 Anna D. Grabowska
Anna D. Grabowska Eliana M. Lacerda
Eliana M. Lacerda Luís Nacul
Luís Nacul Nuno Sepúlveda
Nuno Sepúlveda
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Pediatr. , 12 June 2020
Sec. Pediatric Neurology
Volume 8 - 2020 | https://doi.org/10.3389/fped.2020.00293
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) is a debilitating disease characterized by persistent fatigue and post-exertion malaise, accompanied by other symptoms (1, 2). The direct cause of the disease remains elusive, but it may include genetic factors alongside environmental triggers, such as strong microbial infections and other stressors (3, 4).
With the aim to identify putative genetic factors that could explain the pathophysiological mechanisms of ME/CFS, four genome-wide association studies (GWAS) and two targeted-genome association studies (TGAS) were conducted in the past decade (5–10). In the four GWAS, thousands of genetic markers located across the whole genome were evaluated for their statistical association with ME/CFS (5–8). The two TGAS had the same statistical objective of the four GWAS, but alternatively investigated the association of the disease with numerous genetic markers located in candidate genes related to inflammation and immunity (9) and in genes encoding diverse adrenergic receptors (10). The findings from all these different studies suggested conflicting evidence of genetic association with ME/CFS: from absence of association (7), through mild association (10) up to moderate associations of a relatively small number of genetic markers (5, 6, 9). The most optimistic GWAS suggested more than 5,500 candidate gene-disease associations (8). This inconsistency in the reported findings prompted us to review the respective data. With this purpose, the present opinion paper first revisits the recommended quality control (QC) checks for GWAS and TGAS, and then summarizes which ones were performed by those studies on ME/CFS.
Current GWAS or TGAS of ME/CFS are based on data of the so-called single nucleotide polymorphisms (SNPs) located in specific positions of the human genome. These genetic markers are short nucleotide sequences that differ in a single position from each other. Each possible sequence of a SNP is interpreted as a different allele. In theory, there are up to four alleles of the same SNP given that there are only four possible nucleotides (A, C, G, and T). However, by design, classical genotyping technologies can only assess the two most frequent alleles per SNP. As an alternative to classical GWAS and TGAS, studies using data from next-generation sequencing technologies are able to assess all possible alleles of a given SNP. As far as we know, these alternative studies have been never performed on ME/CFS.
In general, several QC checks should be performed in the genetic data before carrying out the association analysis itself. First, it is important to determine all monomorphic SNPs and to report the respective number. These SNPs are non-informative for the subsequent genetic association analysis, because they show the same allele in all study participants. It is also important to calculate the so-called minor allele frequency (MAF) of each SNP. Statistically speaking, the MAF is defined as the frequency of the least frequent allele of a given SNP. In practice, a very low MAF is in the same order of magnitude of the underlying genotyping error rate and, therefore, SNPs under this condition should be excluded from the study. A typical threshold for a very low MAF ranges from 1 to 5%. Less stringent thresholds for the MAF can be used in studies with smaller sample sizes.
Second, the validity of the Hardy-Weinberg Equilibrium (HWE) should be tested in the observed genotype frequency distribution of each SNP. The HWE is a mathematical expectation for the probability of observing a given genotype under random mating (or panmixia), no selection, no migration, non-overlapping generations, and no genotyping errors. According to the HWE, the frequency of a given genotype is expected to be factorized into the product of the respective allele frequencies. The HWE is usually tested by the popular Pearson's χ2 goodness-of-fit test. In this statistical test, p-values below the specified significance level suggest evidence against the HWE. Since the HWE is supposed to be tested in data of each SNP separately, the significance level of each individual test should be adjusted in order to ensure a global significance level for this QC check. Bonferroni or Sidak-Dunn corrections are two popular methods to make such adjustment. Alternatively, one can use procedures based on the control of the false discovery rate, as proposed by Benjamini and Hochberg (11). In theory, deviations of the HWE can result from the genetic selection of a specific allele in patients. Because of this possibility, some researchers prefer to test the HWE using data from healthy controls alone. However, this preference has the disadvantage to decrease the power of the respective statistical test. On the other hand, a flagrant deviation of the HWE also suggests non-negligible genotype errors associated with a given SNP. Since one cannot distinguish selection from eventual genotyping errors, the SNPs with gross deviations of the HWE are typically excluded from the analysis.
