
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr., 16 September 2020
Sec. Pediatric Hematology and Hematological Malignancies
Volume 8 - 2020 | https://doi.org/10.3389/fped.2020.00586
This article is part of the Research TopicPrevention in Acute Leukemias in ChildrenView all 5 articles
 Carolina Molina Garay1Karol Carrillo Sánchez1Luis Leonardo Flores Lagunes1Marco Jiménez Olivares1Anallely Muñoz Rivas1
Carolina Molina Garay1Karol Carrillo Sánchez1Luis Leonardo Flores Lagunes1Marco Jiménez Olivares1Anallely Muñoz Rivas1 Beatríz Eugenia Villegas Torres1Hilario Flores Aguilar2
Beatríz Eugenia Villegas Torres1Hilario Flores Aguilar2 Juan Carlos Núñez Enríquez3
Juan Carlos Núñez Enríquez3 Elva Jiménez Hernández4
Elva Jiménez Hernández4 Vilma Carolina Bekker Méndez5José Refugio Torres Nava6
Vilma Carolina Bekker Méndez5José Refugio Torres Nava6 Janet Flores Lujano3Jorge Alfonso Martín Trejo7
Janet Flores Lujano3Jorge Alfonso Martín Trejo7 Minerva Mata Rocha3
Minerva Mata Rocha3 Aurora Medina Sansón8Laura Eugenia Espinoza Hernández4José Gabriel Peñaloza Gonzalez9Rosa Martha Espinosa Elizondo10Luz Victoria Flores Villegas11Raquel Amador Sanchez12Maria Luisa Pérez Saldívar3
Aurora Medina Sansón8Laura Eugenia Espinoza Hernández4José Gabriel Peñaloza Gonzalez9Rosa Martha Espinosa Elizondo10Luz Victoria Flores Villegas11Raquel Amador Sanchez12Maria Luisa Pérez Saldívar3 Omar Alejandro Sepúlveda Robles13Haydeé Rosas Vargas13
Omar Alejandro Sepúlveda Robles13Haydeé Rosas Vargas13 Angélica Rangel López14
Angélica Rangel López14 María Lilia Domínguez López15
María Lilia Domínguez López15 Ethel Awilda García Latorre15Elba Reyes Maldonado15
Ethel Awilda García Latorre15Elba Reyes Maldonado15 Patricia Galindo Delgado16Juan Manuel Mejía Aranguré3,14
Patricia Galindo Delgado16Juan Manuel Mejía Aranguré3,14 Carmen Alaez Verson1*
Carmen Alaez Verson1*Background: Acute myeloid leukemia (AML) is the second most frequent leukemia in childhood. The FLT3 gene participates in hematopoietic stem cell proliferation. FLT3 mutations are recurrent in AML and influence prognosis. In Mexican pediatric AML patients, FLT3 mutational profile, and their clinical impact have not been evaluated.
Aim of the study: This study aimed to identify the profile of FLT3 mutations in pediatric patients with de novo AML and to assess their possible influence on overall survival (OS) and other clinical features.
Methods: Massive parallel target sequencing of FLT3 was performed in 80 patients.
Results: FLT3 mutations [internal tandem duplication (ITD) or tyrosine kinase domain (TKD)] were identified in 24% of them. OS was significantly lower in FLT3POS cases than in FLT3NEG (p = 0.03). The average OS for FLT3POS was 1.2 vs. 2.2 years in FLT3NEG. There were no significant differences in the children's sex, age, percentage of blasts in bone marrow aspirate, or white blood cell count in peripheral blood at diagnosis between both groups. No differences were identified stratifying by the mutational load (high > 0.4) or type of mutation. The negative effect of FLT3 mutations was also observed in patients with acute promyelocytic leukemia (APL).
Conclusions: FLT3 mutational profile is described in Mexican pediatric AML patients for the first time. Mutated FLT3 negatively impacts the outcome of AML patients, even considering the APL group. The clinical benefit from treatment with tyrosine kinase inhibitors in the FLT3POS pediatric patients needs to be assessed in clinical trials. FLT3 testing may contribute to better risk stratification in our pediatric AML patients.
Genomic investigations of acute myeloid leukemia (AML) have demonstrated that several genes are recurrently mutated, leading to the identification of new biomarkers. Genetic screening of molecular abnormalities plays a significant role in prognostic categorization and definition of treatment strategies in AML (1, 2). FLT3 is a class III family receptor tyrosine kinase expressed in hematopoietic stem cells and has essential roles in cell survival and proliferation. FLT3 gene mutations are among the most frequently observed in AML, which occurs in approximately one-third of patients with de novo AML. They have been associated with the clinical prognosis, treatment response, and survival of patients (3).
Two types of FLT3 activating mutations have been identified in AML. Internal tandem duplications (ITDs) affect the juxtamembrane (JM) domain or tyrosine kinase domain 1 (TKD1) of the FLT3 receptor and point mutation preset in the TKD2 (4). Both types of mutations promote ligand-independent auto-phosphorylation and constitutive activation of the receptor. This unregulated activation impairs normal hematopoiesis and contributes to leukemogenesis (5).
ITD mutations are the most frequent and are considered drivers in AML. At diagnosis, the FLT3–ITD clone may possess a growth advantage. This clone becomes dominant at relapse through a clonal expansion process (6, 7). Higher relapse rates and more reduced survival have been associated with FLT3–ITD in previous studies, largely depending on the allelic ratio (AR) of the mutations and the presence of mutations in other genes (8). The clinical impact of FLT3–TKD mutations is less clear (9).
FLT3 genetic testing, detecting both ITD and TKD mutations, is recommended by the National Comprehensive Cancer Network (10) and European Leukemia Net (3) for diagnostic workup of patients with AML, to identify those who may benefit from different targeted treatment options. FLT3 screening is not routinely performed in Mexico. Epidemiological data about the FLT3 mutational profile and its clinical impact in Mexican AML patients are minimal. Studies including a small number of de novo and secondary AML patients (children and adults) have reported a prevalence between 13 and 20% of FLT3 mutation using polymerase chain reaction (PCR)-based technologies (11).
The landscape of somatic mutations in pediatric AML was shown to be markedly different from adults, suggesting different biology and etiology for adult and childhood AML (12). These findings stress the importance of analyzing the mutational profile and evaluate the clinical impact of genetic alteration separately in both groups of patients. The purpose of this study was to assess the prevalence and the influence of FLT3 mutations on the outcome of pediatric AML de novo patients. AML is one of the main types of cancer in Mexican children; its incidence has a trend toward an increase in the last year, and mortality is higher than in developed countries despite that the same chemotherapy protocols are used (13).
Eighty patients with de novo AML, coming from eight different public hospitals, were included. Patients were diagnosed between March 2010 and March 2018. AML diagnosis was performed at each institution using bone marrow aspirate and immunophenotype. The bone marrow samples analyzed in the present study were obtained at the time of the diagnosis, before treatment initiation, and submitted to the Mexican Inter-Institutional Group for Identifying Childhood Leukemia Causes in Mexico City.
A predefined set of data was collected from medical charts, including the child's sex, age, white blood cell (WBC) count in peripheral blood, and percentage of blasts in the bone marrow at diagnosis, French-American-British (FAB) classification, and treatment protocol.
The ethics and scientific review boards of the National Institute of Genomic Medicine, Mexico City, Mexico, approved this study (document number 28-2015-1). All human samples and clinical information were approved for its use in the present study. The child's parents signed the informed consent obtained in accordance with the Declaration of Helsinki.
DNA was extracted using Maxwell® 16 Blood DNA Purification Kit (Promega, Madison, WI, USA) according to the manufacturer's recommendations. The purity and concentration of the DNA samples were measured using NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and Qubit fluorometer (Thermo Fisher Scientific, Waltham, MA, USA).
