
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Pediatr. , 15 May 2019
Sec. Genetics of Common and Rare Diseases
Volume 7 - 2019 | https://doi.org/10.3389/fped.2019.00199
This article is part of the Research Topic Genetic Testing in Pediatric Disorders View all 35 articles
 Rita Fischetto1
Rita Fischetto1 Viviana V. Palmieri2*
Viviana V. Palmieri2* Maria E. Tripaldi2Alberto Gaeta3
Maria E. Tripaldi2Alberto Gaeta3 Angela Michelucci4
Angela Michelucci4 Maurizio Delvecchio5
Maurizio Delvecchio5 Ruggiero Francavilla2Paola Giordano2
Ruggiero Francavilla2Paola Giordano2Alagille syndrome is an autosomal dominant multisystem disorder with variable phenotypic penetrance, caused by heterozygous mutations in JAG1 or NOTCH2, encoding for the components of the Notch signaling pathway. In this paper, we described a novel mutation not yet reported in literature. This 3-years old male child was referred to our Clinical Genetics Unit because of delayed psychomotor development, systolic murmur, dysmorphic facial features, and hypertransaminasemia. The novel JAG1 heterozygous c.2026delT variant in exon 16 was found. JAG1 mutations are classified as protein truncating and non-protein truncating, without any genotype-phenotype correlation. The detected mutation determines a stop codon (p.Cys676AlafsTer67) in the gene sequence, encoding a truncated protein. Our report broadens the spectrum of JAG1 gene mutations.
Alagille syndrome (ALGS; OMIM 118450) is an autosomal dominant multisystem disorder with variable phenotypic penetrance first described in 1969 by Daniel Alagille (1). The incidence rate is 1:70,000–100,000 live births (2, 3). Molecular diagnosis has increased the number of cases detected and the true incidence is probably close to 1 in 30,000 (1, 4). Although clinical features may differ significantly, the diagnosis of ALGS is mainly based on clinical findings (5, 6). Initial diagnosis is based on the presence of intrahepatic bile duct paucity and at least of 3 other clinical features: chronic cholestasis, cardiac disease (pulmonary stenosis), ocular abnormalities (posterior embryotoxon), skeletal abnormalities (butterfly-like vertebrae), and peculiar facial features (broad forehead, deep-set eyes, bulbous nose, and small pointed mandible) (7–9). Patients have a high prevalence of renal and vascular disease as well (1).
ALGS is caused by mutations in genes that impair Notch signaling, a highly conserved pathway that is fundamental to the transcription of genes for cell fate and differentiation of multiple organ systems (10–14). Almost all cases of ALGS are caused by mutations in the JAG1 gene (20p12.2), which consists of 26 exons, while a small rate of patients has a heterozygous mutation in the Notch receptor, NOTCH2 gene (1p13) (15–18). Although mutations causing ALGS have now been identified, diagnostic challenges remain because there are not genotype–phenotype correlations (19). Albeit several genotype-phenotype correlation studies have been performed, they have not shown a link between mutation type and clinical manifestation or severity, leading to the hypothesis that a second gene could work to modify the effects of a JAG1 or NOTCH2 mutation (1). The diagnosis is based on clinical features. Liver biopsy typically shows paucity of the intrahepatic bile ducts, but it is no longer considered mandatory to make a diagnosis of ALGS, and the presence of cholestasis is acceptable to fulfill this criterion. Confirmatory diagnosis is based on gene sequencing. However, about 4% of patients with clinical diagnosis of ALGS may not show any genetic variant, suggesting that further genetic mechanisms are still to be elucidated (1).
In this paper, we describe a patient carrying a novel mutation not yet reported in literature. Signed informed consent has been acquired from the patient's parents for the publication of this case report and any potentially identifying information was removed.
