 Elizabeth Whittaker
Elizabeth Whittaker Elisa López-Varela
Elisa López-Varela Claire Broderick
Claire Broderick James A. Seddon
James A. Seddon- 1Department of Paediatrics, Imperial College London, London, United Kingdom
- 2Department of Paediatric Infectious Diseases, Imperial College Healthcare NHS Trust, St. Mary's Campus, London, United Kingdom
- 3Desmond Tutu TB Centre, Department of Paediatrics and Child Health, Faculty of Medicine and Health Sciences, Stellenbosch University, Cape Town, South Africa
Millions of children are exposed to tuberculosis (TB) each year, many of which become infected with Mycobacterium tuberculosis. Most children can immunologically contain or eradicate the organism without pathology developing. However, in a minority, the organism overcomes the immunological constraints, proliferates and causes TB disease. Each year a million children develop TB disease, with a quarter dying. While it is known that young children and those with immunodeficiencies are at increased risk of progression from TB infection to TB disease, our understanding of risk factors for this transition is limited. The most immunologically disruptive process that can happen during childhood is infection with another pathogen and yet the impact of co-infections on TB risk is poorly investigated. Many diseases have overlapping geographical distributions to TB and affect similar patient populations. It is therefore likely that infection with viruses, bacteria, fungi and protozoa may impact on the risk of developing TB disease following exposure and infection, although disentangling correlation and causation is challenging. As vaccinations also disrupt immunological pathways, these may also impact on TB risk. In this article we describe the pediatric immune response to M. tuberculosis and then review the existing evidence of the impact of co-infection with other pathogens, as well as vaccination, on the host response to M. tuberculosis. We focus on the impact of other organisms on the risk of TB disease in children, in particularly evaluating if co-infections drive host immune responses in an age-dependent way. We finally propose priorities for future research in this field. An improved understanding of the impact of co-infections on TB could assist in TB control strategies, vaccine development (for TB vaccines or vaccines for other organisms), TB treatment approaches and TB diagnostics.
Introduction
Each year millions of children are exposed to infectious cases of tuberculosis (TB) and estimates suggest that around 70 million children currently have TB infection globally (1). TB infection is a clinical state in which the child exhibits no symptoms or signs of disease, but, if tested, would have evidence of immunological sensitization to Mycobacterium tuberculosis (M. tuberculosis), as detected through a tuberculin skin test (TST) or interferon (IFN) gamma release assay (IGRA). Each year about a million children develop TB disease (2) a clinical state characterized by symptoms, signs, radiological changes, and in some children, microbiological isolation of M. tuberculosis. Of these one million children, modeling studies suggest that 250,000 children die each year (3).
The majority of children with TB infection do not progress to TB disease. Most children are either able to eradicate the mycobacteria or contain them immunologically so that they do not cause pathology. Understanding which children are at high risk of disease progression, following infection, would be useful to better understand host-mycobacterial interactions which in turn could help with vaccine design, host directed therapies, as well as diagnostics that might assist in predicting which TB-infected children are at high risk of future disease. It would also be important to understand if there are factors that drive disease progression, as it may be possible to modify or eliminate these drivers if they are found to impact. Currently our understanding of why some children progress to disease while others do not is limited. Age, however, is crucially important.
The risk of having a positive test of infection with M. tuberculosis increases in a fairly linear way with age, reflecting cumulative exposure (4). However, the risk of progressing from infection to disease is heavily age-dependent, with very young children at high risk of disease progression, the risk falling to a nadir in the pre-pubertal years, followed by a rise in risk as children enter adolescence (5). In addition the type of TB disease that children develop is age-dependent. The youngest children typically develop either intrathoracic lymph node disease (in which the mycobacteria are generally confined) or widely disseminated disease, including miliary TB and TB meningitis (reflecting poorly contained mycobacteria and unchecked proliferation). As children enter adolescence, the typical adult-type disease begins to manifest, with apical cavities and parenchymal breakdown, reflecting pathology largely caused by the host immune response. Although some societal and behavioral elements may influence these age-related changes in risk of disease progression and resulting disease phenotype, it is likely that to a large extent these result from age-related changes in the host immune system. These changes may be driven by a variety of environmental factors, including co-infection (Figure 1).
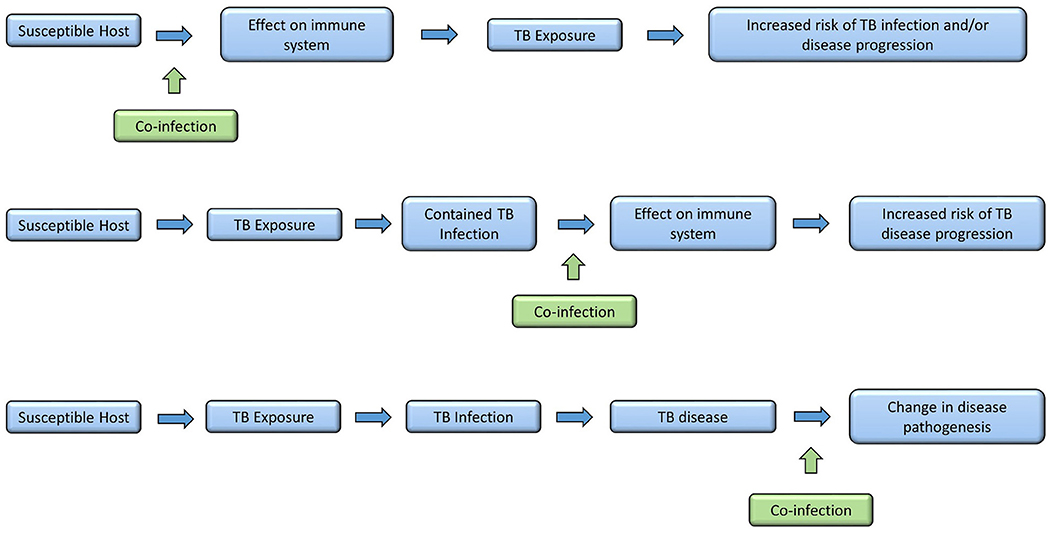
Figure 1. Times in disease spectrum and progression when co-infections may have an influence.
Through childhood, the events that have the most profound impact on a child's immune system are infections with pathogens, and to a lesser extent, vaccinations. Therefore, infection with a variety of pathogens could have a profound influence on the interaction between host immune system and M. tuberculosis. Concurrent infection prior to M. tuberculosis exposure could modify the host immune system so that following M. tuberculosis exposure there is an increased risk of subsequent TB infection or disease. Alternatively, a child may have well-contained TB infection and subsequent co-infection may then disrupt the carefully controlled immunological equilibrium, allowing M. tuberculosis to proliferate with TB disease progression resulting. Finally, a child may develop TB disease and then become co-infected with another organism. This may have an impact on the outcome of disease, in respect to resolution or severity (Figure 2). However, the relationship between infections with other organisms and M. tuberculosis have not been well-described. In this article we aim to bring together all the available evidence into one review. Where evidence is available, we have focused on evidence from co-infection with TB in children. If there are no data specifically in children, we present data on co-infection in adults, recognizing this is imperfect, but preferable to excluding those co-infections. Where appropriate, we have also considered the impact of other infections on a child's immune system and then discussed how this might impact on TB risk. We further acknowledge that for many organisms there is significant epidemiological overlap with TB in children, with challenges therefore in disentangling correlation and causation. We outline the geographical distribution of TB, HIV, helminth infections and malaria in Figure 3 as an example of this. For simplicity, where available, we provide specific evidence for pathogens that have been studied in greater detail, and for others, we group them together according to system, for example, respiratory viruses. Finally we suggest some key research priorities and possible study designs that might address them.
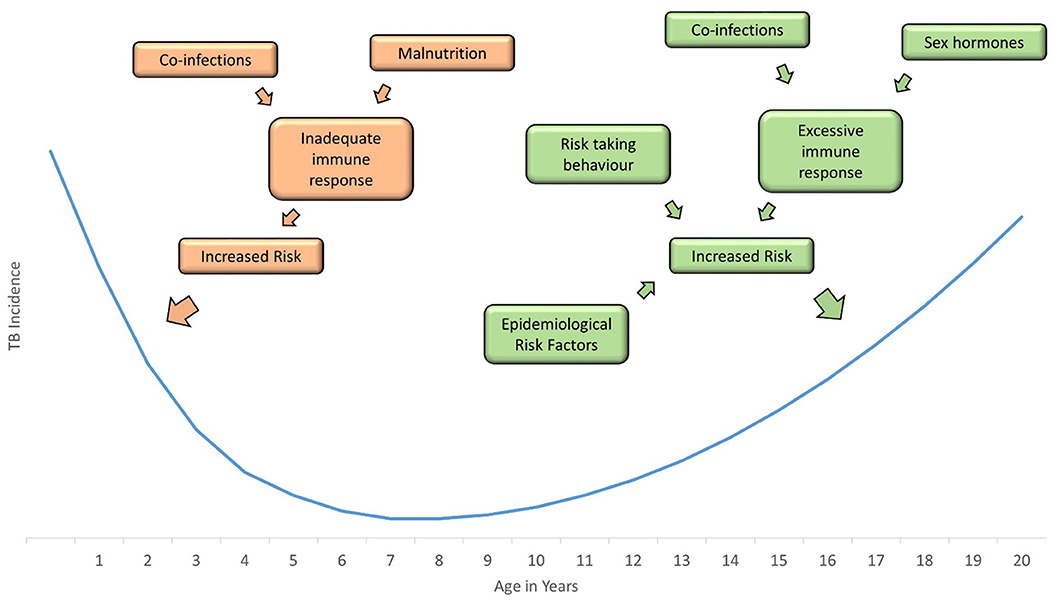
Figure 2. Incidence of childhood tuberculosis is greatest in infants and adolescents; a number of factors increase the risk in these age groups, including co-infections, hormones, behavior, and epidemiological risk factors.
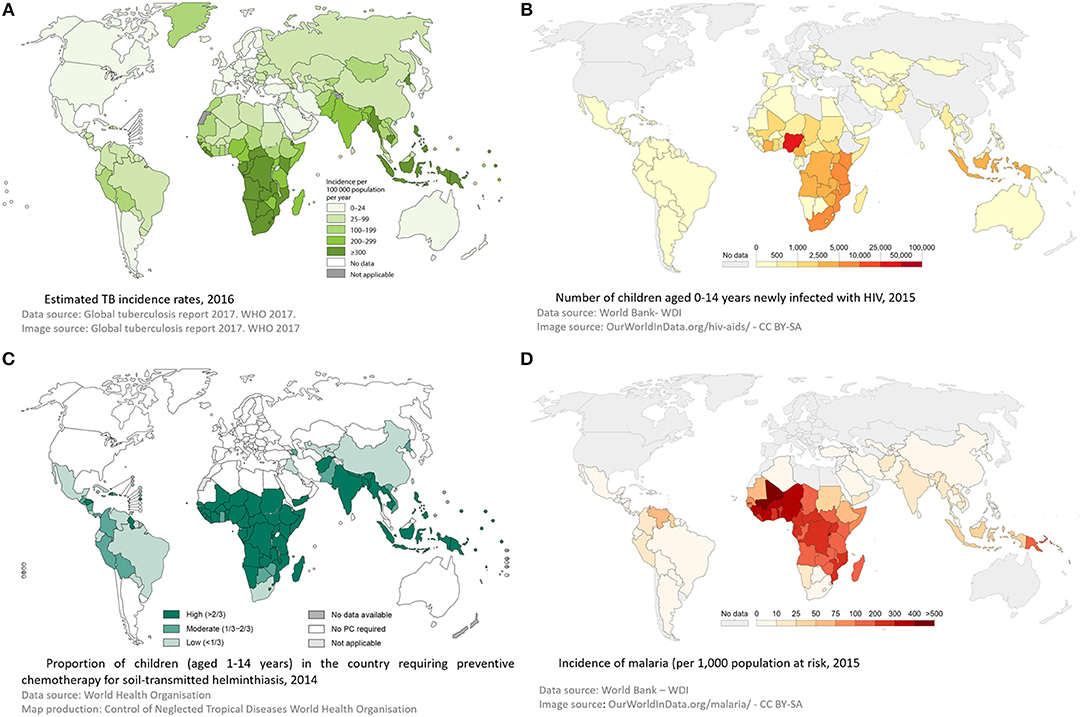
Figure 3. Global incidence of tuberculosis (A), HIV (B), Helminths (C), and malaria (D)—demonstrating geographical overlap between these 4 infections, supporting a role for interplay between them. Panel (A) was reproduced from © World Health Organization (2). Panel (B) was reproduced from © World Bank -WDI (6). Panel (C) was reproduced from © World Health Organization 2015 (7). Panel (D) was reproduced from © World Bank -WDI (8). Data from WHO Malaria Report (9).
Overview of TB Immunology in Children
The host immune response to M. tuberculosis involves both the innate and adaptive immune system, starting with antimicrobial peptides and neutrophils, followed by the interaction between the antigen presenting cells and the bacteria and granuloma formation, followed by a more targeted approach by CD4+ and CD8+ T cells (10–12).
Should the bacilli be successful in traversing the physical and anatomical barriers encountered, M. tuberculosis bacilli are inhaled into the terminal alveoli where they are readily phagocytosed by resident alveolar macrophages and dendritic cells. This process results in activation of antimicrobial mechanisms, which serve to limit the growth of M. tuberculosis and recruit additional immune cells. Bacilli are processed and presented on the cell surface by antigen presenting cells that migrate to regional lymph nodes and present M. tuberculosis antigens to T cells. Secretion of a variety of cytokines, including IL-12 and IL-23, cause CD4+ T cells to proliferate and secrete cytokines, including IL-2, IFN-γ and tumor necrosis factor alpha (TNF-α), which further activate macrophages to become microbicidal if the encounter is successful. TNF-α increases the ability of macrophages to phagocytose and kill mycobacteria, stimulating apoptosis; this leads to increased presentation of mycobacterial antigens by dendritic cells (13). TNF-α also co-ordinates the inflammatory response through induction of IL-1, IL-6, and recruitment of macrophages, Natural Killer (NK), γδ and CD8+ T cells promoting their activation (14). Absence of TNF-α is associated with progression to severe TB disease, as seen following treatment with anti-TNF monoclonal antibodies for autoimmune conditions (15, 16). Yet, excessive TNF-α promotes immunopathology by interfering with cell death processes and induction of a hyper-inflammatory milieu. As with so many factors in the immune response to TB, balance is critical. A neutrophil-driven, IFN-inducible transcript signature in adult whole blood was recently identified that correlated with clinical severity, (17) and neutrophilia has been associated with poorer prognosis. Additionally, T cell activation, as measured by HLA DR+ expression and production of cytokines such as IFN-γ, IL-1β, and TNF-α has been shown to be associated with TB disease (Figure 4) (14, 16, 18, 19).
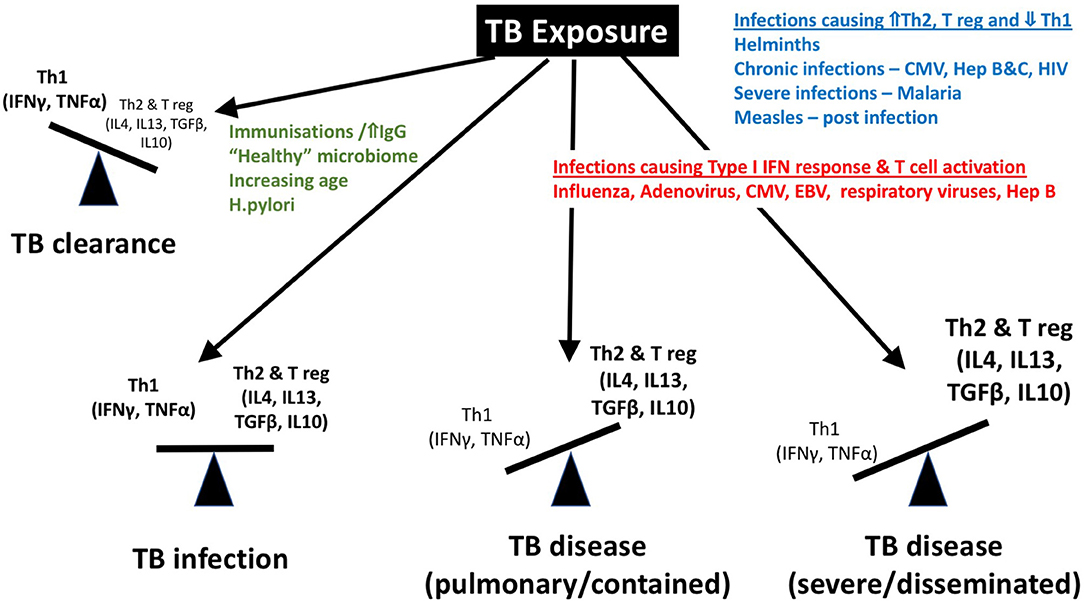
Figure 4. Although a “protective immune response” to tuberculosis remains elusive, a Th1 predominant response is associated with protection, while a Th2 and regulatory T cell predominance has been described in association with severe disease and dissemination. We propose that the balance of these immune responses is influenced by co-infections including helminths, CMV, hepatitis viral infections, malaria, measles, etc. IFN, interferon; IgG, Immunoglobulin G; TNF, tumor necrosis factor; T reg, regulatory T cell; IL, interleukin; TGF, tumor growth factor; HIV, human immunodeficiency virus; CMV, cytomegalovirus.
A variety of age-related immune differences have previously been described in the host response to mycobacteria (11, 12). Most notably, young children have fewer antigen presenting cells, with reduced functional responses, including phagocytosis and recruitment capacity, compared to older children and adults. These functional impairments lead to poor T-cell priming and consequently impaired immunity to M. tuberculosis. The role of non-conventional T cells, such as γδ T cells, NK T cells, Th17 and regulatory T cells, which link or modify the innate-adaptive T cell interaction have recently been explored (20, 21) they are noted to be increased in younger children with TB disease compared to similarly aged healthy controls (20).
