pathway (Yu et al., 2018).
Ischemia Reperfusion Injury
Ischemic heart disease is mainly caused by atherosclerotic lesions of the coronary arteries, with a reduction in blood supply to the heart. The most serious type is myocardial infarction with a high mortality rate. Reperfusion is necessary to improve ischemia, but it also causes irreversible myocardial damage (Heusch, 2015). Therefore, it is necessary to understand the potential mechanisms of myocardial ischemia-reperfusion design, including AMPK, Akt, MAPK, PKA, and NO (Heusch, 2015, 2017) to better treat myocardial ischemia-reperfusion injury.
There are many reports on the role of H2S in cardiac I/R injury in rats. For example, NaHS prevents rat I/R heart damage by reducing the expression of proinflammatory cytokines and inducible nitric oxide synthase and up-regulating Akt/endothelial nitric oxide synthase (eNOS) (Issa et al., 2013). NaHS reduces infarct size of rat heart induced by I/R includes upregulation of heat shock protein 72 (Bliksøen et al., 2008), increased phosphorylated Akt and phosphorylated mTOR (Zhou et al., 2014), inhibition of mitochondria permeability transition (MPT) pore openings and increased cardiac mitochondrial membrane potential (Shymans’ka et al., 2012), and activation of PKC to regulate Intracellular Ca(2+) overload (Pan et al., 2008). Homocysteine levels and endogenous H2S production are primarily regulated by CBS and CSE enzymes. NaHS also improved myocardial recovery after cardiac I/R injury, however, its protective effect was abolished in CSE(-/--), CBS(-/-), and dietary hyperhomocysteinemia mice (Nakano et al., 2015). Hydrosulfide inhibits acute myocardial infarction (AMI)-induced apoptotic cell death by enhancing the phosphorylation of GSK-3β and the concentration of β-catenin (Ge et al., 2016). GYY4137 can prevent myocardial infarct size in rats by enhancing PI3K/Akt signaling (Karwi et al., 2016), reducing oxidative stress and apoptosis (Meng et al., 2015a). H2S prevents myocardial infarct size in rats by enhancing AMPK activity and restoring I/R impaired autophagy (Xie et al., 2015). H2S restores mitochondrial dysfunction, thereby reducing myocardial damage in I/R-impaired rat hearts (Ansari and Kurian, 2016). H2S prevent myocardial infarct size in rats by increasing the mitochondrial KATP channel opening time and the KATP opening frequency (Zhang Z. et al., 2007; Ji et al., 2008). H2S treatment inhibits myocardial infarct size in rats by activating the Akt, PKC, and eNOS pathways (Yong et al., 2008). In addition, H2S significantly reduced the I/R infarct size in isolated hearts, decreased the activity of creatine kinase and lactate dehydrogenase in heart tissue, and restored mitochondrial dysfunction (Ravindran et al., 2016). H2S inhibits myocardial infarct size and myocardial enzyme release in rats by activating Sirt1/PGC1α and JAK2/STAT3 signaling pathways (Luan et al., 2012; Hu et al., 2016), and by inhibiting oxidation and inhibiting the release of inflammatory factors (Zhang et al., 2015). H2S promotes angiogenesis by inhibiting the formation of parstatin (protease-activated receptor-1, a fragment of PAR-1) and promoting VEGF activation, and significantly inhibits the degree of MI injury in male mice that were ligated to the left anterior descending (LAD) (Qipshidze et al., 2012). The novel H2S-donor 4-carboxyphenyl isothiocyanate (4-CPI) significantly attenuates myocardial infarct size and ventricular arrhythmias in isolated rat hearts (Testai et al., 2016). The mitochondria-specific H2S donor AP39 significantly reduced infarct size induced by myocardial I/R injury in rats by inhibiting mitochondrial permeability transition pore (PTP) opening and mitochondrial ROS production (Karwi et al., 2017).
Alpha lipoic acid prevents arrhythmia after cardiac I/R in rats by affecting KATP channels. This effect of alpha lipoic acid may be related to the release of sulfur sulfur and H2S (Dudek et al., 2014). The intake of beetroot juice (BRJ) prevents myocardial infarction and ventricular dysfunction after I/R in adult male CD-1 mice through CSE-mediated endogenous H2S production (Salloum et al., 2015). Na2S administration can reduce infarct size induced by myocardial I/R injury in db/db mice (Peake et al., 2013). Zofenopril reduces myocardial infarct size in mice and pigs after I/R injury by increasing H2S and NO (Donnarumma et al., 2016a).
In addition to protection against rat heart I/R damage, H2S has the effect of improving the adverse physiological changes caused by porcine aortic occlusion (Causey et al., 2015). Infusion of H2S could provide myocardial protection against Yorkshire boar myocardial infarct size induced by I/R injury by increasing the expression of phosphorylated p44/42 MAPK and decreasing Beclin-1 expression, as well as reducing cell necrosis (Osipov et al., 2009). NaHS also reduces myocardial infarct size in myocardial I/R rabbits via the cGMP/PKG pathway (Bibli et al., 2015). Except for studying animal models of cardiac I/R injury, in the hypoxia/reoxygenation injury model of cardiomyocytes, H2S could inhibit the hypoxia/reoxygenation-induced cardiomyocyte apoptosis of rat H9c2 cardiomyocytes by attenuating the endo/sarcoplasmic reticulum (ER/SR) stress (Li et al., 2015e). H2S also regulates autophagy through activation of mTOR (Xiao et al., 2015), regulates PI3K/SGK1/GSK3β signaling pathway (Jiang et al., 2016), down-regulates microRNA-1 (miR-1), and upregulates Bcl-2 to exert resistance in hypoxia-reoxygenation models of neonatal rat cardiomyocytes.
Conclusion and Future Directions
In summary, H2S plays a significant protective role in atherosclerosis, cardiac hypertrophy, heart failure, and myocardial ischemia. Mechanisms responsible for these protective effects include down-regulation of oxidative stress responses, restoration of mitochondrial function, regulation of autophagy, attenuation of apoptosis, and increased blood vessel growth and angiogenesis (Figure 1). However, the evidence for these protective effects comes primarily from animal and cell models and lacks strong clinical evidence. The most urgent task for the future may be to replace H2S cardiac protection research from the laboratory to the clinic. In addition, plasma H2S levels in patients with cardiovascular diseases such as ACS and atherosclerosis are significantly reduced, which may provide strong support for clinical trials of H2S (Ali et al., 2016; Kanagy et al., 2017).
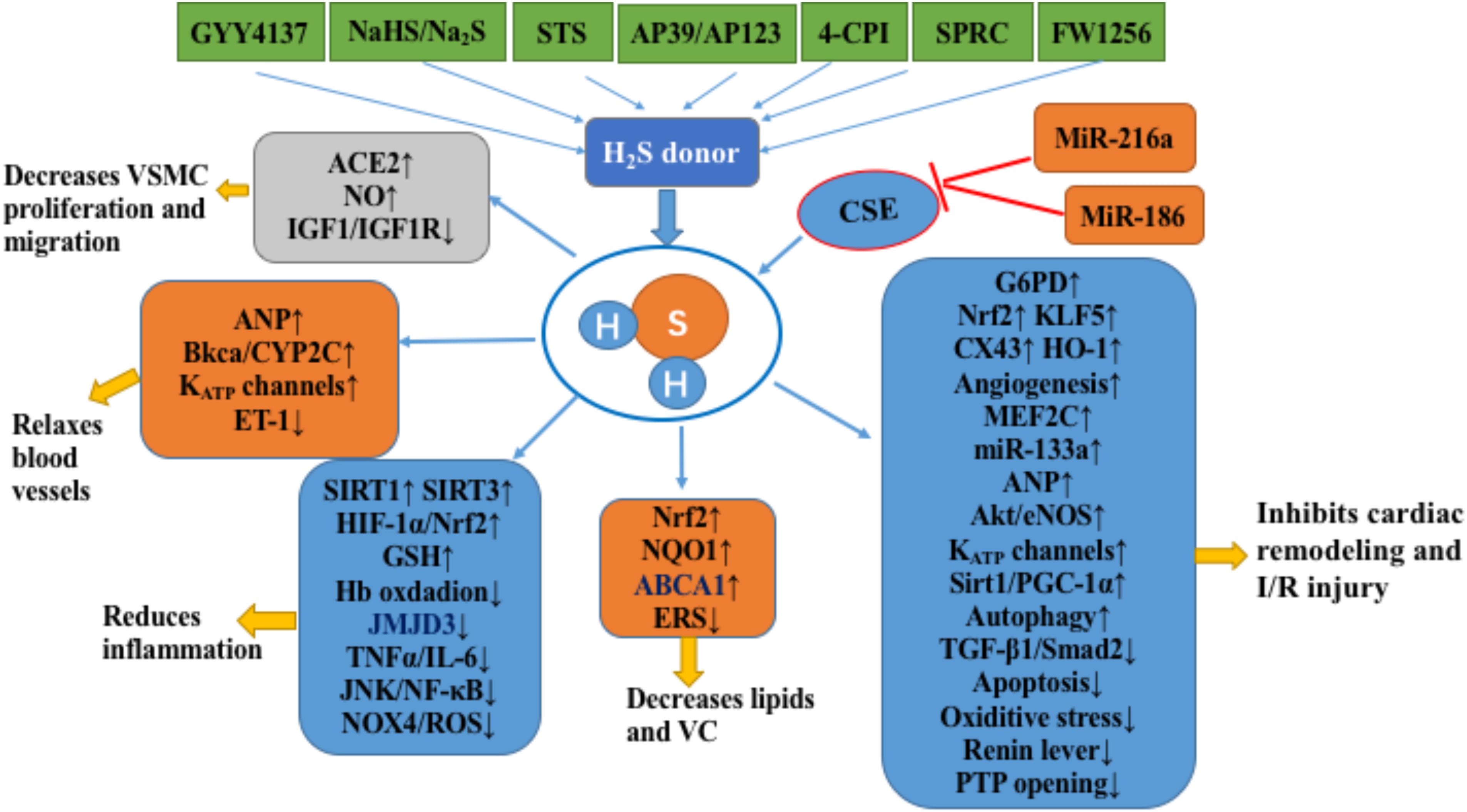
FIGURE 1. H2S in the cardiovascular system. H2S inhibits the occurrence and development of atherosclerosis by inhibiting the proliferation and migration of VSMCs, relaxing blood vessels, inhibiting inflammation, and inhibiting lipid accumulation and vascular calcification. In addition, H2S inhibits cardiac remodeling and cardiac ischemia-reperfusion injury by improving energy metabolism, anti-oxidation, anti-apoptosis, angiogenesis, and enhancing autophagy. The notation ↑ indicates increase or activation, and ↓indicates decrease or suppression. Abbreviations: STS, sodium thiosulfate; 4-CPI, 4-carboxyphenyl isothiocyanate; SPRC, S-propargyl-cysteine (also called ZYZ-802), a novel slow release H2S release compound (FW1256); VSMCs, vascular smooth muscle cells; ACE2, angiotensin converting enzyme 2; IGF-1, insulin-like growth factor-1; ANP, atrial natriuretic peptide; BKCa, large-conductance Ca(2+)-activated potassium channel; Cyp2C, cytochrome P-450 2C; KATP, ATP-sensitive potassium channel; ET-1, endothelin-1; Hb, hemoglobin; JMJD3, Jumonji domain protein 3; TNFα, tumor necrosis factor alpha; IL6, interleukin 6′; JNK, c-Jun NH2-terminal kinase; NF-κB, nuclear factor-kappa B; Nox4, NADPH oxidase member NADPH oxidase 4; ROS, reactive oxygen species; Sirt1, sirtuin 1; Sirt3, sirtuin 3; HIF-1α, hypoxia-inducible factor 1-alpha; GSH, glutathione; NRF2, nuclear factor (erythrocyte-derived 2)-like 2; NQO1, NADPH dehydrogenase 1; ABCA1, ATP-binding cassette transporter A1; ERS, endoplasmic reticulum stress; VC, vascular calcification; G6PD, glucose-6-phosphate dehydrogenase; KLF5, Krüppel-like factor 5; Cx43, connexin 43; HO-1, heme oxygenase-1; MEF2C, myosin enhancer factor-2c; Akt, protein kinase B; eNOS, endothelial nitric oxide synthase; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1 alpha; I/R, ischemia/reperfusion; CSE, cystathionine gamma-lyase; PTP, mitochondrial permeability transition pore.
