 Shahrzad Bakhtiar
Shahrzad Bakhtiar Bella Shadur
Bella Shadur Polina Stepensky
Polina Stepensky- 1Division for Pediatric Stem Cell Transplantation and Immunology, University Hospital Frankfurt, Frankfurt, Germany
- 2Department of Bone Marrow Transplantation and Cancer Immunotherapy, Hadassah Medical Center, Jerusalem, Israel
- 3Department of Immunology, Garvan Institute of Medical Research, Darlinghurst, NSW, Australia
- 4Graduate Research School, University of New South Wales, Kensington, NSW, Australia
Congenital disorders of the immune system affecting maturation and/or function of phagocytic leucocytes can result in severe infectious and inflammatory complications with high mortality and morbidity. Further complications include progression to MDS/AML in some cases. Allogeneic stem cell transplantation is the only curative treatment for most patients with these diseases. In this review, we provide a detailed update on indications and outcomes of alloHSCT for congenital neutrophil disorders, based on data from the available literature.
Introduction
Congenital neutrophil disorders as a category of primary immunodeficiency (PID) can be classified in many ways, but a key point of distinction is whether the disorder is quantitative, or qualitative (1). The 2017 International Union of Immunological Societies (IUIS) Phenotypic Classification for Primary Immunodeficiencies divides neutrophil disorders into four broad categories: congenital neutropenia associated with or without syndromic disease, and functional neutrophil defects with or without syndromic disease (2). Affected patients can present with variable symptoms including recurrent infections, failure to thrive, and overwhelming septic episodes leading to high morbidity and mortality. Early and severe respiratory infections (e.g., Burkholderia cepacia, Aspergillus spp.), visceral abscesses, cellulitis, lymphadenitis, and granulomatous lesions are observed in patients suffering from CGD (3, 4). Some patients develop severe autoinflammatory complications underlining the role of neutrophils in autoinflammatory processes beyond microbial defense (5, 6). In many of these diseases there is a recognized risk of progression to myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) (1, 2, 7, 8). Treatment for neutrophil disorders classically comprises anti-microbial therapy, granulocyte-colony stimulating factor (G-CSF), and allogeneic hematopoietic stem cell transplantation (alloHSCT) (8).
In this review we provide an update on the evidence, indications and modalities of alloHSCT for the various congenital neutrophil disorders based on data from the available literature, excluding CGD which will be discussed elsewhere in this special edition. Also beyond the scope of this manuscript is neutropenia that features in predominantly lymphocyte immune deficiencies (e.g., some forms of severe combined immune deficiency, CD40L deficiency, etc.).
Materials and Methods
We used the 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies as the basis for this review (2). Data were gathered via an English-language Pubmed literature search whereby the name of each disorder in figure five of the IUIS classification was searched individually, and together with the terms “alloHSCT” and “transplantation.” We searched for case reports and case series on each disorder, focusing on the question of treatability of the disease by alloHSCT.
Results
Group 1: Syndrome-Associated Neutropenia
This group of diseases is dominated by conditions defined by bone marrow failure (BMF), predisposition to myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML), as well as additional non-hematological manifestations such as neurodevelopmental delay (Table 1). Disease control can be achieved for some patients with G-CSF in combination with antibiotic prophylaxis (40, 41). However, the refractoriness of the disease and the predisposition to myeloid malignancies raise the question of if and when curative treatment by alloHSCT is indicated. A lack of evidence regarding long-term outcomes both with and without transplantation, the lack of genotype-phenotype correlation, and the persistence of neurodevelopmental disorders, make recommendations difficult for many of these diseases (Table 1). In some cases only case reports are available, such as in the case of successful transplant of Glycogen Storage Disease type 1b using reduced intensity conditioning (18, 42). On the contrary, more data is available on Schwachman Diamond Syndrome and deficiency of VPS45 protein, and is presented here (43).
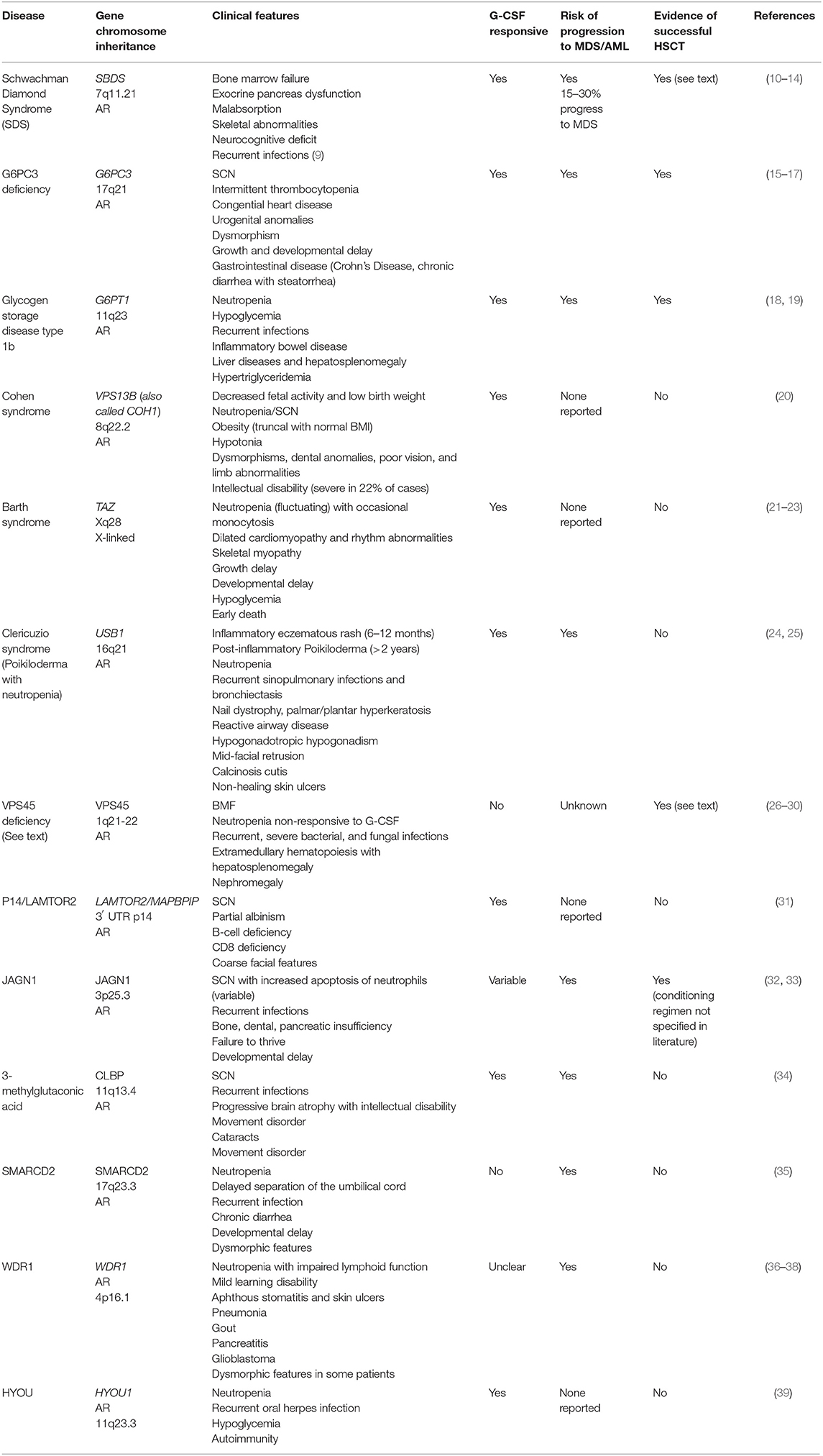
Table 1. Syndrome-associated neutropenia.
