 Susan Hua
Susan Hua Maria B. C. de Matos
Maria B. C. de Matos Josbert M. Metselaar4,5
Josbert M. Metselaar4,5- 1Therapeutic Targeting Research Group, School of Biomedical Sciences and Pharmacy, University of Newcastle, Callaghan, NSW, Australia
- 2Hunter Medical Research Institute, New Lambton Heights, Newcastle, NSW, Australia
- 3Department of Pharmaceutics, Utrecht Institute for Pharmaceutical Sciences, Utrecht University, Utrecht, Netherlands
- 4Department of Biomaterials Science and Technology, MIRA Institute for Biomedical Technology and Technical Medicine, University of Twente, Enschede, Netherlands
- 5Department of Experimental Molecular Imaging, RWTH University Clinic Aachen, Aachen, Germany
- 6Imaging Division, University Medical Centre Utrecht, Utrecht, Netherlands
The use of nanotechnology in medicine has the potential to have a major impact on human health for the prevention, diagnosis, and treatment of diseases. One particular aspect of the nanomedicine field which has received a great deal of attention is the design and development of nanoparticulate nanomedicines (NNMs) for drug delivery (i.e., drug-containing nanoparticles). NNMs are intended to deliver drugs via various mechanisms: solubilization, passive targeting, active targeting, and triggered release. The NNM approach aims to increase therapeutic efficacy, decrease the therapeutically effective dose, and/or reduce the risk of systemic side effects. In order to move a NNM from the bench to the bedside, several experimental challenges need to be addressed. This review will discuss the current trends and challenges in the clinical translation of NNMs as well as the potential pathways for translational development and commercialization. Key issues related to the clinical development of NNMs will be covered, including biological challenges, large-scale manufacturing, biocompatibility and safety, intellectual property (IP), government regulations, and overall cost-effectiveness in comparison to current therapies. These factors can impose significant hurdles limiting the appearance of NNMs on the market, irrelevant of whether they are therapeutically beneficial or not.
Introduction
Nanomedicine applies nanotechnology to highly specific medical interventions for the prevention, diagnosis, and treatment of diseases (Teli et al., 2010). In the last several decades, the application of nanotechnology for medical purposes has received significant attention from researchers, academia, funding agencies, government, and regulatory bodies (Allen and Cullis, 2004; Sercombe et al., 2015; Hare et al., 2017). One particular aspect of the nanomedicine field which has received a great deal of attention is the design and development of nanoparticulate nanomedicines (NNMs) for drug delivery (i.e., drug-containing nanoparticles), which are most often given by parenteral (particularly intravenous) administration. NNMs are intended to increase the therapeutic index of drugs (i.e., increase efficacy and/or reduce toxicity) by delivering them via various mechanisms: solubilization, passive targeting, active targeting, and triggered release (Figure 1). Nanoencapsulation gives the opportunity to protect fragile compounds that degrade easily in biological environments and to provide solubilization, i.e., to deliver compounds which have physicochemical properties that strongly limit their aqueous solubility and therefore systemic bioavailability (Talekar et al., 2015; Kim et al., 2016; Larsson et al., 2017; Mishra et al., 2017; Shajari et al., 2017). Targeted drug delivery and triggered release of NNMs have been shown to be beneficial for increasing the therapeutic index of compounds, by improving the in vivo fate of drug molecules such that more efficient delivery to the target site is achieved (to yield improved therapeutic effects) with less accumulation in many healthy body sites (to reduce toxicity). Also NNMs have been studied for their ability to stimulate target cell uptake and improve intracellular trafficking, processes sometimes required when they have localized in target tissues (Mastrobattista et al., 1999; Hua, 2013; Hua et al., 2015).
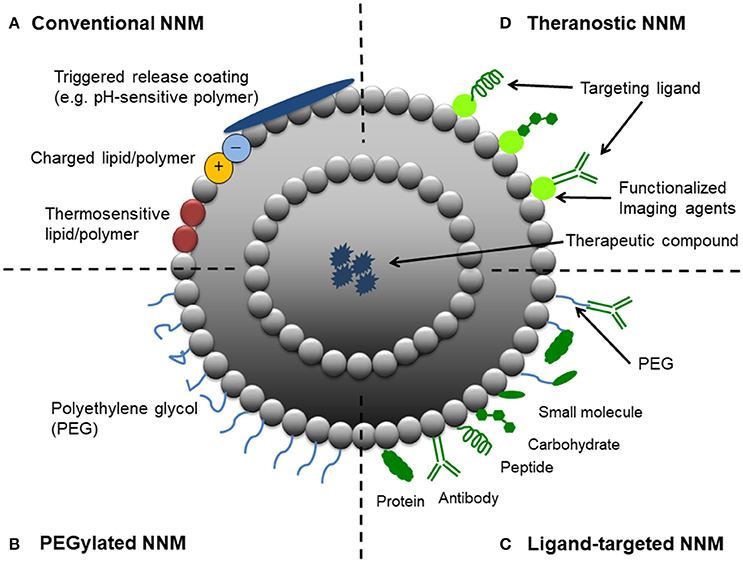
Figure 1. Schematic representation of different strategic designs for nanoparticulate nanomedicines (NNMs). (A) Conventional NNM—These NNMs can be modified with charged lipids/polymers, thermosensitive lipids/polymers and/or components for triggered release (e.g., pH-sensitive coating). (B) PEGylated NNM—Nanoparticle characteristics and behavior in vivo can be modified by the addition of a hydrophilic polymer coating, polyethylene glycol (PEG), to the NNM surface to confer steric stabilization. (C) Ligand-targeted NNM—Nanoparticles can be used for active targeting by attaching ligands (e.g., antibodies, peptides and carbohydrates) to its surface or to the terminal end of the attached PEG chains. (D) Theranostic NNM – These NNM systems consist of an imaging component and a therapeutic component, and may include a targeting element.
Although NNMs have demonstrated significant therapeutic advantages for a multitude of biomedical applications, their clinical translation has not progressed as rapidly as the plethora of positive preclinical results would have suggested (Luxenhofer et al., 2014). In order to move a NNM from the bench to the bedside, several experimental challenges need to be addressed. From a biological perspective, these include studies focused on understanding the in vivo fate and interactions of NNMs with the blood, tissue, cellular, and intracellular compartments in the host in healthy and diseased states (Nehoff et al., 2014; Sercombe et al., 2015; Hare et al., 2017). For NNMs to have clinical translation potential, the complexity in their design and development also needs to be minimized as much as possible to create systems that are able to be reproducibly prepared and characterized (Lammers, 2013; Barz et al., 2015). This review will address the current trends and challenges in the clinical translation of NNMs as well as the potential pathways for translational development and commercialization.
Current Trends in the Clinical Translation of Nanomedicines
NNMs are often studied to improve drug targeting to specific sites of disease (i.e., site-specific drug delivery) and/or attenuate localization in healthy non-target tissues (i.e., site-avoidance drug delivery; Rizzo et al., 2013). The vast majority of NNMs in preclinical and clinical development as well in clinical use are for targeting a wide variety of cancers and tumors (Hare et al., 2017). The application of NNM-based therapies for drug targeting to non-cancer conditions has increased in recent years. In particular, NNMs have been developed to address the clinical challenge of effectively managing inflammatory diseases by exploiting the underlying biology of these conditions (Milane and Amiji, 2017). Non-cancerous inflammatory diseases that have been explored with NNM therapy include rheumatoid arthritis, inflammatory bowel disease, asthma, multiple sclerosis, diabetes, and neurodegenerative diseases (Milane and Amiji, 2017).
Enhanced Permeability and Retention (EPR) Effect and Passive Accumulation of NNMs
The EPR effect refers to the preferential localization of NNMs in pathological tissues due to the enhanced permeability of the vasculature that supplies such tissues (e.g., tumors and inflammatory conditions). Deregulations in angiogenesis and/or the increased expression and activation of vascular permeability factors predominates at these sites, which can lead to fenestrations allowing passage of NNMs (Hashizume et al., 2000; Nehoff et al., 2014). In addition to the enhanced leakiness of tumorous and inflamed blood vessels, the EPR effect also relates to the observation that solid tumors tend to lack functional lymphatic drainage, which limits the removal of extravasated NNMs from the target site (Maeda et al., 2013; Danhier, 2016). These pathological properties allow NNMs to accumulate at pathological sites and is referred to as passive targeting. To achieve this, it is important that NNMs with drug cargo circulate long enough in the bloodstream (i.e., show prolonged circulation kinetics). This can be achieved by conjugating polyethylene glycol (PEG) to the surface of NNMs (Figure 1). Thus, the EPR effect is expected to increase the therapeutic efficacy of NNMs in comparison to small molecules, which often show inferior pharmacokinetic properties (Matsumura and Maeda, 1986; Hobbs et al., 1998; van der Meel et al., 2013). The EPR effect was first observed in 1986 (Matsumura and Maeda, 1986) and has since been exploited particularly for the development of NNMs for passive tumor targeting, leading to NNMs with adequate physicochemical properties and prolonged circulation half-life that accumulate in tumors over time (Maeda et al., 2013; Nakamura et al., 2015; Danhier, 2016). The EPR effect and thus extent of passive targeting is highly dependent on the tumor pathophysiology. Currently, it is recognized that EPR is a very heterogeneous phenomenon as it depends on the type of tumor and can vary significantly within the same tumor type (Lammers et al., 2012; Ojha et al., 2017). The degree of tumor vascularization and passive targeting of NNMs has been observed to be positively correlated (Theek et al., 2014). For example, Doxil® (pegylated liposomal doxorubicin) is the first FDA-approved NNM and has demonstrated superior efficacy in ovarian cancer and AIDS-related Kaposi's sarcoma compared to standard conventional therapies (Nichols and Bae, 2014). When doxorubicin is encapsulated within PEGylated liposomes, it delays and minimizes uptake and clearance by the reticuloendothelial system (RES), thereby prolonging circulation half-life. This allows the NNM to accumulate in the tumor tissue by exploiting the locally increased permeability of the tumor blood vessels, rather than in non-target healthy tissues which do not have such leaky vessels (Rahman et al., 2007). Furthermore, the use of pegylated liposomal doxorubicin avoids high plasma peak levels of free drug (Lyass et al., 2000) and significantly reduces the risk of cardiotoxicity by preventing doxorubicin release through the heart vasculature (Rahman et al., 2007).