Other components of the innate immune response which are likely to be affected by co-infection, and are recognized to be different in neonates and early infancy, include levels of innate defense molecules such as collectins; maturation of Toll-like-receptors (TLRs) and neutrophils.
The immune mechanisms involved in these processes are poorly defined and the dynamic balance that exists between bacterial persistence and host defense, which in children tips in favor of the mycobacteria more than in adults, can be influenced by several factors, including, we hypothesize, age and co-infection with various pathogens (Figure 4).
In longitudinal studies, many acquired immuno-suppressive conditions are known to disrupt this balance and increase the risk of TB disease, including HIV infection, malnutrition, vitamin D deficiency, diabetes, and anti-TNF-α therapy. Here we aim to explore the evidence that co-infections play a role in disrupting this balance (Table 1).
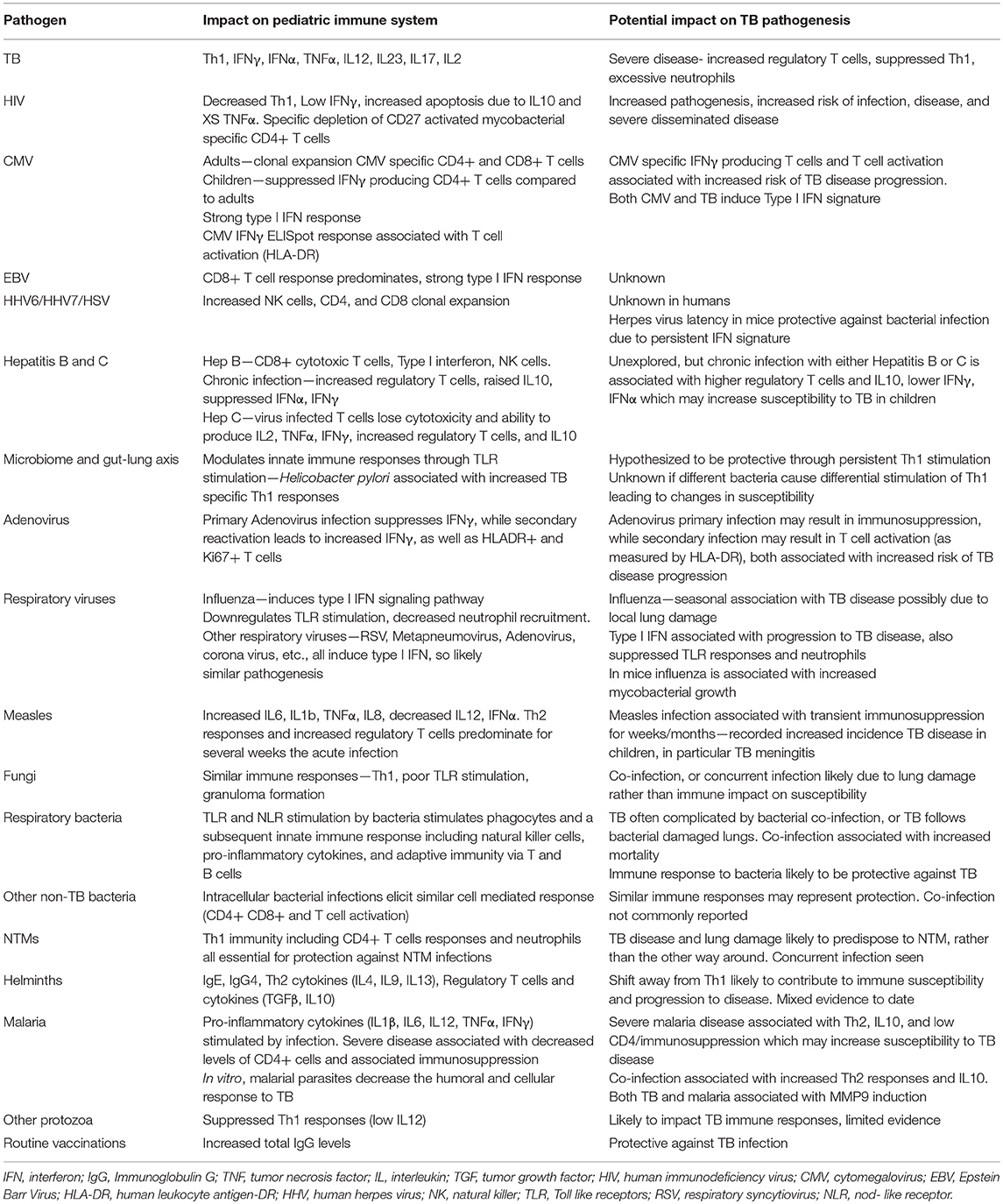
Table 1. A summary of (1) the predominant immune response associated with a variety of pathogens in children and (2) the known impact of that pathogen on tuberculosis pathogenesis.
TB-HIV Co-infection
The “deadly duet” of TB-HIV co-infection has been extensively studied and World Health Organization (WHO) guidelines recommend all children diagnosed with TB should be screened for HIV and conversely, children newly diagnosed with HIV should be screened for TB (22). Dodd et al. in a systematic review of the impact of HIV on TB in children, reported that HIV infection increases the incidence of TB in children by a factor of around eight, increasing with degree of immunosuppression (22). Combination antiretroviral therapy (cART) can restore immune function and has enormously reduced morbidity and mortality among HIV-infected children and is strongly protective against TB, reducing the risk by 70%. However, it takes 2 years for the full potential of protection against TB to be realized. The impact of age on efficacy of antiretroviral therapy is complex, as early treatment initiation, at a better baseline immune status, leads to better immune reconstitution (23). Initiation of cART in children and adults can be complicated by immune reconstitution inflammatory syndrome (IRIS), thought to be a dysregulated immune response to a pathogen, most commonly TB. There is a paucity of data on the epidemiology, risk factors, management, and outcome of TB-IRIS in children (24).
Studies in adults have demonstrated that HIV infection increases susceptibility to TB primarily through decreased numbers of CD4+ T cells and impaired function of CD4+ T cells, in particular in their response to phagocytes (25). As the increased risk of TB is present in HIV-infected individuals prior to significant T cell depletion, this suggests that HIV may alter cellular responses to M. tuberculosis infection. Studies in TB-HIV co-infected adults to characterize functional defects in CD4+ T-cells has increased our understanding of the role of these cells in the immune response to M. tuberculosis. HIV preferentially infects and depletes mycobacterial specific T-cells, most likely due to their activated, CD27 expressing, IL-2 producing state (26). HIV infects other cells including macrophages, dendritic cells and neutrophils, influencing cytokine production, and T cell interactions, which may impact on susceptibility to TB infection, progression to and severity of disease. HIV-infected macrophages act as a reservoir for the virus, leading to TNF-α-induced suppression of apoptosis, thus avoiding immune-mediated clearance by the host (27). HIV1 nef and M. tuberculosis antigen Rv3416 synergistically contributes to anti-apoptotic signaling in macrophages (28, 29). Furthermore, IL-10, produced by macrophages and regulatory T cells, also decreases apoptosis. Plasma IL-10 levels are higher in TB-HIV co-infected adults with pulmonary disease compared to those with HIV infection alone, or those with HIV and TB infection (30). These findings have not been explored in children, although HIV-uninfected children with TB disease have been found to have higher levels of regulatory T cells and IL-10 than healthy controls or children with TB infection (20). Myeloid derived suppressor cells (MDSC), an innate immune cell population known to downregulate T cell proliferation, are increased in adults and children with TB infection and disease. Recently, high levels have also been identified in HIV exposed uninfected infants (HEU), who are known to have increased susceptibility to TB, suggesting a potential mechanism for TB susceptibility. Interestingly HIV infected children on HAART did not have increased levels of MDSC, suggesting that HIV viraemia triggers these regulatory innate immune cells (31). Understanding how HIV increases TB risk in children presents a key research priority.
Herpes Viruses
The herpes viruses that cause pathology in humans include herpes simplex (HSV) 1 and 2, Epstein-Barr virus (EBV), cytomegalovirus (CMV), varicella zoster virus (VZV), and the human herpes viruses (HHV)6-8. Viruses from this family have sophisticated mechanisms for evading the host immune system and consequently establish long-term infections, fluctuating between periods of active disease and periods of inactivity or latency. This is particularly pertinent in children infected in infancy.
CMV infection of the mother during pregnancy can be associated with congenital infection of the fetus, leading to fetal loss and, in surviving infants, neurodevelopmental delay and hearing loss (32). CMV infection has also been well-studied in individuals with various forms of immunosuppression, such as HIV infection, primary immunodeficiencies or post bone marrow transplant (33–35). In these populations it can cause a variety of pathologies, including pneumonitis, retinitis, gastroenteritis and central nervous system dysfunction, among others. CMV infection has also been well-studied in older individuals, causing increased cardiovascular risk and immunosenescence (36, 37). The impact of CMV infection in immunocompetent children, however, is poorly understood.
This is a significant gap in our understanding, as CMV is one of the most prevalent and immunogenic viruses that infect children. In low resource settings, the vast majority acquire CMV in early childhood (38, 39). CMV is acquired from secretions and, in immunocompetent children, usually causes either an asymptomatic viraemia, or viraemia associated with infectious mononucleosis-like symptoms (40). IgG seropositivity usually develops as a consequence of infection. Reactivation, with viraemia and/or symptoms can occur at any point later in life. CMV infection can lead to clonal expansion of differentiated CMV-specific CD4+ and CD8+ T cells, an effect that can last for years (41). Children with asymptomatic CMV infection have been shown to have markedly fewer CMV-specific CD4+ T cells that produced IFN-γ, compared to adults with asymptomatic CMV infection, an effect that lasted for over a year (42). Regarding the relationship with TB, CMV elicits a strong type I IFN response, (43) a response demonstrated to be associated with TB disease (44). In a trial of the MVA85A TB vaccine in infants, Fletcher and colleagues found a significant correlation between CMV IFN-γ ELISpot response at baseline and T-cell activation, in turn associated with future TB disease progression (18). Further analysis of this cohort has demonstrated that a CMV-specific IFN-γ response was associated with increased risk of developing TB disease (45). Groups have also postulated that there is significant overlap in epidemiology between CMV and TB, with new infections of CMV common in early childhood and again in adolescence—potentially impacting on the changes in incidence seen with the TB epidemic (46).
EBV is also a ubiquitous virus, with estimates suggesting that nearly 90% of the global population is infected (47). In resource limited settings, the majority of primary infection is in early childhood, likely as a result of exposure to saliva and resulting in asymptomatic infection (48). Infection in adolescence results in extensive expansion of activated EBV-specific CD8+ T-cells, (49) with an expression profile that suggests uncontrolled inflammation and a strong type I IFN response. Primary infection in young children, however, seems to elicit a virus-specific CD8+ T-cell response that is able to contain the virus without over-expansion (50). Given the overlapping age profiles and the immunological effects of EBV infection, a relationship with TB is very possible.
The other herpes viruses are also very common. Most individuals have serological evidence of exposure to both herpes simplex viruses by adulthood, with more rapid acquisition in resource limited settings (51). Prior to vaccination strategies for varicella, most children had developed chicken pox during childhood (52). Most children are infected with HHV6 by the time they are 2 years of age, many developing a symptomatic illness at the time of primary infection (53). Positive HHV7 serology is almost universal by adulthood (54) and HHV8 is common in low resource settings, mainly acquired during childhood (55). No studies have directly evaluated the relationship between these other herpes viruses and TB. However, the herpes viruses have a marked effect on the host immune system and, dependent on the stage of herpes infection (acute infection, latency, or reactivation) and the timing in relation to TB pathogenesis, could impact on TB risk. Herpes viruses can “arm” NK cells (56) and even during periods of latency, herpes viruses maintain large populations of functional CD4+ and CD8+ T cells. In mouse models herpes virus latency leads to persistent production of IFN-γ and systemic activation of macrophages protects mice from infection with the bacterial pathogens Listeria monocytogenes and Yersinia pestis (57). It is likely that herpes virus infection impacts on the human response to multiple other pathogens, including M. tuberculosis, however whether this leads to disease or is protective requires further research (58–61).
Hepatitis B and C
Hepatitis B and C viruses (HBV, HCV) are important infective causes of chronic hepatitis and cirrhosis. Up to 30% of the world's population show serological evidence of past or current HBV infection and an estimated 257 million people are living with HBV infection (HBV surface antigen positive). Prevalence is highest in the WHO Africa region (6.1%) and the WHO Western Pacific region (6.2%). In highly endemic areas, HBV is acquired perinatally through vertical transmission from the mother to neonate, or during early childhood. In lower endemic settings, typically older susceptible children may become infected through exposure to contaminated blood, through sexual transmission or intravenous drug use. The outcome of acute HBV infection is largely age-dependent with 80–90% of infants and 30–50% of children aged 1–6 years old developing chronic infection, compared to just 5% of adults. Since the introduction of the HBV vaccine, the estimated global prevalence of chronic HBV in children aged 5 years and under has fallen from 4.7 to 1.3% in 2015, though it remains 3% in Africa where TB co-infection is likely to be common (62). Studies have found the prevalence of HBV infection to be higher amongst TB patients than the general population (63–67).
The immune mechanisms of viral clearance are unknown, but both humoral and cellular immune responses are involved. Clearance occurs following the generation of antibodies against viral envelope antigens that clear virus particles, with CD4+ T helper cell interaction playing a vital role. CD8+ cytotoxic T cells eliminate infected cells directly (68). Chronic infection is characterized by a relative immunosuppressive state, perhaps induced directly by the virus. This relative immunosuppressed state in chronic infection in adults is characterized by higher numbers of CD4+CD25+ FOXP3+ regulatory T cell, increased levels of IL-10, and impaired IFN-α and IFN-γ production. Healthy uninfected infants are known to have diminished IFN-γ responses and increased numbers of regulatory T cells—an immune state implicated both in susceptibility to TB infection, TB disease progression and chronicity of hepatitis B infection (69). Further work to determine whether chronic hepatitis B infection in infancy in association with this described immune phenotype is implicated in TB disease progression is required.
Approximately 1% of the global population (71 million) is HCV-infected with an estimated 1.75 million new infections occurring in 2015. An estimated 5 million children under 15 years have chronic viraemic infection. Mother-to-child vertical transmission occurs in 6% of HCV-affected pregnancies, and accounts for up to 60% of pediatric cases of HCV. Among vertically-infected children, up to 80% develop chronic infection (70). Several studies report on the prevalence of anti-HCV antibodies in TB patients, but very few report the prevalence of detectable HCV RNA and thus active infection (63, 67, 71, 72).
Similarly to hepatitis B, the mechanisms of viral clearance are not fully understood, but CD4+ and CD8+ T cell responses seem to be crucial. Those who clear the virus have better T cell proliferation and IL-2, IFN-γ, and TNF-α production than those who develop chronic infection (73). Neutralizing antibodies are not required to clear HCV as demonstrated in hypogammaglobulinaemic patients (74). Like chronic HBV infection, a downregulation of virus-specific T cell responses is observed in chronic HCV infection, with progressive depletion and functional exhaustion of virus-specific CD4+ and CD8+ T cells. Virus-specific T cells lose their cytotoxicity and their ability to produce IL-2, TNF-α and IFN, though production is less impaired than seen in chronic HBV (74). Increased numbers of FOXP3+ regulatory T cells are observed in the blood and the liver (75) and levels of the immunosuppressive cytokine IL-10 are also increased (76). NK cells are activated in chronic HCV infection, though IL-10 and IFN-α suppress NK-production of IFN-γ and TNF-α (74).
Little is known about the effects of HBV and HCV co-infection on TB in children, but given the effects of these viruses on immune responses and their widespread distributions globally, an interaction seems plausible.
Measles
The global burden of measles remains high with an estimated 7 million cases and 89,780 measles-related deaths in 2016, most of which occurred in children under five and in low income countries with poor health systems. Severe disease is most common in poorly nourished young children, particularly those with immunosuppression, or vitamin A deficiency (77).
Young infants are protected against measles by passively acquired maternal anti-measles virus IgG. Maternal antibodies are generally higher in women who had natural measles infection rather than those with vaccine-induced immunity, so children of vaccinated women tend to become susceptible at a younger age (78). The average age of measles cases is dependent on the rate of decline of protective maternal antibodies, the age at which children are vaccinated against measles, and the rate of contact between susceptible and infectious individuals. In densely populated urban centers with poor vaccination coverage, measles is a disease of infants and young children. As measles vaccine coverage increases, the incidence of measles reduces and there is a shift toward cases being in adolescents and adults (79). This may be of relevance in pediatric TB-measles co-infection as it means that measles is increasingly likely to affect older children with TB infection rather than very young children at risk of primary TB.
Measles virus initially infects lymphocytes, dendritic cells, and alveolar macrophages in the respiratory tract, stimulating production of pro-inflammatory cytokines IL6 and IL8, and suppression of IL12 in vitro (80–84). In vivo studies of children with measles demonstrate increased production of pro-inflammatory cytokines IL-1β, TNFα, IL8 (85). The role of type I IFN is less clear with in vitro measles infection leading to variable IFN responses, depending on cell type (86–88). In vivo, expression of IFN-stimulated genes is not increased in the peripheral blood mononuclear cells (PBMCs) of children with measles, though these samples tend to be taken at the time of the rash, when measles is recognized, and therefore may miss IFN production if it occurs earlier in infection (85, 89).
