 Jessie Newville
Jessie Newville Jessie R. Maxwell
Jessie R. Maxwell Yuma Kitase
Yuma Kitase Shenandoah Robinson
Shenandoah Robinson Lauren L. Jantzie
Lauren L. Jantzie
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Pediatr. , 26 June 2020
Sec. Neonatology
Volume 8 - 2020 | https://doi.org/10.3389/fped.2020.00272
This article is part of the Research Topic Experimental and Clinical Approaches in the Pursuit of Novel Therapeutic Strategies for Perinatal Brain Injury and its Neurological Sequelae View all 17 articles
The increased incidence of opioid use during pregnancy warrants investigation to reveal the impact of opioid exposure on the developing fetus. Exposure during critical periods of development could have enduring consequences for affected individuals. Particularly, evidence is mounting that developmental injury can result in immune priming, whereby subsequent immune activation elicits an exaggerated immune response. This maladaptive hypersensitivity to immune challenge perpetuates dysregulated inflammatory signaling and poor health outcomes. Utilizing an established preclinical rat model of perinatal methadone exposure, we sought to investigate the consequences of developmental opioid exposure on in vitro activation of peripheral blood mononuclear cells (PBMCs). We hypothesize that PBMCs from methadone-exposed rats would exhibit abnormal chemokine and cytokine expression at baseline, with exaggerated chemokine and cytokine production following immune stimulation compared to saline-exposed controls. On postnatal day (P) 7, pup PMBCs were isolated and cultured, pooling three pups per n. Following 3 and 24 h, the supernatant from cultured PMBCs was collected and assessed for inflammatory cytokine and chemokine expression at baseline or lipopolysaccharide (LPS) stimulation using multiplex electrochemiluminescence. Following 3 and 24 h, baseline production of proinflammatory chemokine and cytokine levels were significantly increased in methadone PBMCs (p < 0.0001). Stimulation with LPS for 3 h resulted in increased tumor necrosis factor (TNF-α) and C-X-C motif chemokine ligand 1 (CXCL1) expression by 3.5-fold in PBMCs from methadone-exposed PBMCs compared to PBMCs from saline-exposed controls (p < 0.0001). Peripheral blood mononuclear cell hyperreactivity was still apparent at 24 h of LPS stimulation, evidenced by significantly increased TNF-α, CXCL1, interleukin 6 (IL-6), and IL-10 production by methadone PMBCs compared to saline control PBMCs (p < 0.0001). Together, we provide evidence of increased production of proinflammatory molecules from methadone PBMCs at baseline, in addition to sustained hyperreactivity relative to saline-exposed controls. Exaggerated peripheral immune responses exacerbate inflammatory signaling, with subsequent consequences on many organ systems throughout the body, such as the developing nervous system. Enhanced understanding of these inflammatory mechanisms will allow for appropriate therapeutic development for infants who were exposed to opioids during development. Furthermore, these data highlight the utility of this in vitro PBMC assay technique for future biomarker development to guide specific treatment for patients exposed to opioids during gestation.
The incidence of opioid abuse in the United States has steadily increased since 2000, today reaching epidemic proportions (1, 2). The crisis is illustrated by data demonstrating that the number of opioid-related hospitalizations increased by 64%, and the number of deaths due to opioid overdose increased by 27% between the years of 2005 and 2014 (3, 4). Mirroring the national trend, opioid use by pregnant women has escalated to alarming rates (5). Opioid use in this population increased 5-fold from 2000 to 2009 (6), and the prevalence of women with opioid use disorders at delivery hospitalizations quadrupled between 1999 and 2014 (7).
Prenatal opioid exposure results from maternal use or abuse of illicit opioids, such as heroin, and prescription opioids including oxycodone, hydrocodone, morphine, codeine, and fentanyl (8). In addition, the maternal use of buprenorphine or methadone, two synthetic opioids commonly used in opioid maintenance therapy for individuals suffering from opioid use disorder (9, 10), also contribute to prenatal opioid exposure. The growing rate of women using opioids during pregnancy has led to an increase in adverse neonatal outcomes (11, 12). Recent studies showing an association between opioid use during pregnancy and poor health outcomes for both pregnant women and infants highlight prenatal opioid exposure as a serious public health concern (13, 14). Opioid-exposed infants represent an extremely vulnerable patient population (15), with 50–80% experiencing neonatal abstinence syndrome (16). Indeed, prenatal opioid exposure is associated with increased risk of fetal growth restriction, preterm birth, and lifelong motor and cognitive deficits (17–25). The devastating consequences of opioid exposure on the physical health and developmental outcomes of exposed children strengthen the need to advance scientific understanding of the underpinnings of opioid-induced neural injury and to advance biomarker development in this patient population.
Insult during the prenatal period affects ongoing developmental processes in the fetus, leading to lifelong consequences and health challenges. Both the central nervous system (CNS) and the immune system undergo complex and incremental steps toward maturation during gestation (26–28). New advances in molecular neuroscience have begun to elucidate the importance of the multifaceted interplay of central and peripheral immune system in regulating and supporting ongoing brain development. Moreover, these advances highlight the neurodevelopmental consequences of perinatal immune activation following perinatal insult (29–33). The findings from both clinical and preclinical studies implicate perinatal immune activation in the pathophysiology of numerous neurodevelopmental disorders, such as cerebral palsy, autism spectrum disorders, Down syndrome, and fetal alcohol spectrum disorders (33–41).
Previously, we reported evidence of neural injury and reduced cognitive functioning in a model of prenatal opioid exposure, with multiple assays reflecting significant neuroinflammation (42). In the aforementioned study, analysis of serum inflammatory cytokine expression of opioid-exposed animals compared to saline-exposed controls demonstrated elevated levels of interleukin 1β (IL-1β), tumor necrosis factor α (TNF-α), IL-6, and C-X-C motif chemokine ligand 1 (CXCL1), indicating systemic inflammatory response syndrome induced by opioid exposure. Additionally, initial in vitro assessment of isolated PBMC from opioid-exposed animals challenged with lipopolysaccharide (LPS) suggested heightened immune reactivity and immune priming toward exaggerated responses to stimuli (42).
Here, we extend our investigation of opioid-induced inflammation by thoroughly defining the peripheral immune signaling and reactivity of opioid-exposed PBMCs using an established in vitro assay and biomarker platform (35, 37, 43–50). These data enhance the understanding of important inflammatory mechanisms, an essential step to inform future development of appropriate therapeutic interventions for infants who are exposed to opioids during gestation.
Sprague–Dawley rat dams and litters were maintained in a 12-h dark–light cycle (lights on at 0800 h), temperature, and humidity-controlled facility with food and water available ad libitum. All experiments were performed in strict accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of New Mexico Health Sciences Center. Protocols were developed and performed consistent with National Research Council and ARRIVE guidelines (51).
Methadone is a full μ-, δ-, and κ-opioid receptor agonist, similar to heroin, morphine, and fentanyl, whereas buprenorphine is a partial μ-opioid receptor agonist and κ-opioid receptor antagonist (52). The use of methadone in our experiments allows us avoid differential pharmacology related to partial antagonism and study the effects from of full agonism at the predominant opioid receptor subtypes. As previously published (42), osmotic minipumps (model 2004; Alzet, Cupertino, CA, USA) primed with 12 mg/kg of methadone or normal saline were implanted in embryonic day (E) 16 timed-pregnant rat dams (Charles River Laboratories, Wilmington, MA, USA) (Figure 1). Implantation on E16 allows in utero opioid exposure from E16 to birth and postnatal opioid exposure via milk from birth to postnatal day (P) 7 (blood collection). These minipumps allow for continual infusion of methadone or saline at a rate of 0.25 μL per hour for a maximum of 28 days. Under isoflurane-induced anesthesia, dams underwent a minipump placement procedure. Subcutaneous minipump placement was achieved by transverse 1.5-cm incision. The subcutaneous area was opened by careful blunt dissection, and the prefilled, primed osmotic minipump was placed in the opened space. Following closure of the incision with sutures, dams were then returned to their respective home cages, where their recovery was closely monitored. When pups were born on E22, they then received methadone through milk ingestion. Postnatal methadone exposure was confirmed by measuring the concentration of methadone in dam and offspring urine (42). As previously reported, this paradigm of opioid exposure results in significant pup weight loss at the neonatal and perinatal period. Opioid exposure via 12 mg/kg minipump results in a significant 10% reduction in offspring weight at P1 and 23% reduction in weight by P21 compared to saline exposed controls (42). These preclinical data reflect data from clinical studies showing that infants of mothers who exclusively used opioids suffered from a 2 to 10% decrease in birth weight compared to healthy controls (53, 54). Further, another study found that infants of mothers on methadone replacement therapy suffered a 19% reduction in birth weight compared to age-matched controls (55). Thus, this model replicates the systemic consequences of extended prenatal opioid exposure observed in human infants.