The patient is a Caucasian 3-years old male child, late-preterm born (36 weeks) from vaginal delivery, after a pregnancy complicated by placental detachment. Birth weight was 2,490 g (26° centile). He was the first child of an unrelated couple. Family history was negative for cardiac or hepatic disorders. The main stages of psychomotor development were delayed (sitting position at 8 months with hypotonia; walking at 18 months; speaking at 3 years). At 20 months of age a systolic murmur was found at the cardiac auscultation and heart ultrasound was performed, showing a mild stenosis of the pulmonary branches. Screening for metabolic diseases was negative, except for the finding of hypertransaminasemia. Because of dysmorphic facial features, delayed neurological development and elevated liver enzymes, a genetic condition was suspected and the patient was referred to the Clinical Genetics Unit of the Giovanni XXIII Children's Hospital in Bari.
At referral, height, weight and head circumference were normal (>50° centile). He featured prominent frontal bossing, saddle nose with a bulbous tip, 2/VI systolic cardiac murmur, severe psychomotor retardation suggesting an autistic phenotype. His stools were hypocholic with remains of undigested food. Fundus oculi and brain resonance were normal. Karyotype and FRAXA analysis resulted negative. After patient's parents signed the informed consent, gene sequencing of JAG1 (NM_000214) was performed by Next Generation Sequencing. Target enrichment was done by TruSeq custom amplicon (Illumina, San Diego, CA, United States) according to the manufacturer's instructions. Template library was prepared and was sequenced using MiseqIllumina platform (Illumina, San Diego, CA, United States). Annotation and filtering of variants were performed with Illumina Variant Studio version 2.0, following recommended settings. To evaluate the completeness of the method for the screening of the targeted gene, the sequencing coverage of each amplicon was analyzed in detail using Integrative Genome Viewer version 2.3 (Broad Institute, Cambridge, MA). Variants and region with a depth coverage below 30x were confirmed by Sanger sequencing. The heterozygous sequence variant c.2026delT; p.Cys676AlafsTer67 in exon 16 was identified and confirmed using Sanger sequencing (Figure 1), also because the coverage in exon 16 was very low (<30 x). Primers sequences for PCR amplification of the exon 16 were designed using Primer3 software: Forward primer CCTGTCGTGAATGGTCCTG, Reverse primer CCAGGCCCAGAGAAATATCA. The variant was absent in both parents, arisen as a de novo variant, which determines the formation of a stop codon. Variant was checked for previously reported causative mutations in published works and mutation databases: Human Gene Mutation Database (HGMD) and Leiden Open Variation Database (LOVD) and it has never been described before. Moreover, variant was searched indbSNP, 1,000 Genomes and ExACdatbases, to exclude common single nucleotide polymorphism.
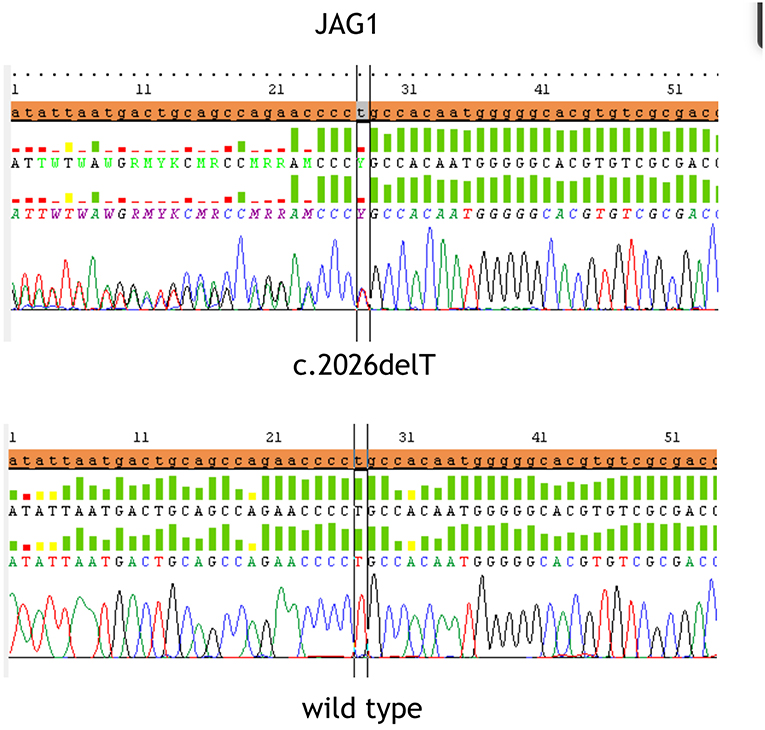
Figure 1. The figure displays the heterozygous sequence variant c.2026delT (p.Cys676AlafsTer67) in exon 16 identified and confirmed by Sanger sequencing in the upper part and the wild type sequence at the bottom.