Donor research that restores the body’s reduced H2S to physiological level is an important area for future research. For example, SG-1002 is a long-acting H2S prodrug to restore as much as possible the reduction of H2S levels in the mouse heart failure model (Kondo et al., 2013). As a potential treatment for reversing H2S deficiency in heart failure, it has now entered clinical research (Polhemus et al., 2015). Various drug-H2S hybrids have been synthesized to enhance drug efficacy and/or reduce adverse drug reactions, which is also a future research direction. For example, sulindac is a chemopreventive agent and its gastrointestinal side effects are common. NOSH-sulindac releases NO and H2S and inhibits 12 human cancer cell lines from 6 different tissue sources, and is 1,000 to 9,000 times more potent than sulindac. In addition, compared with sulindac, NOSH-sulindac significantly increased gastrointestinal safety in rats (Kashfi et al., 2015).
It has been mentioned previously that the role of H2S in platelet aggregation is controversial, and the role of H2S in inflammation is also controversial. For example, C/EBP homologous protein (CHOP) regulates the endoplasmic reticulum stress response and is up-regulated after cecal ligation and puncture (CLP). The CHOP gene knockout improves the survival rate after CLP. H2S plays a protective role by activating Nrf2 to inhibit the expression of CHOP in macrophages (Ferlito et al., 2014). Whereas male Swiss mice undergo CLP, H2S increases the levels of pro-inflammatory mediators through mechanisms involved in NF-κB activation and aggravates systemic inflammation in sepsis (Zhang H. et al., 2007). In addition, H2S circulates freely throughout the body, shuttles between different cells, and acts on a variety of cellular targets (Cirino et al., 2017; Yuan et al., 2017). Therefore, the role of H2S is not limited to cardiovascular, so the use of H2S donors need to consider its impact on the overall physiological and pathological, in order to avoid adverse reactions caused by H2S in some special circumstances.
In the end, due to the potential clinical concern of superphysiological (sub-millimolar to millimolar) concentration of H2S delivered by sulfide salts (such as NaHS, and Na2S), we can anticipate that more and more H2S-enriched natural products or synthetic compounds will be developed for cardiovascular therapeutics. These compounds are pharmacotherapeutically relevant in cardiovascular diseases and are hopeful to stimulate endogenous H2S production or release physiological concentrations of H2S in a sustainable manner (Whiteman et al., 2015). In addition, mitochondria-targeted donors have an >1000-fold increase in potency in inhibiting high glucose-induced endothelial damage (Gerő et al., 2016), and the mitochondria-specific H2S donor AP39 significantly reduced infarct size induced by myocardial I/R injury in rats (Karwi et al., 2017). Therefore, mitochondria-targeted H2S donors may represent an important direction of H2S research.
Author Contributions
LZ and YW contributed to the writing of the manuscript. YL and LL contributed to the revision. XF, SL, and SX conceptualized the manuscript.
Funding
This work was supported by National Nature Science Foundation of China (81603339) and Natural Science Foundation of Anhui Province (1708085QH175). SX is a recipient of Career Development Award from American Heart Association (18CDA34110359).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aghagolzadeh, P., Radpour, R., Bachtler, M., van Goor, H., Smith, E. R., Lister, A., et al. (2017). Hydrogen sulfide attenuates calcification of vascular smooth muscle cells via KEAP1/NRF2/NQO1 activation. Atherosclerosis 265, 78–86. doi: 10.1016/j.atherosclerosis.2017.08.012
Ahmad, A., Sattar, M. A., Azam, M., Khan, S. A., Bhatt, O., and Johns, E. J. (2018). Interaction between nitric oxide and renal α1-adrenoreceptors mediated vasoconstriction in rats with left ventricular hypertrophyin Wistar Kyoto rats. PLoS One 13:e0189386. doi: 10.1371/journal.pone.0189386
Ali, F. F., Abdel-Hamid, H. A., and Toni, N. D. (2018). H2S attenuates acute lung inflammation induced by administration of lipopolysaccharide in adult male rats. Gen. Physiol. Biophys. doi: 10.4149/gpb_2018002 [Epub ahead of print].
Ali, S. E., Farag, M. A., Holvoet, P., Hanafi, R. S., and Gad, M. Z. (2016). A comparative metabolomics approach reveals early biomarkers for metabolic response to acute myocardial infarction. Sci. Rep. 6:36359. doi: 10.1038/srep36359
Altaany, Z., Yang, G., and Wang, R. (2013). Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J. Cell. Mol. Med. 17, 879–888. doi: 10.1111/jcmm.12077
Ansari, S. B., and Kurian, G. A. (2016). Hydrogen sulfide modulates sub-cellular susceptibility to oxidative stress induced by myocardial ischemic reperfusion injury. Chem. Biol. Interact. 252, 28–35. doi: 10.1016/j.cbi.2016.03.036
Bai, L., Gao, J., Wei, F., Zhao, J., Wang, D., and Wei, J. (2018). Therapeutic potential of ginsenosides as an adjuvant treatment for diabetes. Front. Pharmacol. 9:423. doi: 10.3389/fphar.2018.00423
Bankhele, P., Salvi, A., Jamil, J., Njie-Mbye, F., Ohia, S., and Opere, C. A. (2018). Comparative effects of hydrogen sulfide-releasing compounds on [3H]D-aspartate release from bovine isolated retinae. Neurochem. Res. 43, 692–701. doi: 10.1007/s11064-018-2471-5
Barr, L. A., Shimizu, Y., Lambert, J. P., Nicholson, C. K., and Calvert, J. W. (2015). Hydrogen sulfide attenuates high fat diet-induced cardiac dysfunction via the suppression of endoplasmic reticulum stress. Nitric Oxide 46, 145–156. doi: 10.1016/j.niox.2014.12.013
Bartel, D. P. (2009). MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. doi: 10.1016/j.cell.2009.01.002
Baskar, R., Sparatore, A., Del, S. P., and Moore, P. K. (2008). Effect of S-diclofenac, a novel hydrogen sulfide releasing derivative inhibit rat vascular smooth muscle cell proliferation. Eur. J. Pharmacol. 594, 1–8. doi: 10.1016/j.ejphar.2008.07.029
Bełtowski, J. (2015). Hydrogen sulfide in pharmacology and medicine–An update. Pharmacol. Rep. 67, 647–658. doi: 10.1016/j.pharep.2015.01.005
Bentzon, J. F., Otsuka, F., Virmani, R., and Falk, E. (2014). Mechanisms of plaque formation and rupture. Circ. Res. 114, 1852–1866. doi: 10.1161/CIRCRESAHA.114.302721
Bibli, S. I., Andreadou, I., Chatzianastasiou, A., Tzimas, C., Sanoudou, D., Kranias, E., et al. (2015). Cardioprotection by H2S engages a cGMP-dependent protein kinase G/phospholamban pathway. Cardiovasc. Res. 106, 432–442. doi: 10.1093/cvr/cvv129
Bibli, S. I., Hu, J., Sigala, F., Wittig, I., Heidler, J., Zukunft, S., et al. (2018). Cystathionine γ lyase sulfhydrates the RNA binding protein HuR to preserve endothelial cell function and delay atherogenesis. Circulation doi: 10.1161/CIRCULATIONAHA.118.034757 [Epub ahead of print].
Bliksøen, M., Kaljusto, M. L., Vaage, J., and Stensløkken, K. O. (2008). Effects of hydrogen sulphide on ischaemia-reperfusion injury and ischaemic preconditioning in the isolated, perfused rat heart. Eur. J. Cardiothorac. Surg. 34, 344–349. doi: 10.1016/j.ejcts.2008.03.017
Bouillaud, F., and Blachier, F. (2011). Mitochondria and sulfide: a very old story of poisoning, feeding, and signaling. Antioxid. Redox Signal. 15, 379–391. doi: 10.1089/ars.2010.3678
Bryan, S., Yang, G., Wang, R., and Khaper, N. (2011). Cystathionine gamma-lyase-deficient smooth muscle cells exhibit redox imbalance and apoptosis under hypoxic stress conditions. Exp. Clin. Cardiol. 16, e36–e41.
Calvert, J. W., Elston, M., Nicholson, C. K., Gundewar, S., Jha, S., Elrod, J. W., et al. (2010). Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia-induced heart failure in mice. Circulation 122, 11–19. doi: 10.1161/CIRCULATIONAHA.109.920991
Cao, L., Cao, X., Zhou, Y., Vijay, N. B., Wu, Z. Y., Hu, L. F., et al. (2018). Hydrogen sulfide inhibits ATP-induced neuroinflammation and Aβ1-42 synthesis by suppressing the activation of STAT3 and cathepsin S. Brain Behav. Immun. doi: 10.1016/j.bbi.2018.07.005 [Epub ahead of print].
