 Tayfun Güngör
Tayfun Güngör Robert Chiesa
Robert Chiesa
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Pediatr. , 26 June 2020
Sec. Pediatric Immunology
Volume 8 - 2020 | https://doi.org/10.3389/fped.2020.00327
This article is part of the Research Topic HSCT for Primary Immunodeficiencies and Rare Metabolic Diseases View all 16 articles
Allogeneic hematopoietic stem cell transplantation (HSCT) has become the main curative treatment in patients with chronic granulomatous disease (CGD). CGD is caused by inherited defects of the phagolysomal NADPH-oxidase, leading to a lifelong propensity for invasive infections and granulomatous inflammation. After successful allogeneic HSCT, chronic infections and inflammation resolve and quality-of-life improves. Favorable long-term outcome after HSCT is dependent on the prevention of primary and secondary graft failure (GF), including falling myeloid donor chimerism (DC) below 10 %, and chronic graft-vs.-host-disease (cGVHD). The risk of GF and GvHD increases with the use of HLA-incompatible donors and this may outweigh the benefits of HSCT, mainly in patients with severe co-morbidities and in asymptomatic patients with residual NADPH-oxidase function. Seventeen scientific papers have reported on a total of 386 CGD-patients treated by HSCT with HLA-matched family/sibling (MFD/MSD), 9/10-/10/10-matched-unrelated volunteer (MUD) and cord blood donors. The median OS/EFS-rate of these 17 studies was 91 and 82%, respectively. The median rates of GF, cGVHD and de-novo autoimmune diseases were 14, 10, and 12%, respectively. Results after MFD/MSD and 10/10-MUD-transplants were rather similar, but outcome in adults with significant co-morbidities and after transplants with 9/10 HLA-MUD were less successful, mainly due to increased GF and chronic GVHD. Transplantation protocols using T-cell depleted haploidentical donors with post-transplant cyclophosphamide or TCR-alpha/beta depletion have recently reported promising results. Autologous gene-therapy after lentiviral transduction of HSC achieved OS/EFS-rates of 78/67%, respectively. Careful retrospective and prospective studies are mandatory to ascertain the most effective cellular therapies in patients with CGD.
Chronic granulomatous disease is caused by mutations leading to defects in individual subunits of the phagocyte NADPH-oxidase (gp91phox in X-linked-; p22-, p47-, p67-, p40phox, and EROS in autosomal recessive-CGD) (1–4). The NADPH-oxidase-myeloperoxidase system generates microbicidal oxidants required for host defense and control of inflammation. CGD affects ~1:200,000–250,000 live-births (5–7) and X-linked-CGD accounts for approximately two-thirds of patients. P47phox-mutations are the most common AR-defects. Rarely, female carriers of X-CGD with random X-lyonization of <15% of circulating NADPH-oxidase-producing neutrophils present with CGD-symptoms (7–9). Symptoms comprise of invasive infections and chronic autoinflammatory diseases leading to frequent medical interventions, impaired quality-of-life, and increased morbidity/mortality (10–13). The majority of patients are diagnosed in childhood, while some develop symptoms in adulthood (7, 14, 15). Due to residual NADPH-oxidase activity, patients with AR-p47phox-mutations survive longer than X-CGD-patients (survival >40 years: >80 vs. 55%) (16). The clinical course may be unpredictable even in individuals of families with identical CGD-mutations (17). Short stature, osteoporosis, organ failure, and amyloidosis are long-term complications (18). There is still paucity of data on quality-of-life and emotional health in patients with CGD (11, 12, 14, 19, 20). Today, 90% of children with CGD are reaching adulthood and the transition into adult care is challenging (21, 22).
The infections typically affect lungs, lymph nodes, skin, liver, perianal region, gingiva and bone and are mainly caused by Staphylococcus aureus, Burkholderia cepacia, Nocardia, Serratia marcescens, and Aspergillus species. Klebsiella pneumoniae, Salmonella (7, 23), Mycobacteria (21, 24), Actinomyces, Granulibacter bethesthensis (25–27). Infections caused by Chromobacterium violaceum and B. pseudomallei (28–30) are less frequently encountered. The use of life-long antibacterial prophylaxis with trimethoprim-sulfamethoxazole is recommended. Pulmonary Aspergillus-infections are the leading cause of mortality (31). Anti-fungal prophylaxis, mainly with itraconazole (32, 33), can reduce the incidence of fungal infections, but the emergence of azole-resistant aspergillus species and dematiaceous molds is becoming a clinical challenge (34).
Absent or reduced NADPH-oxidase activity in monocytes/macrophages causes impairment of efferocytosis and autophagy (35, 36). Ineffective apoptotic cell clearance increases the risks of developing autoinflammation (37). Progressive granulomatous lung disease (PGLD), Crohn-like enterocolitis (38, 39) and obstructive genitourinary inflammation (40, 41) are relevant autoinflammatory syndromes and their risk increase steadily during life (14). Initial gastrointestinal involvement without infections has often been misdiagnosed as Crohn‘s disease (38, 39). Treatment of autoinflammation includes steroids (42) and more recently IL1- or TNF-alpha inhibitors to replace steroids, however, all of these drugs bear the risk of increasing the risks for invasive infections (43). Thalidomide (44–46), vedolizumab, ustekinumab (47, 48), as well as pioglitazone (36) can be beneficial to reduce autoinflammation in CGD and regular IFN-gamma injections decrease the incidence of bacterial infections with no impact on the incidence of colitis (21, 49–55).
Before HSCT, screening for infections is mandatory in biopsies of infectious lesions and in bronchoalveolar lavage specimens (14, 56). Steroids added to antimicrobials can accelerate the regression of infectious lesions (42, 57, 58) and can help to avoid extensive surgery (59). Granulocyte-transfusions should be strictly indicated to prevent CMV-transmission and sensitization to blood cell antigens (60–62). The McLeod-blood group should be evaluated in X-CGD-patients to minimize the sensitization against Kx-positive red cell transfusions (63–65).
In Europe, the first major survey of the Inborn Errors Working Party of the EBMT reported on 27 patients with CGD who had been transplanted between 1985 and 2000. At HSCT, nine of 27 patients had intractable invasive infections and received antibiotics as well as granulocyte-transfusions (seven of nine). Eighteen of 27 patients were free of infection at HSCT. Seven of the 18 patients without overt infection had signs of active ongoing autoinflammation including enterocolitis and PGLD. Twenty-five of the 27 patients received MSD-transplants (five heterozygous carriers). Two patients with no overt infection or autoinflammation received a MUD-transplant. Conditioning-regimens were mainly myeloablative with full-dose busulfan/cyclophosphamide and mainly without serotherapy (67). Recovery from refractory infection, remission of inflammatory organ dysfunction and catch-up growth were observed (67). Patients without overt autoinflammation/infections had an OS of 100%, whereas patients with ongoing infections at transplant had a TRM of 44% (four of nine) (67). The OS/EFS was 85/81%, respectively. The GF and chronic GVHD rate were 7 and 11%, respectively. The majority of surviving patients had >95% circulating myeloid cells of donor origin. This important paper showed that myeloablative HSCT based on busulfan/cyclophosphamide and no in vivo T-cell depletion was overall efficient in sibling transplants but induced exuberant inflammation in patients suffering from ongoing infections at transplant. The same was observed in a transplantation model in non-infected CGD mice after myeloablative allogeneic HSCT resulting in marked infiltration of the lungs with inflammatory cells, in contrast to normal mice (81). Cultured monocytes from the CGD-mice produced 3-fold TNF-alpha (81), explaining the higher incidence of severe GvHD in patients with pre-existing overt infections treated with HSCT without serotherapy. Myeloablative regimens containing cyclophosphamide were greatly abandoned in Europe after this experience. The authors at that time concluded that all infectious/inflammatory foci had to be detected and treated before HSCT and that HSCT should be mainly restricted to children with MSD/MFD (67).
