 Cédric Boularan*
Cédric Boularan* Céline Gales*
Céline Gales*- Institut des Maladies Métaboliques et Cardiovasculaires, Institut National de la Santé et de la Recherche Médicale, U1048, Université Toulouse III Paul Sabatier, Toulouse, France
Cyclic adenosine 3′,5′-monophosphate (cAMP) modulates a broad range of biological processes including the regulation of cardiac myocyte contractile function where it constitutes the main second messenger for β-adrenergic receptors' signaling to fulfill positive chronotropic, inotropic and lusitropic effects. A growing number of studies pinpoint the role of spatial organization of the cAMP signaling as an essential mechanism to regulate cAMP outcomes in cardiac physiology. Here, we will briefly discuss the complexity of cAMP synthesis and degradation in the cardiac context, describe the way to detect it and review the main pharmacological arsenal to modulate its availability.
Introduction
In cardiomyocytes, the influx of Ca2+ ions through voltage-dependent L-type Ca2+ channels (LTCC) plays an essential role in cardiac excitability and in coupling excitation to contraction of these cells. The depolarizing current through LTCC (ICa) contributes to the plateau phase of the cardiac action potential as well as to pacemaker activity in nodal cells (Shaw and Colecraft, 2013). This influx of Ca2+ triggers the release of intracellular stores of Ca2+ from the sarcoplasmic reticulum via the Ryanodine receptor (RyR), which results in activation of myofilaments contraction. Alterations in density or function of LTCC have been implicated in a variety of cardiovascular diseases, including atrial fibrillation (Van Wagoner et al., 1999) or heart failure (Mukherjee and Spinale, 1998). Cyclic adenosine 3′,5′-monophosphate (cAMP) is the main second messenger of the β-adrenergic receptor signaling inducing phosphorylation of the LTCC and the ryanodine receptor to increase the amount of intracellular Ca2+ necessary for heart contractility (responsible for positive chronotropic and inotropic effects during sympathetic stimulation) (Guellich et al., 2014). Moreover, catecholamine stimulated β-adrenergic receptor not only leads to cAMP effector dependent-troponin I phosphorylation to allow faster force development and shortening during systole and faster force relaxation and re-lengthening during diastole but also mediated cAMP effector dependent-phospholamban phosphorylation responsible for Ca2+ re-uptake in the sarcoplasmic reticulum and myofilament relaxation (lusitropic effects) (Bers, 2008). However sustained stimulation of this pathway may be detrimental thus leading to cardiac remodeling and development of heart failure (Brodde, 1993; Kiuchi et al., 1993). Thus, the proper physiological cardiac function relies on tight control of cellular cAMP concentration by fine-tuning the balance between cAMP synthesis and degradation. In mammalian cells, cAMP is produced by adenylyl cyclases (AC). Extracellular stimuli such as neurotransmitters, hormones, chemokines, lipid mediators, and drugs, can modulate AC activity to increase or decrease cAMP production by binding to a large number of transmembrane G protein-coupled receptors (GPCRs). The degradation of cAMP to AMP is catalyzed by phosphodiesterases (PDE) that are regulated by intracellular nucleotide concentrations, phosphorylation, binding of Ca2+/calmodulin and other regulatory proteins while cAMP efflux out of the cell is mediated by cyclic nucleotide efflux transporters. Over the years, several genetic models have been created to assess the role of the cAMP synthesis and hydrolysis proteins in cardiac physiology (Table 1). Once cAMP is produced it activates a set of diverse proteins, including cAMPdependent-protein kinase (PKA) or cAMP-dependent exchange proteins (Epac), the two main cAMP effectors to mediate downstream signaling as well as cyclic nucleotide-gated ion channels (CNGC) and POPDC proteins (Beavo and Brunton, 2002). From the compartmentation hypothesis proposed by Brunton et al., in which cAMP microdomains are distinctly coupled to cellular functions (Brunton et al., 1981), a variety of technologies has been developed to study in vivo the different localizations and organization around macromolecular complexes to ensure a fine-tuned spatio-temporal compartmentation of cAMP production (for detailed reviews (Baillie, 2009; Edwards et al., 2012; Perera and Nikolaev, 2013). Recently, these tools led to the identification of a β2-adrenergic-dependent cAMP compartmentation defect in failing cardiomyocytes (Nikolaev et al., 2010). In this review, we will focus on cAMP in synthesis and hydrolysis in cardiology, the way to detect it and how to manipulate this cAMP pathway.
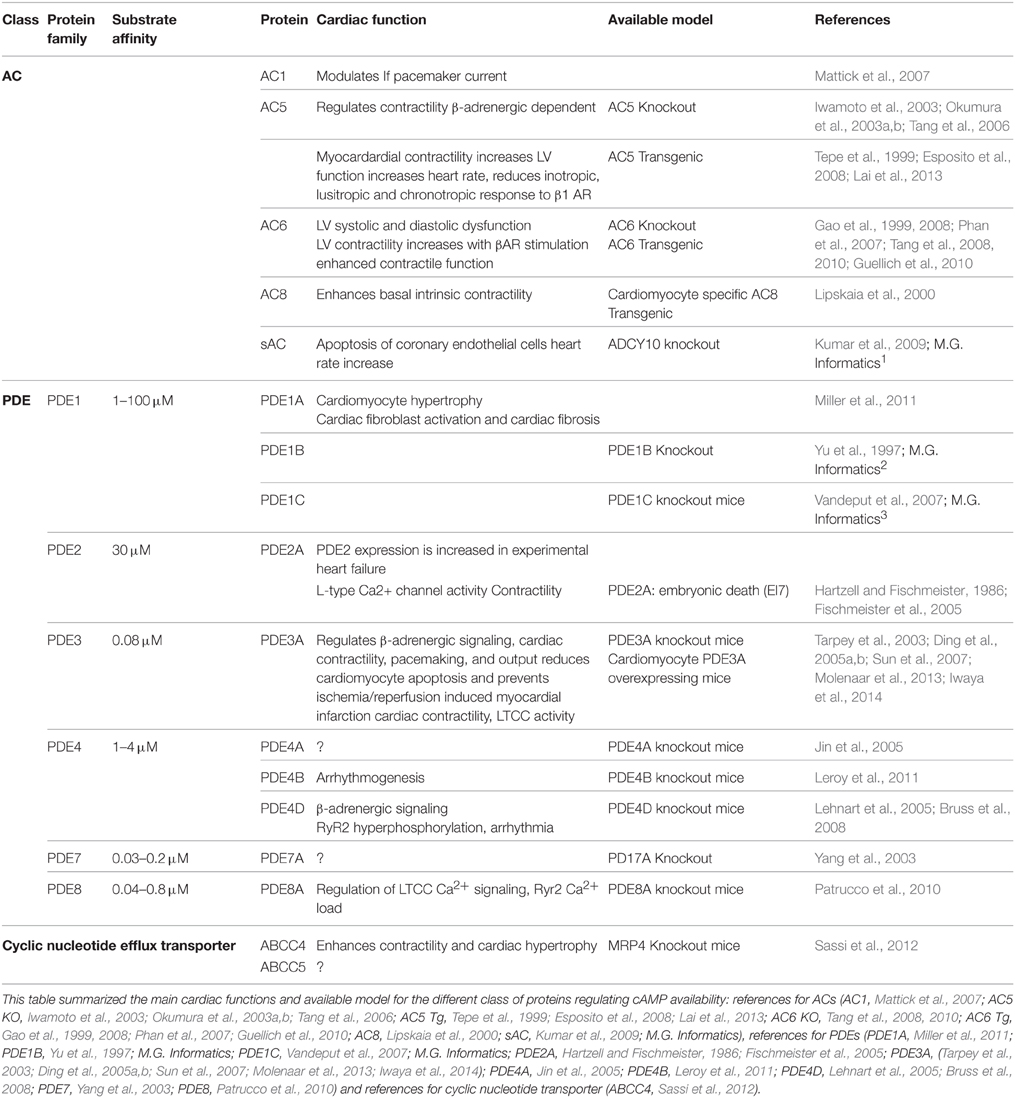
Table 1. Cardiac phenotype for cAMP synthesis, hydrolysis and transporter proteins adapted from Guellich et al. (2014).
cAMP in the Cardiac Tissue
cAMP Synthesis
Adenylyl cyclases (AC) are ubiquitous enzymes that catalyze the conversion of Adenosine triphosphate (ATP) into cAMP and pyrophosphate. ACs structure consists in 12 transmembrane domains divided into 2 hydrophobics domains (6 transmembrane domains each) and 2 main intracellular loops called C1 and C2 that naturally dimerize to form the catalytic domain (Figure 1A). In mammals, 9 transmembrane and 1 soluble AC (sAC) encoded by different genes have been identified and have different regulatory mechanisms (Willoughby and Cooper, 2007). Mammalian ACs are strongly activated by Mn2+ or Mg2+ (Tesmer et al., 1999) and inhibited by millimolar concentrations of free Ca2+ probably acting as a Mg2+ competitor (Mou et al., 2009) but at submicromolar concentrations, Ca2+ can activate AC via calmodulin (CaM) throught its binding to a putative helical structure on the C1b region (Halls and Cooper, 2011). More precisely AC1, 3, and 8 are Ca2+/CaM sensitive isoforms which localize in lipid rafts while AC2, 4, 7, and 9 are Ca2+/CaM insensitive and are excluded from these membrane domains (Willoughby and Cooper, 2007). On the contrary, biochemical studies on membrane preparations revealed that AC5 and AC6 could be inhibited by Ca2+ (independently of CaM) in the submicromolar range (Guillou et al., 1999; Hu et al., 2002). Along with nitric oxide (NO), Hydrogen sulfide (H2S) is a biological gaseous transmitter able to modulate cAMP production. Using NO donors, the gasotransmitter NO is thought to attenuate forskolin-stimulated AC5 and AC6 isoforms activities without altering their basal activity on membrane from rat striatum (Hudson et al., 2001). Even if H2S is a poisonous gas used as a chemical reagent, H2S is endogenously formed in mammalian cells from cysteine by the action of cystathionine β-synthase with serine as a by-product at a concentration around 50–160 μmol/L (Goodwin et al., 1989). In the central nervous system, H2S enhances NMDA receptor-mediated response via cAMP production (Kimura, 2000) while in cardiac context it can also suppressed AC activity and, therefore, decreased forskolin-stimulated cAMP accumulation in different cell lines and tissue (Lim et al., 2008; Yong et al., 2008). In the cardiomyocytes, expression of AC1, AC5, AC6, AC8, and sAC has been detected and most function as modulators of inotropic and chronotropic β-adrenergic receptor (β-AR) signaling output (Table 1) but AC5 and AC6 represent the dominant isoform (Defer et al., 2000). Along with AC distribution within membrane microdomains (Efendiev and Dessauer, 2011), cAMP synthesis is spatially restricted by localization of activating receptors like β1-adrenergic receptors or β2-adrenergic receptors at caveolae or non-caveolae plasma membrane domains (Rybin et al., 2000; Ostrom et al., 2001). Moreover, specific A-kinase anchor proteins (AKAP) complexes (Kapiloff et al., 2014) have been identified as a potential molecular mechanism for the formation of specific cAMP microdomains (Kapiloff et al., 2014). The AKAPs constitute signaling hub proteins that scaffold on a same membrane domain the AC and the regulatory subunit of protein kinase A (PKA) cAMP effector, thus confining the enzyme activity to discrete locations within the cell. Cardiac myocytes exhibit at least four distinct AKAP complexes: AKAP79/150 (aka AKAP5) with AC5/6 (Nichols et al., 2010); mAKAPβ (aka AKAP6) with AC2/5 (Kapiloff et al., 2009), YOTIAO (aka AKAP9) with AC2/9 (Piggott et al., 2008), and AKAP18δ with PKA (Fraser et al., 1998) (Figure 2).
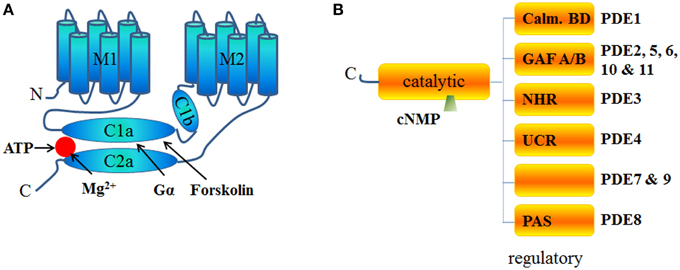
Figure 1. Schematic structure of ACs and PDEs. (A) Structure of Adenylyl cyclase is comprised of 2 transmembrane domains (M1 and M2 6 helixes each) and 2 cytosolic domains (C1 and C2) subdivided into a and b domains. C1 and C2 contain the catalytic core, the Gα and the forskolin binding sites and other regulatory sites. C2b domain is almost inexistent in all AC isoforms. (B) PDEs are homodimers with the exception of PDE1 and PDE6 (usually heterotetramers). PDEs have an NH2-terminal regulatory domain and share a conserved catalytic domain located in the COOH-terminal portion of the protein. The structure of the regulatory domain varies according the PDE isoform. GAF is an acronym for cGMP-specific PDE, Adenylyl cyclases and FhlA, NHR for N-terminal Hydrophobic Region, PAS for Per-ARNT-Sim and Calm. BD for calmodulin binding domain. No known domains are present in PDE7 or PDE9 regulatory C-terminal part. PDE4 proteins are classified as “long” or “short” isoforms, depending on the presence or absence of two highly conserved domains, Upstream Conserved Region 1 (UCR 1) and Upstream Conserved Region 2 (UCR 2) which interact to form a regulatory module that may influence catalytic activity by a PKA-dependent phosphorylation mechanism (Houslay, 2001; MacKenzie et al., 2002).