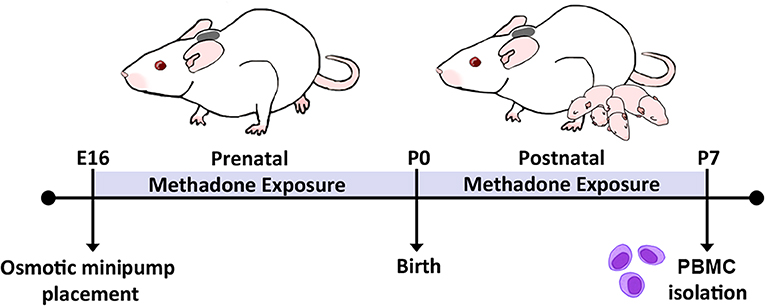
Figure 1. Experimental timeline. Perinatal methadone exposure was accomplished by minipump implantation on E16, permitting pup exposure to methadone during critical stages of immune and neurological maturation. On P7, PBMCs from methadone- or saline-exposed pups were isolated for culture and biochemical analysis.
At P7, rats are developmentally equivalent to human infants between approximately 32 and 38 weeks' gestation (56–62). From P7 to P10 in rats and 36–40 weeks' gestation in humans (term infant), overall brain growth peaks while important neural developmental processes, such as gliogenesis and expansion of axonal and dendritic density, occur (60, 63–65, 65–69). During this same developmental period, consolidation of the immune system in humans and rats occurs, whereby the functional capacity of immune cells evolves, and the number of circulating leukocytes, neutrophils, and monocytes increases (26–28, 60, 70–72). At P7, PBMC isolation was performed as previously published (41). Venous blood was collected from the right atrium of deeply anesthetized P7 pups and pooled across three animals in pyrogen-free K2 EDTA Vacutainer tubes (366643; Becton Dickson, Franklin Lakes, NJ, USA). Each n represents PBMCs isolated from blood pooled across three separate animals. In this study, equal numbers of male and female pups were used throughout. Peripheral blood mononuclear cells were isolated by Ficoll gradient separation (37), whereby equal volumes of peripheral blood and RPMI 1640 media (Gibco, Waltham, MA, USA) were combined and layered atop 3 mL of Ficoll-Plaque Plus (17144002; GE Healthcare, Chicago, IL, USA) within sterile 15-mL conical tubes. Following centrifugation at 400 g for 30 min at room temperature, the PBMC cell layer was transferred to a new centrifuge tube and resuspended in three volumes of RPMI media. Two wash cycles were performed, consisting of centrifugation at 400 g for 10 min at room temperature, disposal of the supernatant, and resuspension of the pellet in three volumes of RPMI media. Isolated PBMCs resuspended in media were plated in triplicates at a density of 1 × 106 cells/mL on 3.5-cm Petri dishes.
Plated PBMCs from saline and methadone groups were stimulated with 10, 50, or 100 ng/mL of LPS to generate a dose response curve. Based on TNF-α secretion levels at 3 h, we determined that stimulation with 100 ng of LPS was ideal to produce a robust PBMC secretory response in both treatment groups. Therefore, consistent with previous studies, 100 ng of LPS was used for LPS challenge experiments (41, 45, 73). Supernatant samples were collected at 3 and 24 h in sterile 2-mL Eppendorf tubes, snap frozen on dry ice, and stored at −80°C until biochemical analysis.
To capture the secretory activity of PBMCs prior to protein synthesis, and then after protein synthesis, the supernatants from plated PBMCs were assayed at 3 and 24 h, respectively. Subsequently, secreted cytokine and chemokine expression was quantified using a V-PLEX Proinflammatory Panel 2 Rat Kit (K15059D; Meso Scale Diagnostics, Rockville, MD, USA) created to detect levels of interferon γ, IL-1β, IL-4, IL-5, IL-10, IL-13, IL-6, CXCL1, and TNF-α. The V-PLEX multielectrochemiluminescent immunoassay (MECI) was performed according to manufacturer instructions with <5% interassay variation. Specifically, supernatants from cultured PBMCs were diluted 1:4 and, together with prepared standards, were loaded in duplicate onto the manufacturer-provided blocked and washed 96-well plates. Then, following a series of washes and incubation with the antibody detection solution, plates were washed and loaded with the manufacturer-provided Read Buffer and read on a Quickplex SQ 120 Imager. Here, we report data on the levels of TNF-α and CXCL1 production at 3 h and TNF-α, CXCL1, IL-6, and IL-10 at 24 h. Of note, levels of IL-6 and IL-10 at 3 h were below the detectable limit in baseline conditions, as were other cytokine levels in the panel.
To determine appropriate sample size, statistical power was calculated using G*Power 3.1.9.7 (Institut für Psychologie, Kiel, Germany) (74), using estimated means from previous studies (33, 42). Here, we prepared an n of three per treatment group, whereby each n represents isolated PBMCs from three animals pooled into one sample. Comparisons to determine statistical significance, defined as p < 0.05, were performed within Prism 7.05 (GraphPad software, San Diego, CA, USA). For comparisons between saline and methadone PMBC secretion, a Student t-test was used to determine significance. For statistical analysis of the dose response to LPS, a one-way analysis of variance (ANOVA) with Bonferroni multiple comparisons was performed. To make comparisons between treatments (baseline secretion vs. LPS-stimulated secretion) and to make comparisons between time points (3- vs. 24-h secretion), two-way ANOVA was performed with Bonferroni post-hoc analyses.
At 3 h, unstimulated PBMCs from methadone-exposed pups produced significantly augmented levels of the inflammatory cytokine TNF-α (methadone: 3.589 pg/mL, saline: 0.8142 pg/mL, t-test, p < 0.0001), as well as CXCL1 (methadone: 1.280 pg/mL, saline: 0.0, t-test, p = 0.0004) (Figure 2A).
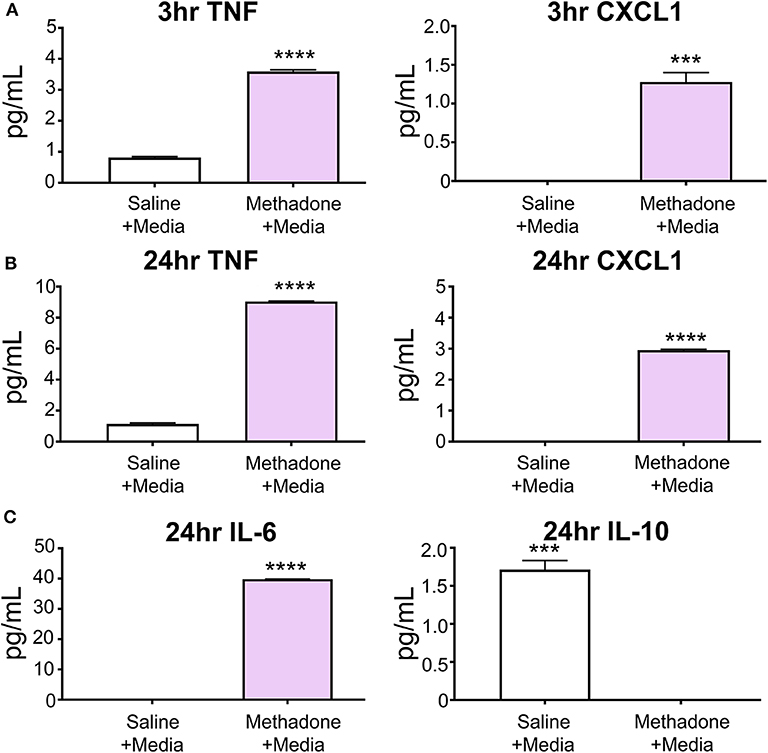
Figure 2. At baseline, PBMCs from methadone-exposed pups demonstrated dysregulated production of inflammatory signaling molecules. (A) At 3 h following isolation from P7 pups and plating in media alone, PBMCs from methadone-exposed pups produced significantly increased levels of TNF-α and CXCL1, compared to PBMCs from saline-exposed control PBMCs. (B) After 24 h, TNF-α and CXCL1 levels from methadone PBMCs compared to saline were further augmented. (C) Additionally, methadone PBMCs produced significantly higher levels of IL-6 and significantly lower levels of IL-10, compared to saline controls (n = 3 per treatment group, ***p < 0.001, ****p < 0.0001).