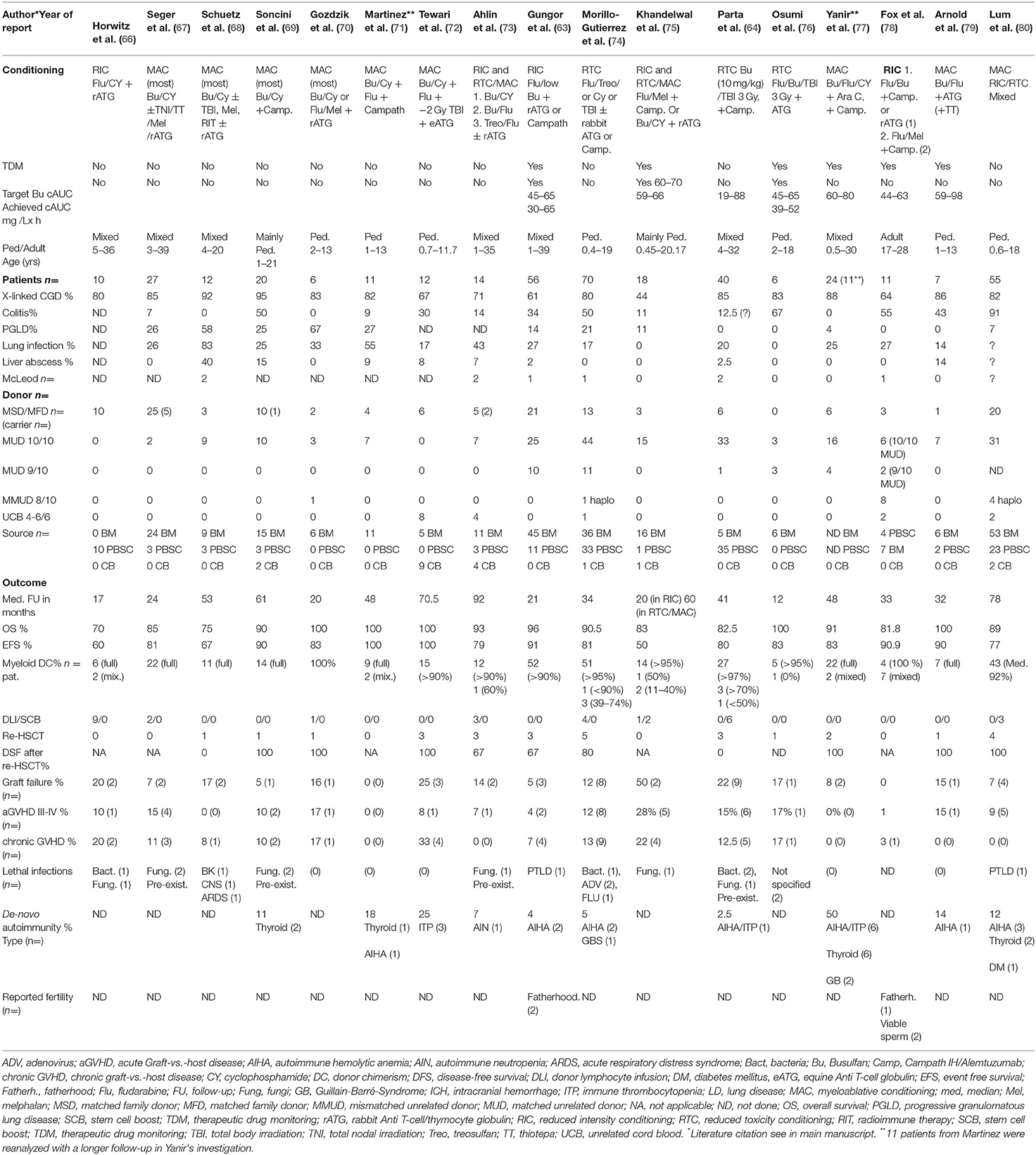
Table 1. Major HSCT studies with HLA-matched donor transplants in CGD between 2001 and 2019 (n >5 patients).
Almost simultaneously to the above mentioned European experience, the NIH in the USA used for the first time a reduced intensity conditioning (RIC) comprising of non-myeloablative fludarabine/cyclophosphamide followed by in-vitro T-cell depleted grafts. This approach resulted in clearly increased GF-rates (20%), even with the use of matched family/sibling donors (66). Donor-lymphocyte infusions were necessary to prevent falling DC but unfortunately induced severe acute GVHD and resulted in a transplant-related mortality rate of 30% (66, 82). RIC-regimens including melphalan and fludarabine were associated with similarly high GF-rates (30%) (75).
RIC-regimens based on reduced or targeted busulfan, fludarabine and serotherapy were more successful and achieved sufficient myeloablation and clearly lower rates of GF and chronic GVHD (38, 63, 83–85). These busulfan-fludarabine-based RIC-regimens were first used in adult high-risk CGD-patients suffering from invasive Aspergillus-infections and/or enterocolitis using MSD/MFD- or MUD transplants. The OS/EFS rates were 100% in these small initial series (38, 84). Administration of anti-T-cell/thymocyte globulins as well as of a humanized monoclonal anti-CD52 antibody (Campath IH; alemtuzumab) were shown to deplete successfully T-cells and allo-stimulatory dendritic cells (86) of recipient origin. The importance of using serotherapy for in-vivo T-cell depletion to reduce both GF and chronic GVHD after HSCT for CGD became obvious. Viral reactivations after serotherapy were fortunately rare or well manageable rendering clinical HSCT outcomes with MUD-donors vastly similar to MSD/MFD-donors (68, 69, 71).
Busulfan-based RIC-conditioning was further refined by investigating the interindividually variable busulfan clearance and exposure in patients (87, 88). Therapeutic drug monitoring (TDM) helped optimize both safety and efficacy of busulfan-administration. The assessment of the cumulative AUC (cAUC) turned out as an appropriate tool to measure the total busulfan-exposure and -toxicity (87, 89). A 10-year prospective study on 56 pediatric/adult CGD-patients (2/3 high-risk patients) treated with submyeloablative busulfan (half-dose or cAUC 45–65 mg/L × h) yielded, indeed, excellent results. Busulfan-dose adjustments (90) were necessary in 14/44 patients (32%) (63). Immunoablation was achieved with fludarabine and serotherapy including rabbit ATG or alemtuzumab. After a follow-up time of 21 months, the OS/EFS-rates were 93 and 89%, respectively. However, GF could not be abolished and occurred in 5% of patients. The cumulative incidences of grade III–IV acute GVHD and chronic GHVD were low with 4 and 7%, respectively. Stable ≥90% myeloid DC was documented in 93% surviving patients leading to resolution of infectious and inflammatory lesions. Equivalent outcomes were observed between MFD/MSD and MUD rendering matched unrelated donors a good donor choice in the absence of matched sibling donors. Outcomes were not different between 9/10-HLA- (n = 10) and 10/10-matched MUD (n = 25), but the numbers were low. Two fatherhoods were documented after successful HSCT. To further reduce the risk of graft failure with this RIC-regimen, some investigators have narrowed the submyeloablative target of the cumulative AUC of busulfan to 55–65 mg/L × h (83) and have started using busulfan starting doses based on a new body weight-dependent busulfan dosing nomogram (91).