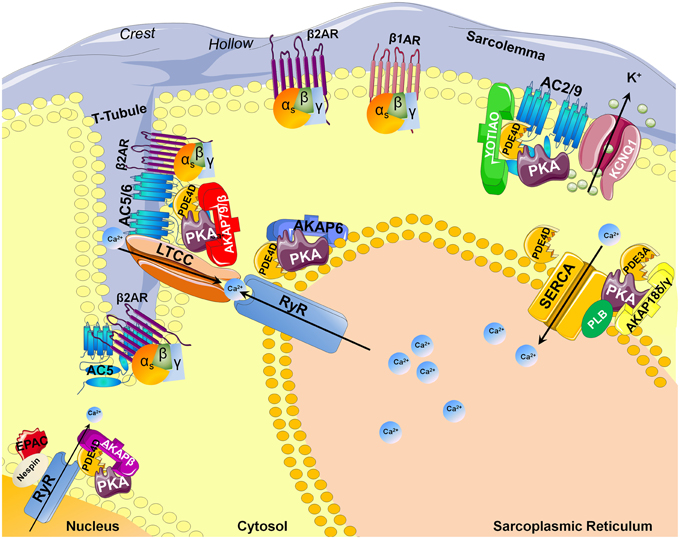
Figure 2. AKAP-dependent AC and PDE compartmentalizations in the cardiomyocyte. Abbreviations stand for: AKAP, A-kinase anchor proteins; PKA, Protein Kinase A; β2AR/β1AR, beta adrenergic receptor; PLN, Phospholamban; EPAC, cAMP-dependent exchange proteins; AC, Adenylyl cyclase; RyR, Ryanodine Receptor; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase; KCNQ1, potassium channel voltage gated KQT-like subfamily Q; PDE, Phosphodiesterase; T-tubule, Transverse tubule; LTCC, L-type calcium channel.
cAMP Elimination: Phosphodiesterases and Cyclic Nucleotide Efflux Transporters
Phosphodiesterases
Cyclic AMP is hydrolyzed exclusively by cyclic nucleotide PDEs classified in 11 families and encoded by at least 21 different genes with the existence of some splice variants (Omori and Kotera, 2007). PDEs are structured around a catalytic domain containing the cyclic nucleotide binding site conserved across all families and a regulatory N-terminus varying according to the different PDEs (Figure 1B). In the heart, 8 PDE families have been described: PDE1; PDE2, PDE3, PDE4, PDE5, PDE7, PDE8, and PDE9. Among them, PDE1, PDE2, and PDE3 are dual-specificity enzymes that can hydrolyze both cAMP and cGMP while PDE4, PDE7, PDE8 selectively hydrolyze cAMP and conversely PDE5, PDE9 selectively hydrolyze cGMP. Of the cAMP-hydrolyzing PDEs expressed in the heart, cGMP inhibits PDE3 and possibly PDE1, whereas PDE2 is activated by cGMP (detailed review in Zaccolo and Movsesian, 2007). Jurevicius and Fischmeister (1996a,b) provided the first direct evidence for PDE-mediated cAMP signaling compartmentation, showing that PDE inhibition allowed local β-adrenergic stimulation to enhance Ca2+ currents in frog ventricular myocytes (Jurevicius and Fischmeister, 1996a,b). Later on, imaging approaches confirmed that PDEs play a key role in shaping the intracellular cAMP gradient in rat neonatal cardiomyocytes (Zaccolo et al., 2000; Zaccolo and Pozzan, 2002). Like ACs, PDEs have also been shown to be compartmented by AKAPs complexes. Thus, specific cAMP hydrolysis-based PDE4 enzyme were shown to interact with mAKAP for PDE4D3 (Dodge et al., 2001); AKAP9 for PDE4D3 (Taskén et al., 2001), AKAP95 (aka AKAP8) for PDE4A (Asirvatham et al., 2004), AKAP149 for PDE4A (Asirvatham et al., 2004) (Figure 2).
Cyclic Nucleotide Efflux Transporters
In addition to PDEs and ACs, the intracellular concentration of cAMP is regulated by its efflux into the extracellular space through a specific transmembrane transport system named multidrug resistance proteins (MRP) (Cheepala et al., 2013) that belongs to the ATP-binding cassette (ABC) transporter superfamily (subfamily C). Three of them (MRP4 aka ABCC4, MRP5 aka ABCC5, and MRP8 aka ABCC11) have the ability to actively extrude cAMP and cGMP from the cell (Kruh and Belinsky, 2003) and in cardiac myocytes, MRP4 has been shown to enhance cAMP formation, contractility, and cardiac hypertrophy (Sassi et al., 2012). The compartimentation of MRPs expression may also play an important role in the intra- and extracellular cAMP signaling processes. For instance, caveolin-rich membrane MRP4 localization (Sassi et al., 2008) could explain the local MRP4-modulated contraction of cardiac myocytes induced by activation of β-adrenoceptor (Sellers et al., 2012).
cAMP in Heart Failure
Heart failure (HF) occurs when the heart is unable to pump sufficiently to maintain blood flow to meet the body's needs. Around 2% of adults have HF and this percentage increases to 6–10% for people over the age of 65 (McMurray and Pfeffer, 2005). The HF syndrome arises as a consequence of an abnormality in cardiac structure, function, rhythm, or conduction. As stated in introduction, cAMP primarily, but not exclusively, controls beating frequency, force of contraction and relaxation, essentially through the β-adrenergic signaling pathway. This pathway is necessary for the beneficial effects of catecholamines on cardiac contractility. During heart failure set up, increased sympathetic activity drives the βAR overstimulation in cardiomyocytes, thus promoting higher intracellular cAMP signals for compensatory cardiac function in the heart (Baker, 2014). However, chronic βAR stimulation and uncontrolled cellular cAMP signals have been shown to affect heart function in a much more detrimental way responses such as cell apoptosis (Zhu et al., 2003) and the loss of pump function (Michel et al., 1990; Engelhardt et al., 1999; Lohse et al., 2003), ultimately leading to HF setup. During the ongoing of the disease, a down-regulation of β1AR expression (Nikolaev et al., 2010) is correlated with a modulation of Gαi proteins (Eschenhagen et al., 1992a,b) expression to attenuate cAMP synthesis. The ratio between β1AR and β2AR converts the latter to be the major βAR subtype in failing hearts. Interestingly, associated with this receptor expression imbalance, the β2AR dominant-induced cAMP signal is broadly distributed in the failing heart (Nikolaev et al., 2010) compare to a compartmentalized cAMP signal in physiological condition. The functional output of this broadly distributed cAMP signal in modulating contractile properties in failing hearts has to be studied. In failing cardiomyocytes, chronic β2AR stimulation also promotes CaMKII-dependent contractile responses which has a pronounced role in promoting the development of cardiac hypertrophy, myocyte apoptosis, cardiac dysfunction and arrhythmias by causing sarcoplasmic reticulum Ca2+ overload (Anderson et al., 2011). Inhibition of CaMKII is able to ameliorate cardiac remodeling and reduce cardiac arrhythmias after myocardial infarction (Zhang et al., 2005). Although the direct link between cAMP and CaMKII is still missing, the detrimental CaMKII activity in cardiomyocytes could be related to dysregulation of distribution of cAMP signals under chronic βAR stimulation. HF is a complex process where the various components in the cAMP signaling pathway constitute potential pharmacological targets.
Modulation of cAMP Concentration in the Cardiac Tissue
Modulation of cAMP Production
Targeting Adenylyl Cyclases
Pharmacological AC activators
The most prominent AC activator is forskolin (FSK). FSK, a diterpene extracted from the plant Coleus forskohlii, directly activates all AC isoforms except AC9. Despite a strong hydrophobic property, its action is not limited to the native membrane-bound form of the enzyme since it can readily stimulate some synthetic soluble ACs. FSK binds to the same cleft that contains the active site of AC (Tesmer et al., 1997) where it glues together its two cytoplasmic domains (Figure 1A) by a combination of hydrophobic and hydrogen-binding interactions (Zhang et al., 1997). Based on equilibrium dialysis experiments of the C1 and C2 domains of type AC5 and AC2, respectively, the C1/C2 complex binds only one Gsα, one ATP, and one FSK molecule (Dessauer et al., 1997). However, FSK has been shown to also inhibit a number of membrane transport proteins and channel proteins like Glucose transporter or voltage dependent K+ channel (Laurenza et al., 1989). As Protein kinase C (PKC) activates AC2 by phosphorylating it on Thr-1057 (Böl et al., 1997), another alternative, but more restrictive one to activate AC, relies on the use of Phorbol 12-myristate 13-acetate (PMA) a phorbol diester and a potent tumor promoter known to activate PKC signaling. Thus, PKC-dependent phosphorylation of AC-C1 domain induces AC activation (Ebina et al., 1997). However, one must be cautious on the use of PMA as a specific AC-activator since PMA has also been reported to have actions on non-kinase proteins including chimaerins, RasGRP, and Unc-13/Munc-13 (Han and Meier, 2009; Kazanietz et al., 1995).
Pharmacological AC inhibitors
As schemed in Figure 1A, ACs are structured around 2 hydrophobic domains and 2 main intracellular loops containing the catalytical domain and the diterpene regulatory site. Based on this structure, ACs inhibitors can be divided into 4 groups (reviewed in Seifert et al., 2012): (i) the inhibitors competing with the ATP at the catalytic site like MANT-GTP (Gille and Seifert, 2003), (ii) the uncompetitive P-site inhibitors like 2′,5′-dideoxyadenosine-3′-tetraphosphate Vidarabine [aka 9-β-D-arabinofuranosyladenine (ara-A)] (Seifert, 2014) or NKY80 (a cell-permeable quinazolinone) to name a few, which work by stabilizing a pyrophosphate-bound transition state (Dessauer et al., 1999; Onda et al., 2001), (iii) the allosteric non-competitive inhibitors targeting the diterpene regulatory site like BODIPY-FS in presence of divalent cations (Erdorf et al., 2011), and (iv) the allosteric non-competitive inhibitors targeting alternated and unknown site like calmidazolium (Haunsø et al., 2003). Even though some specificity has been assigned to some of the molecules listed, to our knowledge, those inhibitors have not been accurately examined at all ACs isoforms, thus preventing any formal conclusion only based on their use to assess the involvement of AC activity.
Targeting G-protein Coupled Receptor Signaling
We previously mentioned that cAMP constitutes the master second messenger of β-adrenergic receptor signaling which belong to the G protein-coupled receptors (GPCRs) family. According to conventional knowledge, 7 transmembrane GPCRs at the plasma membrane convert extracellular signals into intracellular ones through canonical heterotrimeric G proteins which transduce signals from GPCRs to secondary effectors thus leading to the second messengers production and the propagation of the signal through ensuing regulation of numerous downstream intracellular signaling targets (Gilman, 1987). G proteins localized on the cytoplasmic side of the plasma membrane and are composed of a guanine nucleotide binding α subunit (Gα) and a βγ dimer (Gβγ), both constitutively associated in the G protein inactive state. Upon GPCR activation, the Gαβγ protein associates with the receptor thus allowing GDP/GTP exchange on the Gα GTPase domain, leading to subsequent Gα-GTP and Gβγ dissociation both regulating downstream specific signaling targets (Denis et al., 2012). Intrinsic GTPase activity of the Gα then allows GTP hydrolysis and to turn off the G protein activity to its initial inactive Gαβγ associated state. G proteins have been classified into five subfamilies (Gi/o, Gs, Gq/11, and G12/13) according to the secondary effector of the Gα subunit (Denis et al., 2012). Thus, isoforms of the Gαi/o family classically inhibit ACs and cAMP production while, conversely, isoforms from the Gαs family activate ACs to favor cAMP production. It follows that modulation of the activity of cardiac expressed Gαi- or Gαs-coupled receptors either through the use of selective GPCR agonists and antagonists or G proteins activators or inhibitors will directly alter the G protein activity and cAMP availability.
GPCR agonists and antagonists
In the human genome, it is estimated that the GPCR superfamily consists in ~600–1000 receptors (Lander et al., 2001; Vassilatis et al., 2003; Fredriksson and Schioth, 2005) where ≈ 200 have known cognate agonists and the larger part are still “orphan,” i.e., without yet identified agonists (Vassilatis et al., 2003). Evaluation of GPCR expression in vivo has been largely hampered by lack of specific antibodies against this class of receptors. Thus, over the years, microarray technology allowed researchers to monitor the mRNA expression levels of thousands of GPCRs encoding genes. Based on the available genomic data (Hakak et al., 2003; Katugampola and Davenport, 2003; Tang and Insel, 2004; Regard et al., 2008; Moore-Morris et al., 2009), we tried to summarize the different GPCRs detected in the whole cardiac tissue (cardiomyocytes, endothelial cells, fibroblasts…) (Table 2), their classical G protein coupling and a selective agonist/antagonist for most of them. This list is non-exhaustive and selectivity or description of these compounds will not be detailed here. Thus, selective pharmacological targeting of Gαi- or Gαs-coupled cardiac receptors represents a way to modulate intracellular cAMP levels. It is noteworthy that the classical GPCR coupling has to be enlarged as a recent study shows dual agonist occupancy of the AT1-R and α2C-AR heterodimer, two GPCRs known to be coupled to Gαq and Gαi, respectively, created an original conformation different from the active individual protomers and triggered an atypical Gs/cAMP/PKA signaling (Bellot et al., 2015). Thus, co-stimulation or bivalent ligand development might be a new pharmacological area to regulate cAMP signaling (Berque-Bestel et al., 2008; Lezoualc'h et al., 2009).
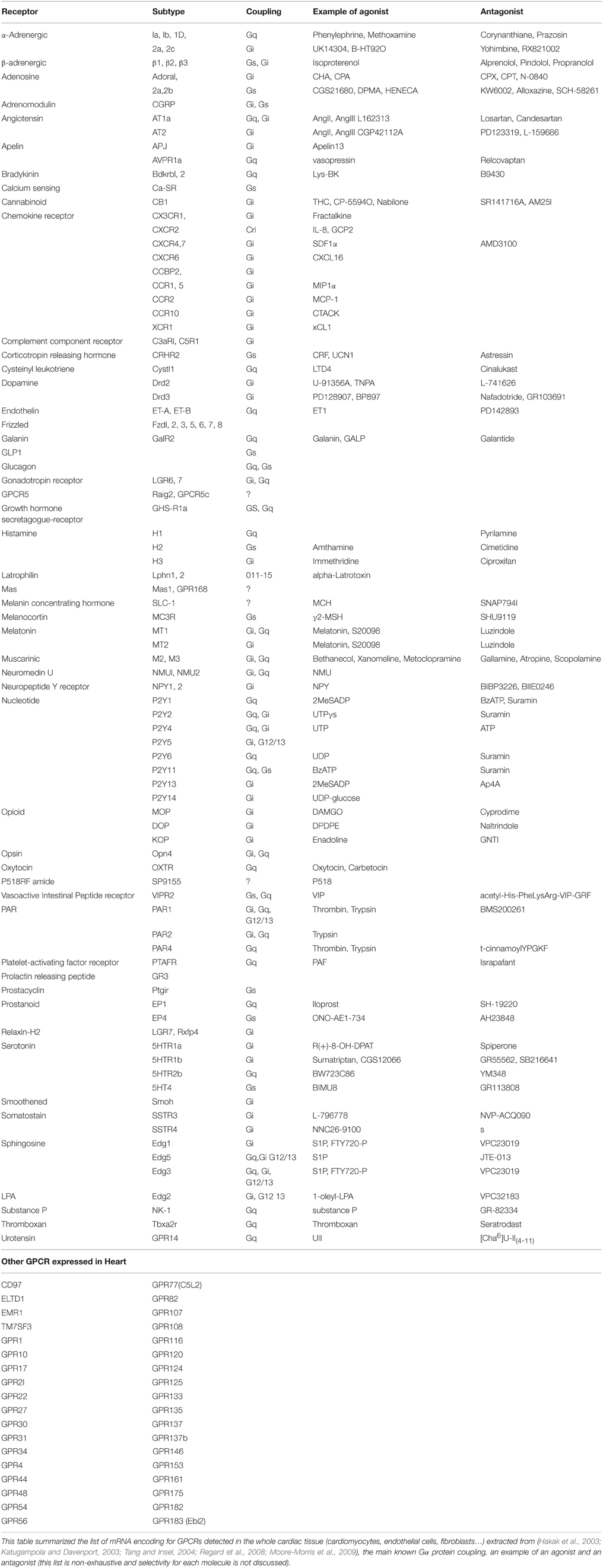
Table 2. GPCR expressed in heart: Gα coupling and pharmacological way to modulate their signaling.
Gα activators
Cholera toxin (CTX) is a specific Gαs potent activator secreted by the bacteria Vibrio cholerae which catalyzed the ADP-ribosylation of the Gαs proteins. The ADP-ribosylation blocks the Gαs catalytic activity and thus prevents the Gαs subunit to hydrolyze the GTP once activated, leading to the ensuing sustained Gs and AC activity (De Haan and Hirst, 2004). CTX administration in non-ischemic or ischemic heart contributes to the genesis of arrhythmia highlighting the essential role for Gαs in the regulation of cardiac physiology (Huang and Wong, 1989). More recently, Pasteurella multocida toxin (PMT), produced by toxigenic strains of the Gram-negative Pasteurella multocida bacteria, was identified as a potent and selective activator of Gαq, Gαi, and Gα13 by deamidating a glutamine residue in the switch II region of the Gα-GTPase domain (Orth et al., 2005, 2008). It was recently shown that, in vivo, PMT treatment in mice increased secretion and expression of connective tissue growth factor (CTGF) in cardiac fibroblasts to aggravate cardiac hypertrophy and fibrosis (Weise et al., 2015).