Sequencing of the exons 13–15 and 20 of the FLT3 gene was performed using the multigene panel “Myeloid solution” by SOPHiA Genetics (Sophia Genetics SA, Saint Sulpice, Switzerland). Library preparation and sequencing were performed according to the manufacturer's protocol. Capture-base enriched libraries were pooled (12 indexed libraries per pool) and sequenced on a MiSeq system using version 3 chemistry (Illumina, San Diego CA, USA). Sequencing data were analyzed with the Sophia DDM® software version 5.2.7.1 (Sophia Genetics SA, Saint Sulpice, Switzerland). Sequencing deep was higher than 1,000 × for all the target regions.
Variant fraction (VF) was computed by dividing the number of mutation-supporting reads by the total number of reads at the mutation site. AR was calculated by dividing the number of reads corresponding to the mutation by the number of reads according to the wild-type allele in the mutation site.
The comparison of proportions between different groups was made using chi2 or two-sided Fisher's exact test for dichotomic variables, when appropriate. A non-parametric Mann–Whitney U-test was used for continuous variables, p < 0.05 was considered as statistically significant. All calculations were performed using the SPSS software package, SPSS Version 21 (Chicago, IL, USA).
The Kaplan–Meier method (14) was used for assessing overall survival (OS). The log-rank test was used to evaluate differences between survival distributions with a 95% confidence interval (CI). The OS time was calculated from the day of diagnosis confirmation until either the last follow-up or patient's death from any cause. Patients who did not experience an event were censored at the time of the last follow-up. Those who did not attend a follow-up were censored at their date of the last known contact.
The demographic, clinical, and main biological features of the patients are shown in Table 1. Fifty-five percent of the cases were male. The median age at diagnosis was 9.3 (range 0.4–17.5) years; the age at diagnosis was very similar in both genders. Acute promyelocytic leukemia (APL), M3, was the most prevalent subtype (36.3%), followed by the M2 (33.8%). M0, M5, and M6 were the less common subtypes. APL seems to occur more frequently in Hispanic populations than in other ethnic groups (15).
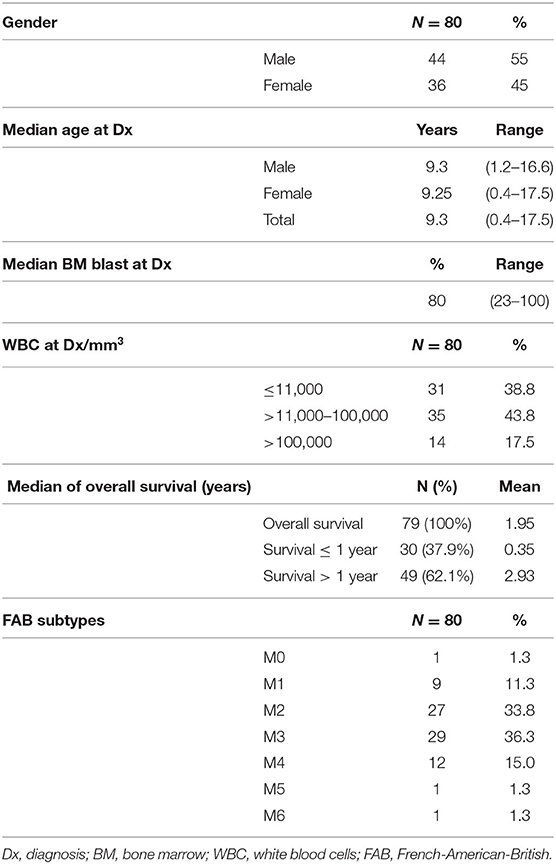
Table 1. Demographic and biological characteristics of the patients.
Almost 38% of the patients do not survive more than 1 year after diagnosis. The median OS in the study population was 1.95 years.
Patients were treated according to one of the following four protocols: BFM-1998, BFM-2001 (Berlin-Frankfurt-Münster 1998 and 2001), NOPHO-AML93 (Nordic Society of Pediatric Hematology and Oncology), or PETHEMA-APL-05 (Spanish Program of Treatments in Hematology). Treatment information is summarized in Supplementary Table 1. Noteworthy, all APL patients were treated according to the PETHEMA-APL-05 protocol. For the M2 group, BFM-1998, NOPHO-AML93, and BFM-2001 were used, while for M4 and M1, only BFM-1998, and NOPHO-AML-93 were applied. None of the patients received allogeneic or autologous bone marrow transplant.
Since chemotherapeutic drugs affect outcomes, we assess the impact of the treatment protocol in the OS (see Supplementary Figures 1A,B). No significant differences were found among patients treated with NOPHO-AML93, BFM-2001, or BFM-1998, although those patients receiving NOPHO-AML93 seem to have the best long-term outcome while BFM1998 the worst. Patients treated with these protocols were considered as a single group for comparison with the PETHEMA-APL-05 treated group. OS was significantly higher in APL patients in contrast to those children from other FAB subtypes and treated with any different therapeutic combination (Supplementary Figure 1B).
Mutations in FLT3 were identified in 23.7% (19/80) of the patients, being more frequent in females (52.6%). The mutational profile is shown in Table 2. ITD mutations were present in 52.6% (10/19) of the FLT3POS cases, with VFs ranging from 2.4 to 64.9%. The range of affected amino acids expanded from position 584 to 613. In one patient, in-frame deletion of amino acid 600 in the JM domain was observed, without any insertions or duplications of nucleotides. This patient was included in the ITD group since the mutation may affect the functioning of the JM domain.
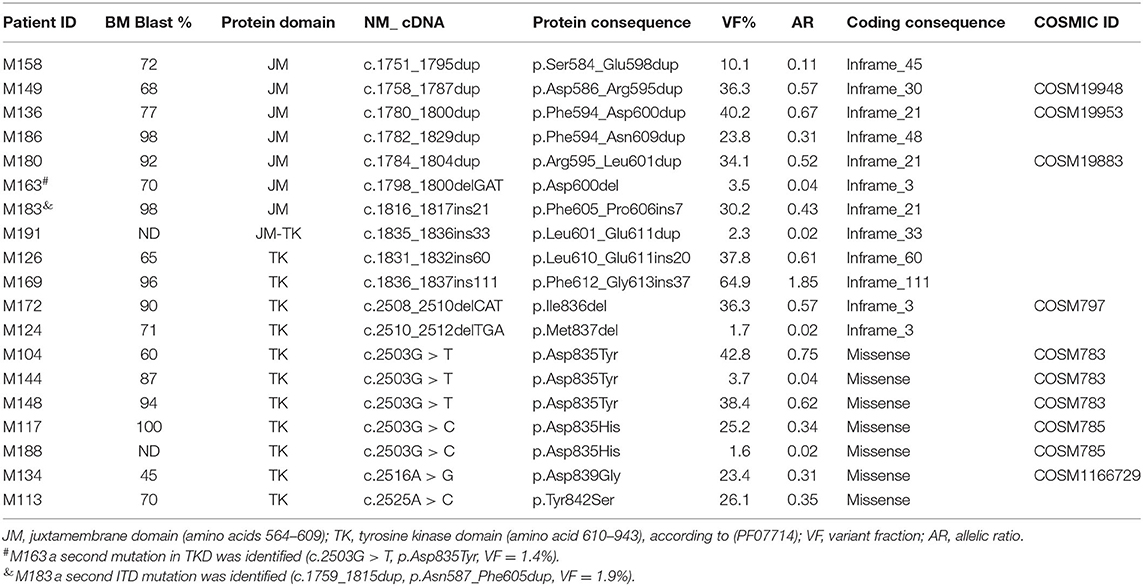
Table 2. Mutational profile of FLT3 in pediatric AML patients.