NNMs and Active Targeting
Active targeting, also termed ligand-targeting or receptor-mediated targeting, involves the use of ligands (e.g., antibodies, peptides or sugar moieties) which are physically or chemically conjugated onto the surface of NNMs to facilitate localization to and/or uptake by target cells (van der Meel et al., 2013; Danhier, 2016; Figure 1). Ligand-targeted NNMs have enormous potential for site-specific delivery of therapeutic compounds to designated cell types in vivo, which selectively express or over-express specific receptors (e.g., cellular receptors or cell adhesion molecules) at the site of disease (Willis and Forssen, 1998; Hua, 2013). For example, three sets of cellular targets are generally considered for active targeting in cancer—(i) targeting of cancer cells, which present overexpression of receptors for transferrin, folate, epidermal growth factor or glycoproteins; (ii) targeting of the tumor endothelium overexpressing vascular endothelial growth factors, integrins, vascular cell adhesion molecule-1 or matrix metalloproteinases; and (iii) targeting of stroma cells (e.g., macrophages, fibroblasts) that can acquire a tumor survival-promoting phenotype in response to cytokines in the tumor microenvironment (Coimbra et al., 2010; Danhier et al., 2010; Kuijpers et al., 2010; Danhier, 2016). There is still much debate about whether ligand-targeted NNMs are capable of significantly enhancing NNM accumulation at target sites over non-targeted NNMs (passive-targeting), with conflicting results reported in the literature (Ferrari, 2005; Puri et al., 2009; Riehemann et al., 2009; van der Meel et al., 2013). Enhanced therapeutic effects have been demonstrated with ligand-targeted NNMs, despite showing no differences in accumulation in target tissues compared to non-targeted NNMs. For example, similar high levels of tumor tissue accumulation were achieved with both non-targeted liposomes and liposomes conjugated with HER2 monoclonal antibody fragments (7–8% injected dose/g tumor tissue) in HER2-overexpressing breast cancer xenografts models (Kirpotin et al., 1997, 2006). However, significantly superior therapeutic results was demonstrated with the doxorubicin-loaded anti-HER2 immunoliposomes in comparison to all other control groups, including recombinant anti-HER2 Mab trastuzumab, non-targeted liposomal doxorubicin, and free doxorubicin (Park et al., 2002). Differences in pharmacodynamics of the targeted NNM formulation in vivo was suggested as the reason for the improved anti-tumor effect, by enhancing intracellular drug delivery to HER2-overexpressing cancer cells (Kirpotin et al., 2006).
NNMs for Triggered Release
A third targeting strategy based on stimuli-responsive NNMs, referred to as triggered drug release, is currently receiving much attention from academia and industry. This class of NNMs is designed with the goal of enhancing drug release in tumors by means of endogenous or exogenous stimuli. Endogenous stimuli-responsive NNMs exploit factors associated with the local environment at the site of disease (Figure 1). For example, low pH, presence of redox gradients or certain enzymes in the tumor microenvironment. Exogenous-responsive NNMs respond to external stimuli to trigger drug release, such as temperature, light, magnetic field or ultrasound. Of these strategies, the use of an external hyperthermic trigger to release therapeutic compounds from NNMs (e.g., thermosensitive liposomal doxorubicin, ThermoDox®) appears to be the most promising to date (Needham et al., 2000). ThermoDox® was shown to be superior to its counterpart Doxil® in an in vivo model of non-resectable hepatocellular carcinoma (Torchilin, 2006; Sawant and Torchilin, 2012; Oude Blenke et al., 2013; Bertrand et al., 2014; Min et al., 2015; Jang et al., 2016; Shi et al., 2017). Thermosensitive liposomes are typically modified with temperature-sensitive lipids (e.g., distearoyl phosphocholine, DSPC) and/or polymers [e.g., poly(N-isopropylacrylamide)]. This composition allows the NNM to remain stable and retain their contents at physiologic temperatures, and undergo a phase change that makes them more permeable upon heating, thereby triggering the release of the cargo (Kono, 2001). The advantages of these NNMs can be further extended with the incorporation of imaging moieties (Figure 1) to enable monitoring of biodistribution, target accumulation and efficacy.
NNMs Approved and in Clinical Trials
A number of NNM products are on the market with more in clinical development. The majority of NNMs in clinical development incorporate already approved drugs and are based on a variety of drug delivery platforms, including polymeric micelles, liposomes, dendrimers, and inorganic nanoparticles (Torchilin, 2006; Wagner et al., 2006; Sercombe et al., 2015). Despite the arsenal of nanoparticulate targeted systems currently under preclinical development or in clinical trials, it is indisputable that liposomes are dominant on the NNM market (Table 1) and were the first FDA-approved NNM (Caster et al., 2017; Shi et al., 2017). In fact, liposomes have all the necessary features to allow formulation of highly toxic and/or poorly soluble drugs, such as paclitaxel and amphotericin B (Min et al., 2015; Caster et al., 2017). Soon after their discovery in 1965 (Sessa and Weissmann, 1968; Deamer, 2010), liposomes were proposed as drug delivery vehicles for both small molecules as well as macromolecular drugs (Gregoriadis and Ryman, 1971; Gregoriadis et al., 1971). Years of research led to the development of the first FDA-approved NNM (Doxil®/Caelyx®) as well as additional therapeutics (Allen and Cullis, 2013). Expectedly, many more NNMs are progressing to clinical investigation every year (Table 2), and again liposomal formulations represent the biggest share of the NNMs under clinical evaluation. The most frequently observed clinical benefit so far has been a reduction in toxicity with little evidence of improved efficacy. However, recently approved liposomal NNM, Vyxeos® (daunorubicin/cytarabine liposomal formulation), demonstrated improved survival and response rates, with tolerable toxicity in phase III clinical trials in older patients with therapy-related acute myeloid leukemia (t-AML) or AML with myelodysplasia-related changes (AML-MRC; Kim and Williams, 2018).
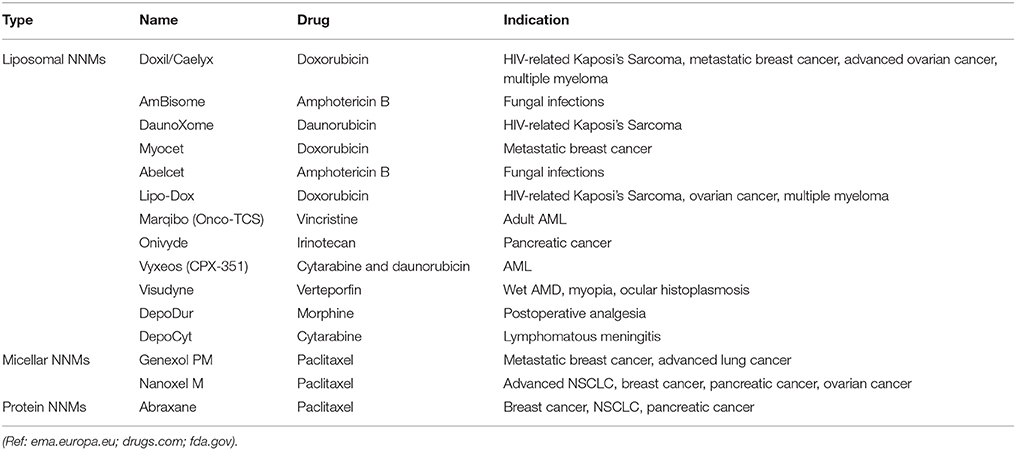
Table 1. NNM formulations currently approved for marketing.
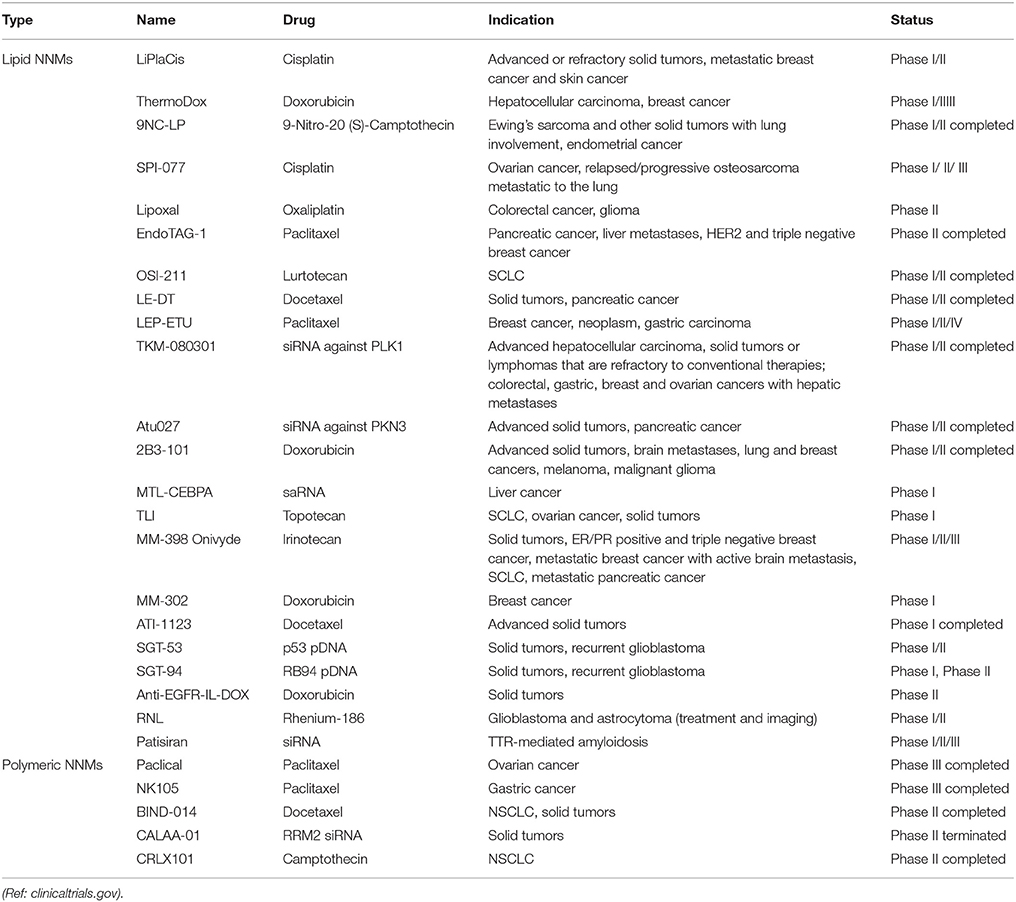
Table 2. NNM formulations in clinical trials.