Gα inhibitors
Basically, all Gα subunits inhibitors share a common molecular mechanism by preventing the GDP/GTP exchange on the Gα-GTPase domain. A famous specific and highly effective Gαi inhibitor is Pertussis Toxin (PTX). PTX is a protein complex released by the bacterium Bordetella pertussis in an inactive form. PTX catalyzes the ADP-ribosylation of the Gαi subunit of the heterotrimeric G protein. The Gαi subunit remains locked in its GDP-bound inactive state, thus unable to interact with the receptor and to inhibit adenylyl cyclase activity (Hsia et al., 1984; Burns, 1988). PTX-pretreatment is classically used to delineate the involvement of Gαi-dependent signaling. It revealed for instance an increase in β-AR dependent inotropic response and cAMP accumulation in isolated ventricular cardiomyocytes (Melsom et al., 2014), confirming the dual coupling of β2-AR to both Gαi and Gαs (Xiao, 2001) in the cardiac tissue. On a purified Gα activity assay, suramin, an antimicrobial drug, was identified as a more selective inhibitor for Gαs (IC50 ≈ 250 nM) than for Gαo (IC50 ≈ 2 μM) or Gαi (IC50 ≈ 5 μM). Suramin exerts its effects by binding the effectors binding site on the Gα proteins (Freissmuth et al., 1996). It has to be noted that suramin is a large highly sulfonated and negatively charged molecule that limits its use to in vitro studies as it cannot cross the cell plasma membrane. Hohenegger and coworkers worked on suramin derivatives to increase specificity toward Gαs and identified two compounds (NF449 and NF503) that suppress the Gαs activation coupled to β-adrenergic receptors, whereas they affect the Gαi/Gαo- and Gαq-coupled receptors (A1-adenosine and angiotensin II receptor, respectively) to a much lesser extent (Hohenegger et al., 1998). Lately, BIM-46174 and BIM-46187 were classified and used as pan Gα inhibitors targeting Gαs, Gαq/11, Gαi/o, and Gα12/13 family (Prévost et al., 2006). However, these cell permeable compounds have not be tested toward all the individual members of Gα subunit family and more recently Kostenis and colleagues found that BIM-46187 was more selective to inhibit Gαq depending on the cellular context (Schmitz et al., 2014).
Gβγ complex inhibitors
Smrcka and coworkers described small molecule Gβγ inhibitors that selectively block Gβγ-binding interactions to their effectors, including M119 and its highly related analog, gallein (Lehmann et al., 2008). These compounds blocked interaction of Gβγ and GRK2 in vitro and reduced β-AR–mediated membrane recruitment of GRK2 in isolated adult mouse cardiomyocytes (Casey et al., 2010). The authors showed M119 enhanced both adenylyl cyclase activity and cardiomyocyte contractility in response to β-AR agonist (Casey et al., 2010). More recently, in a screen for the identification of OXE receptor antagonists, Gue1654 was discovered as a biased inhibitor that selectively prevent Gβγ signaling without affecting the Gα pathway (Blättermann et al., 2012). The molecular mechanism underlying Gue1654 action is still under investigation.
Targeting Tyrosine Kinase Receptor Signaling
It has to be noted that AC have been involved in the mechanisms of action of insulin and other peptides of the insulin superfamily like Insulin-like Growth factor I, relaxin and mollusc insulin-like peptide which are ligands for tyrosine kinase receptors (TKR) (Pertseva et al., 2003). Earlier, it was shown that in the heart EGF, another TKR, triggered some AC mediated effect (Nair and Patel, 1993). At a molecular level, TKR dependent activation of AC can rely on the activation of PI3K, PKCζ, or the Gβγ complex (Wilson et al., 1996; Standaert et al., 1997; Molina-Munoz et al., 2006). Thus, modulating activities of TKRs and their signaling regulators constitute an alternative approach to modulate cAMP but such compounds will not be described in this review.
Modulation of cAMP Degradation
Phosphodiesterases Inhibitors
The cardiostimulatory action of PDE make their inhibition as a promising therapeutic approach for the treatment of heart failure by sustaining cAMP production and action. Methylated xanthines, like theophylline, caffeine, or Iso-butyl-methyl-xanthyl (IBMX), are long known to act as competitive nonselective PDE inhibitors (Hess et al., 1975) but they also exhibit nonselective PDE action like adenosine receptor antagonist activities (Ukena et al., 1986). Over the years, several more specific and selective PDEs inhibitors have been developed. Representative selective inhibitors that can be used in cardiac tissue are listed below. Originally, 8-MM-IBMX was thought to be PDE1 selective (Rybalkin et al., 2002), but an extensive in vitro study characterized more potent and more selective compounds able to inhibit PDE1 activity like SCH51866 (Dunkern and Hatzelmann, 2007). The first specific inhibitor developed for PDE2 was EHNA [erythro-9-(2-hydroxy-3-nonyl)adenine] with an IC50 value of ~1 μM (Podzuweit et al., 1993) but a screen of compounds developed by Bayer showed that BAY60-7550 (an EHNA analog) was 100-fold more potent and 50-fold more selective for PDE2A over other PDEs compared to EHNA (Boess et al., 2004). Cilostamide-dependent PDE inhibition was discovered in 1970's (Hidaka et al., 1979) but cilostamide and its derivative selectivity for PDE3 family was described by Sudo et al. (2000). The prototypical PDE4 inhibitor is rolipram; originally named ZK62711, that was discovered in 1976 (Schwabe et al., 1976) but its use was limited by its associated side effects, particularly those affecting the gastrointestinal tract (Barnette and Underwood, 2000). Thus, in 2010, potency and selectivity of roflumilast and its active metabolite have been studied for all PDE (Hatzelmann et al., 2010). Roflumilast does not affect PDE enzymes apart from PDE4 family, and has a subnanomolar inhibitor activity toward all PDE4 splicing variants tested (Rabe, 2011). As the PDE4 family is encoded by 4 genes (PDE4A, B, C, or D) and 27 splice variants, identification of selective PDE4 subtypes inhibitors has been boosted and especially for PDE4B that can be selectively inhibited for example by triazine derivative (Hagen et al., 2014). ASB16165 was characterized as a specific and highly potent inhibitor for PDE7A with an IC50 value of 15 nM for human PDE7A (Kadoshima-Yamaoka et al., 2009). PDE8s are inhibited by dipyridamole, despite this drug is also known as a relatively nonselective cGMP specific PDE5 inhibitor (Soderling et al., 1998) while two studies have described a newly available PDE8 inhibitor developed by Pfizer, PF-04957325 (Vang et al., 2010; Shimizu-Albergine et al., 2012).
Cyclic Nucleotide Efflux Transporters: Description and Inhibitors
As mentioned earlier, ABCC [ATP-binding cassette (ABC) transporter superfamily (subfamily C)] regulates cAMP efflux into the extracellular space to decrease cAMP availability. Three of them (ABCC4, ABCC5, and ABCC11) are expressed in cardiac tissue but ABCC4 is the most studied and has been shown to enhance cAMP formation, contractility, and cardiac hypertrophy (Sassi et al., 2012). Non selective inhibitors including MK-571, dipyrimamole or indomethacin (Reid et al., 2003) have been described to dually inhibit ABCC transporters and PDEs (Xie et al., 2011). Thus, the interpretation of experiments using those compounds has to take in account their side activities. In 2014, a high throughput screening identified Ceefourin 1 and 2 as highly selective ABCC4 inhibitors (Cheung et al., 2014). The authors described a micromolar inhibition of ABCC4 over other members of ABCC transporter families but no data are available concerning their effect on PDE activity (Cheung et al., 2014).
Optogenetics Methods to Modulate cAMP Availability
The genome of Beggiatoa, a sulfide-oxidizing bacterium, revealed the presence of a DNA sequence encoding for a cytosolic adenylyl cyclase directly linked to a BLUF (blue light receptor using FAD) type light sensor domain. This photoactivatatable adenylyl cyclase (bPAC) shows a low cyclase activity in the dark but that increases about 300-fold upon light activation (Stierl et al., 2011). Efetova et al. pioneered the use of bPAC to distinguish between the functions of alternative cAMP effectors in the in vivo regulation of a Drosophila melanogaster physiological process (Efetova et al., 2013) while Von Zastrow's group recently used the bPAC fused to different targeting sequences to assess the role of cAMP compartmentation in GPCR signaling (Tsvetanova and von Zastrow, 2014). Recently, a red light-activated PDE was engineered by recombining the photosensor module of Deinococcus radiodurans bacterial phytochrome with the effector module of Homo sapiens PDE2A (Gasser et al., 2014). Compare to the bPAC system, the red-shifted activation of this new tool will allow the creation of interesting animal model to study the spatio-temporal cAMP signaling pathway. This concept was declined for multiple targets referenced and collected by the CHROMus project (Shui et al., 2014) and applied to GPCR where the cytosolic part of the Rhodopsin receptor was replaced by the β2-AR receptor part to create a photoactivable Gαs coupled GPCR (Airan et al., 2009).
Modulation of cAMP Effectors
cAMP has four direct intracellular targets: protein kinase A (PKA), the exchange protein activated by cAMP (EPAC), the cyclic nucleotide gated ion channels (CNGC) and the popeye domain containing protein (POPDC). cAMP output signaling can be modulated by targeting its effectors.
PKA Inhibitors and Activators
Inactive PKA relies on an heterotretamer consisting of two regulatory (R) and two catalytic (C) subunits (Figure 3A). Two principal isoforms of the R-subunit (type I and II) each further subclassified into α and β subtypes (Hofmann et al., 1975) and three isoforms of the C-subunit have been described in mammals (Cα, Cβ, and Cγ) (Uhler et al., 1986; Beebe et al., 1990). RIIα is the major isoform expressed in the heart but can also be found in the brain (Skalhegg and Tasken, 2000). RIα is also expressed in the cardiac tissue and the central nervous system while RIβ and RIIβ are respectively found in the spinal cord or brain and liver or fat tissue (reviewed in Skalhegg and Tasken, 2000). In regard to its molecular mechanism of activation, two cAMP molecules bind to each R-subunit and induce a conformational rearrangement of PKA which initiates the functional dissociation of the regulatory from the catalytic subunits (Murray, 2008).
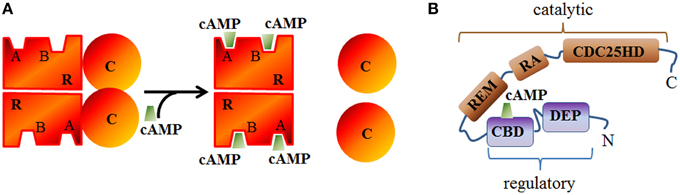
Figure 3. Schematic structure of PKA and EPAC. (A) The catalytic (c) subunit of cAMP-dependent Protein Kinase (PKA) is a serine/threonine protein kinase associated, in the absence of cAMP, with the regulatory (R) subunit to form the inactive PKA holoenzyme. cAMP can bind to A or B sites in the regulatory subunits and induces the dissociation of the catalytic subunits. (B) Epac structure showing the conserved cAMP binding domain (CBD), Disheveled/Egl-10/pleckstrin (DEP) domain, RAS exchange motif (REM) domain, RAS association (RA) domain, and CDC25-homology domain (CDC25HD).
The classically used PKA inhibitors, H89 (isoquinolone derivative) and KT5720 (synthesized from fungus Nocardiopsis sp.) act as competitive antagonists of the cAMP nucleotide for the binding site on the PKA regulatory subunit (Kase et al., 1987; Engh et al., 1996). Studying the specificity for commonly used inhibitors for a range of protein kinases, Davies and coworkers found unspecific effect for H89 and KT5720 as they were found to inhibit other kinases at lower concentrations than those used to prevent PKA activation (Davies et al., 2000). For instance, H89 is able to inhibit ROCK, S6K, PKBα, or MSK1 while KT5720 inhibits PDK1 and PHK (Davies et al., 2000). Alternate PKA inhibitors were developed including Rp-cAMPs and its derivatives. Those inhibitors act as competitive antagonists of the cyclic nucleotide binding domain on the regulatory PKA subunit. A study in Dictyostelium has characterized both selectivity and degradation of such compounds (Schaap et al., 1993) and demonstrated that those molecules can indeed inhibit proteins containing other cAMP binding domain. Finally, the protein kinase inhibitor peptide (PKI) remains likely the most specific way to interfere with PKA as it binds to the free catalytic subunit and prevents phosphorylation of PKA targets (Dalton and Dewey, 2006). However, high concentration of this peptide can also inhibit PKG signaling (Glass et al., 1992). By opposition to PKA inhibitors, 8pcpt-cAMP and its derivative (Sp-5,6-DCl-cBiMPS) are cell permeable cAMP analogs that can bind the PKA-cAMP binding site and promote the activation of PKA downstream effectors (Sandberg et al., 1991).
Epac Inhibitors and Activators
Epac (exchange protein activated by cAMP) constitutes with PKA the main direct cAMP effector and has been identified by two independent group in 1998 (de Rooij et al., 1998; Kawasaki et al., 1998). In mammals, two isoforms of Epac (Epac1 and Epac2), products of independent genes have been identified which contain a cAMP binding domain (that is homologous to that of PKA R subunits) and other conserved domains (Figure 3B). Its activation relies on a conformational rearrangement of the protein promoted by cAMP binding. Epac1 is mostly abundant in the heart, kidney, blood vessels, adipose tissue, central nervous system, ovary, and uterus, whereas Epac2 splice variants (Epac2A and Epac2B) are mostly expressed in the central nervous system (Epac2A), adrenal gland (Epac2B), and pancreas (Epac2A) (de Rooij et al., 1998; Kawasaki et al., 1998; Niimura et al., 2009). Once activated, Epac proteins activate the Ras superfamily small G proteins Rap1 and Rap2 (for review Cheng et al., 2008) by functioning as guanine nucleotide exchange factors. In cardiomyocytes, Epac proteins are involved in the formation of gap junctions to coordinate cardiac contractions through gating ions and small molecules (Somekawa et al., 2005) and enhances intracellular Ca2+ release during cardiac excitation-contraction coupling (Pereira et al., 2007). A High throughput screening assay to identify Epac inhibitors without affecting PKA activity led Cheng and coworkers to the identification of ESI-05 as an isoform specific inhibitor of Epac2 but not Epac1 (Tsalkova et al., 2012) and ESI-09 an pan inhibitor of Epac1 and 2 (Almahariq et al., 2013). CE3F4 compound (Courilleau et al., 2012) was identified as a specific Epac1 inhibitor without influence on PKA activity and its isoform selectivity for EPAC1 toward EPAC2 was demonstrated later (Courilleau et al., 2013). CE3F4 could be of interest in the therapeutic of cardiac pathophysiology as Epac1 is involved in β-adrenergic receptor-induced cardiomyocyte hypertrophy (Métrich et al., 2008). The compound usually named 007 [8-(4-Chloro-phenylthio)-2'-O-methyl-cAMP] is a cAMP analog activating Epac but not PKA (Enserink et al., 2002) but it has to be noted that 007 can behave as an inhibitor of PDEs which may indirectly increases cyclic nucleotide concentration (Poppe et al., 2008).