Missense or small deletion mutations in the TKD region were present in 47.4% (9/19) of the FLT3POS patients with VFs ranging from 1.6 to 42.8%. Changes in position 835 of the TKD domain were the most frequent. Two different amino acid changes were observed: p.Asp835Tyr in three cases and p.Asp835His in two. Additionally, single nucleotide variations affecting positions 839 and 842 of TKD were observed in one patient each. Two in-frame deletions affecting amino acid 836 and 837 were also found. A second FLT3 mutation, at a lower VF, was identified in two patients (M183 and M163) (Table 2, Figure 1).
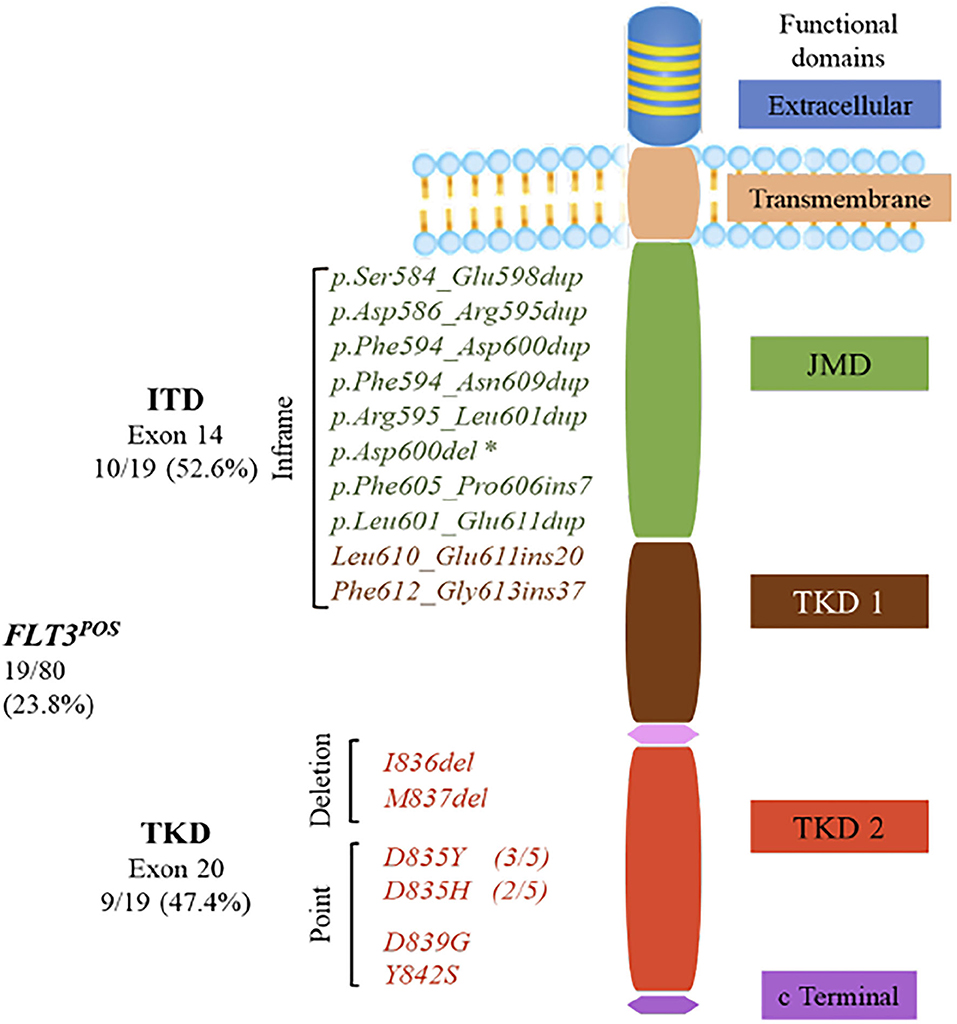
Figure 1. FMS-like tyrosine kinase 3 (FLT3) gene encodes a 993-amino-acid protein, consisting of five functional domains: an immunoglobulin-like extracellular domain, a transmembrane domain, a juxtamembrane domain (JMD), one interrupted tyrosine kinase domain (TKD), and a small C-terminal domain. Internal tandem duplications (ITDs) were the most common mutations in the FLT3POS patients with insertions from 21 to 111 bp that mainly occurred in JMD, although two ITDs were found in the β1 sheet of TKD1. *Asp600del was positioned in exon 14 of the JMD, but it is not considered as an ITD, because no nucleotide duplication occurred. TKD activating mutations were located within the activation loop of the TKD2. Most of them were single-nucleotide variations resulting in amino acid changes at positions 835, 839, and 842. Two activating mutations caused by deletions were also identified in the TKD2.
Age at diagnosis, sex, percentage of blasts in bone marrow, and FAB subtype distribution was compared between FLT3POS and negative patients. The results are displayed in Table 3.
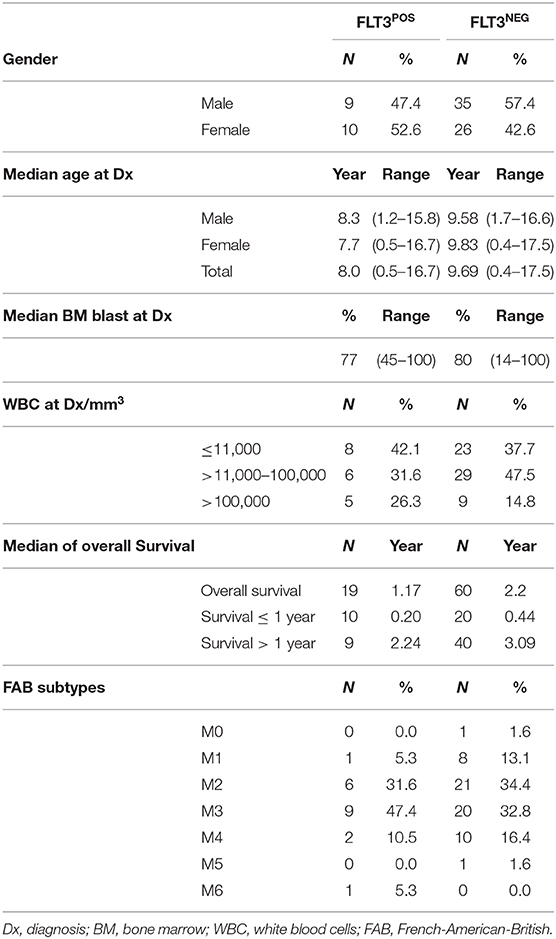
Table 3. Distribution of demographic and biological features between FLT3POS and negative patients.
No significant differences were found. However, the median age at diagnosis was lower in the FLT3POS group. When it was stratified by gender, FLT3POS female patients were 2.1 years younger at diagnosis than FLT3NEG females. In male FLT3POS patients, the disease was diagnosed 0.4 years earlier than in the FLT3NEG negative. None of these differences reached statistical significance.
OS results, according to the FLT3 status, are shown in Figures 2A,B. The OS was significantly decreased among patients with FLT3 mutation (Figure 2A). The median of OS time was 1.2 (0.05–3.1) years in the FLT3POS vs. 2.2 (0.05–7.6) years, in negative children (Table 3). When ITD or TKD mutations were considered separated (Figure 2B), no differences were observed between survival curves from ITD or TKD positive patients vs. FLT3NEG patients. The OS analysis was calculated with 79 patients due to the date for the last contact or death was not available in one patient.