Challenges in the Clinical Translation of Nanomedicines
The clinical translation of NNMs is an expensive and time-consuming process. NNM technology is usually far more complex in comparison to conventional formulation technology containing free drug dispersed in a base (e.g., tablets, capsules and injections; Teli et al., 2010; Tinkle et al., 2014; Sainz et al., 2015). Key issues related to the clinical development of NNMs are listed in Table 3, and include biological challenges, large-scale manufacturing, biocompatibility and safety, intellectual property (IP), government regulations, and overall cost-effectiveness in comparison to current therapies (Allen and Cullis, 2004, 2013; Zhang et al., 2008; Sawant and Torchilin, 2012; Narang et al., 2013). These factors can impose significant hurdles limiting the appearance of NNMs on the market, irrelevant of whether they are therapeutically efficacious or not.

Table 3. Considerations for the translational development of nanomedicines.
Biological Challenges
Traditionally, NNM development has been based on a formulation-driven approach, whereby novel delivery systems are firstly engineered and characterized from a physicochemical perspective. It is only when attempting to align the NNM with a pathological application that limitations in the clinical translation of the system have been identified. Understanding the relationship between biology and technology, including understanding the influence of disease pathophysiology on nanomedicine accumulation, distribution, retention and efficacy, as well as the biopharmaceutical correlation between delivery system properties and in vivo behavior in animals versus humans are important determinants for the successful translation of NNMs. Therefore, applying a disease-driven approach by designing and developing NNMs that are able to exploit pathophysiological changes in disease biology has been suggested to improve clinical translation (Hare et al., 2017).
From the outset in NNM development, it is essential to consider the relationship between disease pathophysiology and the heterogeneity of the disease in humans, and the importance of physicochemical characteristics of different NNMs to overcoming biological barriers to enable improved targeting to diseased tissue and/or reduced accumulation in non-target organs. Considerably less research effort has been dedicated to comprehensively understanding the correlations between NNM behavior and patient biology in specific clinical applications as well as disease heterogeneity in patients—which are likely the major reasons for the failure seen in the translation of promising NNMs in clinical trials (Hare et al., 2017). These biological challenges can be a significant deterrent for pharmaceutical industry investment into nanomedicines. In order to reduce investment risk for NNMs, the preclinical data sets need to comprehensively evaluate therapeutic efficacy, safety, biodistribution, and pharmacokinetics in appropriate animal models of the disease that are relevant to human disease. Evaluation of NNMs in multiple preclinical animal models that represent aspects of the clinical disease is preferred to achieve reproducibility of results for the specific disease and not for a specific animal model. In addition, animal models that reflect only a narrow spectrum of the clinical disease may provide useful data that can predict their suitability for treating a specific patient sub-group (Hare et al., 2017). Differences in the anatomy and/or physiology of the animal species compared to humans should be taken into account based on different routes of administration. Preclinical studies of NNMs should also be conducted under appropriate randomization and blinding to reduce bias, as well be evaluated against proper controls, including the gold standard treatment and not just free drug solution. These factors are currently lacking in many published studies, which makes it difficult to assess clinical applicability and translatability. Other considerations include designing preclinical studies to optimize NNM performance in vivo, dosing schedules, and treatment combinations based on the specific clinical disease, as well as understanding the influence of disease progression and severity on nanomedicine performance. This will determine whether specific patient sub-groups may respond more favorably to NNM-based treatment.
Interestingly, the majority of the NNM formulations in development and clinical trials are focused on cancer targeting, including more than 80% of the publications on nanomedicine in the last two decades alone (Park, 2017). Despite the large number of publications, the translation of the published studies to clinical applications has been disappointing. Cancer targeting of NNMs has generally been universally based on the EPR effect, despite the fact that EPR-mediated accumulation has only been reported for some tumor types (Maeda, 2015). Tumors, like other clinical diseases, can be highly heterogeneous and can show inter-patient and intra-patient variability as the disease progresses. Hence a one-size-fits-all approach when designing NNM-based treatment is unlikely to translate to clinically beneficial outcomes. The EPR effect has increasingly been exploited for NNM targeting in other non-cancer conditions, especially those involving an inflammatory component that causes leakiness of inflamed blood vessels (e.g., rheumatoid arthritis, atherosclerosis, and inflammatory bowel disease; Metselaar et al., 2003, 2004; Maiseyeu et al., 2009; Crielaard et al., 2012; Hua and Cabot, 2013; Hua et al., 2015; Milane and Amiji, 2017). It should be appreciated that not all diseases can be accessed with NNMs due to biological barriers and that the EPR effect is unlikely to be present in all clinical diseases. EPR is also not the only determinant of NNM efficacy. NNM activity is also influenced by the extent of cellular uptake and kinetics of drug release within target tissues (Hare et al., 2017).
Furthermore, the advantages of ligand-targeted NNMs in the clinical research phase have so far been negligible, despite the enhanced accumulation in target sites and therapeutic outcomes in a number of preclinical studies (Sercombe et al., 2015). Potential reasons for this discrepancy have previously been reviewed (Sawant and Torchilin, 2012; Allen and Cullis, 2013), and include factors such as target accessibility and expression, disease-dependent anatomical and physiological barriers, and formulation stability. In addition, the optimal targeting ligand density on the surface of each NNM has yet to be determined and will likely depend on characteristics of the molecular target (e.g., expression, location, internalization rate and immunogenicity; Puri et al., 2009; Hua and Wu, 2013; Kraft et al., 2014). Detailed analysis of the degree of NNM accumulation, cellular internalization, intracellular functionality and intracellular degradation will also be important considerations for clinical validation and translation (Puri et al., 2009). Through extensive experimentation, we are gaining a better understanding of the more appropriate clinical applications for ligand-targeted NNMs. Therefore, by taking a disease-driven approach to NNM development, it will be possible to build comprehensive preclinical data sets that best predict efficacy for patient sub-groups and support translatable clinical development.
Large-Scale Manufacturing
One of the important factors contributing to the slow pace in the clinical translation of NNMs is the structural and physicochemical complexity of the formulation itself. Platforms that require complex and/or laborious synthesis procedures generally have limited clinical translation potential, as they can be quite problematic to pharmaceutically manufacture on a large-scale (Teli et al., 2010; Tinkle et al., 2014; Barz et al., 2015; Sainz et al., 2015). Pharmaceutical manufacturing development is centered on quality and cost. Quality includes the manufacturing process and stability of the formulation, with NNM manufacturing being challenged by potential issues related to: (i) poor quality control; (ii) scalability complexities; (iii) incomplete purification from contaminants (e.g., by-products and starting materials); (iv) high material and/or manufacturing costs; (v) low production yield; (vi) insufficient batch-to-batch reproducibility, consistency and storage stability of the final product (e.g., regarding size distribution, porosity, charge and mass); (vii) lack of infrastructure and/or in-house expertise; (viii) chemical instability or denaturation of the encapsulated compound during the manufacturing process; and (ix) scarcity of venture funds and pharmaceutical industry investment (Teli et al., 2010; Narang et al., 2013; Hafner et al., 2014; Tinkle et al., 2014).
An essential requirement for clinical translation is to have access to a preparation method that allows the production of large scalable quantities of NNMs, which is also consistently manufactured at the same high level of quality and batch-to-batch reproducibility to set specifications (Grainger, 2013; Lammers, 2013; Barz et al., 2015). Suitable methods for the industrial scale production of several basic nanomedicine platforms, such as liposomes, have been successfully developed without the need for numerous manufacturing steps or the use of organic solvents (Jaafar-Maalej et al., 2012; Kraft et al., 2014). The challenges arise when the NNM system becomes more complex. For example, with the addition of surface modification with coatings and/or ligands, inclusion of multiple targeting components, or by encapsulating more than one therapeutic agent. Integration of multiple components into a single nanosized carrier requires multiple steps in the production process, which inevitably poses problems for large-scale good manufacturing (cGMP) production, increases the cost of production, and makes the quality assurance and quality control (QA and QC) evaluation of such products more difficult (Teli et al., 2010; Svenson, 2012; Tinkle et al., 2014).
Characteristics of the manufactured NNM need to be well-defined and reproducibly generated to allow initiation of clinical translation. Chemistry, Manufacturing, and Controls (CMC) information is required for investigational new drugs (IND) at each phase of investigation to ensure proper identity, strength or potency, quality, and purity of the drug substance and drug product (FDA, 2003). The type of information submitted will depend on the phase of the investigation, the extent of the human study, the duration of the investigation, the nature and source of the drug substance, and the drug product dosage form (FDA, 2003). The characterization and validation of more complex NNMs can be particularly challenging due to the sheer number of parameters to address (e.g., size distribution, morphology, charge, purity, drug encapsulation efficiency, coating efficiency, and density of conjugated ligand/s; Teli et al., 2010). Batch-to-batch variation of NNMs can potentially lead to significant changes to their physicochemical properties (e.g., polarity and size), pharmacokinetic parameters (i.e., absorption, distribution, metabolism and excretion), and/or pharmacodynamic interactions (e.g., cellular interaction and activity; Teli et al., 2010; Tinkle et al., 2014; Barz et al., 2015). In addition, NNMs need to be stable after the manufacturing process, during long-term storage, and upon clinical administration (i.e., to avoid massive drug release or aggregation in the bloodstream en route to the site of action).