Schwachman Diamond Syndrome (SDS)
Schwachman Diamond Syndrome is caused by mutations in the SBDS gene at chromosome 7q11.21. Mutations in this gene lead to impaired RNA metabolism and ribosomal function, with clinical features as described in Table 1 (10). One third of patients develop major hematological complications (9), and alloHSCT is indicated in cases of bone marrow failure (with subsequent transfusion-dependent anemia, bleeding and severe, recurrent infections), MDS, or AML (11, 12, 44, 45). Numerous studies have shown that alloHSCT is able to correct hematological abnormalities in patients with SDS, however most studies are limited by small sample numbers and/or short follow-up time. A 2005 study by Cesaro et al. (44) reviewed 26 patients with SDS who underwent alloHSCT and demonstrated overall survival of 64.5% at 1 year follow-up. Fifty-six percent of patients who were transplanted following the development of MDS/AML died post-transplant, compared to 19% who were transplanted for BMF, although the difference was not statistically significant. Patients transplanted with total body irradiation (TBI) appeared to do worse than those transplanted with busulfan and fludarabine conditioning, with 67 vs. 20% mortality (P = 0.03) (44). In 2002 Hsu reviewed 15 cases of SDS alloHSCT patients and found that transplantation following the development of MDS/AML was associated with a worse outcome; overall survival in this series was 40% (12).
A 2005 review of French registry data found 10 patients who had been transplanted for SDS, five because of bone marrow failure (BMF) and five following development of MDS/AML. They found a 5 years event-free survival of 60% with two patients failing to engraft, one dying 17 months post-HSCT from a respiratory illness, and one death from relapsed MDS. This study noted that patients transplanted prior to the development of malignancy had improved transplant outcomes, and that patients transplanted for BMF demonstrated sustained engraftment with myeloablative regimens based on busulfan and cyclophosphamide. The authors felt that more intense conditioning for BMF-SDS is not warranted, but reduced intensity conditioning for patients with MDS/AML would be insufficient (45). In contrast, Burroughs et al. document three SDS patients transplanted from matched unrelated donors (MUD) with treosulfan and fludarabine conditioning regimens and no serotherapy; these patients underwent transplantation for BMF and achieved sustained engraftment with acute graft vs. host disease (GvHD) developing in two of the three (13).
Thus, for patients with SDS, alloHSCT can correct the hematopoietic aspects of the disease with improved outcomes if transplantation is undertaken prior to development of MDS/AML. Regular monitoring for cytogenetic abnormalities is thus recommended, although some appear to be indolent or transient and are not an automatic indication for HSCT [e.g., i7(q10) and del(20q)]. Larger studies are required before recommendations can be made regarding ideal conditioning regimens (treosulfan or busulfan) and need for serotherapy (particularly when using MUDs) (12, 13, 44, 45).
Deficiency of VPS45 Protein
Deficiency of VPS45 protein leads to impaired trafficking of endosomes and lysosomes, with impaired degranulation, release of inflammatory mediators, and neutrophil migration. Patients present very early in life (before 1 year of age) with severe, recurrent, deep-seated bacterial and fungal infections, and a severe neutropenia unresponsive to G-CSF therapy. Bone marrow biopsy classically demonstrates primary myelofibrosis with a dry tap (26–29). HSCT is indicated for severe neutropenia unresponsive to G-CSF and for recurrent, severe infections (27, 30). alloHSCT should be undertaken as early as possible with a myeloablative regimen including busulfan (27). A total of nine transplants have been performed for children with biallelic mutations in the VPS45 gene, with three deaths and six patients surviving. Surviving patients were transfusion-independent with resolution of the extramedullary hematopoiesis if full donor chimerism was achieved (27).
Group 2: Neutropenia Without Syndromic Disease
This group includes congenital neutropenia caused by mutations in ELANE, GFI1, HAX1, WAS (X-linked neutropenia), CSF3R, and SRP54 genes (Table 2) (40, 43, 46). Several reports on successful alloHSCT are available for both reduced intensity conditioning (RIC), and myeloablative conditioning (MAC) transplantation for this group of diseases (Table 2) and patients younger than 10 years of age appear to have favorable outcomes (7).
