 Konstantin A. Petrov
Konstantin A. Petrov Evgeny E. Nikolsky
Evgeny E. Nikolsky Patrick Masson
Patrick Masson
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pharmacol. , 12 July 2018
Sec. Neuropharmacology
Volume 9 - 2018 | https://doi.org/10.3389/fphar.2018.00766
Neuromuscular junctions (NMJs) are directly involved into such indispensable to life processes as respiration and locomotion. However, motor nerve forms only one synaptic contact at each muscle fiber. This unique configuration requires specific properties and constrains to be effective. The very high density of acetylcholine receptors (AChRs) of muscle type in synaptic cleft and an excess of acetylcholine (ACh) released under physiological conditions make this synapse extremely reliable. Nevertheless, under pathological conditions such as myasthenia gravis and congenital myasthenic syndromes, the safety factor can be markedly reduced. Drugs used for short-term symptomatic therapy of these pathological states, cause partial inhibition of cholinesterases (ChEs). These enzymes catalyze the hydrolysis of ACh, thus terminate its action on AChRs. Extension of the lifetime of ACh molecules compensates muscular AChRs abnormalities and, consequently, rescues muscle contractions. In this mini review, we will first outline the functional organization of the NMJ, and then, consider the concept of the safety factor and how it may be changed. This will be followed by a look at autoregulation of ACh release that influences the safety factor of NMJs. Finally, we will consider the morphological features of NMJs as a putative reserve to increase effectiveness of pathological muscle weakness therapy by ChEs inhibitors due to opportunity to use micro-pharmacodynamic mechanisms.
Neuromuscular junction (NMJ) is a synapse made up of motor axon branch (so-called nerve terminal), synaptic cleft, and postsynaptic region of muscle fiber (so-called end-plate), which is a folded structure where primary and secondary folds are distinguished. Such a configuration provides multiple extension of surface area allowing a huge density of receptors, ionic channels, and cholinesterases (ChEs) in a small crowded space (Figure 1). NMJ is a tripartite synapse because three to five terminal Schwann cells (TSCs) covering each nerve terminal actively participate in the process of neuromuscular synaptic transmission (Robitaille, 1995; Robitaille et al., 1997; Rochon et al., 2001; Todd et al., 2007, 2010; Ko and Robitaille, 2015; Arbour et al., 2017; Heredia et al., 2018).
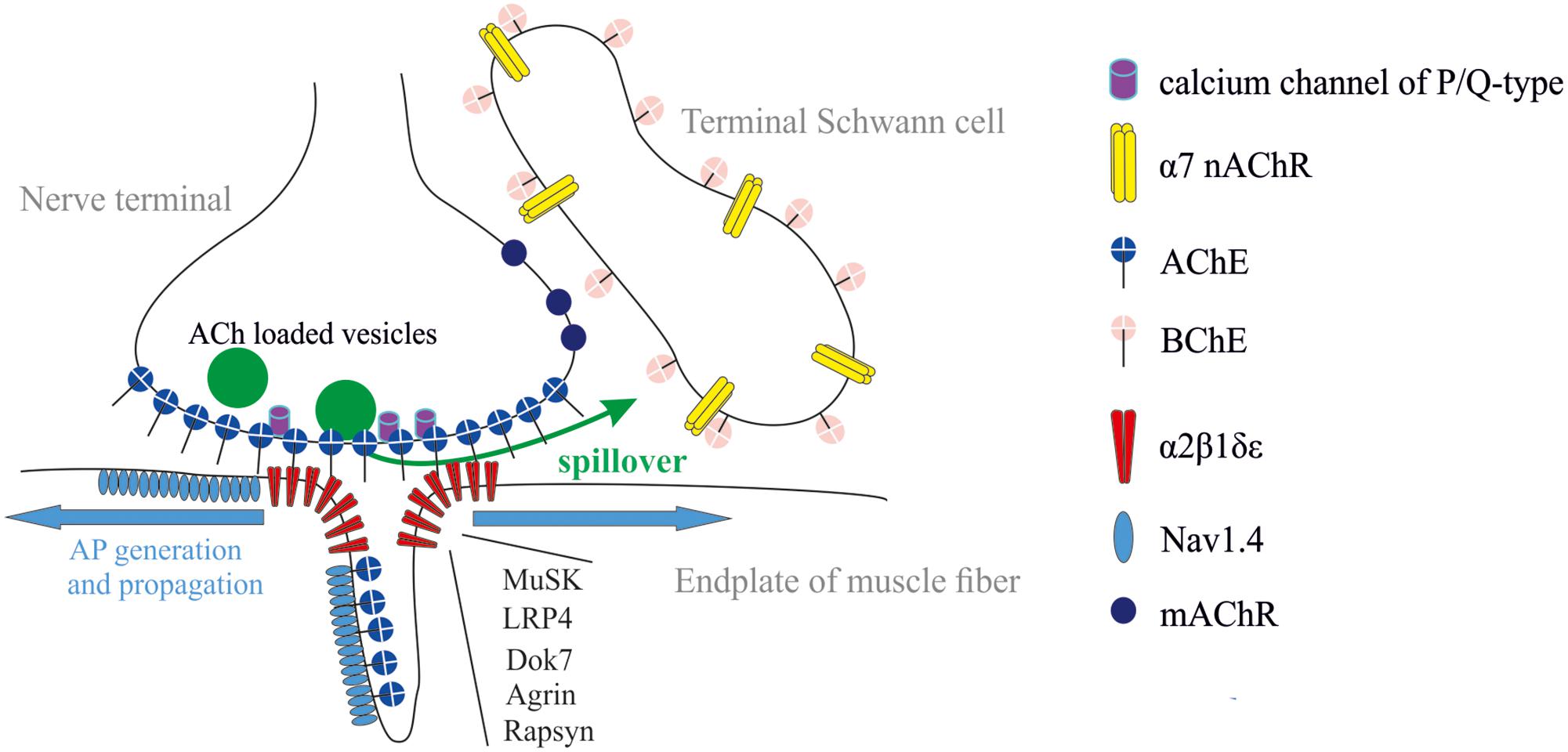
FIGURE 1. Tripartite organization of the NMJ: nerve terminal, muscle fiber, and terminal Schwann cell (TSC). Acetylcholinesterase (AChE) is mainly clustered in synaptic cleft. It controls the activation of muscle type AChR (α2βδ𝜀). Butyrylcholinesterase (BChE) is anchored at the TSC and controls activation of α7 nicotinic AChR (nAChR) and, probably, muscarinic (mAChRs). The most common forms of synaptic abnormalities are caused by autoantibodies to voltage-gated calcium channels of P/Q-type, α2β1δ𝜀, muscle-specific kinase (MuSK) and low-density lipoprotein-related protein 4 (LRP4). The most common congenital myasthenic syndromes are caused by mutations in genes coding for several functional proteins: (i) subunits of α2β1δ𝜀, (ii) proteins necessary for clustering of α2β1δ𝜀 (LRP4, Dok-7, agrin, rapsyn), and (iii) voltage-gated sodium channels (Nav1.4).