CNGC Inhibitors
The family of cyclic nucleotide gated channels (CNGC) comprises two groups: cyclic nucleotide gated (CNG) channels and the hyperpolarization-activated cyclic nucleotide-gated (HCN) channels. Both types are members of the six transmembrane channel superfamily and contain a cyclic nucleotide binding domain in their cytosolic C-Terminus that serves as an activation domain. Upon cyclic nucleotide binding, CNG channels gates the flow of monovalent cations such as Na+ and K+ to cross the plasma membrane and have a greater sensitivity for cGMP than for cAMP (for review Podda and Grassi, 2014). HCN cations channels open upon hyperpolarization and cAMP enhance their activity by shifting the activation curve to more positive voltage (Scicchitano et al., 2012). Four members exist in mammals (HCN1–HCN4) and are known to regulate the If current to control heart rate and rhythm by acting as a pacemaker current in the sinoatrial node (Wahl-Schott et al., 2014). If current is regulated by various neurotransmitters and metabolic stimuli (Pape, 1996) and are promising pharmacological targets in the treatment of cardiac arrhythmias. Thus, the most extensively studied HCN channels blocker is ZD7288 (BoSmith et al., 1993) but If current can also be blocked by ivabradine (Bucchi et al., 2002, 2006), zatebradine, and cilobradine (Van Bogaert and Pittoors, 2003). Ivabradine derivatives led to the discovery of HCN selective blockers with EC18 identified as a selective blocker for HCN4 and MEL57A induced mHCN1 inhibition (Melchiorre et al., 2010; Del Lungo et al., 2012).
POPDC Inhibitor
The Popeye domain-containing gene family consists of 3 genes (podc1, podc2, and popdc3) encoding a 3 transmembrane proteins that bind cAMP through their conserved cytoplasmic Popeye domain with an affinity (IC50) of 120 nM, which is comparable to the affinities reported for PKA (100 nM) (Froese et al., 2012). These proteins are essential for stress mediated modulation of cardiac pacemaking (Froese et al., 2012; Schindler et al., 2012). To our knowledge, no pharmacological inhibitors have been reported to investigate specific POPDC protein function so that the only way to modulate their activities so far is the use of genetic tools (small interfering RNA technology or gene knockout).
Methods for cAMP Detection in the Cardiac Tissue
The number of technologies that enables the functional screening of cAMP production has expanded over the years. Consequently, the choice of the technology will define the scope of the conclusions that can be drawn. Those methods can be divided into two groups: the direct methods allowing an “absolute” cAMP concentration quantification and the indirect methods which give a relative representation of cAMP availability. Thus, as summarized in Table 3, direct methods are generally more sensitive than indirect one since lacking any mediator but cannot accurately sense low cAMP levels produced in subcellular compartments. The advantages and limits of the common systems are listed below and basic principle for each technique is shown on Table 3. As previously pinpointed, cAMP availability is fine-tuned by a tight balance between its synthesis and immediate hydrolysis/efflux/use so that at one time point, cAMP is not enough amenable to quantification assays. Thus, accumulation of cAMP is often mandatory in most of cAMP detection assays with the common use of the pan PDEs inhibitor IBMX (when cAMP production needs to be measured) or FSK pretreatment (when cAMP production inhibitory function wants to be outlined).

Table 3. Comparison of cAMP detection system.
Direct Methods: Biochemical Approaches
cAMP has been long quantified through a radioactive functional assay based on affinity chromatography purification using 3H-ATP-preloaded cells lysates. After lysis and cAMP production can be estimated by measuring the ratio between 3H-cAMP over 3H-ATP + 3H-cAMP separated on affinity column (Piñeyro et al., 2005). Despite its high sensitivity, this technique often requires the presence of PDEs inhibitor and is not suitable for high throughput screening (HTS) strategy. Most of HTS biochemical methods relies on the general principle that cAMP accumulation is being detected by competition for a specific cAMP antibody between free unlabelled cAMP present in the sample to evaluate and a labeled form (radioactive, fluorescent, or enzymatic) of cAMP (Williams, 2004). Radiometric assays allow detection of cAMP using competition with 125-I labeled cAMP for anti-cAMP antibody immobilized on a solid scintillant plate. In those assays, the radiometric signal decreases proportionally to the amount of cAMP present in the sample (Horton and Baxendale, 1995). Fluorescence polarization cAMP assays monitor the light emitted from a fluorescent-tagged cAMP following excitation by a polarized light source. When the labeled cAMP is bound to an antibody more polarized light will be produced upon excitation (Prystay et al., 2001; Huang et al., 2002). To increase the signal to noise and to avoid cell autofluorescence detection, Cisbio developed an HTRF (Homogeneous time resolved fluorescence)-based cAMP assay. This assay is still based on a competitive immunoassay using cryptate-labeled anti-cAMP antibody and d2-labeled cAMP (Degorce et al., 2009). Alpha-screen technology uses acceptor beads conjugated to an antibody that recognizes cAMP and streptavidin-coated donor beads. When brought into close proximity by the presence of biotinylated cAMP, an oxygen radical dependent light is emitted. The cAMP extracted from a cell lysate will compete with the biotinylated cAMP and reduce the emitted light. This kind of assay is also available with an enzymatic based detection method where cAMP found in test sample competes with a fixed amount of Horse Radish Peroxydase-linked cAMP for binding to an anti-cAMP immobilized antibody (Bouchard et al., 2006). The electroluminescence technique (Mesa Scale Discovery) is another competitive immunoassay based on the displacement of ruthenium-labeled cAMP for an anti-cAMP antibody. The electrochemical reaction is initiated upon substrate and electrical charge addition and produce light which is inversely proportional to the cAMP present in the sample (Filip et al., 2004). The immuno-based competition can also be revealed using an enzyme complementation method (DiscoverX). In this assay, a fragment of β-galactosidase (β-gal) is conjugated with cAMP and act as an enzyme donor (ED) (Golla and Seethala, 2002). This ED-cAMP conjugate and cellular cAMP compete for binding to an anti-cAMP antibody. In presence of the enzyme acceptor (EA), the active enzyme will be reconstituted and will be able to subsequently hydrolyze a substrate to produce a chemiluminescent signal that is directly proportional to the amount of cAMP in the cells. Despite being sensitive and specific, all those techniques require cells or tissue disruption making the real-time and sub-cellular analysis of cAMP quantification impossible. Moreover if those assays are highly efficient to measure cAMP production, their use to bring to the fore cAMP inhibition is challenging and require FSK pretreatment.
Indirect Methods
Integrative Methods
Promega developed an assay based on the GloSensor Technology, a genetically modified form of firefly luciferase into which a cAMP-binding protein domain has been inserted (Fan et al., 2008). The firefly luciferase is circularly permuted and upon cAMP binding, a conformational change induces luciferase enzyme reconstitution which produces light in presence of its substrate. This technique is sensitive enough to assess role of endogenous receptors but requires transfection of the biosensor, thus limiting the quantification of the cAMP to the transfection efficiency (expression heterogeneity between cells) which can lead to high results variability. Maintaining advantages of integrative methods (sensitivity and kinetic) but avoiding transfection variability, Rivero-Müller's group developed a derived detection method CANDLES (Cyclic AMP iNdirect Detection by Light Emission from Sensor cells). Briefly, a stable cell line expressing a GloSensor plasmid is co-cultured with cells expressing the receptor to be tested and through cell-cell interaction via gap junctions, cAMP produced by the cell of interest can be transferred to the sensor cells to activate GloSensor plasmid (Trehan et al., 2014). Finally, given that the GloSensor is a cytosolic probe, it could be not appropriate to detect low concentrations of compartmentalized cAMP at the plasma membrane.
Reporter Gene Methods
The reporter gene method is a homogeneous, simple and inexpensive but indirect method to detect cAMP-downstream signaling. This assay is based on the specific activation of the transcription factor CREB (cAMP response element binding protein) upon cAMP production which induces a reporter gene under the control of a CRE element (cAMP Response Element) promoter. Various reporter genes have been used over the years: fluorescent proteins, luciferase, β-galactosidase or β-lactamase. Far downstream of the cAMP activation cascade, this method is sensitive but unable to give kinetics or localization information.
Resonance Energy Transfer Methods
The methods described above are unable to define the cellular localizations of cAMP production. Thus, visualization was achieved using resonance energy transfer (RET) techniques described by the Theodor Förster in 1940's (Forster, 1946). RET is a mechanism relying on an energy transfer between a donor chromophore that may transfer energy to another acceptor chromophore through non-radiative dipole–dipole coupling upon distances proximity conditions (Hebert et al., 2006; Kiyokawa et al., 2006). The name FRET “Förster resonance energy transfer” which includes the commonly used term FRET “Fluorescence resonance energy transfer” and BRET “Bioluminescence resonance energy transfer” is a non-radiative transfer of energy occurring between two fluorescent chromophores for FRET or between an enzyme generating luminescent signal upon addition of its substrate and a fluorescent acceptor partner in the case of BRET technology. The efficiency of this energy transfer is inversely proportional to the sixth power of the distance between donor and acceptor, making RET extremely sensitive to very small changes in distance thus allowing an accurate sensing of change in protein conformations for instance (Hebert et al., 2006). BRET is a first line assay for HTS screening as it avoids the consequences of fluorescence excitation and has a better Stokes' shift over FRET but is not recommended for imaging technique to identify localized cAMP compartmentalization. Thus RET-based methods have been developed to detect cAMP production and extensively reviewed (Williams, 2004; Willoughby and Cooper, 2008; Sprenger and Nikolaev, 2013; Calebiro and Maiellaro, 2014). All the methods rely on the use of the downstream cAMP effectors PKA, Epac or CNGC either that all directly bind cAMP molecules related to the expression of a specific cAMP-binding motif. Briefly, either full-length cAMP-effector probes or single cAMP domain sensor extracted from the different effectors are fused to an energy donor and acceptor allowing the generation of a basal RET signal in the absence of cAMP production. Upon cAMP binding, a conformational rearrangement in the RET-based sensor will lead to a modification of the RET signal. Since the pioneering studies using those RET probes to study cAMP availability in cardiac tissue (Zaccolo et al., 2000; Zaccolo and Pozzan, 2002), many efforts have been made these last years to improve signal to noise ratio, RET efficiency (optimizing donor-acceptor couple, linker optimization), cAMP binding affinity (mutagenesis on single domain or full length protein probes), RET detection methods (e.g., Sensitized emission vs Fluorescence lifetime imaging microscopy for FRET-based probes, Renilla luciferase variants for BRET-based probes) (Willoughby and Cooper, 2008; Sprenger and Nikolaev, 2013). Moreover with the prominently recognized mechanism for cAMP compartmentation, several group restricted the expression of those RET based probes to subcellular localization using for example plasma membrane targeting sequence or endosome localization (Klarenbeek and Jalink, 2014; Sprenger et al., 2015).
Copper Free Azide-alkaline Cycloaddition: a “Click Chemistry”
In chemical synthesis, click chemistry is a process that generates by joining small units together. The azide alkyne Huisgen cycloaddition using a Copper (Cu) catalyst is one of the most popular reactions within the Click chemistry concept between an azide and a terminal or internal alkyne to give a 1,2,3-triazole (Rostovtsev et al., 2002; Tornøe et al., 2002). To avoid Cu toxicity, Baskin et al developed a Cu-free click reaction with comparable kinetics to Cu dependent cycloaddition, but adapted for dynamic in vivo imaging (Baskin et al., 2007). In a recent study, this copper free method was applied to detect cAMP derivative (8-azido cAMP) in vivo after the addition of difluorinated cyclooctyne (DIFO) as a reagent (Ito et al., 2013). If this approach will enable to visualize and quantify derivative cAMP endogenous modulators at the single cell level without exogenous transfection protocol, it has to be noted that the molecule used is a cAMP derivative so the signal observed will be the result of ACs/PDEs activities in competition with cAMP endogenously produced.
Conclusion
The development of optical methods that allow monitoring of cAMP dependent signaling in living cells and the growing list of molecules (summarized in Figure 4) available to modulate cAMP availability played a fundamental role in revealing an unexpected level of cAMP organization in cardiac tissue. It is likely that new optical methods development, with higher temporal and spatial resolution, will improve our knowledge of cAMP dependent signaling microdomains located on the cell surface or other intracellular membranes for individual cells within heart architecture.
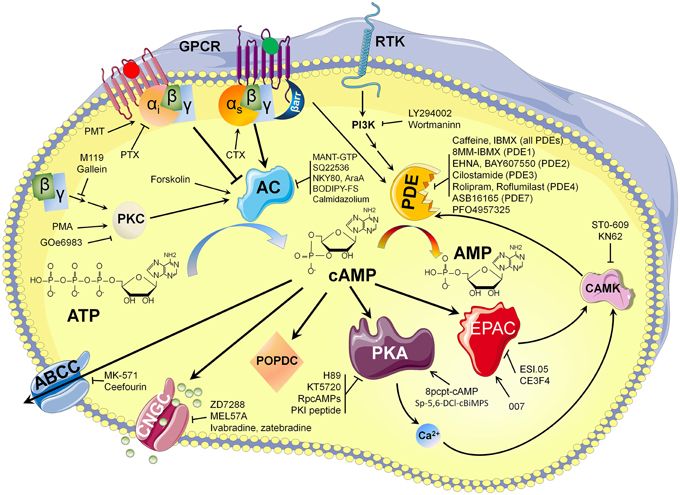
Figure 4. cAMP synthesis and hydrolysis: pharmacological way to modulate its availability.
Funding
This work was supported and funded by “Fondation Bettencourt Schueller and Institut National de la santé et de la Recherche Médicale.”
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
1. ^http://www.informatics.jax.org/marker/MGI:2660854. Informatix JAX.
2. ^http://www.informatics.jax.org/marker/MGI:97523. JAX.
3. ^http://www.informatics.jax.org/reference/marker/MGI:108413. JAX.