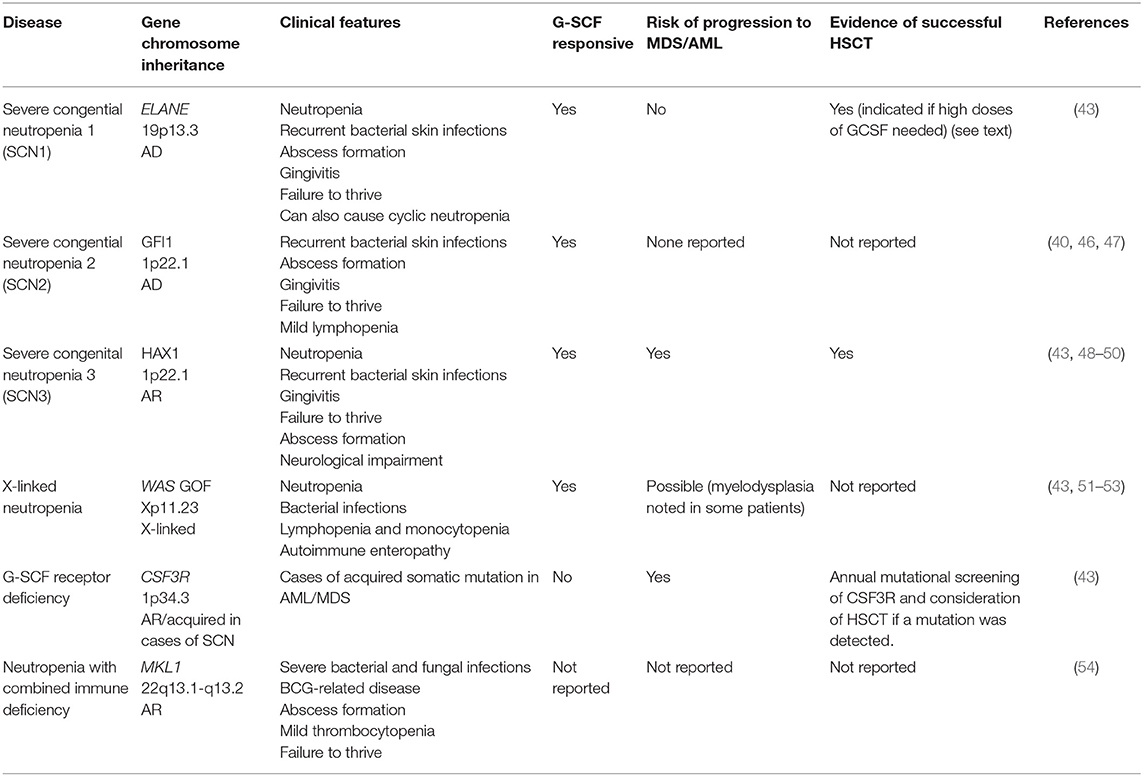
Table 2. Neutropenia without syndromic disease.
Severe infections are seen in ELANE and HAX1 mutations, thus alloHSCT is indicated in these patients, particularly if they require high-dose G-CSF to maintain their neutrophil count, or progress to MDS/AML (it has been found that patients who require more than 8 mcg/kg/day of G-CSF to maintain a neutrophil count above 0.5 × 109/L have an increased risk of sepsis and MDS/AML) (43, 46, 55). With regard to HAX1, patients with mutations in exon 2 encoding isoform A develop isolated congenital neutropenia, whereas other mutations encoding both isoform A and B cause hematological and neurological manifestations (neurological delay, epilepsy). Thus, the degree of neurological impairment should be taken into account when considering alloHSCT (48, 49). Patients with GFI1 and WAS gain of function (GOF) mutations are very rare and seem to present with mild to moderate neutropenia, and there are no reports on alloHSCT in these patients (40, 47). WAS GOF (X-linked neutropenia) is a distinct clinical entity from Wiskott Aldrich Syndrome (caused by WAS loss of function) with male patients exhibiting variable neutropenia, recurrent infections, and lymphopenia, with normal platelet counts and no eczema. Transient myelodysplastic changes have been seen in the bone marrow of some patients, resolving in most cases but progressing to AML at an elderly age in one case (51–53). In contrast, patients with acquired mutations in CSF3R present very often with MDS/AML, therefore yearly screening for receptor mutations is indicated, as is pre-emptive alloHSCT (Table 2) (43).
Group 3: Phagocyte Function With Syndromic Disease
This group of disease is characterized by defective neutrophil function with normal or elevated neutrophil numbers that is part of a broader syndrome (Table 3). For patients with Papillon-Lefevre, localized juvenile periodontitis, and ß-ACTIN-deficiency there is no need and no evidence for alloHSCT. Patients suffering from leucocyte adhesion deficiencies I and III (LAD) can be cured by alloHSCT, however patients with LADII are not candidates for transplantation. For patients with LADI and III, it appears preferable to transplant early, prior to the development of life-threatening infections. Both RIC and MAC regimens have been successful, although it appears myeloablative reduced toxicity regimens (fludaribine-based, combined with treosulfan, or targeted busulfan dosing) are preferable, with RIC regimens more suitable for sicker patients with reduced Lansky score (61–65). However, a recent study of the EBMT/IEWP on 83 transplanted LAD patients shows no significant benefit in patients receiving MAC vs. non-myeloablation. Furthermore, regardless of the conditioning regimen, a relatively high frequency of severe inflammatory complications (graft rejection and severe aGVHD) during alloHSCT was observed. This cohort included a few LAD III patients, and their outcome was not significantly different from that of the LAD I patients. Early transplantation using anti-inflammtory treatment pre-alloHSCT and the additional use of thiotepa in the conditioning protocol might be beneficial [unpublished data of author SB, (66)]. Following on from successful gene therapy trials for other primary immune deficiencies, a gene therapy phase I/II trial for LAD-1 has been announced for the end of 2019 whereby autologous CD34+ stem cells of patients with LAD1 will be transduced using a lentiviral vector; stem cells successfully transduced will be transplanted following conditioning with a low dose of busulfan (Rocket Pharma, USA; https://clinicaltrials.gov/ct2/show/NCT03812263, https://clinicaltrials.gov/ct2/show/NCT03825783) (67).