The AP spreads along motor nerve fiber, leading to the temporal activation of voltage-gated calcium channels. The resulting acetylcholine (ACh) release as quanta, takes place at the active zone. ACh is loaded into presynaptic vesicles which fuse with presynaptic cell membrane and elbow out ACh into synaptic cleft (Slater, 2015, 2017; Jones et al., 2017; Badawi and Nishimune, 2018). It is worth noting that other neurotransmitters (glutamate, GABA, ATP) exist at NMJ where their putative role is fine tuning of ACh release (Silinsky and Redman, 1994; Correia-de-Sá et al., 1996; Petrov et al., 2013; Malomouzh et al., 2015; Nascimento et al., 2017).
Diffusing across synaptic cleft, ACh molecules reach postsynaptic membrane and bind to ACh receptors (AChRs) of muscle type (α2β1δ𝜀). AChRs occupancy, i.e., activation, results in the opening of coupled ionic channels. Inflow of sodium ions according to electrochemical gradient causes transient lowering of membrane potential in the postsynaptic region, i.e., generation of excitatory postsynaptic potential that are known as end-plate potential (EPP). In turn, EPP triggers the opening of voltage-gated sodium channels (Nav1.4) in muscle fiber and muscle AP generation. The time necessary for the whole system to recover determines the lability of synapse, i.e., the ability to reproduce the specific for each muscle pattern of excitation. This time depends on the rate of enzymatic ACh hydrolysis. During the falling phase of EPPs (2–3 ms), high rate of acetylcholinesterase (AChE)-catalyzed hydrolysis of ACh with a turnover approaching 1.5 × 104 ACh molecules per second (Taylor et al., 2009) clears the synaptic cleft of all released ACh and, consequently, guarantees low probability of AChR activation. Thus, the main function of AChE in synaptic cleft is to ensure the rapid destruction of released ACh in the interstimuli intervals, before release of next quanta.
Mammalian ChEs family includes AChE (E.C. 3.1.1.7) and butyrylcholinesterase (BChE; E.C. 3.1.1.8) that are closely related enzymes. Their catalytic site is dedicated to hydrolyze ACh (Dvir et al., 2010; Masson and Lockridge, 2010). At NMJs, the most functional distinctions between these enzymes are associated with their localization. AChE at the NMJ is clustered mainly in synaptic cleft (Bernard et al., 2011; Blotnick-Rubin and Anglister, 2018), whereas BChE is mainly accumulated outside synaptic cleft, around TSCs (Davis and Koelle, 1967; Petrov et al., 2014) (Figure 1). Thus, ACh hydrolysis in synaptic cleft is accomplished by the sole AChE. AChE controls the lifetime of ACh and its occupancy time on AChRs in synaptic cleft. BChE, in turn, controls the spillover and dynamics of ACh outside synaptic cleft.
AP is generated according to the principle “all or nothing.” It means that any depolarization of exceeding threshold results in generation of the same APs. The reliability of neuromuscular transmission results from the release of more ACh molecules than are required to depolarize the muscle fiber to the threshold of AP generation. Thus, the “safety factor” is expressed most often as the ratio of the estimated mean peak amplitude of EPPs to the threshold depolarization required to generate an AP in the muscle fiber (Wood and Slater, 2001; Slater, 2009).
At NMJ, there are two main points at which the safety factor of synaptic transmission can be compromised. These are reduction of ACh release and decreasing in efficiency of postsynaptic depolarization, caused by ACh (Ruff, 2011). Here we describe briefly some of the most common instances in which the safety factor of neuromuscular transmission is impaired.
The most common form of pathological muscle weakness is myasthenia gravis (MG). Weakness in MG is caused by autoantibodies directed against muscle type AChRs (80% of all MG patients), muscle-specific kinase (MuSK), and low-density lipoprotein-related protein 4 (LRP4) (Figure 1). Additional antigenic targets as agrin, collagen Q, titin, and ryanodine receptor have been described recently, but their pathogenicity and clinical significance are unclear so far. The cause of autoimmune response is unknown (Cavalcante et al., 2012; Howard, 2018; Kusner et al., 2018; Plomp, 2018). MG therapy approaches are ChEs inhibitors for short-term symptom control, immunosuppressive drugs for long-term modification of the disease course, and in some cases thymectomy. Treatments of acute exacerbations are plasmapheresis, immunoadsorption, and intravenous immunoglobulin (Verschuuren et al., 2016). A complement inhibitor, eculizumab was recently approved for the treatment of generalized MG. Other treatments including targeted monoclonal antibody agents are currently under investigation (Lee and Jander, 2017).
Lambert–Eaton myasthenic syndrome (LEMS) is the presynaptic disorder of neuromuscular transmission caused by decrease in the number of released ACh quanta. Weakness is caused by autoantibodies to voltage-gated calcium channels of P/Q-type (Figure 1). Around 60% of patients suffering from LEMS have an underlying malignancy, most commonly lung cancer. In other cases the trigger mechanism is unknown (Verschuuren et al., 2016). The most effective symptomatic treatment of LEMS involves administration of 3,4-diaminopyridine (Keogh et al., 2011). This drug blocks voltage-gated potassium channels in nerve terminals (Figure 1), thereby prolonging the AP. This enhances entry of Ca2+ into the nerve terminal and increases ACh release. Treatments proposed for long-term modification of this disease include immunosuppressants (Kesner et al., 2018).
