
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Pediatr., 11 September 2020
Sec. Pediatric Neurology
Volume 8 - 2020 | https://doi.org/10.3389/fped.2020.00550
 Raffaele Falsaperla1
Raffaele Falsaperla1 Xena Giada Pappalardo2,3Catia Romano4
Xena Giada Pappalardo2,3Catia Romano4 Simona Domenica Marino1Giovanni Corsello5Martino Ruggieri4Enrico Parano2
Simona Domenica Marino1Giovanni Corsello5Martino Ruggieri4Enrico Parano2 Piero Pavone4*
Piero Pavone4*Introduction: Mutations in the contactin-associated protein-like 2 (CNTNAP2) gene (MIM#604569) encoding for CASPR2, a cell adhesion protein of the neurexin family, are known to be associated with autism, intellectual disability, and other neuropsychiatric disorders. A set of intronic deletions of CNTNAP2 gene has also been suggested to have a causative role in individuals with a wide phenotypic spectrum, including Pitt-Hopkins syndrome, cortical dysplasia–focal epilepsy syndrome, Tourette syndrome, language dysfunction, and abnormal behavioral manifestations.
Case presentation: A 10-years-old boy was referred to the hospital with mild intellectual disability and language impairment. Moreover, the child exhibited minor facial features, epileptic seizures, and notable behavioral abnormalities including impulsivity, aggressivity, and hyperactivity suggestive of the diagnosis of disruptive, impulse-control and conduct disorder (CD). Array comparative genomic hybridization (CGH) revealed a copy number variant (CNV) deletion in the first intron of CNTNAP2 gene inherited from a healthy father.
Conclusions: A comprehensive description of the phenotypic features of the child is provided, revealing a distinct and remarkable alteration of social behavior not previously reported in individuals affected by disorders related to CNTNAP2 gene disruptions. A possible causative link between the deletion of a non-coding regulatory region and the symptoms presented by the boy has been advanced.
A new strong candidate gene for psychiatric and language disorders is contactin-associated protein-like 2 (CNTNAP2) gene (MIM#604569). CNTNAP2 is a member of the neurexin family and consists of a transcript of 24 exons encoding for CASPR2 protein, which functions as a cell-adhesion molecule in many neuronal activities, such as neuronal migration, dendritic arborization, and synaptic transmission. The main role of CASPR2 is the conduction of axon potentials and the clustering of voltage-gated potassium channels at the juxtaparanodes in both myelinated axons of the spinal cord and of the central nervous system (1, 2). However, the high expression of the protein in Broca's area and other perisylvian regions is consistent with the emerging role in normal language development and social communication (3, 4). By its size, spanning 2.3 Mb at chromosomal region 7q35-36, CNTNAP2 is a target for a wide variety of mutations and structural rearrangements, including copy number variants (CNVs). Some intronic CNV deletions of CNTNAP2 gene have been reported in individuals with Pitt-Hopkins syndrome (PTHS) and cortical dysplasia–focal epilepsy syndrome (CDFES), epilepsy with auditory features (EAF), autism spectrum disorder (ASD) and intellectual disability (ID), speech impairment, Tourette syndrome (TS), and abnormal behavioral manifestations (5–9). Interestingly, variants found in the first intronic region of CNTNAP2 have been shown to lead to the loss of critical regulatory elements of some conserved transcription factor (TF) binding sites (TFBSs) involved in language and social-emotional development.
Here, we report a child presenting with neuropsychiatric disturbances including notable behavioral disorders consisting of frequent and severe outbursts of impulsivity, aggressivity, and hyperactivity suggestive of the diagnosis of “disruptive, impulse-control, and conduct disorder (CD)” and TS. Moreover, the child exhibited minor facial features, mild ID, language impairment, and epileptic seizures. Molecular investigation carried out by array comparative genomic hybridization (aCGH) revealed a CNV deletion spanning 95.4 kb in intron 1 of CNTNAP2 gene inherited from a healthy father. A possible causative link between the deletion of a non-coding regulatory region and the clinical manifestations observed in the boy has been raised.