The child started symptomatic therapy with ursodeoxycholic acid and multidisciplinary follow-up, in particular rehabilitative psychomotor follow-up.
Alagille syndrome is a highly variable, autosomal dominant disorder, which involves multiple organ systems. Table 1 shows a summary of the clinical characteristics and its frequency in patients with ALGS. Its management requires a multidisciplinary team, including mainly specialists in medical genetics, gastroenterology, hepatology, nutrition, cardiology, ophthalmology, and many others (20, 21). The main clinical and pathological features are chronic cholestasis, characteristic facial features, cardiac defect, minor abnormalities of vertebral segmentation, ophthalmologic abnormalities, and dysplastic kidneys (20). The liver disease typically causes severe debilitating pruritus and disfiguring xanthomas, which require treatment with ursodeoxycholic acid and other medications such as cholestyramine, rifampin, and naltrexone (22). Infants and children with ALGS are often diagnosed based on their clinical features: chronic cholestasis is the most common presenting attribute of patients, but dysmorphic facial features are considered one of the best ways to diagnose ALGS (5). The differential diagnosis included Progressive Familiar Cholestasis (PFIC), childhood primary sclerosing cholangitis, congenital hepatic fibrosis, childhood autoimmune hepatitis, childhood primary biliary cirrhosis, alpha-1 antitripsin deficiency, cystic fibrosis and other diseases in which paucity of bile ducts occurs (3).
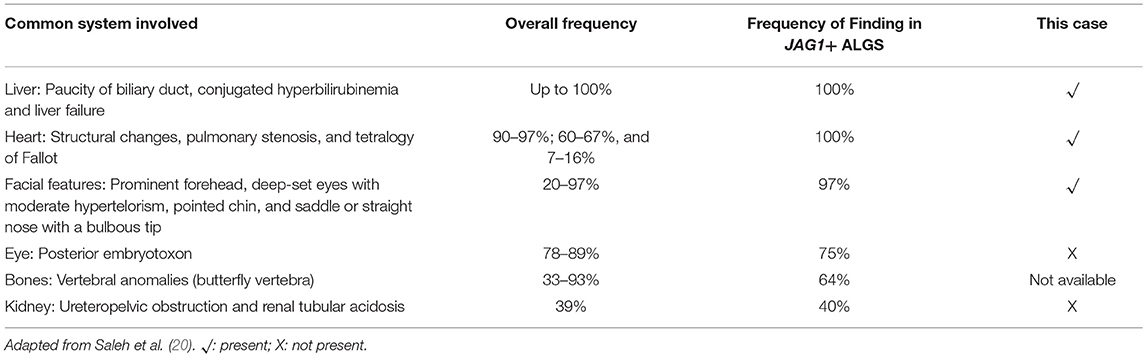
Table 1. A summary of the clinical features and the frequency reported among individuals with ALGS.