References
Airan, R. D., Thompson, K. R., Fenno, L. E., Bernstein, H., and Deisseroth, K. (2009). Temporally precise in vivo control of intracellular signalling. Nature 458, 1025–1029. doi: 10.1038/nature07926
Almahariq, M., Tsalkova, T., Mei, F. C., Chen, H., Zhou, J., Sastry, S. K., et al. (2013). A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol. Pharmacol. 83, 122–128. doi: 10.1124/mol.112.080689
Anderson, M. E., Brown, J. H., and Bers, D. M. (2011). CaMKII in myocardial hypertrophy and heart failure. J. Mol. Cell. Cardiol. 51, 468–473. doi: 10.1016/j.yjmcc.2011.01.012
Asirvatham, A. L., Galligan, S. G., Schillace, R. V., Davey, M. P., Vasta, V., Beavo, J. A., et al. (2004). A-kinase anchoring proteins interact with phosphodiesterases in T lymphocyte cell lines. J. Immunol. 173, 4806–4814. doi: 10.4049/jimmunol.173.8.4806
Baillie, G. S. (2009). Compartmentalized signalling: spatial regulation of cAMP by the action of compartmentalized phosphodiesterases. FEBS J. 276, 1790–1799. doi: 10.1111/j.1742-4658.2009.06926.x
Baker, A. J. (2014). Adrenergic signaling in heart failure: a balance of toxic and protective effects. Pflugers Arch. 466, 1139–1150. doi: 10.1007/s00424-014-1491-5
Barnette, M. S., and Underwood, D. C. (2000). New phosphodiesterase inhibitors as therapeutics for the treatment of chronic lung disease. Curr. Opin. Pulm. Med. 6, 164–169. doi: 10.1097/00063198-200003000-00014
Baskin, J. M., Prescher, J. A., Laughlin, S. T., Agard, N. J., Chang, P. V., Miller, I. A., et al. (2007). Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl. Acad. Sci. U.S.A. 104, 16793–16797. doi: 10.1073/pnas.0707090104
Beavo, J. A., and Brunton, L. L. (2002). Cyclic nucleotide research – still expanding after half a century. Nat. Rev. Mol. Cell Biol. 3, 710–718. doi: 10.1038/nrm911
Beebe, S. J., Oyen, O., Sandberg, M., Froysa, A., Hansson, V., and Jahnsen, T. (1990). Molecular cloning of a tissue-specific protein kinase (C gamma) from human testis–representing a third isoform for the catalytic subunit of cAMP-dependent protein kinase. Mol. Endocrinol. 4, 465–475. doi: 10.1210/mend-4-3-465
Bellot, M., Galandrin, S., Boularan, C., Matthies, H. J., Despas, F., Denis, C., et al. (2015). Dual agonist occupancy of AT1-R-alpha2C-AR heterodimers results in atypical Gs-PKA signaling. Nat. Chem. Biol. 11, 271–279. doi: 10.1038/nchembio.1766
Berque-Bestel, I., Lezoualc'h, F., and Jockers, R. (2008). Bivalent ligands as specific pharmacological tools for G protein-coupled receptor dimers. Curr. Drug Discov. Technol. 5, 312–318. doi: 10.2174/157016308786733591
Bers, D. M. (2008). Calcium cycling and signaling in cardiac myocytes. Annu. Rev. Physiol. 70, 23–49. doi: 10.1146/annurev.physiol.70.113006.100455
Blättermann, S., Peters, L., Ottersbach, P. A., Bock, A., Konya, V., Weaver, C. D., et al. (2012). A biased ligand for OXE-R uncouples Galpha and Gbetagamma signaling within a heterotrimer. Nat. Chem. Biol. 8, 631–638. doi: 10.1038/nchembio.962
Boess, F. G., Hendrix, M., van der Staay, F. J., Erb, C., Schreiber, R., van Staveren, W., et al. (2004). Inhibition of phosphodiesterase 2 increases neuronal cGMP, synaptic plasticity and memory performance. Neuropharmacology 47, 1081–1092. doi: 10.1016/j.neuropharm.2004.07.040
Böl, G. F., Gros, C., Hulster, A., Bösel, A., and Pfeuffer, T. (1997). Phorbol ester-induced sensitisation of adenylyl cyclase type II is related to phosphorylation of threonine 1057. Biochem. Biophys. Res. Commun. 237, 251–256. doi: 10.1006/bbrc.1997.7123
BoSmith, R. E., Briggs, I., and Sturgess, N. C. (1993). Inhibitory actions of ZENECA ZD7288 on whole-cell hyperpolarization activated inward current (If) in guinea-pig dissociated sinoatrial node cells. Br. J. Pharmacol. 110, 343–349. doi: 10.1111/j.1476-5381.1993.tb13815.x
Bouchard, N., Robitaille, E., and Wenham, D. (2006). cAMP AlphaScreen Assay: A Method for the Pharmacological Characterization and Screening of Gi-Coupled Receptors in Whole Cells. Waltham, MA: Perkin Elmer Application Note.
Brodde, O. E. (1993). Beta-adrenoceptors in cardiac disease. Pharmacol. Ther. 60, 405–430. doi: 10.1016/0163-7258(93)90030-H
Brunton, L. L., Hayes, J. S., and Mayer, S. E. (1981). Functional compartmentation of cyclic AMP and protein kinase in heart. Adv. Cyclic Nucleotide Res. 14, 391–397.
Bruss, M. D., Richter, W., Horner, K., Jin, S. L., and Conti, M. (2008). Critical role of PDE4D in beta2-adrenoceptor-dependent cAMP signaling in mouse embryonic fibroblasts. J. Biol. Chem. 283, 22430–22442. doi: 10.1074/jbc.M803306200
Bucchi, A., Baruscotti, M., and DiFrancesco, D. (2002). Current-dependent block of rabbit sino-atrial node I(f) channels by ivabradine. J. Gen. Physiol. 120, 1–13. doi: 10.1085/jgp.20028593
Bucchi, A., Tognati, A., Milanesi, R., Baruscotti, M., and DiFrancesco, D. (2006). Properties of ivabradine-induced block of HCN1 and HCN4 pacemaker channels. J. Physiol. (Lond). 572, 335–346. doi: 10.1113/jphysiol.2005.100776
Burns, D. L. (1988). Subunit structure and enzymic activity of pertussis toxin. Microbiol. Sci. 5, 285–287.
Calebiro, D., and Maiellaro, I. (2014). cAMP signaling microdomains and their observation by optical methods. Front. Cell. Neurosci. 8:350. doi: 10.3389/fncel.2014.00350
Casey, L. M., Pistner, A. R., Belmonte, S. L., Migdalovich, D., Stolpnik, O., Nwakanma, F. E., et al. (2010). Small molecule disruption of G beta gamma signaling inhibits the progression of heart failure. Circ. Res. 107, 532–539. doi: 10.1161/CIRCRESAHA.110.217075
Cheepala, S., Hulot, J. S., Morgan, J. A., Sassi, Y., Zhang, W., Naren, A. P., et al. (2013). Cyclic nucleotide compartmentalization: contributions of phosphodiesterases and ATP-binding cassette transporters. Annu. Rev. Pharmacol. Toxicol. 53, 231–253. doi: 10.1146/annurev-pharmtox-010611-134609
Cheng, X., Ji, Z., Tsalkova, T., and Mei, F. (2008). Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim. Biophys. Sin. (Shanghai). 40, 651–662. doi: 10.1111/j.1745-7270.2008.00438.x
Cheung, L., Flemming, C. L., Watt, F., Masada, N., Yu, D. M., Huynh, T., et al. (2014). High-throughput screening identifies Ceefourin 1 and Ceefourin 2 as highly selective inhibitors of multidrug resistance protein 4 (MRP4). Biochem. Pharmacol. 91, 97–108. doi: 10.1016/j.bcp.2014.05.023
Courilleau, D., Bisserier, M., Jullian, J. C., Lucas, A., Bouyssou, P., Fischmeister, R., et al. (2012). Identification of a tetrahydroquinoline analog as a pharmacological inhibitor of the cAMP-binding protein Epac. J. Biol. Chem. 287, 44192–44202. doi: 10.1074/jbc.M112.422956
Courilleau, D., Bouyssou, P., Fischmeister, R., Lezoualc'h, F., and Blondeau, J. P. (2013). The (R)-enantiomer of CE3F4 is a preferential inhibitor of human exchange protein directly activated by cyclic AMP isoform 1 (Epac1). Biochem. Biophys. Res. Commun. 440, 443–448. doi: 10.1016/j.bbrc.2013.09.107
Dalton, G. D., and Dewey, W. L. (2006). Protein kinase inhibitor peptide (PKI): a family of endogenous neuropeptides that modulate neuronal cAMP-dependent protein kinase function. Neuropeptides 40, 23–34. doi: 10.1016/j.npep.2005.10.002
Davies, S. P., Reddy, H., Caivano, M., and Cohen, P. (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351, 95–105. doi: 10.1042/bj3510095
Defer, N., Best-Belpomme, M., and Hanoune, J. (2000). Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase. American journal of physiology. Ren. Physiol. 279, F400–F416.
Degorce, F., Card, A., Soh, S., Trinquet, E., Knapik, G. P., and Xie, B. (2009). HTRF: a technology tailored for drug discovery—a review of theoretical aspects and recent applications. Curr. Chem. Genomics 3, 22–32. doi: 10.2174/1875397300903010022
De Haan, L., and Hirst, T. R. (2004). Cholera toxin: a paradigm for multi-functional engagement of cellular mechanisms (Review). Mol. Membr. Biol. 21, 77–92. doi: 10.1080/09687680410001663267
Del Lungo, M., Melchiorre, M., Guandalini, L., Sartiani, L., Mugelli, A., Koncz, I., et al. (2012). Novel blockers of hyperpolarization-activated current with isoform selectivity in recombinant cells and native tissue. Br. J. Pharmacol. 166, 602–616. doi: 10.1111/j.1476-5381.2011.01782.x
Denis, C., Saulière, A., Galandrin, S., Sénard, J. M., and Galés, C. (2012). Probing heterotrimeric G protein activation: applications to biased ligands. Curr. Pharm. Des. 18, 128–144. doi: 10.2174/138161212799040466
de Rooij, J., Zwartkruis, F. J., Verheijen, M. H., Cool, R. H., Nijman, S. M., Wittinghofer, A., et al. (1998). Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396, 474–477. doi: 10.1038/24884
Dessauer, C. W., Scully, T. T., and Gilman, A. G. (1997). Interactions of forskolin and ATP with the cytosolic domains of mammalian adenylyl cyclase. J. Biol. Chem. 272, 22272–22277. doi: 10.1074/jbc.272.35.22272
Dessauer, C. W., Tesmer, J. J., Sprang, S. R., and Gilman, A. G. (1999). The interactions of adenylate cyclases with P-site inhibitors. Trends Pharmacol. Sci. 20, 205–210. doi: 10.1016/S0165-6147(99)01310-3
Ding, B., Abe, J., Wei, H., Huang, Q., Walsh, R. A., Molina, C. A., et al. (2005a). Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: implication in heart failure. Circulation 111, 2469–2476. doi: 10.1161/01.CIR.0000165128.39715.87
Ding, B., Abe, J., Wei, H., Xu, H., Che, W., Aizawa, T., et al. (2005b). A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (ICER) leads to cardiomyocyte apoptosis. Proc. Natl. Acad. Sci. U.S.A. 102, 14771–147714776. doi: 10.1073/pnas.0506489102
Dodge, K. L., Khouangsathiene, S., Kapiloff, M. S., Mouton, R., Hill, E. V., Houslay, M. D., et al. (2001). mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 20, 1921–1930. doi: 10.1093/emboj/20.8.1921
Dunkern, T. R., and Hatzelmann, A. (2007). Characterization of inhibitors of phosphodiesterase 1C on a human cellular system. FEBS J. 274, 4812–4824. doi: 10.1111/j.1742-4658.2007.06001.x
Ebina, T., Kawabe, J., Katada, T., Ohno, S., Homcy, C. J., and Ishikawa, Y. (1997). Conformation-dependent activation of type II adenylyl cyclase by protein kinase C. J. Cell. Biochem. 64, 492–498.
Edwards, H. V., Christian, F., and Baillie, G. S. (2012). cAMP: novel concepts in compartmentalised signalling. Semin. Cell Dev. Biol. 23, 181–190. doi: 10.1016/j.semcdb.2011.09.005
Efendiev, R., and Dessauer, C. W. (2011). A kinase-anchoring proteins and adenylyl cyclase in cardiovascular physiology and pathology. J. Cardiovasc. Pharmacol. 58, 339–344. doi: 10.1097/FJC.0b013e31821bc3f0
Efetova, M., Petereit, L., Rosiewicz, K., Overend, G., Haußig, F., Hovemann, B. T., et al. (2013). Separate roles of PKA and EPAC in renal function unraveled by the optogenetic control of cAMP levels in vivo. J. Cell Sci. 126, 778–788. doi: 10.1242/jcs.114140
Engelhardt, S., Hein, L., Wiesmann, F., and Lohse, M. J. (1999). Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 96, 7059–7064. doi: 10.1073/pnas.96.12.7059
Engh, R. A., Girod, A., Kinzel, V., Huber, R., and Bossemeyer, D. (1996). Crystal structures of catalytic subunit of cAMP-dependent protein kinase in complex with isoquinolinesulfonyl protein kinase inhibitors H7, H8, and H89. Structural implications for selectivity. J. Biol. Chem. 271, 26157–26164. doi: 10.1074/jbc.271.42.26157
Enserink, J. M., Christensen, A. E., de Rooij, J., van Triest, M., Schwede, F., Genieser, H. G., et al. (2002). A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat. Cell Biol. 4, 901–906. doi: 10.1038/ncb874
Erdorf, M., Mou, T. C., and Seifert, R. (2011). Impact of divalent metal ions on regulation of adenylyl cyclase isoforms by forskolin analogs. Biochem. Pharmacol. 82, 1673–1681. doi: 10.1016/j.bcp.2011.07.099
Eschenhagen, T., Mende, U., Nose, M., Schmitz, W., Scholz, H., Haverich, A., et al. (1992a). Increased messenger RNA level of the inhibitory G protein alpha subunit Gi alpha-2 in human end-stage heart failure. Circ. Res. 70, 688–696. doi: 10.1161/01.RES.70.4.688
Eschenhagen, T., Mende, U., Nose, M., Schmitz, W., Scholz, H., Schulte am Esch, J., Sempell, R., et al. (1992b). Regulation and possible functional implications of G-protein mRNA expression in nonfailing and failing ventricular myocardium. Basic Res. Cardiol. 87(Suppl. 1), 51–64. doi: 10.1007/978-3-642-72474-9_4
Esposito, G., Perrino, C., Ozaki, T., Takaoka, H., Defer, N., Petretta, M. P., et al. (2008). Increased myocardial contractility and enhanced exercise function in transgenic mice overexpressing either adenylyl cyclase 5 or 8. Basic Res. Cardiol. 103, 22–30. doi: 10.1007/s00395-007-0688-6
Fan, F., Binkowski, B. F., Butler, B. L., Stecha, P. F., Lewis, M. K., and Wood, K. V. (2008). Novel genetically encoded biosensors using firefly luciferase. ACS Chem. Biol. 3, 346–351. doi: 10.1021/cb8000414
Filip, Z. A., Vanhauwe, J., Mathew, A., Eason, P. D., Ly, J. T., Leginus, J., et al. (2004). A Multi-Array™ Technology Based Assay for cAMP. Rockville, MD: Meso Scale Discovery Application note.
Fischmeister, R., Castro, L., Abi-Gerges, A., Rochais, F., and Vandecasteele, G. (2005). Species- and tissue-dependent effects of NO and cyclic GMP on cardiac ion channels. Comparative biochemistry and physiology. Part A Mol. Integr. Physiol. 142, 136–143. doi: 10.1016/j.cbpb.2005.04.012
Forster, T. (1946). Energiewanderung und Fluoreszenz. Naturwissenschaften 33, 166–175. doi: 10.1007/BF00585226
Fraser, I. D., Tavalin, S. J., Lester, L. B., Langeberg, L. K., Westphal, A. M., Dean, R. A., et al. (1998). A novel lipid-anchored A-kinase Anchoring Protein facilitates cAMP-responsive membrane events. EMBO J. 17, 2261–2272. doi: 10.1093/emboj/17.8.2261
Fredriksson, R., and Schiöth, H. B. (2005). The repertoire of G-protein-coupled receptors in fully sequenced genomes. Mol. Pharmacol. 67, 1414–1425. doi: 10.1124/mol.104.009001
Freissmuth, M., Boehm, S., Beindl, W., Nickel, P., Ijzerman, A. P., Hohenegger, M., et al. (1996). Suramin analogues as subtype-selective G protein inhibitors. Mol. Pharmacol. 49, 602–611.