A 10-years-old boy is the second child of healthy unrelated Italian parents. Both older half-siblings, an 18-years-old boy born from the father's previous marriage and a 16-years-old sister are healthy. In the paternal line, there is a history of family members affected by polyposis intestinalis with early deaths.
At gestation, the mother was 34 years old and the father was 40 years old. The mother denied having had complications during the gestation and reported normal fetal movements. Intrauterine ultrasound did not show fetal anomalies. The child was born at 40 weeks of gestation by cesarean section as in the previous delivery. Birth weight was 3.6 kg, length 51 cm, and head circumference 35 cm (all within normal limits). The Apgar scores were eight at one and 10 at 5 min. At birth, neither clinical signs nor notable facial features were noticed, and the child was discharged by the hospital in good condition.
During the 1st months, he exhibited normal motor development but had speech delay, which started at the age of 28 months with the pronunciation of a single word. At the age of 3 years, the parents noted that the child was extremely overactive with mood swings and difficulty in sleeping. The anomalous behavior became progressively more evident, and at the age of 4 years, he was remarkably aggressive. At the age of 5 years, he was hospitalized in North Italy due to developmental delay, frequent insomnia episodes, stereotypic movements (lateral swinging), and phonologic disturbances. During hospitalization in that center, routine examination including thyroid markers, ECG, and brain MRI were performed with normal results. In the Leiter-R test (non-verbal cognitive capacity), brief IQ of 85 was registered, and the “Child Behavioral Check List” (CBCL) showed affective impairment, anxiety, hyperactivity, and obsessive behavior. The child was discharged with the diagnosis of behavioral disturbance with hyperactivity, phonologic disturbances, and EEG alterations. At the age of 8 and 9 years, right-sided focal tonic seizures lasting a few minutes were recorded. Treatment with valproate at 20 mg/kg/day was started with good drug response. During the subsequent years, he was followed up as an outpatient at the Pediatric Unit and Pediatric Emergency Unit, University Hospital “Policlinico-Vittorio Emanuele,” Catania, Italy, since he showed mild speech impairment and episodes of aggressivity against parents, teachers, and peers. In one of these episodes, he caused injury to one of his teachers. Risperidone 1 mg/day was started but irregularly administered with poor results. At the age of 10 years, he was admitted to this institution for a clinical workup. His weight was 51 kg (>90th percentile), length 143 cm (75th percentile), and head circumference 55 cm (90th percentile). On physical examination, minor feature anomalies were noticed and consisted of upslanting palpebral fissures, sparse eyebrows, short nose, flat philtrum, thin lips, and wide earlobes (Figure 1). Hands and feet were short. The heart, thorax, abdomen, and internal organs were normal. Neurological examination was normal, and patellar tendon reflexes were normally elicited. EEG during wakefulness and during sleep showed spike and wave discharges in the fronto-centro-temporal region, and during eye opening, photic stimulation and hyperventilation was unchanged (Figure 2). A fundus examination and hearing exploration were normal. At the age of 10 years, brief IQ was 85. No more seizures were reported, and treatment with valproate was withdrawn. Due to the frequent chronic episodes of diarrhea and familiar history of polyposis intestinalis, an abdominal ultrasound exam and esophagogastroduodenoscopy (EDG) were performed, and tissue samples were examined. The EDG did not show anomalies, and the macroscopic sample analysis of the duodenal tissue was normal. The neuropsychiatric evaluation of the child displayed several dysfunctions. His cognitive and behavioral profile consisted of a mild ID and high score of generalized anxiety and depression with intrusive thoughts. Complex, chronic motor tics were seen in the child in association with signs of obsessive–compulsive disorder (TS). Self-esteem and social relation result compromised. Verbal and physical aggressivity and impulsivity were particularly expressed. The final psychiatric diagnosis was disruptive, impulse-control and CD. The results of the neuropsychiatric evaluation of proband and parents are reported in Table 1(A–J). At last examination, at 11 years old, seizures were not reported, while behavioral impairments were unmodified.