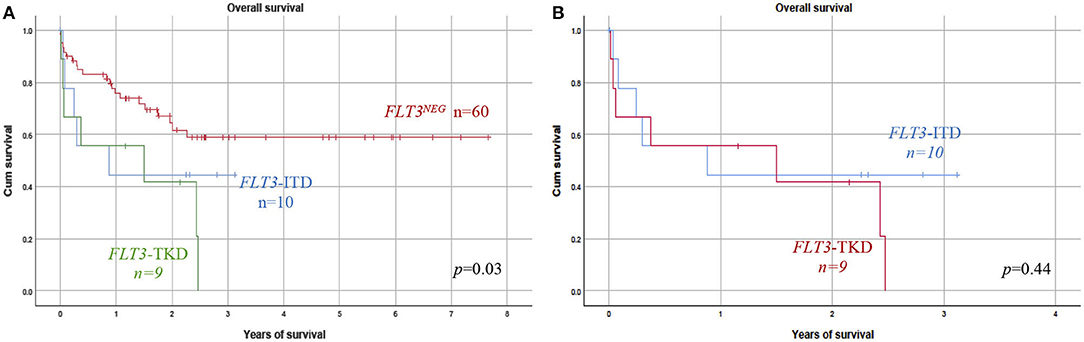
Figure 2. Prognosis impact of FLT3 mutation. (A) Overall survival (OS) was analyzed considering FLT3–ITD or FLT3–TKD patients vs. the FLT3NEG negative group. FLT3POS patients (TKD or ITD) showed a significantly reduced OS as compared with those without FLT3 mutation. (B) No significant difference in OS was found between FLT3–ITD and TKD groups of patients. ITD, internal tandem duplication; TKD, tyrosine kinase domain.
Since the allelic burden of the FLT3 mutation has been shown to influence the outcome in adults, we stratified both FLT3–ITD and FLT3POS (the type of mutation) patients according to their AR, using 0.4 as the cutoff value (16). The mutational burden did not influence OS, considering the FLT3–ITD group or the whole FLT3POS group.
The impact of FLT3 mutations was evaluated in the APL(M3) group of patients. The results are shown in the Supplementary Figure 2. APL-FLT3POS patients had a significantly lower global survival than the APL-FLT3NEG group. The OS of APL-FLT3POS was similar to the OS observed for other FAB subtypes grouped (p = 0.014).
Mutational profiling of patients with AML is part of routine diagnostic workups for both de novo and relapsed AML patients in centers from developed countries. In Mexico, the FLT3 mutational profile is not routinely performed in most public institutions; therefore, little epidemiological data exist about prevalence and heterogeneity of FLT3 mutations and their influence on clinical evolution in both AML adult and pediatric patients.
This analysis of de novo AML cases represents the most extensive cohort of Mexican children and adolescents analyzed for FLT3 mutational status. Eighty pediatric de novo AML patients, diagnosed between March 2010 and March 2018, were examined. The use of next-generation sequencing technology provides additional information about the length, position, and sequence of the identified mutations, revealing details about the structural variability of ITD and TKD mutations and the presence of additional clones.
FLT3–ITD or TKD mutations combined were present in 24% of the studied samples. To the best of our knowledge, there is no other study analyzing only Mexican AML children. Data about FLT3 mutation are scarce for Latin-American AML pediatric patients. The reported prevalence in Costa Rica and Argentina is lower than in our patients (14.3% in 14 patients and 15.2% in 92 patients, respectively) (17). The results from Brazil are very similar to those from Mexico, with 23.3% reported in an extensive collaborative analysis, including a large number of pediatric patients (18).
More extensive analysis has been conducted in pediatric AML patients from the USA. In 2006, 19% of FLT3POS cases (FLT3–ITD with 12% and FLT3-TKD with 6.7%) were detected in a group of 630 de novo AML pediatric patients, belonging to the Children's Cancer Group. The authors suggested that, in general, the frequency of mutated FLT3 is 15% lower in the pediatric population than in adults (19). However, in 2015, Tarlock et al. (20) reported a prevalence of 28% of FLT3POS cases in a group of 799 pediatric AML patients (16.02% with ITDs, 4.6% with TKDs, and 7.6% with novel mutation).
Differences in sensitivity between the FLT3 detection methods used could be contributing to the lower prevalence of mutation observed in some of the previous studies compared with ours. Molecular methods based on PCR were employed in all of them. These methods can provide the length, but not the exact position or the sequence of the insertion, and have lower sensitivity than massive parallel sequencing technology. Mutations with meager allelic fraction could be missed by PCR-based methodologies. Interestingly, in this study, five patients had a VF lower than 4%. If these patients are removed from the prevalence calculation, the frequency of FLT3 mutation turns to be only 17.5%.
FLT3 mutations have been reported with a higher prevalence in adults than in children, although children have a mutational profile not observed in adults (12, 20). The prevalence in adults with AML ranges from ~20% (21) to 30% (22, 23). Reports from Europe, Japan, and China have shown a prevalence of 27.7% (24), 24.9% (23), and 21.23% (21), respectively.
The prevalence of FLT3 mutation seems to be lower in adult AML Hispanic patients than in other groups, although technical limitations may have contributed to the observed differences (22). Ruiz Arguelles et al. reported 13% in 31 patients, while Arana Trejo et al. found 15% of FLT3 mutated in 21 patients; both studies were done in Mexican patients (11). A collaborative study, including patients from referral centers in Mexico and Colombia, reported only 20% of FLT3POS patients. They analyzed 138 patients with de novo or secondary AML (11). This study also included the patients previously reported by Arana Trejo and Ruiz Arguelles (11). In AML adults from Argentina, Sánchez et al. (25) reported 20.6%, while in Brazil, 26.3% was found (26).
Although the frequency of FLT3 mutation was higher in the female group, no statistically significant difference in gender distribution was observed, similar to what has been found in most series (18). No association was seen between age at diagnosis, WBC, or blast percentage at bone marrow and the presence of FLT3 mutations.
ITD mutation was present in 52.6% (10/19) of the patients. The length of the alteration varies from 21 to 111 bp, being 21 bp, the only size observed in more than one patient. The size of the duplicated region ranges from 15 to 174 bp (19), but up to 400 bp has been previously reported (3, 27).
In all cases, FLT3–ITD occurred in multiples of three nucleotides, preserving the reading frame of the transcript, as has been observed in other studies (3). Evaluation of the role of ITD size in clinical prognosis has opposite results. Some authors showed that increasing ITD size was associated with an adverse outcome (28), while others found no association (29). The number of patients in our study was too small to perform this analysis.
Furthermore, it has been suggested that ITD mutations always affect at least one amino acid residue from codon 591–597, with codon 597 being the most duplicated (30, 31). We do not confirm this observation. In our series, ITD mutations expand from codon 584–613. ITDs involving codons 591–597 were present only in 50% of the patients with this type of mutation. Only four of them had the amino acid 597 included in the duplication. For the other 50% of the ITD patients, the mutation affected codons 600–613.
Most of the ITD mutations were located only at the JM domain (6/9 patients). However, in three cases, the mutation was found on the β1 sheet of the first kinase domain, corresponding to amino acids 610–615. In one patient (M191, p.Leu601_Glu611dup), the duplication alters both the JM domain and the first residues of the β1 sheet in TKD. In the other two patients (M126: p.Leu610_Glu611ins20 and M169: p.Phe612_Gly613ins37), the integration occurred exclusively in the region of the β1 sheet of the first kinase domain. ITDs affecting the β1 sheet represent 22.2% of the ITD patients in this study, very similar to the frequency found in other series. No ITDs affecting different regions of the TKD were observed, although they have been described in a proportion of ITD patients (4).