CD8+ T cells are important for viraemic clearance which occurs within a few days of the onset of the rash (90). After viraemic clearance, numbers of circulating activated CD8+ T cells and plasma levels of IFN-γ fall rapidly. Circulating activated CD4+ T cells reduce in number much more slowly, possibly due to the continued presence of measles virus-infected cells (91). Early in the immune response, CD4+ Th1 responses predominate, with IL-2 and IFN-γ production. As the virus is cleared and CD8+ cells and IFN-γ levels decline, there is a switch to CD4+ Th2 cell responses with production of IL-4, IL-10, and IL-13 which lasts for several weeks. Regulatory T cells are also prominent (92). This is thought to promote B cell maturation and establishment of humoral memory.
Measles infection is associated with transient immunosuppression lasting weeks to months, though the underlying mechanisms are incompletely understood (90). Secondary infections, particularly in the respiratory and gastrointestinal tracts, are important causes of measles-associated mortality (93, 94). Transient lymphopenia, (95) disappearance of tuberculin skin reactivity, (96) impaired lymphocyte proliferative responses (97), and impaired dendritic functions (98–100) have all been described. The Th2 and regulatory T cell predominance also depresses macrophage activation and suppresses lymphoproliferation and induction of Th1 responses in response to other pathogens, including M. tuberculosis.
The clinical effect of measles co-infection on TB has been studied since at least the early 20th century with the observation that the TST becomes transiently negative during measles infection, (101, 102) returning to previous levels of reactivity 2–4 weeks later. Several reports from measles epidemics have suggested that measles co-infection has a deleterious effect on TB containment in both children and adults. (103, 104) TB typically appears 2 months after measles infection and an American hospital noted that almost 10% of their pediatric TB meningitis cases experienced their first symptoms while they were convalescing from measles (105). In the 1960s, increased TB relapses in those with wild measles compared to those with vaccine measles or no measles, were reported. The relapses were observed 2 weeks to 3 months after the measles, at a similar time to their regaining tuberculin skin reactivity (105). However, a later review by Flick reported deficiencies with many of these studies, concluding there was inadequate data to support a significant interaction (106). More recently, a study of the 2000–2001 measles outbreak in Korea estimated the overall incidence of TB cases following measles to be lower than the general population (107). An Iraqi study reported increased anti-measles IgG antibody titres in adults with TB compared to controls, the authors suggesting that immunosuppression induced by recent measles infection or reactivation may have triggered reactivation of TB infection (108). The impact of measles on TB in children, is therefore far from clear and further well-designed studies, both of the impact of measles infection and measles vaccination, are warranted.
Human Adenovirus
Human adenoviruses are a common cause of childhood infections worldwide. Their ability to infect many cell types makes them useful vectors for gene delivery (109) and their immunogenic properties mean they are also used as vaccine vectors (110). The majority of primary infections occur during the first 5 years of life. In children, adenoviral infections account for up to 15% of upper respiratory tract infections and about 5% of lower respiratory tract infections (LRTI) (111). Latent infection may follow primary infection with latency described in tonsillar tissue, T lymphocytes and lung epithelial cells. (112–115). Reactivation is important in the severely immunosuppressed (116, 117).
The immunological effects of adenovirus infection are complex (118) and may vary depending on past exposures. Different adenovirus species share a common hexon protein which is a key T cell target (119). Exposure to one human adenovirus therefore generates cytotoxic CD4+ and CD8+ T cells which cross-react with multiple adenovirus species. This is thought to contribute toward broad immunity in adults (120). In rhesus macaques, T cell and cytokine responses to human adenoviruses have been shown to vary with repeated exposures, with increased IFN-γ mRNA expression in PBMCs and increased frequencies of Ki67+, HLADR+, and CD95+/CCR5+ CD4+ T cells in blood recorded after the secondary, but not the primary exposure, mRNA expression of CCL20, TNF-α, and IL-1β in PBMCs was reduced after primary exposure and further suppressed on repeat exposures (121). Given the frequency of adenovirus exposure in young children, cross-reactivity between different virus types, and potential for latency and reactivation, an interaction between TB and adenovirus could be important and further studies are warranted.
Respiratory Viruses
Influenza
The global burden of influenza disease in young children is high, with an estimated 90 million new cases of influenza and 20 million cases of influenza-associated acute LRTI in children <5 years old each year. Of the estimated annual 28,000-111,500 pediatric deaths attributable to influenza LRTIs, 99% occur in developing countries (122).
Epidemiological and modeling studies of previous influenza pandemics suggest an interaction between influenza and TB. An analysis of Swiss historical data found that TB-associated mortality increased during the 1889 and 1918 influenza pandemics and declined thereafter (123). This selective mortality of TB patients is suggested to have contributed to the subsequent decline in TB mortality observed in USA, Japan and the Netherlands, by killing people with TB and reducing transmission (124–126). Studies of more recent influenza outbreaks have found that patients co-infected with TB are at greater risk of dying from influenza than those without TB, even when adjusted for HIV status (127, 128). Amongst patients with TB, those with influenza co-infection have an increased risk of death compared to those without. However, the evidence is mixed (129) and these studies cannot ascertain the mechanisms underlying these observations. It has been hypothesized that lung pathology due to respiratory viral infections predisposes to TB pathogenesis.
Evidence suggests that TB-influenza interactions in children may be complex. In South Africa, where there is seasonal variability in pediatric TB hospitalisations, a temporal association has been observed in hospitalized children between cases of influenza, pulmonary TB, and invasive pneumococcal disease (IPD) (130). A seasonal pattern of influenza activity is followed by a peak in pulmonary TB cases 2–3 months later and IPD 3 months later. This 2–3-month window between influenza and TB peaks corresponds to the time it takes for a young TB-exposed child to develop primary TB, and for TB infection to progress to TB disease. The authors speculated that during influenza outbreaks, young children living with adults with infectious pulmonary TB could be at enhanced risk of TB exposure (from increased aerosolization of TB by an influenza-co-infected adult) and enhanced risk of developing primary TB infection (due to influenza-induced immunomodulation). In older children, this immunomodulation could precipitate reactivation of TB infection (130). However, results from a single study comparing anti-influenza antibody titres in adults without and without pulmonary TB were inconclusive, and further studies, including in children, are warranted (131).
Mouse studies support the hypothesis that influenza virus induces immunological changes which may reduce the host's ability to contain TB infection, as has been observed for secondary bacterial infections (132). More rapid proliferation of mycobacteria and impaired mycobacterial-specific T cell responses are observed in mice co-infected with BCG and influenza compared to mice infected with BCG alone (133). In mouse models of M. tuberculosis infection, influenza co-infection enhances mycobacterial growth, (134, 135) through a type I IFN-signaling pathway. Prior exposure of mice to influenza type A before infection with M. tuberculosis also leads to enhanced mycobacterial growth and decreased survival. Influenza, like many viruses, induces a strong type I IFN response in humans. Influenza infection also downregulates certain TLRs, resulting in reduced neutrophil recruitment (136).
Other Respiratory Viruses
Non-influenza respiratory viruses account for an even greater burden of morbidity and mortality in children than influenza. Respiratory syncytial virus is the most important, causing an estimated 33 million new episodes of acute LRTI per year in children age 5 years and under, 3.2 million of which necessitate hospital admission (137). The literature on non-influenza respiratory viral co-infection and TB is very limited. An Tanzanian study reported no difference in the frequency of observed influenza and non-influenza respiratory viral infections in adults with and without pulmonary TB (138). A South African study comparing children with “definite,” “unconfirmed,” and “unlikely TB” found no clear association between TB categorization and detection of specific respiratory pathogens (139).
Respiratory Bacteria
Although less frequent than viral infections, childhood bacterial respiratory infections are common, particularly in the developing world. Streptococcus pneumoniae is the most common respiratory pathogen, Haemophilus influenzae and Staphylococcus aureus are also important causes of pneumonia. There were an estimated 3.7 million episodes of severe pneumococcal disease and 340,000 episodes of severe Haemophilus influenza type b (Hib) infection globally in children in 2015, with the highest incidences observed in Africa, South East Asia and Western Pacific (140).
Innate immune responses play a pivotal role in early host defense against extracellular bacteria including S. pneumoniae and H. influenzae. Bacteria are initially recognized by pattern recognition receptors consisting of TLRs, the cytosolic NOD-like receptors and DNA sensors. Recognition by these pattern recognition receptors triggers the release of pro-inflammatory mediators and stimulates the recruitment and activation of phagocytic cells. The resulting innate immune response involves complement (particularly C3), acute phase proteins (e.g., C-reactive protein, serum amyloid protein), neutrophils, macrophages, NK cells, and pro-inflammatory cytokines (TNF-α, IL-1, -6, -12, -17, -18) (141). Type I IFNs also appear to have an immunoregulatory role (142). Adaptive immunity is also important, particularly the synthesis of IgM, IgA, and IgG antibodies by B cells activated by bacterial antigens (143).
Several studies report on pneumonia and TB in children. In studies of pediatric pneumonia in TB-endemic countries, TB was diagnosed in 1.8–23% of cases and HIV co-infection was common (144–147). It is often challenging to determine what proportion of these cases represent TB pneumonia and what proportion are TB cases complicated by bacterial co-infection. A study of South African children aged 5 years and under with severe LRTI, found that 10% of HIV-infected and uninfected children with culture-proven TB had concurrent bacteraemia (148). Autopsy studies in children suggest that bacterial co-infections are important causes of death in children with TB (149–151). A recent study by Shimazaki et al. in the Philippines found that 29% of HIV-uninfected adults with pulmonary TB had purulent sputum with detectable bacterial DNA, most commonly H. influenzae (21.2%) and S. pneumonia (7.9%) (152). Bacterial co-infection was associated with an increased risk of 2 week mortality among confirmed TB cases.
These studies suggest that TB-bacterial pneumonia co-infection may be common in TB-endemic areas but there are few studies which investigate a possible interaction between them and the evidence is mixed. A systematic review of pneumonia in children from TB endemic countries suggested that TB might increase the risk of secondary bacterial pneumonia (149). On the other hand, a Tanzanian study which compared smear positive adult TB patients and household contact uninfected controls, found that respiratory bacteria were less frequently detected in the nasopharyngeal swabs of TB patients compared to controls. TB disease severity was higher only in those in whom both viruses and bacteria were detected (138). Evidence from vaccination studies is also mixed as described below.
Other Important Bacteria
Most of the existing literature on TB-bacterial co-infection focuses on pneumonia-related bacteria. However, bacterial bloodstream infection (particularly Salmonella enterica serovar Typhi, Staphylcoccus aureus, and enterobacteriaceae), as well as bacterial zoonosis (brucellosis, leptospirosis, Q fever, and rickettsiosis) have also been reported as a common cause of febrile illness in children, particularly among hospitalized patients (153). Immunity to intracellular bacterial infections (ICBIs) such as salmonella, listeria or chlamydia, is mediated by host responses similar to those observed in M. tuberculosis infection. ICBIs are characterized by their ability to survive within macrophages and elicit a cell mediated response, stimulating CD4+ and CD8+ T cells through expression of the antigen epitope associated to either MHC class II or MHC class I, respectively. Activation of CD4+ T helper cells, specifically Th1 cells, leads to the secretion of IFN-γ which stimulates killing mechanisms inside the infected macrophage, liberating ICBI antigen epitopes, and increasing antigen presentation by bystander dendritic cells. It seems possible therefore that ICBIs could impact the immune response to TB. Case reports of co-infections with TB and ICBIs are present in the literature (154–157). However, an American population-based study found no difference in the risk of the ICBIs Salmonella spp, Yersinia spp, and Listeria monocytogenes in persons who developed TB vs. those in the general population, and found there were actually fewer Chlamydia trachomatis infections observed within the first year post-TB diagnosis compared to the non-TB population. Extrapulmonary TB was linked to a higher rate of salmonella infections compared to pulmonary TB but all 8 patients with salmonellosis and TB were also HIV co-infected (158).
Besides ICBI, multiple other bacteria have been reported to be associated with TB, including S. aureus, Streptococcus milleri, (159) enterococcus and klebsiella (160). Besides presenting in the lung, these concurrent tuberculous and bacterial infections have been described in other anatomical locations in children including the retropharynx (161) and central nervous system (162, 163).
Non-tuberculous Mycobacteria
Non-tuberculous mycobacteria (NTM) include many environmental mycobacterial species other than M. tuberculosis and M. leprae. NTM and M. tuberculosis share microbiological attributes, induce similar immune responses, and have overlapping clinical manifestations (164). However, unlike M. tuberculosis, NTM are not always pathogenic and a major challenge with the diagnosis and management of NTM is to differentiate environmental contamination, colonization, and disease.
In the absence of primary or secondary immunodeficiencies, the host immune system is usually capable of containing and possibly eradicating NTM via established innate and acquired immune mechanisms. As for TB, containment of NTM relies on the integrity of the helper T cell type 1-cytokine pathway and cellular immune mechanisms. NTM disease has been associated with cystic fibrosis, mendelian susceptibility to mycobacterial disease and HIV infection. However, while the two latter are also risk factors of TB, there exists an inverse correlation between cystic fibrosis carriership and TB incidence, suggesting a lower susceptibility of cystic fibrosis carriers to TB (165). Exposure to NTM strains in the environment is thought to contribute to the variability of BCG vaccine, although the relationship is complex and unproven; both blocking and masking mechanisms have been proposed.
The epidemiology of NTM varies by world region (166). Attempts to understand the burden of NTM disease and identify risk factors in the pediatric population are hampered by inadequate mandatory NTM reporting and by the overlap of clinical presentation with TB. An association between increased disease incidence of mycobacterial disease caused by NTM and decreased incidence of TB has been suggested in adults although no causal relation has been proven. No estimates exist for the incidence of clinical syndromes caused by NTM in children or adults, and the available data is usually a by-product of studies assessing the burden of pulmonary TB in children, from whom NTM were isolated from respiratory specimens. The rate of NTM isolation in high TB burden settings varies between countries; from 2.7–26.3%, and increasing age is associated with a higher proportion (167–170). Mycobacterium avium complex species, M. fortuitum, scrofulaceum, and gordonae, are among the most frequently identified NTM in these studies.
Although concurrent TB and NTM is thought to be common, only a few reports have addressed this issue, (171–174) and the prevalence of co-infection depends on the sensitivity of the assay in detecting multiple species. In a recent study of HIV-infected children from African and South East Asian settings, NTM was isolated in 46/427(10.8%) of children, including 5 (1.2%) with both NTM and M. tuberculosis (170).
Whether or not concurrent infection is identified, the clinical significance of an NTM isolate in a patient receiving TB treatment is unknown (174, 175). Even if the isolate is likely to be clinically insignificant, it is plausible that co-infection plays a part in TB pathogenesis or time to sputum clearance (172). In adults, a previous history of TB disease is a risk factor for NTM pulmonary disease, probably due to structural damage to the lung (such as in bronchiectasis) altering mucociliary clearance and thereby predisposing the lung tissue to NTM isolation and disease (176, 177). Although the same phenomenon has not been described in children, bronchiectasis is often seen in children with HIV and chronic lung disease, (178) past TB, recurrent pneumonia, severe immunosuppression, and lymphoid interstitial pneumonitis (179).
Leprosy
Leprosy, a disease caused by M. leprae has similar geographic endemicity as TB. A total of 14 adult co-infected cases have been published to date (180). There is no clear consensus on whether prior exposure to one mycobacteria offers protection or predisposition to the other, but several authors have suggested that impaired cell mediated immunity in patients with multibacillary leprosy may predispose them to TB co-infection (181, 182).
Fungi
Mycobacteria share several features with pathogenic fungi including infection site, metabolic features, the composition and display of cell surface molecules, the range of innate immune receptors engaged during infection, and the ability to form granulomas (183). A number of immunodeficiencies, including chronic granulomatous disease, T cell disorders and deficiencies in the IL-17 pathways lead to increased susceptibility to mycobacterial and fungal infections, suggesting their immunopathogenesis is similar (184, 185). The innate immune response is defined by an array of pattern recognition receptors, such as the TLRs, which recognize bacterial targets such as lipopolysaccharide, flagellin etc. M. tuberculosis and fungi are both “poor agonists” of the TLRs, and virulent strains of M. tuberculosis have even been reported to down-regulate MyD88, a key TLR signaling molecule (186). Virulent strains of both mycobacteria and fungi appear to induce a Th2 rather than a Th1 response in order to evade the host immune response. The key immunopathological feature of TB is the formation of granuloma in the lungs—a condition that can be considered mutually beneficial for host and pathogen, as it constrains the pathogen, while providing a microenvironment for replication of the organism. Granuloma are also noted in fungal, but not other bacterial infections. Both mycobacterial and fungal disease dissemination depends on the ability of the host to maintain the granuloma, a balance influenced both by virulence factors of the invading organism, as well as a variety of external factors including age, malnutrition, and co-infections.
Mycobacterial-fungal co-infection most frequently occurs in the context of immunodeficiency, such as HIV, bone marrow transplant, and primary immunodeficiencies, such as severe combined immunodeficiency or chronic granulomatous disease. Pneumocystis jirovecii pneumonia (PCP) is one of the commonest opportunistic infections in HIV-infected patients in the developed world and although less common in low and middle income countries, still poses a threat. PCP-TB co-infection has been described in these patients, but not in HIV-uninfected populations (146).
A number of adult studies have suggested that TB-fungal co-infection with agents such as Candida and Aspergillus may occur in between 6.5 and 40% of cases of pulmonary TB (187, 188). Co-infection was more common in adult patients with multidrug-resistant-TB, who were more likely to have significant lung pathology. Chronic pulmonary aspergillosis affects patients without obvious immune compromise, but with concurrent or prior TB disease. Chronic pulmonary aspergillosis has recently been recognized as an important global health problem, associated with significant morbidity and mortality. The most common predisposing factor is previously-treated TB, independent of HIV infection (189). Although this is not seen frequently in children, identifying, and treating TB promptly in childhood may prevent chronic lung disease in adulthood.