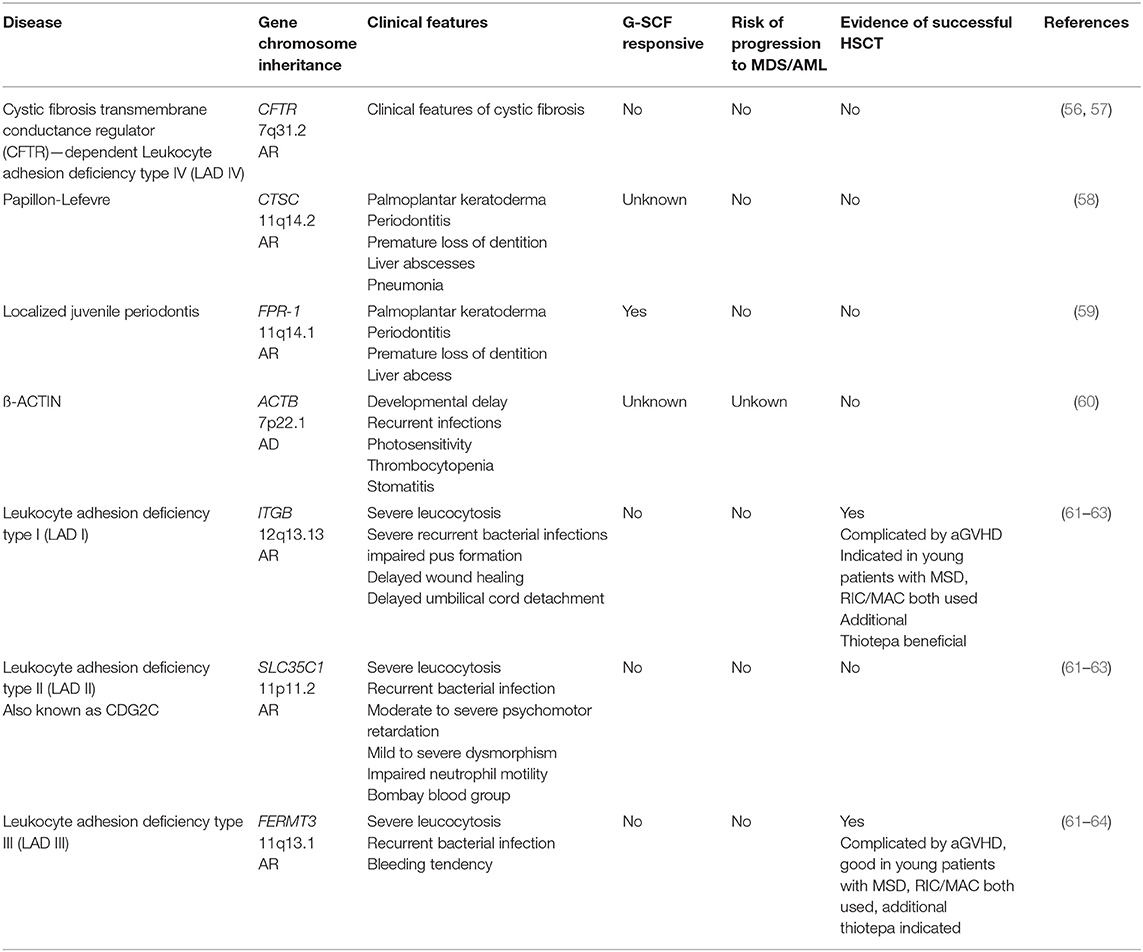
Table 3. Phagocyte dysfunction with syndromic disease.
Group 4: Phagocyte Dysfunction Without Syndromic Disease
Excluding CGD, the only disease in this group with evidence of successful bone marrow transplantation is MonoMAC (monocytopenia and mycobacterial infections syndrome) (Table 4). This disease is characterized by profoundly decreased or absent monocytes, B lymphocytes, natural killer (NK) cells, and circulating and tissue dendritic cells (DCs), with little, or no effect on T-lymphocyte numbers. Patients are susceptible to mycobacterial, viral and opportunistic fungal infections. Bone marrow dysfunction is prominent and variable with progressive aplastic and dysplastic changes. Stem cell transplantation is curative and can be performed pre-emptively in cases where a matched donor is available (68). Anti-inflammatory pre-treatment and proper infection control might result in lower transplant related mortality in these patients.
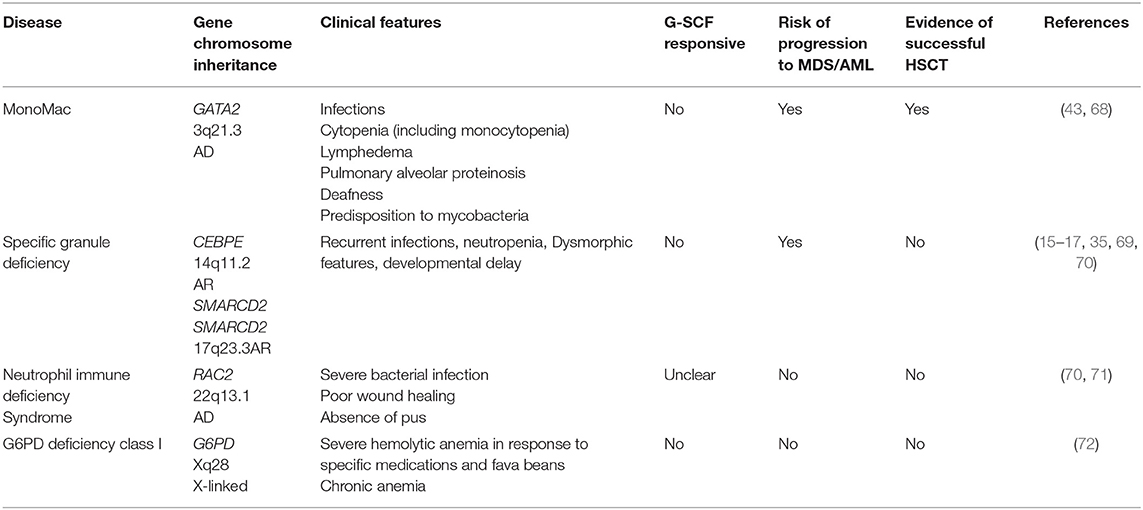
Table 4. Phagocyte dysfunction without syndromic disease (excluding CGD).
Discussion
The list of neutrophil disorders is varied and expanding rapidly with the increasing availability of next generation gene sequencing. As our ability to establish a genetic diagnosis is improved, the challenge of linking genotype to phenotype arises, a challenge that is made difficult by the small numbers of patients diagnosed with each individual disease. There is much we still do not understand about the pathomechanism of these diseases, and this makes prognostication and treatment decisions difficult. Serious infectious complications and neutropenia are life threatening but can be treated with anti-microbial therapy and G-CSF, respectively, however inflammatory complications of these diseases are likely underappreciated and undertreated (5, 6). Furthermore, the decision to undertake alloHSCT, with all its inheritent risks and potential complications, is made difficult by the lack of published data for most diseases.