Biocompatibility and Safety
Detailed toxicology is essential for the clinical translation of NNMs to determine the overall safety for human use (Nystrom and Fadeel, 2012). Pharmaceutical regulatory authorities generally recommend that the sponsor carefully assess for any changes in the drug substance and drug product manufacturing process or drug product formulation at any phase of clinical development, in order to determine if the changes can directly or indirectly affect the safety of the product. CMC modifications throughout the IND process that can affect safety include: (i) changes in the synthetic pathway or reagents used to manufacture the drug substance, product or formulation; (ii) changes resulting in a different impurity profile; (iii) changes in the actual manufacturing method (e.g., chemical synthesis, fermentation, or derivation from a natural source); (iv) changes in the source material; (v) changes in the method of sterilization of the drug substance or drug product; (vi) changes in the route of administration; (vii) changes in the composition and/or dosage form of the drug product; (viii) changes in the drug product manufacturing process that can affect product quality; and (ix) changes in the drug product container closure system that can affect product quality (e.g., dose delivery; FDA, 2003). If any changes are identified, stringent procedures are in place to ensure appropriate comparison testing of the drug substance and/or drug product produced from the previous manufacturing process with the changed manufacturing process to evaluate product equivalency, quality, and safety (FDA, 2003). When analytical data demonstrate that the materials manufactured before and after are not comparable, sponsors should perform additional qualification and/or bridging studies to support the safety and bioavailability of the material to be used in the proposed trials (FDA, 2003).
Knowledge of the activity and toxicities of the free drug, the behavior of different NNM delivery systems and their interaction with biological components, and the influence of drug release rate on target and off-target concentrations of bioavailable drug allow the ability to predict potential side effects or toxicities in vivo (Hare et al., 2017). In particular, the rational design of NNMs from the early phase of material selection, production method optimization, and product purification is of fundamental importance to increase their clinical translation potential (Accomasso et al., 2018). Although the safety of some common materials such as phospholipids and biodegradable polymers have been studied previously (Storm et al., 1993), increasing the complexity of NNMs, such as the use of different synthetic compositions, coatings and ligands, can have a significant effect on the biocompatibility, biodistribution and toxicology profile of nanomedicines following in vivo administration (Allen and Cullis, 2004, 2013; Zhang et al., 2008; Sawant and Torchilin, 2012; Narang et al., 2013; Tinkle et al., 2014). For example, complement activation-related pseudoallergy (CARPA) is an acute adverse immune reaction caused by many NNMs (Szebeni, 2005; Sercombe et al., 2015; Szebeni and Storm, 2015; Jackman et al., 2016). The complement system is part of the innate immune response and is involved in a range of inflammatory and immunological processes (Moghimi and Hunter, 2001). CARPA is an immediate, non-IgE-mediated hypersensitivity reaction that can cause symptoms, including anaphylaxis, facial swelling, facial flushing, chills, headache, and cardiopulmonary distress (Szebeni, 2005). This adverse reaction is generally managed by slowing the infusion rate or ceasing therapy, as well as the use of standard allergy medications (e.g., antihistamines, corticosteroids and epinephrine; Sercombe et al., 2015; Szebeni and Storm, 2015). The development of immunogenic reactions to NNM-based therapies may lead to altered pharmacokinetics, loss of efficacy, and the rise of potentially serious toxicities (e.g., anaphylaxis; Szebeni and Moghimi, 2009; Szebeni and Storm, 2015).
There is a regulatory need for validated, sensitive and standardizable assays incorporating in vitro, ex vivo and in vivo protocols to appropriately assess the nanotoxicology of NNMs during the early stages of clinical development (Dobrovolskaia and McNeil, 2013; Jackman et al., 2016; Accomasso et al., 2018). Comprehensive in vitro or ex vivo assays for nanosafety testing are essential to screen for potential hazards prior to preclinical evaluation in animal models (Gaspar, 2007). For example, standardized in vitro protocols using different cell culture models (i.e., blood, liver, lung, brain, placenta, gastrointestinal system) to assess potential risk of cytotoxicity, immunotoxicity, and genotoxicity of NNMs (Accomasso et al., 2018). This is particularly important with the development of NNMs incorporating many new materials with the goal for use in the clinical setting. In order to do this effectively across the board, standardized reference materials would need to be established and the testing would also need to be relevant for the intended route of administration (Tinkle et al., 2014). Although current testing approaches are limited and insufficient for nanotoxicology evaluations for clinical translation, a number of techniques that are more specific for nanomedicines are under development. This includes alternative test strategies, high-throughput screening techniques, high-content screening, and computational modeling (Nel et al., 2013; Oomen et al., 2014; Dusinska et al., 2015; Accomasso et al., 2018). These techniques have the potential to analyze in a comparative way many NNMs simultaneously.
There is also a need to perform specialized toxicology studies in animal models to assess both short-term and long-term toxicity, as circulation half-lives and drug retention times are generally significantly increased with nanoencapsulation. A thorough understanding of the absorption, distribution, metabolism, and excretion of emerging nanomaterials in vivo is important to predict the toxicological responses to NNMs (Dobrovolskaia and McNeil, 2013; Tinkle et al., 2014). Adequate assessment protocols are needed to monitor various aspects of the NNM drug delivery process, including pharmacokinetics, biodistribution, target site accumulation, local distribution at the target site, localization in healthy tissues, kinetics of drug release, and therapeutic efficacy (Kunjachan et al., 2015). Incorporation of real-time imaging techniques have enabled better understanding of the interaction of NNMs with biological organs and tissues following in vivo administration (Gaspar, 2007; Nystrom and Fadeel, 2012; Dobrovolskaia and McNeil, 2013; Kunjachan et al., 2015).
In addition, biocompatibility, immunotoxicological, and inflammatory potential should be assessed, with functional outcomes correlated with mechanisms of tissue uptake and clearance (Gaspar, 2007). These parameters need to be well-investigated based on dose, dosage form and route of administration to establish safe limits prior to clinical trials (Gaspar, 2007; Nystrom and Fadeel, 2012). This is of particular importance for NNMs composed of materials that have never been used before in clinical applications. Even in the clinical trial phase, regulatory protocols should be in place to detect any toxicity caused not only by the encapsulated therapeutic compounds, but also novel mechanisms unique to nanotechnology (Gaspar, 2007; Nystrom and Fadeel, 2012). For example, short- and long-term effects of NNM accumulation in RES organs (esp. liver, kidneys, spleen, lungs, lymph nodes, and bone marrow; Senior, 1987; Szebeni and Barenholz, 2009; Szebeni and Moghimi, 2009), which are the main sites for NNM accumulation following systemic administration (Poste et al., 1976; Senior, 1987). The cells of the RES are also part of the innate immune system, which has raised concerns regarding whether macrophage saturation by NNMs can cause immunosuppression and increase the risk of infections (Sercombe et al., 2015; Liu et al., 2017). There have been no reports of clinically significant immunosuppression at therapeutic doses of non-cytotoxic NNMs, despite suggestions that excessive NNM deposition in macrophages may impair their phagocytic capacity or modulate other cellular functions (Szebeni and Barenholz, 2009; Szebeni and Moghimi, 2009). However, NNMs that contain cytotoxic compounds are capable of inducing macrophage destruction following uptake (Szebeni and Barenholz, 2009; Szebeni and Moghimi, 2009), with indirect signs that suggest the possibility of some immune suppression (Storm et al., 1998; Szebeni and Barenholz, 2009; Szebeni and Moghimi, 2009). For example, administration of Doxil® in mice was reported to interfere with the clearance of bacteria from the blood due to macrophage suppression (Storm et al., 1998; Szebeni and Barenholz, 2009). Addressing these issues are necessary to safeguard the application of emerging NNMs in the clinical setting.
Intellectual Property (IP)
Given the complexities of incorporating nanotechnology into biomedical and clinical applications, there needs to be more precise definitions on what constitutes novel IP of a nanomedicine (Satalkar et al., 2015). Nanomedicines are complex as they have a number of variable components, and bridge between the field of medicine and medical device (Paradise et al., 2009). Generally, the control of a NNM product requires an IP position on: (i) the encapsulated cargo; (ii) the carrier technology; and (iii) the characteristics of the drug and carrier together. Although this definition is straightforward, it does open up a number of problems with the issuing of patents to date (Bawa, 2007; Bawa et al., 2008). For example, NNMs that incorporate existing drugs with novel carrier technology, or those that incorporate existing drugs with existing carrier technology for a new biomedical or disease application. The IP situation becomes even more confusing with more complex drug delivery systems, such as those which incorporate commercially available targeting ligands (e.g., antibodies) or coatings (e.g., Eudragit®) that are owned by other companies. IP strategies may likely involve multiple patents associated with any given technology and the need for cross-licensing arrangements (Murday et al., 2009). Therefore, new IP practices and protocols are required to simplify the pathway from invention to commercialization to reduce the time and expense required for negotiating collaboration and licensing agreements (Murday et al., 2009).
With the significant increase in the number of nanotechnology patent applications over the last few decades, other key issues that need to be addressed include patent review delays, patent thickets, and issuance of invalid patents (Bawa, 2005, 2007; Bawa et al., 2005). There needs to be a universal nano-nomenclature on identical or similar nanostructures or nanomaterials, and more refined search tools and commercial databases to avoid the issuing of multiple nanopatents on the same invention (Bawa et al., 2005; Bawa, 2007). Databases used by the Patent and Trademark Office (PTO) need to be able to search through nanotech-related prior art that resided in scientific publications world-wide, including earlier publications that preceded the emergence of online publication databases (Tinkle et al., 2014). Patent examiners also require expertise and training with respect to the emerging fields of nanotechnology and nanomedicine. The complexities with nanotechnology have led to the so called “patent thickets”, which can lead to costly litigation and halt commercialization efforts (Tinkle et al., 2014). Therefore, improved clarity on IP and patenting surrounding nanotechnology in health and medicine is required, and will need to involve implementation of universal regulations and policies that are tailored toward this niche commercialization field.