Third, the proportion of heterozygous genotypes (i.e., heterozygosity rate) across all SNPs should be calculated for each individual sample. Excessive heterozygosity rate suggests a possible contamination of the respective biological sample, while reduced heterozygosity rate indicates genetic inbreeding. The usual practice is to exclude samples from individuals whose heterozygosity rates are not falling into a “confidence” band. This confidence band is usually defined by the average heterozygosity rate of all the samples plus/minus a given number of times the standard deviation of the heterozygosity rate. The heterozygosity of SNPs located in the X chromosome is also used to confirm the gender of a sample and to detect putative label swaps.
Fourth, data of SNPs or of individuals with low genotyping rates should be excluded from the analysis. The genotyping rate of a given SNP is the proportion of individuals with fully determined genotypes of that SNP, whereas the genotyping rate of a given individual is the proportion of SNPs with a fully determined genotype of that individual. A low genotyping rate of a given SNP suggests that the genomic site associated with that SNP includes another type of genetic variation (e.g., deletion or insertion). A low genotyping rate of a given individual indicates a low quality of the DNA material used for genotyping. Again, researchers must decide what is considered a reasonable genotyping rate for their study. In addition, different exclusion criteria can be applied to the genotyping rates of SNPs and individuals.
Additional QC checks (e.g., assessing the genetic distance between sampled individuals or checking their ancestry) can also be performed in GWAS and TGAS, as reviewed elsewhere (12). However, they are more relevant for large-scale population genetic studies.
Table 1 summarizes the QC checks performed by each GWAS and TGAS on ME/CFS. On the one hand, the study of Perez et al. (8) only performed the QC check based on the MAF. This study also used a non-standard criterium for selecting SNPs: those with MAF <0.10 in either patients or reported in the Kaviar database were excluded from the analysis. On the other hand, Herrera et al. (7) performed all QC checks recommended for a GWAS. The remaining studies performed almost all standard QC checks with the exception of the one based on the heterozygosity rate. Interestingly, Johnston et al. (10) mentioned this QC check in the Materials & Methods of their study. However, they neither provided any specific information about how this QC was actually performed nor showed any statistical summary of the heterozygosity rate. Finally, Smith et al. (5) did not exclude any SNP based on a too-low MAF.
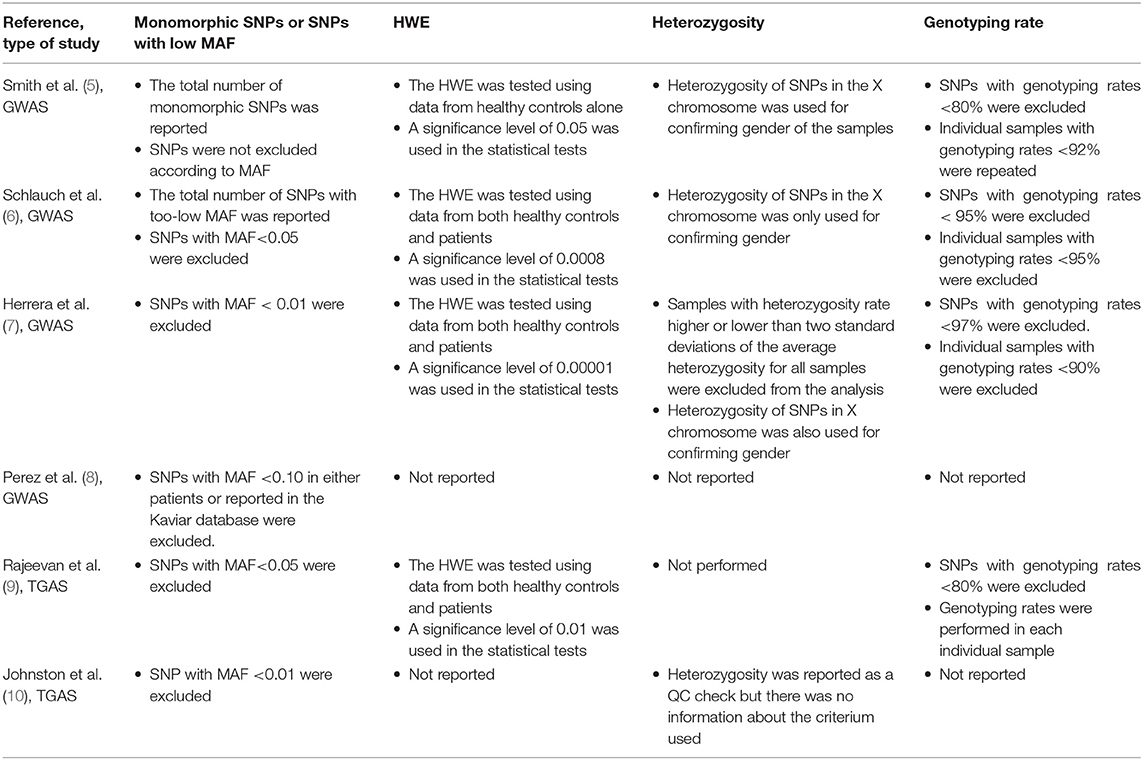
Table 1. Summary of the QC checks performed in published GWAS and TGAS on ME/CFS.