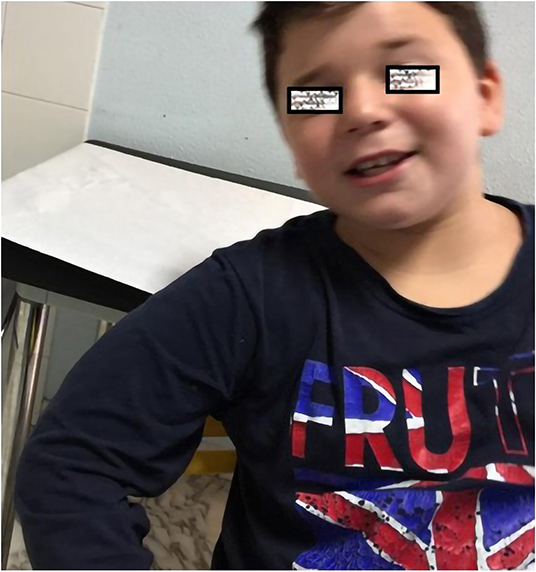
Figure 1. Photo showing a 10-years-old boy with sparse eyebrows, short nose, long flat philtrum and thin lips.
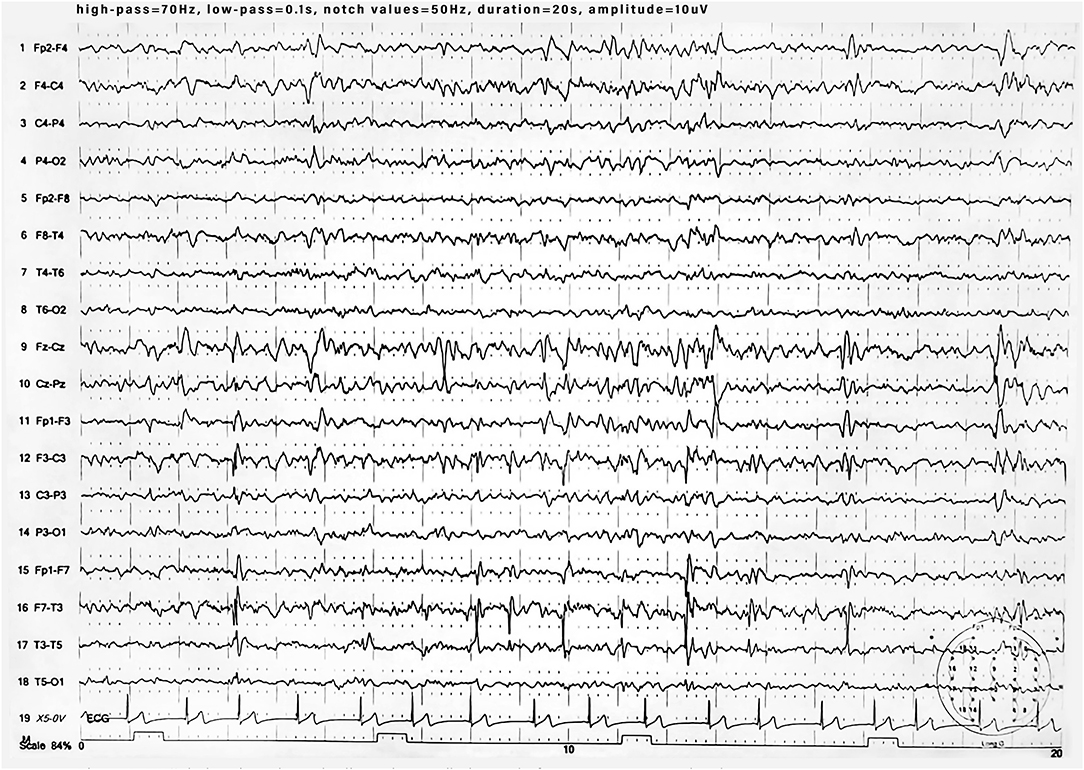
Figure 2. EEG during sleep showing spike and wave discharges in fronto-centro-temporal region.