Microbiome and Gastrointestinal Infections
Between ten trillion and a hundred trillion organisms live within, or on the surface of, each human being, termed the human microbiota (190). The genetic material within these organisms is referred to as the human microbiome. Although the majority of the microbiome is within the gut, an important population resides in the lung and cross-talk between the two, termed the gut-lung axis, is emerging as a central concept in our understanding of the microbiome (191, 192). Bacteria, bacterial toxins, cytokines, metabolites, and hormones move between the two populations through the bloodstream, with the two communicating to each other and each influencing the composition of the other.
The microbiome can impact on host immunology in a number of ways (193). First, it can act as a barrier to the overgrowth of other organisms, through what has been termed colonization resistance. In this situation, competition for limited resources limits the growth of non-microbiota organisms. Second, it can impact on the innate immune system, both through the stimulation of TLRs to produce antimicrobial peptides (194) as well as through modulation of innate immune cell development. Finally, in addition to its impact on the innate system, the microbiota can also prime the adaptive immune system, particularly in its mucosal T-cell response.
Our understanding of the microbiome is rapidly progressing, and there is increasing interest in the role of the microbiome in TB pathogenesis, (195–197) and specific interest in the gut-lung axis (198, 199). While a comprehensive review of the microbiome and potential TB risk is beyond the scope of this article, it is possible that the microbiome impacts on risk of TB infection following exposure, risk of disease progression following infection and also disease outcome. Although there may be some mycobacterial exposure in the gut (mainly with M. bovis), most mucosal interaction with M. tuberculosis is within the lung. Wu et al. used 16S RNA sequencing to analyse and compare the sputum microbiota of adults with new TB, recurrent TB, TB treatment failure and healthy controls, demonstrating significant differences in the abundance of commensals between the groups (200).
The lung microbiota may influence the host response at the initial point of M. tuberculosis exposure, through colonization resistance and stimulation of TLR-mediated responses (201). However, as TB infection develops, stimulation of host immune cells and cytokines is required to contain proliferation of the mycobacteria, and it is likely that this is influenced by both the lung and gut microbiomes. It has been demonstrated that individuals infected with H. pylori, a bacteria that resides in the stomach of nearly 50% of the world's population, had higher TB antigen-induced IFN-γ responses, compared to those with negative H. pylori serology. Those with positive H. pylori serology were also less likely to progress to TB disease compared to those with negative serology (199).
The composition of the gut bacterial microbiome changes with age (202). Soon after birth the neonatal gut is dominated by Enterobacteriaceae. This is soon replaced by predominately Bifidobacteria which continue to be the most commonly represented class of bacteria until the child is weaned onto solids. Following weaning, the adult pattern is seen with Bacteroides, Prevotella, Ruminococcus, Clostridium, and Veillonella occupying the gut (203).
In addition to the alterations in the microbiota seen with age, the organisms that cause enteric infections, and frequency of infections, also changes (204). Infants experience infections with rotavirus, cryptosporidium, E. coli, Shigella and adenovirus most commonly. Children 12–23 months have fewer infections but with similar pathogens. Children over 2 years, have fewer infections still mainly due to Shigella and rotavirus (205). As rotavirus vaccination becomes more widespread, this distribution will likely change. The relationship between malnutrition, microbiome, intestinal infections, and host immunological status is complex as interplay exists between each of these and is beyond the scope of this review (206–209). It is likely that this combination will impact on TB pathogenesis.
Helminths
Studies of the immune interactions between helminths and TB have largely focused on the effect of co-infection on: the efficacy of BCG vaccination; diagnostic tests for TB infection (TST and IGRA); and the role of anti-helminthic treatment on TB outcome, as measured by progression from TB infection to disease (210). Results of these studies have been variable, likely reflecting the diversity of environmental influences and possibly differing immune responses induced by different helminth species.
The most common helminth infections globally are the soil-transmitted intestinal helminths, including the roundworm Ascaris lumbricoides, the hookworms Necator americanus and Ancylostoma duodenale, the whipworm Trichuris trichiura, and Enterobius vermicularis, and Strongyloides spp. Tissue damage by these intestinal helminths during feeding and migration results in production of danger associated molecular patterns and induce an immune response characterized by IgE secretion, IgG4 production, eosinophilia, production of Th2 cytokines (IL-4, IL-9, IL-10, IL-13) and induction of FOXP3+ regulatory T cells and the associated regulatory cytokines Tumor Growth Factor-β and IL-10. As with measles, it is hypothesized that this shift away from Th1 responses is responsible for making individuals infected with helminths more susceptible to TB infection and disease. This regulation can affect not only responses to helminth antigens, but also responses to unrelated antigens such as TB or the BCG vaccine.
There is variability in studies of the influence of helminth infection on diagnostic tests for TB infection, mostly focusing on the TST. A recent South African study showed no impact of deworming on either the TST or IGRA in children (211) however Thomas and colleagues found an increase in indeterminate results of IGRA assays (212). The influence of helminth infection on progression to TB disease is similarly unclear. In one study on recent immigrants to the UK, those patients with helminth infection showed a significant increase in CD4+ FOXP3+ regulatory T cells compared to those without helminth infection. Following anti-helminth treatment, the frequency of regulatory T cells decreased, with an associated increase in IFN-γ producing CD4+ T cells, demonstrating a potential mechanism for susceptibility to TB disease (213). Although Indonesian children with helminth infections had similar frequencies of regulatory T cells in comparison with those without infections, in vitro T-cell functional studies demonstrated suppressed IFN-γ responses to whole blood stimulation with BCG and Plasmodium falciparum, an effect that was reversed with depletion of regulatory T cells (214). Furthermore, children with ascaris or schistosomiasis infections showed significantly increased Th2 responses to mycobacterial stimulation compared to uninfected children, and these responses persisted for up to 6 months following confirmed successful anti-helminth treatment in children with schistosomiasis, but not ascaris (215). In addition, these children had also evidence of epigenetic changes due to helminth infection, as measured by corresponding DNA methylation signatures.
However, no longitudinal studies to date have been powered to determine whether helminth infection influences progression from TB infection to disease, or to confirm a direct relationship between helminth infection and TB disease severity. While in vitro studies clearly demonstrate that pre-exposure or co-incident infection with filarial, hookworm, strongyloides and schistosoma infections is associated with downregulated Th1 and Th17 responses and upregulate Th2 and Regulatory T cells to mycobacterial antigens, (210, 213–215) these findings have not been replicated in patients with TB disease, although increased regulatory and Th2 responses have been shown in patients with asymptomatic helminth infections and TB disease (216). Adults from helminth endemic countries with TB disease demonstrate a mixed Th1/Th2 picture, with reduced CD8+ T cells, while those from low helminth settings have a Th1, IFN (type I and IFN-γ) predominant phenotype (217). In Brazil and Ethiopia, patients with TB disease were more likely to have intestinal helminths than those unaffected and co-infection was associated with more advanced TB disease (218, 219). However, a randomized control trial showed no benefit of anti-helminthic treatment on clinical improvement of TB (220). An Indian study of more than 5000 patients over 10 years showed no effect of helminth co-infection on progression to disease nor severity of disease in an adult population although as helminth status was only measured at baseline, it is difficult to draw definitive conclusions about this (221). In endemic regions, the burden of helminthic infection peaks by adolescence, suggesting at least partial protective immunity with increasing age. Larger longitudinal studies in young children could elucidate the impact of helminth co-infection on TB disease progression.
Malaria
Malaria kills more individuals each year than any other parasitic disease, responsible for ~445,000 deaths in 2016, most of them young children in sub-Saharan Africa (9) Of the estimated 216 million cases of malaria worldwide in 2016, 90% took place in the WHO African Region, followed by the WHO South-East Asia region.
Protection against malaria is postulated to be mediated by both cell mediated and humoral immune responses. P. falciparum infection is characterized by the production of pro-inflammatory cytokines including IL-1β, IL-6, IL-12, TNF-α, IFN-γ, and associated Th1 cytokines (222). The variability in the TNF-α response to endotoxins could mediate differential host responses which contribute to severe malaria when TNF-α levels are high (223). High levels of IFN-γ have also been associated with severe malaria infection, but evidence is inconsistent (224). Moreover, malaria has several immunomodulating effects during acute infection including lymphopenia, decreased levels of CD4+ T cells, and a functional immunosuppression greater than can be attributed to the quantitative fall in CD4+ cells (225, 226).
Although the first published report of malaria TB co-infection was in 1945, (227) there are only few papers published to date, including a case of perinatal malaria and TB in an infant (228), and several epidemiological surveys with prevalence varying from 37% in hospitalized adult and pediatric TB patients in Angola, to 4.3% among adult pulmonary TB patients in Tanzania (229, 230). Co-infections have been studied in both animal models and humans (226, 231–234), and biological interactions seem to exist between P. falciparum, M. tuberculosis, and a shared human host.
Malarial parasites decrease the host's effective humoral and cellular immune responses to M. tuberculosis, and in experimental models co-infection exacerbates acute and chronic mycobacterial infection (235, 236). In humans, co-infection with malaria and TB seems to modulate the immune response to confer immunological protection against malaria while weakening response to TB (231). Chukwuanukwu et al. recently showed that patients co-infected with malaria had an increase in the production of Th2-associated cytokine IL-4 and anti-inflammatory IL-10 in tuberculin-stimulated cells of TB patients >12 years of age compared to malaria-free TB patients, suggesting that malaria co-infection diverts immune response toward a Th2/anti-inflammatory response (231). However, the true impact on risk of infection and disease progression remains unclear. Some authors suggest that TB co-infection has no impact on the outcome of induced experimental cerebral malaria in mice and attribute this to the induction of the inflammatory response which rapidly dominates (233).
Malaria may also impact the ability to diagnose TB infection. Drabe et al. evaluated the performance of IGRA and IP-10 release assays in adult patients with concurrent malaria in Tanzania and found that during malaria infection, IP-10 and IFN-γ levels in the unstimulated samples were elevated, mitogen responsiveness was impaired and CD4 cell counts were decreased (225). These alterations reverted rapidly after malaria treatment.
In terms of the impact of M. tuberculosis on malaria immunity, animal models suggest that M. tuberculosis offers some non-specific protection against rodent plasmodium, reflected by reduced parasitaemia due to mycobacterium-induced pro-inflammatory response (IFN-γ and TNF-α mediated activation of macrophages) (235, 237).
Beyond the complex immunological interactions described above, both diseases seem to share common pathogenesis pathways and genetic factors affecting susceptibility. First, matrix metalloproteinases (MMP), a family of proteolytic enzymes involved in modulating inflammatory response, disrupting tight junctions and degrading sub-endothelial basal lamina, seem to play a critical role in both TB and cerebral malaria pathogenesis (238–240). Data from in vitro and in vivo studies suggest that MMP might be involved in the pathogenesis of cerebral malaria through blood brain barrier damage and leakage as well as through induced inflammatory response. In addition, MMP-1 and MMP-9, as well as other MMP, have been implicated in lung matrix destruction in TB through degradation of fibrillar collagens and other matrix components. MMP-9 concentrations are also increased in the cerebrospinal fluid of patients with TB meningitis and correlated with extent of neurological compromise (238). Moreover, it has recently been suggested that, through complement-mediated lysis, α-Gal immunity might protect against malaria, TB, as well as other NTM, leishmania and trypanosoma; all of which express α-Gal on their surface (241, 242). Although the direct association between blood type, low α-Gal antibody titres and susceptibility to pathogens containing α-Gal still remains to be verified, Cabezas-Cruz et al. suggest that blood type B decreases the anti-α-Gal antibody levels increasing the risk of malaria or TB (242). It has been shown that the incidence of blood type B is positively correlated with the incidence of malaria and TB, but not dengue, which does not produce α-Gal antigen.
Other Protozoa
Besides malaria, multiple other protozoa, including leishmania, trichomona, toxoplasma, giardia and entamoeba, have been described in co-infection with TB although the literature is limited to several case reports, few epidemiological surveys and some immunological research (236). Among non-malaria protozoa, leishmaniasis, a vector borne zoonosis caused by an intracellular pathogen, is reported to co-exist more frequently with TB. In Sudan, up to 77% of TB patients (adult and children) were positive for the leishmania skin test in the community (243). In a systematic review of TB and parasites co-infection published in 2013, the authors found that TB and parasitic diseases were reported as risk factors for each other (236). Two studies conducted in Sudan and Ethiopia evaluated the inter-relationship of TB and parasitic diseases and reported that the risk of TB was higher in patients who were Leishmania donovani positive or Giardia lamblia positive (243, 244). Likewise, TB patients were more easily infected by Leishmania donovani and Giardia lamblia than those without pulmonary TB.
Several reports (245–247), including a pediatric case (248), have described the potential immunological changes observed during co-infection of leishmania and TB, the development of both depends on impaired cell-mediated immunity. A case report on the triple infection of leishmania, M. tuberculosis, and M. lepra showed that the adult patient, with no recognized immunodeficiency, was unable to mount a Th-cell response to upregulate the IL-12 receptor expression after stimulation of the triple infection (245). It has recently been suggested that TB could be a decisive contributing factor to the high mortality observed in patients with visceral leishmaniasis and HIV, although the authors conclude that the true prevalence and impact of co-infection remain unclear (246).
Vaccinations
As discussed above, infants, and young children are highly susceptible to both TB infection and disease and we have described how a variety of childhood infections, many of which are most common in the early years, may impact on immune responses essential to protection from TB. During these early years, infants also receive several routine vaccinations, both live and inactivated, that may also have an immunomodulatory effect on TB susceptibility (Table 2).
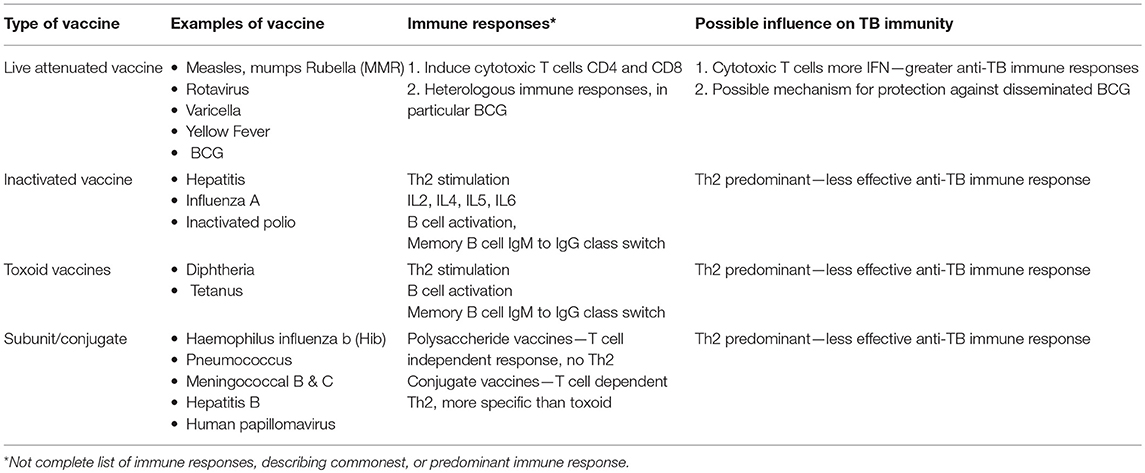
Table 2. Type of vaccines and how they may influence susceptibility and protection to tuberculosis in children.
Antigen specific induction of T and B lymphocytes and the “adaptive” immune system have traditionally been ascribed the role of vaccine-induced protection. However, cells in the innate immune system, such as monocytes, macrophages, dendritic cells, and NK cells, appear to be influenced by contact with a variety of antigens, leading to functional reprogramming, which facilitates rapid, enhanced responses to future, non-specific threats—termed “trained immunity.” The duration and mechanisms by which this long-term innate immunity is induced are the subject of extensive research (249).
BCG is a live, attenuated vaccine that is widely administered to infants in most areas endemic for TB and although it provides imperfect protection against TB infection and disease, it does result in at least partial protection against severe manifestations of TB disease during the first years of life (250, 251). The BCG vaccine unfortunately does not protect all infants from dissemination of M. tuberculosis, and why it works for some and not for others is still not sufficiently explained through immune mechanisms (252). Recently, in a study of adult BCG vaccinated healthy volunteers, cytokines associated with trained immunity, TNF-α, IL-1β, and IL-6 were found to be induced, and associated with control of mycobacterial growth in an inhibition assay (253). Novel markers of trained immunity, CXCL-9 and CXCL-10 were also identified in this study. Both helminth infection and NTM infection have been implicated in variable BCG responses, although the evidence is variable and no definitive study has confirmed a mechanism to date.
Most efficacious vaccines induce antibody mediated protection, and although there is some evidence that antibody responses may mediate some protection against TB, this has not been explored, or understood to the same extent as cell-mediated responses (254, 255). Recently, total IgG levels were found to be protective against TB infection as measured by IGRA positivity in infants. There was a trend toward a protective effect of BCG and measles IgG to Quantiferon positivity, and the authors concluded that BCG and measles vaccination may provide heterologous protection against M. tuberculosis infection, however this is speculative, and numbers were small. Conversely, as mentioned above, measles vaccination has previously been associated with hypo-responsiveness to the TST (256, 257).
A relationship between S. pneumoniae and TB has been explored. In a clinical trial of a 9-valent polysaccharide pneumococcal vaccine, South African children who received the vaccine were 43% less likely to be hospitalized with culture-confirmed pulmonary TB than those who received placebo. In HIV-infected children, vaccines were 47% less likely to be diagnosed with culture-confirmed pulmonary TB compared to placebo recipients. These observations were attributed to pneumococcal infections precipitating hospitalisations and diagnosis of pulmonary TB (258). However, a subsequent study by the same group looking at the trends in pediatric pulmonary TB hospitalisations found no evidence for an effect of the vaccine (259). Limited studies exist on whether other vaccines may increase or decrease the susceptibility of infants and young children to TB and further studies are warranted.