This opinion paper shows partial QC checks in the majority of the published genetic association studies on ME/CFS, the exception being the study carried out by Herrera et al. (7). The assessment of the performed QC checks is essential to ascertain the quality of the respective genetic data. In this regard, the genetic data from Perez et al. (8) deserves to be further analyzed to ascertain the validity of the reported findings. Such assessment can follow the QC steps outlined here and exemplary performed by Herrera et al. (7). The remaining studies can also benefit by an additional quality check related to heterozygosity rate so that possible sample contaminations can be ruled out. The absence of this check does not immediately invalidate the genetic data of these studies. We could have done such check if the corresponding genetic data were available either in an open-access repository or as a Supplementary File within the respective publication, a data-sharing practice followed by several ME/CFS researchers (13–15). Consequently, it is unclear whether aberrant heterozygosity rates (due to sample contamination) are one of the explanations for the conflicting evidence of genetic associations reported by these studies. In this regard, Herrera et al. (7) excluded five out of their 109 samples (5%) based on the heterozygosity rate. In simple statistical applications using large sample sizes, a 5% sample contamination might be too low to have a substantial impact on the respective findings. However, in the specific context of GWAS and TGAS where stringent significance levels are used to control for multiple testing, such a level of sample contamination could reduce the underlying statistical power and leave relevant disease-gene associations undetected.
Besides the partial QC checks, the investigated genetic data on ME/CFS suffer from the curse of not having an objective biomarker for disease diagnosis. Similar problem can be envisioned for other complex diseases lacking a biomarker, such as Fibromyalgia and the Gulf War Syndrome. The absence of a biomarker is likely to introduce a possible misclassification of the true disease status of the recruited patients (16). To illustrate this putative problem, Herrera et al. (7) recruited nine obese (with body mass indexes equal or higher than 35 kg/m2) out of 61 patients based on the 1994 Center for Diseases Control Criteria (1) and Canadian Consensus Criteria (2). Notwithstanding controlling for the body mass index in the respective association analysis and the exclusion of known diseases, it is unclear whether the obesity observed in these patients was a direct consequence of ME/CFS or instead caused by another ongoing disease strongly associated with fatigue. A solution to this problem is to use more advanced statistical methodology where misclassification can be directly included in the data analysis (17, 18). However, given the complexity of this methodology, we argue that a stronger collaboration between the ME/CFS research community and statistical geneticists should be reached. In principle, this collaboration is expected to promote better statistical analyses, to improve data interpretations and, ultimately, a better assessment of the genetic component in ME/CFS.