Government Regulations
Nanomedicines have significant potential to increase the growth of the pharmaceutical market and improve health benefits, however the current scientific and regulatory gap for nanomedicines is large and challenging. Commercialization of nanomedicines is highly dependent on a number of regulatory factors based on government policies in the area of manufacturing practice, quality control, safety, and patent protection (Gaspar, 2007; Tinkle et al., 2014; Sainz et al., 2015). The lack of clear regulatory and safety guidelines has affected the development of NNM products toward timely and effective clinical translation (Gaspar, 2007; Tinkle et al., 2014; Sainz et al., 2015). For example, polymers have been widely investigated as an effective platform for NNM strategies; however, their safety and efficacy is highly dependent on the polymer molecular weight, polydispersity, molecular structure, and conjugation chemistry (Gaspar and Duncan, 2009; Diab et al., 2012). Due to the increased number of novel polymeric materials and complex polymeric-based NNM formulations, there is an urgent need for an appropriate regulatory framework to assist in evaluation (Gaspar and Duncan, 2009). As each polymer-based NNM is different, it is important to consider each individually based on doses, administration routes, dosing frequency, and proposed clinical use. This would be the same for most other NNM platforms.
NNMs are currently regulated within the conventional framework governed by the key regulatory authority of each country (e.g., FDA, TGA, and EMA). Although NNMs have been on the market for nearly two decades, the first generation of NNM products passed regulatory approval by only having to meet general standards, applicable to medicinal compounds. These regulations are no longer appropriate to confirm the quality, safety, and efficacy of NNMs for clinical use (Gaspar, 2007; Tinkle et al., 2014; Sainz et al., 2015). Reasons for this are based on the complex structure of NNMs, their unclear interaction with cells and tissues within the human body, increased complexity of clinical use, and the multifunctional nature of some formulations (e.g., integration of therapeutics with imaging diagnostics; Gaspar, 2007; Tinkle et al., 2014; Sainz et al., 2015). Regulatory standards and protocols validated specifically for nanoparticles are needed that bridge both medicine and medical devices regulations. This should take into account a NNM's complexity, route of administration, pharmacokinetics, pharmacodynamics and safety profile, as well as provide information on the most appropriate clinical trial design and patient selection (Tinkle et al., 2014). There needs to be a fine balance to ensure the safety and quality of NNMs without over-regulation, which can negatively affect the progress of innovative products to the market, by escalating costs for achieving regulatory approval and/or consuming a significant portion of the life of a patent.
Development of global regulatory standards for NNMs should be established alongside key countries with invested interest. Although major steps have been taken in the last 5 years, a closer collaboration between regulatory agencies, academia, research and industry is needed (Gaspar, 2007; Murday et al., 2009; Hafner et al., 2014). This is of particular importance due to the limited availability of contract manufacturing organizations world-wide that specialize in producing NNM products in accordance with the requirements for good manufacturing practice (GMP; Hafner et al., 2014). It should be noted that this limited number of manufacturing organizations may be further divided based on their infrastructure capabilities of producing specific NNM platforms (e.g., liposomes, polymeric nanoparticles, dendrimers and drug-polymer conjugates). Therefore, NNMs produced in these manufacturing organizations will likely be marketed in multiple countries and thus should be governed under the same regulatory standards (Hafner et al., 2014). There will need to be complete evaluation and documentation of production processes for NNMs, incorporating appropriate industrial standards for both quality control and prevention of environmental issues (Gaspar, 2007). Manufactured NNMs will still need to meet general pharmaceutical standards such as purity, sterility, stability, manufacturing operations, and related industrial control standards (Gaspar, 2007). In addition, new analytical tools and standardized methods will need to be implemented to evaluate key physical characteristics of NNMs that can affect in vivo performance such as particle size and size distribution, surface chemistry, morphology, surface area, surface coating, hydrophilicity, porosity, and surface charge density (Gaspar, 2007; Tinkle et al., 2014; Sainz et al., 2015). These methods will vary for different nanomaterials and nanostructures. Thus, regulatory authorities should work together to develop the testing methods and appropriate standardized protocols for toxicity studies and regulatory requirements, which will be needed to ensure the efficacy and safety of current and emerging NNMs.
Perspectives on the Translational Development of Nanomedicines
From a therapeutic perspective, increasing drug accumulation at target tissues and minimizing systemic adverse effects are still the biggest design challenges to meet when developing new drug delivery systems. Even though promising NNMs may demonstrate significant efficacy in in vitro or ex vivo studies, it is important to evaluate the platforms in vivo using appropriate animal models of the disease. It is here where many of the current NNM platforms have shown limited specificity, accumulation and/or stability, therefore providing unsatisfactory results to warrant progression in the R&D process (Hua et al., 2015). Efficacy in an animal model also does not necessarily equate to efficacy in humans, as drug delivery within the human body is complex and can be highly variable, especially when associated with disease (Hare et al., 2017). Therefore, this concept of designing nanomedicines that act like a “magic bullet,” which refers to the exclusive delivery of active compounds to specific organs, tissues or cells, is just not realistic when taking into account the pharmacokinetic and pharmacodynamic processes that occur following administration into the body (Barz et al., 2015). This term should refer to the development of realistic therapeutic platforms, in which therapeutic effects are maximized, doses are minimized, and complexity in dosage form design is reduced (Barz et al., 2015).
Complexity in dosage form design is a key factor in the ability for a NNM formulation to be translated to the clinic, irrelevant of its therapeutic efficacy. Simplification in formulation design is required to allow efficient and reproducible large-scale manufacturing (Grainger, 2013; Lammers, 2013; Barz et al., 2015). Any added complexities to the basic NNM platform would need to show significantly improved benefits that is reliable and reproducible in animal models and patients, due to the added costs and complexity in the manufacturing process. For example, further studies are required to examine the benefits of ligand-targeted delivery systems over basic NNM platforms, in particular the reliability and consistency of the expression of the target across disease severity and in different patients (Hua et al., 2015; Sercombe et al., 2015; Hare et al., 2017). In addition, when translating findings from animal models to humans, we need to determine how to modify these formulations so that they are appropriate for human administration (Hua et al., 2015). In vivo studies are typically conducted in animal models of experimental diseases, especially in mice and rats, which can place limitations on the size and consistency of the dosage form that can be administered—for example, via oral, topical or intraperitoneal delivery (Hua et al., 2015; Sercombe et al., 2015). The practicability of designing dosage forms that are both acceptable to humans and efficacious should be further explored for clinical studies. Thus, there needs to be a balance between complexity, therapeutic efficacy, and clinical translation.
To transition NNMs to the clinic, attention should be given to nanosized carriers that are stable following in vivo administration, easily able to be up-scaled for manufacturing with high control over their physicochemical properties (e.g., size and polydispersity, morphology, drug encapsulation efficiency, and charge), as well as being composed of materials that are biocompatible, biodegradable, and non-toxic. As nanoparticles are able to enter cells and interfere with molecular pathways, synthetic polymers and lipids should be carefully evaluated for potential short-term and long-term toxicity for clinical application (Gaspar and Duncan, 2009). For example, potentially toxic in vitro and in vivo effects have been identified with the use of cationic polymers and lipids, including reduced number of mitoses, cell shrinking, detrimental effects on key cellular proteins (e.g., protein kinase C), and vacuolization of the cytoplasm (Lv et al., 2006).
Pathway to Translation and Commercialization
The experimental development of NNMs is progressing at a fast pace, however significant challenges still exist in promoting these platforms into clinically feasible therapies (Table 3). The majority of NNMs in the clinic are for the treatment of cancer, predominantly by the parenteral route of administration. They are structurally based on simple nanomedicine platforms, in particular basic nanoparticles, surface charge-modified nanoparticles, and PEGylated nanoparticles (Hafner et al., 2014; Sainz et al., 2015). Although clinical applications of nanotechnology for non-cancer diseases are increasing based on promising experimental results, there are several barriers that have slowed progress in the preclinical and, especially, clinical stages of development. This includes issues surrounding complexity in manufacturing and characterization, lack of understanding of in vivo pharmacokinetics and pharmacodynamics, acute and chronic toxicity, and cost-effectiveness (Gaspar, 2007; Teli et al., 2010; Hafner et al., 2014; Tinkle et al., 2014; Sainz et al., 2015). These challenges are even greater with increasing complexity of the NNM design.
The pace for the clinical translation of NNMs has been relatively slow as the development trajectory is very costly, complex and time-consuming, which has affected the attitudes of the pharmaceutical industry and capital investors. There has to be a clear positive benefit-to-risk ratio that will accompany the clinical implementation of products and procedures based on nanotechnology. In particular, the cost-benefit analysis may be a limitation to the clinical translation of some NNMs when compared to an approved counterpart or existing therapies. This analysis has to be clear before starting the development process. Emerging NNM products, which are more complex in structure and more expensive than conventional therapies, need to provide an overall reduction in health care costs and provide a worthwhile opportunity for the pharmaceutical industry to invest its R&D budgets (Hafner et al., 2014). This reduction in health care costs is likely to be obtained by increasing therapeutic efficacy, improving quality of life, reducing adverse effects or toxicities in non-target organs, and/or reducing the need for surgical or other high-risk interventions (Gandjour and Chernyak, 2011). Nanopharmaceuticals can offer the ability to extend the economic life of proprietary drugs and create additional revenue streams (Tinkle et al., 2014). In addition, market analysis, investment risk, potential profit margins, and value proposition of novel NNMs are important factors for the pharmaceutical industry and investors. Typically, pharmaceutical products that are developed to address larger disease populations with treatment expected in a primary or secondary care setting are preferred by the pharmaceutical industry. From a business perspective, the necessary infrastructure, understanding of NNMs, and skill set required for the commercial development of NNMs are not currently well represented at most pharmaceutical companies. These factors should be taken into account when assessing the overall cost-effectiveness of NNMs in comparison to existing therapies.