After 24 h, the level of TNF-α secreted by saline PBMCs was 41% increased from levels measured at 3 h, whereas methadone PBMCs demonstrated a 152% increase from levels measured at 3 h (two-way ANOVA, p < 0.0001). Increased production of TNF-α by methadone PBMCs at 24 h (methadone: 9.047 pg/mL, saline: 1.148 pg/mL, t-test, p < 0.0001) was accompanied by increased secretion of CXCL1 by methadone PBMCs relative to saline PBMCs (methadone: 2.950 pg/mL, saline: 0.0 pg/mL, t-test, p = 0.0004) (Figure 2B). By 24 h, CXCL1 secretion by methadone PBMCs was 130% increased from measurement at 3 h (t-test, p < 0.0002), whereas levels of CXCL1 production by saline PBMCs remained below detectable levels at both the 3- and 24-h time points. Moreover, after 24 h in culture, methadone PBMCs demonstrated additional evidence of dysregulated immune signaling, evident by increased IL-6 (methadone: 39.82 pg/mL, saline: 0.0 pg/mL, t-test, p < 0.0001) and decreased IL-10 expression (methadone: 0.0 pg/mL, saline: 1.712 pg/mL, t-test, p = 0.0001) (Figure 2C). Together, these data show significantly altered baseline production of inflammatory signaling molecules by methadone-derived PBMCs at 3 and 24 h compared to controls.
To illuminate potential discrepancies in reactivity to immune stimulus between treatment groups, PBMCs from methadone and saline exposed pups were challenged with LPS in vitro. By increasing the dose of LPS from 10, to 50, to 100 ng/mL, we observed a dose-dependent increase in TNF-α secretion from both saline and methadone PBMCs, compared to PMBCs in media alone (one-way ANOVA, p < 0.0001) (Figure 3). In this assay, addition of media alone resulted in levels of TNF-α production that were not statistically different from baseline levels of TNF-α measured at 3 h, as reported earlier in this study (one-way ANOVA, p > 0.05). With each dose of LPS, PBMCs derived from methadone-exposed animals produced greater levels of TNF-α compared to saline PBMCs (one-way ANOVA, p < 0.001). As 100 ng/mL LPS elicited a significant response in PBMCs from both methadone- and saline-exposed pups, we stimulated a separate cohort of PBMCs with this dose and measured cytokine and chemokine levels at 3 and 24 h.
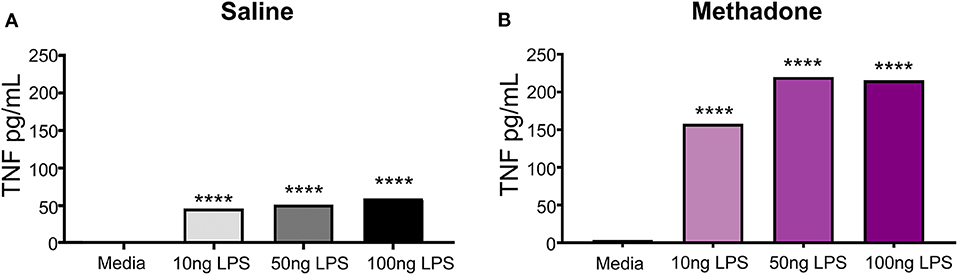
Figure 3. Peripheral blood mononuclear cells isolated from P7 pups exhibit LPS induced dose-responsive increases in TNF-α production. (A) Increasing doses of LPS (10, 50, or 100 ng/mL) elicited significantly augmented secretion of TNF-α from saline and (B) methadone PBMCs, compared to PMBCs in media alone (n = 3 per treatment group, 1 way ANOVA, ****p < 0.0001).
At 3 h following stimulation with 100 ng/mL LPS, TNF-α (methadone: 215.8 pg/mL, saline: 59.08 pg/mL, t-test, p < 0.0001) and CXCL1 (methadone: 53.29 pg/mL, saline: 14.33 pg/mL, t-test, p < 0.0001) production by PBMCs from P7 methadone-exposed pups was increased compared to PBMCs from saline-exposed controls, representing a 265 and 272% increase, respectively (Figure 4A). Compared to baseline levels of unstimulated PBMCS, LPS-stimulated PBMCs derived from both saline and methadone animals produced significantly increased levels of TNF-α and CXCL1 3 h (two-way ANOVA, p < 0.0001).
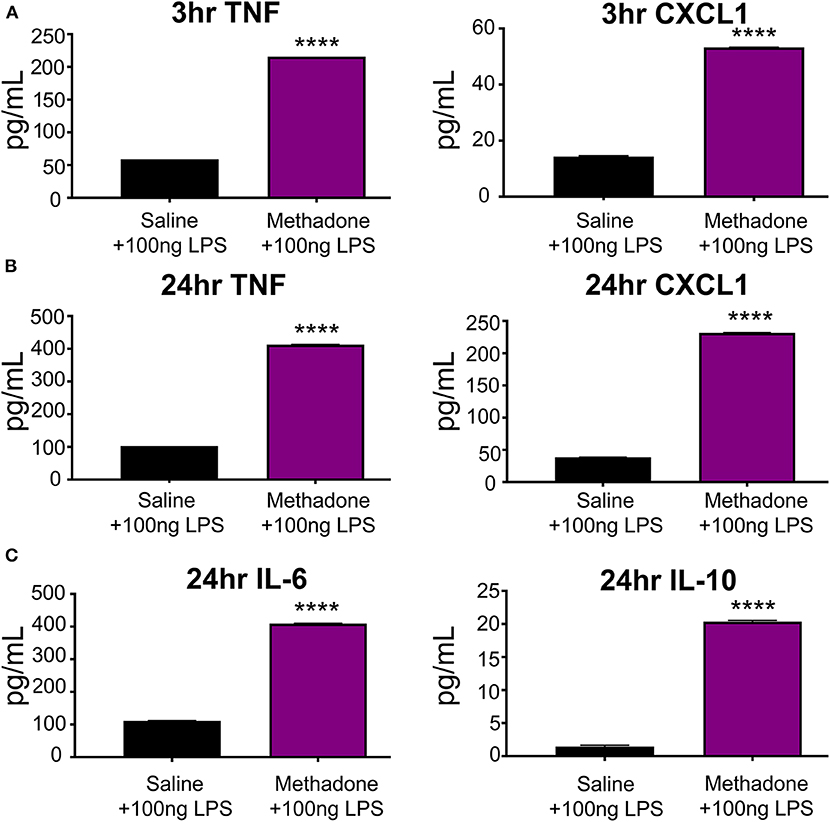
Figure 4. Stimulation with LPS revealed hyperreactivity of PBMCs from methadone-exposed pups. (A) At 3 h, TNF-α and CXCL1 production by PBMCs from P7 methadone-exposed pups was significantly increased compared to PBMCs from saline exposed controls. (B) After 24 h, TNF-α and CXCL1 levels from methadone PBMCs were significantly increased compared to saline levels. (C) Additionally, production of IL-6 and IL-10 by methadone PBMCs was significantly augmented, compared to saline controls (n = 3 per treatment group, unpaired t-test, ****p < 0.0001).
Following 24 h of LPS stimulation, levels of TNF-α (methadone: 412.6 pg/mL, saline: 102.8 pg/mL, t-test, p < 0.0001) and CXCL1 (methadone: 231.6 pg/mL, saline: 38.34 pg/mL, t-test, p < 0.0001) produced by methadone PBMCs were increased by 301 and 504%, respectively, compared to levels from saline PBMCs (Figure 4B). Compared to measurements taken at 3 h following LPS stimulation, TNF-α secretion at 24 h increased by 74% in saline PBMCs, whereas methadone PBMCs demonstrated a 91% increase (two-way ANOVA, p < 0.0001). A similar pattern arose with CXCL1 production. By 24 h, CXCL1 production by saline PBMCs increased 164% from levels measured at 3 h, whereas CXCL1 produced by LPS-challenged methadone PBMCs rose 334% (two-way ANOVA, p < 0.0001). At 24 h, we also found significantly increased levels of IL-6 (methadone: 409.4 pg/mL, saline: 111.7 pg/mL, t-test, p < 0.0001) and IL-10 (methadone: 20.35 pg/mL, saline: 1.442 pg/mL, t-test, p < 0.0001) from methadone PBMCs, compared to saline controls (Figure 4C).
As the opioid crisis continues to grow, increasing numbers of pregnant women and infants are affected. While mounting evidence indicates that prenatal opioid exposure is associated with significant and long-lasting neurological injury (17–19, 25), information on the pathophysiology of opioid exposure during the perinatal period is limited. Increasing our understanding of the cellular and molecular mechanisms that are impacted in circumstances of in utero opioid exposure is important for the development and implementation of informed clinical practices, in addition to the improvement of therapeutic options to support opioid-exposed infants.