Morillo-Gutierrez et al. (74) showed in a large retrospective European study of the EBMT on 70 CGD-children that HSCT after treosulfan-based conditioning was well tolerated and achieved OS/EFS-rates of 91.4/81.4%, respectively. Treosulfan, an alkylating drug with both myeloablative and immunosuppressive effects, exhibited an overall low acute toxicity in CGD transplants. If used as a single alkylator, treosulfan may be less gonadotoxic than other alkylators, however, there is no study yet available convincingly proving this assumption (92–94). Excellent myeloid DC (>95%) was documented in 80% of surviving patients. With this paper, treosulfan-based RTC was shown to be an alternative conditioning to targeted busulfan-based-RIC, although it remained unclear which treosulfan systemic exposure was more likely to be myeloablative or submyeloablative. Graft failure remained a problem occurring in 12% of the patients (74). Some centers have therefore started to add thiotepa to treosulfan to further reduce the risk of GF (80), probably at the expense of augmented gonadotoxicity (94).
The experience with unrelated 4/6–6/6-HLA-compatible cord blood transplants (CBT) in CGD is scarce, but there are a few examples of successful transplants using cord blood in patients lacking MSD or MUD (72, 95, 96). Due to low HSC-numbers in CB, CBT is usually restricted to patients with low body weight (<20 kg) and viral reactivations may be of concern. CBT usually requires myeloablative and therefore more gonadotoxic conditioning, e.g., busulfan (cAUC 80–100 mg/L × h) or treosulfan/thiotepa, to achieve sufficient myeloid engraftment.
For this review, we have analyzed the results of the two above mentioned major European studies together with 15 other relevant international papers published between 2010 and 2019. We have summarized the results of 386 CGD-patients receiving transplants from mainly MSD/MFD- and MUD-donors in Table 1. The median overall incidences of OS, EFS, graft failure, chronic GVHD and de-novo autoimmune disease in these 17 papers were 92, 81, 14, 9, and 15%, respectively. The most important secondary problems were graft failure including patients with slowly falling myeloid DC <10% (DHR/NBT-tests <10%), de-novo autoimmunity and chronic GVHD. Graft failure or low donor myeloid DC was associated with reappearance of CGD associated symptoms, and chronic GVHD clearly impacted negatively on quality-of-life and life expectancy (Table 1).
Hoenig et al. demonstrated for the first time that haploidentical HSCT was curative CGD (97). They used myeloablative conditioning (full-dosed busulfan, thiotepa and alemtuzumab) and in-vitro selected peripheral HSCs and achieved full donor donor cell engraftment and complete resolution of pulmonary aspergillosis. More recently, haploidentical TCR alpha-beta -/CD19-depleted grafts were shown to successfully achieve myeloid donor cell engraftment without inducing relevant GVHD (103–105). The advantage of these in-vitro T-cell depletion techniques is that chronic GVHD is rare (14, 80, 106). In-vivo T-cell depletion strategies in haploidentical transplants include the use post-transplant cyclophosphamide (PT/CY) (50 mg/kg/day), administered on day+3 and day +4 (107). PT/CY is non-toxic to donor HSCs, but efficiently eliminates activated alloreactive donor-derived CD3+T-cells while sparing resting CD3+T-cells with potential anti-infective properties. The first successful haplo HSCT with PT/CY in CGD was reported in the USA after the administration of targeted busulfan (cAUC 40 mg/L × h), fludarabine, cyclophosphamide and 2 Gy TBI (98). However, in a very recent follow-up paper by Parta et al. on seven patients with CGD a rather high rate of severe GVHD was observed leading to death in two patients (OS and EFS 71%, respectively). The estimated total cumulative of busulfan ranged from 30–52 mg/L × h (2,461–4,250 min × micromol/L × 3 days). They used a protocol with mainly PBSC grafts and sirolimus for GVHD-prophylaxis (101). Patients' age ranged between 14 and 26 years and comprised of mainly adults. Severe grade III acute GVHD were observed in three patients with enterocolitis.
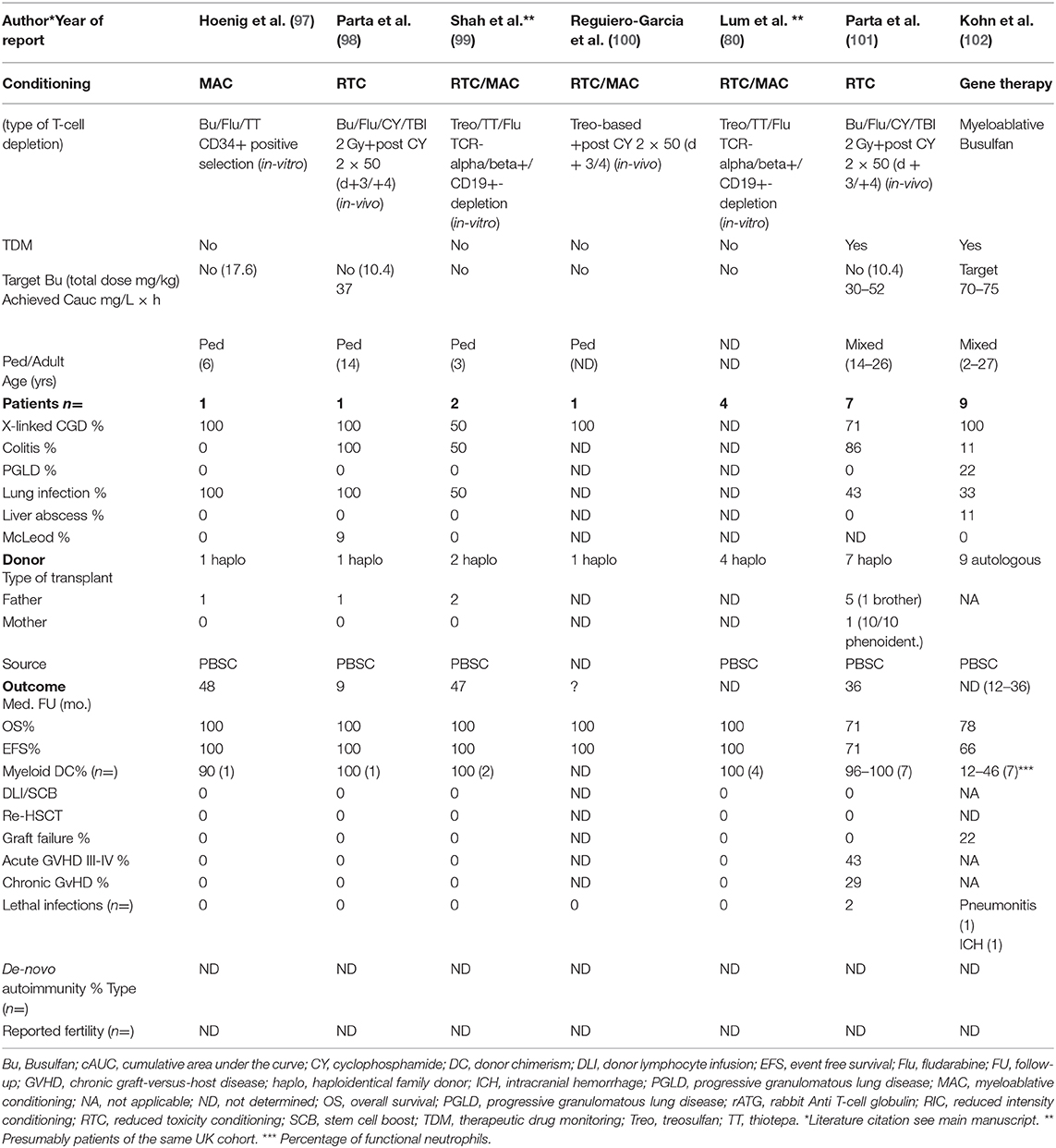
Table 2. Haploidentical HSCT and autologous gene-therapy in CGD between 2014 and 2020.