The congenital myasthenic syndromes (CMS) are rare genetic disorders that are characterized by abnormal neuromuscular synaptic transmission. The most common forms of CMS are due to mutations in the genes coding for the different subunits of AChR. Other forms of CMS include mutations coding for different postsynaptic proteins (Dok-7, rapsyn, voltage-gated sodium channels), proteins present in the synaptic cleft (collagen Q, forming a tail that anchors AChE) (Legay, 2018), and presynaptic terminal proteins such as choline acetyltransferase (Ohno et al., 2001; Engel et al., 2015; Nicole et al., 2017) (Figure 1). For most of CMS syndromes that can be characterized physiologically as having “underactive” synapses, trial of a ChE inhibitor, and/or 3,4-diaminopyridine may be appropriate. The exception is for patients carrying a Dok-7 mutation who do not respond to ChE inhibitors. Some of CMS syndromes can be described physiologically as having “overactive” synapses (e.g., collagen Q deficiency and slow channel syndrome). In these cases, AChE inhibitors are contraindicated. Patients with slow channel syndrome may benefit from treatment of long-acting agents that block the AChR ion pore, such as quinidine or fluoxetine. Finally, albuterol or one of the β-agonists have been empirically found to benefit patients with Dok-7 and collagen Q deficiency (Engel, 2007; Engel et al., 2015).
Toxicant-induced NMJ pathologies include poisoning by inhibitors of ChEs and nicotinic AChR (nAChR) blockers (Pope et al., 2005; Pope and Brimijoin, 2018; Renew et al., 2018).
At the moment, processes accompanying inhibition of ChEs at NMJ are very well studied at the level of single quantum release effects, by recording so-called miniature EPPs (mEPPs). These mEPPs are the result of release of a single ACh vesicle, which under resting conditions (absence of a nerve APs or in between two nerve APs) every few seconds fuse with the nerve terminal membrane so that small spontaneous EPPs can be recorded.
In this case, the sequences of inhibited synaptic AChE are: (i) more ACh molecules reach the postsynaptic membrane without being catalytically hydrolyzed by AChE during their diffusion across the synaptic cleft, and therefore, more AChRs are activated; (ii) because of prolonged lifespan in synaptic cleft, ACh molecules activate AChRs sustainably. Electrophysiological recordings show that prolonged lifetime of ACh in synaptic cleft due to AChE inactivation results in the increase of amplitude and duration of mEPPs (Katz and Miledi, 1975; Petrov et al., 2006, 2009, 2011). This fact is in agreement with idea that the main function of AChE in synaptic cleft is to control the duration of ACh action on postsynaptic AChRs.
Butyrylcholinesterase inhibition does not influence the mEPP amplitude and duration either when AChE is active or when this enzyme is inactivated (Minic et al., 2003). Thus, BChE inhibition does not potentiate the effect of ACh on muscle type AChRs. The absence of effects on mEPPs parameters due to BChE inhibition can be explained by localization of this enzyme outside the synaptic cleft on the surface of TSCs.
Since each EPP is the sum of the effect of individual mEPPs released simultaneously, it can be expected that the effects of AChE and BChE inhibition on mEPP and EPP are similar. However, electrophysiological recordings show that after complete inhibition of ChEs, EPPs amplitude varies, but in most cases, below the level of EPPs amplitude, recorded when ChEs are fully active. At the same time, mEPPs amplitude recorded in interstimuli intervals after ChEs inhibition is higher than mEPPs amplitude under conditions of active AChE and BChE. This suggests that when ChEs are inhibited, despite the amplification of postsynaptic effect of each quantum, fewer quanta of ACh are secreted in response to nerve AP. In other words, excess of ACh in synaptic cleft, resulting from ChEs inhibition, depresses ACh release.
Autoregulation of ACh release, i.e., the capability of ACh to modulate parameters of own secretion was described long time ago (Ciani and Edwards, 1963; Duncan and Publicover, 1979). It is known that presynaptic AChRs are of both types, ionotropic (nAChR) and metabotropic [muscarinic (mAChRs)]. It was shown that mAChRs, at least of four subtypes (Ì1, M2, M3, and Ì4), are present at NMJ (Garcia et al., 2005), although their physiological role is not fully established. It was shown that in some cases exogenous mAChRs agonists can suppress ACh release, while in other situations these agonists may enhance ACh release (Abbs and Joseph, 1981; Wessler et al., 1987; Arenson, 1991; Robitaille et al., 1997; Slutsky et al., 2001; Oliveira et al., 2002, 2009; Santafé et al., 2003, 2004, 2006, 2007; Dudel, 2007; Kovyazina et al., 2010, 2015).
It was shown that specific inhibition of AChE increases the probability of ACh release through activation of M1 mAChR subtype (Minic et al., 2002). On the contrary, it was shown that inhibition of both AChE and BChE decreases the probability of ACh release regardless the type of mAChRs (Minic et al., 2003).
The regulation of ACh release at NMJs by nAChRs is less documented compared to mAChRs. It was shown that α7 nAChRs can be localized at the TSC and act as a sensor for spillover of ACh adjusted by BChE (Figure 1). It was shown that ACh release was significantly depressed through the activation of α7 nAChR when BChE was specifically inhibited (Petrov et al., 2014). When both AChE in the synaptic cleft and BChE at TSC were inhibited, the spillover is increased. This induces a dramatic reduction of ACh release that compromises the muscle twitch triggered by the nerve stimulation.
It was shown that MG triggers homeostatic synaptic plasticity, resulting in increased ACh release. However, this pool of vesicles is small. Vesicles are rapidly depleted, leading to a larger depression in EPP amplitude during repetitive stimulations. This depression may contribute to the reduction of safety factor in patients with MG (Wang and Rich, 2018).
Under conditions of reduced safety factor, even slight downregulation in intensity of ACh release may influence muscle contraction. Because, the activation of AChRs, working as sensors at the input of different autoregulation pathways, is also controlled by AChE and BChE, the use of ChEs inhibitors does not only potentiate the effect of ACh on postsynaptic nAChRs, but also causes additional activation of presynaptic autoregulation pathways.
The use of selective AChE or non-selective (AChE and BChE) inhibitors to treat pathological muscle weakness was previously discussed (Komloova et al., 2010; Petrov et al., 2018a). In this review, we provide new evidences that selective AChE inhibitors could be even better than non-specific inhibitors to improve the muscle function. AChE inhibition increases the life-time of ACh molecules in the synaptic cleft and thus the number of nAChRs opened upon ACh binding. Indeed, AChE greatly contributes to make possible the jumping of EPP amplitude above the threshold of AP generation, and thus, the occurrence of muscle fibers twitch. With non-selective inhibitors, if BChE is also inhibited, the negative loop is stimulated. Then, less ACh quanta are released and EPP amplitude is reduced.