Using the same model of perinatal opioid exposure employed in the current study, we previously provided evidence of neuroinflammation, microstructural brain injury, persistent cognitive deficits, and peripheral immune activation following perinatal opioid exposure (42). Here, we expand our understanding of the systemic immune dysfunction through an in-depth characterization of peripheral immune cell activity and reactivity following perinatal methadone exposure. Utilizing a clinically applicable in vitro cell culture protocol and translational multiplex immunoassay inflammatory biomarker panel (36, 37, 75–80), we found that at baseline PBMCs derived from methadone-exposed P7 pups hypersecreted proinflammatory molecules. Consistent with our previous findings that PBMCs derived from methadone-exposed rats secreted increased levels of TNF-α at 3 and 24 h after collection (42), here, we demonstrate that methadone PBMCs also secrete elevated levels of CXCL1 at 3 h. Increased expression of TNF-α and CXCL1 relative to PBMCs derived from saline-exposed controls was still evident at 24 h, demonstrating sustained peripheral immune activation of methadone-derived PBMCs at P7. Interestingly, after 24 h in culture, we observe additional evidence of dysregulated immune signaling with elevated IL-6 and diminished IL-10 in methadone-exposed animals relative to saline controls, in addition to the increased TNF-α and CXCL1 levels from methadone PBMCs. Despite probing for the presence of IL-6 and IL-10 at 3 h, the levels from both saline- and methadone-derived PBMCs remained below the detectable limits of the MECI. In summary, this evidence of elevated PBMC baseline production of known proinflammatory molecules TNF-α, CXCL1, and IL-6, with decreased levels of anti-inflammatory IL-10 from methadone-exposed animals, likely contributes to a proinflammatory environment within the systemic circulation (81).
To further characterize altered immune function, we stimulated cultured PBMCs with 100 ng/mL of LPS to represent an immune challenge and then quantified the chemokine and cytokine profile signature. While the addition of 100 ng/mL of LPS is a supraphysiological dose, it was formulated in consideration of our previous work (41, 42) and based on the dose response measured in TNF-α secretion to 10, 50, and 100 ng/mL of LPS that we performed in this study. We demonstrate that 100 ng/mL produces a robust PBMC response in the in vitro PBMC culture assay we performed, allowing us to examine chemokine and cytokine production by PBMCs from both saline and methadone treatment groups. Following 3 h of LPS stimulation, we detected levels of TNF-α and CXCL1 that were elevated from baseline in both methadone- and saline-exposed groups. Lipopolysaccharide-stimulated methadone PBMCs produced significantly more TNF-α and CXCL1 compared to PBMCs from stimulated saline control PBMCs. At 24 h, hyperreactivity of methadone PBMCs was evident from significantly elevated levels of TNF-α, CXCL1, IL-6, and IL-10 compared to saline controls. Taken together, these data provide evidence of increased sensitivity and a priming effect to subsequent inflammatory challenge in PBMCs from methadone-exposed pups. This suggests a mechanism of deleterious feed-forward inflammatory pathophysiology and fetal programming of immune system activation induced by methadone. Indeed, immune plasticity altered by methadone exposure may have long-lasting effects on the inflammatory responses of circulating leukocytes later in life. Future studies that assess the secretome and reactivity of PBMCs derived from opioid-exposed subjects at later developmental time points beyond P7 would be important to answer these questions.
Importantly, our in vitro approach allowed us to study PBMC responsiveness and sensitivity in isolation of potential confounders such as Toll-like receptor–stimulating agents in the peripheral circulation (82). Our in vitro data showing increased proinflammatory signaling at baseline from methadone-exposed PBMCs, without any immune stimulation, are distinct to this paradigm of perinatal injury and highlight this in vitro assay for use as a potential biomarker. The in vitro LPS challenge we perform here is a method that has been used clinically in children with developmental disorders and brain injury (35, 37). For instance, in a clinical study of children born preterm with cerebral palsy (37) and preclinical model reminiscent of preterm CNS injury (41), PBMCs were assayed consistent with the in vitro approach employed in the current study. Interestingly, in these studies, baseline secretion of PBMCs did not differ between treatment groups (37, 41). Only after stimulation with LPS did appreciable differences in PBMC chemokine and cytokine production appear in subjects with cerebral palsy (37), similar to preclinical studies (41, 45, 73). Perinatal insult–specific PBMC properties, revealed using this in vitro approach, support the potential use of secreted protein profiles from isolated PBMCs as a biomarker to discern distinct pathologies and potentially guide clinical treatment. Indeed, elucidating these profiles of immune signaling molecules holds potential for use as a biomarker to determine vulnerability to sustained peripheral immune hyperreactivity. Specifically, biomarkers in neonates could provide estimation of extent of immune system abnormalities and CNS injury and provide pharmacodynamic support to guide duration or degree of treatment for neonatal opioid withdrawal syndrome or supportive care in neonatal intensive care units. In this context, durable changes in PBMC reactivity may be an effective biomarker, and clinical utility may prove high given the ease of access to these cells and well-defined clinical stimulation protocols (37, 43). However, while these in vitro PBMC assays are clinically relevant, they are distinctly different than studying the complex, multidimensional in vivo response to inflammation, sepsis, and systemic sensitization catalyzed by an LPS challenge. Unquestionably, further study is required to validate how circulating leukocytes respond to LPS immune challenges in vivo and in the context of complex inflammatory networks, systemic circulating factors, and all cells that express TLR4.
Peripheral blood mononuclear cell hypersecretion of proinflammatory molecules and PBMC hyperreactivity resultant of gestational opioid exposure have important implications for the developing CNS. Our previous preclinical report strongly implicates brain injury secondary to opioid-induced systemic and neuroinflammation (42). In alignment with the aforementioned study, we now provide evidence of PBMC hypersection and hyperreactivity, which could contribute to increased systemic inflammation during the term equivalent developmental time point, coinciding with the brain growth spurt, peak myelination and gliogenesis, and astrocyte production (60, 63, 65–69, 83). Increased chemokine and cytokine production by PBMCs during the perinatal period jeopardizes proper neural cell development and circuitry maturation. Indeed, inflammation during perinatal development results in lasting neurological impairment (77, 80, 84, 85). While future studies are needed to clarify if methadone elicits PBMC systemic inflammation via PBMC activation throughout the methadone exposure, it is well-established that elevated levels of circulating inflammatory proteins during later stages in brain development (late third trimester, term equivalent) are associated with brain injury, characterized by increased structural and functional neurological deficits (80, 84, 86–89). Specifically, in a recent study of systemic TNF-α inhibition in preterm fetal sheep exposed to LPS-induced inflammation, researchers identified circulating TNF-α as a critical contributor to neuroinflammation and pathogenesis of impaired neurodevelopment (90). Cytokine and chemokines produced by circulating leukocytes are able to cross the blood–brain barrier via selective transporters (91). Furthermore, as in the specific case of TNF-α, increased levels can contribute to impairment of the blood–brain barrier function (92–94), allowing for increased proinflammatory molecule access to the developing CNS. Opioids are able to cross from maternal circulation through the placenta to fetal circulation owing to their low molecular weight, moderate lipid solubility, and low protein binding (95). Once in fetal circulation, opioids are able to cross the fetal blood-brain barrier by means of numerous transporters (96, 97). Thus, not only are developing neural cells and circuitry exposed to elevated levels of proinflammatory molecules in the context of opioid exposure, but they are directly exposed to opioids as well. In the developing CNS, neurons in addition to oligodendrocytes, astrocytes, and microglia express opioid receptors (98). Intriguingly, oligodendrocytes express opioid receptors in a maturation-dependent manner, in which immature stages of oligodendrocytes have increased opioid receptor expression (98), rendering this population more vulnerable to opioid exposure. Ultimately, exposure to opioids in conjunction with the proinflammatory profile produced by opioid exposure characterized in the current investigation could contribute to observed brain injury in preclinical studies (42), as well as clinical studies indicating particular vulnerability to major white matter tracts in infants exposed to opioids during brain development (20, 22, 24, 25).
Similar to the CNS, the immune system develops and matures over the course of gestation and the perinatal period. Dysregulated chemokines and cytokine production, and changes in immune cells themselves, culminate in impaired immune function that can last decades (35, 37). Similar to neural cells, leukocytes are uniquely responsive to their environment. Indeed, immune plasticity altered by prenatal insults may have long-term effects on the inflammatory responses of circulating leukocytes, which may serve as a biomarker of persistent or prior neuroinflammation and brain injury (99, 100). Infants exposed to intrauterine inflammation are at an increased risk of neurodevelopmental disorders (101). Notably, newborns that have elevated levels of biomarkers of systemic inflammation on two occasions 1 week apart are at a higher risk of brain injury and impaired neurodevelopment (77, 80, 84) Thus, understanding the homeostatic regulation of central and peripheral inflammatory cells in infants following opioid exposure, and the long-term consequences of their dysregulation, is essential (102). Significantly, an increase in chemokines/cytokines can contribute to perinatal brain injury by multiple overlapping mechanisms, including direct initiation of programed cell death pathways, microglial activation, immune cell recruitment, mitochondrial damage, and endoplasmic reticulum stress (85, 103, 104).