ALGS is caused by heterozygous mutations in 1 of 2 genes that are fundamental components of the Notch signaling pathway, JAGGED1 and Notch 2. Mutations in JAG1 gene were first identified more than 20 years ago, and above 500 mutations have been reported (1, 13, 16, 23, 24). The spectrum of JAG1 mutations includes more frequently protein-truncating mutations (75%), and non-protein truncating mutations (25%) (Tables 2A,B) (1, 24). Sequencing all exons and the immediately adjacent intronic regions to identify splice site mutations allows to identify the majority of JAG1 and NOTCH2 mutations. Because mutations in JAG1 are predominant, sequencing of this gene occurs first followed by deletion or duplication analysis via multiplex ligation-dependent probe amplification, chromosomal microarray, or fluorescence in situ hybridization. Sequencing of JAG1 identifies approximately 85% of ALGS mutations, and deletion/duplication analysis yields an additional approximately 9% of molecular diagnoses. In the absence of an identified mutation in JAG1, sequencing of NOTCH2 identifies another 2 to 3% of mutations in ALGS. Up to date large deletion or duplication mutations in NOTCH2 have not been reported. A causative mutation for the remaining 2 to 4% of clinically diagnosed ALGS patients has not been identified yet, and the application of various next-generation sequencing techniques could help identify a molecular origin in this population (1). Unfortunately no genotype-phenotype correlation exists between clinical manifestations and the specific JAG1 pathogenic variant or the location of the mutation within gene (19). Although genetics of ALGS is well-defined, there is a very variable expressivity of the disease. Individuals with the same mutations, including patients belonging to the same family, show discordance in the phenotype (1). In support of this concept, the genotype-phenotype correlation studies did not identify a link between the mutation type and clinical manifestation or severity (18). In view of the absence of an identified environmental factor that influences the severity of the disease or presentation, scientists hypothesized that a second gene could work modifying the effects of a JAG1 or NOTCH2 mutation to worsen or improve the disease features. Several studies have identified putative genetic modifiers to help explain the variable expressivity of this disease (1).
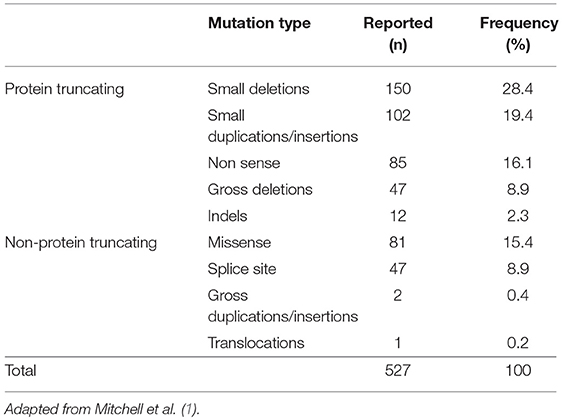
Table 2A. Type and frequency of JAG1 mutations found in ALGS.
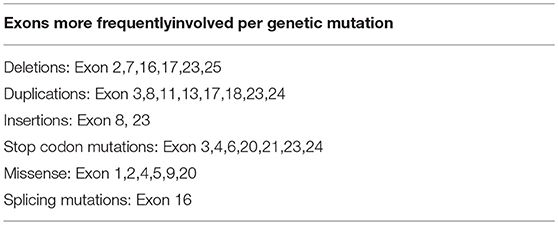
Table 2B. Exons more frequently involved per genetic mutation (Results for variants in the JAG1 gene reported in the ClinVar database).
The patient described in this paper was referred to our Unit because of psycho-motor delay, autistic pattern, facial dysmorphic features, and hypertransaminasemia. Thus, at referral beside the typical liver involvement which is the main feature of ALGS, the clinical features of our patient were facial features, heart disease, and severe neurological involvement; the less frequent clinical features in ALGS were not present in our case (Table 1).