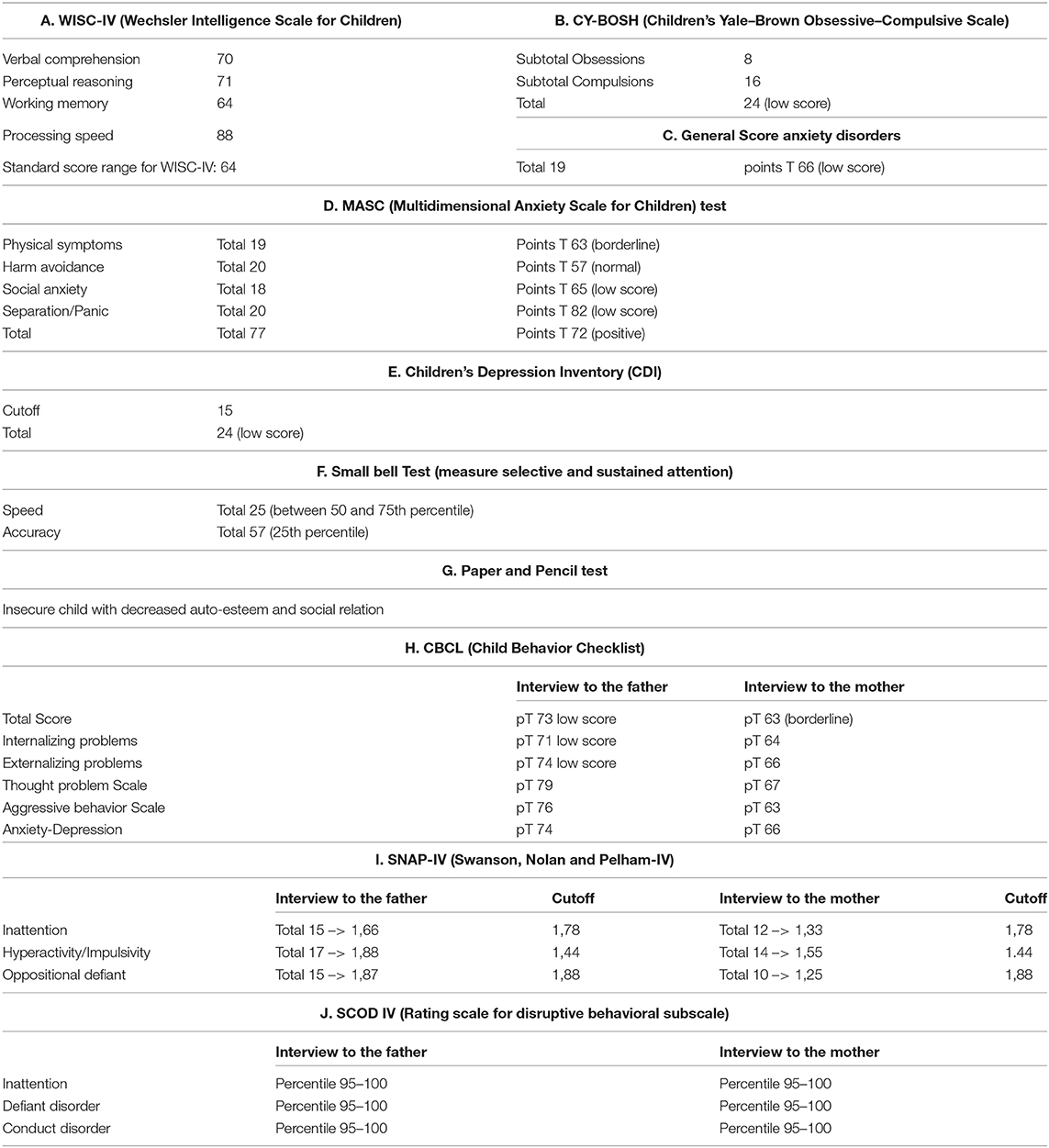
Table 1. (A–J). Neuropsychological assessment of proband (A–G) and both parents (H–J).