In one patient, the in-frame loss of three nucleotides, producing Asp600 deletion in the JM domain, was observed. That mutation had a low VF. It does not correspond to the classic insertion/duplication occurring in the JM domain and has not been previously reported. It has been suggested that even a minimal mutation would be able to disrupt the intrinsic negative regulatory effects of the JM domain in preventing dimerization without ligand stimulation and would be sufficient to lead to auto-phosphorylation in cis (32). It could be hypothesized that this small deletion originates gain of function of the FLT3 receptor, similar to ITD involving a more significant number of amino acid residues.
Revealing the heterogeneity of ITD mutations seems to be important in the clinical setting since it could be one of the factors leading to variability in the treatment response of the FLT3–ITD AML patients. Location of the ITD in FLT3 influences the sensitivity to tyrosine kinase inhibitors as well as disease progression in mice (33). TKD-ITDs have been associated with a worse survival prognosis and to chemotherapy resistance in comparison with those with JM-ITD (30). In this study, only two patients with ITD-TKD were found; both had a high mutational burden. One of them had the highest mutational burden observed in our study (1.85 in patients M169). Marhäll et al. (34) showed that TKD-ITD and JM-ITD mutations display a similar oncogenic potential and that it is higher than the one from the D835Y point mutation located in the activation loop of the TKD.
FLT3-TKD mutations represent 47.4% (9/19) of the FLT3POS cases, and 11.25% (9/80) of the total of patients, a higher prevalence than the 7.7% reported for AML adult patients (35). Six different mutations were observed; two of them were not listed in the COSMIC database (as of June 2020): p.Tyr842Ser, and, p.Met837del. Mutations within the activation loop of the second TKD were the most frequent (five of nine TKD cases). Asp835Tyr, considered the most common substitution in position 835, was found in only three patients, while Asp835His was identified in the other two cases.
FLT3-Asp835 mutations lead to constitutive activation of FLT3 (36) and have been reported in 7–14% of AML (3), 3% of myelodysplastic syndromes, and 3% of acute lymphoblastic leukemia cases (37). Other reported substitutions, including Asp835Val, Asp835Glu, and Asp835Asn, were not found in our patients. The activating mutation p.Ile836del, identified in one patient, was described for the first time by Thiede et al. (35) in 13/979 AML adult patients.
The mutation p.Tyr842Ser was identified in one patient with VF = 26%. p.Tyr842Cys affecting the same codon has been associated with resistance to midostaurin, sunitinib, sorafenib, lestaurtinib, KW2449, and AGS324 (38). The main challenge with FLT3 inhibitors and other target therapies is to overcome primary and secondary resistance. The possibility that Tyr842Ser could be related to primary resistance should be considered in the clinical setting. Since none of our patients were treated with FLT3 inhibitors, the possible role of this mutation in response cannot be evaluated.
In an extensive series of pediatric AML patients, Bolouri et al. (12) reported mutations not found in adults with AML. New FLT3 mutations affecting residues 451, 444, 491, 676, 680, and 941 were described, some of them with functional implications and influencing response to conventional therapy (20). If these mutations exist in our patients, they could not be detected due to target sequencing strategy limitations. Only mutations located between amino acids 534–647 and amino acids 807–847 could be detected. A full gene sequencing strategy is the best option to explore somatic mutations in FLT3 and other genes too.
Two patients (22.2% of the FLT3POS group) presented a second mutation at a lower VF, suggesting the presence of additional clones at diagnosis. A similar observation was reported by Kottaridis et al. (39) who found more than one mutation in 23% of FLT3–ITD AML patients, with up to five different FLT3–ITD clones of various sizes, insertion sites, and ARs identified. A worse OS has been observed in patients with more than one clone (39).
OS was analyzed considering ITD and TKD mutations separated; both groups of patients showed lower survival than did the FLT3NEG group (Figure 2). The influence of TKD or ITD on prognosis was very similar; no significant difference in OS between both groups of patients was observed.
The FLT3–ITD mutations have been previously related to a poor outcome in both adults (40, 41) and pediatric patients (42, 43). Wu et al. (44) performed a meta-analysis, including 1661 pediatric patients with AML. Patients with FLT3–ITD mutation had an inferior OS [hazard ratio (HR) = 2.19 (1.60–3.01), p < 0.001] in comparison with patients without FLT3 mutation. The impact of TKD mutations was not analyzed in this paper. The author suggested that FLT3–ITDs produce a significantly negative prognostic effect in pediatric patients with AML.
There is no clear explanation of why FLT3–ITD could have such a negative impact. FLT3–ITD mutations contribute to increased production of reactive oxygen species (45), which leads to DNA oxidative damage, increases DNA double-strand breaks, and mistakes in the repair mechanisms starting a cycle of genomic instability. The existence of genomic instability leading to a cytogenetic evolution in AML is supported by the acquisition of new structural chromosomal abnormalities by leukemic cells between diagnosis and relapse. McCormick et al. (46) showed that cytogenetic evolution was more frequent among FLT3–ITD AML patients. Ten out of 14 FLT3–ITD AML patients acquired new cytogenetic and structural chromosomal abnormalities, compared with seven out of 21 FLT3 wild-type AML patients. This observation has been confirmed by others analyzing FLT3–ITD AML patients at relapse (47). Other genomic features supporting genomic instability in FLT3–ITD patients have been shown, including microsatellite instability (48, 49), shorter telomeres (50), and high frequency of somatic mutations (51). We speculate that acquisition of additional poor-prognosis mutation is favored by the presence of genomic instability in FLT3–ITD patients and might be one of the factors contributing to the detrimental effect of FLT3 mutations on survival (52).
Genomic instability is a hallmark of cancer, and high rates of genomic instability are present in other myeloid malignancies containing activated tyrosine kinase pathways, such as BCR/ABL in chronic myeloid leukemia, JAK2 in myeloproliferative neoplasias, and RAS mutations in myelodysplastic syndromes (45).
The effect of TKD mutations on prognosis has been mainly evaluated in adult patients and is considered controversial (3). Mead et al. (53) showed that patients with FLT3-TKD have a significantly more favorable prognosis than FLT3–ITD AML patients. In contrast, Whitman et al. (54) found that FLT3 D835/I836 mutations were associated with poor disease-free survival and a distinct gene-expression signature among younger adults. Our results suggest that both FLT3-TKD and ITD mutations have a similar negative effect on OS in pediatric patients. Still, additional factors, like cytogenetic background, mutations in other genes, and treatment, may be contributing to the negative outcome in the TKD group. For example, the number of patients no-APL is significantly higher in TKD than in the ITD group (77.8% TKD vs. 30% ITD; p Fisher = 0.046). Patients with other FAB subtypes had OS significantly lower than the APL group, treated with the PETHEMA-APL-05 (see Supplementary Figure 1B).
When the OS was considered, regardless of the subtype of FLT3 mutation, patients with FLT3 mutation had significantly lower OS than the FLT3NEG patients, supporting that constitutive activation of the FLT3 receptor, for any subtype of mutation, predisposes to high-risk disease. The negative effect of an FLT3 mutation was additionally supported by the analysis in the APL group of patients. All APL patients were treated with the same protocol. However, APL-FLT3POS patients had significantly worse OS that M3-FLT3NEG patients. Other studies have shown an adverse effect of mutated FLT3 in APL patients. Analyses of 205 adults and children found a significantly higher risk of death among APL patients with FLT3–ITD than in those without (HR = 11.74; 95% CI = 1.03–134.5), a reduced OS was also observed (15). Additionally, a higher relapse rate and a more reduced post-relapse survival have also been reported in the APL FLT3–ITD as compared with patients with APL FLT3 wild-type (55, 56).
Both subtypes of FLT3 mutation yield proteins that spontaneously dimerize, bypassing ligand-mediated activation. When they were transfected into murine cell lines, factor-independent growth originated (57). The analysis of 979 AML adult patients showed that both aberrations are also associated with similar clinical features: higher WBC and higher numbers of bone marrow blasts (35).