Causey, M. W., Miller, S., Singh, N., and Martin, M. (2015). Pharmacologic attenuation of the hyperdynamic response to supraceliac aortic clamping. J. Vasc. Surg. 61, 224–230. doi: 10.1016/j.jvs.2013.08.033
Cerda, M. M., and Pluth, M. D. (2018). S marks the spot: linking the antioxidant activity of N-acetyl cysteine to H2S and sulfane sulfur species. Cell Chem. Biol. 25, 353–355. doi: 10.1016/j.chembiol.2018.04.001
Chao, C., Zatarain, J. R., Ding, Y., Coletta, C., Mrazek, A. A., Druzhyna, N., et al. (2016). Cystathionine-beta-synthase inhibition for colon cancer: enhancement of the efficacy of aminooxyacetic acid via the prodrug approach. Mol. Med. doi: 10.2119/molmed.2016.00102 [Epub ahead of print].
Chen, G. P., Zhang, X. Q., Wu, T., Han, J., and Ye, D. (2017). Inhibition of farnesyl pyrophosphate synthase attenuates high glucose-induced vascular smooth muscle cells proliferation. Mol. Med. Rep. 15, 3153–3160. doi: 10.3892/mmr.2017.6360
Cheung, S. H., Kwok, W. K., To, K. F., and Lau, J. Y. (2014). Anti-atherogenic effect of hydrogen sulfide by over-expression of cystathionine gamma-lyase (CSE) gene. PLoS One 9:e113038. doi: 10.1371/journal.pone.0113038
Cheung, S. H., and Lau, J. Y. W. (2018). Hydrogen sulfide mediates athero-protection against oxidative stress via S-sulfhydration. PLoS One 13:e0194176. doi: 10.1371/journal.pone.0194176
Chhabra, A., Mishra, S., Kumar, G., Gupta, A., Keshri, G. K., Bharti, B., et al. (2018). Glucose-6-phosphate dehydrogenase is critical for suppression of cardiac hypertrophy by H2S. Cell Death Discov. 4:6. doi: 10.1038/s41420-017-0010-9
Chiku, T., Padovani, D., Zhu, W., Singh, S., Vitvitsky, V., and Banerjee, R. (2009). H2S biogenesis by human cystathionine gamma-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem. 284, 11601–11612. doi: 10.1074/jbc.M808026200
Cirino, G., Vellecco, V., and Bucci, M. (2017). Nitric oxide and hydrogen sulfide: the gasotransmitter paradigm of the vascular system. Br. J. Pharmacol. 174, 4021–4031. doi: 10.1111/bph.13815
Danik, S. B., Liu, F., Zhang, J., Suk, H. J., Morley, G. E., Fishman, G. I., et al. (2004). Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ. Res. 95, 1035–1041. doi: 10.1161/01.RES.0000148664.33695.2a
d’Emmanuele, D. V. B. R., Mitidieri, E., Di, M. N., Kirkby, N. S., Warner, T. D., Di, M. G., et al. (2013). Hydrogen sulphide pathway contributes to the enhanced human platelet aggregation in hyperhomocysteinemia. Proc. Natl. Acad. Sci. U.S.A. 110, 15812–15817. doi: 10.1073/pnas.1309049110
Donnarumma, E., Ali, M. J., Rushing, A. M., Scarborough, A. L., Bradley, J. M., Organ, C. L., et al. (2016a). Zofenopril protects against myocardial ischemia-reperfusion injury by increasing nitric oxide and hydrogen sulfide bioavailability. J. Am. Heart Assoc. 5:e003531. doi: 10.1161/JAHA.116.003531
Donnarumma, E., Bhushan, S., Bradley, J. M., Otsuka, H., Donnelly, E. L., Lefer, D. J., et al. (2016b). Nitrite therapy ameliorates myocardial dysfunction via H2S and nuclear factor-erythroid 2-related factor 2 (Nrf2)-dependent signaling in chronic heart failure. J. Am. Heart Assoc. 5:e003551. doi: 10.1161/JAHA.116.003551
Du, C., Lin, X., Xu, W., Zheng, F., Cai, J., Yang, J., et al. (2018). Sulfhydrated sirtuin-1 increasing its deacetylation activity is an essential epigenetics mechanism of anti-atherogenesis by hydrogen sulfide. Antioxid. Redox Signal. doi: 10.1089/ars.2017.7195 [Epub ahead of print].
Du, H. P., Li, J., You, S. J., Wang, Y. L., Wang, F., Cao, Y. J., et al. (2016). DNA methylation in cystathionine-γ-lyase (CSE) gene promoter induced by ox-LDL in macrophages and in apoE knockout mice. Biochem. Biophys. Res. Commun. 469, 776–782. doi: 10.1016/j.bbrc.2015.11.132
Du, J., Huang, Y., Yan, H., Zhang, Q., Zhao, M., Zhu, M., et al. (2014). Hydrogen sulfide suppresses oxidized low-density lipoprotein (ox-LDL)-stimulated monocyte chemoattractant protein 1 generation from macrophages via the nuclear factor κB (NF-κB) pathway. J. Biol. Chem. 289, 9741–9753. doi: 10.1074/jbc.M113.517995
Dudek, M., Knutelska, J., Bednarski, M., Nowiński, L., Zygmunt, M., Bilska-Wilkosz, A., et al. (2014). Alpha lipoic acid protects the heart against myocardial post ischemia-reperfusion arrhythmias via KATP channel activation in isolated rat hearts. Pharmacol. Rep. 66, 499–504. doi: 10.1016/j.pharep.2013.11.001
Elies, J., Johnson, E., Boyle, J. P., Scragg, J. L., and Peers, C. (2015). H2S does not regulate proliferation via T-type Ca2+ channels. Biochem. Biophys. Res. Commun. 461, 659–664. doi: 10.1016/j.bbrc.2015.04.087
Escárcega, R. O., Lipinski, M. J., García-Carrasco, M., Mendoza-Pinto, C., Galvez-Romero, J. L., and Cervera, R. (2018). Inflammation and atherosclerosis: cardiovascular evaluation in patients with autoimmune diseases. Autoimmun. Rev. 17, 703–708. doi: 10.1016/j.autrev.2018.01.021
Ezeriņa, D., Takano, Y., Hanaoka, K., Urano, Y., and Dick, T. P. (2018). N-acetyl cysteine functions as a fast-acting antioxidant by triggering intracellular H2S and sulfane sulfur production. Cell Chem. Biol. 25, 447–459.e4. doi: 10.1016/j.chembiol.2018.01.011
Fan, D. C., Qi, J. Y., and Zhang, M. Z. (2017). Insights of Chinese medicine on ventricular remodeling: multiple-targets, individualized-treatment. Chin. J. Integr. Med. 23, 643–647. doi: 10.1007/s11655-017-2415-y
Feng, S. J., Li, H., and Wang, S. X. (2015). Lower hydrogen sulfide is associated with cardiovascular mortality, which involves cPKCβII/akt pathway in chronic hemodialysis patients. Blood Purif. 40, 260–269. doi: 10.1159/000439580
Ferlito, M., Wang, Q., Fulton, W. B., Colombani, P. M., Marchionni, L., Fox-Talbot, K., et al. (2014). Hydrogen sulfide [corrected] increases survival during sepsis: protective effect of CHOP inhibition. J. Immunol. 192, 1806–1814. doi: 10.4049/jimmunol.1300835
Fonseca, M. D., Cunha, F. Q., Kashfi, K., and Cunha, T. M. (2015). NOSH-aspirin (NBS-1120), a dual nitric oxide and hydrogen sulfide-releasing hybrid, reduces inflammatory pain. Pharmacol. Res. Perspect. 3:e00133. doi: 10.1002/prp2.133
Fujimoto, H., Ohno, M., Ayabe, S., Kobayashi, H., Ishizaka, N., Kimura, H., et al. (2004). Carbon monoxide protects against cardiac ischemia–reperfusion injury in vivo via MAPK and Akt–eNOS pathways. Arterioscler. Thromb. Vasc. Biol. 24, 1848–1853. doi: 10.1161/01.ATV.0000142364.85911.0e
Gadalla, M. M., and Snyder, S. H. (2010). Hydrogen sulfide as a gasotransmitter. J. Neurochem. 113, 14–26. doi: 10.1111/j.1471-4159.2010.06580.x
Gao, L., Cheng, C., Sparatore, A., Zhang, H., and Wang, C. (2015a). Hydrogen sulfide inhibits human platelet aggregation in vitro in part by interfering gap junction channels: effects of ACS14, a hydrogen sulfide-releasing aspirin. Heart Lung Circ. 24, 77–85. doi: 10.1016/j.hlc.2014.05.019
Gao, L., Xu, Z., Yin, Z., Chen, K., Wang, C., and Zhang, H. (2015b). Association of hydrogen sulfide with alterations of monocyte chemokine receptors, CCR2 and CX3CR1 in patients with coronary artery disease. Inflamm. Res. 64, 627–635. doi: 10.1007/s00011-015-0844-7
Ge, N., Liu, C., Li, G., Xie, L., Zhang, Q., Li, L., et al. (2016). Hydrosulfide attenuates acute myocardial ischemic injury through the glycogen synthase kinase-3β/β-catenin signaling pathway. Int. J. Mol. Med. 37, 1281–1289. doi: 10.3892/ijmm.2016.2538
Gerő, D., Torregrossa, R., Perry, A., Waters, A., Le-Trionnaire, S., Whatmore, J. L., et al. (2016). The novel mitochondria-targeted hydrogen sulfide (H2S) donors AP123 and AP39 protect against hyperglycemic injury in microvascular endothelial cells in vitro. Pharmacol. Res. 113, 186–198. doi: 10.1016/j.phrs.2016.08.019
Givvimani, S., Munjal, C., Gargoum, R., Sen, U., Tyagi, N., Vacek, J. C., et al. (2011). Hydrogen sulfide mitigates transition from compensatory hypertrophy to heart failure. J. Appl. Physiol. 110, 1093–1100. doi: 10.1152/japplphysiol.01064.2010
Go, Y. M., and Jones, D. P. (2005). Intracellular proatherogenic events and cell adhesion modulated by extracellular thiol/disulfide redox state. Circulation 111, 2973–2980. doi: 10.1161/CIRCULATIONAHA.104.515155
Go, Y. M., Lee, H. R., and Park, H. (2012). H(2)S inhibits oscillatory shear stress-induced monocyte binding to endothelial cells via nitric oxide production. Mol. Cells 34, 449–455. doi: 10.1007/s10059-012-0200-5
Gong, D., Cheng, H. P., Xie, W., Zhang, M., Liu, D., Lan, G., et al. (2016). Cystathionine γ-lyase(CSE)/hydrogen sulfide system is regulated by miR-216a and influences cholesterol efflux in macrophages via the PI3K/AKT/ABCA1 pathway. Biochem. Biophys. Res. Commun. 470, 107–116. doi: 10.1016/j.bbrc.2016.01.003
Grambow, E., Leppin, C., Leppin, K., Kundt, G., Klar, E., Frank, M., et al. (2017). The effects of hydrogen sulfide on platelet-leukocyte aggregation and microvascular thrombolysis. Platelets 28, 509–517. doi: 10.1080/09537104.2016.1235693
Grambow, E., Mueller-Graf, F., Delyagina, E., Frank, M., Kuhla, A., and Vollmar, B. (2014). Effect of the hydrogen sulfide donor GYY4137 on platelet activation and microvascular thrombus formation in mice. Platelets 25, 166–174. doi: 10.3109/09537104.2013.786823
Guo, R., Lin, J., Xu, W., Shen, N., Mo, L., Zhang, C., et al. (2013). Hydrogen sulfide attenuates doxorubicin-induced cardiotoxicity by inhibition of the p38 MAPK pathway in H9c2 cells. Int. J. Mol. Med. 31, 644–650. doi: 10.3892/ijmm.2013.1246