The JAG1 gene sequencing showed the novelc.2026delT variant in exon 16, which determines a stop codon (p.Cys676AlafsTer67) in the gene sequence. As a result, this mutation produces a truncated protein. Although exon 16 has already been described as a site of disease-causing mutations, this variant has not been reported yet in literature. Further studies could clarify the effect of this mutation, but the stop codon causes the premature truncation of the amino acids sequence. In the same position, it was previously reported the de novo c.2026T>G variant leading to the p.C676G missense mutation which was disease-causing according to the Mutation Tester and probably damaging according to the PolyPhen-2 (25). The mutation was found in a male with 4 clinical features (cholestasis, cardiac murmur, skeletal abnormalities, characteristic face). The majority of point and frameshift mutations could be detected by sequencing 11 exons (exons 3, 5, 6, 11, 14, 16, 18, 21, and 23–25). It has been reported that exon 16 is involved mostly by deletions and splicing mutations, due to a single nucleotide substitution (such c.2071T>A, c.2078G>A, and c.2091G>A) or small duplications (c.2070_2073dupCTGT) (25, 26). In this paper we describe a patient with Alagille syndrome carrying a frameshift mutation in the exon 16 of JAG1 gene. This exon is frequently involved by disease-causing mutations in Alagille syndrome.
Our report broadens the spectrum of mutations in the JAG1 gene. Broadening the mutations spectrum may help genetists to better identify etiological mutations leading to ALGS and overall it could provide further insights to identify the hotspots exons where mutations happen more frequently.
Signed informed consent has been acquired from the patient's parents for the publication of this case report and any potentially identifying information was removed. Gene sequencing was performed after informed consent was signed by the patient's parents.
RiF and RuF were in charge of clinical follow-up of the patient. VP and MT drafted the paper. AG is the radiologist who performed the imaging. AM is the biologist who ran the gene sequencing. MD and PG reviewed the paper and gave substantial contribution to data interpretation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Mitchell E, Gilbert M, Loomes KM. Alagille syndrome. Clin Liver Dis. (2018) 22:625–41. doi: 10.1016/j.cld.2018.06.001
2. Vajro P, Ferrante L, Paolella G. Alagille syndrome: an overview. Clin Res Hepatol Gastroenterol. (2012) 36:275–7. doi: 10.1016/j.clinre.2012.03.019
3. Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet. (2012) 20:251–7. doi: 10.1038/ejhg.2011.181
4. Kamath BM, Bason L, Piccoli DA, Krantz ID, Spinner NB. Consequences of JAG1 mutations. J Med Genet. (2003) 40:891–5. doi: 10.1136/jmg.40.12.891
5. Jesina D. Alagille syndrome: an overview. Neonatal Netw. (2017). 36:343–7. doi: 10.1891/0730-0832.36.6.343
6. Subramaniam P, Knisely A, Portmann B, Qureshi SA, Aclimandos WA, Karani JB, et al. Diagnosis of Alagille syndrome−25 years of experience at King's college hospital. J Pediatr Gastroenterol Nutr. (2011) 52:84–9. doi: 10.1097/MPG.0b013e3181f1572d
7. Reyes-de la Rosa ADP, Varela-Fascinetto G, García-Delgado C, Vázquez-Martínez ER, Valencia-Mayoral P, Cerbón M, et al. A Novel c.91dupG JAG1 gene mutation is associated with early onset and severe alagille syndrome. Case Rep Genet. (2018). 2018:369413. doi: 10.1155/2018/1369413
8. Adams JM, Jafar-Nejad H. A new model of alagille syndrome with broad phenotypic representation. Gastroenterology. (2018) 154:803–6. doi: 10.1053/j.gastro.2018.02.007
9. Ahn KJ, Yoon JK, Kim GB, Kwon BS, Go JM, Moon JS, et al. Alagille syndrome and a JAG1mutation: 41 cases of experience at a single center. Korean J Pediatr. (2015) 58:392–7. doi: 10.3345/kjp.2015.58.10.392