The NMJ architecture containing high density of AChE (5000 AChE monomers/μm2) (Anglister et al., 1994) and α2β1δ𝜀 nAChR (10,000 receptors/μm2) (Sine, 2002) in a small crowded space determines a sub-compartment at the origin of particular PK/PD mechanisms (Figure 2). The micro-anatomical structure of NMJ determines high target occupancy of ligands and increases residence time of these ligands on targets due possible rebinding. As a consequence, binding kinetics controls the duration of drug action in micro-anatomical compartments. Therefore, potent slow-binding ligands with slow rate of dissociation from targets display long-lasting action. The latter is important for the medical use AChE inhibitors, since pharmacological effect persists only while AChE at the NMJ is inhibited. In addition, faster elimination of inhibitors from myocardium and smooth muscles where the architecture of synapses is different from NMJ of skeletal muscles can reduce the duration of unwanted side effects.
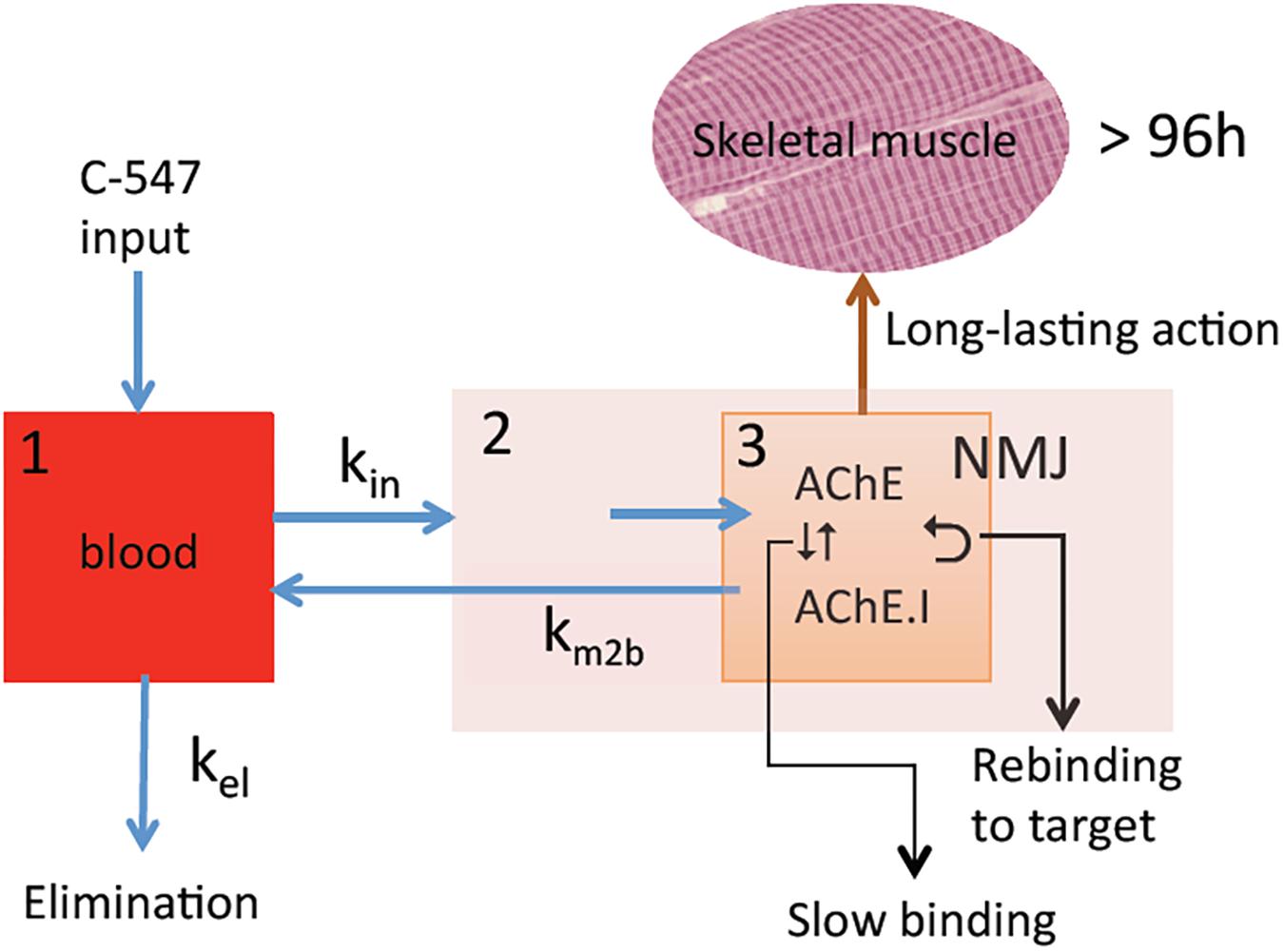
FIGURE 2. PK/PD model for the action of C-547 (I) at NMJ. (1) Central compartment (blood stream); (2) striatal muscle compartment; (3) NMJ sub-muscular compartment with high concentration of AChE (physiological target) in contained space. Slow-binding inhibition of AChE, long residence time of I on target, and slow elimination of I from NMJ lead to possibility of re-binding to AChE. This determines long-lasting action of C-547.
Concepts and methodology for analysis of micro-PK/PD mechanisms and drug discovery have been developed in the past decade (see Copeland et al., 2006; Vauquelin, 2010, 2015, 2016; Vauquelin and Charlton, 2010; Copeland, 2011; Walkup et al., 2015; Tonge, 2018).
Very few slow-binding inhibitors of AChE have been used for their pharmacological properties and they have not been analyzed in terms of micro-PK/PD mechanisms. However, a recent PK/PD study of a potent and highly selective inhibitor of AChE, C-547 (Kharlamova et al., 2016), revealed micro-pharmacodynamic mechanisms taking place in NMJ (Petrov et al., 2018b).
C-547 is a slow-binding inhibitor of AChE of type B (for a review about slow-binding inhibitors, see Masson and Lushchekina, 2016). Slow-binding inhibitors of type B are ligands that bind rapidly to the enzyme, making a complex EI that slowly “isomerizes” to EI∗ (Scheme 1).
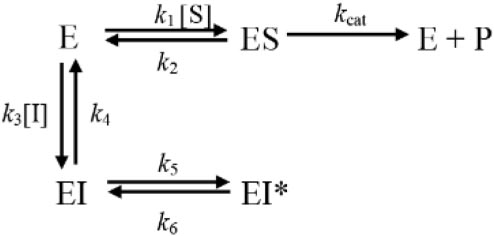
SCHEME 1. Mechanistic scheme for slow-binding inhibition of AChE by C-547.