In 2015, the EBMT and SCETIDE released the findings of the largest retrospective cohort of patients with severe congenital neutropenia to undergo alloHSCT. The 136 patients in that study demonstrated an overall 3 years survival of 82%, with transplant related mortality at 17%. It concluded that transplantation should be considered in patients with severe infections or unresponsiveness to G-CSF or requiring high doses of G-CSF (over 8 mcg/kg/day to maintain an absolute neutrophil count over 0.5 × 109/L). Both MAC and RIC conditioning was effective and transplant outcomes were improved if patients were transplanted before 10 years of age and before the development of MDS/AML (7). Despite the wide range of disease we have covered in this review, those findings appear to hold true, although in diseases where evidence is lacking family choice and center-specific expertise must also be taken into account.
In diseases where the need to transplant has been established (e.g., deficiency of VPS45 protein, LAD, SDS), there is little to guide decision making as to the method of transplantation. The need to achieve stable myeloid engraftment, often into an impaired bone marrow environment, may well call for the use of toxicity reduced MAC over the RIC regimens that have become established practice for PIDs characterized by loss-of-function lymphoid deficiencies (e.g., severe combined immunodeficiency, or SCID) (27). Again, evidence is lacking regarding side effects, long-term outcomes, and the degree of chimerism required to maintain cure. Secondary graft failure or graft insufficiency in the case of mixed chimerism with a reappearance of post-transplant neutropenia has been reported in the literature and observed by the authors in some of the quantitative phagocytic disorders (73–75).
We recommend that all physicians treating patients with neutrophil disorders submit their data to the EBMT and SCETIDE, as it is only through the ordered collection of data that clinical experience can be gathered, studied and shared. It is our hope that in the coming years many disease mechanisms will be uncovered and long-term treatment data accumulated, which will be of great benefit to physicians, patients, and their families.
Author Contributions
SB, BS, and PS designed the study, collected data, performed review, wrote the manuscript.
Funding
BS position was supported by the Australian Government Research Training Program Scholarship and Hadassah Australia. PS was supported by DFG grant W-A 1597/4-2.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor, AG, declared a collaboration with one of the authors, SB.
Acknowledgments
We would like to thank our patients and their families, as well as departmental nursing and administrative staff for their tireless commitment to patient care. We would also like to thank Prof. Zeev Rotstein, Director of The Hadassah Medical Center for his support of the department and the patients.
Abbreviations
AD, Autosomal Dominant; aGVHD, Acute Graft vs. Host Disease; alloHSCT, Allogeneic Hematopoietic Stem Cell Transplant; AML, Acute Myeloid Leukemia; AR, Autosomal Recessive; BMF, Bone Marrow Failure; CGD, Chronic Granulomatous Disease; cGVHD, Chronic Graft vs. Host Disease; CHD, Congenital Heart Disease; EBMT, European Society for Bone Marrow Transplantation; ESID, European Society for Immunodeficiencies; G-CSF, Granulocyte Colony Stimulating Factor; GOF, Gain of Function; GVHD, Graft vs. Host Disease; HSCT, Hematopoietic Stem Cell Transplantation; IEWP, Inborn Errors Working Party; IUIS, International Union of Immunological Studies; LAD, Leukocyte Adhesion Deficiency; MAC, Myeloablative Conditioning; MDS, Myelodysplastic Syndrome; MUD, Matched Unrelated Donor; PID, Primary Immune Deficiency; RIC, Reduced Intensity Conditioning; SAA, Severe Aplastic Anemia; SCETIDE, Stem Cell Transplant for Immunodeficiencies in Europe; SCN, Severe Congenital Neutropenia; SDS, Schwachman Diamond Syndrome; TBI, Total Body Irradiation.
References
1. Sullivan K. Neutropenia as a sign of immunodeficiency. J Allergy Clin Immunol. (2019) 143:96–100. doi: 10.1016/j.jaci.2018.09.018
2. Bousfiha A, Jeddane L, Picard C, Ailal F, Gaspar H, Al-Herz W, et al. The 2017 IUIS phenotypic classification for primary immunodeficiencies. J Clin Immunol. (2018) 38:129–43. doi: 10.1007/s10875-017-0465-8
3. Adinoff A, Johnston R, Dolen J, South M. Chronic granulomatous disease and pneumocystis carinii pneumonia. Pediatrics. (1982) 69:133–4.