The impact of constitutive FLT3 activation has been evaluated in several murine models. FLT3–ITD expression originated a myeloproliferative disorder having different phenotypes and severity depending on the murine model. In a bone marrow transplant model, a fatal myeloproliferative neoplasm occurred (57), while in a knock-in FLT3–ITD model, a less severe human chronic myelomonocytic leukemia like disease resulted. FLT3 constitutive activation alone does not produce a full AML genotype; additional cooperating genetic alterations are needed in human and mouse for AML development (58).
FLT3–ITD is not expressed to the same level in all patients; differences in expression measured using the FLT3–ITD to wild-type AR also impact prognosis (3). It has been retrospectively shown that patients with a higher FLT3–ITD mutant-to-wild-type ratio have significantly worse outcomes than patients with a lower ratio, being 0.4 the cut-point in children and 0.5 in adults (59). In our ITD patients, the allelic burden ranged from 0.02 to 1.85; it was higher than 0.4 in 66.7% (6/9) of them. This study did not find that the mutational burden influences OS in the FLT3–ITD group or the whole FLT3POS group. A significant limitation is the small sample size impairing the statistical power of the analysis. The role of the FLT3 AR on the patient's outcome needs further clarification in prospective analysis with larger number of patients.
It has been suggested that when the FLT3–ITD AR is low, the presence of NPM1 mutation may mitigate the adverse prognostic effects of the FLT3–ITD mutation (60). However, all FLT3POS patients had wild-type NPM1. Mutation in this gene was identified only in one of the cases (unpublished data), supporting the observation that this alteration is more common in adults than in pediatric AML patients (8).
Several limitations for this study must be considered: the results were not based on a randomized controlled trial but a retrospective analysis of a heterogeneously treated group of patients coming from eight different institutions. Patients were treated according to four different protocols that had different effects on the outcome. Besides, cytogenetic/fluorescence in situ hybridization (FISH) characterization, considered an independent prognostic factor in AML, was not performed in these patients. This study was not able to assess the potential effects of chromosomal aberration and mutations in other genes and its possible interaction with FLT3 alterations. Since a full FLT3 gene sequencing was not performed, mutation occurring outside exons 13–15 and exon 20 were not detected. Therefore, the frequency of FLT3 mutation in Mexican pediatric AML patients may be even higher than the one obtained in the present work.
This is the first study evaluating the FLT3 mutational profile in pediatric de novo AML Mexican patients and its impact on the prognostic. The results suggest that FLT3 mutation could be considered as an independent biomarker for high-risk disease in pediatric AML patients and highlights the need for testing FLT3 routinely in the clinical setting. A risk-adapted management decision may help to improve the clinical outcome in AML patients, similar to what has been achieved in adult AML patients.
The trend toward better response rates has seen in patients who received chemotherapy as first-line therapy in combination with FLT3 inhibitors compared with those who did not receive it (3). Stone et al. (61) reported the results of an international randomized phase III study, including adult AML patients. Induction therapy (daunorubicin and cytarabine) and consolidation therapy (high-dose cytarabine), plus either midostaurin or placebo, revealed improved 4-year survival in the midostaurin arm compared with the placebo. These results were independent of allelic burden (high vs. low) or the type of FLT3 mutation present (ITD or TKD). The benefit of midostaurin was observed among patients who underwent transplantation during the first remission but not among those who underwent transplantation at a later time (61). Even after intensive chemotherapy and stem cell transplant, FLT3–ITD is an adverse prognostic factor that remains prognostically relevant (3).
A mutant FLT3 is present in approximately one out of four pediatric AML patients in Mexico, and it is related to adverse outcomes. OS was impaired even in the APL subgroup that nowadays has higher complete remission and cure rates than other FAB subgroups due to more efficient therapeutic combinations.
All patients with newly diagnosed AML should have the FLT3 tested using whole-genome sequencing approach better than a specific exon-target enrichment method to be able to detect some clinically relevant children with particular mutations. Earlier identification of FLT3 mutational status will allow identification of patients with a more aggressive disease that could benefit from modification in the treatment strategy, including entering clinical trials, receiving focus therapy, or be considered for bone marrow transplant.
Increasing awareness about the importance and clinical utility of FLT3 testing in AML is the first step to make FLT3 testing part of routine diagnostic workups for these patients in Mexico.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by National Institute of Genomic Medicine, Mexico City, Mexico (number of approval 28-2015-1). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
CM obtained the results, analyzed data, and drafted and approved the manuscript. KC, LLF, MJ, AMR, and BV obtained and validated the results and approved the manuscript. HF validated the results, analyzed statistical data, and approved the manuscript. JN, EJ, VB, JT, JF, JAM, MM, AMS, LE, JP, RE, LVF, RA, MP, OS, HR, AR, MD, EG, PG, and JMM collected clinical data, cared for patients, and approved the manuscript. MD, EG, and ER, validated the results and approved the manuscript. CA carried out the general oversight and the acquisition of funds, designed the study, obtained and validated the results, analyzed the data, and drafted and approved the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by a grant from CONACYT, Fondo Sectorial de Investigación en Salud y Seguridad Social SS/IMSS/ISSSTE-CONACYT number 000000000273210.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to all patients, their families, and the clinical and technical personnel who participated in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fped.2020.00586/full#supplementary-material
1. Bullinger L, Döhner K, Dohner H. Genomics of acute myeloid leukemia diagnosis and pathways. J Clin Oncol. (2017) 35:934–46. doi: 10.1200/JCO.2016.71.2208
2. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the world health organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–406. doi: 10.1182/blood-2016-03-643544
3. Patnaik MM. The importance of FLT3 mutational analysis in acute myeloid leukemia. Leuk Lymphoma. (2018) 59:2273–86. doi: 10.1080/10428194.2017.1399312
4. Breitenbuecher F, Schnittger S, Grundler R, Markova B, Carius B, Brecht A, et al. Identification of a novel type of ITD mutations located in nonjuxtamembrane domains of the FLT3 tyrosine kinase receptor. Blood. (2009) 113:4074–7. doi: 10.1182/blood-2007-11-125476
5. Lagunas-Rangel FA, Chávez-Valencia V. FLT3–ITD and its current role in acute myeloid leukaemia. Med Oncol. (2017) 34:114. doi: 10.1007/s12032-017-0970-x
6. Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. (2019) 33:299–312. doi: 10.1038/s41375-018-0357-9
7. Metzeler KH, Herold T, Rothenberg-Thurley M, Amler S, Sauerland MC, Gorlich D, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. (2016) 128:686–98. doi: 10.1182/blood-2016-01-693879
8. Chauhan PS, Ihsan R, Singh LC, Gupta DK, Mittal V, Kapur S. Mutation of NPM1 and FLT3 genes in acute myeloid leukemia and their association with clinical and immunophenotypic features. Dis Markers. (2013) 35:581–8. doi: 10.1155/2013/582569
9. Janke H, Pastore F, Schumacher D, Herold T, Hopfner KP, Schneider S, et al. Activating FLT3 mutants show distinct gain-of-function phenotypes in vitro and a characteristic signaling pathway profile associated with prognosis in acute Myeloid Leukemia. PLoS ONE. (2014) 9:e89560. doi: 10.1371/journal.pone.0089560
10. NCCN NNCC. Clinical Practice Guidelines in Oncology. Acute Myeloid Leukemia. Natl Compr Cancer Netw. (2017) 3:2017. doi: 10.6004/jnccn.2017.0116
11. Cuervo-Sierra J, Jaime-Pérez JC, Martínez-Hernández RA, García-Sepúlveda RD, Sánchez-Cárdenas M, Gómez-Almaguer D, et al. Prevalence and clinical significance of FLT3 mutation status in acute myeloid leukemia patients: a multicenter study. Arch Med Res. (2016) 47:172–9. doi: 10.1016/j.arcmed.2016.06.003
12. Bolouri H, Farrar JE, Triche TJ, Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. (2018) 24:526. doi: 10.1038/nm0418-526b
13. Mejía-Aranguré JM, Núñez-Enríquez JC, Fajardo-Gutiérrez A, Rodríguez-Zepeda MDC, Martín-Trejo JA, Duarte-Rodríguez DA, et al. Epidemiología descriptiva de la leucemia mieloide aguda (LMA) en niños residentes de la Ciudad de México: reporte del Grupo Mexicano Interinstitucional para la Identificación de las Causas de la Leucemia en Niños. Gac Med Mex. (2016) 152(Suppl. 2):66–77.
14. Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J ASA. (1958) 53:457–81. doi: 10.1080/01621459.1958.10501452
15. Kamath GR, Tremblay D, Coltoff A, Caro J, Lancman G, Bhalla S, et al. Comparing the epidemiology, clinical characteristics and prognostic factors of acute myeloid leukemia with and without acute promyelocytic leukemia. Carcinogenesis. (2019) 40:651–60. doi: 10.1093/carcin/bgz014
16. Schlenk RF, Kayser S, Bullinger L, Kobbe G, Casper J, Ringhoffer M, et al. Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood. (2014) 124:3441–9. doi: 10.1182/blood-2014-05-578070
17. Alonso DCN, Rubio longo PL, Luppo S, Medina A, Gallego MS, Scopinaro MJ, et al. Nuevos blancos terapéuticos en leucemia aguda pediátrica: caracterización de las mutaciones del gen FLT3. Med Infant. (2007) 14:116–23.
18. Andrade FG, Noronha EP, Brisson GD, dos Santos Vicente Bueno F, Cezar IS, Terra-Granado E. Molecular characterization of pediatric acute myeloid leukemia: results of a multicentric study in Brazil. Arch Med Res. (2016) 47:656–67. doi: 10.1016/j.arcmed.2016.11.015
19. Meshinchi S, Alonzo TA, Stirewalt DL, Zwaan M, Zimmerman M, Kaspers GJL, et al. Clinical implications of FLT3 mutations in pediatric AML. Blood. (2006) 108:3654–61. doi: 10.1182/blood-2006-03-009233
20. Tarlock K, Hansen ME, Hylkema T, Rhonda Ries M, Farrar JE, Auvil JG, et al. Discovery and functional validation of novel pediatric specific FLT3 activating mutations in acute myeloid leukemia: results from the COG/NCI target initiative. Blood. (2015) 126:87. doi: 10.1182/blood.V126.23.87.87
21. Zhang Y, Zhang Y, Wang F, Wang M, Liu H, Chen X, et al. The mutational spectrum of FLT3 gene in acute lymphoblastic leukemia is different from acute myeloid leukemia. Cancer Gene Ther. (2020) 27:81–8. doi: 10.1038/s41417-019-0120-z
22. Kindler T, Lipka DB, Fischer T. FLT3 as a therapeutic target in AML: still challenging after all these years. Blood. (2010) 116:5089–102. doi: 10.1182/blood-2010-04-261867
23. Sakaguchi M, Yamaguchi H, Kuboyama M, Najima Y, Usuki K, Ueki T, et al. Significance of FLT3-tyrosine kinase domain mutation as a prognostic factor for acute myeloid leukemia. Int J Hematol. (2019) 110:566–74. doi: 10.1007/s12185-019-02720-z
24. Jiménez-mejía AM, Muskus C, Torres JD, Cuéllar-Ambrossi F, Camargo-guerrero M, Vásquez-palacio G. Caracterización molecular de las mutaciones FLT3-ITD en pacientes colombianos con leucemia mieloide aguda. Rev Hematol Mex. (2013) 14:166–72.
25. Sanchez D, Gargallo P, Romano V, Cabrerizo R. Determinación de la mutación FLT3-ITD por dos métodos en pacientes con leucemia mieloide aguda : comparación e implementación de un nuevo método. Hematologia. (2018) 22:134–43.
26. Lucena-Araujo AR, Souza DL, De Oliveira FM, Benicio MTL, Figueiredo-Pontes LL, Santana-Lemos BA, et al. Results of FLT3 mutation screening and correlations with immunophenotyping in 169 Brazilian patients with acute myeloid leukemia. Ann Hematol. (2010) 89:225–8. doi: 10.1007/s00277-009-0817-4
27. Levis M, Small D. FLT3: ITDoes matter in leukemia. Leukemia. (2003) 17:1738–52. doi: 10.1038/sj.leu.2403099
28. Stirewalt DL, Kopecky KJ, Meshinchi S, Engel JH, Pogosova-agadjanyan EL, Linsley J, et al. Size of FLT3 internal tandem duplication has prognostic significance in patients with acute myeloid leukemia. Blood. (2006) 107:3724–7. doi: 10.1182/blood-2005-08-3453
29. Ponziani V, Gianfaldoni G, Mannelli F, Leoni F, Ciolli S, Guglielmelli P, et al. The size of duplication does not add to the prognostic significance of FLT3 internal tandem duplication in acute myeloid leukemia patients. Leukemia. (2006) 20:2074–6. doi: 10.1038/sj.leu.2404368
30. Kayser S, Schlenk RF, Londono MC, Breitenbuecher F, Wittke K, Du J, et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood. (2009) 114:2386–92. doi: 10.1182/blood-2009-03-209999
31. Meshinchi S, Appelbaum FR. Structural and functional alterations of FLT3 in acute myeloid leukemia. Clin Cancer Res. (2009) 15:4263–70. doi: 10.1158/1078-0432.CCR-08-1123
32. Chan PM. Differential signaling of Flt3 activating mutations in acute myeloid leukemia: a working model. Protein Cell. (2011) 2:108–15. doi: 10.1007/s13238-011-1020-7
33. Arreba-Tutusaus P, Mack T, Bullinger L, Schnöder T, Polanetzki A, Weinert S, et al. Impact of FLT3-ITD location on sensitivity to TKI-therapy in vitro and in vivo. Leukemia. (2016) 30:1220–5. doi: 10.1038/leu.2015.292
34. Marhäll A, Heidel F, Fischer T, Rönnstrand L. Internal tandem duplication mutations in the tyrosine kinase domain of FLT3 display a higher oncogenic potential than the activation loop D835Y mutation. Ann Hematol. (2018) 97:773–80. doi: 10.1007/s00277-018-3245-5
35. Thiede C, Steudel C, Mohr B, Schaich M, Schäkel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. (2002) 99:4326–35. doi: 10.1182/blood.V99.12.4326
36. Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F, et al. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell. (2004) 13:169–78. doi: 10.1016/S1097-2765(03)00505-7
37. Yamamoto Y, Kiyoi H, Nakano Y, Suzuki R, Kodera Y, Miyawaki S, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. (2001) 97:2434–9. doi: 10.1182/blood.V97.8.2434
38. Williams A, Nguyen B, Li L, Brown P, Levis M, Leahy D, et al. Mutations of FLT3/ITD confer resistance to multiple tyrosine kinase inhibitors. Leukemia. (2013) 27:233–6. doi: 10.1038/leu.2012.191
39. Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy : analysis of 854 patients from the United K. Blood. (2001) 98:1752–9. doi: 10.1182/blood.V98.6.1752
40. Abu-Duhier FM, Goodeve AC, Wilson GA, Gari MA, Peake IR, Rees DC, et al. FLT3 internal tandem duplication mutations in adult acute myeloid leukaemia define a high-risk group. Br J Haematol. (2000) 111:190–5. doi: 10.1046/j.1365-2141.2000.02317.x
41. Rombouts W, Blokland I, Löwenberg B, Ploemacher R. Biological characteristics and prognosis of adult acute myeloid leukemia with internal tandem duplication in the FLT3 gene. Leukemia. (2000) 14:675–83. doi: 10.1038/sj.leu.2401731