There are important limitations to this present study. For instance, here PBMCs were isolated from pooled peripheral blood from term equivalent male and female rats, limiting the ability to elucidate differences between individual animals and between male and female rat pups. Evidence from studies examining PBMCs isolated from adult humans suggests that sex differences in stimulated PBMC properties and secretion exist (105–107). While sex differences in secretion of PBMCs isolated at neonatal time points are not well-defined (35, 37), evidence exists demonstrating sex-specific differences in brain inflammation following circulating myeloid cells depletion in neonatal mice (108) and that inflammatory responses following immune cell activation in the immature brain differ between males and females, as reviewed by Mallard et al. (109). Thus, separate pooling of males and female peripheral blood from P7 pups for sex-specific analysis represents an important future direction. Additionally, although pooling of blood from multiple P7 rat pups was necessary in these experiments to collect an adequate PBMC fraction following differential centrifugation, analysis at later time points with larger animals would not require pooling, allowing for analysis of individual animal PBMC secretion and reactivity. Peripheral blood mononuclear cells represent a heterogeneous population of mononuclear cells in the peripheral circulation composed of T cells, T regulatory cells, T helper cells, B cells, and natural killer/dendritic cells/monocytes (110). Undoubtedly, flow cytometric studies beyond the scope of the present investigation are needed to define the precise immune cell population composition of PBMCs isolated from animals exposed to opioids during development.
In the current study, pregnant ram dams were implanted with methadone administering osmotic minipumps on E16 prior to complete oligodendrocyte, microglial, and astrocyte maturation (60), limiting rat pup opioid exposure to E16 through P7 when PBMCs were collected. This prenatal and postnatal opioid exposure paradigm accomplishes opioid exposure up until the equivalent end of the human third trimester. Future studies should now aim to commence opioid exposure from the onset of pregnancy (E0), thereby encompassing the entirety of brain and immune development.
In conclusion, we provide evidence in support of a systemic inflammatory response to perinatal opioid exposure, characterized by immune cell reprogramming and priming. This evidence may in part contribute to the neurological injury following developmental opioid exposure characterized in our previous preclinical study (42). The current study with the study by Jantzie et al. (42) joins a host of new and intriguing investigations that link developmental neurological injuries including cerebral palsy (37) and Down syndrome (35) with underlying systemic inflammation resultant of abnormal PBMC activity. Treatments that reduce inflammation or support developing neural cells in the context of inflammation could rescue the poor neural outcomes observed in preclinical and clinical investigations of perinatal opioid exposure. Our future studies will aim to identify appropriate therapies that target these proinflammatory mechanisms underlying brain injury associated with in utero opioid exposure (33, 111–114).
The datasets generated for this study are available on request to the corresponding author.
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of New Mexico Health Sciences Center.
LJ conceptualized the hypothesis and supervised the experiments. JN, JM, YK, SR, and LJ designed and performed the experiments. SR and LJ interpreted the data. JN, LJ, and SR wrote the manuscript. All authors contributed to manuscript revision and approved the final version.
This study was supported by generous funding from the National Institutes of Health 1R01HL139492 to LJ, Dedicated Health Research Funds from the University of New Mexico, and the Department of Pediatrics at the University of New Mexico Health Sciences Center. The authors are grateful for support from Departments of Pediatrics and Neurosciences at the University of New Mexico, and from the Departments of Neurosurgery, Neurology and Pediatrics at Johns Hopkins University and the Kennedy Krieger Institute.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful for the technical expertise of Tracylyn Yellowhair and Dr.Ksenia Matlowska.
1. Lyden J, Binswanger IA. The United States opioid epidemic. Semin Perinatol. (2019) 43:123–31. doi: 10.1053/j.semperi.2019.01.001
2. US Department of Health and Human Services. HHS Acting Secretary Declares Public Health Emergency to Address National Opioid Crisis (2017). Available online at: https://www.hhs.gov/about/news/2017/10/26/hhs-acting-secretary-declares-public-health-emergency-address-national-opioid-crisis.html (accessed February 22, 2019).
3. Seth P, Rudd RA, Noonan RK, Haegerich TM. Quantifying the epidemic of prescription opioid overdose deaths. Am J Public Health. (2018) 108:500–2. doi: 10.2105/AJPH.2017.304265
4. Weiss AJ, Bailey MK, O'Malley LO, Barrett ML, Elixhauser A, Steiner CA. Patient Characteristics of Opioid-Related Inpatient Stays and Emergency Department Visits Nationally and by State, 2014. (2017). Available online at: https://www.hcup-usahrqgov/faststats/landingjsp (accessed April 22, 2019).
5. Krans EE, Patrick SW. Opioid use disorder in pregnancy: health policy and practice in the midst of an epidemic. Obstet Gynecol. (2016) 128:4–10. doi: 10.1097/AOG.0000000000001446
6. Patrick SW, Schumacher RE, Benneyworth BD, Krans EE, McAllister JM, Davis MM. Neonatal abstinence syndrome and associated health care expenditures: United States, 2000-2009. JAMA. (2012) 307:1934–40. doi: 10.1001/jama.2012.3951
7. Haight SC, Ko JY, Tong VT, Bohm MK, Callaghan WM. Opioid use disorder documented at delivery hospitalization — United States, 1999–2014. MMWR Morb Mortal Wkly Rep. (2018) 67:845–9. doi: 10.15585/mmwr.mm6731a1
8. Reddy UM, Davis JM, Ren Z, Greene MF. Opioid use in pregnancy, neonatal abstinence syndrome, and childhood outcomes: executive summary of a joint workshop by the eunice kennedy shriver national institute of child health and human development, american congress of obstetricians and gynecologists, american academy of pediatrics, society for maternal-fetal medicine, centers for disease control and prevention, and the march of dimes foundation. Obstet Gynecol. (2017) 130:10–28. doi: 10.1097/AOG.0000000000002054
9. Mattick RP, Breen C, Kimber J, Davoli M. Methadone maintenance therapy versus no opioid replacement therapy for opioid dependence. Cochrane Database Syst Rev. (2009) 2009:CD002209. doi: 10.1002/14651858.CD002209.pub2
10. Zedler BK, Man AL, Kim MM, Amick HR, Joyce AR, Murrelle EL, et al. Buprenorphine compared with methadone to treat pregnant women with opioid use disorder: a systematic review and meta-analysis of safety in the mother, fetus and child. Addiction. (2016) 111:2115–28. doi: 10.1111/add.13462
11. Monnelly VJ, Hamilton R, Chappell FM, Mactier H, Boardman JP. Childhood neurodevelopment after prescription of maintenance methadone for opioid dependency in pregnancy: a systematic review and meta-analysis. Dev Med Child Neurol. (2019) 61:750–60. doi: 10.1111/dmcn.14117
12. Patrick SW, Davis MM, Lehmann CU, Cooper WO. Increasing incidence and geographic distribution of neonatal abstinence syndrome: United States 2009 to 2012. J Perinatol. (2015) 35:650–5. doi: 10.1038/jp.2015.36
13. Maeda A, Bateman BT, Clancy CR, Creanga AA, Leffert LR. Opioid abuse and dependence during pregnancy: temporal trends and obstetrical outcomes. Anesthesiology. (2014) 121:1158–65. doi: 10.1097/ALN.0000000000000472
14. Ross EJ, Graham DL, Money KM, Stanwood GD. Developmental consequences of fetal exposure to drugs: what we know and what we still must learn. Neuropsychopharmacology. (2015) 40:61–87. doi: 10.1038/npp.2014.147
15. Osborn DA, Jeffery HE, Cole MJ. Opiate treatment for opiate withdrawal in newborn infants. Cochrane Database Syst Rev. (2010) 10:CD002059. doi: 10.1002/14651858.CD002059.pub3
16. Ko JY, Patrick SW, Tong VT. Incidence of neonatal abstinence syndrome - 28 states, 1999-2013. MMWR Morb Mortal Wkly Rep. (2016) 65:799–802. doi: 10.15585/mmwr.mm6531a2