In summary, given the partial QC checks performed in current GWAS and TGAS, the question of a genetic component in ME/CFS remains open for investigation. To accelerate the discovery of promising disease-gene association, future genetic studies of ME/CFS should set data and methodological standards as high as those followed by the 1,000 Human Genome Project and the UK10K project (19, 20). Data sharing should also be a general practice to provide the researcher community the opportunity to perform additional checks or alternative analyses of the same data.
NS conceptualized this research. AG and NS performed the literature review. EL and LN helped in the interpretation and discussion of all the results. All authors read, revised, and approved the final draft of the manuscript.
NS acknowledges partial funding from Fundação para a Ciência e Tecnologia, Portugal (grant ref. UID/MAT/00006/2019). LN and EL acknowledge the funding support from the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH-Award Number: R01AI103629), and from the the ME Association (Award number: PF8947) for their studies on ME/CFS. The funding agencies did not have any role in the designing, data collection, data analysis, and interpretation or writing-up of the present manuscript.
The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Dr. Francisco Westermeier for proof-reading the manuscript.
GWAS, Genome-wide association study; HWE, Hardy-Weinberg Equilibrium; MAF, minor allele frequency; ME/CFS, myalgic encephalomyelitis/chronic fatigue syndrome; QC, quality control; SNP, single nucleotide polymorphism; TGAS, targeted-genome association study.
1. Fukuda K, Straus SE, Hickie I, Sharpe MC, Dobbins JG, Komaroff A. The chronic fatigue syndrome: a comprehensive approach to its definition and study. Ann Intern Med. (1994) 121:953–9. doi: 10.7326/0003-4819-121-12-199412150-00009
2. Carruthers BM. Definitions and aetiology of myalgic encephalomyelitis: how the Canadian consensus clinical definition of myalgic encephalomyelitis works. J Clin Pathol. (2007) 60:117–9. doi: 10.1136/jcp.2006.042754
3. Rasa S, Nora-Krukle Z, Henning N, Eliassen E, Shikova E, Harrer T, et al. Chronic viral infections in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). J Transl Med. (2018) 16:268. doi: 10.1186/s12967-018-1644-y
4. Blomberg J, Gottfries C-G, Elfaitouri A, Rizwan M, Rosén A. Infection elicited autoimmunity and myalgic encephalomyelitis/chronic fatigue syndrome: an explanatory model. Front Immunol. (2018) 9:229. doi: 10.3389/fimmu.2018.00229
5. Smith AK, Fang H, Whistler T, Unger ER, Rajeevan MS. Convergent genomic studies identify association of GRIK2 and NPAS2 with chronic fatigue syndrome. Neuropsychobiology. (2011) 64:183–94. doi: 10.1159/000326692
6. Schlauch KA, Khaiboullina SF, De Meirleir KL, Rawat S, Petereit J, Rizvanov AA, et al. Genome-wide association analysis identifies genetic variations in subjects with myalgic encephalomyelitis/chronic fatigue syndrome. Transl Psychiatry. (2016) 6:e730. doi: 10.1038/tp.2015.208
7. Herrera S, de Vega WC, Ashbrook D, Vernon SD, McGowan PO. Genome-epigenome interactions associated with myalgic encephalomyelitis/chronic fatigue syndrome. Epigenetics. (2018) 13:1174–90. doi: 10.1080/15592294.2018.1549769
