 Fei Zhou Zhang
Fei Zhou Zhang Jie Xin Yuan
Jie Xin Yuan Lu Qin
Lu Qin Lan Fang Tang
Lan Fang Tang- Department of Pneumology, Zhejiang University School of Medicine of Children's Hospital, Hangzhou, China
Background: Pulmonary alveolar proteinosis (PAP) is a rare diffuse lung disease. Reports of rare cases of PAP due to Pneumocystis jirovecii (P. jirovecii) exist in infants with immunodeficiency diseases, but no cases have been reported to date in pediatric patients with type 1 hyper-IgM syndrome (HIGM1).
Case Presentation: Herein, we present a case of PAP secondary to P. jirovecii on an infant with HIGM1. He was admitted to our unit because of cough and tachypnea. Lung biopsy confirmed the diagnosis of PAP, whereas hexamine-silver staining of the bronchoalveolar lavage fluid identified P. jirovecii infection. No other probable cause of PAP was observed. Whole exome sequencing indicated a novel c.511dupA (p.I171N*30) hemizygous mutation in the CD40 ligand (CD40LG) gene. He was cured with bronchoalveolar lavage and compound sulfamethoxazole tablets.
Conclusions: To our knowledge, this is the first reported case of P. jirovecii infection as a reversible cause of PAP in an infant with HIGM1.
Background
Pulmonary alveolar proteinosis (PAP), first described by Rosen et al. (1) in 1958, is a rare diffuse lung disease of idiopathic etiology. Its estimated prevalence is about one in 3.7–6.9 × 106 with a male/female ratio of 1:1 to 2:1 (2–5). Most patients are diagnosed between 20 and 50 years of age (2, 4–6). PAP is pathologically characterized by the presence of massive, amorphous, insoluble phospholipid-rich protein deposition in the alveolar and bronchial cavities. The first description of PAP and the vast majority of data on treatment and prognosis were connected with autoimmune PAP (1). The etiology of PAP includes surfactant-dysfunction syndromes, impaired granulocyte macrophage-colony stimulating factor signaling, hematological disorders and other malignancies, immunological diseases, infections, drugs, dust exposure, and systemic diseases (7). Etiologically, PAP is commonly classified into three types including congenital, autoimmune, and secondary PAP. Formerly, the term congenital alveolar proteinosis was used to describe surfactant dysfunction syndromes in neonates (8). Furthermore, secondary PAP primarily occurs in patients with immunodeficiency or under immunosuppressive therapy for immunodeficiency syndromes, chronic inflammatory disorders, malignancies, infections, toxic dust, and fumes (9). In 2014, Raj et al. (10) reported the first case of infant PAP secondary to Pneumocystis jirovecii (P. jirovecii) infection, and in 2016, Gallagher et al. (11) first reported a case of PAP in children with type 1 hyper-IgM syndrome but without P. jirovecii infection. Herein, we report P. jirovecii infection as a reversible cause of PAP in an infant with type 1 hyper-IgM syndrome [HIGM1, Online Mendelian Inheritance in Man (OMIM) 308230] to highlight this rare condition.
Case Presentation
A 9-month-old boy was admitted to our unit due to dry cough and wheezing for 2 weeks, and tachypnea for 5 days. He is the second child of nonconsanguineous parents with an uneventful birth history. Medical history revealed three prior episodes of lower respiratory tract infections necessitating hospitalization and one instance of methylprednisolone treatment before referral to our hospital.