17. Azuine RE, Ji Y, Chang H, Kim Y, Ji H, DiBari J, et al. Prenatal risk, factors, and perinatal and postnatal outcomes associated with maternal opioid exposure in an urban, low-income, multiethnic, U. S population. JAMA Netw Open. (2019) 2:e196405. doi: 10.1001/jamanetworkopen.2019.6405
18. Feder KA, Letourneau EJ, Brook J. Children in the opioid epidemic: addressing the next generation's public health crisis. Pediatrics. (2019) 144:e20181656. doi: 10.1542/peds.2018-1656
19. Hudak ML, Tan RC, The Committee on Drugs and the Committee on Fetus and Newborn. Neonatal drug withdrawal. Pediatrics. (2012) 129:e540–60. doi: 10.1542/peds.2011-3212
20. Monnelly VJ, Anblagan D, Quigley A, Cabez MB, Cooper ES, Mactier H, et al. Prenatal methadone exposure is associated with altered neonatal brain development. NeuroImage Clin. (2018) 18:9–14. doi: 10.1016/j.nicl.2017.12.033
21. Salzwedel AP, Grewen KM, Vachet C, Gerig G, Lin W, Gao W. Prenatal drug exposure affects neonatal brain functional connectivity. J Neurosci. (2015) 35:5860–9. doi: 10.1523/JNEUROSCI.4333-14.2015
22. Sirnes E, Oltedal L, Bartsch H, Eide GE, Elgen IB, Aukland SM. Brain morphology in school-aged children with prenatal opioid exposure: a structural MRI study. Early Hum Dev. (2017) 106–7:33–9. doi: 10.1016/j.earlhumdev.2017.01.009
23. Sirnes E, Griffiths ST, Aukland SM, Eide GE, Elgen IB, Gundersen H. Functional MRI in prenatally opioid-exposed children during a working memory-selective attention task. Neurotoxicol Teratol. (2018) 66:46–54. doi: 10.1016/j.ntt.2018.01.010
24. Walhovd KB, Westlye LT, Moe V, Slinning K, Due-Tonnessen P, Bjornerud A, et al. White matter characteristics and cognition in prenatally opiate- and polysubstance- exposed children: a diffusion tensor imaging study. Am J Neuroradiol. (2010) 31:894–900. doi: 10.3174/ajnr.A1957
25. Yeoh SL, Eastwood J, Wright IM, Morton R, Melhuish E, Ward M, et al. Cognitive and motor outcomes of children with prenatal opioid exposure: a systematic review and meta-analysis. JAMA Netw Open. (2019) 2:e197025. doi: 10.1001/jamanetworkopen.2019.7025
26. Davies NP, Buggins AG, Snijders RJ, Jenkins E, Layton DM, Nicolaides KH. Blood leucocyte count in the human fetus. Arch Dis Child. (1992) 67:399–403. doi: 10.1136/adc.67.4_Spec_No.399
27. Madse-Bouterse SA, Romero R, Tarca AL, Kusanovic JP, Espinoza J, Kim CJ, et al. The transcriptome of the fetal inflammatory response syndrome. Am J Reprod Immunol. (2009) 63:73–92. doi: 10.1111/j.1600-0897.2009.00791.x
28. Zakharova LA, Malyukova IV, Proshlyakova EV, Potapova AA, Sapronova AY, Ershov PV, et al. Hypothalamo-pituitary control of the cell-mediated immunity in rat embryos: role of LHRH in regulation of lymphocyte proliferation. J Reprod Immunol. (2000) 47:17–32. doi: 10.1016/S0165-0378(00)00057-7
29. Hasegawa K, Ichiyama T, Isumi H, Nakata M, Sase M, Furukawa S. NF-κB activation in peripheral blood mononuclear cells in neonatal asphyxia. Clin Exp Immunol. (2003) 132:261–4. doi: 10.1046/j.1365-2249.2003.02127.x
30. Knuesel I, Chicha L, Britschgi M, Schobel SA, Bodmer M, Hellings JA, et al. Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol. (2014) 10:643–60. doi: 10.1038/nrneurol.2014.187
31. Marc T. Brain development and the immune system: an introduction to inflammatory and infectious diseases of the child's brain. Handb Clin Neurol. (2013) 112:1087–9. doi: 10.1016/B978-0-444-52910-7.00026-X
32. Tanabe S, Yamashita T. The role of immune cells in brain development and neurodevelopmental diseases. Int Immunol. (2018) 30:437–44. doi: 10.1093/intimm/dxy041
33. Yellowhair TR, Noor S, Maxwell JR, Anstine CV, Oppong AY, Robinson S, et al. Preclinical chorioamnionitis dysregulates CXCL1/CXCR2 signaling throughout the placental-fetal-brain axis. Exp Neurol. (2019) 301:110–9. doi: 10.1016/j.expneurol.2017.11.002
34. Eliwan HO, Watson WRG, Regan I, Philbin B, O'Hare FM, Strickland T, et al. Pediatric intensive care: immunomodulation with activated protein c ex vivo. Front Pediatr. (2019) 7:386. doi: 10.3389/fped.2019.00386
35. Huggard D, McGrane F, Lagan N, Roche E, Balfe J, Leahy TR, et al. Altered endotoxin responsiveness in healthy children with down syndrome. BMC Immunol. (2018) 19:31. doi: 10.1186/s12865-018-0270-z
36. Kuban KC, Joseph RM, O'Shea TM, Heeren T, Fichorova RN, Douglass L, et al. Circulating inflammatory-associated proteins in the first month of life and cognitive impairment at age 10 years in children born extremely preterm. J Pediatr. (2017) 180:116–23.e1. doi: 10.1016/j.jpeds.2016.09.054
37. Lin CY, Chang YC, Wang ST, Lee TY, Lin CF, Huang C-C. Altered inflammatory responses in preterm children with cerebral palsy. Ann Neurol. (2010) 68:204–12. doi: 10.1002/ana.22049
38. Noor S, Milligan E. Lifelong impacts of moderate prenatal alcohol exposure on neuroimmune function. Front Immunol. (2018) 9:1107. doi: 10.3389/fimmu.2018.01107
39. Parker-Athill EC, Tan J. Maternal immune activation and autism spectrum disorder: interleukin-6 signaling as a key mechanistic pathway. Neurosignals. (2010) 18:113–28. doi: 10.1159/000319828
40. Rossignol DA. A review of research trends in physiological abnormalities in autism spectrum disorders: immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol Psychiatry. (2012) 17:389–401. doi: 10.1038/mp.2011.165
41. Yellowhair TR, Noor S, Mares B, Jose C, Newville JC, Maxwell JR, et al. Chorioamnionitis in rats precipitates extended postnatal inflammatory lymphocyte hyperreactivity. Dev Neurosci. (2019) 40:523–33. doi: 10.1159/000497273
42. Jantzie LL, Maxwell JR, Newville JC, Yellowhair TR, Kitase Y, Madurai N, et al. Prenatal opioid exposure: the next neonatal neuroinflammatory disease. Brain Behav Immun. (2020)84:45–58. doi: 10.1016/j.bbi.2019.11.007
43. Huggard D, Koay WJ, Kelly L, McGrane F, Ryan E, Lagan N, et al. Altered toll-like receptor signalling in children with down syndrome. Mediators Inflamm. (2019) 2019:4068734. doi: 10.1155/2019/4068734
44. Jyonouchi H, Sun S, Le H. Proinflammatory and regulatory cytokine production associated with innate and adaptive immune responses in children with autism spectrum disorders and developmental regression. J Neuroimmunol. (2001) 120:170–9. doi: 10.1016/S0165-5728(01)00421-0
45. Kowalski ML, Wolska A, Grzegorczyk J, Hilt J, Jarzebska M, Drobniewski M, et al. Increased responsiveness to toll-like receptor 4 stimulation in peripheral blood mononuclear cells from patients with recent onset rheumatoid arthritis. Mediators Inflamm. (2008) 2008:132732. doi: 10.1155/2008/132732
46. Maes OC, Chertkow HM, Wang E, Schipper HM. Stress gene deregulation in alzheimer peripheral blood mononuclear cells. In: Basu S, Wiklund L, editors. Studies on Experimental Models. Oxidative Stress in Applied Basic Research and Clinical Practice. Totowa, NJ: Humana Press (2011). p. 251–63.
47. Moore DF, Li H, Jefferies N, Wright V, Cooper RA, Elkahloun A, et al. Using peripheral blood mononuclear cells to determine a gene expression profile of acute ischemic stroke: a pilot investigation. Circulation. (2005) 111:212–21. doi: 10.1161/01.CIR.0000152105.79665.C6
48. Nagabhushan M, Mathews HL, Witek-Janusek L. Aberrant nuclear expression of ap-1 and nfkb in lymphocytes of women stressed by the experience of breast biopsy. Brain Behav Immun. (2001) 15:78–84. doi: 10.1006/brbi.2000.0589
49. Segman RH, Shefi N, Goltser-Dubner T, Friedman N, Kaminski N, Shalev AY. Peripheral blood mononuclear cell gene expression profiles identify emergent post-traumatic stress disorder among trauma survivors. Mol Psychiatry. (2005) 10:500–13. doi: 10.1038/sj.mp.4001636
50. Witek-Janusek L, Gabram S, Mathews HL. Psychologic stress, reduced NK cell activity, and cytokine dysregulation in women experiencing diagnostic breast biopsy. Psychoneuroendocrinology. (2007) 32:22–35. doi: 10.1016/j.psyneuen.2006.09.011
51. Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. (2010) 8:e1000412. doi: 10.1371/journal.pbio.1000412
52. Nelson LS, Olsen D. Opioids. In: Hoffman RS, Howland M, Lewin NA, Nelson LS, Goldfrank LR, editors. Goldfrank's Toxicologic Emergencies. 10th ed. New York, NY: McGraw-Hill (2015). p. 559–78.