Research Priorities
A great deal of further work is required to better understand the relationship between TB and other infections. We need more details of the epidemiology of co-infections to understand how commonly they occur and in which patient populations they take place. We need to know more about how co-infections impact on a child's immune system, specifically in relation to the way that the host responds to M. tuberculosis, and also how this changes with age. A more comprehensive understanding of the effect of co-infections on the diagnosis and treatment of children with TB would help manage children with both illnesses and finally it would be useful to understand how vaccination strategies for co-infections might impact on the pathogenesis of TB in children. We outline some of these issues in Table 3.
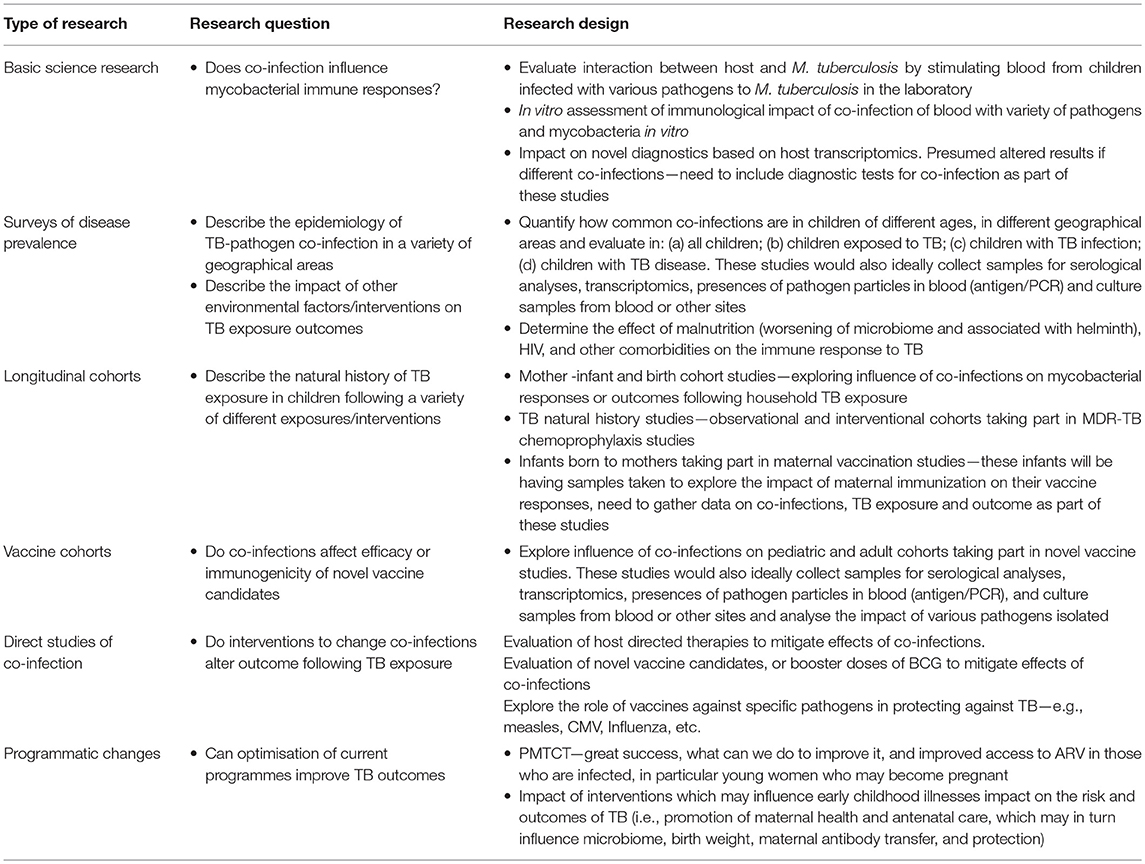
Table 3. Potential study designs to answer the priority research questions in the field of tuberculosis co-infections.
Conclusions
This review suggests that for many children in low resource settings co-infections are common and it is likely that many childhood illnesses impact on the host response to M. tuberculosis, affecting risk of TB infection and disease. Vaccinations are also important, both for direct protection from the pathogens vaccines are designed for, but also for the indirect protection from vaccines such as BCG and measles. Better understanding the relationship between co-infections and TB may allow clinicians to improve the care of children at every stage of TB pathogenesis, through early treatment of co-infections, immunomodulation, or vaccination.
Author Contributions
EW and JS devised the manuscript. EW, EL-V, CB, and JS contributed to the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
JS is supported by a Clinician Scientist Fellowship jointly funded by the UK Medical Research Council (MRC) and the UK Department of International Development (DFID) under the MRC/DFID Concordat agreement (MR/R007942/1). CB is supported by a Wellcome Trust 4i/National Institute for Health Research (NIHR) Imperial Biomedical Research Centre (BRC) Clinical Ph.D. Fellowship. EL-V is supported by a Ramon Areces Foundation fellowship and by the Spanish Society of Pediatrics.
References
1. Dodd PJ, Gardiner E, Coghlan R, Seddon JA. Burden of childhood tuberculosis in 22 high-burden countries: a mathematical modelling study. Lancet Global Health. (2014) 2:e453–9. doi: 10.1016/S2214-109X(14)70245-1
2. World Health Organization Geneva. Global Tuberculosis Report 2017. (2017). Available online at: http://apps.who.int/iris/bitstream/10665/259366/1/9789241565516-eng.pdf?ua=1
3. Dodd PJ, Yuen CM, Sismanidis C, Seddon JA, Jenkins HE. The global burden of tuberculosis mortality in children: a mathematical modelling study. Lancet Global Health. (2017) 5:e898–906. doi: 10.1016/S2214-109X(17)30289-9
4. Wood R, Liang H, Wu H, Middelkoop K, Oni T, Rangaka MX, et al. Changing prevalence of tuberculosis infection with increasing age in high-burden townships in South Africa. Int J Tuberc Lung Dis. (2010) 14:406–12.
5. Marais BJ, Gie RP, Schaaf HS, Hesseling AC, Obihara CC, Starke JJ, et al. The natural history of childhood intra-thoracic tuberculosis: a critical review of literature from the pre-chemotherapy era. Int J Tuberc Lung Dis. (2004) 8:392–402.
6. Roser M, Ritchie H. HIV/AIDS (2019). Available online at: https://ourworldindata.org/hiv-aids
7. World Health Organization. Map: Proportion of Children (1-14 Years of Age) in the Country Requiring Preventive Chemotherapy for Soil-Transmitted Helminthiases. Geneva (2014). Available online at: https://www.who.int/intestinal_worms/en/ (accessed June 04, 2019).
8. Roser M, Ritchie H. Malaria (2019). Available online at: https://ourworldindata.org/malaria
9. World Health Organization. World Malaria Report 2017. (2017), 1–196. Available online at: http://www.who.int/malaria/publications/world-malaria-report-2017/en/
10. Scriba TJ, Coussens AK, Fletcher HA. Human immunology of tuberculosis. Microbiol. Spectr. (2017) 5. doi: 10.1128/microbiolspec.TBTB2-0016-2016
11. Basu Roy R, Whittaker E, Kampmann B. Current understanding of the immune response to tuberculosis in children. Curr Opin Infect Dis. (2012) 25:250–7. doi: 10.1097/QCO.0b013e3283529af9
12. Jones C, Whittaker E, Bamford A, Kampmann B. Immunology and pathogenesis of childhood TB. Paediatr Respir Rev. (2011) 12:3–8. doi: 10.1016/j.prrv.2010.09.006
13. Keane J, Balcewicz-Sablinska MK, Remold HG, Chupp GL, Meek BB, Fenton MJ, et al. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect Immun. (1997) 65:298–304.
14. Roach DR, Bean AGD, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol. (2002) 168:4620–7. doi: 10.4049/jimmunol.168.9.4620
15. Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, et al. Tuberculosis associated with infliximab, a tumor necrosis factor α-neutralizing agent. N Engl J Med. (2001) 345:1098–104. doi: 10.1056/NEJMoa011110
16. Solovic I, Sester M, Gomez-Reino JJ, Rieder HL, Ehlers S, Milburn HJ, et al. The risk of tuberculosis related to tumour necrosis factor antagonist therapies: a TBNET consensus statement. Eur Respir J. (2010) 36:1185–206. doi: 10.1183/09031936.00028510
17. Berry MPR, Graham CM, Mcnab FW, Xu Z, Bloch SAA, Oni T, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. (2010) 466:973–7. doi: 10.1038/nature09247
18. Fletcher HA, Snowden MA, Landry B, Rida W, Satti I, Harris SA, et al. T-cell activation is an immune correlate of risk in BCG vaccinated infants. Nat Commun. (2016) 7:11290. doi: 10.1038/ncomms11290
19. Bustamante J, Boisson-Dupuis S, Abel L, Casanova J-L. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin Immunol. (2014) 26:454–70. doi: 10.1016/j.smim.2014.09.008
20. Whittaker E, Nicol M, Zar HJ, Kampmann B. Regulatory T cells and pro-inflammatory responses predominate in children with tuberculosis. Front Immunol. (2017) 8:448. doi: 10.3389/fimmu.2017.00448
21. Scriba TJ, Kalsdorf B, Abrahams D-A, Isaacs F, Hofmeister J, Black G, et al. Distinct, specific IL-17- and IL-22-producing CD4+ T cell subsets contribute to the human anti-mycobacterial immune response. J Immunol. (2008) 180:1962–70. doi: 10.4049/jimmunol.180.3.1962
22. Dodd PJ, Prendergast AJ, Beecroft C, Kampmann B, Seddon JA. The impact of HIV and antiretroviral therapy on TB risk in children: a systematic review and meta-analysis. Thorax. (2017) 72:559–75. doi: 10.1136/thoraxjnl-2016-209421
23. Bamford A, Turkova A, Lyall H, Foster C, Klein N, Bastiaans D, et al. Paediatric european network for treatment of AIDS (PENTA) guidelines for treatment of paediatric HIV-1 infection 2015: optimizing health in preparation for adult life. HIV Med. (2018) 19:e1–42. doi: 10.1111/hiv.12217
24. Link-Gelles R, Moultrie H, Sawry S, Murdoch D, Van Rie A. Tuberculosis immune reconstitution inflammatory syndrome in children initiating antiretroviral therapy for HIV infection. Pediatr Infect Dis J. (2014) 33:499–503. doi: 10.1097/INF.0000000000000142
25. Diedrich CR, Flynn JL. HIV-1/Mycobacterium tuberculosis coinfection immunology: how does HIV-1 exacerbate tuberculosis? Infect Immun. (2011) 79:1407–17. doi: 10.1128/IAI.01126-10
26. Geldmacher C, Ngwenyama N, Schuetz A, Petrovas C, Reither K, Heeregrave EJ, et al. Preferential infection and depletion of Mycobacterium tuberculosis-specific CD4 T cells after HIV-1 infection. J Exp Med. (2010) 207:2869–81. doi: 10.1084/jem.20100090
27. Patel NR, Zhu J, Tachado SD, Zhang J, Wan Z, Saukkonen J, et al. HIV impairs TNF- mediated macrophage apoptotic response to Mycobacterium tuberculosis. J Immunol. (2007) 179:6973–80. doi: 10.4049/jimmunol.179.10.6973
28. Bruyn Du E, Wilkinson RJ. The Immune Interaction between HIV-1 Infection and Mycobacterium tuberculosis. Microbiol Spectr. (2016) 4:TBTB2-0012-2016. doi: 10.1128/microbiolspec.TBTB2-0012-2016
29. Mehto S, Antony C, Khan N, Arya R, Selvakumar A, Tiwari BK, et al. Mycobacterium tuberculosis and human immunodeficiency virus type 1 cooperatively modulate macrophage apoptosis via toll like receptor 2 and calcium homeostasis. PLoS ONE. (2015) 10:e0131767. doi: 10.1371/journal.pone.0131767
30. Patel NR, Swan K, Li X, Tachado SD, Koziel H. Impaired M. tuberculosis-mediated apoptosis in alveolar macrophages from HIV+ persons: potential role of IL-10 and BCL-3. J Leukoc Biol. (2009) 86:53–60. doi: 10.1189/jlb.0908574
31. Plessis Du N, Jacobs R, Gutschmidt A, Fang Z, van Helden PD, Lutz MB, et al. Phenotypically resembling myeloid derived suppressor cells are increased in children with HIV and exposed/infected with Mycobacterium tuberculosis. Eur J Immunol. (2017) 47:107–18. doi: 10.1002/eji.201646658
32. Revello MG, Tibaldi C, Masuelli G, Frisina V, Sacchi A, Furione M, et al. Prevention of primary cytomegalovirus infection in pregnancy. EBioMedicine. (2015) 2:1205–10. doi: 10.1016/j.ebiom.2015.08.003
33. Bhat V, Joshi A, Sarode R, Chavan P. Cytomegalovirus infection in the bone marrow transplant patient. World J Transplant. (2015) 5:287–91. doi: 10.5500/wjt.v5.i4.287
35. Ruffner MA, Sullivan KE, Henrickson SE. Recurrent and sustained viral infections in primary immunodeficiencies. Front Immunol. (2017) 8:281. doi: 10.3389/fimmu.2017.00665
36. Wang H, Peng G, Bai J, He B, Huang K, Hu X, et al. Cytomegalovirus infection and relative risk of cardiovascular disease (Ischemic Heart Disease, Stroke, and Cardiovascular Death): a meta-analysis of prospective studies up to 2016. J Am Heart Assoc. (2017) 6:e005025. doi: 10.1161/JAHA.116.005025
37. Kallemeijn MJ, Boots AMH, van der Klift MY, Brouwer E, Abdulahad WH, Verhaar JAN, et al. Ageing and latent CMV infection impact on maturation, differentiation and exhaustion profiles of T-cell receptor gammadelta T-cells. Sci Rep. (2017) 7:5509. doi: 10.1038/s41598-017-05849-1
38. Bates M, Brantsaeter AB. Human cytomegalovirus (CMV) in Africa: a neglected but important pathogen. J Virus Erad. (2016) 2:136–42.
39. Evans C, Chasekwa B, Rukobo S, Govha M, Mutasa K, Ntozini R, et al. Cytomegalovirus acquisition and inflammation in human immunodeficiency virus-exposed uninfected zimbabwean infants. J Infect Dis. (2017) 215:698–702. doi: 10.1093/infdis/jiw630
40. Tembo J, Kabwe M, Chilukutu L, Chilufya M, Mwaanza N, Chabala C, et al. Prevalence and risk factors for betaherpesvirus DNAemia in children >3 weeks and <2 years of age admitted to a large referral hospital in sub-Saharan Africa. Clin Infect Dis. (2015) 60:423–31. doi: 10.1093/cid/ciu853
41. Miles DJC, Sande MVD, Kaye S, Crozier S, Ojuola O, Palmero MS, et al. CD4(+) T cell responses to cytomegalovirus in early life: a prospective birth cohort study. J Infect Dis. (2008) 197:658–62. doi: 10.1086/527418
42. Tu W, Chen S, Sharp M, Dekker C, Manganello AM, Tongson EC, et al. Persistent and selective deficiency of CD4+ T cell immunity to cytomegalovirus in immunocompetent young children. J Immunol. (2004) 172:3260–7. doi: 10.4049/jimmunol.172.5.3260
43. Fodil-Cornu N, Vidal SM. Type I interferon response to cytomegalovirus infection: the kick-start. Cell Host Microbe. (2008) 3:59–61. doi: 10.1016/j.chom.2008.01.005
44. O'garra A, Redford PS, Mcnab FW, Bloom CI, Wilkinson RJ, Berry MPR. The immune response in tuberculosis. Annu Rev Immunol. (2013) 31:475–527. doi: 10.1146/annurev-immunol-032712-095939
45. Muller J, Matsumiya M, Snowden MA, Landry B, Satti I, Harris SA, et al. Cytomegalovirus infection is a risk factor for TB disease in Infants. bioRxiv [Preprint]. (2017). doi: 10.1101/222646
46. Cobelens F, Nagelkerke N, Fletcher H. The convergent epidemiology of tuberculosis and human cytomegalovirus infection. F1000Research. (2018) 7:280. doi: 10.12688/f1000research.14184.1