4. Arnold D, Heimall J. A review of chronic granulomatous disease. Adv Ther. (2017) 34:2543–57. doi: 10.1007/s12325-017-0636-2
5. Henrickson S, Jongco A, Thomsen K, Garabedian E, Thomsen I. Non-infectious manifestations and complications of chronic granulomatous disease. J Pediatric Infect Dis Soc. (2018) 7(S1):S18–24. doi: 10.1093/jpids/piy014
6. Rosales C. Neutrophil: a cell with many roles in inflammation or several cell types? Front Physiol. (2018) 9:113. doi: 10.3389/fphys.2018.00113
7. Fioredda F, Iacobelli S, van Biezen A, Gaspar B, Ancliff P, Donadieu J, et al. Stem cell transplantation in severe congential neutropenia: an analysis from the European Society for Blood and Bone Marrow Transplantation. Blood. (2015) 126:1885–92. doi: 10.1182/blood-2015-02-628859
8. Newburger P. Disorders of neutrophil number and function. Hematol Am Soc Hematol Educ Program. (2006) 2006:104–10. doi: 10.1182/asheducation-2006.1.104
9. Donadieu J, Fenneteau O, Beaupain B, Beaufils S, Bellanger F, Mahlaoui N, et al. Classification of and risk factors for hematologic complications in a French national cohort of 102 patients with Shwachman-Diamond syndrome. Haematologica. (2012) 97:1312–9. doi: 10.3324/haematol.2011.057489
10. Huang J, Shimamura A. Clinical spectrum and molecular pathophysiology of Shwachman-Diamond syndrome. Curr Opin Hematol. (2011) 18:30–5. doi: 10.1097/MOH.0b013e32834114a5
11. Alter B. Inherited bone marrow failure syndromes: considerations pre-and posttransplant. Blood. (2017) 130:2257–64. doi: 10.1182/blood-2017-05-781799
12. Hsu J, Vogelsang G, Jones R, Brodsky R. Bone marrow transplantation in shwachman-diamond syndrome. Bone Marrow Transplant. (2002) 30:255–8. doi: 10.1038/sj.bmt.1703631
13. Burroughs L, Shimamura A, Talano J, Domm J, Baker K, Delaney C, et al. Allogeneic hematopoietic cell transplantation using treosulfan-based conditioning for treatment of marrow failure disorders. Biol Blood Marrow Transplant. (2017) 23:1669–77. doi: 10.1016/j.bbmt.2017.06.002
14. Bezzerri V, Vella A, Di Gennaro G, Ortolani R, Nicolis E, Cesaro S, et al. Peripheral blood immunophenotyping in a large cohort of patients with Shwachman-Diamond syndrome. Pediatr Blood Cancer. (2019) 66:e27597. doi: 10.1002/pbc.27597
15. Boztug K, Appaswamy G, Ashikov A, Schaffer A, Salzer U, Diestelhorst J, et al. A novel syndrome with congenital neutropenia caused by mutations in G6PC3. N Engl J Med. (2009) 360:32–43. doi: 10.1056/NEJMoa0805051
16. Desplantes C, Fremond M, Beaupain B, Harousseau J, Buzyn A, Pellier I, et al. Clinical spectrum and long-term follow-up of 14 cases with G6PC3 mutations from the French severe congenital neutropenia registry. Orph J Rare Dis. (2014) 9:183. doi: 10.1186/s13023-014-0183-8
17. Notarangelo L, Savoldi G, Cavagnini S, Bennato V, Vasile S, Pilotta A, et al. Severe congenital neutropenia due to G6PC3 deficiency: early and delayed phenotype in two patients with two novel mutations. Italian J Pediatrics. (2014) 40:80. doi: 10.1186/s13052-014-0080-8
18. Mehyar L, Abu-Arja R, Rangarajan H, Pai V, Bartholomew D, Rose M, et al. Matched unrelated donor transplantation in glycogen storage disease type 1b patient corrects severe neutropenia and recurrent infections. Bone Marrow Transplant. (2018) 53:1076–8. doi: 10.1038/s41409-018-0147-z
19. Dale D, Bolyard A, Marrero T, Kelley M, Makaryan V, Tran E, et al. Neutropenia in glycogen storage disease 1b: outcomes for patients treated with granulocyte colony-stimulating factor. Curr Opin Hematol. (2019) 26:16–21. doi: 10.1097/MOH.0000000000000474
20. Rodrigues J, Fernandes H, Caruthers C, Braddock S, Knutsen A. Cohen syndrome: review of the literature. Cureus. (2018) 10:e3330. doi: 10.7759/cureus.3330
21. Steward C, Groves S, Taylor C, Maisenbacher M, Versluys B, Newbury-Ecob R, et al. Neutropenia in Barth syndrome: characteristics, risks, and management. Curr Opin Hematol. (2019) 26:6–15. doi: 10.1097/MOH.0000000000000472
22. Rigaud C, Lebre A, Touraine R, Beaupain B, Ottolenghi C, Chabli A, et al. Natural history of Barth Syndrome: a national cohort study of 22 patients. Orphanet J Rare Dis. (2013) 8:70. doi: 10.1186/1750-1172-8-70
23. Saric A, Andreau K, Armand A, Moller I, Petit P. Barth syndrome: from mitochondrial dysfunctions associated with aberrant production of reactive oxygen species to pluripotent stem cell studies. Front Genet. (2016) 6:359. doi: 10.3389/fgene.2015.00359
24. Patiroglu T, Akar H. Clericuzio-type poikiloderma with neutropenia syndrome. Iran J Allergy Asthma Immunol. (2015) 14:331–7.
25. Wang L, Clericuzio C, Larizza L. Poikiloderma with neutropenia. In: Adam M, Ardinger H, Pagon R, Wallace S, Bean L, Stephens K, et al., editors. GeneReviews. Seattle, WA: University of Washington (2017). p. 1993–2019.
26. Meerschaut I, Bordon V, Dhooge C, Delbeke P, Vanlander A, Simon A, et al. Severe congenital neutropenia with neurological impairment due to a homozygous VPS45 p.E238K mutation: a case report suggesting a genotype–phenotype correlation. Am J Med Genet Part A. (2015) 167A:3214–8. doi: 10.1002/ajmg.a.37367
27. Shadur B, Asherie A, Newburger P, Stepensky P. How we approach: severe congeital neutropenia and myelofibrosis due to mutations in VPS45. Pediatr Blood Cancer. (2019) 66:e27473. doi: 10.1002/pbc.27473
28. Stepensky P, Saada A, Cowan M, Tabib A, Fischer U, Berkun Y, et al. The Thr224Asn mutation in the VPS45 gene is associated with the congenital neutropenia and primary myelofibrosis of infancy. Blood. (2013) 121:5078–87. doi: 10.1182/blood-2012-12-475566