The isomerization step, in Scheme 1, corresponds to the crossing of the bottleneck in the active center gorge of the enzyme to reach its final position (Kharlamova et al., 2016; Masson and Lushchekina, 2016). The overall slow rate of dissociation of the enzyme inhibitor complex, koff, is (Eq. 1):
The reciprocal of koff is the residence time, τ, of ligand on AChE. The importance of this parameter has been emphasized as it determines the temporal duration of drug-target complex and leads to sustained pharmacology (Copeland, 2011).
Thus, in confined anatomical space such as NMJ where there is a high density of AChE subunits, binding kinetics of C-547 to AChE is characterized by long residence time on target (τ = 20 min) (Kharlamova et al., 2016) and slow diffusion rate of C-547 out of NMJ (Petrov et al., 2018b). This makes possible re-binding of C-547 to AChE, and therefore slow elimination from NMJ. In addition, binding of C-547 to albumin determines a slow distribution in tissues and long PK in the bloodstream (t1/2 = 3 h in rat). As the result of thermodynamic and kinetic selectivity, C-547 has a long-lasting action on skeletal muscles, higher than 72 h in rat model of MG. Compared to current drugs used for palliative treatment of MG, that display lower affinity and selectivity for AChE, short PK (t1/2 < 30 min) and short residence time (except for carbamates that form transient covalent adducts with AChE), binding kinetics and PK/PD of C-547 make this compound as a promising leader drug for improving sustained treatment of MG and related diseases. Therefore, the discovery of new drugs for treatment of NMJ diseases depends on identification of highly selective molecules that fast associate to targets (fast-on rates), show long residence times on target (slow-off rates) and display long PK in the bloodstream.
Pharmacology of ChE inhibitors used for the treatment of muscle weakness is still poor. At the moment, only the carbamylating agent pyridostigmine bromide is used as ChEs inhibitor for treatment of muscle weakness symptoms. However, the selectivity of pyridostigmine for AChE versus BChE is low (6 against >10,000 for C-547; Petrov et al., 2018b). Taking into account ACh release down regulation under conditions of BChE inhibition, we can also conclude that selective inhibitors of AChE vs. BChE may have an advantage for MG treatment over non-selective ChE inhibitors, like pyridostigmine.
Moreover, symptomatic drug therapy often requires continuing sustained administration for maintaining high levels of target occupancy. Based on long-lasting target binding and rebinding, it is therefore possible to increase in vivo duration of drug action. Micro-anatomical properties of NMJ with high density of AChE could be helpful to design such long-lasting drugs that may greatly improve MG therapy. Design of new drugs, including selective AChE inhibitors, must be oriented in that direction.
KP wrote the manuscript in part associated with autoregulation of ACh release. PM wrote the manuscript in part associated with micro-pharmacodynamic mechanisms. EN suggested important information in order to improve the manuscript in part associated with NMJ architecture.
This work was supported by Russian Science Foundation project number 17-14-01097 for PM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbs, E. T., and Joseph, D. N. (1981). The effects of atropine and oxotremorine on acetylcholine release in rat phrenic nerve-diaphragm preparations. Br. J. Pharmacol. 73, 481–483. doi: 10.1111/j.1476-5381.1981.tb10446.x
Anglister, L., Stiles, J. R., and Salpetert, M. M. (1994). Acetylcholinesterase density and turnover number at frog neuromuscular junctions, with modeling of their role in synaptic function. Neuron 12, 783–794. doi: 10.1016/0896-6273(94)90331-X
Arbour, D., VandeVelde, C., and Robitaille, R. (2017). New perspectives on amyotrophic lateral sclerosis: the role of glial cells at the neuromuscular junction. J. Physiol. 595, 647–661. doi: 10.1113/JP270213
Arenson, M. (1991). The effects of muscarine and atropine reveal that inhibitory autoreceptors are present on frog motor nerve terminals but are not activated during transmission. Naunyn Schmiedebergs Arch. Pharmacol. 343, 128–133. doi: 10.1007/BF00168599
Badawi, Y., and Nishimune, H. (2018). Presynaptic active zones of mammalian neuromuscular junctions: nanoarchitecture and selective impairments in aging. Neurosci. Res. 127, 78–88. doi: 10.1016/j.neures.2017.11.014
Bernard, V., Girard, E., Hrabovska, A., Camp, S., Taylor, P., Plaud, B., et al. (2011). Distinct localization of collagen Q and PRiMA forms of acetylcholinesterase at the neuromuscular junction. Mol. Cell. Neurosci. 46, 272–281. doi: 10.1016/j.mcn.2010.09.010
Blotnick-Rubin, E., and Anglister, L. (2018). Fine localization of acetylcholinesterase in the synaptic cleft of the vertebrate neuromuscular junction. Front. Mol. Neurosci. 11:123. doi: 10.3389/fnmol.2018.00123
Cavalcante, P., Bernasconi, P., and Mantegazza, R. (2012). Autoimmune mechanisms in myasthenia gravis. Curr. Opin. Neurol. 25, 621–629. doi: 10.1097/WCO.0b013e328357a829
Ciani, S., and Edwards, C. (1963). The effect of acetylcholine on neuromuscular transmission in the frog. J. Pharmacol. Exp. Ther. 142, 21–23.