10. Gilbert MA, Spinner NB. Alagille syndrome: Genetics and Functional Models. Curr Pathobiol Rep. (2017) 5:233–41. doi: 10.1007/s40139-017-0144-8
11. Penton AL, Leonard LD, Spinner NB. Notch signaling in human development and disease. Semin Cell Dev Biol. (2012) 23:450–7. doi: 10.1016/j.semcdb.2012.01.010
12. Bray SJ. Notch signaling in context. Nat Rev Mol Cell Biol. (2016) 17:722–35. doi: 10.1038/nrm.2016.94
13. Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. (1997) 16:243–51. doi: 10.1038/ng0797-243
14. Guegan K, Stals K, Day M, Turnpenny P, Ellard S. JAG1 mutations are found in approximately one third of patients presenting with only one or two clinical features of Alagille syndrome. Clin Genetics. (2012) 82:33–40. doi: 10.1111/j.1399-0004.2011.01749.x
16. Krantz ID, Piccoli DA, Spinner NB. Alagille syndrome. J Med Genet. (1997) 34:152–7. doi: 10.1136/jmg.34.2.152
17. McDaniell R, Warthen DM, Sanchez-Lara PA, Pai A, Krantz ID, Piccoli DA, et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the Notch signaling pathway. Am J Hum Genet. (2006) 79:169–73. doi: 10.1086/505332
18. Spinner NB, Colliton RP, Crosnier C, Krantz ID, Hadchouel M, Meunier-Rotival M. Jagged1 mutations in Alagille syndrome. Hum Mutat. (2001) 17:18–33. doi: 10.1002/1098-1004(2001)17:1<18::AID-HUMU3>3.0.CO;2-T
19. Kamath BM, Baker A, Houwen R, Todorova L, Kerkar N. Systematic Review: The Epidemiology, Natural History, and Burden of Alagille Syndrome. J Pediatr Gastroenterol Nutr. (2018) 67:148–56. doi: 10.1097/MPG.0000000000001958
20. Saleh M, Kamath BM, Chitayat D. Alagille syndrome: clinical perspectives. Appl Clin Genetics. (2016) 9:75–82. doi: 10.2147/TACG.S86420
21. Lee HP, Kang B, Choi SY, Lee S, Lee SK, Choe YH. Outcome of alagille syndrome patients who had previously received kasai operation during infancy: a single center study. Ped Gastroenter Hepat Nutr. (2015) 18:175–9. doi: 10.5223/pghn.2015.18.3.175
22. Mouzaki M, Bass LM, Sokol RJ, Piccoli DA, Quammie C, Loomes KM, et al. Early life predictive markers of liver disease outcome in an International, Multicentre Cohort of children with Alagille syndrome. Liver Int. (2016) 36:755–60. doi: 10.1111/liv.12920
23. Togawa T, Sugiura T, Ito K, Endo T, Aoyama K, Ohashi K, et al. Molecular genetic dissection and neonatal/infantile intrahepatic cholestasis using targeted next-generation sequencing. J Pediatr. (2016) 171:171–7. doi: 10.1016/j.jpeds.2016.01.006
24. Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, et al. The human gene mutation database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. (2017) 136:665–77. doi: 10.1007/s00439-017-1779-6
25. Li L, Dong J, Wang X, Guo H, Wang H, Zhao J, et al. JAG1 mutation spectrum and origin in chinese children with clinical features of alagille syndrome. PLoS ONE. (2015) 10:e0130355. doi: 10.1371/journal.pone.0130355
Keywords: Alagille syndrome, JAG1, stop codon mutation, Next Generation Sequencing, hypertransaminasemia
Citation: Fischetto R, Palmieri VV, Tripaldi ME, Gaeta A, Michelucci A, Delvecchio M, Francavilla R and Giordano P (2019) Alagille Syndrome: A Novel Mutation in JAG1 Gene. Front. Pediatr. 7:199. doi: 10.3389/fped.2019.00199
Received: 10 January 2019; Accepted: 30 April 2019;
Published: 15 May 2019.
Edited by:
Merlin Gene Butler, University of Kansas Medical Center, United StatesReviewed by:
Zohreh Talebizadeh, Children's Mercy Hospital, United StatesCopyright © 2019 Fischetto, Palmieri, Tripaldi, Gaeta, Michelucci, Delvecchio, Francavilla and Giordano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Viviana V. Palmieri, dml2aWFuYSYjeDAwMDVGO3BhbG1pZXJpQGxpYmVyby5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.