Another currently investigated RTC-protocol is currently used in our institution. It comprises of up-front rabbit ATG (30–40 mg/kg), fludarabine (180 mg/sqm) and targeted busulfan (cAUC 65–75 mg/L × h) followed by haplo-HSCT with PT/CY and GVHD prophylaxis with CSA and MMF (starting at day +5). We believe that both haploidentical HSCT with PT/CY and with antibodies containing magnetic beads are promising alternatives in high-risk patients with CGD when HLA-matched related or unrelated donors are unavailable (105). To further explore the rates of graft failure and cGVHD after haploidentical HSCT comparative studies of both techniques are urgently needed (Table 2).
Autologous gene-therapy (GT) of HSCs leads to partially functional correction of defective phagocytes and is a potentially curative treatment approach in CGD. Graft failure may occur after GT, but the risk of GVHD is zero (102, 108, 109). While early studies with unconditioned transfusions of retrovirally transduced HSCs were unsuccessful (110), autologous infusion of HSCs transduced with a gamma-retroviral vector after busulfan-based myeloablative conditioning helped to successfully engraft 4 CGD-patients (2 adults, 2 children) (109). Approximately 15% of gp91phox-expressing neutrophils had been detectable within the first 5 months after GT leading to resolution of life-threatening invasive fungal infections. Unfortunately, methylation with downregulation of the transduced gene and clonal expansion of transduced myeloid cells due to random viral integrations were observed, leading to activation of endogenous oncogenes and development of MDS with or without monosomy 7. Both children treated with GT survived after subsequent allogeneic HSCT (65), while 2 adult patients died due to secondary MDS and AML, respectively. Recently, nine X-CGD-patients (age 2–27 years) received GT using a self-inactivating lentiviral vector designed to limit the risk of mutagenesis (102). Patients were pretreated with myeloablative busulfan exposures (cAUC 70–75 mg/L × h). Two patients died within 3 months from GT due to severe pulmonary disease and hemorrhage. At 12 months, 6/7 surviving patients demonstrated persistence of sufficiently NADPH-oxidase-expressing neutrophils (16–46%) and stable vector copy numbers. One patient had graft failure with a decline <5% enzyme-producing neutrophils. There was no evidence of clonal dysregulation or transgene silencing. Surviving patients did not develop new CGD-related infections, and six have been able to discontinue antibiotic prophylaxis (OS/EFS >12 mo.: 78/66%, respectively) (Table 2).
Traditionally, indications for HSCT in CGD had been the following: (1) > 1 invasive life-threatening infections, (2) non-tolerability of prophylactic drugs, (3) non-compliance, (4) severe autoinflammation, or (5) unavailability of a CGD-experienced physician (14, 21, 22, 111). Due to the above mentioned favorable results, there is nowadays agreement that HLA-matched HSCT is indicated in any CGD-patient with absent NADPH-oxidase enzyme activity (16). Small children with CGD may clearly benefit from 5/6- or 6/6-HLA-matched CBT in experienced centers. Less than 10/10-HLA-MUD should probably not be offered to asymptomatic CGD-patients since the rates of graft failure and chronic GVHD are higher than in completely matched transplants. The indication for HSCT in adults should be carefully assessed by the treating physician, although the results in recent years have been encouraging (38, 63, 67, 78, 84, 112). We believe that haploidentical transplants and GT in X-CGD should only be offered to high-risk CGD-patients suffering from severe infectious and/or autoinflammatory complications with no other treatment alternatives. Ideally, high-risk CGD-patients without matched donors should be prospectively investigated in trials comparing GT vs. haploidentical HSCT.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor AL declared a collaboration with the authors RC, TG.
aGVHD, acute graft-vs. -host disease; AIHA, autoimmune hemolytic anemia; AR-CGD, autosomal recessive CGD; BM, bone marrow; Bu, busulfan; CGD, chronic granulomatous disease; cAUC, cumulative area under the curve; CBT, cord blood transplantation; cGVHD, chronic graft-vs.-host disease; CY, cyclophosphamide; DHR, Dihydrorhodamin test; DC, donor chimerism; DLI, donor lymphocyte infusion; EBMT, European Group For Bone and Marrow Transplantation; EFS, event-free survival; Flu, fludarabine; FU, follow-up; GBS, Guillain Barré syndrome; GT, gene therapy; GVHD, graft-vs.-host disease; HSC, hematopoietic stem cell; HSCT, hematopoietic stem cell transplantation; IFI, invasive fungal infection; IBD, inflammatory bowel disease; ITP, idiopathic thrombocytopenic purpura; MAC, myeloablative conditioning; MUD, matched unrelated donor; Haplo, haploidentical; Mel, melphalan; MMUD, mismatched unrelated donor; MSD, matched sibling donor; Ped, pediatric; NBT, Nitroblue tetrazolium test; OS, overall survival; PB, peripheral blood; PGLD, progressive granulomatous lung disease; PTLD, post-transplant lymphoproliferative disorder; RIC, reduced intensity conditioning; TBI, total body irradiation; TNI, total nodal irradiation; TDM, therapeutic drug monitoring; Treo, treosulfan; TRM, transplant-related mortality; TT, thiotepa; UCB, umbilical cord blood; X-CGD, X-linked CGD.
1. Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine. (2000) 79:170–200. doi: 10.1097/00005792-200005000-00004
2. van de Geer A, Nieto-Patlan A, Kuhns DB, Tool At, Arias AA, Bouaziz M, et al. Inherited p40phox deficiency differs from classic chronic granulomatous disease. J Clin Invest. (2018) 128:3957–75. doi: 10.1172/JCI97116
3. Dinauer MC. Chronic granulomatous disease and other disorders of phagocyte function. Hematology Am Soc Hematol Educ Program. (2005) 2005:89–95. doi: 10.1182/asheducation-2005.1.89
4. Thomas DC, Charbonnier LM, Schejtman A, Aldhekri H, Coomber EL, Dufficy ER, et al. EROS/CYBC1 mutations: decreased NADPH oxidase function and chronic granulomatous disease. J Allergy Clin Immunol. (2019) 143:782–5.e781. doi: 10.1016/j.jaci.2018.09.019
5. Ahlin A, Fasth A. Chronic granulomatous disease - conventional treatment vs. hematopoietic stem cell transplantation: an update. Curr Opin Hematol. (2015) 22:41–5. doi: 10.1097/MOH.0000000000000097
6. Winkelstein JA, Marino MC, Johnston RB Jr, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine. (2000) 79:155–69. doi: 10.1097/00005792-200005000-00003
7. van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic granulomatous disease: the European experience. PLoS ONE. (2009) 4:e5234. doi: 10.1371/journal.pone.0005234
8. Magnani A, Brosselin P, Beaute J, de Vergnes N, Mouy R, Debré M, et al. Inflammatory manifestations in a single-center cohort of patients with chronic granulomatous disease. J Allergy Clin Immunol. (2014) 134:655–62.e658. doi: 10.1016/j.jaci.2014.04.014
9. Marciano BE, Zerbe CS, Falcone EL, Ding L, DeRavin SS, Daub J, et al. X-linked carriers of chronic granulomatous disease: illness, lyonization, and stability. J Allergy Clin Immunol. (2018) 141:365–71. doi: 10.1016/j.jaci.2017.04.035