Copeland, R. (2011). Conformational adaptation in drug-target interactions and residence time. Future Med. Chem. 3, 1491–1501. doi: 10.4155/fmc.11.112
Copeland, R., Pompliano, D., and Meek, T. (2006). Drug-target residence time and its implications for lead optimization. Nat. Rev. Drug Discov. 5, 730–739. doi: 10.1038/nrd2082
Correia-de-Sá, P., Timóteo, M., and Ribeiro, J. (1996). Presynaptic A1 inhibitory/A2A facilitatory adenosine receptor activation balance depends on motor nerve stimulation paradigm at the rat hemidiaphragm. J. Neurophysiol. 76, 3910–3919. doi: 10.1152/jn.1996.76.6.3910
Davis, R., and Koelle, G. (1967). Electron microscopic localization of acetylcholinesterase and nonspecific cholinesterase at the neuromuscular junction by the gold-thiocholine and gold-thiolacetic acid methods. J. Cell Biol. 34, 157–171. doi: 10.1083/jcb.34.1.157
Dudel, J. (2007). The time course of transmitter release in mouse motor nerve terminals is differentially affected by activation of muscarinic M1 or M2 receptors. Eur. J. Neurosci. 26, 2160–2168. doi: 10.1111/j.1460-9568.2007.05770.x
Duncan, C., and Publicover, S. (1979). Inhibitory effects of cholinergic agents on the release of transmitter at the frog neuromuscular junction. J. Physiol. 294, 91–103. doi: 10.1113/jphysiol.1979.sp012917
Dvir, H., Silman, I., Harel, M., Rosenberry, T., and Sussman, J. (2010). Acetylcholinesterase: from 3D structure to function. Chem. Biol Interact. 187, 10–22. doi: 10.1016/j.cbi.2010.01.042
Engel, A. (2007). The therapy of congenital myasthenic syndromes. Neurotherapeutics 4, 252–257. doi: 10.1016/j.nurt.2007.01.001
Engel, A., Shen, X., Selcen, D., and Sine, S. (2015). Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol. 14, 420–434. doi: 10.1016/S1474-4422(14)70201-7
Garcia, N., Santafé, M., Salon, I., Lanuza, M., and Tomàs, J. (2005). Expression of muscarinic acetylcholine receptors (M1-, M2-, M3- and M4-type) in the neuromuscular junction of the newborn and adult rat. Histol. Histopathol. 20, 733–743. doi: 10.14670/HH-20.733
Heredia, D., Feng, C., Hennig, G., Renden, R., and Gould, T. (2018). Activity-induced Ca2+ signaling in perisynaptic Schwann cells of the early postnatal mouse is mediated by P2Y1 receptors and regulates muscle fatigue. eLife 7:e30839. doi: 10.7554/eLife.30839
Howard, J. (2018). Myasthenia gravis: the role of complement at the neuromuscular junction. Ann. N. Y. Acad. Sci. 1412, 113–128. doi: 10.1111/nyas.13522
Jones, R., Harrison, C., Eaton, S., Llavero Hurtado, M., Graham, L., Alkhammash, L., et al. (2017). Cellular and molecular anatomy of the human neuromuscular junction. Cell Rep. 21, 2348–2356. doi: 10.1016/j.celrep.2017.11.008
Katz, B., and Miledi, R. (1975). The nature of the prolonged endplate depolarization in anti-esterase treated muscle. Proc. R. Soc. Lond. B Biol. Sci. 192, 27–38. doi: 10.1098/rspb.1975.0149
Keogh, M., Sedehizadeh, S., and Maddison, P. (2011). Treatment for Lambert-Eaton myasthenic syndrome. Cochrane Database Syst. Rev. 2:CD003279. doi: 10.1002/14651858.CD003279.pub3
Kesner, V., Oh, S., Dimachkie, M., and Barohn, R. (2018). Lambert-eaton myasthenic syndrome. Neurol. Clin. 36, 379–394. doi: 10.1016/j.ncl.2018.01.008
Kharlamova, A., Lushchekina, S., Petrov, K., Kots, E., Nachon, F., Villard-Wandhammer, M., et al. (2016). Slow-binding inhibition of acetylcholinesterase by an alkylammonium derivative of 6-methyluracil: mechanism and possible advantages for myasthenia gravis treatment. Biochem. J. 473, 1225–1236. doi: 10.1042/BCJ20160084
Ko, C., and Robitaille, R. (2015). Perisynaptic Schwann cells at the neuromuscular synapse: adaptable, multitasking glial cells. Cold Spring Harb. Perspect. Biol. 7:a020503. doi: 10.1101/cshperspect.a020503
Komloova, M., Musilek, K., Dolezal, M., Gunn-Moore, F., and Kuca, K. (2010). Structure-activity relationship of quaternary acetylcholinesterase inhibitors - outlook for early myasthenia gravis treatment. Curr. Med. Chem. 17, 1810–1824. doi: 10.2174/092986710791111198
Kovyazina, I., Tsentsevitsky, A., and Nikolsky, E. (2015). Identification of the muscarinic receptor subtypes involved in autoregulation of acetylcholine quantal release from frog motor nerve endings. Dokl. Biol. Sci. 460, 5–7. doi: 10.1134/S0012496615010081
Kovyazina, I., Tsentsevitsky, A., Nikolsky, E., and Bukharaeva, E. (2010). Kinetics of acetylcholine quanta release at the neuromuscular junction during high-frequency nerve stimulation. Eur. J. Neurosci. 32, 1480–1489. doi: 10.1111/j.1460-9568.2010.07430.x
Kusner, L., Sengupta, M., and Kaminski, H. (2018). Acetylcholine receptor antibody-mediated animal models of myasthenia gravis and the role of complement. Ann. N. Y. Acad. Sci. 1413, 136–142. doi: 10.1111/nyas.13555
Lee, J., and Jander, S. (2017). Myasthenia gravis: recent advances in immunopathology and therapy. Expert Rev. Neurother. 17, 287–299. doi: 10.1080/14737175.2017.1241144
Legay, C. (2018). Congenital myasthenic syndromes with acetylcholinesterase deficiency, the pathophysiological mechanisms. Ann. N. Y. Acad. Sci. 1413, 104–110. doi: 10.1111/nyas.13595
Malomouzh, A., Petrov, K., Nurullin, L., and Nikolsky, E. (2015). Metabotropic GABAB receptors mediate GABA inhibition of acetylcholine release in the rat neuromuscular junction. J. Neurochem. 135, 1149–1160. doi: 10.1111/jnc.13373