42. Iwai T, Nakao M, Yokota S, Kaneko H, Horiike S, Kashima K, et al. Internal tandem duplication of the FLT3 gene and clinical evaluation in childhood acute myeloid leukemia. Leukemia. (1999) 13:38–43. doi: 10.1038/sj.leu.2401241
43. Meshinchi S, Woods WG, Stirewalt DL, Sweetser DA, Buckley JD, Tjoa TK, et al. Prevalence and prognostic significance of Flt3 internal tandem duplication in pediatric acute myeloid leukemia. Blood. (2001) 97:89–94. doi: 10.1182/blood.V97.1.89
44. Wu X, Feng X, Zhao X, Ma F, Liu N, Guo H, et al. Prognostic significance of FLT3-ITD in pediatric acute myeloid leukemia: a meta-analysis of cohort studies. Mol Cell Biochem. (2016) 420:121–8. doi: 10.1007/s11010-016-2775-1
45. Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. (2008) 270:1–9. doi: 10.1016/j.canlet.2008.03.036
46. Mccormick SR, Mccormick MJ, Grutkoski PS, Ducker GS, Banerji N, Higgins RR, et al. FLT3 mutations at diagnosis and relapse in acute myeloid leukemia. Arch Pathol Lab Med. (2010) 134:1143–51. doi: 10.1043/2009-0292-OA.1
47. Rebechi M, Hand W, Smith BD, Gojo I, Showel MM, Prince GT, et al. Structural Chromosomal changes are common manifestation of FLT3 ITD relapse and presence of chromosomal progression is independent of normal karyotype at diagnosis. Blood. (2016) 128:2868. doi: 10.1182/blood.V128.22.2868.2868
48. Das-Gupta EP, Seedhouse CH, Russell NH. Microsatellite instability occurs in defined subsets of patients with acute myeloblastic leukaemia. Br J Haematol. (2001) 114:307–12. doi: 10.1046/j.1365-2141.2001.02920.x
49. Nomdedéu JF, Perea G, Estivill C, Lasa A, Carnicer MJ, Brunet S, et al. Microsatellite instability is not an uncommon finding in adult de novo acute myeloid leukemia. Ann Hematol. (2005) 84:368–75. doi: 10.1007/s00277-005-1035-3
50. Watts JM, Dumitriu B, Hilden P, Chen C, Rapaport F, Kishtagari A, et al. Telomere length is associated with specific mutations and mutation classes in patients with acute myeloid leukemia. Blood. (2014) 124:2280. doi: 10.1182/blood.V124.21.2280.2280
51. Garg M, Nagata Y, Kanojia D, Mayakonda A, Yoshida K, Keloth SH, et al. Profiling of somatic mutations in acute myeloid leukemia with FLT3 -ITD at diagnosis and relapse. Blood. (2015) 126:2491–501. doi: 10.1182/blood-2015-05-646240
52. Rebechi MT, Pratz KW. Genomic instability is a principle pathologic feature of FLT3 ITD kinase activity in acute myeloid leukemia leading to clonal evolution and disease progression. Leukemia. (2017) 58:2040–50. doi: 10.1080/10428194.2017.1283031
53. Mead AJ, Linch DC, Hills RK, Wheatley K, Burnett AK, Gale RE. FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood. (2007) 110:1262–70. doi: 10.1182/blood-2006-04-015826
54. Whitman SP, Ruppert AS, Radmacher MD, Mrózek K, Paschka P, Langer C, et al. FLT3 D835/I836 mutations are associated with poor disease-free survival and a distinct gene-expression signature among younger adults with de novo cytogenetically normal acute myeloid leukemia lacking FLT3 internal tandem duplications. Blood. (2008) 111:1552–9. doi: 10.1182/blood-2007-08-107946
55. Callens C, Chevret S, Cayuela JM, Cassinat B, Raffoux E, de Botton S, et al. Prognostic implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL): a retrospective study from the European APL Group. Leukemia. (2005) 19:1153–60. doi: 10.1038/sj.leu.2403790
56. Singh H, Werner L, DeAngelo D, Ballen K, Amrein P, Wadleigh M, et al. Clinical outcome of patients with acute promyelocytic leukemia and FLT3 mutations. Am J Hematol. (2010) 85:956–7. doi: 10.1002/ajh.21867
57. Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. (2002) 99:310–8. doi: 10.1182/blood.V99.1.310
58. Lee BH, Tothova Z, Levine RL, Anderson K, Cullen DE, Mcdowell EP, et al. FLT3 mutations confer enhanced proliferation and survival properties to multipotent progenitors in a murine model of chronic myelomonocytic leukemia. Cancer Cell. (2007) 12:367–80. doi: 10.1016/j.ccr.2007.08.031
59. Blau O, Berenstein R, Sindram A, Blau IW. Molecular analysis of different FLT3-ITD mutations in acute myeloid leukemia. Leuk Lymphoma. (2013) 54:145–52. doi: 10.3109/10428194.2012.704999
60. Schnittger S, Bacher U, Kern W, Alpermann T, Haferlach C, Haferlach T. Prognostic impact of FLT3-ITD load in NPM1 mutated acute myeloid leukemia. Leukemia. (2011) 25:1297–304. doi: 10.1038/leu.2011.97
Keywords: FLT3, pediatric, Mexican, AML, survival, risk-stratification
Citation: Molina Garay C, Carrillo Sánchez K, Flores Lagunes LL, Jiménez Olivares M, Muñoz Rivas A, Villegas Torres BE, Flores Aguilar H, Núñez Enríquez JC, Jiménez Hernández E, Bekker Méndez VC, Torres Nava JR, Flores Lujano J, Martín Trejo JA, Mata Rocha M, Medina Sansón A, Espinoza Hernández LE, Peñaloza Gonzalez JG, Espinosa Elizondo RM, Flores Villegas LV, Amador Sanchez R, Pérez Saldívar ML, Sepúlveda Robles OA, Rosas Vargas H, Rangel López A, Domínguez López ML, García Latorre EA, Reyes Maldonado E, Galindo Delgado P, Mejía Aranguré JM and Alaez Verson C (2020) Profiling FLT3 Mutations in Mexican Acute Myeloid Leukemia Pediatric Patients: Impact on Overall Survival. Front. Pediatr. 8:586. doi: 10.3389/fped.2020.00586
Received: 14 April 2020; Accepted: 07 August 2020;
Published: 16 September 2020.
Edited by:
Birgit Knoechel, Dana–Farber Cancer Institute, United StatesReviewed by:
Concetta Micalizzi, Giannina Gaslini Institute, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS), ItalyCopyright © 2020 Molina Garay, Carrillo Sánchez, Flores Lagunes, Jiménez Olivares, Muñoz Rivas, Villegas Torres, Flores Aguilar, Núñez Enríquez, Jiménez Hernández, Bekker Méndez, Torres Nava, Flores Lujano, Martín Trejo, Mata Rocha, Medina Sansón, Espinoza Hernández, Peñaloza Gonzalez, Espinosa Elizondo, Flores Villegas, Amador Sanchez, Pérez Saldívar, Sepúlveda Robles, Rosas Vargas, Rangel López, Domínguez López, García Latorre, Reyes Maldonado, Galindo Delgado, Mejía Aranguré and Alaez Verson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carmen Alaez Verson, Y2FsYWV6QGlubWVnZW4uZ29iLm14
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.