Froese, A., Breher, S. S., Waldeyer, C., Schindler, R. F., Nikolaev, V. O., Rinné, S., et al. (2012). Popeye domain containing proteins are essential for stress-mediated modulation of cardiac pacemaking in mice. J. Clin. Invest. 122, 1119–1130. doi: 10.1172/JCI59410
Gao, M. H., Lai, N. C., Roth, D. M., Zhou, J., Zhu, J., Anzai, T., et al. (1999). Adenylylcyclase increases responsiveness to catecholamine stimulation in transgenic mice. Circulation 99, 1618–1622. doi: 10.1161/01.CIR.99.12.1618
Gao, M. H., Tang, T., Guo, T., Miyanohara, A., Yajima, T., Pestonjamasp, K., et al. (2008). Adenylyl cyclase type VI increases Akt activity and phospholamban phosphorylation in cardiac myocytes. J. Biol. Chem. 283, 33527–33535. doi: 10.1074/jbc.M805825200
Gasser, C., Taiber, S., Yeh, C. M., Wittig, C. H., Hegemann, P., Ryu, S., et al. (2014). Engineering of a red-light-activated human cAMP/cGMP-specific phosphodiesterase. Proc. Natl. Acad. Sci. U.S.A. 111, 8803–8808. doi: 10.1073/pnas.1321600111
Gille, A., and Seifert, R. (2003). MANT-substituted guanine nucleotides: a novel class of potent adenylyl cyclase inhibitors. Life Sci. 74, 271–279. doi: 10.1016/j.lfs.2003.09.014
Gilman, A. G. (1987). G proteins: transducers of receptor-generated signals. Annu. Rev. Biochem. 56, 615–649. doi: 10.1146/annurev.bi.56.070187.003151
Glass, D. B., Feller, M. J., Levin, L. R., and Walsh, D. A. (1992). Structural basis for the low affinities of yeast cAMP-dependent and mammalian cGMP-dependent protein kinases for protein kinase inhibitor peptides. Biochemistry 31, 1728–1734. doi: 10.1021/bi00121a021
Golla, R., and Seethala, R. (2002). A homogeneous enzyme fragment complementation cyclic AMP screen for GPCR agonists. J. Biomol. Screen. 7, 515–525. doi: 10.1177/1087057102238625
Goodwin, L. R., Francom, D., Dieken, F. P., Taylor, J. D., Warenycia, M. W., Reiffenstein, R. J., et al. (1989). Determination of sulfide in brain tissue by gas dialysis/ion chromatography: postmortem studies and two case reports. J. Anal. Toxicol. 13, 105–109. doi: 10.1093/jat/13.2.105
Guellich, A., Gao, S., Hong, C., Yan, L., Wagner, T. E., Dhar, S. K., et al. (2010). Effects of cardiac overexpression of type 6 adenylyl cyclase affects on the response to chronic pressure overload. American journal of physiology. Heart Circul. Physiol. 299, H707–H712. doi: 10.1152/ajpheart.00148.2010
Guellich, A., Mehel, H., and Fischmeister, R. (2014). Cyclic AMP synthesis and hydrolysis in the normal and failing heart. Pflugers Arch. 466, 1163–1175. doi: 10.1007/s00424-014-1515-1
Guillou, J. L., Nakata, H., and Cooper, D. M. (1999). Inhibition by calcium of mammalian adenylyl cyclases. J. Biol. Chem. 274, 35539–35545. doi: 10.1074/jbc.274.50.35539
Hagen, T. J., Mo, X., Burgin, A. B., Fox, D. I. I. I., Zhang, Z., and Gurney, M. E. (2014). Discovery of triazines as selective PDE4B versus PDE4D inhibitors. Bioorg. Med. Chem. Lett. 24, 4031–4034. doi: 10.1016/j.bmcl.2014.06.002
Hakak, Y., Shrestha, D., Goegel, M. C., Behan, D. P., and Chalmers, D. T. (2003). Global analysis of G-protein-coupled receptor signaling in human tissues. FEBS Lett. 550, 11–17. doi: 10.1016/S0014-5793(03)00762-2
Halls, M. L., and Cooper, D. M. (2011). Regulation by Ca2+-signaling pathways of adenylyl cyclases. Cold Spring Harb. Perspect. Biol. 3:a004143. doi: 10.1101/cshperspect.a004143
Han, S., and Meier, K. E. (2009). Integrated modulation of phorbol ester-induced Raf activation in EL4 lymphoma cells. Cell. Signal. 21, 793–800. doi: 10.1016/j.cellsig.2009.01.025
Hartzell, H. C., and Fischmeister, R. (1986). Opposite effects of cyclic GMP and cyclic AMP on Ca2+ current in single heart cells. Nature 323, 273–275. doi: 10.1038/323273a0
Hatzelmann, A., Morcillo, E. J., Lungarella, G., Adnot, S., Sanjar, S., Beume, R., et al. (2010). The preclinical pharmacology of roflumilast–a selective, oral phosphodiesterase 4 inhibitor in development for chronic obstructive pulmonary disease. Pulm. Pharmacol. Ther. 23, 235–256. doi: 10.1016/j.pupt.2010.03.011
Haunsø, A., Simpson, J., and Antoni, F. A. (2003). Small ligands modulating the activity of mammalian adenylyl cyclases: a novel mode of inhibition by calmidazolium. Mol. Pharmacol. 63, 624–631. doi: 10.1124/mol.63.3.624
Hébert, T. E., Galés, C., and Rebois, R. V. (2006). Detecting and imaging protein-protein interactions during G protein-mediated signal transduction in vivo and in situ by using fluorescence-based techniques. Cell Biochem. Biophys. 45, 85–109. doi: 10.1385/CBB:45:1:85
Hess, S. M., Chasin, M., Free, C. A., and Harris, D. N. (1975). Modulators of cyclic AMP systems. Psychopharmacol. Bull. 11, 57–58.
Hidaka, H., Hayashi, H., Kohri, H., Kimura, Y., Hosokawa, T., Igawa, T., et al. (1979). Selective inhibitor of platelet cyclic adenosine monophosphate phosphodiesterase, cilostamide, inhibits platelet aggregation. J. Pharmacol. Exp. Ther. 211, 26–30.
Hofmann, F., Beavo, J. A., Bechtel, P. J., and Krebs, E. G. (1975). Comparison of adenosine 3′:5′-monophosphate-dependent protein kinases from rabbit skeletal and bovine heart muscle. J. Biol. Chem. 250, 7795–7801.
Hohenegger, M., Waldhoer, M., Beindl, W., Böing, B., Kreimeyer, A., Nickel, P., et al. (1998). Gsalpha-selective G protein antagonists. Proc. Natl. Acad. Sci. U.S.A. 95, 346–351. doi: 10.1073/pnas.95.1.346
Horton, J. K., and Baxendale, P. M. (1995). Mass measurements of cyclic AMP formation by radioimmunoassay, enzyme immunoassay, and scintillation proximity assay. Methods Mol. Biol. 41, 91–105. doi: 10.1385/0-89603-298-1:91
Houslay, M. D. (2001). PDE4 cAMP-specific phosphodiesterases. Prog. Nucleic Acid Res. Mol. Biol. 69, 249–315. doi: 10.1016/S0079-6603(01)69049-4
Hsia, J. A., Moss, J., Hewlett, E. L., and Vaughan, M. (1984). Requirement for both choleragen and pertussis toxin to obtain maximal activation of adenylate cyclase in cultured cells. Biochem. Biophys. Res. Commun. 119, 1068–1074. doi: 10.1016/0006-291X(84)90883-0
Hu, B., Nakata, H., Gu, C., De Beer, T., and Cooper, D. M. (2002). A critical interplay between Ca2+ inhibition and activation by Mg2+ of AC5 revealed by mutants and chimeric constructs. J. Biol. Chem. 277, 33139–33147. doi: 10.1074/jbc.M112373200
Huang, W., Zhang, Y., and Sportsman, J. R. (2002). A fluorescence polarization assay for cyclic nucleotide phosphodiesterases. J. Biomol. Screen. 7, 215–222. doi: 10.1177/108705710200700305
Huang, X. D., and Wong, T. M. (1989). Cholera toxin enhances ischemia-induced arrhythmias in the isolated rat heart–involvement of a guanine nucleotide binding protein (Gs). Life Sci. 45, 679–683. doi: 10.1016/0024-3205(89)90085-4
Hudson, T. Y., Corbett, J. A., Howlett, A. C., and Klein, C. (2001). Nitric oxide regulates adenylyl cyclase activity in rat striatal membranes. J. Neurochem. 77, 1279–1284. doi: 10.1046/j.1471-4159.2001.00331.x
Ito, K., Liu, H., Komiyama, M., Hayashi, T., and Xu, Y. (2013). Direct light-up of cAMP derivatives in living cells by click reactions. Molecules 18, 12909–12915. doi: 10.3390/molecules181012909
Iwamoto, T., Okumura, S., Iwatsubo, K., Kawabe, J., Ohtsu, K., Sakai, I., et al. (2003). Motor dysfunction in type 5 adenylyl cyclase-null mice. J. Biol. Chem. 278, 16936–16940. doi: 10.1074/jbc.C300075200
Iwaya, S., Oikawa, M., Chen, Y., and Takeishi, Y. (2014). Phosphodiesterase 3A1 protects the heart against angiotensin II-induced cardiac remodeling through regulation of transforming growth factor-beta expression. Int. Heart J. 55, 165–168. doi: 10.1536/ihj.13-268
Jin, S. L., Lan, L., Zoudilova, M., and Conti, M. (2005). Specific role of phosphodiesterase 4B in lipopolysaccharide-induced signaling in mouse macrophages. J. Immunol. 175, 1523–1531. doi: 10.4049/jimmunol.175.3.1523
Jurevicius, J., and Fischmeister, R. (1996a). Acetylcholine inhibits Ca2+ current by acting exclusively at a site proximal to adenylyl cyclase in frog cardiac myocytes. J. Physiol. 491(Pt 3), 669–675. doi: 10.1113/jphysiol.1996.sp021248
Jurevicius, J., and Fischmeister, R. (1996b). cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by beta-adrenergic agonists. Proc. Natl. Acad. Sci. U.S.A. 93, 295–299. doi: 10.1073/pnas.93.1.295
Kadoshima-Yamaoka, K., Murakawa, M., Goto, M., Tanaka, Y., Inoue, H., Murafuji, H., et al. (2009). ASB16165, a novel inhibitor for phosphodiesterase 7A (PDE7A), suppresses IL-12-induced IFN-gamma production by mouse activated T lymphocytes. Immunol. Lett. 122, 193–197. doi: 10.1016/j.imlet.2009.01.004
Kapiloff, M. S., Piggott, L. A., Sadana, R., Li, J., Heredia, L. A., Henson, E., et al. (2009). An adenylyl cyclase-mAKAPbeta signaling complex regulates cAMP levels in cardiac myocytes. J. Biol. Chem. 284, 23540–23546. doi: 10.1074/jbc.M109.030072
Kapiloff, M. S., Rigatti, M., and Dodge-Kafka, K. L. (2014). Architectural and functional roles of A kinase-anchoring proteins in cAMP microdomains. J. Gen. Physiol. 143, 9–15. doi: 10.1085/jgp.201311020
Kase, H., Iwahashi, K., Nakanishi, S., Matsuda, Y., Yamada, K., Takahashi, M., et al. (1987). K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem. Biophys. Res. Commun. 142, 436–440. doi: 10.1016/0006-291X(87)90293-2
Katugampola, S., and Davenport, A. (2003). Emerging roles for orphan G-protein-coupled receptors in the cardiovascular system. Trends Pharmacol. Sci. 24, 30–35. doi: 10.1016/S0165-6147(02)00007-X
Kawasaki, H., Springett, G. M., Mochizuki, N., Toki, S., Nakaya, M., Matsuda, M., et al. (1998). A family of cAMP-binding proteins that directly activate Rap1. Science 282, 2275–2279. doi: 10.1126/science.282.5397.2275
Kazanietz, M. G., Lewin, N. E., Bruns, J. D., and Blumberg, P. M. (1995). Characterization of the cysteine-rich region of the Caenorhabditis elegans protein Unc-13 as a high affinity phorbol ester receptor. Analysis of ligand-binding interactions, lipid cofactor requirements, and inhibitor sensitivity. J. Biol. Chem. 270, 10777–10783. doi: 10.1074/jbc.270.18.10777
Kimura, H. (2000). Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochem. Biophys. Res. Commun. 267, 129–133. doi: 10.1006/bbrc.1999.1915
Kiuchi, K., Shannon, R. P., Komamura, K., Cohen, D. J., Bianchi, C., Homcy, C. J., et al. (1993). Myocardial beta-adrenergic receptor function during the development of pacing-induced heart failure. J. Clin. Invest. 91, 907–914. doi: 10.1172/JCI116312
Kiyokawa, E., Hara, S., Nakamura, T., and Matsuda, M. (2006). Fluorescence (Forster) resonance energy transfer imaging of oncogene activity in living cells. Cancer Sci. 97, 8–15. doi: 10.1111/j.1349-7006.2006.00141.x
Klarenbeek, J., and Jalink, K. (2014). Detecting cAMP with an EPAC-based FRET sensor in single living cells. Methods Mol. Biol. 1071, 49–58. doi: 10.1007/978-1-62703-622-1_4
Kruh, G. D., and Belinsky, M. G. (2003). The MRP family of drug efflux pumps. Oncogene 22, 7537–7552. doi: 10.1038/sj.onc.1206953
Kumar, S., Kostin, S., Flacke, J. P., Reusch, H. P., and Ladilov, Y. (2009). Soluble adenylyl cyclase controls mitochondria-dependent apoptosis in coronary endothelial cells. J. Biol. Chem. 284, 14760–14768. doi: 10.1074/jbc.M900925200
Lai, L., Yan, L., Gao, S., Hu, C. L., Ge, H., Davidow, A., et al. (2013). Type 5 adenylyl cyclase increases oxidative stress by transcriptional regulation of manganese superoxide dismutase via the SIRT1/FoxO3a pathway. Circulation 127, 1692–1701. doi: 10.1161/CIRCULATIONAHA.112.001212
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., et al. (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921. doi: 10.1038/35057062
Laurenza, A., Sutkowski, E. M., and Seamon, K. B. (1989). Forskolin: a specific stimulator of adenylyl cyclase or a diterpene with multiple sites of action? Trends Pharmacol. Sci. 10, 442–447. doi: 10.1016/S0165-6147(89)80008-2
Lehmann, D. M., Seneviratne, A. M., and Smrcka, A. V. (2008). Small molecule disruption of G protein beta gamma subunit signaling inhibits neutrophil chemotaxis and inflammation. Mol. Pharmacol. 73, 410–418. doi: 10.1124/mol.107.041780
Lehnart, S. E., Wehrens, X. H., Reiken, S., Warrier, S., Belevych, A. E., Harvey, R. D., et al. (2005). Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell 123, 25–35. doi: 10.1016/j.cell.2005.07.030
Leroy, J., Richter, W., Mika, D., Castro, L. R., Abi-Gerges, A., Xie, M., et al. (2011). Phosphodiesterase 4B in the cardiac L-type Ca(2)(+) channel complex regulates Ca(2)(+) current and protects against ventricular arrhythmias in mice. J. Clin. Invest. 121, 2651–2661. doi: 10.1172/JCI44747
Lezoualc'h, F., Jockers, R., and Berque-Bestel, I. (2009). Multivalent-based drug design applied to serotonin 5-HT(4) receptor oligomers. Curr. Pharm. Des. 15, 719–729. doi: 10.2174/138161209787315602
Lim, J. J., Liu, Y. H., Khin, E. S., and Bian, J. S. (2008). Vasoconstrictive effect of hydrogen sulfide involves downregulation of cAMP in vascular smooth muscle cells. American journal of physiology. Cell Physiol. 295, C1261–C1270. doi: 10.1152/ajpcell.00195.2008