Physical examination revealed that the Z-score of weight for age and weight for height was 0.60; respiratory rate, 66 bpm; heart rate, 156 bpm, and temperature, 36.6°C. A depression of the suprasternal and supraclavicular fossa with coarse breath sounds without rale were noted in both lungs. The liver and spleen were non-palpable below the costal margin. Laboratory data showed a high white blood cell count, 30.61 × 109 cells/L, with 41.1% neutrophils; hemoglobin, 130 g/L; hypersensitive C-reactive protein, <1 mg/L; erythrocyte sedimentation rate, 8.0 mm/h, and serum lactate dehydrogenase, 323 U/L (normal range, 135.0–215.0 U/L). Additionally, low levels of immunoglobulin A, 0.04 g/L (normal range, 0.10–0.56 g/L) and immunoglobulin G, 0.15 g/L (normal range, 3.60–9.20 g/L), and normal levels of immunoglobulin M, 0.48 g/L (normal range 0.40–1.28 g/L), were found. T cell subsets—CD19+, CD20+, CD4+, CD4+, and CD4+/CD8+–were in their normal ranges. Screening tests for HIV, syphilis, hepatitis C, mycobacteria, nocardia, Epstein-Barr virus, and fungal pathogens were negative or within normal range. Antinuclear, anti-double-stranded DNA, anti-smooth muscle actin, anti-ribonucleoprotein, and anti-neutrophil cytoplasmic antibodies were negative, respectively.
Computed tomography (CT) scan of the chest showed ground-glass density images in both lungs (Figure 1). On the 7th day of hospitalization, bronchoscopy with bronchoalveolar lavage and lung biopsy was performed under general anesthesia. The bronchoalveolar lavage fluid appeared milky (Figure 2A), while hematoxylin-eosin staining showed extensive macrophage infiltration in the alveolar and bronchial cavities without evidence of fibrosis and hexamine-silver staining for bronchoalveolar lavage fluid revealed the presence of P. jirovecii (Figure 2B). Moreover, large amount of periodic acid-Schiff (PAS) stain-positive and diastase-Periodic acid-Schiff (D-PAS) stain-positive substance was found in the alveolar and bronchial cavities (Figures 3A,B), consistent with pathological characteristics of PAP and non-specific interstitial pneumonia.
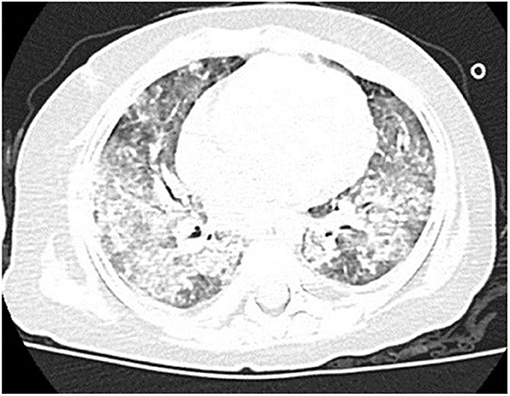
Figure 1. Clinical characteristics of our patient. Chest CT showed disclosed extensive progressive interstitial changes in both lungs on August 2016.
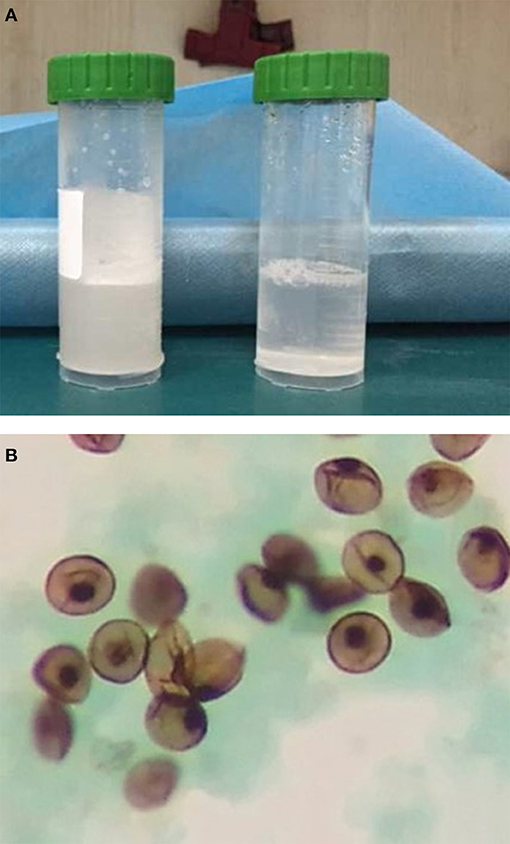
Figure 2. (A) The “milky” bronchoalveolar lavage fluid. (B) P. jirovecii was detected in the bronchoalveolar lavage fluid by silver hexamine staining.