29. Vilboux T, Lev A, Malicdan M, Simon A, Jarvinen P, Racek T, et al. A congenital neutrophil defect syndrome associated with mutations in VPS45. NEJM. (2013) 369:54–65. doi: 10.1056/NEJMoa1301296
30. Stepensky P, Simanovsky N, Averbuch D, Gross M, Yanir A, Mevorach D, et al. VPS45-associated primary infantile myelofibrosis - successful treatment with hematopoietic stem cell transplantation. Pediatr Transplant. (2013) 17:820–5. doi: 10.1111/petr.12169
31. Bohn G, Allroth A, Brandes G, Thiel J, Glocker E, Schaffer A, et al. A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14. Nat Med. (2007) 13:38–45. doi: 10.1038/nm1528
32. Boztug K, Jarvinen P, Salzer E, Racek T, Monch S, Garncarz W, et al. JAGN1 deficiency causes aberrant myeloid cell homeostasis and congenital neutropenia. Nat Gent. (2014) 46:1021–7. doi: 10.1038/ng.3069
33. Baris S, Karakoc-Aydiner E, Ozen A, Delil K, Kiykim A, Ogulur I, et al. JAGN1 deficient severe congenital neutropenia: two cases from the same family. J Clin Immunol. (2015) 35:339–43. doi: 10.1007/s10875-015-0156-2
34. Wortmann S, Zietkiewicz S, Kousi M, Szklarczyk R, Haack T, Gersting S, et al. CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder. Am J Hum Genet. (2015) 96:245–57. doi: 10.1016/j.ajhg.2014.12.013
35. Witzel M, Petersheim D, Fan Y, Bahrami E, Racek T, Rohlfs M, et al. Chromatin-remodeling factor SMARCD2 regulates transcriptional networks controlling differentiation of neutrophil granulocytes. Nat Genet. (2017) 49:742–52. doi: 10.1038/ng.3833
36. Kuhns D, Fink D, Choi U, Lau K, Priel D, Riva D, et al. Cytoskeletal abnormalities and neutrophil dysfunction in WDR1 deficiency. Blood. (2016) 128:2135–43. doi: 10.1182/blood-2016-03-706028
37. Kile B, Panopoulos A, Stirzaker R, Hacking D, Tahtamouni L, Willson T, et al. Mutations in the cofilin partner Aip1/Wdr1 cause autoinflammatory disease and macrothrombocytopenia. Blood. (2007) 110:2371–80. doi: 10.1182/blood-2006-10-055087
38. Pfajfer L, Mair N, Jimenez-Heredia R, Genel F, Gulez N, Ardeniz O, et al. Mutations affecting the actin regulator WD repeat-containing protein 1 lead to aberrant lymphoid immunity. J Allergy Clin Immunol. (2018) 142:1589–604. doi: 10.1016/j.jaci.2018.04.023
39. Chinen J, Cowan M. Advances and highlights in primary immunodeficiencies in 2017. J Allergy Clin Immunol. (2018) 142:1041–51. doi: 10.1016/j.jaci.2018.08.016
40. Donadieu J, Fenneteau O, Beaupain B, Mahlaoui N, Chantelot C. Congenital neutropenia: diagnosis, molecular bases and patient management. Orph J Rare Dis. (2011) 6:26. doi: 10.1186/1750-1172-6-26
41. Rosenberg P, Alter B, Bolyard A, Bonilla M, Boxer L, Cham B, et al. The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood. (2006) 107:4628–35. doi: 10.1182/blood-2005-11-4370
42. Pierre G, Chakupurakal G, Mckiernan P, Hendriksz C, Lawson S, Chakrapani A. Bone marrow transplantation in glycogen storage disease type 1b. J Pediatr. (2008) 152:286–8. doi: 10.1016/j.jpeds.2007.09.031
43. Donadieu J, Beaupain B, Fenneteau O, Bellanne-Chantelot C. Congenital neutropenia in the era of genomics: classification, diagnosis, and natural history. Br J Haematol. (2017) 179:557–74. doi: 10.1111/bjh.14887
44. Cesaro S, Oneto R, Messina C, Gibson B, Buzyn A, Steward C, et al. Haematopoietic stem cell transplantation for Shwachman-Diamond disease: a study from the European Group for blood and marrow transplantation. Br J Haematol. (2005) 131:231–6. doi: 10.1111/j.1365-2141.2005.05758.x
45. Donadieu J, Michel G, Merlin E, Bordigoni P, Monteux B, Beaupain B, et al. Hematooietic stem cell transplantation for shwachman-diamond syndrome: experience of the french neutropenia registry. Bone Marrow Transplant. (2005) 36:787–92. doi: 10.1038/sj.bmt.1705141
46. Skokowa J, Dale D, Touw I, Zeidler C, Welte K. Severe congenital neutropenias. Nat Rev Dis Primers. (2017) 3:17032. doi: 10.1038/nrdp.2017.32
47. Person R, Li F, Duan Z, Benson K, Wachsler J, Papadaki H, et al. Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat Genet. (2003) 34:308–12. doi: 10.1038/ng1170
48. Germeshausen M, Grudzien M, Zeidler C, Abdollahpour H, Yetgin S, Rezaei N, et al. Novel HAX1 mutations in patients with severe congenital neutropenia reveal isoform-dependent genotype-phenotype associations. Blood. (2008) 111:4954–7. doi: 10.1182/blood-2007-11-120667
49. Boztug K, Ding X-Q, Hartmann H, Ziesenitz L, Schaffer A, Diestelhorst J, et al. HAX1 mutations causing SCN and neurological disease lead to microstructural abnormalities revealed by quantitative MRI. Am J Med Genet. (2010) 152A:3157–63. doi: 10.1002/ajmg.a.33748
50. Klein C. Kostmann's disease and HCLS1-associated protein X-1 (HAX1). J Clin Immunol. (2017) 37:117–22. doi: 10.1007/s10875-016-0358-2
51. Devriendt K, Kim A, Mathijs G, Frints S, Schwartz M, Van den Oord J, et al. Constitutively activating mutation in WASP causes X-linked severe congential neutropenia. Nat Genet. (2001) 27:313–7. doi: 10.1038/85886
52. Ancliff P, Blundell M, Cory G, Calle Y, Worth A, Kempski H, et al. Two novel activating mutations in the Wiskott-Aldrich syndrome protein result in congenital neutropenia. Blood. (2006) 108:2182–9. doi: 10.1182/blood-2006-01-010249
53. Beel K, Cotter M, Blatny J, Bond J, Lucas G, Green F, et al. A large kindred with X-linked neutropenia with an I294T mutation of the Wiskott-Aldrich syndrome gene. Br J Haematol. (2008) 144:120–6. doi: 10.1111/j.1365-2141.2008.07416.x