10. Pulvirenti F, Sangerardi M, Plebani A, Soresina A, Finocchi A, Pignata C, et al. Health-related quality of life and emotional difficulties in chronic granulomatous disease: data on adult and pediatric patients from italian network for primary immunodeficiency (IPINet). J Clin Immunol. (2020) 40:289–98. doi: 10.1007/s10875-019-00725-1
11. Cole TS, McKendrick F, Cant AJ, Pearce MS, Cale CM, Goldblatt DR, et al. Cognitive ability in children with chronic granulomatous disease: a comparison of those managed conservatively with those who have undergone hematopoietic stem cell transplant. Neuropediatrics. (2013) 44:230–2. doi: 10.1055/s-0033-1333875
12. Cole T, McKendrick F, Titman P, Cant AJ, Pearce MS, Cale CM, et al. Health related quality of life and emotional health in children with chronic granulomatous disease: a comparison of those managed conservatively with those that have undergone haematopoietic stem cell transplant. J Clin Immunol. (2013) 33:8–13. doi: 10.1007/s10875-012-9758-0
13. Cole T, Pearce MS, Cant AJ, Cale CM, Goldblatt D, Gennery AR. Clinical outcome in children with chronic granulomatous disease managed conservatively or with hematopoietic stem cell transplantation. J Allergy Clin Immunol. (2013) 132:1150–5. doi: 10.1016/j.jaci.2013.05.031
14. Gennery A. Recent advances in understanding and treating chronic granulomatous disease. F1000Res. (2017) 6:1427. doi: 10.12688/f1000research.11789.1
15. Holland S, Seger R, Sullivan KE. The chronicles of chronic granulomatous disease. Clin Immunol. (2005) 116:99–100. doi: 10.1016/j.clim.2005.04.001
16. Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. (2010) 363:2600–10. doi: 10.1056/NEJMoa1007097
18. Kaltenis P, Mudeniene V, Maknavicius S, Seinin D. Renal amyloidosis in a child with chronic granulomatous disease and invasive aspergillosis. Pediatr Nephrol. (2008) 23:831–4. doi: 10.1007/s00467-007-0702-0
19. Cole TS, Jones LK, McGrogan P, Pearce MS, Flood TJ, Cant AJ, et al. Emotional and behavioural difficulties in chronic granulomatous disease. Arch Dis Child. (2012) 97:87. doi: 10.1136/archdischild-2011-300780
20. Battersby AC, Braggins H, Pearce MS, McKendrick F, Campbell M, Burns S, et al. Health-related quality of life and emotional health in x-linked carriers of chronic granulomatous disease in the United Kingdom. J Clin Immunol. (2019) 39:195–9. doi: 10.1007/s10875-019-00607-6
21. Thomsen IP, Smith MA, Holland SM, Creech CB. A comprehensive approach to the management of children and adults with chronic granulomatous disease. J Allergy Clin Immunol Pract. (2016) 4:1082–8. doi: 10.1016/j.jaip.2016.03.021
22. Connelly JA, Marsh R, Parikh S, Talano JA. Allogeneic hematopoietic cell transplantation for chronic granulomatous disease: controversies and state of the art. J Pediatric Infect Dis Soc. (2018) 7(Suppl_1):S31–9. doi: 10.1093/jpids/piy015
23. Wolach B, Gavrieli R, de Boer M, van Leeuwen K, Wolach O, Grisaru-Soen G, et al. Analysis of chronic granulomatous disease in the kavkazi population in israel reveals phenotypic heterogeneity in patients with the same NCF1 mutation (c.579G>A). J Clin Immunol. (2018) 38:193–203. doi: 10.1007/s10875-018-0475-1
24. Bustamante J, Aksu G, Vogt G, De Beaucoudrey L, Genel F, Chapgier A, et al. BCG-osis and tuberculosis in a child with chronic granulomatous disease. J Allergy Clin Immunol. (2007) 120:32–8. doi: 10.1016/j.jaci.2007.04.034
25. Reichenbach J, Lopatin U, Mahlaoui N, Beovic B, Siler U, Zbinden R, et al. Actinomyces in chronic granulomatous disease: an emerging and unanticipated pathogen. Clin Infect Dis. (2009) 49:1703–10. doi: 10.1086/647945
26. Greenberg DE, Shoffner AR, Zelazny AM, Fenster ME, Zarember KA, Stock F, et al. Recurrent Granulibacter bethesdensis infections and chronic granulomatous disease. Emerg Infect Dis. (2010) 16:1341–8. doi: 10.3201/eid1609.091800
27. Greenberg DE, Ding L, Zelazny AM, Stock F, Wong A, Anderson VL, et al. A novel bacterium associated with lymphadenitis in a patient with chronic granulomatous disease. PLoS Pathog. (2006) 2:e28. doi: 10.1371/journal.ppat.0020028
28. Tarlow MJ, Lloyd J. Melioidosis and chronic granulomatous disease. Proc R Soc Med. (1971) 64:19–20. doi: 10.1177/003591577106400111
29. Meher-Homji Z, Mangalore RP, P DRJ, K YLC. Chromobacterium violaceum infection in chronic granulomatous disease: a case report and review of the literature. JMM Case Rep. (2017) 4:e005084. doi: 10.1099/jmmcr.0.005084
30. Renella R, Perez JM, Chollet-Martin S, Sarnacki S, Fischer A, Blanche S, et al. Burkholderia pseudomallei infection in chronic granulomatous disease. Eur J Pediatr. (2006) 165:175–7. doi: 10.1007/s00431-005-0022-y
31. Henriet S, Verweij PE, Holland SM, Warris A. Invasive fungal infections in patients with chronic granulomatous disease. Adv Exp Med Biol. (2013) 764:27–55. doi: 10.1007/978-1-4614-4726-9_3
32. Segal BH, Barnhart LA, Anderson VL, Walsh TJ, Malech HL, Holland SM. Posaconazole as salvage therapy in patients with chronic granulomatous disease and invasive filamentous fungal infection. Clin Infect Dis. (2005) 40:1684–8. doi: 10.1086/430068
33. Mouy R, Veber F, Blanche S, Donadieu J, Brauner R, Levron JC, et al. Long-term itraconazole prophylaxis against Aspergillus infections in thirty-two patients with chronic granulomatous disease. J Pediatr. (1994) 125(6 Pt 1):998–1003. doi: 10.1016/S0022-3476(05)82023-2
34. Mortensen KL, Mellado E, Lass-Florl C, Rodriguez-Tudela JL, Johansen HK, Arendrup MC. Environmental study of azole-resistant Aspergillus fumigatus and other aspergilli in Austria, Denmark, and Spain. Antimicrob Agents Chemother. (2010) 54:4545–9. doi: 10.1128/AAC.00692-10
35. de Luca A, Smeekens SP, Casagrande A, Iannitti R, Conway KL, Gresnigt MS, et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc Natl Acad Sci USA. (2014) 111:3526–31. doi: 10.1073/pnas.1322831111
36. Fernandez-Boyanapalli RF, Falcone EL, Zerbe CS, Marciano BE, Frasch SC, Henson PM, et al. Impaired efferocytosis in human chronic granulomatous disease is reversed by pioglitazone treatment. J Allergy Clin Immunol. (2015) 136:1399–401 e1393. doi: 10.1016/j.jaci.2015.07.034
37. Bagaitkar J, Huang J, Zeng MY, Pech NK, Monlish DA, Perez-Zapata LJ, et al. NADPH oxidase activation regulates apoptotic neutrophil clearance by murine macrophages. Blood. (2018) 131:2367–78. doi: 10.1182/blood-2017-09-809004
38. Freudenberg F, Wintergerst U, Roesen-Wolff A, Albert MH, Prell C, Strahm B, et al. Therapeutic strategy in p47-phox deficient chronic granulomatous disease presenting as inflammatory bowel disease. J Allergy Clin Immunol. (2010) 125:943–6.e941. doi: 10.1016/j.jaci.2010.01.035
39. Schappi MG, Smith VV, Goldblatt D, Lindley KJ, Milla PJ. Colitis in chronic granulomatous disease. Arch Dis Childhood. (2001) 84:147–51. doi: 10.1136/adc.84.2.147
40. Marciano BE, Rosenzweig SD, Kleiner DE, Anderson VL, Darnell DN, Anaya-O'Brien S, et al. Gastrointestinal involvement in chronic granulomatous disease. Pediatrics. (2004) 114:462–8. doi: 10.1542/peds.114.2.462
41. De Ravin SS, Naumann N, Robinson MR, Barron KS, Kleiner DE, Ulrick J, et al. Sarcoidosis in chronic granulomatous disease. Pediatrics. (2006) 117:e590–5. doi: 10.1542/peds.2005-1349
42. Freeman AF, Marciano BE, Anderson VL, Uzel G, Costas C, Holland SM. Corticosteroids in the treatment of severe nocardia pneumonia in chronic granulomatous disease. Pediatr Infect Dis J. (2011) 30:806–8. doi: 10.1097/INF.0b013e318218181d