Lipskaia, L., Defer, N., Esposito, G., Hajar, I., Garel, M. C., Rockman, H. A., et al. (2000). Enhanced cardiac function in transgenic mice expressing a Ca(2+)-stimulated adenylyl cyclase. Circ. Res. 86, 795–801. doi: 10.1161/01.RES.86.7.795
Lohse, M. J., Engelhardt, S., and Eschenhagen, T. (2003). What is the role of beta-adrenergic signaling in heart failure? Circ. Res. 93, 896–906. doi: 10.1161/01.RES.0000102042.83024.CA
MacKenzie, S. J., Baillie, G. S., McPhee, I., MacKenzie, C., Seamons, R., McSorley, T., et al. (2002). Long PDE4 cAMP specific phosphodiesterases are activated by protein kinase A-mediated phosphorylation of a single serine residue in Upstream Conserved Region 1 (UCR1). Br. J. Pharmacol. 136, 421–433. doi: 10.1038/sj.bjp.0704743
Mattick, P., Parrington, J., Odia, E., Simpson, A., Collins, T., and Terrar, D. (2007). Ca2+-stimulated adenylyl cyclase isoform AC1 is preferentially expressed in guinea-pig sino-atrial node cells and modulates the I(f) pacemaker current. J. Physiol. (Lond). 582, 1195–1203. doi: 10.1113/jphysiol.2007.133439
McMurray, J. J., and Pfeffer, M. A. (2005). Heart failure. Lancet 365, 1877–1889. doi: 10.1016/S0140-6736(05)66621-4
Melchiorre, M., Del Lungo, M., Guandalini, L., Martini, E., Dei, S., Manetti, D., et al. (2010). Design, synthesis, and preliminary biological evaluation of new isoform-selective f-current blockers. J. Med. Chem. 53, 6773–6777. doi: 10.1021/jm1006758
Melsom, C. B., Hussain, R. I., Ørstavik, Ø., Aronsen, J. M., Sjaastad, I., Skomedal, T., et al. (2014). Non-classical regulation of beta1- and beta 2-adrenoceptor-mediated inotropic responses in rat heart ventricle by the G protein Gi. Naunyn Schmiedebergs Arch. Pharmacol. 387, 1177–1186. doi: 10.1007/s00210-014-1036-7
Métrich, M., Lucas, A., Gastineau, M., Samuel, J. L., Heymes, C., Morel, E., et al. (2008). Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ. Res. 102, 959–965. doi: 10.1161/CIRCRESAHA.107.164947
Michel, M. C., Maisel, A. S., and Brodde, O. E. (1990). Mitigation of beta 1- and/or beta 2-adrenoceptor function in human heart failure. British journal of clinical pharmacology 30(Suppl. 1), 37S–42S. doi: 10.1111/j.1365-2125.1990.tb05466.x
Miller, C. L., Cai, Y., Oikawa, M., Thomas, T., Dostmann, W. R., Zaccolo, M., et al. (2011). Cyclic nucleotide phosphodiesterase 1A: a key regulator of cardiac fibroblast activation and extracellular matrix remodeling in the heart. Basic Res. Cardiol. 106, 1023–1039. doi: 10.1007/s00395-011-0228-2
Molenaar, P., Christ, T., Hussain, R. I., Engel, A., Berk, E., Gillette, K. T., et al. (2013). PDE3, but not PDE4, reduces beta(1) - and beta(2)-adrenoceptor-mediated inotropic and lusitropic effects in failing ventricle from metoprolol-treated patients. Br. J. Pharmacol. 169, 528–538. doi: 10.1111/bph.12167
Molina-Muñoz, T., Romero-Avila, M. T., and García-Sáinz, J. A. (2006). Insulin-like growth factor-I induces alpha(1B)-adrenergic receptor phosphorylation through G beta gamma and epidermal growth factor receptor transactivation. Mol. Endocrinol. 20, 2773–2783. doi: 10.1210/me.2006-0090
Moore-Morris, T., Varrault, A., Mangoni, M. E., Le Digarcher, A., Negre, V., Dantec, C., et al. (2009). Identification of potential pharmacological targets by analysis of the comprehensive G protein-coupled receptor repertoire in the four cardiac chambers. Mol. Pharmacol. 75, 1108–1116. doi: 10.1124/mol.108.054155
Mou, T. C., Masada, N., Cooper, D. M., and Sprang, S. R. (2009). Structural basis for inhibition of mammalian adenylyl cyclase by calcium. Biochemistry 48, 3387–3397. doi: 10.1021/bi802122k
Mukherjee, R., and Spinale, F. G. (1998). L-type calcium channel abundance and function with cardiac hypertrophy and failure: a review. J. Mol. Cell. Cardiol. 30, 1899–1916. doi: 10.1006/jmcc.1998.0755
Murray, A. J. (2008). Pharmacological PKA inhibition: all may not be what it seems. Sci. Signal. 1:re4. doi: 10.1126/scisignal.122re4
Nair, B. G., and Patel, T. B. (1993). Regulation of cardiac adenylyl cyclase by epidermal growth factor (EGF). Role of EGF receptor protein tyrosine kinase activity. Biochem. Pharmacol. 46, 1239–1245. doi: 10.1016/0006-2952(93)90473-A
Nichols, C. B., Rossow, C. F., Navedo, M. F., Westenbroek, R. E., Catterall, W. A., Santana, L. F., et al. (2010). Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of L-type calcium channels. Circ. Res. 107, 747–756. doi: 10.1161/CIRCRESAHA.109.216127
Niimura, M., Miki, T., Shibasaki, T., Fujimoto, W., Iwanaga, T., and Seino, S. (2009). Critical role of the N-terminal cyclic AMP-binding domain of Epac2 in its subcellular localization and function. J. Cell. Physiol. 219, 652–658. doi: 10.1002/jcp.21709
Nikolaev, V. O., Moshkov, A., Lyon, A. R., Miragoli, M., Novak, P., Paur, H., et al. (2010). Beta2-adrenergic receptor redistribution in heart failure changes cAMP compartmentation. Science 327, 1653–1657. doi: 10.1126/science.1185988
Okumura, S., Kawabe, J., Yatani, A., Takagi, G., Lee, M. C., Hong, C., et al. (2003a). Type 5 adenylyl cyclase disruption alters not only sympathetic but also parasympathetic and calcium-mediated cardiac regulation. Circ. Res. 93, 364–371. doi: 10.1161/01.RES.0000086986.35568.63
Okumura, S., Takagi, G., Kawabe, J., Yang, G., Lee, M. C., Hong, C., et al. (2003b). Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc. Natl. Acad. Sci. U.S.A. 100, 9986–9990. doi: 10.1073/pnas.1733772100
Omori, K., and Kotera, J. (2007). Overview of PDEs and their regulation. Circ. Res. 100, 309–327. doi: 10.1161/01.RES.0000256354.95791.f1
Onda, T., Hashimoto, Y., Nagai, M., Kuramochi, H., Saito, S., Yamazaki, H., et al. (2001). Type-specific regulation of adenylyl cyclase. Selective pharmacological stimulation and inhibition of adenylyl cyclase isoforms. J. Biol. Chem. 276, 47785–47793. doi: 10.1074/jbc.M107233200
Orth, J. H., Fester, I., Preuss, I., Agnoletto, L., Wilson, B. A., and Aktories, K. (2008). Activation of Galpha (i) and subsequent uncoupling of receptor-Galpha(i) signaling by Pasteurella multocida toxin. J. Biol. Chem. 283, 23288–23294. doi: 10.1074/jbc.M803435200
Orth, J. H., Lang, S., Taniguchi, M., and Aktories, K. (2005). Pasteurella multocida toxin-induced activation of RhoA is mediated via two families of G{alpha} proteins, G{alpha}q and G{alpha}12/13. J. Biol. Chem. 280, 36701–36707. doi: 10.1074/jbc.M507203200
Ostrom, R. S., Gregorian, C., Drenan, R. M., Xiang, Y., Regan, J. W., and Insel, P. A. (2001). Receptor number and caveolar co-localization determine receptor coupling efficiency to adenylyl cyclase. J. Biol. Chem. 276, 42063–42069. doi: 10.1074/jbc.M105348200
Pape, H. C. (1996). Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu. Rev. Physiol. 58, 299–327. doi: 10.1146/annurev.ph.58.030196.001503
Patrucco, E., Albergine, M. S., Santana, L. F., and Beavo, J. A. (2010). Phosphodiesterase 8A (PDE8A) regulates excitation-contraction coupling in ventricular myocytes. J. Mol. Cell. Cardiol. 49, 330–333. doi: 10.1016/j.yjmcc.2010.03.016
Pereira, L., Métrich, M., Fernández-Velasco, M., Lucas, A., Leroy, J., Perrier, R., et al. (2007). The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J. Physiol. (Lond). 583, 685–694. doi: 10.1113/jphysiol.2007.133066
Perera, R. K., and Nikolaev, V. O. (2013). Compartmentation of cAMP signalling in cardiomyocytes in health and disease. Acta Physiol. 207, 650–662. doi: 10.1111/apha.12077
Pertseva, M. N., Shpakov, A. O., Plesneva, S. A., and Kuznetsova, L. A. (2003). A novel view on the mechanisms of action of insulin and other insulin superfamily peptides: involvement of adenylyl cyclase signaling system. Comparative biochemistry and physiology. Part B Biochem. Mol. Biol. 134, 11–36. doi: 10.1016/S1096-4959(02)00160-4
Phan, H. M., Gao, M. H., Lai, N. C., Tang, T., and Hammond, H. K. (2007). New signaling pathways associated with increased cardiac adenylyl cyclase 6 expression: implications for possible congestive heart failure therapy. Trends Cardiovasc. Med. 17, 215–221. doi: 10.1016/j.tcm.2007.07.001
Piggott, L. A., Bauman, A. L., Scott, J. D., and Dessauer, C. W. (2008). The A-kinase anchoring protein Yotiao binds and regulates adenylyl cyclase in brain. Proc. Natl. Acad. Sci. U.S.A. 105, 13835–13840. doi: 10.1073/pnas.0712100105
Piñeyro, G., Azzi, M., deLéan, A., Schiller, P. W., and Bouvier, M. (2005). Reciprocal regulation of agonist and inverse agonist signaling efficacy upon short-term treatment of the human delta-opioid receptor with an inverse agonist. Mol. Pharmacol. 67, 336–348. doi: 10.1124/mol.104.004549
Podda, M. V., and Grassi, C. (2014). New perspectives in cyclic nucleotide-mediated functions in the CNS: the emerging role of cyclic nucleotide-gated (CNG) channels. Pflugers Arch. 466, 1241–1257. doi: 10.1007/s00424-013-1373-2
Podzuweit, T., Müller, A., and Opie, L. H. (1993). Anti-arrhythmic effects of selective inhibition of myocardial phosphodiesterase II. Lancet 341, 760. doi: 10.1016/0140-6736(93)90534-N
Poppe, H., Rybalkin, S. D., Rehmann, H., Hinds, T. R., Tang, X. B., Christensen, A. E., et al. (2008). Cyclic nucleotide analogs as probes of signaling pathways. Nat. Methods 5, 277–278. doi: 10.1038/nmeth0408-277
Prévost, G. P., Lonchampt, M. O., Holbeck, S., Attoub, S., Zaharevitz, D., Alley, M., et al. (2006). Anticancer activity of BIM-46174, a new inhibitor of the heterotrimeric Galpha/Gbetagamma protein complex. Cancer Res. 66, 9227–9234. doi: 10.1158/0008-5472.CAN-05-4205
Prystay, L., Gagné, A., Kasila, P., Yeh, L. A., and Banks, P. (2001). Homogeneous cell-based fluorescence polarization assay for the direct detection of cAMP. J. Biomol. Screen. 6, 75–82. doi: 10.1177/108705710100600203
Rabe, K. F. (2011). Update on roflumilast, a phosphodiesterase 4 inhibitor for the treatment of chronic obstructive pulmonary disease. Br. J. Pharmacol. 163, 53–67. doi: 10.1111/j.1476-5381.2011.01218.x
Regard, J. B., Sato, I. T., and Coughlin, S. R. (2008). Anatomical profiling of G protein-coupled receptor expression. Cell 135, 561–571. doi: 10.1016/j.cell.2008.08.040
Reid, G., Wielinga, P., Zelcer, N., van der Heijden, I., Kuil, A., de Haas, M., et al. (2003). The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc. Natl. Acad. Sci. U.S.A. 100, 9244–9249. doi: 10.1073/pnas.1033060100
Rostovtsev, V. V., Green, L. G., Fokin, V. V., and Sharpless, K. B. (2002). A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angewandte Chemie 41, 2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4
Rybalkin, S. D., Rybalkina, I., Beavo, J. A., and Bornfeldt, K. E. (2002). Cyclic nucleotide phosphodiesterase 1C promotes human arterial smooth muscle cell proliferation. Circ. Res. 90, 151–157. doi: 10.1161/hh0202.104108
Rybin, V. O., Xu, X., Lisanti, M. P., and Steinberg, S. F. (2000). Differential targeting of beta -adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. A mechanism to functionally regulate the cAMP signaling pathway. J. Biol. Chem. 275, 41447–41457. doi: 10.1074/jbc.M006951200
Sandberg, M., Butt, E., Nolte, C., Fischer, L., Halbrügge, M., Beltman, J., et al. (1991). Characterization of Sp-5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole- 3′,5′-monophosphorothioate (Sp-5,6-DCl-cBiMPS) as a potent and specific activator of cyclic-AMP-dependent protein kinase in cell extracts and intact cells. Biochem. J. 279(Pt 2), 521–527. doi: 10.1042/bj2790521
Sassi, Y., Abi-Gerges, A., Fauconnier, J., Mougenot, N., Reiken, S., Haghighi, K., et al. (2012). Regulation of cAMP homeostasis by the efflux protein MRP4 in cardiac myocytes. FASEB J. 26, 1009–1017. doi: 10.1096/fj.11-194027
Sassi, Y., Lipskaia, L., Vandecasteele, G., Nikolaev, V. O., Hatem, S. N., Cohen Aubart, F., et al. (2008). Multidrug resistance-associated protein 4 regulates cAMP-dependent signaling pathways and controls human and rat SMC proliferation. J. Clin. Invest. 118, 2747–2757. doi: 10.1172/JCI35067
Schaap, P., van Ments-Cohen, M., Soede, R. D., Brandt, R., Firtel, R. A., Dostmann, W., et al. (1993). Cell-permeable non-hydrolyzable cAMP derivatives as tools for analysis of signaling pathways controlling gene regulation in Dictyostelium. J. Biol. Chem. 268, 6323–6331.
Schindler, R. F., Poon, K. L., Simrick, S., and Brand, T. (2012). The Popeye domain containing genes: essential elements in heart rate control. Cardiovasc. Diagn. Ther. 2, 308–319. doi: 10.3978/j.issn.2223-3652.2012.12.01
Schmitz, A. L., Schrage, R., Gaffal, E., Charpentier, T. H., Wiest, J., Hiltensperger, G., et al. (2014). A cell-permeable inhibitor to trap Galphaq proteins in the empty pocket conformation. Chem. Biol. 21, 890–902. doi: 10.1016/j.chembiol.2014.06.003
Schwabe, U., Miyake, M., Ohga, Y., and Daly, J. W. (1976). 4-(3-Cyclopentyloxy-4-methoxyphenyl)-2-pyrrolidone (ZK 62711): a potent inhibitor of adenosine cyclic 3',5'-monophosphate phosphodiesterases in homogenates and tissue slices from rat brain. Mol. Pharmacol. 12, 900–910.