Genomic DNA was isolated from peripheral blood of the proband, together with the proband's mother and father. aCGH was performed by CytoSure ISCA 8 × 60 k array from Oxford Gene Technology (OGT) according to the manufacturer's recommendations (Agilent Technologies, Santa Clara, CA). aCGH data were analyzed and interpreted using Cytosure software (GRCh38 assembly) provided by OGT.
Aiming at exploring and better interpreting the phenotype linked with the proband's CNV, we used the publicly available patient data on the Database of Genomic Variants (DGV) (dgv.tcag.ca), DECIPHER web-based resource (decipher.sanger.ac.uk), CNV dataset from Clinical Genome Resource (ClinGen), and Morbidity Map of Developmental Delay displayed at UCSC Genome Browser (genome.ucsc.edu).
The molecular karyotype revealed that the proband carries a CNV deletion of 95.4 kb in 7q35(146,271,924-146,367,324)x1 inherited from the healthy father. The microdeletion detected in the first intronic region of the CNTNAP2 gene has been previously reported in the DGV database.
The young boy presented with minor facial dysmorphism, mild ID, speech impairment, sleep disorders, and severe behavioral disturbances and TS. Two episodes of partial epileptic seizures were also recorded. Behavioral disturbances were impressive, and the child exhibited frequent and severe outbursts of aggressivity, impulsivity, and hyperactivity, which led to the diagnosis of disruptive, impulse-control and CD. Much of the clinical signs presented in the child have been also reported in patients affected by CASPR2-deficiency disorder (CDD) associated with language impairment, notable impaired behavior and CD, TS, epileptic seizures, moderate ID, and poor social interactions (8, 10, 11). aCGH analysis revealed a small CNV deletion in intron 1 of CNTNAP2 gene inherited from the unaffected father. The incomplete penetrance of the CNV variant may not exclude the pathogenic link between the microdeletion and disruption of gene-regulatory and protein interactions underlying some neural and behavioral pathways involved in learning ability and language and social behaviors (11–13). Deletions with similar size (e.g., essv12997739) of the variant in question have been reported in population, but their pathogenicity and phenotypic contribution are still uncertain. A wide variety of intronic variants of CNTNAP2 gene has been reported in patients with severe ID, autistic behavior, epilepsy, and breathing anomalies that phenotypically overlap with PTHS (14, 15). Additional reports have also been found in individuals with epilepsy (16), EAF (7), epilepsy, and schizophrenia in three non-related Caucasian patients (17). Moreover, Strauss et al. (18) reported children with cortical dysplasia, focal epilepsy, relative macrocephaly, and diminished deep-tendon reflexes in association with language regression, hyperactivity, and impulsive and aggressive behavior. Speech disorders, behavioral disturbances, and other neuropsychiatric disorders have been identified in patients with alterations in the CNTNAP2 gene (12, 19–22). Verkerk et al. (23) described a family with members affected by TS and obsessive–compulsive disorder (OCD), in which mutations in CNTNAP2 gene expression have been found to alter the distribution of the K(+) channels in the nervous system with abnormal conduction and/or repolarization of active potential, leading to cause anomalous motor movements observed in individuals affected by TS. In contrast with this finding, no individuals with clinical evidence of TS have been reported by Belloso et al. (24) in a family with a balanced reciprocal translocation (t7;15)(q35:q26.1). The first intron of CNTNAP2 is known to be a susceptible locus for structural rearrangements that may influence the genetic modulation of the gene since it contains some conserved regulatory regions implicated in the transcriptional network of neurodevelopmental processes regulating language skills and social-emotional functioning. Some studies have experimentally identified consensus binding sequences for a selected set of TFs, such as STOX1A (8), TCF4 (25), FOXP1, and FOXP2 (26, 27), which can influence the transcriptional control of the gene and interact with CNTNAP2 to co-regulate the developmental pathways of human speech and social behavior.