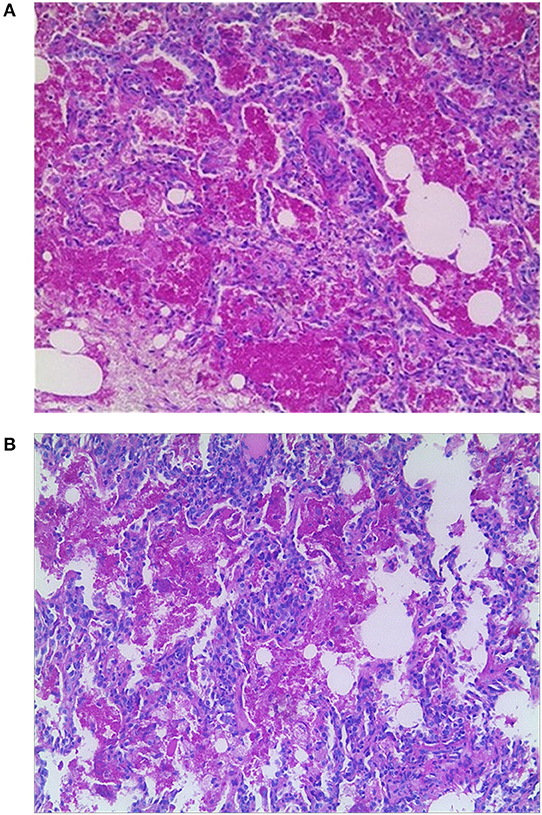
Figure 3. (A) Pathology showed large amounts of PAS-positive lipoproteins in alveolar and bronchial cavities (×100). (B) Pathology showed large amounts of D-PAS-positive fine granular lipoproteins in alveolar and bronchial cavities (×100).
Whole exome sequencing was conducted by Huada Inspection Center (Shenzhen, China) to detect gene mutation related to immunodeficiency, alveolar surface-active substance or inherited disorder associated with PAP. A c.511dupA hemizygous mutation in the EX5E region of the CD40 ligand (CD40LG, OMIM 300386) gene, present on Xq26.3 and coding for CD40L protein, was noted. Bioinformatic analysis suggested a frameshift mutation inducing the early termination of the amino acid-encoded protein (p.I171N). Genetic analysis of the patient's parents indicated maternal inheritance of the mutation.
Following the confirmation of P. jirovecii infection, imipenem, sulperazone, and erythromycin were administered intravenously, and sulfamethoxazole was given orally. Meanwhile, sodium bicarbonate tablets were given to alkalize urine and methylprednisolone and human immunoglobulin was used for anti-inflammatory and immune support therapy. Bronchoscopy with bronchoalveolar lavage was repeated 10 days later, and significant clearing of the lavage fluid was observed. The patient displayed gradual improvement and was eventually discharged on the 48th day of hospitalization.
Over the course of the longitudinal follow-up, at the age of 3 years old, he was readmitted to the Department of Urology with voiding symptom persistent for more than a month. The physical examination at that time suggested that the Z-score of weight for age and weight for height were −0.30 and −0.40, respectively. Pelvic enhancement CT revealed a 3.5 × 3.5 × 2.5 cm calculi in the urinary bladder, subsequently extracted by cystolithotomy. The patient displayed normal growth and development since, comparable to his peers, and had no recurrence of PAP or infection during the 2-year follow-up. A chest CT showed significant improvement in both lungs in the latest follow-up (Figure 4). The parents, however, refused the proposal of hematopoietic stem cell transplantation for managing HIGM1.