Hartle, M. D., and Pluth, M. D. (2016). A practical guide to working with H2S at the interface of chemistry and biology. Chem. Soc. Rev. 45, 6108–6117. doi: 10.1039/c6cs00212a
Heusch, G. (2015). Molecular basis of cardioprotection: signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 116, 674–699. doi: 10.1161/CIRCRESAHA.116.305348
Heusch, G. (2017). Cardioprotection is alive but remains enigmatic: the nitric oxide-protein kinases-mitochondria signaling axis. Circulation 136, 2356–2358. doi: 10.1161/CIRCULATIONAHA.117.031978
Hosoki, R., Matsuki, N., and Kimura, H. (1997). The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 237, 527–531. doi: 10.1006/bbrc.1997.6878
Hu, M. Z., Zhou, B., Mao, H. Y., Sheng, Q., Du, B., Chen, J. L., et al. (2016). Exogenous hydrogen sulfide postconditioning protects isolated rat hearts from ischemia/reperfusion injury through Sirt1/PGC-1α signaling pathway. Int. Heart J. 57, 477–482. doi: 10.1536/ihj.15-506
Huang, C., Kan, J., Liu, X., Ma, F., Tran, B. H., Zou, Y., et al. (2013). Cardioprotective effects of a novel hydrogen sulfide agent-controlled release formulation of S-propargyl-cysteine on heart failure rats and molecular mechanisms. PLoS One 8:e69205. doi: 10.1371/journal.pone.0069205
Huang, C. W., Feng, W., Peh, M. T., Peh, K., Dymock, B. W., and Moore, P. K. (2016). A novel slow-releasing hydrogen sulfide donor, FW1256, exerts anti-inflammatory effects in mouse macrophages and in vivo. Pharmacol. Res. 113, 533–546. doi: 10.1016/j.phrs.2016.09.032
Huang, J., Wang, D., Zheng, J., Huang, X., and Jin, H. (2012). Hydrogen sulfide attenuates cardiac hypertrophy and fibrosis induced by abdominal aortic coarctation in rats. Mol. Med. Rep. 5, 923–928. doi: 10.3892/mmr.2012.748
Iciek, M., Bilska-Wilkosz, A., Górny, M., Sokołowska-Jeżewicz, M., and Kowalczyk-Pachel, D. (2016). The effects of different garlic-derived allyl sulfides on anaerobic sulfur metabolism in the mouse kidney. Antioxidants 5:E46. doi: 10.3390/antiox5040046
Iciek, M., Kowalczyk-Pachel, D., Bilska-Wilkosz, A., Kwiecień, I., Górny, M., and Włodek, L. (2015). S-sulfhydration as a cellular redox regulation. Biosci. Rep. 36:e00304. doi: 10.1042/BSR20150147
Immenschuh, S., and Schröder, H. (2006). Heme oxygenase-1 and cardiovascular disease. Histol. Histopathol. 21, 679–685. doi: 10.14670/HH-21.679
Issa, K., Kimmoun, A., Collin, S., Ganster, F., Fremont-Orlowski, S., Asfar, P., et al. (2013). Compared effects of inhibition and exogenous administration of hydrogen sulphide in ischaemia-reperfusion injury. Crit. Care 17:R129. doi: 10.1186/cc12808
Jackson-Weaver, O., Osmond, J. M., Riddle, M. A., Naik, J. S., Gonzalez, B. L. V., Walker, B. R., et al. (2013). Hydrogen sulfide dilates rat mesenteric arteries by activating endothelial large-conductance Ca2 +-activated K+ channels and smooth muscle Ca2 + sparks. Am. J. Physiol. Heart Circ. Physiol. 304, H1446–H1454. doi: 10.1152/ajpheart.00506.2012
Jain, S. K., Huning, L., and Micinski, D. (2014). Hydrogen sulfide upregulates glutamate-cysteine ligase catalytic subunit, glutamate-cysteine ligase modifier subunit, and glutathione and inhibits interleukin-1β secretion in monocytes exposed to high glucose levels. Metab. Syndr. Relat. Disord. 12, 299–302. doi: 10.1089/met.2014.0022
Ji, Y., Pang, Q. F., Xu, G., Wang, L., Wang, J. K., and Zeng, Y. M. (2008). Exogenous hydrogen sulfide postconditioning protects isolated rat hearts against ischemia-reperfusion injury. Eur. J. Pharmacol. 587, 1–7. doi: 10.1016/j.ejphar.2008.03.044
Jiang, H., Xiao, J., Kang, B., Zhu, X., Xin, N., and Wang, Z. (2016). PI3K/SGK1/GSK3β signaling pathway is involved in inhibition of autophagy in neonatal rat cardiomyocytes exposed to hypoxia/reoxygenation by hydrogen sulfide. Exp. Cell Res. 345, 134–140. doi: 10.1016/j.yexcr.2015.07.005
Johnson, R. A., Colombari, E., Colombari, D. S., Lavesa, M., Talman, W. T., and Nasjletti, A. (1997). Role of endogenous carbon monoxide in central regulation of arterial pressure. Hypertension 30, 962–967. doi: 10.1161/01.HYP.30.4.962
Kabil, O., and Banerjee, R. (2010). Redox biochemistry of hydrogen sulfide. J. Biol. Chem. 285, 21903–21907. doi: 10.1074/jbc.R110.128363
Kanagy, N. L., Szabo, C., and Papapetropoulos, A. (2017). Vascular biology of hydrogen sulfide. Am. J. Physiol. Cell Physiol. 312, C537–C549. doi: 10.1152/ajpcell.00329.2016
Kang, B., Hong, J., Xiao, J., Zhu, X., Ni, X., Zhang, Y., et al. (2014). Involvement of miR-1 in the protective effect of hydrogen sulfide against cardiomyocyte apoptosis induced by ischemia/reperfusion. Mol. Biol. Rep. 41, 6845–6853. doi: 10.1007/s11033-014-3570-2
Karwi, Q. G., Bornbaum, J., Boengler, K., Torregrossa, R., Whiteman, M., Wood, M. E., et al. (2017). AP39, a mitochondria-targeting hydrogen sulfide (H2S) donor, protects against myocardial reperfusion injury independently of salvage kinase signalling. Br. J. Pharmacol. 174, 287–301. doi: 10.1111/bph.13688
Karwi, Q. G., Whiteman, M., Wood, M. E., Torregrossa, R., and Baxter, G. F. (2016). Pharmacological postconditioning against myocardial infarction with a slow-releasing hydrogen sulfide donor, GYY4137. Pharmacol. Res. 111, 442–451. doi: 10.1016/j.phrs.2016.06.028
Kashfi, K., Chattopadhyay, M., and Kodela, R. (2015). NOSH-sulindac (AVT-18A) is a novel nitric oxide- and hydrogen sulfide-releasing hybrid that is gastrointestinal safe and has potent anti-inflammatory, analgesic, antipyretic, anti-platelet, and anti-cancer properties. Redox Biol. 6, 287–296. doi: 10.1016/j.redox.2015.08.012
Kashfi, K., and Olson, K. R. (2013). Biology and therapeutic potential of hydrogen sulfide and hydrogen sulfide-releasing chimeras. Biochem. Pharmacol. 85, 689–703. doi: 10.1016/j.bcp.2012.10.019
Kesherwani, V., Nandi, S. S., Sharawat, S. K., Shahshahan, H. R., and Mishra, P. K. (2015). Hydrogen sulfide mitigates homocysteine-mediated pathological remodeling by inducing miR-133a in cardiomyocytes. Mol. Cell. Biochem. 404, 241–250. doi: 10.1007/s11010-015-2383-5
Kodela, R., Chattopadhyay, M., and Kashfi, K. (2012). NOSH-aspirin: a novel nitric oxide-hydrogen sulfide-releasing hybrid: a new class of anti-inflammatory pharmaceuticals. ACS Med. Chem. Lett. 3, 257–262. doi: 10.1021/ml300002m
Kondo, K., Bhushan, S., King, A. L., Prabhu, S. D., Hamid, T., Koenig, S., et al. (2013). H2S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation 127, 1116–1127. doi: 10.1161/CIRCULATIONAHA.112.000855
Konstam, M. A., Kramer, D. G., Patel, A. R., Maron, M. S., and Udelson, J. E. (2011). Left ventricular remodeling in heart failure: current concepts in clinical significance and assessment. JACC Cardiovasc. Imaging 4, 98–108. doi: 10.1016/j.jcmg.2010.10.008
Kuiper, K. K., Muna, Z. A., Erga, K. S., Dyrøy, E., Svendsen, E., Berge, R. K., et al. (2001). Tetradecylthioacetic acid reduces stenosis development after balloon angioplasty injury of rabbit iliac arteries. Atherosclerosis 158, 269–275. doi: 10.1016/S0021-9150(01)00417-8
Kulkarni, K. H., Monjok, E. M., Zeyssig, R., Kouamou, G., Bongmba, O. N., Opere, C. A., et al. (2009). Effect of hydrogen sulfide on sympathetic neurotransmission and catecholamine levels in isolated porcine iris-ciliary body. Neurochem. Res. 34, 400–406. doi: 10.1007/s11064-008-9793-7
Lertratanangkoon, K., Scimeca, J. M., and Wei, J. N. (1999). Inhibition of glutathione synthesis with propargylglycine enhances N-acetylmethionine protection and methylation in bromobenzene-treated Syrian hamsters. J. Nutr. 129, 649–656. doi: 10.1093/jn/129.3.649
Leucker, T. M., Nomura, Y., Kim, J. H., Bhatta, A., Wang, V., Wecker, A., et al. (2017). Cystathionine γ-lyase protects vascular endothelium: a role for inhibition of histone deacetylase 6. Am. J. Physiol. Heart Circ. Physiol. 312, H711–H720. doi: 10.1152/ajpheart.00724.2016
Levitt, M. D., Abdel-Rehim, M. S., and Furne, J. (2011). Free and acid-labile hydrogen sulfide concentrations in mouse tissues: anomalously high free hydrogen sulfide in aortic tissue. Antioxid. Redox Signal. 15, 373–378. doi: 10.1089/ars.2010.3525
Li, D., Xiong, Q., Peng, J., Hu, B., Li, W., Zhu, Y., et al. (2016). Hydrogen sulfide up-regulates the expression of ATP-binding cassette transporter A1 via promoting nuclear translocation of PPARα. Int. J. Mol. Sci. 17:635. doi: 10.3390/ijms17050635
Li, H., Mani, S., Wu, L., Fu, M., Shuang, T., Xu, C., et al. (2017a). The interaction of estrogen and CSE/H2S pathway in the development of atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 312, H406–H414. doi: 10.1152/ajpheart.00245.2016
Li, X. H., Xue, W. L., Wang, M. J., Zhou, Y., Zhang, C. C., Sun, C., et al. (2017b). H2S regulates endothelial nitric oxide synthase protein stability by promoting microRNA-455-3p expression. Sci. Rep. 7:44807. doi: 10.1038/srep44807
Li, H., Teng, X., Yang, R., Guo, Q., Xue, H., Xiao, L., et al. (2017c). Hydrogen sulfide facilitates the impaired sensitivity of carotid sinus baroreflex in rats with vascular calcification. Front. Pharmacol. 8:629. doi: 10.3389/fphar.2017.00629
Li, L., Rose, P., and Moore, P. K. (2011). Hydrogen sulfide and cell signaling. Annu. Rev. Pharmacol. Toxicol. 51, 169–187. doi: 10.1146/annurev-pharmtox-010510-100505
Li, X. H., Zhang, C. Y., and Zhang, T. (2011). [Sodium hydrosulfide improves cardiac functions and structures in rats with chronic heart failure]. Zhonghua Yi Xue Za Zhi 91, 3044–3049.