8. Perez M, Jaundoo R, Hilton K, Del Alamo A, Gemayel K, Klimas NG, et al. Genetic predisposition for immune system, hormone, and metabolic dysfunction in myalgic encephalomyelitis/chronic fatigue syndrome: a pilot study. Front Pediatr. (2019) 7:206. doi: 10.3389/fped.2019.00206
9. Rajeevan MS, Dimulescu I, Murray J, Falkenberg VR, Unger ER. Pathway-focused genetic evaluation of immune and inflammation related genes with chronic fatigue syndrome. Hum Immunol. (2015) 76:553–60. doi: 10.1016/j.humimm.2015.06.014
10. Johnston S, Staines D, Klein A, Marshall-Gradisnik S. A targeted genome association study examining transient receptor potential ion channels, acetylcholine receptors, and adrenergic receptors in chronic fatigue syndrome/myalgic encephalomyelitis. BMC Med Genet. (2016) 17:79. doi: 10.1186/s12881-016-0342-y
11. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. (1995) 57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
12. Marees AT, de Kluiver H, Stringer S, Vorspan F, Curis E, Marie-Claire C, et al. A tutorial on conducting genome-wide association studies: quality control and statistical analysis. Int J Methods Psychiatr Res. (2018) 27:e1608. doi: 10.1002/mpr.1608
13. Loebel M, Eckey M, Sotzny F, Hahn E, Bauer S, Grabowski P, et al. Serological profiling of the EBV immune response in chronic fatigue syndrome using a peptide microarray. PLoS ONE. (2017) 12:e0179124. doi: 10.1371/journal.pone.0179124
14. Trivedi MS, Oltra E, Sarria L, Rose N, Beljanski V, Fletcher MA, et al. Identification of myalgic encephalomyelitis/ chronic fatigue syndrome-associated DNA methylation patterns. PLoS ONE. (2018) 13:e0201066. doi: 10.1371/journal.pone.0201066
15. Saiki T, Kawai T, Morita K, Ohta M, Saito T, Rokutan K, et al. Identification of marker genes for differential diagnosis of chronic fatigue syndrome. Mol Med. (2008) 14:599–607. doi: 10.2119/2007-00059.Saiki
16. Nacul L, Lacerda EM, Kingdon CC, Curran H, Bowman EW. How have selection bias and disease misclassification undermined the validity of myalgic encephalomyelitis/chronic fatigue syndrome studies? J Health Psychol. (2017) 24:1765–69. doi: 10.1177/1359105317695803
17. Paulino CD, Soares P, Neuhaus J. Binomial regression with misclassification. Biometrics. (2003) 59:670–5. doi: 10.1111/1541-0420.00077
18. Luo S, Chan W, Detry MA, Massman PJ, Doody RS. Binomial regression with a misclassified covariate and outcome. Stat Methods Med Res. (2016) 25:101–17. doi: 10.1177/0962280212441965
19. Auton A, Abecasis GR, Altshuler DM, Durbin RM, Bentley DR, Chakravarti A, et al. A global reference for human genetic variation. Nature. (2015) 526:68–74. doi: 10.1038/nature15393
Keywords: myalgic encephalomyelitis chronic fatigue syndrome, quality control, targeted-genome association study, reproducibility, genome-wide association study
Citation: Grabowska AD, Lacerda EM, Nacul L and Sepúlveda N (2020) Review of the Quality Control Checks Performed by Current Genome-Wide and Targeted-Genome Association Studies on Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Front. Pediatr. 8:293. doi: 10.3389/fped.2020.00293
Received: 27 January 2020; Accepted: 07 May 2020;
Published: 12 June 2020.
Edited by:
Marco Carotenuto, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Massimiliano Valeriani, Bambino Gesù Children Hospital (IRCCS), ItalyCopyright © 2020 Grabowska, Lacerda, Nacul and Sepúlveda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nuno Sepúlveda, bnVuby5zZXB1bHZlZGFAbHNodG0uYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.