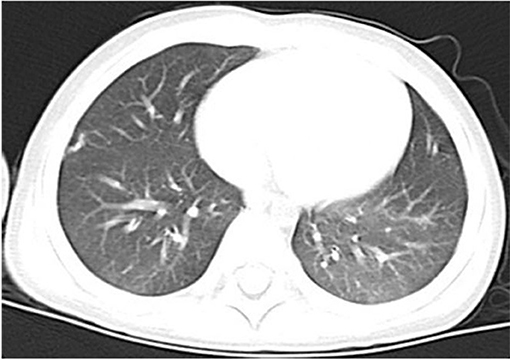
Figure 4. Chest CT showed significant improvement in both lungs on August 2018.
Discussion and Conclusion
PAP is an intractable interstitial lung disease involving bilateral sedimentation of lipoprotein acetous material in the lungs, affecting gas exchange and impairing the pulmonary defense against pathogenic bacteria. The diagnosis of PAP is based on clinical symptoms, imaging data, characteristics of bronchoalveolar lavage fluid, and results of lung biopsy (12, 13). The present case of PAP diagnosis was confirmed by evaluation of the bronchoalveolar lavage and pathology results. P. jirovecii was positively identified in the bronchoalveolar lavage fluid, although PAP secondary to P. jirovecii has only been reported in adults (14). Moreover, anti-P. jirovecii treatment is effective for treating PAP, with P. jirovecii being the cause of PAP in our case. Although the precise mechanism of P. jirovecii-induced PAP remains unclear, P. jirovecii should be considered as a potential etiology in pediatric cases of PAP.
P. jirovecii is an opportunistic pathogen, and its incidences are mainly reported in patients with immunodeficiency or under immunosuppressive therapy (15). As our case had three prior history of pneumonia with low IgA and IgG levels, immunodeficiency was considered. The patient was reported to have a heterozygous CD40LG gene mutation (c.511dupA), a frameshift mutation resulting in premature termination of the protein. CD40L dysfunction is one of the causes of rare primary immunodeficiency diseases accounting for 65–70% of HIGM1 cases (16–18). Hence, in P. jirovecii-infected children with or without PAP, immunodeficiency should be considered, and genetics analysis should be undertaken.
HIGM1, an X-linked immunodeficiency disorder (also named X-liked hyper IgM syndrome, XHIGM), typically presents with reduced or absent levels of IgG, IgA, and IgE, and normal to elevated levels of IgM. Clinical symptoms are complex and variable, with more than 50% of the male infants, less than a year old, developing the symptoms, whereas the figure rises to more than 90% in those younger than 4 years (19). The primary symptoms include recurrent respiratory tract bacterial infections, opportunistic infections, and recurrent or refractory diarrhea accompanied by growth retardation (20). P. jirovecii infection is the main cause of deaths in opportunistic pneumonia infections (21).
It remains unclear whether HIGM1 causes PAP or progresses to PAP following P. jirovecii infection. PAP secondary to P. jirovecii infection is another commonly reported variant of immunodeficiency. To our knowledge, this is the first case report of P. jirovecii-induced PAP in children with HIGM1. CD40L, the ligand of CD40, is a member of the tumor necrosis factor family and mainly expressed on activated CD4+ T cells (22). The CD40–CD40L interaction contributes to a majority of humoral immune responses and especially the B-cell proliferation and germinal center formation (23). Mutation in the CD40LG gene causes serious impairment of T-cell-dependent antibody responses because of the lack of memory B-cells and deficient induction of somatic mutations, and results in immunodeficiency with little or no circulating IgG, IgA, and IgE antibodies (24). Reports indicate that humoral immunodeficiency patients are more likely to develop opportunistic infections (e.g., P. jirovecii) that weaken the phagocytic function of macrophages and reduce the clearance of pulmonary surfactant, eventually leading to its overabundance, which is the most essential characteristic of PAP (7, 25, 26). Furthermore, our case displayed no recurrence of PAP during the longitudinal follow-up, supporting the opinion that HIGM1 does not directly lead to PAP.