Scicchitano, P., Carbonara, S., Ricci, G., Mandurino, C., Locorotondo, M., Bulzis, G., et al. (2012). HCN channels and heart rate. Molecules 17, 4225–4235. doi: 10.3390/molecules17044225
Seifert, R. (2014). Vidarabine is neither a potent nor a selective AC5 inhibitor. Biochem. Pharmacol. 87, 543–546. doi: 10.1016/j.bcp.2013.12.025
Seifert, R., Lushington, G. H., Mou, T. C., Gille, A., and Sprang, S. R. (2012). Inhibitors of membranous adenylyl cyclases. Trends Pharmacol. Sci. 33, 64–78. doi: 10.1016/j.tips.2011.10.006
Sellers, Z. M., Naren, A. P., Xiang, Y., and Best, P. M. (2012). MRP4 and CFTR in the regulation of cAMP and beta-adrenergic contraction in cardiac myocytes. Eur. J. Pharmacol. 68, 80–87. doi: 10.1016/j.ejphar.2012.02.018
Shaw, R. M., and Colecraft, H. M. (2013). L-type calcium channel targeting and local signalling in cardiac myocytes. Cardiovasc. Res. 98, 177–186. doi: 10.1093/cvr/cvt021
Shimizu-Albergine, M., Tsai, L. C., Patrucco, E., and Beavo, J. A. (2012). cAMP-specific phosphodiesterases 8A and 8B, essential regulators of Leydig cell steroidogenesis. Mol. Pharmacol. 81, 556–566. doi: 10.1124/mol.111.076125
Shui, B., Lee, J. C., Reining, S., Lee, F. K., and Kotlikoff, M. I. (2014). Optogenetic sensors and effectors: CHROMus-the Cornell Heart Lung Blood Institute Resource for Optogenetic Mouse Signaling. Front. Physiol. 5:428. doi: 10.3389/fphys.2014.00428
Skalhegg, B. S., and Tasken, K. (2000). Specificity in the cAMP/PKA signaling pathway. Differential expression,regulation, and subcellular localization of subunits of PKA. Front. Biosci. 5, D678–D693.
Soderling, S. H., Bayuga, S. J., and Beavo, J. A. (1998). Cloning and characterization of a cAMP-specific cyclic nucleotide phosphodiesterase. Proc. Natl. Acad. Sci. U.S.A. 95, 8991–8996. doi: 10.1073/pnas.95.15.8991
Somekawa, S., Fukuhara, S., Nakaoka, Y., Fujita, H., Saito, Y., and Mochizuki, N. (2005). Enhanced functional gap junction neoformation by protein kinase A-dependent and Epac-dependent signals downstream of cAMP in cardiac myocytes. Circ. Res. 97, 655–662. doi: 10.1161/01.RES.0000183880.49270.f9
Sprenger, J. U., and Nikolaev, V. O. (2013). Biophysical Techniques for Detection of cAMP and cGMP in Living Cells. Int. J. Mol. Sci. 14, 8025–8046. doi: 10.3390/ijms14048025
Sprenger, J. U., Perera, R. K., Steinbrecher, J. H., Lehnart, S. E., Maier, L. S., Hasenfuss, G., et al. (2015). In vivo model with targeted cAMP biosensor reveals changes in receptor-microdomain communication in cardiac disease. Nat. Commun. 6, 6965. doi: 10.1038/ncomms7965
Standaert, M. L., Galloway, L., Karnam, P., Bandyopadhyay, G., Moscat, J., and Farese, R. V. (1997). Protein kinase C-zeta as a downstream effector of phosphatidylinositol 3-kinase during insulin stimulation in rat adipocytes. Potential role in glucose transport. J. Biol. Chem. 272, 30075–30082. doi: 10.1074/jbc.272.48.30075
Stierl, M., Stumpf, P., Udwari, D., Gueta, R., Hagedorn, R., Losi, A., et al. (2011). Light modulation of cellular cAMP by a small bacterial photoactivated adenylyl cyclase, bPAC, of the soil bacterium Beggiatoa. J. Biol. Chem. 286, 1181–1188. doi: 10.1074/jbc.M110.185496
Sudo, T., Tachibana, K., Toga, K., Tochizawa, S., Inoue, Y., Kimura, Y., et al. (2000). Potent effects of novel anti-platelet aggregatory cilostamide analogues on recombinant cyclic nucleotide phosphodiesterase isozyme activity. Biochem. Pharmacol. 59, 347–356. doi: 10.1016/S0006-2952(99)00346-9
Sun, B., Li, H., Shakur, Y., Hensley, J., Hockman, S., Kambayashi, J., et al. (2007). Role of phosphodiesterase type 3A and 3B in regulating platelet and cardiac function using subtype-selective knockout mice. Cell. Signal. 19, 1765–1771. doi: 10.1016/j.cellsig.2007.03.012
Tang, C. M., and Insel, P. A. (2004). GPCR expression in the heart; “new” receptors in myocytes and fibroblasts. Trends Cardiovasc. Med. 14, 94–99. doi: 10.1016/j.tcm.2003.12.007
Tang, T., Gao, M. H., Lai, N. C., Firth, A. L., Takahashi, T., Guo, T., et al. (2008). Adenylyl cyclase type 6 deletion decreases left ventricular function via impaired calcium handling. Circulation 117, 61–69. doi: 10.1161/CIRCULATIONAHA.107.730069
Tang, T., Lai, N. C., Hammond, H. K., Roth, D. M., Yang, Y., Guo, T., et al. (2010). Adenylyl cyclase 6 deletion reduces left ventricular hypertrophy, dilation, dysfunction, and fibrosis in pressure-overloaded female mice. J. Am. Coll. Cardiol. 55, 1476–1486. doi: 10.1016/j.jacc.2009.11.066
Tang, T., Lai, N. C., Roth, D. M., Drumm, J., Guo, T., Lee, K. W., et al. (2006). Adenylyl cyclase type V deletion increases basal left ventricular function and reduces left ventricular contractile responsiveness to beta-adrenergic stimulation. Basic Res. Cardiol. 101, 117–126. doi: 10.1007/s00395-005-0559-y
Tarpey, S. B., Sawmiller, D. R., Kelly, C., Thompson, W. J., and Townsley, M. I. (2003). Phosphodiesterase 3 activity is reduced in dog lung following pacing-induced heart failure. American journal of physiology. Lung Cell. Mol. Physiol. 284, L766–L773. doi: 10.1152/ajplung.00373.2002
Taskén, K. A., Collas, P., Kemmner, W. A., Witczak, O., Conti, M., and Tasken, K. (2001). Phosphodiesterase 4D and protein kinase a type II constitute a signaling unit in the centrosomal area. J. Biol. Chem. 276, 21999–22002. doi: 10.1074/jbc.C000911200
Tepe, N. M., Lorenz, J. N., Yatani, A., Dash, R., Kranias, E. G., Dorn, G. W., et al. (1999). Altering the receptor-effector ratio by transgenic overexpression of type V adenylyl cyclase: enhanced basal catalytic activity and function without increased cardiomyocyte beta-adrenergic signalling. Biochemistry 38, 16706–16713. doi: 10.1021/bi991619k
Tesmer, J. J., Sunahara, R. K., Gilman, A. G., and Sprang, S. R. (1997). Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsalpha.GTPgammaS. Science 278, 1907–1916. doi: 10.1126/science.278.5345.1907
Tesmer, J. J., Sunahara, R. K., Johnson, R. A., Gosselin, G., Gilman, A. G., and Sprang, S. R. (1999). Two-metal-Ion catalysis in adenylyl cyclase. Science 285, 756–760. doi: 10.1126/science.285.5428.756
Tornøe, C. W., Christensen, C., and Meldal, M. (2002). Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 67, 3057–3064. doi: 10.1021/jo011148j
Trehan, A., Rotgers, E., Coffey, E. T., Huhtaniemi, I., and Rivero-Müller, A. (2014). CANDLES, an assay for monitoring GPCR induced cAMP generation in cell cultures. Cell Commun. Signal. 12:70. doi: 10.1186/s12964-014-0070-x
Tsalkova, T., Mei, F. C., Li, S., Chepurny, O. G., Leech, C. A., Liu, T., et al. (2012). Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc. Natl. Acad. Sci. U.S.A. 109, 18613–18618. doi: 10.1073/pnas.1210209109
Tsvetanova, N. G., and von Zastrow, M. (2014). Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat. Chem. Biol. 10, 1061–1065. doi: 10.1038/nchembio.1665
Uhler, M. D., Chrivia, J. C., and McKnight, G. S. (1986). Evidence for a second isoform of the catalytic subunit of cAMP-dependent protein kinase. J. Biol. Chem. 261, 15360–15363.
Ukena, D., Shamim, M. T., Padgett, W., and Daly, J. W. (1986). Analogs of caffeine: antagonists with selectivity for A2 adenosine receptors. Life Sci. 39, 743–750. doi: 10.1016/0024-3205(86)90023-8
Van Bogaert, P. P., and Pittoors, F. (2003). Use-dependent blockade of cardiac pacemaker current (If) by cilobradine and zatebradine. Eur. J. Pharmacol. 478, 161–171. doi: 10.1016/j.ejphar.2003.08.083
Vandeput, F., Wolda, S. L., Krall, J., Hambleton, R., Uher, L., McCaw, K. N., et al. (2007). Cyclic nucleotide phosphodiesterase PDE1C1 in human cardiac myocytes. J. Biol. Chem. 282, 32749–32757. doi: 10.1074/jbc.M703173200
Vang, A. G., Ben-Sasson, S. Z., Dong, H., Kream, B., DeNinno, M. P., Claffey, M. M., et al. (2010). PDE8 regulates rapid Teff cell adhesion and proliferation independent of ICER. PLoS ONE 5:e12011. doi: 10.1371/journal.pone.0012011
Van Wagoner, D. R., Pond, A. L., Lamorgese, M., Rossie, S. S., McCarthy, P. M., and Nerbonne, J. M. (1999). Atrial L-type Ca2+ currents and human atrial fibrillation. Circ. Res. 85, 428–436. doi: 10.1161/01.RES.85.5.428
Vassilatis, D. K., Hohmann, J. G., Zeng, H., Li, F., Ranchalis, J. E., Mortrud, M. T., et al. (2003). The G protein-coupled receptor repertoires of human and mouse. Proc. Natl. Acad. Sci. U.S.A. 100, 4903–4908. doi: 10.1073/pnas.0230374100
Wahl-Schott, C., Fenske, S., and Biel, M. (2014). HCN channels: new roles in sinoatrial node function. Curr. Opin. Pharmacol. 15, 83–90. doi: 10.1016/j.coph.2013.12.005
Weise, M., Vettel, C., Spiger, K., Gilsbach, R., Hein, L., Lorenz, K., et al. (2015). A systemic Pasteurella multocida toxin aggravates cardiac hypertrophy and fibrosis in mice. Cell. Microbiol. 17, 1320–1331. doi: 10.1111/cmi.12436
Williams, C. (2004). cAMP detection methods in HTS: selecting the best from the rest. Nat. Rev. Drug Discov. 3, 125–135. doi: 10.1038/nrd1306
Willoughby, D., and Cooper, D. M. (2007). Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains. Physiol. Rev. 87, 965–1010. doi: 10.1152/physrev.00049.2006
Willoughby, D., and Cooper, D. M. (2008). Live-cell imaging of cAMP dynamics. Nat. Methods 5, 29–36. doi: 10.1038/nmeth1135
Wilson, M., Burt, A. R., Milligan, G., and Anderson, N. G. (1996). Wortmannin-sensitive activation of p70s6k by endogenous and heterologously expressed Gi-coupled receptors. J. Biol. Chem. 271, 8537–8540. doi: 10.1074/jbc.271.15.8537
Xiao, R. P. (2001). Beta-adrenergic signaling in the heart: dual coupling of the beta2-adrenergic receptor to G(s) and G(i) proteins. Sci. STKE 2001:re15. doi: 10.1126/stke.2001.104.re15
Xie, M., Rich, T. C., Scheitrum, C., Conti, M., and Richter, W. (2011). Inactivation of multidrug resistance proteins disrupts both cellular extrusion and intracellular degradation of cAMP. Mol. Pharmacol. 80, 281–293. doi: 10.1124/mol.111.071134
Yang, G., McIntyre, K. W., Townsend, R. M., Shen, H. H., Pitts, W. J., Dodd, J. H., et al. (2003). Phosphodiesterase 7A-deficient mice have functional T cells. J. Immunol. 171, 6414–6420. doi: 10.4049/jimmunol.171.12.6414
Yong, Q. C., Pan, T. T., Hu, L. F., and Bian, J. S. (2008). Negative regulation of beta-adrenergic function by hydrogen sulphide in the rat hearts. J. Mol. Cell. Cardiol. 44, 701–710. doi: 10.1016/j.yjmcc.2008.01.007
Yu, J., Wolda, S. L., Frazier, A. L., Florio, V. A., Martins, T. J., Snyder, P. B., et al. (1997). Identification and characterisation of a human calmodulin-stimulated phosphodiesterase PDE1B1. Cell. Signal. 9, 519–529. doi: 10.1016/S0898-6568(97)00046-6
Zaccolo, M., De Giorgi, F., Cho, C. Y., Feng, L., Knapp, T., Negulescu, P. A., et al. (2000). A genetically encoded, fluorescent indicator for cyclic AMP in living cells. Nat. Cell Biol. 2, 25–29. doi: 10.1038/71345
Zaccolo, M., and Movsesian, M. A. (2007). cAMP and cGMP signaling cross-talk: role of phosphodiesterases and implications for cardiac pathophysiology. Circ. Res. 100, 1569–1578. doi: 10.1161/CIRCRESAHA.106.144501
Zaccolo, M., and Pozzan, T. (2002). Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science 295, 1711–1715. doi: 10.1126/science.1069982
Zhang, G., Liu, Y., Ruoho, A. E., and Hurley, J. H. (1997). Structure of the adenylyl cyclase catalytic core. Nature 386, 247–253. doi: 10.1038/386247a0
Zhang, R., Khoo, M. S., Wu, Y., Yang, Y., Grueter, C. E., Ni, G., et al. (2005). Calmodulin kinase II inhibition protects against structural heart disease. Nat. Med. 11, 409–417. doi: 10.1038/nm1215
Keywords: GPCR, resonance energy transfer, phosphodiesterase, protein kinase A (PKA), Cyclic AMP
Citation: Boularan C and Gales C (2015) Cardiac cAMP: production, hydrolysis, modulation and detection. Front. Pharmacol. 6:203. doi: 10.3389/fphar.2015.00203
Received: 31 July 2015; Accepted: 03 September 2015;
Published: 01 October 2015.
Edited by:
Apostolos Zarros, University of Glasgow, UKReviewed by:
Jinsong Bian, National University of Singapore, SingaporeJoshua B. Rubin, Washington University School of Medicine, USA
Copyright © 2015 Boularan and Gales. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cédric Boularan and Céline Gales, Institut des Maladies Métaboliques et Cardiovasculaires - I2MC, Institut National de la Santé et de la Recherche Médicale UMR1048, 1, Avenue Jean-Poulhès - BP84225, 31432 Toulouse, France, cedric.boularan@inserm.fr; celine.gales@inserm.fr