Our study has some limitations. First is that the described CNV may be a combined effect of additional rare variants, which are not excluded in the present case, and should be investigated by whole-exome sequencing (WES). Secondly, with regard to the quantitative PCR validation, CNTNAP2 gene dosage variation was not included. However, we neglected to correlate the effect of CNV loss with the gene expression level to examine alterations in mRNA synthesis and the activity of Caspr2 protein. The impact of the intronic deletion in the transcriptional regulatory network of CNTNAP2 that correlated with the analysis of FOXP interactome warrants additional investigations. Our study might serve as a first step to identify a non-coding regulatory variant enriched for potentially important TFs implicated in neurodevelopment. We believe that it would be important to determine whether (i) the effect of CNV loss may affect variations in the CNTNAP2-encoded CASPR2 protein (e.g., splice site mutations, posttranslational modifications) and whether (ii) the clinical phenotype may depend on the association between CNV loss and DNA methylation pattern of gene promoter and intron 1.
The present study deems that regardless of sufficient evidence to conclude that the intronic deletion of CNTNAP2 has a potential contribution in the diagnosis, there are some findings for believing in a likely positive correlation between the clinical presentation of the child and individuals affected by CDD.
The data used to support the findings of this study may be released upon application to the corresponding author who can be contacted at cHBhdm9uZUB1bmljdC5pdA==.
The study was conducted ethically in accordance with the World Medical Association Declaration of Helsinki and was approved by the ethic committee of the University of Catania, Italy (Ethical Committee Catania 1 Clinical Registration n. 95/2018/PO). Informed consent was obtained from parents of the proband.
RF and PP worked with and helped gather patient data and drafted and redrafted the present manuscript. XP helped analyze the genetic data and interpret the literature relevant to the mutation. CR performed the neuropsychological assessment. CR and SM contributed to the clinical understanding of the case and revised the manuscript. GC, MR, and EP were called as consultants regarding the clinical diagnosis and reviewed the manuscript. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
aCGH, array comparative genome hybridization; ASD, autism spectrum disorder; CCD, CASPR2-deficiency disorder; CD, conduct disorder; CNTNAP2, contactin-associated protein-like 2; CNV, copy number variant; CDFES, cortical dysplasia–focal epilepsy syndrome; EAFs, epilepsy and auditory features; EDG, esophagogastroduodenoscopy; ESR1, estrogen receptor alpha gene; FOXP1, forkhead box P1; FOXP2, forkhead box P2; ID, intellectual disability; OCD, obsessive–compulsive disorder; PTHS, Pitt-Hopkins syndrome; STOX1A, storkhead box 1A; TFBS, transcription factor binding site; TF, transcription factor; TCF4, transcription factor 4; TS, Tourette syndrome.