Li, L., Whiteman, M., Guan, Y. Y., Neo, K. L., Cheng, Y., Lee, S. W., et al. (2008). Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): new insights into the biology of hydrogen sulfide. Circulation 117, 2351–2360. doi: 10.1161/CIRCULATIONAHA.107.753467
Li, X. J., Li, C. K., Wei, L. Y., Lu, N., Wang, G. H., Zhao, H. G., et al. (2015a). Hydrogen sulfide intervention in focal cerebral ischemia/reperfusion injury in rats. Neural Regen. Res. 10, 932–937. doi: 10.4103/1673-5374.158353
Li, J. J., Li, Q., Du, H. P., Wang, Y. L., You, S. J., Wang, F., et al. (2015b). Homocysteine triggers inflammatory responses in macrophages through inhibiting CSE-H2S signaling via DNA hypermethylation of CSE promoter. Int. J. Mol. Sci. 16, 12560–12577. doi: 10.3390/ijms160612560
Li, W., Du, J. B., and Jin, H. F. (2015c). [Effects of hydrogen sulfide donor on production of adrenomedullin and atrial natriuretic peptide in rats with atherosclerosis]. Zhongguo Dang Dai Er Ke Za Zhi 17, 1119–1123.
Li, W., Du, J., and Jin, H. (2015d). [Suppressive effect of hydrogen sulfide donor on endothelin-1 production in aorta of atherosclerotic rats]. Zhonghua Er Ke Za Zhi 53, 448–452.
Li, C., Hu, M., Wang, Y., Lu, H., Deng, J., and Yan, X. (2015e). Hydrogen sulfide preconditioning protects against myocardial ischemia/reperfusion injury in rats through inhibition of endo/sarcoplasmic reticulum stress. Int. J. Clin. Exp. Pathol. 8, 7740–7751.
Liang, D., Huang, A., Jin, Y., Lin, M., Xia, X., Chen, X., et al. (2018). Protective effects of exogenous NaHS against sepsis-induced myocardial mitochondrial injury by enhancing the PGC-1α/NRF2 pathway and mitochondrial biosynthesis in mice. Am. J. Transl. Res. 10, 1422–1430.
Liang, D., Wu, H., Wong, M. W., and Huang, D. (2015). Diallyl trisulfide is a fast H2S donor, but diallyl disulfide is a slow one: the reaction pathways and intermediates of glutathione with polysulfides. Org. Lett. 17, 4196–4199. doi: 10.1021/acs.orglett.5b01962
Liang, M., Jin, S., Wu, D. D., Wang, M. J., and Zhu, Y. C. (2015). Hydrogen sulfide improves glucose metabolism and prevents hypertrophy in cardiomyocytes. Nitric Oxide 46, 114–122. doi: 10.1016/j.niox.2014.12.007
Lima, C. P., Davis, S. R., Mackey, A. D., Scheer, J. B., Williamson, J., and Gregory, J. F. (2006). Vitamin B-6 deficiency suppresses the hepatic transsulfuration pathway but increases glutathione concentration in rats fed AIN-76A or AIN-93G diets. J. Nutr. 136, 2141–2147. doi: 10.1093/jn/136.8.2141
Lin, S., Lian, D., Liu, W., Haig, A., Lobb, I., Hijazi, A., et al. (2018). Daily therapy with a slow-releasing H2S donor GYY4137 enables early functional recovery and ameliorates renal injury associated with urinary obstruction. Nitric Oxide 76, 16–28. doi: 10.1016/j.niox.2018.03.002
Lin, J., Chen, M., Liu, D., Guo, R., Lin, K., Deng, H., et al. (2018). Exogenous hydrogen sulfide protects human umbilical vein endothelial cells against high glucose-induced injury by inhibiting the necroptosis pathway. Int. J. Mol. Med. 41, 1477–1486. doi: 10.3892/ijmm.2017.3330
Lin, Z., Altaf, N., Li, C., Chen, M., Pan, L., Wang, D., et al. (2018). Hydrogen sulfide attenuates oxidative stress-induced NLRP3 inflammasome activation via S-sulfhydrating c-Jun at Cys269 in macrophages. Biochim. Biophys. Acta 1864, 2890–2900. doi: 10.1016/j.bbadis.2018.05.023
Lin, Y., Chen, Y., Zhu, N., Zhao, S., Fan, J., and Liu, E. (2016). Hydrogen sulfide inhibits development of atherosclerosis through up-regulating protein S-nitrosylation. Biomed. Pharmacother. 83, 466–476. doi: 10.1016/j.biopha.2016.07.003
Lin, Y., Zeng, H., Gao, L., Gu, T., Wang, C., and Zhang, H. (2017). Hydrogen sulfide attenuates atherosclerosis in a partially ligated carotid artery mouse model via regulating angiotensin converting enzyme 2 expression. Front. Physiol. 8:782. doi: 10.3389/fphys.2017.00782
Lisjak, M., Teklic, T., Wilson, I. D., Whiteman, M., and Hancock, J. T. (2013). Hydrogen sulfide: environmental factor or signalling molecule. Plant Cell Environ. 36, 1607–1616. doi: 10.1111/pce.12073
Liu, C., Gu, X., and Zhu, Y. Z. (2010). Synthesis and biological evaluation of novel leonurine-SPRC conjugate as cardioprotective agents. Bioorg. Med. Chem. Lett. 20, 6942–6946. doi: 10.1016/j.bmcl.2010.09.135
Liu, C., Guo, W., Shi, X., Kaium, M. A., Gu, X., and Zhu, Y. Z. (2011). Leonurine-cysteine analog conjugates as a new class of multifunctional anti-myocardial ischemia agent. Eur. J. Med. Chem. 46, 3996–4009. doi: 10.1016/j.ejmech.2011.05.073
Liu, S., Wang, X., Pan, L., Wu, W., Yang, D., Qin, M., et al. (2018). Endogenous hydrogen sulfide regulates histone demethylase JMJD3-mediated inflammatory response in LPS-stimulated macrophages and in a mouse model of LPS-induced septic shock. Biochem. Pharmacol. 149, 153–162. doi: 10.1016/j.bcp.2017.10.010
Liu, X., Pachori, A. S., Ward, C. A., Davis, J. P., Gnecchi, M., Kong, D., et al. (2006). Heme oxygenase-1 (HO-1) inhibits postmyocardial infarct remodeling and restores ventricular function. FASEB J. 20, 207–216. doi: 10.1096/fj.05-4435com
Liu, Y. H., Lu, M., Xie, Z. Z., Hua, F., Xie, L., Gao, J. H., et al. (2014). Hydrogen sulfide prevents heart failure development via inhibition of renin release from mast cells in isoproterenol-treated rats. Antioxid. Redox Signal. 20, 759–769. doi: 10.1089/ars.2012.4888
Liu, Z., Han, Y., Li, L., Lu, H., Meng, G., Li, X., et al. (2013). The hydrogen sulfide donor, GYY4137, exhibits anti-atherosclerotic activity in high fat fed apolipoprotein E(-/-) mice. Br. J. Pharmacol. 169, 1795–1809. doi: 10.1111/bph.12246
Lohninger, L., Tomasova, L., Praschberger, M., Hintersteininger, M., Erker, T., Gmeiner, B. M., et al. (2015). Hydrogen sulphide induces HIF-1α and Nrf2 in THP-1 macrophages. Biochimie 112, 187–195. doi: 10.1016/j.biochi.2015.03.009
Lu, F., Xing, J., Zhang, X., Dong, S., Zhao, Y., Wang, L., et al. (2013). Exogenous hydrogen sulfide prevents cardiomyocyte apoptosis from cardiac hypertrophy induced by isoproterenol. Mol. Cell. Biochem. 381, 41–50. doi: 10.1007/s11010-013-1686-7
Luan, H. F., Zhao, Z. B., Zhao, Q. H., Zhu, P., Xiu, M. Y., and Ji, Y. (2012). Hydrogen sulfide postconditioning protects isolated rat hearts against ischemia and reperfusion injury mediated by the JAK2/STAT3 survival pathway. Braz. J. Med. Biol. Res. 45, 898–905. doi: 10.1590/S0100-879X2012007500090
Luo, Y., Yang, X., Zhao, S., Wei, C., Yin, Y., Liu, T., et al. (2013). Hydrogen sulfide prevents OGD/R-induced apoptosis via improving mitochondrial dysfunction and suppressing an ROS-mediated caspase-3 pathway in cortical neurons. Neurochem. Int. 63, 826–831. doi: 10.1016/j.neuint.2013.06.004
Martelli, A., Testai, L., Citi, V., Marino, A., Bellagambi, F. G., Ghimenti, S., et al. (2014). Pharmacological characterization of the vascular effects of aryl isothiocyanates: is hydrogen sulfide the real player. Vascul. Pharmacol. 60, 32–41. doi: 10.1016/j.vph.2013.11.003
Martelli, A., Testai, L., Citi, V., Marino, A., Pugliesi, I., Barresi, E., et al. (2013). Arylthioamides as H2S donors: l-cysteine-activated releasing properties and vascular effects in vitro and in vivo. ACS Med. Chem. Lett. 4, 904–908. doi: 10.1021/ml400239a
Maytin, M., and Colucci, W. S. (2002). Molecular and cellular mechanisms of myocardial remodeling. J. Nucl. Cardiol. 9, 319–327. doi: 10.1067/mnc.2002.123207
McConnell, B. B., and Yang, V. W. (2010). Mammalian Krüppel-like factors in health and diseases. Physiol. Rev. 90, 1337–1381. doi: 10.1152/physrev.00058.2009
Meng, G., Xiao, Y., Ma, Y., Tang, X., Xie, L., Liu, J., et al. (2016). Hydrogen sulfide regulates krüppel-like factor 5 transcription activity via specificity protein 1 S-sulfhydration at Cys664 to prevent myocardial hypertrophy. J. Am. Heart Assoc. 5:e004160. doi: 10.1161/JAHA.116.004160
Meng, G., Wang, J., Xiao, Y., Bai, W., Xie, L., Shan, L., et al. (2015a). GYY4137 protects against myocardial ischemia and reperfusion injury by attenuating oxidative stress and apoptosis in rats. J. Biomed. Res. 29, 203–213. doi: 10.7555/JBR.28.20140037
Meng, G., Zhu, J., Xiao, Y., Huang, Z., Zhang, Y., Tang, X., et al. (2015b). Hydrogen sulfide donor GYY4137 protects against myocardial fibrosis. Oxid. Med. Cell. Longev. 2015:691070. doi: 10.1155/2015/691070
Mishra, P. K., Tyagi, N., Sen, U., Givvimani, S., and Tyagi, S. C. (2010). H2S ameliorates oxidative and proteolytic stresses and protects the heart against adverse remodeling in chronic heart failure. Am. J. Physiol. Heart Circ. Physiol. 298, H451–H456. doi: 10.1152/ajpheart.00682.2009
Monti, M., Terzuoli, E., Ziche, M., and Morbidelli, L. (2016). H2S dependent and independent anti-inflammatory activity of zofenoprilat in cells of the vascular wall. Pharmacol. Res. 113, 426–437. doi: 10.1016/j.phrs.2016.09.017