43. Deffert C, Olleros ML, Huiping Y, Herrmann FR, Zekry D, Garcia I, et al. TNF-alpha blockade in chronic granulomatous disease-induced hyperinflammation: patient analysis and murine model. J Allergy Clin Immunol. (2011) 128:675–7. doi: 10.1016/j.jaci.2011.04.028
44. Noel N, Mahlaoui N, Blanche S, Suarez F, Coignard-Biehler H, Durieu I, et al. Efficacy and safety of thalidomide in patients with inflammatory manifestations of chronic granulomatous disease: a retrospective case series. J Allergy Clin Immunol. (2013) 132:997–1000.e1001–1004. doi: 10.1016/j.jaci.2013.04.059
45. Uzel G, Orange JS, Poliak N, Marciano BE, Heller T, Holland SM. Complications of tumor necrosis factor-alpha blockade in chronic granulomatous disease-related colitis. Clin Infect Dis. (2010) 51:1429–34. doi: 10.1086/657308
46. Hahn KJ, Ho N, Yockey L, Kreuzberg S, Daub J, Rump A, et al. Treatment with anakinra, a recombinant IL-1 receptor antagonist, unlikely to induce lasting remission in patients with CGD colitis. Am J Gastroenterol. (2015) 110:938–9. doi: 10.1038/ajg.2015.135
47. Hahn L, Beggs A, Wahaib K, Kodali L, Kirkwood V. Vedolizumab: an integrin-receptor antagonist for treatment of Crohn's disease and ulcerative colitis. Am J Health Syst Pharm. (2015) 72:1271–8. doi: 10.2146/ajhp140449
48. Butte MJ, Park KT, Lewis DB. Treatment of CGD-associated Colitis with the IL-23 Blocker Ustekinumab. J Clin Immunol. (2016) 36:619–20. doi: 10.1007/s10875-016-0318-x
49. Frazao JB, Colombo M, Simillion C, Bilican A, Keller I, Wüthrich D, et al. Gene expression in chronic granulomatous disease and interferon-gamma receptor-deficient cells treated in vitro with interferon-gamma. J Cell Biochem. (2019) 120:4321–32. doi: 10.1002/jcb.27718
50. Fernandez-Boyanapalli R, McPhillips KA, Frasch SC, Janssen WJ, Dinauer MC, Riches DW, et al. Impaired phagocytosis of apoptotic cells by macrophages in chronic granulomatous disease is reversed by IFN-gamma in a nitric oxide-dependent manner. J Immunol. (2010) 185:4030–41. doi: 10.4049/jimmunol.1001778
51. Mouy R, Seger R, Bourquin JP, Veber F, Blanche S, Griscelli C, et al. Interferon gamma for chronic granulomatous disease. N Engl J Med. (1991) 325:1516–7. doi: 10.1056/NEJM199111213252115
52. Ezekowitz RA, Sieff CA, Dinauer MC, Nathan DG, Orkin SH, Newburger PE. Restoration of phagocyte function by interferon-gamma in X-linked chronic granulomatous disease occurs at the level of a progenitor cell. Blood. (1990) 76:2443–8. doi: 10.1182/blood.V76.12.2443.2443
53. Alimchandani M, Lai JP, Aung PP, Khangura S, Kamal N, Gallin JI, et al. Gastrointestinal histopathology in chronic granulomatous disease: a study of 87 patients. Am J Surg Pathol. (2013) 37:1365–72. doi: 10.1097/PAS.0b013e318297427d
54. Yang Y, Xiang Z, Ertl HC, Wilson JM. Upregulation of class I major histocompatibility complex antigens by interferon gamma is necessary for T-cell-mediated elimination of recombinant adenovirus-infected hepatocytes in vivo. Proc Natl Acad Sci USA. (1995) 92:7257–61. doi: 10.1073/pnas.92.16.7257
55. Ezekowitz RA, Dinauer MC, Jaffe HS, Orkin SH, Newburger PE. Partial correction of the phagocyte defect in patients with X-linked chronic granulomatous disease by subcutaneous interferon gamma. N Engl J Med. (1988) 319:146–51. doi: 10.1056/NEJM198807213190305
56. Gungor T, Engel-Bicik I, Eich G, Willi UV, Nadal D, Hossle JP, et al. Diagnostic and therapeutic impact of whole body positron emission tomography using fluorine-18-fluoro-2-deoxy-D-glucose in children with chronic granulomatous disease. Arch Dis Child. (2001) 85:341–5. doi: 10.1136/adc.85.4.341
57. Straughan DM, McLoughlin KC, Mullinax JE, Marciano BE, Freeman AF, Anderson VL, et al. The changing paradigm of management of liver abscesses in chronic granulomatous disease. Clin Infect Dis. (2018) 66:1427–34. doi: 10.1093/cid/cix1012
58. Siddiqui S, Anderson VL, Hilligoss DM, Abinun M, Kuijpers TW, Masur H, et al. Fulminant mulch pneumonitis: an emergency presentation of chronic granulomatous disease. Clin Infect Dis. (2007) 45:673–81. doi: 10.1086/520985
59. Leiding JW, Freeman AF, Marciano BE, Anderson VL, Uzel G, Malech HL, et al. Corticosteroid therapy for liver abscess in chronic granulomatous disease. Clin Infect Dis. (2012) 54:694–700. doi: 10.1093/cid/cir896
60. Marciano BE, Allen ES, Conry-Cantilena C, Kristosturyan E, Klein HG, Fleisher TA, et al. Granulocyte transfusions in patients with chronic granulomatous disease and refractory infections: The NIH experience. J Allergy Clin Immunol. (2017) 140:622–5. doi: 10.1016/j.jaci.2017.02.026
61. Heim KF, Fleisher TA, Stroncek DF, Holland SM, Gallin JI, Malech HL, et al. The relationship between alloimmunization and posttransfusion granulocyte survival: experience in a chronic granulomatous disease cohort. Transfusion. (2011) 51:1154–62. doi: 10.1111/j.1537-2995.2010.02993.x
62. Shigemura T, Nakazawa Y, Yoshikawa K, Hirabayashi K, Saito S, Kobayashi N, et al. Successful cord blood transplantation after repeated transfusions of unmobilized neutrophils in addition to antifungal treatment in an infant with chronic granulomatous disease complicated by invasive pulmonary aspergillosis. Transfusion. (2014) 54:516–21. doi: 10.1111/trf.12325
63. Gungor T, Teira P, Slatter M, Stussi G, Stepensky P, Moshous D, et al. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet. (2014) 383:436–48. doi: 10.1016/S0140-6736(13)62069-3
64. Parta M, Kelly C, Kwatemaa N, Theobald N, Hilligoss D, Qin J, et al. Allogeneic reduced-intensity hematopoietic stem cell transplantation for chronic granulomatous disease: a single-center prospective trial. J Clin Immunol. (2017) 37:548–58. doi: 10.1007/s10875-017-0422-6
65. Siler U, Paruzynski A, Holtgreve-Grez H, Kuzmenko E, Koehl U, D Renner E, et al. Successful combination of sequential gene therapy and rescue allo-HSCT in two children with X-CGD - importance of timing. Curr Gene Ther. (2015) 15:416–27. doi: 10.2174/1566523215666150515145255
66. Horwitz ME, Barrett AJ, Brown MR, Carter CS, Childs R, Gallin JI, et al. Treatment of chronic granulomatous disease with nonmyeloablative conditioning and a T-cell-depleted hematopoietic allograft. N Engl J Med. (2001) 344:881–8. doi: 10.1056/NEJM200103223441203
67. Seger RA, Gungor T, Belohradsky BH, Blanche S, Bordigoni P, Di Bartolomeo P, et al. Treatment of chronic granulomatous disease with myeloablative conditioning and an unmodified hemopoietic allograft: a survey of the European experience, 1985-2000. Blood. (2002) 100:4344–50. doi: 10.1182/blood-2002-02-0583
68. Schuetz C, Hoenig M, Gatz S, Speth F, Benninghoff U, Schulz A, et al. Hematopoietic stem cell transplantation from matched unrelated donors in chronic granulomatous disease. Immunol Res. (2009) 44:35–41. doi: 10.1007/s12026-008-8068-3
69. Soncini E, Slatter MA, Jones LB, Hughes S, Hodges S, Flood TJ, et al. Unrelated donor and HLA-identical sibling haematopoietic stem cell transplantation cure chronic granulomatous disease with good long-term outcome and growth. Br J Haematol. (2009) 145:73–83. doi: 10.1111/j.1365-2141.2009.07614.x