47. Henle G, Henle W. Immunofluorescence in cells derived from Burkitt's lymphoma. J Bacteriol. (1966) 91:1248–56.
48. Chan KH, Tam JS, Peiris JS, Seto WH, Ng MH. Epstein-Barr virus (EBV) infection in infancy. J. Clin. Virol. (2001) 21:57–62. doi: 10.1016/S1386-6532(01)00149-4
49. Rickinson AB, Long HM, Palendira U, Münz C, Hislop AD. Cellular immune controls over Epstein-Barr virus infection: new lessons from the clinic and the laboratory. Trends Immunol. (2014) 35:159–69. doi: 10.1016/j.it.2014.01.003
50. Jayasooriya S, de Silva TI, Njie-Jobe J, Sanyang C, Leese AM, Bell AI, et al. Early virological and immunological events in asymptomatic epstein-barr virus infection in African children. PLoS Pathog. (2015) 11:e1004746. doi: 10.1371/journal.ppat.1004746
51. Smith JS, Robinson NJ. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: a global review. J Infect Dis. (2002) 186(Suppl. 1):S3–28. doi: 10.1086/343739
52. Bollaerts K, Riera-Montes M, Heininger U, Hens N, Souverain A, Verstraeten T, et al. A systematic review of varicella seroprevalence in European countries before universal childhood immunization: deriving incidence from seroprevalence data. Epidemiol Infect. (2017) 145:2666–77. doi: 10.1017/S0950268817001546
53. Zerr DM, Meier AS, Selke SS, Frenkel LM, Huang M-L, Wald A, et al. A population-based study of primary human herpesvirus 6 infection. N Engl J Med. (2005) 352:768–76. doi: 10.1056/NEJMoa042207
54. Clark DA, Freeland ML, Mackie LK, Jarrett RF, Onions DE. Prevalence of antibody to human herpesvirus 7 by age. J Infect Dis. (1993) 168:251–2. doi: 10.1093/infdis/168.1.251
55. Dow DE, Cunningham CK, Buchanan AM. A review of human herpesvirus 8, the kaposi's sarcoma-associated herpesvirus, in the pediatric population. J Pediatric Infect Dis Soc. (2014) 3:66–76. doi: 10.1093/jpids/pit051
56. White DW, Keppel CR, Schneider SE, Reese TA, Coder J, Payton JE, et al. Latent herpesvirus infection arms NK cells. Blood. (2010) 115:4377–83. doi: 10.1182/blood-2009-09-245464
57. Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, et al. Herpesvirus latency confers symbiotic protection from bacterial infection. Nature. (2007) 447:326–9. doi: 10.1038/nature05762
58. Reese TA, Goodrum F. Coinfections: another variable in the herpesvirus latency-reactivation dynamic. J Virol. (2016) 90:5534–7. doi: 10.1128/JVI.01865-15
59. Cruz-Muñoz ME, Fuentes-Pananá EM. Beta and gamma human herpesviruses: agonistic and antagonistic interactions with the host immune system. Front Microbiol. (2017) 8:2521. doi: 10.3389/fmicb.2017.02521
60. Sehrawat S, Kumar D, Rouse BT. Herpesviruses: harmonious pathogens but relevant cofactors in other diseases? Front Cell Infect Microbiol. (2018) 8:3450. doi: 10.3389/fcimb.2018.00177
61. White DW, Suzanne Beard R, Barton ES. Immune modulation during latent herpesvirus infection. Immunol Rev. (2012) 245:189–208. doi: 10.1111/j.1600-065X.2011.01074.x
63. Nooredinvand HA, Connell DW, Asgheddi M, Abdullah M, O'Donoghue M, Campbell L, et al. Viral hepatitis prevalence in patients with active and latent tuberculosis. WJG. (2015) 21:8920–6. doi: 10.3748/wjg.v21.i29.8920
64. Kuniholm MH, Mark J, Aladashvili M, Shubladze N, Khechinashvili G, Tsertsvadze T, et al. Risk factors and algorithms to identify hepatitis C, hepatitis B, and HIV among Georgian tuberculosis patients. Int J Infect Dis. (2008) 12:51–6. doi: 10.1016/j.ijid.2007.04.015
65. Blal CA, Passos SRL, Horn C, Georg I, Bonecini-Almeida MG, Rolla VC, et al. High prevalence of hepatitis B virus infection among tuberculosis patients with and without HIV in Rio de Janeiro, Brazil. Eur J Clin Microbiol Infect Dis. (2005) 24:41–3. doi: 10.1007/s10096-004-1272-8
66. Aires RS, Matos MAD, Lopes CLR, Teles SA, Kozlowski AG, Silva AMC, et al. Prevalence of hepatitis B virus infection among tuberculosis patients with or without HIV in Goiânia City, Brazil. J Clin Virol. (2012) 54:327–31. doi: 10.1016/j.jcv.2012.04.006
67. Sirinak C, Kittikraisak W, Pinjeesekikul D, Charusuntonsri P, Luanloed P, Srisuwanvilai L-O, et al. Viral hepatitis and HIV-associated tuberculosis: risk factors and TB treatment outcomes in Thailand. BMC Public Health. (2008) 8:245. doi: 10.1186/1471-2458-8-245
68. Busca A, Kumar A. Innate immune responses in hepatitis B virus (HBV) infection. Virol J. (2014) 11:22. doi: 10.1186/1743-422X-11-22
69. Morikawa K, Shimazaki T, Takeda R, Izumi T, Umumura M, Sakamoto N. Hepatitis B: progress in understanding chronicity, the innate immune response, and cccDNA protection. Ann Transl Med. (2016) 4:337. doi: 10.21037/atm.2016.08.54
70. Sokal E, Nannini P. Hepatitis C virus in children: the global picture. Arch Dis Child. (2017) 102:668–71. doi: 10.1136/archdischild-2017-312708
71. Richards DC, Mikiashvili T, Parris JJ, Kourbatova EV, Wilson JCE, Shubladze N, et al. High prevalence of hepatitis C virus but not HIV co-infection among patients with tuberculosis in Georgia. Int J Tuberc Lung Dis. (2006) 10:396–401.
72. Agha MA, El-Mahalawy II, Seleem HM, Helwa MA. Prevalence of hepatitis C virus in patients with tuberculosis and its impact in the incidence of anti-tuberculosis drugs induced hepatotoxicity. Egypt. J. Chest Dis. Tubercul. (2015) 64:91–6. doi: 10.1016/j.ejcdt.2014.09.009
73. Park S-H, Rehermann B. Immune responses to HCV and other hepatitis viruses. Immunity. (2014) 40:13–24. doi: 10.1016/j.immuni.2013.12.010
74. Rehermann B. Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nat Med. (2013) 19:859–68. doi: 10.1038/nm.3251
75. Sugimoto K, Ikeda F, Stadanlick J, Nunes FA, Alter HJ, Chang K-M. Suppression of HCV-specific T cells without differential hierarchy demonstrated ex vivo in persistent HCV infection. Hepatology. (2003) 38:1437–48. doi: 10.1053/jhep.2003.09026
76. Accapezzato D, Francavilla V, Paroli M, Casciaro M, Chircu LV, Cividini A, et al. Hepatic expansion of a virus-specific regulatory CD8 T cell population in chronic hepatitis C virus infection. J. Clin. Invest. (2004) 113:963–72. doi: 10.1172/JCI200420515
77. World Health Organization. WHO Health Topic on Measles Geneva. (2019). Available online at: http://www.who.int/news-room/fact-sheets/detail/measles (accessed August 15, 2018).
78. Leuridan E, Hens N, Hutse V, Ieven M, Aerts M, Van Damme P. Early waning of maternal measles antibodies in era of measles elimination: longitudinal study. BMJ. (2010) 340:c1626. doi: 10.1136/bmj.c1626
79. Durrheim DN, Crowcroft NS, Strebel PM. Measles - the epidemiology of elimination. Vaccine. (2014) 32:6880–3. doi: 10.1016/j.vaccine.2014.10.061
80. Ludlow M, McQuaid S, Milner D, de Swart RL, Duprex WP. Pathological consequences of systemic measles virus infection. J Pathol. (2015) 235:253–65. doi: 10.1002/path.4457
81. de Swart RL, Ludlow M, de Witte L, Yanagi Y, van Amerongen G, McQuaid S, et al. Predominant infection of CD150+ lymphocytes and dendritic cells during measles virus infection of macaques. PLoS Pathog. (2007) 3:e178. doi: 10.1371/journal.ppat.0030178
82. Bieback K, Lien E, Klagge IM, Avota E, Schneider-Schaulies J, Duprex WP, et al. Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J Virol. (2002) 76:8729–36. doi: 10.1128/JVI.76.17.8729-8736.2002
83. Karp CL, Wysocka M, Wahl LM, Ahearn JM, Cuomo PJ, Sherry B, et al. Mechanism of suppression of cell-mediated immunity by measles virus. Science. (1996) 273:228–31. doi: 10.1126/science.273.5272.228
84. Sato H, Miura R, Kai C. Measles virus infection induces interleukin-8 release in human pulmonary epithelial cells. Comp Immunol Microbiol Infect Dis. (2005) 28:311–20. doi: 10.1016/j.cimid.2005.08.003
85. Zilliox MJ, Moss WJ, Griffin DE. Gene expression changes in peripheral blood mononuclear cells during measles virus infection. Clin Vacc Immunol. (2007) 14:918–23. doi: 10.1128/CVI.00031-07
86. Sato H, Honma R, Yoneda M, Miura R, Tsukiyama-Kohara K, Ikeda F, et al. Measles virus induces cell-type specific changes in gene expression. Virology. (2008) 375:321–30. doi: 10.1016/j.virol.2008.02.015
87. Druelle J, Sellin CI, Waku-Kouomou D, Horvat B, Wild FT. Wild type measles virus attenuation independent of type I IFN. Virol J. (2008) 5:22. doi: 10.1186/1743-422X-5-22
88. Schlender J, Hornung V, Finke S, Günthner-Biller M, Marozin S, Brzózka K, et al. Inhibition of toll-like receptor 7- and 9-mediated alpha/beta interferon production in human plasmacytoid dendritic cells by respiratory syncytial virus and measles virus. J Virol. (2005) 79:5507–15. doi: 10.1128/JVI.79.9.5507-5515.2005
89. Griffin DE, Ward BJ, Jauregui E, Johnson RT, Vaisberg A. Natural killer cell activity during measles. Clin Exp Immunol. (1990) 81:218–24. doi: 10.1111/j.1365-2249.1990.tb03321.x
90. Griffin DE. Measles virus-induced suppression of immune responses. Immunol Rev. (2010) 236:176–89. doi: 10.1111/j.1600-065X.2010.00925.x
91. Ryon JJ, Moss WJ, Monze M, Griffin DE. Functional and phenotypic changes in circulating lymphocytes from hospitalized zambian children with measles. Clin Vacc Immunol. (2002) 9:994–1003. doi: 10.1128/CDLI.9.5.994-1003.2002
92. Griffin DE, Ward BJ. Differential CD4 T cell activation in measles. J Infect Dis. (1993) 168:275–81. doi: 10.1093/infdis/168.2.275
93. Akramuzzaman SM, Cutts FT, Wheeler JG, Hossain MJ. Increased childhood morbidity after measles is short-term in urban Bangladesh. Am J Epidemiol. (2000) 151:723–35. doi: 10.1093/oxfordjournals.aje.a010267
94. Shanks GD, Lee S-E, Howard A, Brundage JF. Extreme mortality after first introduction of measles virus to the polynesian island of Rotuma, 1911. Am J Epidemiol. (2011) 173:1211–22. doi: 10.1093/aje/kwq504
95. de Vries RD, Mesman AW, Geijtenbeek TBH, Duprex WP, de Swart RL. The pathogenesis of measles. Curr Opin Virol. (2012) 2:248–55. doi: 10.1016/j.coviro.2012.03.005
96. Tamashiro VG, Perez HH, Griffin DE. Prospective study of the magnitude and duration of changes in tuberculin reactivity during uncomplicated and complicated measles. Pediatr Infect Dis J. (1987) 6:451–3. doi: 10.1097/00006454-198705000-00007
97. Hirsch RL, Griffin DE, Johnson RT, Cooper SJ, Lindo de Soriano I, Roedenbeck S, et al. Cellular immune responses during complicated and uncomplicated measles virus infections of man. Clin Immunol Immunopathol. (1984) 31:1–12. doi: 10.1016/0090-1229(84)90184-3
98. Abt M, Gassert E, Schneider-Schaulies S. Measles virus modulates chemokine release and chemotactic responses of dendritic cells. J. Gen. Virol. (2009) 90:909–14. doi: 10.1099/vir.0.008581-0
99. Schnorr JJ, Xanthakos S, Keikavoussi P, Kämpgen E, Meulen ter V, Schneider-Schaulies S. Induction of maturation of human blood dendritic cell precursors by measles virus is associated with immunosuppression. Proc Natl Acad Sci USA. (1997) 94:5326–31.
100. Grosjean I, Caux C, Bella C, Berger I, Wild F, Banchereau J, et al. Measles virus infects human dendritic cells and blocks their allostimulatory properties for CD4+ T cells. J Exp Med. (1997) 186:801–12. doi: 10.1084/jem.186.6.801
101. Pirquet C. Das Verhalten der kutanen Tuberkulinreaktion während der Masern. DMW. (1908) 34:1297–300. doi: 10.1055/s-0028-1135624
102. Nalbant J. The effect of contagious diseases on pulmonary tuberculosis and on the tuberculin reaction in children. Am. Rev. Tuberculosis. (1937) 36:773.
103. Christensen PE, Schmidt H, Bang HO, Andersen V, Jordal B, Jensen O. An epidemic of measles in southern Greenland, 1951; measles in virgin soil. III. Measles and tuberculosis. Acta Med Scand. (1953) 144:450–4. doi: 10.1111/j.0954-6820.1953.tb15718.x
105. Starr S, Berkovich S. The effect of measles, gamma globulin modified measles, and attenuated measles vaccine on the course of treated tuberculosis in children. Pediatrics. (1965) 35:97–102.
106. Flick JA. Does measles really predispose to tuberculosis? Am Rev Respir Dis. (1976) 114:257–65.
107. Lee CH, Lee EG, Lee JY, Park K, Lee BH, Han H, et al. The incidence of tuberculosis after a measles outbreak. Clin Infect Dis. (2008) 46:902–4. doi: 10.1086/528856
108. Al-Saadi Mohammad AK, Muhsin Mohammad A, Al-Jubouri Ashwaq M. Effect of measles infection on cellular immunity in tuberculosis patients. J Clin Immunol Immunopathol Res. (2011) 3:22–24.