Nanomedicines generally face a number of regulatory approval hurdles. The control of materials in the nanosize range often presents greater scientific and technical challenges compared to conventional formulations (Gaspar, 2007; Teli et al., 2010; Hafner et al., 2014; Tinkle et al., 2014; Sainz et al., 2015). NNMs encompass a number of different types of nanomaterials and nanostructures, which make it even more challenging to establish appropriate regulatory protocols and tools to ensure standardized GMP manufacturing and characterization, safety and toxicology evaluation, and clinical trial design. These procedures are paramount to confirming therapeutic efficacy and safety prior to marketing approval for use in patients on a larger scale. Effective clinical translation will require an interdisciplinary approach to develop novel protocols, assays and infrastructure for the manufacturing and characterization of NNMs (Gaspar, 2007; Teli et al., 2010; Hafner et al., 2014; Tinkle et al., 2014; Sainz et al., 2015). This will need to involve experts from academia and industry with specialty in pharmaceutics, engineering, biology, medicine, and toxicology. Potential approaches to fast-track promising novel NNMs to clinical trials include the establishment or coordination of laboratories and centers that have expertise in (i) characterizing NNM platforms, (ii) conducting preclinical studies on NNMs for submission to regulatory agencies, (iii) scale up laboratory preparation of nanomaterials according to regulatory and industry standards for early clinical trials, and (iv) designing and conducting clinical trials of NNM platforms (Hafner et al., 2014).
Conclusion
Overall, the use of nanotechnology in medicine has the potential to have a major impact on human health. It has been suggested to facilitate the development of personalized medicine for specific patient sub-groups, in which therapy is tailored by the patient's individual genetic and disease profile (Teli et al., 2010; Mura and Couvreur, 2012; Laroui et al., 2013). For example, disease-specific characteristics such as capillary permeability (Calcagno et al., 2015), cellular receptor expression and molecular pathway activation could be analyzed and used to design personalized nanomedicines (Teli et al., 2010; Mura and Couvreur, 2012; Laroui et al., 2013). The physicochemical properties (e.g., size and structure) of the delivery system can also be modified according to the severity of the disease for optimal therapeutic benefits (Hua et al., 2015). This concept would significantly advance the way in which we treat patients. However, for this to occur, there are still a number of issues that need to be addressed as detailed in this review—from our basic understanding of the biology of specific diseases and the biological interaction of NNMs in patients, to commercialization hurdles related to manufacturing, costs, and regulatory standards. Finally, researchers need to consider minimizing the complexity of NNMs and take into account the final dosage form for human use, in order for a formulation to have the potential to be translated into a clinically applicable therapeutic. Reducing complexity to the minimum required for pathophysiological or medical need is paramount in nanoparticle design and synthesis to generate clinically translatable nanosized therapeutics.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors wish to thank The Pharmacy Research Trust of New South Wales, The Rebecca L. Cooper Medical Research Foundation, Gladys M Brawn Fellowship, ausEE Research Foundation, and The University of Newcastle for providing financial support for our research.
References
Accomasso, L., Cristallini, C., and Giachino, C. (2018). Risk Assessment and Risk Minimization in Nanomedicine: A Need for Predictive, Alternative, and 3Rs Strategies. Frontiers in Pharmacology. 9:228. doi: 10.3389/fphar.2018.00228
Allen, T. M., and Cullis, P. R. (2004). Drug delivery systems: entering the mainstream. Science 303, 1818–1822. doi: 10.1126/science.1095833
Allen, T. M., and Cullis, P. R. (2013). Liposomal drug delivery systems: from concept to clinical applications. Adv. Drug Del. Rev. 65, 36–48. doi: 10.1016/j.addr.2012.09.037
Barz, M., Luxenhofer, R., and Schillmeier, M. (2015). Quo vadis nanomedicine? Nanomedicine 10, 3089–3091. doi: 10.2217/nnm.15.156
Bawa, R. (2005). Will the nanomedicine “patent land grab” thwart commercialization? Nanomedicine 1, 346–350. doi: 10.1016/j.nano.2005.10.009
Bawa, R., Bawa, S. R., Maebius, S. B., Flynn, T., and Wei, C. (2005). Protecting new ideas and inventions in nanomedicine with patents. Nanomedicine 1, 150–158. doi: 10.1016/j.nano.2005.03.009
Bawa, R., Melethil, S., Simmons, W. J., and Harris, D. (2008). Nanopharmaceuticals - patenting issues and FDA regulatory challenges. Am. Bar Assoc. SciTech Lawyer. 5, 10–15.
Bertrand, N., Wu, J., Xu, X., Kamaly, N., and Farokhzad, O. C. (2014). Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 66, 2–25. doi: 10.1016/j.addr.2013.11.009
Calcagno, C., Lobatto, M. E., Dyvorne, H., Robson, P. M., Millon, A., Senders, M. L., et al. (2015). Three-dimensional dynamic contrast-enhanced MRI for the accurate, extensive quantification of microvascular permeability in atherosclerotic plaques. NMR Biomed. 28, 1304–1314. doi: 10.1002/nbm.3369
Caster, J. M., Patel, A. N., Zhang, T., and Wang, A. (2017). Investigational nanomedicines in 2016: a review of nanotherapeutics currently undergoing clinical trials. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 9:e1416. doi: 10.1002/wnan.1416
Coimbra, M., Banciu, M., Fens, M. H., de Smet, L., Cabaj, M., Metselaar, J. M., et al. (2010). Liposomal pravastatin inhibits tumor growth by targeting cancer-related inflammation. J. Control Release. 148, 303–310. doi: 10.1016/j.jconrel.2010.09.011
Crielaard, B. J., Lammers, T., Schiffelers, R. M., and Storm, G. (2012). Drug targeting systems for inflammatory disease: one for all, all for one. J. Control Release. 161, 225–234. doi: 10.1016/j.jconrel.2011.12.014
Danhier, F. (2016). To exploit the tumor microenvironment: since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release. 244 (Pt A), 108–121. doi: 10.1016/j.jconrel.2016.11.015
Danhier, F., Feron, O., and Preat, V. (2010). To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control Release. 148, 135–146. doi: 10.1016/j.jconrel.2010.08.027
Deamer, D. W. (2010). From “banghasomes” to liposomes: a memoir of Alec Bangham, 1921-2010. FASEB J. 24, 1308–1310. doi: 10.1096/fj.10-0503
Diab, R., Jaafar-Maalej, C., Fessi, H., and Maincent, P. (2012). Engineered nanoparticulate drug delivery systems: the next frontier for oral administration? AAPS J. 14, 688–702. doi: 10.1208/s12248-012-9377-y
Dobrovolskaia, M. A., and McNeil, S. E. (2013). Understanding the correlation between in vitro and in vivo immunotoxicity tests for nanomedicines. J Control Release. 172, 456–466. doi: 10.1016/j.jconrel.2013.05.025
Dusinska, M., Boland, S., Saunders, M., Juillerat-Jeanneret, L., Tran, L., Pojana, G., et al. (2015). Towards an alternative testing strategy for nanomaterials used in nanomedicine: lessons from NanoTEST. Nanotoxicology 9 (Suppl. 1), 118–132. doi: 10.3109/17435390.2014.991431
FDA (2003). Guidance for Industry - INDs for Phase 2 and Phase 3 Studies: Chemistry, Manufacturing, and Control (CMC) Information
Ferrari, M. (2005). Nanovector therapeutics. Curr. Opin. Chem. Biol. 9, 343–346. doi: 10.1016/j.cbpa.2005.06.001
Gandjour, A., and Chernyak, N. (2011). A new prize system for drug innovation. Health Policy. 102, 170–177. doi: 10.1016/j.healthpol.2011.06.001
Gaspar, R. (2007). Regulatory issues surrounding nanomedicines: setting the scene for the next generation of nanopharmaceuticals. Nanomedicine 2, 143–147. doi: 10.2217/17435889.2.2.143
Gaspar, R., and Duncan, R. (2009). Polymeric carriers: preclinical safety and the regulatory implications for design and development of polymer therapeutics. Adv. Drug Deliv. Rev. 61, 1220–1231. doi: 10.1016/j.addr.2009.06.003
Grainger, D. W. (2013). Connecting drug delivery reality to smart materials design. Int. J. Pharm. 454, 521–524. doi: 10.1016/j.ijpharm.2013.04.061
Gregoriadis, G., Leathwood, P. D., and Ryman, B. E. (1971). Enzyme entrapment in liposomes. FEBS Lett. 14, 95–99. doi: 10.1016/0014-5793(71)80109-6
Gregoriadis, G., and Ryman, B. E. (1971). Liposomes as carriers of enzymes or drugs: a new approach to the treatment of storage diseases. Biochem. J. 124:58P. doi: 10.1042/bj1240058P
Hafner, A., Lovric, J., Lakos, G. P., and Pepic, I. (2014). Nanotherapeutics in the EU: an overview on current state and future directions. Int. J. Nanomedicine. 9, 1005–1023. doi: 10.2147/IJN.S55359
Hare, J. I., Lammers, T., Ashford, M. B., Puri, S., Storm, G., and Barry, S. T. (2017). Challenges and strategies in anti-cancer nanomedicine development: an industry perspective. Adv. Drug. Deliv. Rev. 108, 25–38. doi: 10.1016/j.addr.2016.04.025
Hashizume, H., Baluk, P., Morikawa, S., McLean, J. W., Thurston, G., Roberge, S., et al. (2000). Openings between defective endothelial cells explain tumor vessel leakiness. Am. J. Pathol. 156, 1363–1380. doi: 10.1016/S0002-9440(10)65006-7
Hobbs, S. K., Monsky, W. L., Yuan, F., Roberts, W. G., Griffith, L., Torchilin, V. P., et al. (1998). Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc. Natl. Acad. Sci. U.S.A. 95, 4607–4612. doi: 10.1073/pnas.95.8.4607
Hua, S. (2013). Targeting sites of inflammation: intercellular adhesion molecule-1 as a target for novel inflammatory therapies. Front. Pharmacol. 4:127. doi: 10.3389/fphar.2013.00127
Hua, S., and Cabot, P. J. (2013). Targeted nanoparticles that mimic immune cells in pain control inducing analgesic and anti-inflammatory actions: a potential novel treatment of acute and chronic pain condition. Pain Physician. 16, E199–E216.