Unlike the poor prognosis associated with congenital PAP, secondary PAP caused by infections may be reversed. However, no guideline exists for the treatment of secondary PAP in pediatric patients. Bronchial alveolar lavage is regarded as the gold standard for the effective therapy for PAP (27–29). In our case, the pulmonary symptoms resolved immediately following bronchoscopy-assisted whole lung lavage under general anesthesia. For treating P. jirovecii infection, we followed the European council on infections in leukemia guidelines for P. jirovecii pneumonia in non-HIV-infected hematology patients (30). The clinical symptoms improved after sulfamethoxazole therapy. Notably, a bladder stone was noted 3 months after discharge, which might be a renal side effect of sulfamethoxazole, although oral bicarbonate tablets to alkalize urine was administered. Therefore, alkalinity of urine and hydration should not be overlooked during sulfamethoxazole therapy. Although therapies with intravenous immunoglobulins, trimethoprim–sulfamethoxazole, and granulocyte colony-stimulating factor were reported to reduce the opportunistic infections, human leukocyte antigen-matched hematopoietic stem cell transplantation was the only radical treatment for treating HIGM1 (31, 32). However, the transplantation should be restricted to children younger than 5 years because the risk of various complications and organ damage may increase in those older than 6 years (33, 34).
To our knowledge, this is the first report of P. carinii-associated PAP in a HIGM1 pediatric patient. Our observations suggest that P. jirovecii infection should be considered as a potentially reversible cause of PAP in immunodeficient children, including infants with HIGM1.
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Ethics Statement
The Ethics Committee of Zhejiang University Medical College has agreed to the retrospective case report, arguing that there is no ethical dispute in the study.
Author Contributions
LT conceptualized and designed the study, and reviewed and revised the manuscript. FZ drafted the initial manuscript. JY collected data. LQ was responsible for the follow-up. All authors approved the final manuscript as submitted and agreed to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation (81170016 and 81470214) and Zhejiang Provincial Program for the Cultivation of High-Level Innovative Health Talents. The funding provided support for the cost of gene sequencing and publication for the patient.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the patient and his parents for permitting us to use the data and the National Natural Science Foundation (81170016 and 81470214) and Zhejiang Provincial Program for the Cultivation of High-Level Innovative Health Talents.
Abbreviations
PAP, Pulmonary alveolar proteinosis; P. jirovecii, Pneumocystis jirovecii; HIGM1, type 1 hyper-IgM syndrome.
References
1. Rosen SH, Castleman B, Liebow AA. Pulmonary alveolar proteinosis. N Engl J Med. (1958) 258:1507–15. doi: 10.1056/NEJM195806052582301
2. Ben-Dov I, Kishinevski Y, Roznman J, Soliman A, Bishara H, Zelligson E, et al. Pulmonary alveolar proteinosis in Israel: ethnic clustering. Isr Med Assoc J. (1999) 1:75–8.
3. Mccarthy C, Avetisyan R, Carey BC, Chalk C, Trapnell BC. Prevalence and healthcare burden of pulmonary alveolar proteinosis. Orphanet J Rare Dis. (2018) 13:129. doi: 10.1186/s13023-018-0846-y
4. Ishii H, Tazawa R, Kaneko C, Saraya T, Inoue Y, Hamano E, et al. Clinical features of secondary pulmonary alveolar proteinosis: pre-mortem cases in Japan. Eur Respir J. (2011) 37:465–8. doi: 10.1183/09031936.00092910