109. Yamasaki S, Miura Y, Davydova J, Vickers SM, Yamamoto M. Intravenous genetic mesothelin vaccine based on human adenovirus 40 inhibits growth and metastasis of pancreatic cancer. Int J Cancer. (2012) 133:88–97. doi: 10.1002/ijc.27983
110. Ertl HC. Viral vectors as vaccine carriers. Curr Opin Virol. (2016) 21:1–8. doi: 10.1016/j.coviro.2016.06.001
111. Hong JY, Lee HJ, Piedra PA, Choi EH, Park KH, Koh YY, et al. Lower respiratory tract infections due to adenovirus in hospitalized Korean children: epidemiology, clinical features, and prognosis. Clin Infect Dis. (2001) 32:1423–9. doi: 10.1086/320146
112. Garnett CT, Talekar G, Mahr JA, Huang W, Zhang Y, Ornelles DA, et al. Latent species C adenoviruses in human tonsil tissues. J Virol. (2009) 83:2417–28. doi: 10.1128/JVI.02392-08
113. Garnett CT, Erdman D, Xu W, Gooding LR. Prevalence and quantitation of species C adenovirus DNA in human mucosal lymphocytes. J Virol. (2002) 76:10608–16. doi: 10.1128/JVI.76.21.10608-10616.2002
114. Roy S, Calcedo R, Medina-Jaszek A, Keough M, Peng H, Wilson JM. Adenoviruses in lymphocytes of the human gastro-intestinal tract. PLoS ONE. (2011) 6:e24859–9. doi: 10.1371/journal.pone.0024859
115. Hogg JC. Role of latent viral infections in chronic obstructive pulmonary disease and asthma. Am J Respir Crit Care Med. (2001) 164:S71–5. doi: 10.1164/ajrccm.164.supplement_2.2106063
116. Veltrop-Duits LA, van Vreeswijk T, Heemskerk B, Thijssen JCP, Seady El R, Jol-van der Zijde EM, et al. High titers of pre-existing adenovirus serotype-specific neutralizing antibodies in the host predict viral reactivation after allogeneic stem cell transplantation in children. Clin Infect Dis. (2011) 52:1405–13. doi: 10.1093/cid/cir231
117. van Tol MJD, Kroes ACM, Schinkel J, Dinkelaar W, Claas ECJ, Jol-van der Zijde CM, et al. Adenovirus infection in paediatric stem cell transplant recipients: increased risk in young children with a delayed immune recovery. Bone Marrow Transplant. (2005) 36:39–50. doi: 10.1038/sj.bmt.1705003
118. Lion T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin Microbiol Rev. (2014) 27:441–62. doi: 10.1128/CMR.00116-13
119. Serangeli C, Bicanic O, Scheible MH, Wernet D, Lang P, Rammensee H-G, et al. Ex vivo detection of adenovirus specific CD4+ T-cell responses to HLA-DR-epitopes of the Hexon protein show a contracted specificity of T(HELPER) cells following stem cell transplantation. Virology. (2010) 397:277–84. doi: 10.1016/j.virol.2009.10.049
120. Hutnick NA, Carnathan D, Demers K, Makedonas G, Ertl HCJ, Betts MR. Adenovirus-specific human T cells are pervasive, polyfunctional, and cross-reactive. Vaccine. (2010) 28:1932–41. doi: 10.1016/j.vaccine.2009.10.091
121. Qureshi H, Genescà M, Fritts L, McChesney MB, Robert-Guroff M, Miller CJ. Infection with host-range mutant adenovirus 5 suppresses innate immunity and induces systemic CD4+ T cell activation in rhesus macaques. PLoS ONE. (2014) 9:e106004–10. doi: 10.1371/journal.pone.0106004
122. Nair H, Brooks WA, Katz M, Roca A, Berkley JA, Madhi SA, et al. Global burden of respiratory infections due to seasonal influenza in young children: a systematic review and meta-analysis. Lancet. (2011) 378:1917–30. doi: 10.1016/S0140-6736(11)61051-9
123. Zürcher K, Ballif M, Zwahlen M, Rieder HL, Egger M, Fenner L. Tuberculosis mortality and living conditions in Bern, Switzerland, 1856–1950. PLoS ONE. (2016) 11:e0149195. doi: 10.1371/journal.pone.0149195
124. Noymer A. Testing the influenza–tuberculosis selective mortality hypothesis with Union Army data. Soc Sci Med. (2009) 68:1599–608. doi: 10.1016/j.socscimed.2009.02.021
125. Noymer A. The 1918 influenza pandemic hastened the decline of tuberculosis in the United States: an age, period, cohort analysis. Vaccine. (2011) 29:B38–41. doi: 10.1016/j.vaccine.2011.02.053
126. Oei W, Nishiura H. The Relationship between Tuberculosis and Influenza Death during the Influenza (H1N1) Pandemic from 1918-19. Comput Mathemat Methods Med. (2012) 2012:1–9. doi: 10.1155/2012/124861
127. Archer B, Cohen C, Naidoo D, Thomas J, Makunga C, Blumberg L, et al. Interim report on pandemic H1N1 influenza virus infections in South Africa, April to October 2009: epidemiology and factors associated with fatal cases. Euro Surveill. (2009) 14:19369. doi: 10.2807/ese.14.42.19369-en
128. Walaza S. Influenza virus infection is associated with increased risk of death amongst patients hospitalized with confirmed pulmonary tuberculosis in South Africa, 2010Ł2011. BMC Infect Dis. (2015) 15:26. doi: 10.1186/s12879-015-0746-x
129. Roth S, Whitehead S, Thamthitiwat S, Chittaganpitch M, Maloney SA, Baggett HC, et al. Concurrent influenza virus infection and tuberculosis in patients hospitalized with respiratory illness in Thailand. Influen Other Resp Viruses. (2012) 7:244–8. doi: 10.1111/j.1750-2659.2012.00413.x
130. Dangor Z, Izu A, Moore DP, Nunes MC, Solomon F, Beylis N, et al. Temporal association in hospitalizations for tuberculosis, invasive pneumococcal disease and influenza virus illness in South African children. PLoS ONE. (2014) 9:e91464. doi: 10.1371/journal.pone.0091464
131. de Paus RA, van Crevel R, van Beek R, Sahiratmadja E, Alisjahbana B, Marzuki S, et al. The influence of influenza virus infections on the development of tuberculosis. Tuberclosis. (2013) 93:338–42. doi: 10.1016/j.tube.2013.02.006
132. Brundage JF. Interactions between influenza and bacterial respiratory pathogens: implications for pandemic preparedness. Lancet Infect Dis. (2006) 6:303–12. doi: 10.1016/S1473-3099(06)70466-2
133. Florido M, Grima MA, Gillis CM, Xia Y, Turner SJ, Triccas JA, et al. Influenza A virus infection impairs mycobacteria-specific T cell responses and mycobacterial clearance in the lung during pulmonary coinfection. J Immunol. (2013) 191:302–11. doi: 10.4049/jimmunol.1202824
134. Volkert M. The enhancing effect of concurrent infection with pneumotropic viruses on pulmonary tuberculosis in mice. J Exp Med. (1947) 86:203–14. doi: 10.1084/jem.86.3.203
135. Redford PS, Mayer-Barber KD, Mcnab FW, Stavropoulos E, Wack A, Sher A, et al. Influenza A virus impairs control of Mycobacterium tuberculosis coinfection through a type I interferon receptor–dependent pathway. J Infect Dis. (2013) 209:270–4. doi: 10.1093/infdis/jit424
136. Short KR, Habets MN, Hermans PWM, Diavatopoulos DA. Interactions between Streptococcus pneumoniae and influenza virus: a mutually beneficial relationship? Future Microbiol. (2012) 7:609–24. doi: 10.2217/fmb.12.29
137. Shi T, McAllister DA, O'Brien KL, Simões EAF, Madhi SA, Gessner BD, et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet. (2017) 390:946–58. doi: 10.1016/S0140-6736(17)30938-8
138. Mhimbira F, Hiza H, Mbuba E, Hella J, Kamwela L, Sasamalo M, et al. Prevalence and clinical significance of respiratory viruses and bacteria detected in tuberculosis patients compared to household contact controls in Tanzania: a cohort study. Clin Microbiol Infect. (2019) 25:107.e1–7. doi: 10.1016/j.cmi.2018.03.019
139. Dube FS, Kaba M, Robberts FJL, Ah Tow L, Lubbe S, Zar HJ, et al. Respiratory microbes present in the nasopharynx of children hospitalised with suspected pulmonary tuberculosis in Cape Town, South Africa. BMC Infect Dis. (2016) 16:597. doi: 10.1186/s12879-016-1934-z
140. Wahl B, O'Brien KL, Greenbaum A, Majumder A, Liu L, Chu Y, et al. Burden of Streptococcus pneumoniae and Haemophilus influenzae type B disease in children in the era of conjugate vaccines: global, regional, and national estimates for 2000–15. Lancet Global Health. (2018) 6:e744–57. doi: 10.1016/S2214-109X(18)30247-X
141. Paterson GK, Orihuela CJ. Pneumococci: immunology of the innate host response. Respirology. (2010) 15:1057–63. doi: 10.1111/j.1440-1843.2010.01814.x
142. Koppe U, Suttorp N, Opitz B. Recognition of Streptococcus pneumoniae by the innate immune system. Cell Microbiol. (2012) 14:460–6. doi: 10.1111/j.1462-5822.2011.01746.x
143. Zhang L, Li Z, Wan Z, Kilby A, Kilby JM, Jiang W. Humoral immune responses to Streptococcus pneumoniae in the setting of HIV-1 infection. Vaccine. (2015) 33:4430–6. doi: 10.1016/j.vaccine.2015.06.077
144. Moore DP, Dagan R, Madhi SA. Respiratory viral and pneumococcal coinfection of the respiratory tract: implications of pneumococcal vaccination. Expert Rev Respir Med. (2014) 6:451–65. doi: 10.1586/ers.12.32
145. McNally LM, Jeena PM, Gajee K, Thula SA, Sturm AW, Cassol S, et al. Effect of age, polymicrobial disease, and maternal HIV status on treatment response and cause of severe pneumonia in South African children: a prospective descriptive study. Lancet. (2007) 369:1440–51. doi: 10.1016/S0140-6736(07)60670-9
146. Gray DM, Zar HJ. Community-acquired pneumonia in HIV-infected children: a global perspective. Curr Opin Pulm Med. (2010) 16:1–9. doi: 10.1097/MCP.0b013e3283387984
147. Graham SM, Mankhambo L, Phiri A, Kaunda S, Chikaonda T, Mukaka M, et al. Impact of human immunodeficiency virus infection on the etiology and outcome of severe pneumonia in Malawian children. Pediatr Infect Dis J. (2011) 30:33–8. doi: 10.1097/INF.0b013e3181fcabe4
148. Madhi SA, Petersen K, Madhi A, Khoosal M, Klugman KP. Increased disease burden and antibiotic resistance of bacteria causing severe community-acquired lower respiratory tract infections in human immunodeficiency virus type 1-infected children. Clin Infect Dis. (2000) 31:170–6. doi: 10.1086/313925
149. Oliwa JN, Karumbi JM, Marais BJ, Madhi SA, Graham SM. Tuberculosis as a cause or comorbidity of childhood pneumonia in tuberculosis-endemic areas: a systematic review. Lancet Respir Med. (2015) 3:235–43. doi: 10.1016/S2213-2600(15)00028-4
150. Rennert WP, Kilner D, Hale M, Stevens G, Stevens W, Crewe-Brown H. Tuberculosis in children dying with HIV-related lung disease: clinical-pathological correlations. Int J Tuberc Lung Dis. (2002) 6:806–13.
151. Chintu C, Mudenda V, Lucas S, Nunn A, Lishimpi K, Maswahu D, et al. Lung diseases at necropsy in African children dying from respiratory illnesses: a descriptive necropsy study. Lancet. (2002) 360:985–90. doi: 10.1016/S0140-6736(02)11082-8
152. Shimazaki T, Taniguchi T, Saludar NRD, Gustilo LM, Kato T, Furumoto A, et al. Bacterial co-infection and early mortality among pulmonary tuberculosis patients in Manila, The Philippines. Int J Tuberc Lung Dis. (2018) 22:65–72. doi: 10.5588/ijtld.17.0389
153. Maze MJ, Bassat Q, Feasey NA, Mandomando I, Musicha P, Crump JA. The epidemiology of febrile illness in sub-Saharan Africa: implications for diagnosis and management. Clin Microbiol Infect. (2018) 24:808–14. doi: 10.1016/j.cmi.2018.02.011
154. Trauner M, Grasmug E, Stauber RE, Hammer HF, Hoefler G, Reisinger EC. Recurrent Salmonella enteritidis sepsis and hepatic tuberculosis. Gut. (1995) 37:136–9. doi: 10.1136/gut.37.1.136
155. Kindo AJ, Mathew R, Ravi A, Varadrajan M. Rare co-existence of Salmonella typhi and mycobacteria tuberculosis in a psoas abscess–a case report. Indian J Pathol Microbiol. (2001) 44:493–4.
156. Monno R, Maggi P, Carbonara S, Sibilio G, D'Aprile A, Costa D, et al. Chlamydia trachomatis and Mycobacterium tuberculosis lung infection in an HIV-positive homosexual man. AIDS Patient Care STDs. (2001) 15:607–10. doi: 10.1089/108729101753354590
157. Wheeler RR, Peacock JE, Alford PT, McLean RL. Atypical community-acquired pneumonia: concurrent infection with Chlamydia psittaci and Mycobacterium tuberculosis. South Med J. (1987) 80:402–3. doi: 10.1097/00007611-198703000-00038
158. Huaman MA, Fiske CT, Jones TF, Warkentin J, Shepherd BE, Ingram LA, et al. Tuberculosis and the risk of infection with other intracellular bacteria: a population-based study. Epidemiol Infect. (2015) 143:951–9. doi: 10.1017/S0950268814002131
159. Brook MG, Lucas RE, Pain AK. Clinical features and management of two cases of Streptococcus milleri chest infection. Scand J Infect Dis. (1988) 20:345–6. doi: 10.3109/00365548809032463
160. Arora AA, Krishnaswamy UM, Moideen RP, Padmaja MS. Tubercular and bacterial coinfection: a case series. Lung India. (2015) 32:172–4. doi: 10.4103/0970-2113.152645
161. Shin JH, Sung SI, Kim JK, Jung JM, Kim ES, Choi SH, et al. Retropharyngeal abscess coinfected with Staphylococcus aureus and Mycobacterium tuberculosis after rhinoviral infection in a 1-month-old infant. Korean J Pediatr. (2013) 56:86–9. doi: 10.3345/kjp.2013.56.2.86
162. Manigandan G, Venkatesh C, Gunasekaran D, Soundararajan P. Tuberculous otitis media and staphylococcus aureus coinfection in a five-year-old boy with miliary tuberculosis. J Glob Infect Dis. (2013) 5:26–8. doi: 10.4103/0974-777X.107171
163. Levinsky RJ, Wilkinson A. Mixed pneumococcal and tuberculous meningitis. Arch Dis Child. (1974) 49:325–8. doi: 10.1136/adc.49.4.325-a
164. López-Varela E, García-Basteiro AL, Santiago B, Wagner D, van Ingen J, Kampmann B. Non-tuberculous mycobacteria in children: muddying the waters of tuberculosis diagnosis. Lancet Respir Med. (2015) 3:244–56. doi: 10.1016/S2213-2600(15)00062-4
165. Bosch L, Bosch B, De Boeck K, Nawrot T, Meyts I, Vanneste D, et al. Cystic fibrosis carriership and tuberculosis: hints toward an evolutionary selective advantage based on data from the Brazilian territory. BMC Infect Dis. (2017) 17:340. doi: 10.1186/s12879-017-2448-z
166. Brode SK, Daley CL, Marras TK. The epidemiologic relationship between tuberculosis and non-tuberculous mycobacterial disease: a systematic review. Int J Tuberc Lung Dis. (2014) 18:1370–7. doi: 10.5588/ijtld.14.0120
167. Asiimwe BB, Bagyenzi GB, Ssengooba W, Mumbowa F, Mboowa G, Wajja A, et al. Species and genotypic diversity of non-tuberculous mycobacteria isolated from children investigated for pulmonary tuberculosis in rural Uganda. BMC Infect Dis. (2013) 13:88. doi: 10.1186/1471-2334-13-88
168. Hatherill M, Hawkridge T, Whitelaw A, Tameris M, Mahomed H, Moyo S, et al. Isolation of non-tuberculous mycobacteria in children investigated for pulmonary tuberculosis. PLoS ONE. (2006) 1:e21. doi: 10.1371/journal.pone.0000021
169. López-Varela E, García-Basteiro AL, Augusto OJ, Fraile O, Bulo H, Ira T, et al. High rates of non-tuberculous mycobacteria isolation in mozambican children with presumptive tuberculosis. PLoS ONE. (2017) 12:e0169757. doi: 10.1371/journal.pone.0169757
170. Borand L, de Lauzanne A, Inghammar M, Ung V, Pham TH, Msellati P, et al. High Prevalence of Respiratory Non-Tuberculous Mycobacteria Respiratory Infections in Children Living with HIV in South-East Asia. Available online at: http://programme.ias2017.org/Abstract/Print/?abstractid=1251
171. Libanore M, Bicocchi R, Ghinelli F. Mixed bronchial infection due to Mycobacterium tuberculosis and Mycobacterium avium-intracellulare in an AIDS patient. Infection. (1992) 20:298–9. doi: 10.1007/BF01710804
172. Maiga M, Siddiqui S, Diallo S, Diarra B, Traoré B, Shea YR, et al. Failure to recognize nontuberculous mycobacteria leads to misdiagnosis of chronic pulmonary tuberculosis. PLoS ONE. (2012) 7:e36902. doi: 10.1371/journal.pone.0036902
173. Hwang SM, Lim MS, Hong YJ, Kim TS, Park KU, Song J, et al. Simultaneous detection of Mycobacterium tuberculosis complex and nontuberculous mycobacteria in respiratory specimens. Tuberculosis (Edinb). (2013) 93:642–6. doi: 10.1016/j.tube.2013.07.007
174. Kendall BA, Varley CD, Hedberg K, Cassidy PM, Winthrop KL. Isolation of non-tuberculous mycobacteria from the sputum of patients with active tuberculosis. Int J Tuberc Lung Dis. (2010) 14:654–6.
175. Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. (2007) 175:367–416. doi: 10.1164/rccm.200604-571ST
176. Okoi C, Anderson STB, Antonio M, Mulwa SN, Gehre F, Adetifa IMO. Non-tuberculous mycobacteria isolated from Pulmonary samples in sub-Saharan Africa - a systematic review and meta analyses. Sci Rep. (2017) 7:12002. doi: 10.1038/s41598-017-12175-z
177. Mirsaeidi M, Hadid W, Ericsoussi B, Rodgers D, Sadikot RT. Non-tuberculous mycobacterial disease is common in patients with non-cystic fibrosis bronchiectasis. Int J Infect Dis. (2013) 17:e1000–4. doi: 10.1016/j.ijid.2013.03.018
178. Zar HJ. Chronic lung disease in human immunodeficiency virus (HIV) infected children. Pediatr Pulmonol. (2008) 43:1–10. doi: 10.1002/ppul.20676
179. Amorosa JK, Miller RW, Laraya-Cuasay L, Gaur S, Marone R, Frenkel L, et al. Bronchiectasis in children with lymphocytic interstitial pneumonia and acquired immune deficiency syndrome. Plain film and CT observations. Pediatr Radiol. (1992) 22:603–6. discussion: 606–7. doi: 10.1007/BF02015366
180. Mangum L, Kilpatrick D, Stryjewska B, Sampath R. Tuberculosis and leprosy co infection: a perspective on diagnosis and treatment. Open Forum Infect Dis. (2018) 5:ofy133. doi: 10.1093/ofid/ofy133
181. Hasan Z, Jamil B, Zaidi I, Zafar S, Khan AA, Hussain R. Elevated serum CCL2 concomitant with a reduced mycobacterium-induced response leads to disease dissemination in leprosy. Scand J Immunol. (2006) 63:241–7. doi: 10.1111/j.1365-3083.2006.01733.x
182. Nigam P, Dubey AL, Dayal SG, Goyal BM, Saxena HN, Samuel KC. The association of leprosy and pulmonary tuberculosis. Lepr India. (1979) 51:65–73.