Nakano, S., Ishii, I., Shinmura, K., Tamaki, K., Hishiki, T., Akahoshi, N., et al. (2015). Hyperhomocysteinemia abrogates fasting-induced cardioprotection against ischemia/reperfusion by limiting bioavailability of hydrogen sulfide anions. J. Mol. Med. 93, 879–889. doi: 10.1007/s00109-015-1271-5
Nandi, S. S., and Mishra, P. K. (2017). H2S and homocysteine control a novel feedback regulation of cystathionine beta synthase and cystathionine gamma lyase in cardiomyocytes. Sci. Rep. 7:3639. doi: 10.1038/s41598-017-03776-9
Ndisang, J. F., Zhao, W., and Wang, R. (2002). Selective regulation of blood pressure by heme oxygenase-1 in hypertension. Hypertension 40, 315–321. doi: 10.1161/01.HYP.0000028488.71068.16
Nicholson, C. K., Lambert, J. P., Molkentin, J. D., Sadoshima, J., and Calvert, J. W. (2013). Thioredoxin 1 is essential for sodium sulfide-mediated cardioprotection in the setting of heart failure. Arterioscler. Thromb. Vasc. Biol. 33, 744–751. doi: 10.1161/ATVBAHA.112.300484
Ning, J. Z., Li, W., Cheng, F., Rao, T., Yu, W. M., Ruan, Y., et al. (2018). The protective effects of GYY4137 on ipsilateral testicular injury in experimentally varicocele-induced rats. Exp. Ther. Med. 15, 433–439. doi: 10.3892/etm.2017.5417
Nishikawa, H., Hayashi, H., Kubo, S., Tsubota-Matsunami, M., Sekiguchi, F., and Kawabata, A. (2013). Inhibition by hydrogen sulfide of rabbit platelet aggregation and calcium mobilization. Biol. Pharm. Bull. 36, 1278–1282. doi: 10.1248/bpb.b13-00018
Nowak, W. N., Deng, J., Ruan, X. Z., and Xu, Q. (2017). Reactive oxygen species generation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 37, e41–e52. doi: 10.1161/ATVBAHA.117.309228
Osipov, R. M., Robich, M. P., Feng, J., Liu, Y., Clements, R. T., Glazer, H. P., et al. (2009). Effect of hydrogen sulfide in a porcine model of myocardial ischemia-reperfusion: comparison of different administration regimens and characterization of the cellular mechanisms of protection. J. Cardiovasc. Pharmacol. 54, 287–297. doi: 10.1097/FJC.0b013e3181b2b72b
Otterbein, L. E., Zuckerbraun, B. S., Haga, M., Liu, F., Song, R., Usheva, A., et al. (2003). Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat. Med. 9, 183–190. doi: 10.1038/nm817
Pan, T. T., Neo, K. L., Hu, L. F., Yong, Q. C., and Bian, J. S. (2008). H2S preconditioning-induced PKC activation regulates intracellular calcium handling in rat cardiomyocytes. Am. J. Physiol. Cell Physiol. 294, C169–C177. doi: 10.1152/ajpcell.00282.2007
Peake, B. F., Nicholson, C. K., Lambert, J. P., Hood, R. L., Amin, H., Amin, S., et al. (2013). Hydrogen sulfide preconditions the db/db diabetic mouse heart against ischemia-reperfusion injury by activating Nrf2 signaling in an Erk-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 304, H1215–H1224. doi: 10.1152/ajpheart.00796.2012
Peng, J., Tang, Z. H., Ren, Z., He, B., Zeng, Y., Liu, L. S., et al. (2017). TET2 protects against oxLDL-induced HUVEC dysfunction by upregulating the CSE/H2S system. Front. Pharmacol. 8:486. doi: 10.3389/fphar.2017.00486
Pfeffer, M. A., and Braunwald, E. (1990). Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation 81, 1161–1172. doi: 10.1161/01.CIR.81.4.1161
Polhemus, D., Kondo, K., Bhushan, S., Bir, S. C., Kevil, C. G., Murohara, T., et al. (2013). Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ. Heart Fail. 6, 1077–1086. doi: 10.1161/CIRCHEARTFAILURE.113.000299
Polhemus, D. J., Li, Z., Pattillo, C. B., Gojon, G., Gojon, G., Giordano, T., et al. (2015). A novel hydrogen sulfide prodrug, SG1002, promotes hydrogen sulfide and nitric oxide bioavailability in heart failure patients. Cardiovasc. Ther. 33, 216–226. doi: 10.1111/1755-5922.12128
Potor, L., Nagy, P., Méhes, G., Hendrik, Z., Jeney, V., Pethő, D., et al. (2018). Hydrogen sulfide abrogates hemoglobin-lipid interaction in atherosclerotic lesion. Oxid. Med. Cell. Longev. 2018:3812568. doi: 10.1155/2018/3812568
Powell, C. R., Dillon, K. M., Wang, Y., Carrazzone, R. J., and Matson, J. B. (2018). A persulfide donor responsive to reactive oxygen species: insights into reactivity and therapeutic potential. Angew. Chem. Int. Ed. Engl. 57, 6324–6328. doi: 10.1002/anie.201803087
Predmore, B. L., Kondo, K., Bhushan, S., Zlatopolsky, M. A., King, A. L., Aragon, J. P., et al. (2012). The polysulfide diallyl trisulfide protects the ischemic myocardium by preservation of endogenous hydrogen sulfide and increasing nitric oxide bioavailability. Am. J. Physiol. Heart Circ. Physiol. 302, H2410–H2418. doi: 10.1152/ajpheart.00044.2012
Qian, X., Li, X., Ma, F., Luo, S., Ge, R., and Zhu, Y. (2016). Novel hydrogen sulfide-releasing compound, S-propargyl-cysteine, prevents STZ-induced diabetic nephropathy. Biochem. Biophys. Res. Commun. 473, 931–938. doi: 10.1016/j.bbrc.2016.03.154
Qipshidze, N., Metreveli, N., Mishra, P. K., Lominadze, D., and Tyagi, S. C. (2012). Hydrogen sulfide mitigates cardiac remodeling during myocardial infarction via improvement of angiogenesis. Int. J. Biol. Sci. 8, 430–441. doi: 10.7150/ijbs.3632
Qiu, Y., Wu, Y., Meng, M., Luo, M., Zhao, H., Sun, H., et al. (2018). GYY4137 protects against myocardial ischemia/reperfusion injury via activation of the PHLPP-1/Akt/Nrf2 signaling pathway in diabetic mice. J. Surg. Res. 225, 29–39. doi: 10.1016/j.jss.2017.12.030
Raggi, P., Genest, J., Giles, J. T., Rayner, K. J., Dwivedi, G., Beanlands, R. S., et al. (2018). Role of inflammation in the pathogenesis of atherosclerosis and therapeutic interventions. Atherosclerosis 276, 98–108. doi: 10.1016/j.atherosclerosis.2018.07.014
Ravindran, S., Ansari, B. S., and Kurian, G. A. (2016). Hydrogen sulfide preconditioning shows differential protection towards interfibrillar and subsarcolemmal mitochondria from isolated rat heart subjected to revascularization injury. Cardiovasc. Pathol. 25, 306–315. doi: 10.1016/j.carpath.2016.04.005
Rios, E. C., Szczesny, B., Soriano, F. G., Olah, G., and Szabo, C. (2015). Hydrogen sulfide attenuates cytokine production through the modulation of chromatin remodeling. Int. J. Mol. Med. 35, 1741–1746. doi: 10.3892/ijmm.2015.2176
Rizzello, V., Poldermans, D., Biagini, E., Schinkel, A. F., Boersma, E., Boccanelli, A., et al. (2009). Prognosis of patients with ischaemic cardiomyopathy after coronary revascularisation: relation to viability and improvement in left ventricular ejection fraction. Heart 95, 1273–1277. doi: 10.1136/hrt.2008.163972
Salloum, F. N., Sturz, G. R., Yin, C., Rehman, S., Hoke, N. N., Kukreja, R. C., et al. (2015). Beetroot juice reduces infarct size and improves cardiac function following ischemia-reperfusion injury: possible involvement of endogenous H2S. Exp. Biol. Med. 240, 669–681. doi: 10.1177/1535370214558024
Salvi, A., Bankhele, P., Jamil, J., Chitnis, M. K., Njie-Mbye, Y. F., Ohia, S. E., et al. (2016a). Effect of hydrogen sulfide donors on intraocular pressure in rabbits. J. Ocul. Pharmacol. Ther. 32, 371–375. doi: 10.1089/jop.2015.0144
Salvi, A., Bankhele, P., Jamil, J. M., Kulkarni-Chitnis, M., Njie-Mbye, Y. F., Ohia, S. E., et al. (2016b). Pharmacological actions of hydrogen sulfide donors on sympathetic neurotransmission in the bovine anterior uvea, in vitro. Neurochem. Res. 41, 1020–1028. doi: 10.1007/s11064-015-1784-x
Sen, U., Vacek, T. P., Hughes, W. M., Kumar, M., Moshal, K. S., Tyagi, N., et al. (2008). Cardioprotective role of sodium thiosulfate on chronic heart failure by modulating endogenous H2S generation. Pharmacology 82, 201–213. doi: 10.1159/000156486
Shao, M., Zhuo, C., Jiang, R., Chen, G., Shan, J., Ping, J., et al. (2017). Protective effect of hydrogen sulphide against myocardial hypertrophy in mice. Oncotarget 8, 22344–22352. doi: 10.18632/oncotarget.15765
Shimizu, Y., Nicholson, C. K., Lambert, J. P., Barr, L. A., Kuek, N., Herszenhaut, D., et al. (2016). Sodium sulfide attenuates ischemic-induced heart failure by enhancing proteasomal function in an Nrf2-dependent manner. Circ. Heart Fail. 9:e002368. doi: 10.1161/CIRCHEARTFAILURE.115.002368
Shimizu, Y., Polavarapu, R., Eskla, K. L., Nicholson, C. K., Koczor, C. A., Wang, R., et al. (2018). Hydrogen sulfide regulates cardiac mitochondrial biogenesis via the activation of AMPK. J. Mol. Cell. Cardiol. 116, 29–40. doi: 10.1016/j.yjmcc.2018.01.011
Shindo, T., Manabe, I., Fukushima, Y., Tobe, K., Aizawa, K., Miyamoto, S., et al. (2002). Krüppel-like zinc-finger transcription factor KLF5/BTEB2 is a target for angiotensin II signaling and an essential regulator of cardiovascular remodeling. Nat. Med. 8, 856–863. doi: 10.1038/nm738
Shuang, T., Fu, M., Yang, G., Wu, L., and Wang, R. (2018). The interaction of IGF-1/IGF-1R and hydrogen sulfide on the proliferation of mouse primary vascular smooth muscle cells. Biochem. Pharmacol. 149, 143–152. doi: 10.1016/j.bcp.2017.12.009
Shymans’ka, T. V., IuV, H., Semenikhina, O. M., and Sahach, V. F. (2012). [Effect of hydrogen sulfide on isolated rat heart reaction under volume load and ischemia-reperfusion]. Fiziol. Zh. 58, 57–66.