54. Record J, Malinova D, Zenner H, Plagnol V, Nowak K, Syed F, et al. Immunodeficiency and severe susceptibility to bacterial infection associated with a loss-of-function homozygous mutation of MKL1. Blood. (2015) 126:1527–35. doi: 10.1182/blood-2014-12-611012
55. Rosenberg P, Zeidler C, Bolyard A, Alter B, Bonilla M, Boxer L, et al. Stable long-term risk of leukaemia in patients with severe congenital neutropenia maintained on G-CSF therapy. Br J Haematol. (2010) 150:196–9. doi: 10.1111/j.1365-2141.2010.08216.x
56. Sorio C, Montresor A, Bolomini-Vittori M, Caldrer S, Rossi B, Dusi S, et al. Mutations of cystic fibrosis transmembrane conductance regulator gene cause a monocyte-selective adhesion deficiency. Am J Respir Crit Care Med. (2016) 193:1123–33. doi: 10.1164/rccm.201510-1922OC
57. Fan Z, Ley K. Leukocyte adhesion deficiency IV. Am J Respir Crit Care Med. (2016) 193:1075–7. doi: 10.1164/rccm.201512-2454ED
58. Sreeramulu B, Shyam N, Ajay P, Suman P. Papillon-Lefevre syndrome: clinical presentation and management options. Clin Cosmet Invest Dent. (2015) 7:75–81. doi: 10.2147/CCIDE.S76080
59. Maney P, Walters J. Formylpeptide receptor single nucleotide polymorphism 348T>C and its relationship to polymorphonuclear leukocyte chemotaxis in aggressive periodontitis. J Periodontol. (2009) 80:1498–505. doi: 10.1902/jop.2009.090103
60. Nunoi H, Yamazaki T, Tsuchiya H, Kato S, Malech H, Matsuda I, et al. A heterozygous mutation of B-actin associated with neutrophil dysfunction and recurrent infection. Proc Natl Acad Sci USA. (1999) 96:8693–8. doi: 10.1073/pnas.96.15.8693
61. Almarza N, Kasbekar S, Thrasher A, Kohn D, Sevilla J, Nguyen T, et al. Leukocyte adhesion deficiency-1: a comprehensive review of all published cases. J Allergy Clin Immunol Pract. (2018) 6:1418–20. doi: 10.1016/j.jaip.2017.12.008
62. Hirikoshi Y, Umeda K, Imai K, Yabe H, Sasahara Y, Watanabe K, et al. Allogeneic hematopoietic stem cell transplantation for leukocyte adhesion deficiency. J Pediatr Hematol Oncol. (2018) 40:137–40. doi: 10.1097/MPH.0000000000001028
63. Qasim W, Cavazzana-Calvo M, Davies E, Davis J, Duval M, Earmes G, et al. Allogeneic hematopoietic stem-cell transplantation for leukocyte adhesion deficiency. Pediatrics. (2009) 123:836–40. doi: 10.1542/peds.2008-1191
64. Stepensky P, Wolach B, Gavrieli R, Rousso S, Ben Ami T, Goldman V, et al. Leukocyte adhesion deficiency type III: clinical features and treatment with stem cell transplantation. J Pediatr Hematol Oncol. (2015) 37:264–8. doi: 10.1097/MPH.0000000000000228
65. van de Vijver E, van den Berg T, Kuijpers T. Leukocyte adhesion deficiencies. Hematol Oncol Clin North Am. (2013) 27:101–16. doi: 10.1016/j.hoc.2012.10.001
66. A Clinical Trial to Evaluate the Safety and Efficacy of RP-L201 in Subjects With Leukocyte Adhesion Deficiency-I. Clinical Trial Rocket Pharmaceuticals;ClinicalTrials.gov Identifier: NCT03812263.
67. Qasim W, Gennery A. Gene therapy for primary immunodeficiencies: current status and future prospects. Drugs. (2014) 74:963–9. doi: 10.1007/s40265-014-0223-7
68. Parta M, Shah N, Baird K, Rafei H, Calvo K, Hughes T, et al. Allogeneic hematopoietic stem cell transplantation for GATA2 deficiency using a busulfan-based regimen. Biol Blood Marrow Transplant. (2018) 24:1250–9. doi: 10.1016/j.bbmt.2018.01.030
69. Gombart A, Shiohara M, Kwok S, Agematsu K, Komiyama A, Koeffler H. Neutrophil-specific granule deficiency: homozygous recessive inheritance of a frameshift mutation in the gene encoding transcription factor CCAAT/enhancer binding protein-epsilon. Blood. (2001) 97:2561–7. doi: 10.1182/blood.V97.9.2561
70. Ambruso D, Knall C, Abell A, Panepinto J, Kurkchubasche A, Thurman G, et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc Natl Acad Sci USA. (2000) 97:4654–9. doi: 10.1073/pnas.080074897
71. Williams D, Tao W, Yang F, Kim C, Gu Y, Mansfield P, et al. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood. (2000) 96:1646–54.
72. Luzzatto L, Seneca E. G6PD deficiency: a classic example of pharmacogenetics with on-going clinical implications. Br J Haematol. (2014) 164:469–80. doi: 10.1111/bjh.12665
73. Hashem H, Abu-Arja R, Auletta J, Rangarajan H, Varga E, Rose M, et al. Successful second hematopoietic cell transplantation in severe congenital neutropenia. Pediatr Transplant. (2018) 22:e13289. doi: 10.1111/petr.13078
74. Markel M, Haut P, Renbarger J, Robertson K, Goebel W. Unrelated cord blood transplantation for severe congenital neutropenia: report of two cases with very different transplant courses. Pediatr Transplant. (2008) 12:896–901. doi: 10.1111/j.1399-3046.2008.00951.x
Keywords: neutrophils, neutropenia, leukemia, granulocyte colony-stimulating factor, hematopoietic stem cell transplantation
Citation: Bakhtiar S, Shadur B and Stepensky P (2019) The Evidence for Allogeneic Hematopoietic Stem Cell Transplantation for Congenital Neutrophil Disorders: A Comprehensive Review by the Inborn Errors Working Party Group of the EBMT. Front. Pediatr. 7:436. doi: 10.3389/fped.2019.00436
Received: 10 July 2019; Accepted: 07 October 2019;
Published: 24 October 2019.
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Suhag Parikh, Duke University, United StatesSimone Cesaro, Integrated University Hospital Verona, Italy
Pere Soler-Palacín, Vall d'Hebron University Hospital, Spain
Copyright © 2019 Bakhtiar, Shadur and Stepensky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Polina Stepensky, cG9saW5hQGhhZGFzc2FoLm9yZy5pbA==
†These authors have contributed equally to this work