Hua, S., Marks, E., Schneider, J. J., and Keely, S. (2015). Advances in oral nano-delivery systems for colon targeted drug delivery in inflammatory bowel disease: Selective targeting to diseased versus healthy tissue. Nanomedicine 11, 1117–1132. doi: 10.1016/j.nano.2015.02.018
Hua, S., and Wu, S. Y. (2013). The use of lipid-based nanocarriers for targeted pain therapies. Front. Pharmacol. 4:143. doi: 10.3389/fphar.2013.00143
Jaafar-Maalej, C., Elaissari, A., and Fessi, H. (2012). Lipid-based carriers: manufacturing and applications for pulmonary route. Expert Opin. Drug Deliv. 9, 1111–1127. doi: 10.1517/17425247.2012.702751
Jackman, J. A., Meszaros, T., Fulop, T., Urbanics, R., Szebeni, J., and Cho, N. J. (2016). Comparison of complement activation-related pseudoallergy in miniature and domestic pigs: foundation of a validatable immune toxicity model. Nanomedicine 12, 933–943. doi: 10.1016/j.nano.2015.12.377
Jang, B., Kwon, H., Katila, P., Lee, S. J., and Lee, H. (2016). Dual delivery of biological therapeutics for multimodal and synergistic cancer therapies. Adv. Drug Deliv. Rev. 98, 113–133. doi: 10.1016/j.addr.2015.10.023
Kim, H. J., Kim, A., Miyata, K., and Kataoka, K. (2016). Recent progress in development of siRNA delivery vehicles for cancer therapy. Adv. Drug. Deliv. Rev. 104, 61–77. doi: 10.1016/j.addr.2016.06.011
Kim, M., and Williams, S. (2018). Daunorubicin and cytarabine liposome in newly diagnosed therapy-related Acute Myeloid Leukemia (AML) or AML with myelodysplasia-related changes. Ann. Pharmacother. 1:1060028018764923. doi: 10.1177/1060028018764923
Kirpotin, D. B., Drummond, D. C., Shao, Y., Shalaby, M. R., Hong, K., Nielsen, U. B., et al. (2006). Antibody targeting of long-circulating lipidic nanoparticles does not increase tumor localization but does increase internalization in animal models. Cancer Res. 66, 6732–6740. doi: 10.1158/0008-5472.CAN-05-4199
Kirpotin, D., Park, J. W., Hong, K., Zalipsky, S., Li, W. L., Carter, P., et al. (1997). Sterically stabilized anti-HER2 immunoliposomes: design and targeting to human breast cancer cells in vitro. Biochemistry 36, 66–75. doi: 10.1021/bi962148u
Kono, K. (2001). Thermosensitive polymer-modified liposomes. Adv. Drug Deliv. Rev. 53, 307–319. doi: 10.1016/S0169-409X(01)00204-6
Kraft, J. C., Freeling, J. P., Wang, Z., and Ho, R. J. (2014). Emerging research and clinical development trends of liposome and lipid nanoparticle drug delivery systems. J. Pharm. Sci. 103, 29–52. doi: 10.1002/jps.23773
Kuijpers, S. A., Coimbra, M. J., Storm, G., and Schiffelers, R. M. (2010). Liposomes targeting tumour stromal cells. Mol. Membr. Biol. 27, 328–340. doi: 10.3109/09687688.2010.522204
Kunjachan, S., Ehling, J., Storm, G., Kiessling, F., and Lammers, T. (2015). Noninvasive imaging of nanomedicines and nanotheranostics: principles, progress, and prospects. Chem. Rev. 115, 10907–10937. doi: 10.1021/cr500314d
Lammers, T. (2013). Smart drug delivery systems: back to the future vs. clinical reality. Int. J. Pharm. 454, 527–529. doi: 10.1016/j.ijpharm.2013.02.046
Lammers, T., Kiessling, F., Hennink, W. E., and Storm, G. (2012). Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J. Control Release. 161, 175–187. doi: 10.1016/j.jconrel.2011.09.063
Laroui, H., Rakhya, P., Xiao, B., Viennois, E., and Merlin, D. (2013). Nanotechnology in diagnostics and therapeutics for gastrointestinal disorders. Dig. Liver Dis. 45, 995–1002. doi: 10.1016/j.dld.2013.03.019
Larsson, M., Huang, W. T., Liu, D. M., and Losic, D. (2017). Local co-administration of gene-silencing RNA and drugs in cancer therapy: State-of-the art and therapeutic potential. Cancer Treat. Rev. 55, 128–135. doi: 10.1016/j.ctrv.2017.03.004
Liu, Y., Hardie, J., Zhang, X., and Rotello, V. M. (2017). Effects of engineered nanoparticles on the innate immune system. Semin. Immunol. 34, 25–32. doi: 10.1016/j.smim.2017.09.011
Luxenhofer, R., Barz, M., and Schillmeier, M. (2014). Quo vadis nanomedicine? Nanomedicine 9, 2083–2086. doi: 10.2217/nnm.14.140
Lv, H., Zhang, S., Wang, B., Cui, S., and Yan, J. (2006). Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control Release. 114, 100–109. doi: 10.1016/j.jconrel.2006.04.014
Lyass, O., Uziely, B., Ben-Yosef, R., Tzemach, D., Heshing, N. I., Lotem, M., et al. (2000). Correlation of toxicity with pharmacokinetics of pegylated liposomal doxorubicin (Doxil) in metastatic breast carcinoma. Cancer 89, 1037–1047. doi: 10.1002/1097-0142(20000901)89:5<1037::AID-CNCR13>3.0.CO;2-Z
Maeda, H. (2015). Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv. Drug Deliv. Rev. 91, 3–6. doi: 10.1016/j.addr.2015.01.002
Maeda, H., Nakamura, H., and Fang, J. (2013). The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv. Drug. Deliv. Rev. 65, 71–79. doi: 10.1016/j.addr.2012.10.002
Maiseyeu, A., Mihai, G., Kampfrath, T., Simonetti, O. P., Sen, C. K., Roy, S., et al. (2009). Gadolinium-containing phosphatidylserine liposomes for molecular imaging of atherosclerosis. J. Lipid. Res. 50, 2157–2163. doi: 10.1194/jlr.M800405-JLR200
Mastrobattista, E., Koning, G. A., and Storm, G. (1999). Immunoliposomes for the targeted delivery of antitumor drugs. Adv. Drug. Deliv. Rev. 40, 103–127. doi: 10.1016/S0169-409X(99)00043-5
Matsumura, Y., and Maeda, H. (1986). A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 46 (12 Pt 1), 6387–6392.
Metselaar, J. M., Van den Berg, W. B., Holthuysen, A. E., Wauben, M. H., Storm, G., and Van Lent, P. L. (2004). Liposomal targeting of glucocorticoids to synovial lining cells strongly increases therapeutic benefit in collagen type II arthritis. Ann. Rheum. Dis. 63, 348–353. doi: 10.1136/ard.2003.009944
Metselaar, J. M., Wauben, M. H., Wagenaar-Hilbers, J. P., Boerman, O. C., and Storm, G. (2003). Complete remission of experimental arthritis by joint targeting of glucocorticoids with long-circulating liposomes. Arthritis Rheum. 48, 2059–66. doi: 10.1002/art.11140
Milane, L. S., and Amiji, M. (2017). Nanomedicine for Inflammatory Diseases. New York, NY: CRC Press
Min, Y., Caster, J. M., Eblan, M. J., and Wang, A. Z. (2015). Clinical translation of nanomedicine. Chem. Rev. 115, 11147–11190. doi: 10.1021/acs.chemrev.5b00116
Mishra, D. K., Balekar, N., and Mishra, P. K. (2017). Nanoengineered strategies for siRNA delivery: from target assessment to cancer therapeutic efficacy. Drug. Deliv. Transl. Res. 7, 346–358. doi: 10.1007/s13346-016-0352-5
Moghimi, S. M., and Hunter, A. C. (2001). Capture of stealth nanoparticles by the body's defences. Crit. Rev. Ther. Drug Carrier Syst. 18, 527–550. doi: 10.1615/CritRevTherDrugCarrierSyst.v18.i6.30
Mura, S., and Couvreur, P. (2012). Nanotheranostics for personalized medicine. Adv. Drug Deliv. Rev. 64, 1394–1416. doi: 10.1016/j.addr.2012.06.006
Murday, J. S., Siegel, R. W., Stein, J., and Wright, J. F. (2009). Translational nanomedicine: status assessment and opportunities. Nanomedicine 5, 251–273. doi: 10.1016/j.nano.2009.06.001
Nakamura, H., Fang, J., and Maeda, H. (2015). Development of next-generation macromolecular drugs based on the EPR effect: challenges and pitfalls. Expert Opin Drug Deliv. 12, 53–64. doi: 10.1517/17425247.2014.955011
Narang, A. S., Chang, R. K., and Hussain, M. A. (2013). Pharmaceutical development and regulatory considerations for nanoparticles and nanoparticulate drug delivery systems. J. Pharm. Sci. 102, 3867–3882. doi: 10.1002/jps.23691
Needham, D., Anyarambhatla, G., Kong, G., and Dewhirst, M. W. (2000). A new temperature-sensitive liposome for use with mild hyperthermia: characterization and testing in a human tumor xenograft model. Cancer Res. 60, 1197–1201.