Snijder, P. M., Frenay, A. R., de Boer, R. A., Pasch, A., Hillebrands, J. L., Leuvenink, H. G., et al. (2015). Exogenous administration of thiosulfate, a donor of hydrogen sulfide, attenuates angiotensin II-induced hypertensive heart disease in rats. Br. J. Pharmacol. 172, 1494–1504. doi: 10.1111/bph.12825
Sone, K., Mori, A., Sakamoto, K., and Nakahara, T. (2018). GYY4137, an extended-release hydrogen sulfide donor, reduces NMDA-induced neuronal injury in the murine retina. Biol. Pharm. Bull. 41, 657–660. doi: 10.1248/bpb.b17-01032
Sun, Q., Collins, R., Huang, S., Holmberg-Schiavone, L., Anand, G. S., Tan, C. H., et al. (2009). Structural basis for the inhibition mechanism of human cystathionine gamma-lyase, an enzyme responsible for the production of H(2)S. J. Biol. Chem. 284, 3076–3085. doi: 10.1074/jbc.M805459200
Sun, Y., Tian, Z., Liu, N., Zhang, L., Gao, Z., Sun, X., et al. (2018). Exogenous H2S switches cardiac energy substrate metabolism by regulating SIRT3 expression in db/db mice. J. Mol. Med. 96, 281–299. doi: 10.1007/s00109-017-1616-3
Swynghedauw, B. (1999). Molecular mechanisms of myocardial remodeling. Physiol. Rev. 79, 215–262. doi: 10.1152/physrev.1999.79.1.215
Szabó, C. (2007). Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 6, 917–935. doi: 10.1038/nrd2425
Szabo, C. (2012). Roles of hydrogen sulfide in the pathogenesis of diabetes mellitus and its complications. Antioxid. Redox Signal. 17, 68–80. doi: 10.1089/ars.2011.4451
Szabo, C. (2016). Gasotransmitters in cancer: from pathophysiology to experimental therapy. Nat. Rev. Drug Discov. 15, 185–203. doi: 10.1038/nrd.2015.1
Szczesny, B., Módis, K., Yanagi, K., Coletta, C., Le, T. S., Perry, A., et al. (2014). AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide 41, 120–130. doi: 10.1016/j.niox.2014.04.008
Tain, Y. L., Hsu, C. N., Lee, C. T., Lin, Y. J., and Tsai, C. C. (2016). N-acetylcysteine prevents programmed hypertension in male rat offspring born to suramin-treated mothers. Biol. Reprod. 95:8. doi: 10.1095/biolreprod.116.139766
Tang, G., Wu, L., Liang, W., and Wang, R. (2005). Direct stimulation of K(ATP) channels by exogenous and endogenous hydrogen sulfide in vascular smooth muscle cells. Mol. Pharmacol. 68, 1757–1764. doi: 10.1124/mol.105.017467
Testai, L., Marino, A., Piano, I., Brancaleone, V., Tomita, K., Di, C. M. L., et al. (2016). The novel H2S-donor 4-carboxyphenyl isothiocyanate promotes cardioprotective effects against ischemia/reperfusion injury through activation of mitoKATP channels and reduction of oxidative stress. Pharmacol. Res. 113, 290–299. doi: 10.1016/j.phrs.2016.09.006
Teunissen, B. E., Jongsma, H. J., and Bierhuizen, M. F. (2004). Regulation of myocardial connexins during hypertrophic remodelling. Eur. Heart J. 25, 1979–1989. doi: 10.1016/j.ehj.2004.08.007
Tsai, C. Y., Wen, S. Y., Shibu, M. A., Yang, Y. C., Peng, H., Wang, B., et al. (2015). Diallyl trisulfide protects against high glucose-induced cardiac apoptosis by stimulating the production of cystathionine gamma-lyase-derived hydrogen sulfide. Int. J. Cardiol. 195, 300–310. doi: 10.1016/j.ijcard.2015.05.111
Tyagi, N., Moshal, K. S., Sen, U., Vacek, T. P., Kumar, M., Hughes, W. M., et al. (2009). H2S protects against methionine-induced oxidative stress in brain endothelial cells. Antioxid. Redox Signal. 11, 25–33. doi: 10.1089/ars.2008.2073
Vannini, F., Chattopadhyay, M., Kodela, R., Rao, P. P., and Kashfi, K. (2015). Positional isomerism markedly affects the growth inhibition of colon cancer cells by NOSH-aspirin: COX inhibition and modeling. Redox Biol. 6, 318–325. doi: 10.1016/j.redox.2015.08.014
Velmurugan, G. V., Huang, H., Sun, H., Candela, J., Jaiswal, M. K., Beaman, K. D., et al. (2015). Depletion of H2S during obesity enhances store-operated Ca2+ entry in adipose tissue macrophages to increase cytokine production. Sci. Signal. 8:ra128. doi: 10.1126/scisignal.aac7135
Wang, M., Tang, W., Xin, H., and Zhu, Y. Z. (2016). S-propargyl-cysteine, a novel hydrogen sulfide donor, inhibits inflammatory hepcidin and relieves anemia of inflammation by inhibiting IL-6/STAT3 pathway. PLoS One 11:e0163289. doi: 10.1371/journal.pone.0163289
Wang, Q., Wang, X. L., Liu, H. R., Rose, P., and Zhu, Y. Z. (2010). Protective effects of cysteine analogues on acute myocardial ischemia: novel modulators of endogenous H(2)S production. Antioxid. Redox Signal. 12, 1155–1165. doi: 10.1089/ars.2009.2947
Wang, R. (2002). Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter. FASEB J. 16, 1792–1798. doi: 10.1096/fj.02-0211hyp
Wang, R. (2010). Hydrogen sulfide: the third gasotransmitter in biology and medicine. Antioxid. Redox Signal. 12, 1061–1064. doi: 10.1089/ars.2009.2938
Wang, R. (2012). Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev. 92, 791–896. doi: 10.1152/physrev.00017.2011
Wang, X. H., Wang, F., You, S. J., Cao, Y. J., Cao, L. D., Han, Q., et al. (2013). Dysregulation of cystathionine γ-lyase (CSE)/hydrogen sulfide pathway contributes to ox-LDL-induced inflammation in macrophage. Cell. Signal. 25, 2255–2262. doi: 10.1016/j.cellsig.2013.07.010
Wang, X. L., Pan, L. L., Long, F., Wu, W. J., Yan, D., Xu, P., et al. (2018). Endogenous hydrogen sulfide ameliorates NOX4 induced oxidative stress in LPS-stimulated macrophages and mice. Cell. Physiol. Biochem. 47, 458–474. doi: 10.1159/000489980
Wang, Y., Zhao, X., Jin, H., Wei, H., Li, W., Bu, D., et al. (2009). Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 29, 173–179. doi: 10.1161/ATVBAHA.108.179333
Wang, Y., Zhao, Z., Shi, S., Gao, F., Wu, J., Dong, S., et al. (2017). Calcium sensing receptor initiating cystathionine-gamma-lyase/hydrogen sulfide pathway to inhibit platelet activation in hyperhomocysteinemia rat. Exp. Cell Res. 358, 171–181. doi: 10.1016/j.yexcr.2017.06.013
Wen, Y. D., and Zhu, Y. Z. (2015). The pharmacological effects of S-propargyl-cysteine, a novel endogenous H2S-producing compound. Handb. Exp. Pharmacol. 230, 325–336. doi: 10.1007/978-3-319-18144-8_16
Whiteman, M., Perry, A., Zhou, Z., Bucci, M., Papapetropoulos, A., Cirino, G., et al. (2015). Phosphinodithioate and phosphoramidodithioate hydrogen sulfide donors. Handb. Exp. Pharmacol. 230, 337–363. doi: 10.1007/978-3-319-18144-8_17
Wu, T., Li, H., Wu, B., Zhang, L., Wu, S. W., Wang, J. N., et al. (2017). Hydrogen sulfide reduces recruitment of CD11b+Gr-1+ cells in mice with myocardial infarction. Cell Transplant. 26, 753–764. doi: 10.3727/096368917X695029
Wu, D., Hu, Q., Xiong, Y., Zhu, D., Mao, Y., and Zhu, Y. Z. (2018). Novel H2S-NO hybrid molecule (ZYZ-803) promoted synergistic effects against heart failure. Redox Biol. 15, 243–252. doi: 10.1016/j.redox.2017.11.020
Wu, T., Qiao, S., Shi, C., Wang, S., and Ji, G. (2018). Metabolomics window into diabetic complications. J. Diabetes Investig. 9, 244–255. doi: 10.1111/jdi.12723
Wu, W. J., Jia, W. W., Liu, X. H., Pan, L. L., Zhang, Q. Y., Yang, D., et al. (2016). S-propargyl-cysteine attenuates inflammatory response in rheumatoid arthritis by modulating the Nrf2-ARE signaling pathway. Redox Biol. 10, 157–167. doi: 10.1016/j.redox.2016.08.011
Xiao, J., Zhu, X., Kang, B., Xu, J., Wu, L., Hong, J., et al. (2015). Hydrogen sulfide attenuates myocardial hypoxia-reoxygenation injury by inhibiting autophagy via mTOR activation. Cell. Physiol. Biochem. 37, 2444–2453. doi: 10.1159/000438597
Xie, H., Xu, Q., Jia, J., Ao, G., Sun, Y., Hu, L., et al. (2015). Hydrogen sulfide protects against myocardial ischemia and reperfusion injury by activating AMP-activated protein kinase to restore autophagic flux. Biochem. Biophys. Res. Commun. 458, 632–638. doi: 10.1016/j.bbrc.2015.02.017