53. Garrison L, Leeman L, Savich RD, Gutierrez H, Rayburn WF, Bakhireva LN. Fetal growth outcomes in a cohort of polydrug- and opioid-dependent patients. J Reprod Med. (2016) 16:311–9.
54. Jansson LM, Pietro JA, Elko A, Williams EL, Milio L, Velez M. Pregnancies exposed to methadone, methadone and other illicit substances, and poly-drugs without methadone: a comparison of fetal neurobehaviors and infant outcomes. Drug Alcohol Depend. (2012) 122:213–9. doi: 10.1016/j.drugalcdep.2011.10.003
55. Greig E, Ash A, Douiri A. Maternal and neonatal outcomes following methadone substitution during pregnancy. Arch Gynecol Obstet. (2012) 286:843–51. doi: 10.1007/s00404-012-2372-9
56. Clancy B, Darlington RB, Finlay BL. Translating developmental time across mammalian species. Neuroscience. (2001) 105:7–17. doi: 10.1016/S0306-4522(01)00171-3
57. Mallard C, Vexler Z. Modeling ischemia in the immature brain: how translational are animal models? Stroke. (2015) 46:3006–11. doi: 10.1161/STROKEAHA.115.007776
58. Patel SD, Pierce L, Ciardiello AJ, Vannucci SJ. Neonatal encephalopathy: pre-clinical studies in neuroprotection. Biochem Soc Trans. (2014) 42:564–8. doi: 10.1042/BST20130247
59. Salmaso N, Jablonska B, Scafidi J, Vaccarino FM, Gallo V. Neurobiology of premature brain injury. Nat Neurosci. (2014) 17:341–6. doi: 10.1038/nn.3604
60. Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ. Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol. (2013) 106–7:1–16. doi: 10.1016/j.pneurobio.2013.04.001
61. Tucker AM, Aquilina K, Chakkarapani E, Hobbs CE, Thoresen M. Development of amplitude-integrated electroencephalography and interburst interval in the rat. Pediatr Res. (2009) 65:62–6. doi: 10.1203/PDR.0b013e3181891316
62. Vannucci RC, Connor JR, Mauger DT, Palmer C, Smith MB, Towfighi J, et al. Rat model of perinatal hypoxic-ischemic brain damage. J Neurosci Res. (1999) 55:158–63. doi: 10.1002/(SICI)1097-4547(19990115)55:2<158::AID-JNR3>3.0.CO;2-1
63. Dobbing J, Sands J. Comparative aspects of brain growth spurt. Early Hum Dev. (1979) 311:79–83. doi: 10.1016/0378-3782(79)90022-7
64. Downes N, Mullins P. The development of myelin in the brain of the juvenile rat. Toxicol Pathol. (2013) 42:913–22. doi: 10.1177/0192623313503518
65. Bockhorst KH, Narayana PA, Liu R, Ahobila-Vijjula P, Ramu J, Kamel M, et al. Early postnatal development of rat brain: in vivo diffusion tensor imaging. J Neurosci Res. (2008) 86:1520–8. doi: 10.1002/jnr.21607
66. Catalani A, Sabbatini M, Consoli C, Cinque C, Tomassoni D, Azmitia E, et al. Glial fibrillary acidic protein immunoreactive astrocytes in developing rat hippocampus. Development. (2002) 123:481–90. doi: 10.1016/S0047-6374(01)00356-6
67. Cowan WM. The development of the brain. Sci Am. (1979) 241:113–33. doi: 10.1038/scientificamerican0979-112
68. Dean JM, Moravec MD, Grafe M, Abend N, Ren J, Gong X, et al. Strain-specific differences in perinatal rodent oligodendrocyte lineage progression and its correlation with human. Dev Neurosci. (2011) 33:251–60. doi: 10.1159/000327242
69. Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. (2009) 32:149–84. doi: 10.1146/annurev.neuro.051508.135600
70. Hagberg H, Mallard C, Ferriero DM, Vannucci SJ, Levison SW, Vexler ZS, et al. The role of inflammation in perinatal brain injury. Nat Rev Neurol. (2015) 11:192–208. doi: 10.1038/nrneurol.2015.13
71. Holsapple MP, West LJ, Landreth KS. Species comparison of anatomical and functional immune system development. Birth Defects Res B Dev Reprod Toxicol. (2003) 68:321–34. doi: 10.1002/bdrb.10035
72. Lai JCY, Rocha-Ferreira E, Ek CJ, Wang X, Hagberg H, Mallard C. Immune responses in perinatal brain injury. Brain Behav Immun. (2017) 63:210–23. doi: 10.1016/j.bbi.2016.10.022
73. Ayer JG, Song C, Steinbeck K, Celermajer DS, Ben Freedman S. Increased tissue factor activity in monocytes from obese young adults. Clin Exp Pharmacol Physiol. (2010) 37:1049–54. doi: 10.1111/j.1440-1681.2010.05430.x
74. Faul F, Erdfelder E, Lang AG, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. (2007) 39:175–91. doi: 10.3758/bf03193146
75. Dammann O, Phillips TM, Allred EN, O'Shea TM, Paneth N, Van Marter LJ, et al. Mediators of fetal inflammation in extremely low gestational age newborns. Cytokine. (2001) 13:234–9. doi: 10.1006/cyto.2000.0820
76. Kuban KC, Jara H, O'Shea TM, Heeren T, Joseph RM, Fichorova RN, et al. Association of circulating proinflammatory and anti-inflammatory protein biomarkers in extremely preterm born children with subsequent brain magnetic resonance imaging volumes and cognitive function at age 10 years. J Pediatr. (2019) 210:81–90.e3. doi: 10.1016/j.jpeds.2019.03.018
77. Leviton A, Kuban KC, Allred EN, Fichorova RN, O'Shea TM, Paneth N, et al. Early postnatal blood concentrations of inflammation-related proteins and microcephaly two years later in infants born before the 28th post-menstrual week. Early Hum Dev. (2011) 87:325–30. doi: 10.1016/j.earlhumdev.2011.01.043
78. Leviton A, Allred EN, Fichorova RN, VanderVeen DK, O'Shea TM, Kuban K, et al. Early postnatal IGF-1 and IGFBP-1 blood levels in extremely preterm infants: relationships with indicators of placental insufficiency and with systemic inflammation. Am J Perinatol. (2019) 36:1442–52. doi: 10.1055/s-0038-1677472
79. O'Shea TM, Joseph RM, Kuban KC, Allred EN, Ware J, Coster T, et al. Elevated blood levels of inflammation-related proteins are associated with an attention problem at age 24 mo in extremely preterm infants. Pediatr Res. (2014) 75:781–7. doi: 10.1038/pr.2014.41
80. Leviton A, Kuban K, O'Shea TM, Paneth N, Fichorova R, Allred EN, et al. The relationship between early concentrations of 25 blood proteins and cerebral white matter injury in preterm newborns: the ELGAN study. J Pediatr. (2011) 158:897–903 e1–5. doi: 10.1016/j.jpeds.2010.11.059
81. Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochem Biophys Acta. (2014) 1843:2563–82. doi: 10.1016/j.bbamcr.2014.05.014
82. O'Neill LAJ, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. (2007) 7:353–64. doi: 10.1038/nri2079
83. Baloch S, Verma R, Huang H, Khurd P, Clark S, Yarowsky P, et al. Quantification of brain maturation and growth patterns in C57Bl/6 mice via computational neuroanatomy of diffusion tensor images. Cerebral Cortex. (2009) 19:675–87. doi: 10.1093/cercor/bhn112
84. O'Shea TM, Allred EN, Kuban KC, Dammann O, Paneth N, Fichorova R, et al. Elevated concentrations of inflammation-related proteins in postnatal blood predict severe developmental delay at 2 years of age in extremely preterm infants. J Pediatr. (2012) 160:395–401 e4. doi: 10.1016/j.jpeds.2011.08.069
85. McAdams RM, Juul SE. The role of cytokines and inflammatory cells in perinatal brain injury. Neurol Res Int. (2015) 2012:561494. doi: 10.1155/2012/561494
86. Leviton A, Fichorova R, Yamamoto Y, Allred EN, Dammann O, Hecht J, et al. Inflammation-related proteins in the blood of extremely low gestational age newborns. The contribution of inflammation to the appearance of developmental regulation. Cytokine. (2011) 53:66–73. doi: 10.1016/j.cyto.2010.09.003