183. Willcocks S, Wren BW. Shared characteristics between Mycobacterium tuberculosis and fungi contribute to virulence. Future Microbiol. (2014) 9:657–68. doi: 10.2217/fmb.14.29
184. Song E, Jaishankar GB, Saleh H, Jithpratuck W, Sahni R, Krishnaswamy G. Chronic granulomatous disease: a review of the infectious and inflammatory complications. Clin Mol Allergy. (2011) 9:10. doi: 10.1186/1476-7961-9-10
185. Welch EZ, Anderson KL, Feldman SR. Interleukin 17 deficiency and implications in cutaneous and systemic diseases. J Dermatol Dermatol Surg. (2015) 19:73–9. doi: 10.1016/j.jdds.2015.03.004
186. Rahman MA, Sobia P, Gupta N, Van Kaer L, Das G. Mycobacterium tuberculosis subverts the TLR-2 - MyD88 pathway to facilitate its translocation into the cytosol. PLoS ONE. (2014) 9:e86886. doi: 10.1371/journal.pone.0086886
187. Kali A, Charles MP, Noyal MJ, Sivaraman U, Kumar S, Easow JM. Prevalence of Candida co-infection in patients with pulmonary tuberculosis. Australas Med J. (2013) 6:387–91. doi: 10.4066/AMJ.2013.1709
188. Amiri MRJ, Aghili SR, Shokohi T, Hedayati MT, Abastabar M, Aliyali M, et al. Invasive forms of Candida and Aspergillus in sputum samples of pulmonary tuberculosis patients attending the tuberculosis reference laboratory in Ghaemshahr, Northern Iran: an analysis of samples collected during the past 10 years. Int J Mycobacteriol. (2016) 5:S179–80. doi: 10.1016/j.ijmyco.2016.08.010
189. Kosmidis C, Denning DW. The clinical spectrum of pulmonary aspergillosis. Thorax. (2015) 70:270–7. doi: 10.1136/thoraxjnl-2014-206291
190. Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. (2007) 449:804–10. doi: 10.1038/nature06244
191. Budden KF, Gellatly SL, Wood DLA, Cooper MA, Morrison M, Hugenholtz P, et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Micro. (2017) 15:55–63. doi: 10.1038/nrmicro.2016.142
192. Marsland BJ, Trompette A, Gollwitzer ES. The gut-lung axis in respiratory disease. Ann Am Thorac Soc. (2015) 12(Suppl. 2):S150–6. doi: 10.1513/AnnalsATS.201503-133AW
193. Hand TW. The role of the microbiota in shaping infectious immunity. Trends Immunol. (2016) 37:647–58. doi: 10.1016/j.it.2016.08.007
194. Brandl K, Plitas G, Mihu CN, Ubeda C, Jia T, Fleisher M, et al. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature. (2008) 455:804–7. doi: 10.1038/nature07250
195. Wood MR, Yu EA, Mehta S. The human microbiome in the fight against tuberculosis. Am J Trop Med Hyg. (2017) 96:1274–84. doi: 10.4269/ajtmh.16-0581
196. Waldman AJ, Balskus EP. The human microbiota, infectious disease, and global health: challenges and opportunities. ACS Infect Dis. (2018) 4:14–26. doi: 10.1021/acsinfecdis.7b00232
197. Gupta N, Kumar R, Agrawal B. New players in immunity to tuberculosis: the host microbiome, lung epithelium, and innate immune cells. Front Immunol. (2018) 9:709. doi: 10.3389/fimmu.2018.00709
198. Cervantes J, Hong B-Y. The gut–lung axis in tuberculosis. Pathog Dis. (2017) 75:ftx097. doi: 10.1093/femspd/ftx097
199. Perry S, De Jong BC, Solnick JV, la Luz Sanchez de M, Yang S, Lin PL, et al. Infection with Helicobacter pylori is associated with protection against tuberculosis. PLoS ONE. (2010) 5:e8804. doi: 10.1371/journal.pone.0008804
200. Wu J, Liu W, He L, Huang F, Chen J, Cui P, et al. Sputum microbiota associated with new, recurrent and treatment failure tuberculosis. PLoS ONE. (2013) 8:e83445. doi: 10.1371/journal.pone.0083445
201. Lin PL, Flynn JL. Understanding latent tuberculosis: a moving target. J Immunol. (2010) 185:15–22. doi: 10.4049/jimmunol.0903856
202. Yassour M, Vatanen T, Siljander H, Hämäläinen A-M, Härkönen T, Ryhänen SJ, et al. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci Transl Med. (2016) 8:343ra81. doi: 10.1126/scitranslmed.aad0917
203. Tanaka M, Nakayama J. Development of the gut microbiota in infancy and its impact on health in later life. Allergol Int. (2017) 66:515–22. doi: 10.1016/j.alit.2017.07.010
204. Kolling G, Wu M, Guerrant RL. Enteric pathogens through life stages. Front Cell Infect Microbiol. (2012) 2:114. doi: 10.3389/fcimb.2012.00114
205. Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet. (2013) 382:209–22. doi: 10.1016/S0140-6736(13)60844-2
206. Walson JL, Berkley JA. The impact of malnutrition on childhood infections. Curr Opin Infect Dis. (2018) 31:231–6. doi: 10.1097/QCO.0000000000000448
207. Prendergast AJ, Kelly P. Interactions between intestinal pathogens, enteropathy and malnutrition in developing countries. Curr Opin Infect Dis. (2016) 29:229–36. doi: 10.1097/QCO.0000000000000261
208. Dinh DM, Volpe GE, Duffalo C, Bhalchandra S, Tai AK, Kane AV, et al. Intestinal microbiota, microbial translocation, and systemic inflammation in chronic HIV infection. J Infect Dis. (2015) 211:19–27. doi: 10.1093/infdis/jiu409
209. Kane AV, Dinh DM, Ward HD. Childhood malnutrition and the intestinal microbiome. Pediatr Res. (2015) 77:256–62. doi: 10.1038/pr.2014.179
210. Babu S, Nutman TB. Helminth-tuberculosis co-infection: an immunologic perspective. Trends Immunol. (2016) 37:597–607. doi: 10.1016/j.it.2016.07.005
211. Van der Zalm MM, van Soelen N, Mandalakas AM, Jacobsen M, Detjen AK, Marx FM, et al. The effect of deworming on tests of tuberculosis infection in children with recent tuberculosis exposure. Pediatr Infect Dis J. (2016) 35:622–7. doi: 10.1097/INF.0000000000001115
212. Thomas TA, Mondal D, Noor Z, Liu L, Alam M, Haque R, et al. Malnutrition and helminth infection affect performance of an interferon gamma-release assay. Pediatrics. (2010) 126:e1522–9. doi: 10.1542/peds.2010-0885
213. Toulza F, Tsang L, Ottenhoff THM, Brown M, Dockrell HM. Mycobacterium tuberculosis-specific CD4+ T-cell response is increased, and Treg cells decreased, in anthelmintic-treated patients with latent TB. Eur J Immunol. (2016) 46:752–61. doi: 10.1002/eji.201545843
214. Wammes LJ, Hamid F, Wiria AE, De Gier B, Sartono E, Maizels RM, et al. Regulatory T cells in human geohelminth infection suppress immune responses to BCG and Plasmodium falciparum. Eur J Immunol. (2010) 40:437–42. doi: 10.1002/eji.200939699
215. DiNardo AR, Nishiguchi T, Mace EM, Rajapakshe K, Mtetwa G, Kay A, et al. Schistosomiasis induces persistent DNA methylation and tuberculosis-specific immune changes. J Immunol. (2018) 201:124–33. doi: 10.4049/jimmunol.1800101
216. Abate E, Belayneh M, Idh J, Diro E, Elias D, Britton S, et al. Asymptomatic helminth infection in active tuberculosis is associated with increased regulatory and Th-2 responses and a lower sputum smear positivity. PLoS Negl Trop Dis. (2015) 9:e0003994. doi: 10.1371/journal.pntd.0003994
217. Amelio P, Portevin D, Reither K, Mhimbira F, Mpina M, Tumbo A, et al. Mixed Th1 and Th2 Mycobacterium tuberculosis-specific CD4 T cell responses in patients with active pulmonary tuberculosis from Tanzania. PLoS Negl Trop Dis. (2017) 11:e0005817–26. doi: 10.1371/journal.pntd.0005817
218. Tristão-Sá R, Ribeiro-Rodrigues R, Johnson LT, Pereira FEL, Dietze R. Intestinal nematodes and pulmonary tuberculosis. Rev Soc Bras Med Trop. (2002) 35:533–5. doi: 10.1590/S0037-86822002000500020
219. Elias D, Mengistu G, Akuffo H, Britton S. Are intestinal helminths risk factors for developing active tuberculosis? Trop Med Int Health. (2006) 11:551–8. doi: 10.1111/j.1365-3156.2006.01578.x
220. Abate E, Elias D, Getachew A, Alemu S, Diro E, Britton S, et al. Effects of albendazole on the clinical outcome and immunological responses in helminth co-infected tuberculosis patients: a double blind randomised clinical trial. Int J Parasitol. (2015) 45:133–40. doi: 10.1016/j.ijpara.2014.09.006
221. Chatterjee S, Kolappan C, Subramani R, Gopi PG, Chandrasekaran V, Fay MP, et al. Incidence of active pulmonary tuberculosis in patients with coincident filarial and/or intestinal helminth infections followed longitudinally in south India. PLoS ONE. (2014) 9:e94603. doi: 10.1371/journal.pone.0094603
222. Torre D, Speranza F, Giola M, Matteelli A, Tambini R, Biondi G. Role of Th1 and Th2 Cytokines in immune response to uncomplicated Plasmodium falciparum malaria. Clin Diagn Lab Immunol. (2002) 9:348–51. doi: 10.1128/CDLI.9.2.348-351.2002
223. Phillips M, Basa-Dalay V, Bothamley G, Cataneo RN, Lam PK, Natividad MPR, et al. Breath biomarkers of active pulmonary tuberculosis. Tuberculosis (Edinb). (2010) 90:145–51. doi: 10.1016/j.tube.2010.01.003
224. King T, Lamb T. Interferon-γ: the Jekyll and Hyde of malaria. PLoS Pathog. (2015) 11:e1005118. doi: 10.1371/journal.ppat.1005118
225. Drabe CH, Vestergaard LS, Helleberg M, Nyagonde N, Rose MV, Francis F, et al. Performance of interferon-gamma and IP-10 release assays for diagnosing latent tuberculosis infections in patients with concurrent malaria in Tanzania. Am J Trop Med Hyg. (2016) 94:728–35. doi: 10.4269/ajtmh.15-0633
226. Enwere GC, Ota MO, Obaro SK. The host response in malaria and depression of defence against tuberculosis. Ann Trop Med Parasitol. (1999) 93:669–78. doi: 10.1080/00034983.1999.11813470
227. Chatterjee Bahadur GC. How to treat pulmonary tuberculosis case running concurrently with malaria. Indian Med Rec. (1945) 65:260–3.
228. Thapa R, Mallick D, Biswas B. Perinatal malaria and tuberculosis co-infection: a case report. Int J Infect Dis. (2010) 14:e254–6. doi: 10.1016/j.ijid.2009.04.005
229. Range N, Magnussen P, Mugomela A, Malenganisho W, Changalucha J, Temu MM, et al. HIV and parasitic co-infections in tuberculosis patients: a cross-sectional study in Mwanza, Tanzania. Ann Trop Med Parasitol. (2007) 101:343–51. doi: 10.1179/136485907X176373
230. Valadas E, Gomes A, Sutre A, Brilha S, Wete A, Hänscheid T, et al. Tuberculosis with malaria or HIV co-infection in a large hospital in Luanda, Angola. J Infect Dev Ctries. (2013) 7:269–72. doi: 10.3855/jidc.2703
231. Chukwuanukwu RC, Onyenekwe CC, Martinez-Pomares L, Flynn R, Singh S, Amilo GI, et al. Modulation of the immune response to Mycobacterium tuberculosis during malaria/M. tuberculosis co-infection. Clin Exp Immunol. (2016) 187:259–68. doi: 10.1111/cei.12861
232. Wiwanitkit V. Co-infection between tuberculosis and malaria: a consideration on interaction of molecules and pathogenesis. J Vector Borne Dis. (2006) 43:195–7.
233. Blank J, Behrends J, Jacobs T, Schneider BE. Mycobacterium tuberculosis co infection has no impact on Plasmodium berghei ANKA-induced experimental cerebral malaria in C57BL/6 mice. Infect Immun. (2016) 84:502–10. doi: 10.1128/IAI.01290-15
234. Scott JAG, Berkley JA, Mwangi I, Ochola L, Uyoga S, Macharia A, et al. Relation between falciparum malaria and bacteraemia in Kenyan children: a population-based, case-control study and a longitudinal study. Lancet. (2011) 378:1316–23. doi: 10.1016/S0140-6736(11)60888-X
235. Mueller A-K, Behrends J, Blank J, Schaible UE, Schneider BE. An experimental model to study tuberculosis-malaria coinfection upon natural transmission of Mycobacterium tuberculosis and Plasmodium berghei. J Vis Exp. (2014) e50829. doi: 10.3791/50829
236. Li X-X, Zhou X-N. Co-infection of tuberculosis and parasitic diseases in humans: a systematic review. Parasit Vectors. (2013) 6:79. doi: 10.1186/1756-3305-6-79
237. Page KR, Jedlicka AE, Fakheri B, Noland GS, Kesavan AK, Scott AL, et al. Mycobacterium-induced potentiation of type 1 immune responses and protection against malaria are host specific. Infect Immun. (2005) 73:8369–80. doi: 10.1128/IAI.73.12.8369-8380.2005
238. Elkington PT, Ugarte-Gil CA, Friedland JS. Matrix metalloproteinases in tuberculosis. Eur Respir J. (2011) 38:456–64. doi: 10.1183/09031936.00015411
239. Polimeni M, Prato M. Host matrix metalloproteinases in cerebral malaria: new kids on the block against blood-brain barrier integrity? Fluids Barriers CNS. (2014) 11:1. doi: 10.1186/2045-8118-11-1
240. Andrade BB, Pavan Kumar N, Amaral EP, Riteau N, Mayer-Barber KD, Tosh KW, et al. Heme Oxygenase-1 regulation of matrix metalloproteinase-1 expression underlies distinct disease profiles in tuberculosis. J Immunol. (2015) 195:2763–73. doi: 10.4049/jimmunol.1500942
241. Cabezas-Cruz A, la Fuente de J. Immunity to α-Gal: the opportunity for malaria and tuberculosis control. Front Immunol. (2017) 8:1733. doi: 10.3389/fimmu.2017.01733
242. Cabezas-Cruz A, Mateos-Hernández L, Alberdi P, Villar M, Riveau G, Hermann E, et al. Effect of blood type on anti-α-Gal immunity and the incidence of infectious diseases. Exp Mol Med. (2017) 49:e301. doi: 10.1038/emm.2016.164
243. el-Safi SH, Hamid N, Omer A, Abdel-Haleem A, Hammad A, Kareem HG, et al. Infection rates with Leishmania donovani and Mycobacterium tuberculosis in a village in eastern Sudan. Trop Med Int Health. (2004) 9:1305–11. doi: 10.1111/j.1365-3156.2004.01337.x
244. Manuel Ramos J, Reyes F, Tesfamariam A. Intestinal parasites in adults admitted to a rural Ethiopian hospital: relationship to tuberculosis and malaria. Scand J Infect Dis. (2006) 38:460–2. doi: 10.1080/00365540500525187
245. Delobel P, Launois P, Djossou F, Sainte-Marie D, Pradinaud R. American cutaneous leishmaniasis, lepromatous leprosy, and pulmonary tuberculosis co infection with down regulation of the T-helper 1 cell response. Clin Infect Dis. (2003) 37:628–33. doi: 10.1086/376632
246. van Griensven J, Mohammed R, Ritmeijer K, Burza S, Diro E. Tuberculosis in visceral leishmaniasis-human immunodeficiency virus coinfection: an evidence gap in improving patient outcomes? Open Forum Infect Dis. (2018) 5:ofy059. doi: 10.1093/ofid/ofy059
247. Colomba C, Adamoli L, Trizzino M, Siracusa L, Bonura S, Tolomeo M, et al. A case of visceral leishmaniasis and pulmonary tuberculosis in a post-partum woman. Int J Infect Dis. (2015) 33:5–6. doi: 10.1016/j.ijid.2014.12.020
248. Karimi A, Amanati A, Ghanaie RM, Mohajerzadeh L. Co-infection of leishmania and tuberculosis. Arch Pediatric Infect Dis. (2013) 2:183–7. doi: 10.5812/pedinfect.12785
249. Dominguez-Andres J, Netea MG. Long-term reprogramming of the innate immune system. J Leukoc Biol. (2019) 105:329–38. doi: 10.1002/JLB.MR0318-104R
250. Roy A, Eisenhut M, Harris RJ, Rodrigues LC, Sridhar S, Habermann S, et al. Effect of BCG vaccination against Mycobacterium tuberculosis infection in children: systematic review and meta-analysis. BMJ. (2014) 349:g4643. doi: 10.1136/bmj.g4643
251. Colditz GA, Berkey CS, Mosteller F, Brewer TF, Wilson ME, Burdick E, et al. The efficacy of bacillus Calmette-Guérin vaccination of newborns and infants in the prevention of tuberculosis: meta-analyses of the published literature. Pediatrics. (1995) 96:29–35.
252. Lalor MK, Ben-Smith A, Gorak-Stolinska P, Weir RE, Floyd S, Blitz R, et al. Population differences in immune responses to bacille Calmette-Guérin vaccination in infancy. J Infect Dis. (2009) 199:795–800. doi: 10.1086/597069
253. Joosten SA, Van Meijgaarden KE, Arend SM, Prins C, Oftung F, Korsvold GE, et al. Mycobacterial growth inhibition is associated with trained innate immunity. J Clin Invest. (2018) 128:1837–51. doi: 10.1172/JCI97508
254. Phuah J, Wong EA, Gideon HP, Maiello P, Coleman MT, Hendricks MR, et al. Effects of B cell depletion on early Mycobacterium tuberculosis infection in cynomolgus macaques. Infect Immun. (2016) 84:1301–11. doi: 10.1128/IAI.00083-16
255. de Vallière S, Abate G, Blazevic A, Heuertz RM, Hoft DF. Enhancement of innate and cell-mediated immunity by antimycobacterial antibodies. Infect Immun. (2005) 73:6711–20. doi: 10.1128/IAI.73.10.6711-6720.2005
256. Mellman WJ, Wetton R. Depression of the tuberculin reaction by attenuated measles virus vaccine. J Lab Clin Med. (1963) 61:453–8.
257. Brody JA, Overfield T, Hammes LM. Depression of the tuberculin reaction by viral vaccines. N Engl J Med. (1964) 271:1294–6. doi: 10.1056/NEJM196412172712505
258. Moore DP, Klugman KP, Madhi SA. Role of Streptococcus pneumoniae in hospitalization for acute community-acquired pneumonia associated with culture-confirmed Mycobacterium tuberculosis in children. (2010) 29:1099–104. doi: 10.1097/INF.0b013e3181eaefff
Keywords: tuberculosis, co-infection, risk, immunology, children, pediatric, mycobacteria
Citation: Whittaker E, López-Varela E, Broderick C and Seddon JA (2019) Examining the Complex Relationship Between Tuberculosis and Other Infectious Diseases in Children. Front. Pediatr. 7:233. doi: 10.3389/fped.2019.00233
Received: 29 September 2018; Accepted: 22 May 2019;
Published: 25 June 2019.
Edited by:
Manuela Zlamy, Innsbruck Medical University, AustriaReviewed by:
Lim Boon Huat, University of Science, MalaysiaMegan B. Murray, Harvard University, United States
Copyright © 2019 Whittaker, López-Varela, Broderick and Seddon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeth Whittaker, ZS53aGl0dGFrZXJAaW1wZXJpYWwuYWMudWs=