Nehoff, H., Parayath, N. N., Domanovitch, L., Taurin, S., and Greish, K. (2014). Nanomedicine for drug targeting: strategies beyond the enhanced permeability and retention effect. Int. J. Nanomedicine 9, 2539–2555. doi: 10.2147/IJN.S47129
Nel, A., Xia, T., Meng, H., Wang, X., Lin, S., Ji, Z., et al. (2013). Nanomaterial toxicity testing in the 21st century: use of a predictive toxicological approach and high-throughput screening. Acc. Chem. Res. 46, 607–621. doi: 10.1021/ar300022h
Nichols, J. W., and Bae, Y. H. (2014). EPR: evidence and fallacy. J. Control Release. 190, 451–464. doi: 10.1016/j.jconrel.2014.03.057
Nystrom, A. M., and Fadeel, B. (2012). Safety assessment of nanomaterials: implications for nanomedicine. J. Control Release. 161, 403–408. doi: 10.1016/j.jconrel.2012.01.027
Ojha, T., Pathak, V., Shi, Y., Hennink, W. E., Moonen, C. T. W., Storm, G., et al. (2017). Pharmacological and physical vessel modulation strategies to improve EPR-mediated drug targeting to tumors. Adv. Drug Deliv. Rev. 119, 44–60. doi: 10.1016/j.addr.2017.07.007
Oomen, A. G., Bos, P. M., Fernandes, T. F., Hund-Rinke, K., Boraschi, D., Byrne, H. J., et al. (2014). Concern-driven integrated approaches to nanomaterial testing and assessment–report of the NanoSafety Cluster Working Group 10. Nanotoxicology 8, 334–348. doi: 10.3109/17435390.2013.802387
Oude Blenke, E., Mastrobattista, E., and Schiffelers, R. M. (2013). Strategies for triggered drug release from tumor targeted liposomes. Expert. Opin. Drug Deliv. 10, 1399–1410. doi: 10.1517/17425247.2013.805742
Paradise, J., Wolf, S. M., Kuzma, J., Kuzhabekova, A., Tisdale, A. W., Kokkoli, E., et al. (2009). Developing, U.S. oversight strategies for nanobiotechnology: learning from past oversight experiences. J. Law Med. Ethics. 37, 688–705. doi: 10.1111/j.1748-720X.2009.00441.x
Park, J. W., Hong, K., Kirpotin, D. B., Colbern, G., Shalaby, R., Baselga, J., et al. (2002). Anti-HER2 immunoliposomes: enhanced efficacy attributable to targeted delivery. Clin. Cancer Res. 8, 1172–1181.
Park, K. (2017). The drug delivery field at the inflection point: Time to fight its way out of the egg. J. Control Release. 267, 2–14. doi: 10.1016/j.jconrel.2017.07.030
Poste, G., Papahadjopoulos, D., and Vail, W. J. (1976). Lipid vesicles as carriers for introducing biologically active materials into cells. Methods Cell Biol. 14, 33–71. doi: 10.1016/S0091-679X(08)60468-9
Puri, A., Loomis, K., Smith, B., Lee, J. H., Yavlovich, A., Heldman, E., et al. (2009). Lipid-based nanoparticles as pharmaceutical drug carriers: from concepts to clinic. Crit. Rev. Ther. Drug Carrier Syst. 26, 523–580. doi: 10.1615/CritRevTherDrugCarrierSyst.v26.i6.10
Rahman, A. M., Yusuf, S. W., and Ewer, M. S. (2007). Anthracycline-induced cardiotoxicity and the cardiac-sparing effect of liposomal formulation. Int. J. Nanomedicine 2, 567–583.
Riehemann, K., Schneider, S. W., Luger, T. A., Godin, B., Ferrari, M., and Fuchs, H. (2009). Nanomedicine–challenge and perspectives. Angew. Chem. Int. Ed. Engl. 48, 872–897. doi: 10.1002/anie.200802585
Rizzo, L. Y., Theek, B., Storm, G., Kiessling, F., and Lammers, T. (2013). Recent progress in nanomedicine: therapeutic, diagnostic and theranostic applications. Curr. Opin. Biotechnol. 24, 1159–1166. doi: 10.1016/j.copbio.2013.02.020
Sainz, V., Conniot, J., Matos, A. I., Peres, C., Zupancic, E., Moura, L., et al. (2015). Regulatory aspects on nanomedicines. Biochem. Biophys. Res. Commun. 468, 504–510, doi: 10.1016/j.bbrc.2015.08.023
Satalkar, P., Elger, B. S., and Shaw, D. M. (2015). Defining Nano, Nanotechnology and Nanomedicine: Why Should It Matter? Sci. Eng. Ethics 22, 1255–1276. doi: 10.1007/s11948-015-9705-6
Sawant, R. R., and Torchilin, V. P. (2012). Challenges in development of targeted liposomal therapeutics. AAPS J. 14, 303–315. doi: 10.1208/s12248-012-9330-0
Senior, J. H. (1987). Fate and behavior of liposomes in vivo: a review of controlling factors. Crit. Rev. Ther. Drug Carrier Syst. 3, 123–193.
Sercombe, L., Veerati, T., Moheimani, F., Wu, S. Y., Sood, A. K., and Hua, S. (2015). Advances and challenges of liposome assisted drug delivery. Front. Pharmacol. 6:286. doi: 10.3389/fphar.2015.00286
Sessa, G., and Weissmann, G. (1968). Phospholipid spherules (liposomes) as a model for biological membranes. J. Lipid. Res. 9, 310–318.
Shajari, N., Mansoori, B., Davudian, S., Mohammadi, A., and Baradaran, B. (2017). Overcoming the challenges of siRNA delivery: nanoparticle strategies. Curr. Drug. Deliv. 14, 36–46. doi: 10.2174/1567201813666160816105408
Shi, J., Kantoff, P. W., Wooster, R., and Farokhzad, O. C. (2017). Cancer nanomedicine: progress, challenges and opportunities. Nat. Rev. Cancer 17, 20–37. doi: 10.1038/nrc.2016.108
Storm, G., Oussoren, C., Peeters, P., and Barenholz, C. (1993). “Tolerability of liposomes in vivo,” in Liposome Technology. 3, ed G. Gregoriadis (Florida: CRC Press), 345–83.
Storm, G., ten Kate, M. T., Working, P. K., and Bakker-Woudenberg, I. A. (1998). Doxorubicin entrapped in sterically stabilized liposomes: effects on bacterial blood clearance capacity of the mononuclear phagocyte system. Clin. Cancer Res. 4, 111–115.
Svenson, S. (2012). Clinical translation of nanomedicines. Curr. Opin. Solid. State Mater. Sci. 16, 287–294. doi: 10.1016/j.cossms.2012.10.001
Szebeni, J. (2005). Complement activation-related pseudoallergy: a new class of drug-induced acute immune toxicity. Toxicology 216, 106–121. doi: 10.1016/j.tox.2005.07.023
Szebeni, J., and Barenholz, Y. (2009). “Adverse immune effects of liposomes: complement activation, immunogenicity and immune suppression,” in Harnessing biomaterials for nanomedicine: preparation, toxicity and applications, ed P. S. Publishing (Singapore: Pan Stanford Publishing), 1–19.
Szebeni, J., and Moghimi, S. M. (2009). Liposome triggering of innate immune responses: a perspective on benefits and adverse reactions. J. Liposome Res. 19, 85–90. doi: 10.1080/08982100902792855
Szebeni, J., and Storm, G. (2015). Complement activation as a bioequivalence issue relevant to the development of generic liposomes and other nanoparticulate drugs. Biochem. Biophys. Res. Commun. 468, 490–497. doi: 10.1016/j.bbrc.2015.06.177
Talekar, M., Tran, T. H., and Amiji, M. (2015). Translational nano-medicines: targeted therapeutic delivery for cancer and inflammatory diseases. AAPS J. 17, 813–827. doi: 10.1208/s12248-015-9772-2
Teli, M. K., Mutalik, S., and Rajanikant, G. K. (2010). Nanotechnology and nanomedicine: going small means aiming big. Curr. Pharm. Des. 16, 1882–1892. doi: 10.2174/138161210791208992
Theek, B., Gremse, F., Kunjachan, S., Fokong, S., Pola, R., Pechar, M., et al. (2014). Characterizing EPR-mediated passive drug targeting using contrast-enhanced functional ultrasound imaging. J. Control Release. 182, 83–89. doi: 10.1016/j.jconrel.2014.03.007
Tinkle, S., McNeil, S. E., Muhlebach, S., Bawa, R., Borchard, G., Barenholz, Y. C., et al. (2014). Nanomedicines: addressing the scientific and regulatory gap. Ann. N. Y. Acad. Sci. 1313, 35–56. doi: 10.1111/nyas.12403
Torchilin, V. P. (2006). Multifunctional nanocarriers. Adv. Drug Deliv. Rev. 58, 1532–1555. doi: 10.1016/j.addr.2006.09.009
van der Meel, R., Vehmeijer, L. J., Kok, R. J., Storm, G., and van Gaal, E. V. (2013). Ligand-targeted particulate nanomedicines undergoing clinical evaluation: current status. Adv. Drug Deliv. Rev. 65, 1284–1298. doi: 10.1016/j.addr.2013.08.012
Wagner, V., Dullaart, A., Bock, A. K., and Zweck, A. (2006). The emerging nanomedicine landscape. Nat. Biotechnol. 24, 1211–1217. doi: 10.1038/nbt1006-1211
Willis, M., and Forssen, E. (1998). Ligand-targeted liposomes. Adv. Drug Deliv. Rev. 29, 249–271. doi: 10.1016/S0169-409X(97)00083-5
Keywords: nanomedicine, nanoparticles, drug delivery systems, clinical translation, challenges, commercialization, biological, regulations
Citation: Hua S, de Matos MBC, Metselaar JM and Storm G (2018) Current Trends and Challenges in the Clinical Translation of Nanoparticulate Nanomedicines: Pathways for Translational Development and Commercialization. Front. Pharmacol. 9:790. doi: 10.3389/fphar.2018.00790
Received: 31 May 2018; Accepted: 28 June 2018;
Published: 17 July 2018.
Edited by:
Xinhua Qu, Shanghai Ninth People's Hospital, Shanghai Jiao-Tong University School of Medicine, ChinaReviewed by:
Christoph Eugen Hagemeyer, Monash University, AustraliaNazareno Paolocci, Johns Hopkins University, United States
Copyright © 2018 Hua, de Matos, Metselaar and Storm. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Susan Hua, U3VzYW4uSHVhQG5ld2Nhc3RsZS5lZHUuYXU=