5. Zhang D, Tian X, Feng R, Guo X, Wang P, Situ Y, et al. Secondary pulmonary alveolar proteinosis: a single-center retrospective study (a case series and literature review). BMC Pulm Med. (2018) 18:15. doi: 10.1186/s12890-018-0590-z
6. Kiani A, Parsa T, Adimi NP, Dutau H, Razavi F, Farzanegan B, et al. An eleven-year retrospective cross-sectional study on pulmonary alveolar proteinosis. Adv Respir Med. (2018) 86:7–12. doi: 10.5603/ARM.2018.0003
7. Griese M. Pulmonary alveolar proteinosis: a comprehensive clinical perspective. Pediatrics. (2017) 140:e20170610. doi: 10.1542/peds.2017-0610
8. Nogee LM, Mello DE, De Dehner LP, Colten HR. Brief report: deficiency of pulmonary surfactant protein B in congenital alveolar proteinosis. N Engl J Med. (1993) 328:406–10. doi: 10.1056/NEJM199302113280606
9. Ishii H, Seymour JF, Tazawa R, Inoue Y, Uchida N, Nishida A, et al. Secondary pulmonary alveolar proteinosis complicating myelodysplastic syndrome results in worsening of prognosis: a retrospective cohort study in Japan. BMC Pulm Med. (2014) 14:37. doi: 10.1186/1471-2466-14-37
10. Raj D, Bhutia TD, Mathur S, Kabra SK, Lodha R. Pulmonary alveolar proteinosis secondary to Pneumocystis jirovecii infection in an infant with common variable immunodeficiency. Indian J Pediatr. (2014) 81:929–31. doi: 10.1007/s12098-013-1027-6
11. Gallagher J, Adams J, Hintermeyer M, Torgerson TR. X-linked hyper IgM syndrome presenting as pulmonary alveolar proteinosis. J Clin Immunol. (2016) 36:564–70. doi: 10.1007/s10875-016-0307-0
12. Wang BM, Pierson DJ, Stern EJ, Schmidt RA. Diagnosing pulmonary alveolar proteinosis: a review and an update. Chest. (1997) 111:460–6. doi: 10.1378/chest.111.2.460
13. Rab A, Arimura FE, Kairalla RA, Crr C, Baldi BG. Characterization and outcomes of pulmonary alveolar proteinosis in Brazil: a case series. J Bras Pneumol. (2018) 44:231–6. doi: 10.1590/s1806-37562017000000168
14. Petio K, Vinod S. Alveolar proteinosis in a patient recovering from Pneumocystis carinii infection: a case report with a review of literature. Cytojournal. (2006) 3:22. doi: 10.1186/1742-6413-3-22
15. Nhieu JTV, Vojtek AM, Bernaudin JF, Escudier E, Fleury-Feith J. Pulmonary alveolar proteinosis associated with Pneumocystis carinii: ultrastructural identification in bronchoalveolar lavage in AIDS and immunocompromised non-AIDS patients. Chest. (1990) 98:801–5. doi: 10.1378/chest.98.4.801
16. Allen RC, Armitage RJ, Conley ME, Rosenblatt H, Jenkins NA, Copeland NG, et al. CD40 ligand gene defects responsible for X-linked hyper-IgM syndrome. Science. (1993) 259:990–3. doi: 10.1126/science.7679801
17. Burcin U, Francisco B, Howard L. Evaluation of a patient with hyper-IgM syndrome. J Allergy Clin Immunol. (2012) 129:1692–3.e1694. doi: 10.1016/j.jaci.2012.03.043
18. França TT, Barreiros LA, Al-Ramadi BK, Ochs HD, Cabral-Marques O, Condino-Neto A. CD40 ligand deficiency: treatment strategies and novel therapeutic perspectives. Expert Rev Clin Immunol. (2019) 15:529–40. doi: 10.1080/1744666X.2019.1573674
19. Jerry AW, Mary CM, Hans O, Ramsey F, Paul RS, Raif G, et al. The X-linked hyper-IgM syndrome: clinical and immunologic features of 79 patients. Medicine. (2003) 82:373–84. doi: 10.1097/01.md.0000100046.06009.b0
20. Etzioni A, Ochs HD. The hyper IgM syndrome–an evolving story. Pediatr Res. (2004) 56:519–25. doi: 10.1203/01.PDR.0000139318.65842.4A
21. Davies EG, Thrasher AJ. Update on the hyper immunoglobulin M syndromes. Br J Haematol. (2010) 149:167–80. doi: 10.1111/j.1365-2141.2010.08077.x
22. Lederman S, Yellin MJ, Cleary AM, Pernis A, Inghirami G, Cohn LE, et al. T-BAM/CD40-L on helper T lymphocytes augments lymphokine-induced B cell Ig isotype switch recombination and rescues B cells from programmed cell death. J Immunol. (1994) 152:2163–71.