70. Gozdzik J, Pituch-Noworolska A, Skoczen S, Czogala W, Wedrychowicz A, Baran J, et al. Allogeneic haematopoietic stem cell transplantation as therapy for chronic granulomatous disease–single centre experience. J Clin Immunol. (2011) 31:332–7. doi: 10.1007/s10875-011-9513-y
71. Martinez CA, Shah S, Shearer WT, Rosenblatt HM, Paul ME, Chinen J, et al. Excellent survival after sibling or unrelated donor stem cell transplantation for chronic granulomatous disease. J Allergy Clin Immunol. (2012) 129:176–83. doi: 10.1016/j.jaci.2011.10.005
72. Tewari P, Martin PL, Mendizabal A, Parikh SH, Page KM, Driscoll TA, et al. Myeloablative transplantation using either cord blood or bone marrow leads to immune recovery, high long-term donor chimerism and excellent survival in chronic granulomatous disease. Biol Blood Marrow Transplant. (2012) 18:1368–77. doi: 10.1016/j.bbmt.2012.02.002
73. Ahlin A, Fugelang J, de Boer M, Ringden O, Fasth A, Winiarski J. Chronic granulomatous disease-haematopoietic stem cell transplantation versus conventional treatment. Acta Paediatr. (2013) 102:1087–94. doi: 10.1111/apa.12384
74. Morillo-Gutierrez B, Beier R, Rao K, Burroughs L, Schulz A, Ewins AM, et al. Treosulfan-based conditioning for allogeneic HSCT in children with chronic granulomatous disease: a multicenter experience. Blood. (2016) 128:440–8. doi: 10.1182/blood-2016-03-704015
75. Khandelwal P, Bleesing JJ, Davies SM, Marsh RA. A single-center experience comparing alemtuzumab, fludarabine, and melphalan reduced-intensity conditioning with myeloablative busulfan, cyclophosphamide, and antithymocyte globulin for chronic granulomatous disease. Biol Blood Marrow Transplant. (2016) 22:2011–8. doi: 10.1016/j.bbmt.2016.08.013
76. Osumi T, Tomizawa D, Kawai T, Sako M, Inoue E, Takimoto T, et al. A prospective study of allogeneic transplantation from unrelated donors for chronic granulomatous disease with target busulfan-based reduced-intensity conditioning. Bone Marrow Transplant. (2019) 54:168–72. doi: 10.1038/s41409-018-0271-9
77. Yanir AD, Hanson IC, Shearer WT, Noroski LM, Forbes LR, Seeborg FO, et al. High Incidence of Autoimmune Disease after Hematopoietic Stem Cell Transplantation for Chronic Granulomatous Disease. Biol Blood Marrow Transplant. (2018) 24:1643–50. doi: 10.1016/j.bbmt.2018.03.029
78. Fox TA, Chakraverty R, Burns S, Carpenter B, Thomson K, Lowe D, et al. Successful outcome following allogeneic hematopoietic stem cell transplantation in adults with primary immunodeficiency. Blood. (2018) 131:917–31. doi: 10.1182/blood-2017-09-807487
79. Arnold DE, Seif AE, Jyonouchi S, Sullivan KE, Bunin NJ, Heimall JR. Allogeneic hematopoietic stem cell transplantation in adolescent patients with chronic granulomatous disease. J Allergy Clin Immunol Pract. (2019) 7:1052–4. doi: 10.1016/j.jaip.2018.10.034
80. Lum SH, Flood T, Hambleton S, McNaughton P, Watson H, Abinun M, et al. Two decades of excellent transplant survival for chronic granulomatous disease: a supraregional immunology transplant center report. Blood. (2019) 133:2546–9. doi: 10.1182/blood.2019000021
81. Yang S, Panoskaltsis-Mortari A, Shukla M, Blazar BR, Haddad IY. Exuberant inflammation in nicotinamide adenine dinucleotide phosphate-oxidase-deficient mice after allogeneic marrow transplantation. J Immunol. (2002) 168:5840–7. doi: 10.4049/jimmunol.168.11.5840
82. Jacobsohn DA, Duerst R, Tse W, Kletzel M. Reduced intensity haemopoietic stem-cell transplantation for treatment of non-malignant diseases in children. Lancet. (2004) 364:156–62. doi: 10.1016/S0140-6736(04)16628-2
83. Gungor T. Favourable outcome in 47 patients with chronic granulomatous disease after busulfan-based reduced toxicity conditioning and allogeneic hematopoietic stem cell transplantation. J Clin Immunol. (2012) 32:138.
84. Gungor T, Halter J, Klink A, Junge S, Stumpe KD, Seger R, et al. Successful low toxicity hematopoietic stem cell transplantation for high-risk adult chronic granulomatous disease patients. Transplantation. (2005) 79:1596–606. doi: 10.1097/01.TP.0000163466.73485.5E
85. Gungor T, Albert M, Schanz U, Slatter M, Gennery A, Waver A, et al. Low-dose busulfan/full-dose fludarabine-based reduced-intensity conditioning in 30 high-risk paediatric and adult chronic granulomatous disease patients. Bone Marrow Transplant. (2011) 46:S16. doi: 10.1016/j.bbmt.2009.12.094
86. Ratzinger G, Reagan JL, Heller G, Busam KJ, Young JW. Differential CD52 expression by distinct myeloid dendritic cell subsets: implications for alemtuzumab activity at the level of antigen presentation in allogeneic graft-host interactions in transplantation. Blood. (2003) 101:1422–9. doi: 10.1182/blood-2002-04-1093
87. Bartelink IH, Lalmohamed A, van Reij EM, Dvorak CC, Savic RM, Zwaveling J, et al. Association of busulfan exposure with survival and toxicity after haemopoietic cell transplantation in children and young adults: a multicentre, retrospective cohort analysis. Lancet Haematol. (2016) 3:e526–36. doi: 10.1016/S2352-3026(16)30114-4
88. Hassan M, Ljungman P, Bolme P, Ringden O, Syruckova Z, Bekassy A, et al. Busulfan bioavailability. Blood. (1994) 84:2144–50. doi: 10.1182/blood.V84.7.2144.2144
89. Bartelink IH, van Reij EM, Gerhardt CE, Van Maarseveen EM, De Wildt A, Versluys B, et al. Fludarabine and exposure-targeted busulfan compares favorably with busulfan/cyclophosphamide-based regimens in pediatric hematopoietic cell transplantation: maintaining efficacy with less toxicity. Biol Blood Marrow Transplant. (2014) 20:345–53. doi: 10.1016/j.bbmt.2013.11.027
90. Malar R, Sjoo F, Rentsch K, Hassan M, Gungor T. Therapeutic drug monitoring is essential for intravenous busulfan therapy in pediatric hematopoietic stem cell recipients. Pediatric Transplant. (2011) 15:580–8. doi: 10.1111/j.1399-3046.2011.01529.x
91. Bartelink IH, Boelens JJ, Bredius RG, Wang C, Bierings MB, Shaw PJ, et al. Body weight-dependent pharmacokinetics of busulfan in paediatric haematopoietic stem cell transplantation patients: towards individualized dosing. Clin Pharmacokinet. (2012) 51:331–45. doi: 10.2165/11598180-000000000-00000
92. Levi M, Stemmer SM, Stein J, Shalgi R, Ben-Aharon I. Treosulfan induces distinctive gonadal toxicity compared with busulfan. Oncotarget. (2018) 9:19317–27. doi: 10.18632/oncotarget.25029
93. Haskologlu S, Kostel Bal S, Islamoglu C, Altun D, Kendirli T, Dogu EF, et al. Outcome of treosulfan-based reduced-toxicity conditioning regimens for HSCT in high-risk patients with primary immune deficiencies. Pediatr Transplant. (2018) 22:e13266. doi: 10.1111/petr.13266
94. Boztug H, Zecca M, Sykora KW, Veys P, Lankester A, Slatter M, et al. Treosulfan-based conditioning regimens for allogeneic HSCT in children with acute lymphoblastic leukaemia. Ann Hematol. (2015) 94:297–306. doi: 10.1007/s00277-014-2196-8
95. Morio T, Atsuta Y, Tomizawa D, Nagamura-Inoue T, Kato K, Ariga T, et al. Outcome of unrelated umbilical cord blood transplantation in 88 patients with primary immunodeficiency in Japan. Br J Haematol. (2011) 154:363–72. doi: 10.1111/j.1365-2141.2011.08735.x
96. Gungor T, Fraser C, Duffner U, Savasan S, Ballhausen D, Hauri-Hohl M, et al. Targeted busulfan conditioning and unrelated cord blood transplantation in pediatric primary immunodeficiencies and neurometabolic diseases other than Hurler's disease. Bone Marrow Transplant. (2015) 50:S467–8.