1. Varea O, Martin-de-Saavedra MD, Kopeikina KJ, Schurmann B, Fleming HJ, Fawcett-Patel JM, et al. Synaptic abnormalities and cytoplasmic glutamate receptor aggregates in contactin associated protein-like 2/Caspr2 knockout neurons. Proc Natl Acad Sci USA. (2015) 112:6176–81. doi: 10.1073/pnas.1423205112
2. Flaherty E, Deranieh RM, Artimovich E, Lee IS, Siegel AJ, Levy DL. Patient-derived hiPSC neurons with heterozygous CNTNAP2 deletions display altered neuronal gene expression and network activity. NPJ Schizophr. (2017) 3:35. doi: 10.1038/s41537-017-0033-5
3. Abrahams BS, Tentler D, Perederiy JV, Oldham MC, Coppola G, Geschwind DH. Genome-wide analyses of human perisylvian cerebral cortical patterning. Proc Natl Acad Sci USA. (2007) 104:17849–54. doi: 10.1073/pnas.0706128104
4. Bakkaloglu B, O'Roak BJ, Louvi A, Gupta AR, Abelson JF, Morgan TM, et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am J Hum Genet. (2008) 82:165–73. doi: 10.1016/j.ajhg.2007.09.017
5. Penagarikano O, Abrahams BS, Herman EI, Winden KD, Gdalyahu A, Dong H, et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. (2011) 147:235–46. doi: 10.1016/j.cell.2011.08.040
6. Chiocchetti AG, Kopp M, Waltes R, Haslinger D, Duketis E, Jarczok TA, et al. Variants of the CNTNAP2 5' promoter as risk factors for autism spectrum disorders: a genetic and functional approach. Mol Psychiatry. (2015) 20:839–49. doi: 10.1038/mp.2014.103
7. Pippucci T, Licchetta L, Baldassari S, Palombo F, Menghi V, D'Aurizio R, et al. Epilepsy with auditory features: a heterogeneous clinico-molecular disease. Neurol Genet. (2015) 1:e5. doi: 10.1212/NXG.0000000000000005
8. Poot M. Connecting the CNTNAP2 networks with neurodevelopmental disorders. Mol Syndromol. (2015) 6:7–22. doi: 10.1159/000371594
9. Werling AM, Bobrowski E, Taurines R, Gundelfinger R, Romanos M, Grunblatt E, et al. CNTNAP2 gene in high functioning autism: no association according to family and meta-analysis approaches. J Neural Transm. (2016) 123:353–63. doi: 10.1007/s00702-015-1458-5
10. Rodenas-Cuadrado P, Pietrafusa N, Francavilla T, La Neve A, Striano P, Vernes SC. Characterisation of CASPR2 deficiency disorder–a syndrome involving autism, epilepsy and language impairment. BMC Med Genet. (2016) 17: 8. doi: 10.1186/s12881-016-0272-8
11. Poot M. Intragenic CNTNAP2 deletions: a bridge too far? Mol Syndromol. (2017) 8:118–30. doi: 10.1159/000456021
12. Rodenas-Cuadrado P, Ho J, Vernes SC. Shining a light on CNTNAP2: complex functions to complex disorders. Eur J Hum Genet. (2014) 22:171–8. doi: 10.1038/ejhg.2013.100
13. Toma C, Pierce KD, Shaw AD, Heath A, Mitchell PB, Schofield PR, et al. Comprehensive cross-disorder analyses of CNTNAP2 suggest it is unlikely to be a primary risk gene for psychiatric disorders. PLoS Genet. (2018) 14:e1007535. doi: 10.1371/journal.pgen.1007535
14. Zweier C, de Jong EK, Zweier M, Orrico A, Ousager LB, Collins AL, et al. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am J Hum Genet. (2009) 85:655–66. doi: 10.1016/j.ajhg.2009.10.004
15. Gregor A, Albrecht B, Bader I, Bijlsma EK, Ekici AB, Engels H, et al. Expanding the clinical spectrum associated with defects in CNTNAP2 and NRXN1. BMC Med Genet. (2011) 12:106. doi: 10.1186/1471-2350-12-106