23. Van KC, Banchereau J. CD40-CD40 ligand. J Leukocyte Biol. (2000) 67:2–17. doi: 10.1002/jlb.67.1.2
24. Laman JD, Claassen E, Noelle RJ. Functions of CD40 and its ligand, gp39 (CD40L). Crit Rev Immunol. (1996) 16:59–108. doi: 10.1615/CritRevImmunol.v16.i1.40
25. Golde DW. Alveolar proteinosis and the overfed macrophage. Chest. (1979) 76:119–20. doi: 10.1378/chest.76.2.119
26. Nugent KM, Pesanti EL. Macrophage function in pulmonary alveolar proteinosis. Am Rev Respir Dis. (1983) 127:780–1. doi: 10.1164/arrd.1983.127.6.780
27. Beccaria M, Luisetti M, Rodi G, Corsico A, Zoia MC, Colato S, et al. Long-term durable benefit after whole lung lavage in pulmonary alveolar proteinosis. Eur Respir J. (2004) 23:526–31. doi: 10.1183/09031936.04.00102704
28. Dogru D, Yalcin E, Aslan AT, Ocal T, Ozcelik U, Gucer S, et al. Successful unilateral partial lung lavage in a child with pulmonary alveolar proteinosis. J Clin Anesth. (2009) 21:127–30. doi: 10.1016/j.jclinane.2008.06.035
29. Umuroglu T, Altintaş M, Abdullayev T, Kiyan G, Ayanoglu HÖ. An alternative lung isolation technique in paediatric pulmonary alveolar proteinosis. Turk J Anaesthesiol Reanim. (2017) 45:310–2. doi: 10.5152/TJAR.2017.97523
30. Maschmeyer G, Helweglarsen J, Pagano L, Robin C, Cordonnier C, Schellongowski P. ECIL guidelines for treatment of Pneumocystis jirovecii pneumonia in non-HIV-infected haematology patients. J Antimicrob Chemother. (2016) 71:2405–13. doi: 10.1093/jac/dkw158
31. Gennery AR, Khulood K, Paul V, Bredius RGM, Notarangelo LD, Evelina M, et al. Treatment of CD40 ligand deficiency by hematopoietic stem cell transplantation: a survey of the European experience, 1993-2002. Blood. (2004) 103:1152–7. doi: 10.1182/blood-2003-06-2014
32. Petrovic A, Dorsey M, Miotke J, Shepherd C, Day N. Hematopoietic stem cell transplantation for pediatric patients with primary immunodeficiency diseases at All Children's Hospital/University of South Florida. Immunol Res. (2009) 44:169–78. doi: 10.1007/s12026-009-8111-z
33. Heather A, Martin PL, Paul S, Nelson C, Rebecca B, Suhag P. Hematopoietic stem cell transplantation for cd40 ligand deficiency: single institution experience. Pediatr Blood Cancer. (2015) 62:2216–22. doi: 10.1002/pbc.25711
Keywords: pulmonary alveolar proteinosis (PAP), Pneumocystis jirovecii (P. jirovecii), hyper IgM syndrome, type 1 (HIGM1), CD40 ligand (CD40LG), infant
Citation: Zhang FZ, Yuan JX, Qin L and Tang LF (2020) Pulmonary Alveolar Proteinosis Due to Pneumocystis carinii in Type 1 Hyper-IgM Syndrome: A Case Report. Front. Pediatr. 8:264. doi: 10.3389/fped.2020.00264
Received: 17 January 2020; Accepted: 27 April 2020;
Published: 11 June 2020.
Edited by:
Kostas N. Priftis, National and Kapodistrian University of Athens, GreeceReviewed by:
Paulo Camargos, Federal University of Minas Gerais, BrazilDiane M. Gray, University of Cape Town, South Africa
Copyright © 2020 Zhang, Yuan, Qin and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lan Fang Tang, NjE5NTAwNyYjeDAwMDQwO3pqdS5lZHUuY24=