Masson, P., and Lockridge, O. (2010). Butyrylcholinesterase for protection from organophosphorus poisons: catalytic complexities and hysteretic behavior. Arch. Biochem. Biophys. 494, 107–120. doi: 10.1016/j.abb.2009.12.005
Masson, P., and Lushchekina, S. (2016). Slow-binding inhibition of cholinesterases, pharmacological and toxicological relevance. Arch. Biochem. Biophys. 593, 60–68. doi: 10.1016/j.abb.2016.02.010
Minic, J., Chatonnet, A., Krejci, E., and Molgó, J. (2003). Butyrylcholinesterase and acetylcholinesterase activity and quantal transmitter release at normal and acetylcholinesterase knockout mouse neuromuscular junctions. Br. J. Pharmacol. 138, 177–187. doi: 10.1038/sj.bjp.0705010
Minic, J., Molgó, J., Karlsson, E., and Krejci, E. (2002). Regulation of acetylcholine release by muscarinic receptors at the mouse neuromuscular junction depends on the activity of acetylcholinesterase. Eur. J. Neurosci. 15, 439–448. doi: 10.1046/j.0953-816x.2001.01875.x
Nascimento, F., Sebastião, A., and Ribeiro, J. (2017). Presymptomatic and symptomatic ALS SOD1(G93A) mice differ in adenosine A1 and A2A receptor-mediated tonic modulation of neuromuscular transmission. Purinergic Signal. 11, 471–480. doi: 10.1007/s11302-015-9465-4
Nicole, S., Azuma, Y., Bauché, S., Eymard, B., Lochmüller, H., and Slater, C. (2017). Congenital myasthenic syndromes or inherited disorders of neuromuscular transmission: recent discoveries and open questions. J. Neuromuscul. Dis. 4, 269–284. doi: 10.3233/JND-170257
Ohno, K., Tsujino, A., Brengman, J., Harper, C., Bajzer, Z., Udd, B., et al. (2001). Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc. Natl. Acad. Sci. U.S.A. 98, 2017–2022. doi: 10.1073/pnas.98.4.2017
Oliveira, L., Timóteo, M., and Correia-de-Sá, P. (2002). Modulation by adenosine of both muscarinic M1-facilitation and M2-inhibition of [3H]-acetylcholine release from the rat motor nerve terminals. Eur. J. Neurosci. 15, 1728–1736. doi: 10.1046/j.1460-9568.2002.02020.x
Oliveira, L., Timóteo, M., and Correia-de-Sá, P. (2009). Negative crosstalk between M1 and M2 muscarinic autoreceptors involves endogenous adenosine activating A1 receptors at the rat motor endplate. Neurosci. Lett. 459, 127–131. doi: 10.1016/j.neulet.2009.05.001
Petrov, K., Girard, E., Nikitashina, A., Colasante, C., Bernard, V., Nurullin, L., et al. (2014). Schwann cells sense and control acetylcholine spillover at the neuromuscular junction by α7 nicotinic receptors and butyrylcholinesterase. J. Neurosci. 34, 11870–11883. doi: 10.1523/JNEUROSCI.0329-14.2014
Petrov, K., Kharlamova, A., Lenina, O., Nurtdinov, A., Sitdykova, M., Ilyin, V., et al. (2018a). Specific inhibition of acetylcholinesterase as an approach to decrease muscarinic side effects during myasthenia gravis treatment. Sci. Rep. 8:304. doi: 10.1038/s41598-017-18307-9
Petrov, K., Kovyazina, I., Zobov, V., Bukharaeva, E., Nikolsky, E., and Vyskocil, F. (2006). Different sensitivity of miniature endplate currents of the rat extensor digitorum longus, soleus and diaphragm muscles to a novel acetylcholinesterase inhibitor C-547. Physiol. Res. 55, 585–589.
Petrov, K., Kovyazina, I., Zobov, V., Bukharaeva, E., Nikolsky, E., and Vyskocil, F. (2009). Different sensitivity of miniature endplate currents in rat external and internal intercostal muscles to the acetylcholinesterase inhibitor C-547 as compared with diaphragm and extensor digitorum longus. Physiol. Res. 58, 149–153.
Petrov, K., Malomouzh, A., Kovyazina, I., Krejci, E., Nikitashina, A., Proskurina, S., et al. (2013). Regulation of acetylcholinesterase activity by nitric oxide in rat neuromuscular junction via N-methyl-D-aspartate receptor activation. Eur. J. Neurosci. 37, 181–189. doi: 10.1111/ejn.12029
Petrov, K., Yagodina, L., Valeeva, G., Lannik, N., Nikitashina, A., Rizvanov, A., et al. (2011). Different sensitivities of rat skeletal muscles and brain to novel anti-cholinesterase agents, alkylammonium derivatives of 6-methyluracil (ADEMS). Br. J. Pharmacol. 163, 732–744. doi: 10.1111/j.1476-5381.2011.01211.x
Petrov, K., Zueva, I., Kovyazina, I., Sedov, I., Lushchekina, S., Kharlamova, A., et al. (2018b). C-547, a 6-methyluracil derivative with long-lasting binding and rebinding on acetylcholinesterase: pharmacokinetic and pharmacodynamic studies. Neuropharmacology 131, 304–315. doi: 10.1016/j.neuropharm.2017.12.034
Plomp, J. J. (2018). “Neuromuscular junction physiology and pathophysiology,” in Myasthenia Gravis and Related Disorders, eds H. Kaminski, and L. Kusner (Cham: Humana Press), 1–12.
Pope, C., and Brimijoin, S. (2018). Cholinesterases and the fine line between poison and remedy. Biochem. Pharmacol. 153, 205–216. doi: 10.1016/j.bcp.2018.01.044
Pope, C., Karanth, S., and Liu, J. (2005). Pharmacology and toxicology of cholinesterase inhibitors: uses and misuses of a common mechanism of action. Environ. Toxicol. Pharmacol. 19, 433–446. doi: 10.1016/j.etap.2004.12.048
Renew, J. R., Brull, S. J., and Naguib, M. (2018). “Clinical pharmacology of drugs acting at the neuromuscular junction,” in Basic Sciences in Anesthesia, eds E. Farag, M. Argalious, J. Tetzlaff, D. Sharma (Cham: Springer), 139–159.