16. Smogavec M, Cleall A, Hoyer J, Lederer D, Nassogne MC, Palmer EE, et al. Eight further individuals with intellectual disability and epilepsy carrying bi-allelic CNTNAP2 aberrations allow delineation of the mutational and phenotypic spectrum. J Med Genet. (2016) 53:820–7. doi: 10.1136/jmedgenet-2016-103880
17. Friedman JI, Vrijenhoek T, Markx S, Janssen IM, van der Vliet WA, Faas BH, et al. CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol Psychiatry. (2008) 13:261–6. doi: 10.1038/sj.mp.4002049
18. Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, et al. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. (2006) 354:1370–7. doi: 10.1056/NEJMoa052773
19. Petrin AL, Giacheti CM, Maximino LP, Abramides DV, Zanchetta S, Rossi NF, et al. Identification of a microdeletion at the 7q33-q35 disrupting the CNTNAP2 gene in a Brazilian stuttering case. Am J Med Genet A. (2010) 152A:3164–72. doi: 10.1002/ajmg.a.33749
20. Whitehouse AJ, Bishop DV, Ang QW, Pennell CE, Fisher SE. CNTNAP2 variants affect early language development in the general population. Genes Brain Behav. (2011) 10:451–6. doi: 10.1111/j.1601-183X.2011.00684.x
21. Veerappa AM, Saldanha M, Padakannaya P, Ramachandra NB. Family-based genome-wide copy number scan identifies five new genes of dyslexia involved in dendritic spinal plasticity. J Hum Genet. (2013) 58:539–47. doi: 10.1038/jhg.2013.47
22. Centanni TM, Sanmann JN, Green JR, Iuzzini-Seigel J, Bartlett C, Sanger WG, et al. The role of candidate-gene CNTNAP2 in childhood apraxia of speech and specific language impairment. Am J Med Genet B Neuropsychiatr Genet. (2015) 168:536–43. doi: 10.1002/ajmg.b.32325
23. Verkerk AJ, Mathews CA, Joosse M, Eussen BH, Heutink P, Oostra BA, et al. CNTNAP2 is disrupted in a family with Gilles de la Tourette syndrome and obsessive compulsive disorder. Genomics. (2003) 82:1–9. doi: 10.1016/S0888-7543(03)00097-1
24. Belloso JM, Bache I, Guitart M, Caballin MR, Halgren C, Kirchhoff M, et al. Disruption of the CNTNAP2 gene in a t(7;15) translocation family without symptoms of Gilles de la Tourette syndrome. Eur J Hum Genet. (2007) 15:711–3. doi: 10.1038/sj.ejhg.5201824
25. Forrest MP, Hill MJ, Kavanagh DH, Tansey KE, Waite AJ, Blake DJ. The psychiatric risk gene transcription factor 4 (TCF4) regulates neurodevelopmental pathways associated with Schizophrenia, Autism, and Intellectual Disability. Schizophr Bull. (2018) 44:1100–10. doi: 10.1093/schbul/sbx164
26. Vernes SC, Newbury DF, Abrahams BS, Winchester L, Nicod J, Groszer M, et al. A functional genetic link between distinct developmental language disorders. N Engl J Med. (2008) 359:2337–45. doi: 10.1056/NEJMoa0802828
Keywords: CNTNAP2 gene, intronic copy number variant, conduct disorder (CD), epilepsy, intellectual disability (ID)
Citation: Falsaperla R, Pappalardo XG, Romano C, Marino SD, Corsello G, Ruggieri M, Parano E and Pavone P (2020) Intronic Variant in CNTNAP2 Gene in a Boy With Remarkable Conduct Disorder, Minor Facial Features, Mild Intellectual Disability, and Seizures. Front. Pediatr. 8:550. doi: 10.3389/fped.2020.00550
Received: 27 April 2020; Accepted: 30 July 2020;
Published: 11 September 2020.
Edited by:
Pasquale Parisi, Sapienza University of Rome, ItalyReviewed by:
Thea Giacomini, Istituto Giannina Gaslini (IRCCS), ItalyCopyright © 2020 Falsaperla, Pappalardo, Romano, Marino, Corsello, Ruggieri, Parano and Pavone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Piero Pavone, cHBhdm9uZUB1bmljdC5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.