Xie, L., Gu, Y., Wen, M., Zhao, S., Wang, W., Ma, Y., et al. (2016). Hydrogen sulfide induces Keap1 S-sulfhydration and suppresses diabetes-accelerated atherosclerosis via Nrf2 activation. Diabetes Metab. Res. Rev. 65, 3171–3184. doi: 10.2337/db16-0020
Xu, S., Liu, Z., and Liu, P. (2014). Targeting hydrogen sulfide as a promising therapeutic strategy for atherosclerosis. Int. J. Cardiol. 172, 313–317. doi: 10.1016/j.ijcard.2014.01.068
Xu, S., Ogura, S., Chen, J., Little, P. J., Moss, J., and Liu, P. (2013). LOX-1 in atherosclerosis: biological functions and pharmacological modifiers. Cell. Mol. Life Sci. 70, 2859–2872. doi: 10.1007/s00018-012-1194-z
Yang, F., Liu, Z., Wang, Y., Li, Z., Yu, H., and Wang, Q. (2014). Hydrogen sulfide endothelin-induced myocardial hypertrophy in rats and the mechanism involved. Cell Biochem. Biophys. 70, 1683–1686. doi: 10.1007/s12013-014-0113-3
Yang, G., Cao, K., Wu, L., and Wang, R. (2004). Cystathionine gamma-lyase overexpression inhibits cell proliferation via a H2S-dependent modulation of ERK1/2 phosphorylation and p21Cip/WAK-1. J. Biol. Chem. 279, 49199–49205. doi: 10.1074/jbc.M408997200
Yang, G., Wu, L., Jiang, B., Yang, W., Qi, J., Cao, K., et al. (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 322, 587–590. doi: 10.1126/science.1162667
Yang, R., Teng, X., Li, H., Xue, H. M., Guo, Q., Xiao, L., et al. (2016). Hydrogen sulfide improves vascular calcification in rats by inhibiting endoplasmic reticulum stress. Oxid. Med. Cell. Longev. 2016:9095242. doi: 10.1155/2016/9095242
Yao, Y., Zhang, X., Chen, H. P., Li, L., Xie, W., Lan, G., et al. (2016). MicroRNA-186 promotes macrophage lipid accumulation and secretion of pro-inflammatory cytokines by targeting cystathionine γ-lyase in THP-1 macrophages. Atherosclerosis 250, 122–132. doi: 10.1016/j.atherosclerosis.2016.04.030
Yet, S. F., Layne, M. D., Liu, X., Chen, Y. H., Ith, B., Sibinga, N. E., et al. (2003). Absence of heme oxygenase-1 exacerbates atherosclerotic lesion formation and vascular remodeling. FASEB J. 17, 1759–1761. doi: 10.1096/fj.03-0187fje
Yong, Q. C., Lee, S. W., Foo, C. S., Neo, K. L., Chen, X., and Bian, J. S. (2008). Endogenous hydrogen sulphide mediates the cardioprotection induced by ischemic postconditioning. Am. J. Physiol. Heart Circ. Physiol. 295, H1330–H1340. doi: 10.1152/ajpheart.00244.2008
Yu, L., Park, B. M., Ahn, Y. J., Lee, G. J., and Kim, S. H. (2018). Hydrogen sulfide donor, NaHS, stimulates ANP secretion via the KATP channel and the NOS/sGC pathway in rat atria. Peptides doi: 10.1016/j.peptides.2018.04.005 [Epub ahead of print].
Yuan, S., Shen, X., and Kevil, C. G. (2017). Beyond a gasotransmitter: hydrogen sulfide and polysulfide in cardiovascular health and immune response. Antioxid. Redox Signal. 27, 634–653. doi: 10.1089/ars.2017.7096
Zagli, G., Patacchini, R., Trevisani, M., Abbate, R., Cinotti, S., Gensini, G. F., et al. (2007). Hydrogen sulfide inhibits human platelet aggregation. Eur. J. Pharmacol. 559, 65–68. doi: 10.1016/j.ejphar.2006.12.011
Zaichko, N. V., and Pentiuk, O. O. (2009). [Influence of hydrogen sulfide, dithionite, sulfite, thiosulfate and sulfate anions on human platelet aggregation]. Ukr. Biokhim. Zh. 81, 105–113.
Zhang, C. Y., Li, X. H., Zhang, T., Fu, J., and Cui, X. D. (2013). Hydrogen sulfide upregulates heme oxygenase-1 expression in rats with volume overload-induced heart failure. Biomed. Rep. 1, 454–458. doi: 10.3892/br.2013.87
Zhang, H., Guo, C., Wu, D., Zhang, A., Gu, T., Wang, L., et al. (2012). Hydrogen sulfide inhibits the development of atherosclerosis with suppressing CX3CR1 and CX3CL1 expression. PLoS One 7:e41147. doi: 10.1371/journal.pone.0041147
Zhang, Y., Peng, L., and Yu, X. (2015). [Protective effect of hydrogen sulfide on rats with myocardial ischemia/reperfusion injury and its mechanism]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 31, 316–320.
Zhang, H., Zhi, L., Moochhala, S., Moore, P. K., and Bhatia, M. (2007). Hydrogen sulfide acts as an inflammatory mediator in cecal ligation and puncture-induced sepsis in mice by upregulating the production of cytokines and chemokines via NF-kappaB. Am. J. Physiol. Lung Cell. Mol. Physiol. 292, L960–L971. doi: 10.1152/ajplung.00388.2006
Zhang, Z., Huang, H., Liu, P., Tang, C., and Wang, J. (2007). Hydrogen sulfide contributes to cardioprotection during ischemia-reperfusion injury by opening K ATP channels. Can. J. Physiol. Pharmacol. 85, 1248–1253. doi: 10.1139/Y07-120
Zhao, W., Zhang, J., Lu, Y., and Wang, R. (2001). The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 20, 6008–6016. doi: 10.1093/emboj/20.21.6008
Zhao, X., Zhang, L. K., Zhang, C. Y., Zeng, X. J., Yan, H., Jin, H. F., et al. (2008). Regulatory effect of hydrogen sulfide on vascular collagen content in spontaneously hypertensive rats. Hypertens. Res. 31, 1619–1630. doi: 10.1291/hypres.31.1619
Zhao, Z., Liu, X., Shi, S., Li, H., Gao, F., Zhong, X., et al. (2018). Exogenous hydrogen sulfide protects from endothelial cell damage, platelet activation, and neutrophils extracellular traps formation in hyperhomocysteinemia rats. Exp. Cell Res. 370, 434–443. doi: 10.1016/j.yexcr.2018.07.007
Zhao, Z. Z., Wang, Z., Li, G. H., Wang, R., Tan, J. M., Cao, X., et al. (2011). Hydrogen sulfide inhibits macrophage-derived foam cell formation. Exp. Biol. Med. (Maywood) 236, 169–176. doi: 10.1258/ebm.2010.010308
Zhong, L., Lv, L., Yang, J., Liao, X., Yu, J., Wang, R., et al. (2014). Inhibitory effect of hydrogen sulfide on platelet aggregation and the underlying mechanisms. J. Cardiovasc. Pharmacol. 64, 481–487. doi: 10.1097/FJC.0000000000000142
Zhou, K., Gao, Q., Zheng, S., Pan, S., Li, P., Suo, K., et al. (2013). 17β-estradiol induces vasorelaxation by stimulating endothelial hydrogen sulfide release. Mol. Hum. Reprod. 19, 169–176. doi: 10.1093/molehr/gas044
Zhou, X., An, G., and Lu, X. (2015a). Hydrogen sulfide attenuates the development of diabetic cardiomyopathy. Clin. Sci. 128, 325–335. doi: 10.1042/CS20140460
Zhou, X., Zhao, L., Mao, J., Huang, J., and Chen, J. (2015b). Antioxidant effects of hydrogen sulfide on left ventricular remodeling in smoking rats are mediated via PI3K/Akt-dependent activation of Nrf2. Toxicol. Sci. 144, 197–203. doi: 10.1093/toxsci/kfu272
Zhou, X., Tang, S., Hu, K., Zhang, Z., Liu, P., Luo, Y., et al. (2018). DL-Propargylglycine protects against myocardial injury induced by chronic intermittent hypoxia through inhibition of endoplasmic reticulum stress. Sleep Breath. doi: 10.1007/s11325-018-1656-0 [Epub ahead of print].
Keywords: H2S, cardiovascular diseases, cardiac hypertrophy/heart failure, ischemia reperfusion injury, atherosclerosis, donor
Citation: Zhang L, Wang Y, Li Y, Li L, Xu S, Feng X and Liu S (2018) Hydrogen Sulfide (H2S)-Releasing Compounds: Therapeutic Potential in Cardiovascular Diseases. Front. Pharmacol. 9:1066. doi: 10.3389/fphar.2018.01066
Received: 21 June 2018; Accepted: 03 September 2018;
Published: 21 September 2018.
Edited by:
Lei Xi, Virginia Commonwealth University, United StatesReviewed by:
Sai S. Koka, University of Houston, United StatesGuanglong He, University of Wyoming, United States
Ting C. Zhao, Boston University, United States
Copyright © 2018 Zhang, Wang, Li, Li, Xu, Feng and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suowen Xu, c3Vvd2VuLnh1QGdtYWlsLmNvbQ== Xiaojun Feng, anVueGlhb2ZlbmcxMzJAMTI2LmNvbQ== Sheng Liu, bHNsY2NsaGxAMTYzLmNvbQ==
†These authors have contributed equally to this work