97. Hoenig M, Niehues T, Siepermann K, Jacobsen EM, Schütz C, Furlan I, et al. Successful HLA haploidentical hematopoietic SCT in chronic granulomatous disease. Bone Marrow Transplant. (2014) 49:1337–8. doi: 10.1038/bmt.2014.125
98. Parta M, Hilligoss D, Kelly C, Kwatemaa N, Theobald N, Malech H, et al. Haploidentical hematopoietic cell transplantation with post-transplant cyclophosphamide in a patient with chronic granulomatous disease and active infection: a first report. J Clin Immunol. (2015) 35:675–80. doi: 10.1007/s10875-015-0204-y
99. Shah RM, Elfeky R, Nademi Z, Qasim W, Amrolia P, Chiesa R, et al. T-cell receptor alpha beta(+) and CD19(+) cell-depleted haploidentical and mismatched hematopoietic stem cell transplantation in primary immune deficiency. J Allergy Clin Immun. (2018) 141:1417. doi: 10.1016/j.jaci.2017.07.008
100. Regueiro-Garcia A, Farina-Nogueira S, Porto-Arceo JA, Couselo-Sanchez JM. Haploidentical stem cell transplantation in a boy with chronic granulomatous disease. Allergol Immunopathol (Madr). (2018) 46:385–8.
101. Parta M, Hilligoss D, Kelly C, Kwatemaa N, Theobald N, Zerbe CS, et al. Failure to prevent severe graft-versus-host disease in haploidentical hematopoietic cell transplantation with post-transplant cyclophosphamide in chronic granulomatous disease. J Clin Immunol. (2020) 40:619–4. doi: 10.1007/s10875-020-00772-z
102. Kohn DB, Booth C, Kang EM, Pai SY, Shaw KL, Santilli G, et al. Lentiviral gene therapy for X-linked chronic granulomatous disease. Nat Med. (2020) 26:200–6. doi: 10.1038/s41591-019-0735-5
103. Balashov D, Shcherbina A, Maschan M, Trakhtman P, Skvortsova Y, Shelikhova L, et al. Single-center experience of unrelated and haploidentical stem cell transplantation with TCRalphabeta and CD19 depletion in children with primary immunodeficiency syndromes. Biol Blood Marrow Transplant. (2015) 21:1955–62. doi: 10.1016/j.bbmt.2015.07.008
104. Bertaina A, Merli P, Rutella S, Pagliara D, Bernardo ME, Masetti R, et al. HLA-haploidentical stem cell transplantation after removal of alphabeta+ T and B cells in children with nonmalignant disorders. Blood. (2014) 124:822–6. doi: 10.1182/blood-2014-03-563817
105. Kharya G, Nademi Z, Leahy TR, Dunn J, Barge D, Schulz A, et al. Haploidentical T-cell alpha beta receptor and CD19-depleted stem cell transplant for Wiskott-Aldrich syndrome. J Allergy Clin Immunol. (2014) 134:1199–201. doi: 10.1016/j.jaci.2014.04.041
106. Slatter MA, Rao K, Abd Hamid IJ, Nademi Z, Chiesa R, Elfeky R, et al. Treosulfan and fludarabine conditioning for hematopoietic stem cell transplantation in children with primary immunodeficiency: UK experience. Biol Blood Marrow Transplant. (2018) 24:529–36. doi: 10.1016/j.bbmt.2017.11.009
107. Luznik L, Bolanos-Meade J, Zahurak M, Chen AR, Smith BD, Brodsky R, et al. High-dose cyclophosphamide as single-agent, short-course prophylaxis of graft-versus-host disease. Blood. (2010) 115:3224–30. doi: 10.1182/blood-2009-11-251595
108. Malech HL, Booth C, Kang EM, Pai SY, Shaw KL, Santilli G, et al. Lentiviral vector gene therapy for X-linked chronic granulomatous disease corrects neutrophil function. J Clin Immunol. (2019) 39:S45–6.
109. Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. (2006) 12:401–9. doi: 10.1038/nm1393
110. Malech HL, Maples PB, Whiting-Theobald N, Linton GF, Sekhsaria S, Vowells SJ, et al. Prolonged production of NADPH oxidase-corrected granulocytes after gene therapy of chronic granulomatous disease. Proc Natl Acad Sci USA. (1997) 94:12133–8. doi: 10.1073/pnas.94.22.12133
111. Chiriaco M, Salfa I, Di Matteo G, Rossi P, Finocchi A. Chronic granulomatous disease: clinical, molecular, and therapeutic aspects. Pediatr Allergy Immunol. (2016) 27:242–53. doi: 10.1111/pai.12527
112. Chiesa R, Wang JF, Blok HJ, Neven B, Moshous D, Schulz A, et al. Allogeneic hematopoietic stem cell transplantation in children and adults with chronic granulomatous disease (CGD): a study of the inborn errors working party (IEWP) of the EBMT. Blood. (2018) 132:970. doi: 10.1182/blood-2018-99-114841
Keywords: chronic granulomatous disease, CGD, hematopoietic stem cell transplantation, conditioning, therapeutic drug monitoring, serotherapy, gene therapy
Citation: Güngör T and Chiesa R (2020) Cellular Therapies in Chronic Granulomatous Disease. Front. Pediatr. 8:327. doi: 10.3389/fped.2020.00327
Received: 14 December 2019; Accepted: 19 May 2020;
Published: 26 June 2020.
Edited by:
Arjan C. Lankester, Leiden University, NetherlandsReviewed by:
Nancy Bunin, Children's Hospital of Philadelphia, United StatesCopyright © 2020 Güngör and Chiesa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tayfun Güngör, dGF5ZnVuLmd1ZW5nb2VyQGtpc3BpLnV6aC5jaA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.