Robitaille, R. (1995). Purinergic receptors and their activation by endogenous purines at perisynaptic glial cells of the frog neuromuscular junction. J. Neurosci. 15, 7121–7131. doi: 10.1523/JNEUROSCI.15-11-07121.1995
Robitaille, R., Jahromi, B., and Charlton, M. (1997). Muscarinic Ca2+ responses resistant to muscarinic antagonists at perisynaptic Schwann cells of the frog neuromuscular junction. J. Physiol. 504, 337–347. doi: 10.1111/j.1469-7793.1997.337be.x
Rochon, D., Rousse, I., and Robitaille, R. (2001). Synapse-glia interactions at the mammalian neuromuscular junction. J. Neurosci. 21, 3819–3829. doi: 10.1523/JNEUROSCI.21-11-03819.2001
Ruff, R. L. (2011). Endplate contributions to the safety factor for neuromuscular transmission. Muscle Nerve 44, 854–861. doi: 10.1002/mus.22177
Santafé, M., Lanuza, M., Garcia, N., and Tomàs, J. (2006). Muscarinic autoreceptors modulate transmitter release through protein kinase C and protein kinase A in the rat motor nerve terminal. Eur. J. Neurosci. 23, 2048–2056. doi: 10.1111/j.1460-9568.2006.04753.x
Santafé, M., Lanuza, M., Garcia, N., Tomàs, M., and Tomàs, J. (2007). Coupling of presynaptic muscarinic autoreceptors to serine kinases in low and high release conditions on the rat motor nerve terminal. Neuroscience 148, 432–440. doi: 10.1016/j.neuroscience.2007.06.017
Santafé, M., Salon, I., Garcia, N., Lanuza, M., Uchitel, O., and Tomàs, J. (2003). Modulation of ACh release by presynaptic muscarinic autoreceptors in the neuromuscular junction of the newborn and adult rat. Eur. J. Neurosci. 17, 119–127. doi: 10.1046/j.1460-9568.2003.02428.x
Santafé, M., Salon, I., Garcia, N., Lanuza, M., Uchitel, O., and Tomàs, J. (2004). Muscarinic autoreceptors related with calcium channels in the strong and weak inputs at polyinnervated developing rat neuromuscular junctions. Neuroscience 123, 61–73. doi: 10.1016/j.neuroscience.2003.09.012
Silinsky, E., and Redman, R. (1994). ATP released together with acetylcholine as the mediator of neuromuscular depression at frog motor nerve endings. J. Physiol. 477, 117–127. doi: 10.1113/jphysiol.1994.sp020176
Sine, S. M. (2002). The nicotinic receptor ligand binding domain. J. Neurobiol. 53, 431–446. doi: 10.1002/neu.10139
Slater, C. (2009). “Reliability of neuromuscular transmission and how it is maintained,” in Neuromuscular Junction Disorders: Handbook of Clinical Neurology, Vol. 91, ed. A. G. Engel (New York, NY: Elsevier), 27–101.
Slater, C. (2015). The functional organization of motor nerve terminals. Prog. Neurobiol. 134, 55–103. doi: 10.1016/j.pneurobio.2015.09.004
Slater, C. (2017). The structure of human neuromuscular junctions: some unanswered molecular questions. Int. J. Mol. Sci. 18:E2183. doi: 10.3390/ijms18102183
Slutsky, I., Silman, I., Parnas, I., and Parnas, H. (2001). Presynaptic M(2) muscarinic receptors are involved in controlling the kinetics of ACh release at the frog neuromuscular junction. J. Physiol. 536(Pt 3), 717–725. doi: 10.1111/j.1469-7793.2001.00717.x
Taylor, P., Camp, S., and Radic, Z. (2009). “Acetylcholinesterase,” in Encyclopedia of Neuroscience, Vol. 1, eds R. Larry and L. R. Squire (New York, NY: Elsevier), 5–7. doi: 10.1016/B978-008045046-9.01132-3
Todd, K., Auld, D., and Robitaille, R. (2007). Neurotrophins modulate neuron-glia interactions at a vertebrate synapse. Eur. J. Neurosci. 25, 1287–1296. doi: 10.1111/j.1460-9568.2007.05385.x
Todd, K., Darabid, H., and Robitaille, R. (2010). Perisynaptic glia discriminate patterns of motor nerve activity and influence plasticity at the neuromuscular junction. J. Neurosci 30, 11870–11882. doi: 10.1523/JNEUROSCI.3165-10.2010
Tonge, P. (2018). Drug-target kinetics in drug discovery. ACS Chem. Neurosci. 9, 29–39. doi: 10.1021/acschemneuro.7b00185
Vauquelin, G. (2010). Rebinding: or why drugs may act longer in vivo than expected from their in vitro target residence time. Expert Opin. Drug Discov. 5, 927–941. doi: 10.1517/17460441.2010.512037
Vauquelin, G. (2015). On the ‘micro’-pharmacodynamic and pharmacokinetic mechanisms that contribute to long-lasting drug action. Expert Opin. Drug Discov. 10, 1085–1098. doi: 10.1517/17460441.2015.1067196
Vauquelin, G. (2016). Effects of target binding kinetics on in vivo drug efficacy: koff, kon and rebinding. Br. J. Pharmacol. 173, 2319–2334. doi: 10.1111/bph.13504
Vauquelin, G., and Charlton, S. (2010). Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Br. J. Pharmacol. 161, 488–508. doi: 10.1111/j.1476-5381.2010.00936.x
Verschuuren, J., Strijbos, E., and Vincent, A. (2016). “Neuromuscular junction disorders,” in Autoimmune Neurology: Handbook of Clinical Neurology, Vol. 133, eds J. Sean, S. J. Pittock, and A. Vincent (New York, NY: Elsevier), 447–466. doi: 10.1016/B978-0-444-63432-0.00024-4
Walkup, G., You, Z., Ross, P., Allen, E., Daryaee, F., Hale, M., et al. (2015). Translating slow-binding inhibition kinetics into cellular and in vivo effects. Nat. Chem. Biol. 11, 416–423. doi: 10.1038/nchembio.1796
Wang, X., and Rich, M. (2018). Homeostatic synaptic plasticity at the neuromuscular junction in myasthenia gravis. Ann. N. Y. Acad. Sci. 1412, 170–177. doi: 10.1111/nyas.13472
Wessler, I., Karl, M., Mai, M., and Diener, A. (1987). Muscarine receptors on the rat phrenic nerve, evidence for positive and negative muscarinic feedback mechanisms. Naunyn Schmiedebergs Arch. Pharmacol. 1987, 605–612.
Keywords: neuromuscular junction, myasthenia gravis, congenital myasthenic syndromes, cholinesterases, micro-pharmacodynamic mechanisms
Citation: Petrov KA, Nikolsky EE and Masson P (2018) Autoregulation of Acetylcholine Release and Micro-Pharmacodynamic Mechanisms at Neuromuscular Junction: Selective Acetylcholinesterase Inhibitors for Therapy of Myasthenic Syndromes. Front. Pharmacol. 9:766. doi: 10.3389/fphar.2018.00766
Received: 10 April 2018; Accepted: 25 June 2018;
Published: 12 July 2018.
Edited by:
Francisco Lopez-Munoz, Universidad Camilo José Cela, SpainReviewed by:
Renato Mantegazza, Istituto Neurologico Carlo Besta (IRCCS), ItalyCopyright © 2018 Petrov, Nikolsky and Masson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Konstantin A. Petrov, a3BldHJvdjIwMDVAbWFpbC5ydQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.