87. Kuban KC, O'Shea TM, Allred EN, Fichorova RN, Heeren T, Paneth N, et al. The breadth and type of systemic inflammation and the risk of adverse neurological outcomes in extremely low gestation newborns. Pediatr Neurol. (2015) 52:42–8. doi: 10.1016/j.pediatrneurol.2014.10.005
88. Leviton A, Allred EN, Fichorova RN, Kuban KC, Michael O'Shea T, Dammann O, et al. Systemic inflammation on postnatal days 21 and 28 and indicators of brain dysfunction 2years later among children born before the 28th week of gestation. Early Hum Dev. (2016) 93:25–32. doi: 10.1016/j.earlhumdev.2015.11.004
89. Dammann O, Allred EN, Fichorova RN, Kuban K, O'Shea TM, Leviton A, et al. Duration of systemic inflammation in the first postnatal month among infants born before the 28th week of gestation. Inflammation. (2016) 39:672–7. doi: 10.1007/s10753-015-0293-z
90. Galinsky R, Dhillon SK, Dean JM, Davidson JO, Lear CA, Wassink G, et al. Tumor necrosis factor inhibition attenuates white matter gliosis after systemic inflammation in preterm fetal sheep. J Neuroinflamm. (2020) 17:1–16. doi: 10.1186/s12974-020-01769-6
91. Pan W, Stone KP, Hsuchou H, Manda VK, Kastin AJ. Cytokine signaling modulates blood-brain barrier function. Curr Pharm Des. (2011) 17:3729–40. doi: 10.2174/138161211798220918
92. Rosenberg GA, Estrada EY, Dencoff JE, Stetler-Stevenson WG. Tumor necrosis factor-alpha-induced gelatinase B causes delayed opening of the blood-brain barrier: an expanded therapeutic window. Brain Res. (1995) 703:151–5. doi: 10.1016/0006-8993(95)01089-0
93. Tsao N, Hsu HP, Wu CM, Liu CC, Lei HY. Tumour necrosis factor-alpha causes an increase in blood-brain barrier permeability during sepsis. J Med Microbiol. (2001) 50:812–21. doi: 10.1099/0022-1317-50-9-812
94. Zhao C, Ling Z, Newman MB, Bhatia A, Carvey PM. TNF-alpha knockout and minocycline treatment attenuates blood-brain barrier leakage in MPTP-treated mice. Neurobiol Dis. (2007) 26:36–46. doi: 10.1016/j.nbd.2006.11.012
95. Griffiths SK, Campbell JP. Placental structure, function and drug transfer. BJA Educ. (2014) 15:84–9. doi: 10.1093/bjaceaccp/mku013
96. Chaves C, Remiao F, Cisternino S, Decleves X. Opioids and the blood-brain barrier: a dynamic interaction with consequences on drug disposition in brain. Curr Neuropharmacol. (2017) 15:1156–73. doi: 10.2174/1570159X15666170504095823
97. Gharavi R, Hedrich W, Wang H, Hassan HE. Transporter-mediated disposition of opioids: implications for clinical drug interactions. Pharm Res. (2015) 32:2477–502. doi: 10.1007/s11095-015-1711-5
98. Hutchinson MR, Shavit Y, Grace PM, Rice KC, Maier SF, Watkins LR. Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol Rev. (2011) 63:772–810. doi: 10.1124/pr.110.004135
99. Bilbo SD, Biedenkapp JC, Der-Avakian A, Watkins LR, Rudy JW, Maier SF. Neonatal infection-induced memory impairment after lipopolysaccharide in adulthood is prevented via caspase-1 inhibition. J Neurosci. (2005) 25:8000–9. doi: 10.1523/JNEUROSCI.1748-05.2005
100. Bilbo SD, Levkoff LH, Mahoney JH, Watkins LR, Rudy JW, Maier SF. Neonatal infection induces memory impairments following an immune challenge in adulthood. Behav Neurosci. (2005) 119:293–301. doi: 10.1037/0735-7044.119.1.293
101. Yanni D, Korzeniewski SJ, Allred EN, Fichorova RN, O'Shea TM, Kuban K, et al. Both antenatal and postnatal inflammation contribute information about the risk of brain damage in extremely preterm newborns. Pediatr Res. (2017) 82:691–6. doi: 10.1038/pr.2017.128
102. Claus CP, Tsuru-Aoyagi K, Adwanikar H, Walker B, Whetstone W, Noble-Haeusslein LJ. Age is a determinant of the inflammatory response and loss of cortical volume after traumatic brain injury. Dev Neurosci. (2010) 32:454–65. doi: 10.1016/j.nbd.2014.12.003
103. Fleiss B, Gressens P. Tertiary mechanisms of brain damage: a new hope for treatment of cerebral palsy? Lancet Neurol. (2012) 11:556–66. doi: 10.1016/S1474-4422(12)70058-3
104. Thornton C, Rousset CI, Kichev A, Miyakuni Y, Vontell R, Baburamani AA, et al. Molecular mechanisms of neonatal brain injury. Neurol Res Int. (2012) 2012:506320. doi: 10.1155/2012/506320
105. Asai K, Hiki N, Mimura Y, Ogawa T, Unou K, Kaminishi M. Gender differences in cytokine secretion by human peripheral blood mononuclear cells: role of estrogen in modulating LPS-induced cytokine secretion in an ex vivo septic model. Shock. (2001) 16:340–3. doi: 10.1097/00024382-200116050-00003
106. Da Pozzo E, Giacomelli C, Cavallini C, Martini C. Cytokine secretion responsiveness of lymphomonocytes following cortisol cell exposure: sex differences. PLoS ONE. (2018) 13:e0200924. doi: 10.1371/journal.pone.0200924
107. Silaidos C, Pilatus U, Grewal R, Matura S, Lienerth B, Pantel J, et al. Sex-associated differences in mitochondrial function in human peripheral blood mononuclear cells (PBMCs) and brain. Biol Sex Dif. (2018) 9:34. doi: 10.1186/s13293-018-0193-7
108. Smith PLP, Mottahedin A, Svedin P, Mohn CJ, Hagberg H, Ek J, et al. Peripheral myeloid cells contribute to brain injury in male neonatal mice. J Neuroinflammation. (2018) 15:301. doi: 10.1186/s12974-018-1344-9
109. Mallard C, Tremblay ME, Vexler ZS. Microglia and neonatal brain injury. Neuroscience. (2019) 405:68–76. doi: 10.1016/j.neuroscience.2018.01.023
110. Finak G, Langweiler M, Jaimes M, Malek M, Taghiyar J, Korin Y, et al. Standardizing flow cytometry immunophenotyping analysis from the human immunophenotyping consortium. Sci Rep. (2016) 6:20686. doi: 10.1038/srep20686
111. Carloni S, Proietti F, Longini M, Marseglia L, D'Angelo G, Balduini W, et al. Melatonin pharmacokinetics following oral administration in preterm neonates. Molecules. (2017) 22:2115. doi: 10.3390/molecules22122115
112. Jantzie LL, Oppong AY, Conteh FS, Yellowhair TR, Kim J, Fink G, et al. Repetitive neonatal erythropoietin and melatonin combinatorial treatment provides sustained repair of functional deficits in a rat model of cerebral palsy. Front Neurol. (2018) 9:233. doi: 10.3389/fneur.2018.00233
113. Juul SE, Comstock BA, Heagerty PJ, Mayock DE, Goodman AM, Hauge S, et al. High-dose erythropoietin for asphyxia and encephalopathy (HEAL): a randomized controlled trial - background, aims, and study protocol. Neonatology. (2018) 133:331–8. doi: 10.1159/000486820
Keywords: methadone, lymphocyte, PBMC, SPIHR, blood mononuclear cell, neuroinflammation, neonatal abstinence syndrome, prenatal
Citation: Newville J, Maxwell JR, Kitase Y, Robinson S and Jantzie LL (2020) Perinatal Opioid Exposure Primes the Peripheral Immune System Toward Hyperreactivity. Front. Pediatr. 8:272. doi: 10.3389/fped.2020.00272
Received: 07 January 2020; Accepted: 29 April 2020;
Published: 26 June 2020.
Edited by:
Lewis Phillip Rubin, MedStar Georgetown University Hospital, United StatesReviewed by:
Robert Galinsky, Hudson Institute of Medical Research, AustraliaCopyright © 2020 Newville, Maxwell, Kitase, Robinson and Jantzie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lauren L. Jantzie, bGphbnR6aWVAamhtaS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.